
Abstract
Brain tumors, although rare, contribute to distinct mortality and morbidity at all ages. Although there are few therapeutic options for brain tumors, enhanced biological understanding and unexampled innovations in targeted therapies and immunotherapies have considerably improved patients’ prognoses. Nonetheless, the reduced response rates and unavoidable drug resistance of currently available treatment approaches have become a barrier to further improvement in brain tumor (glioma, meningioma, CNS germ cell tumors, and CNS lymphoma) treatment. Previous literature data revealed that several different signaling pathways are dysregulated in brain tumor. Importantly, a better understanding of targeting signaling pathways that influences malignant behavior of brain tumor cells might open the way for the development of novel targeted therapies. Thus, there is an urgent need for a more comprehensive understanding of the pathogenesis of these brain tumors, which might result in greater progress in therapeutic approaches. This paper began with a brief description of the epidemiology, incidence, risk factors, as well as survival of brain tumors. Next, the major signaling pathways underlying these brain tumors’ pathogenesis and current progress in therapies, including clinical trials, targeted therapies, immunotherapies, and system therapies, have been systemically reviewed and discussed. Finally, future perspective and challenges of development of novel therapeutic strategies in brain tumor were emphasized.
Similar content being viewed by others

Introduction
Brain tumors and other central nervous systems (CNS) tumors have a complicated classification according to histological and molecular findings. Based on the developments in the prior five publications from 1979, 1993, 2000, 2007, and 2016, and the recommendations of the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy (cIMPACT-NOW),1,2,3,4,5,6,7,8,9,10,11 the fifth edition of the World Health Organization Classification of Tumors of the Central Nervous System (WHO CNS5) in 2021 advance the role of molecular diagnostics in CNS tumor classification.11 Since the great development of cancer genomics revolutionizing the diagnostic criteria, the discovery of impactful and experimental molecular-targeted therapies provides new insights for current management and prognosis. In addition, the treatment of brain tumors suggests multi-disciplinary treatment (MDT) and individualized therapy to improve patients’ survival and quality of life.
This article briefly introduces the contemporary incidence, survival, as well as mortality of brain tumors and other CNS tumors, and focuses on the significant molecular signal pathway and currently considered therapeutic options (clinical trials, targeted therapies, immunotherapies, and system therapies) in glioma, meningioma, primary CNS lymphoma, and CNS germ cell tumors, setting by the WHO CNS5.
Epidemiology of brain tumors
Based on the data from the Central Brain Tumor Registry of the United States, the overall incidence of malignant brain tumors in patients of all ages decreased by about 0.8% per year from 2008 to 2017,12 while it has been elevated in non-malignant tumors.13 The brain tumors have a total incidence of 24.25/100,000, with 7.06/100,000 for malignant brain tumors and 17.18/100,000 for non-malignant ones between 2014 and 2018.14 Compared with 15 years ago (14.4/100,000), brain tumors’ overall incidence has almost doubled.15 By 2021, 88,190 new brain and other CNS tumors would be diagnosed in the U.S. population, including 25,690 malignant brain tumors and 62,500 nonmalignant brain tumors.14 Malignant brain tumors compromise no more than 1/3 of all brain tumors but are the causes of most disease deaths. The annual mortality rate is about 4.43/100,000, with an average of 16,606 annual deaths from primary malignant brain together with other CNS tumors,14 with gliomas accounting for 78.3% of malignant brain tumors and exceeding 50% of glioblastomas (GBM). Meningioma was the most frequent nonmalignant brain tumor, followed by pituitary tumors and nerve sheath tumors (Fig. 1).

Distribution of Brain together with Other Central Nervous System (CNS) Tumors by Behavior and Major Histology Type, 2014 to 2018. a Includes histology with ICD-O-3 behavior code of /3 from neuronal and mixed neuronal–glial tumors, choroid plexus tumors, tumors of the pineal region, mesenchymal tumors, embryonal tumors, nerve sheath tumors, primary melanocytic lesions, lymphoma, germ cell tumors, tumors of the pituitary, other hematopoietic neoplasms, neoplasm unspecified, craniopharyngioma, hemangioma, as well as all other. b Includes histology with ICD-O-3 behavior code of /0 or /1 from tumors of the pineal region, neuronal and mixed neuronal–glial tumors, embryonal tumors, mesenchymal tumors, primary melanocytic lesions, craniopharyngioma, hemangioma, other tumors of cranial and spinal nerves, other hematopoietic neoplasms, germ cell tumors, neoplasm unspecified, as well as all other
Incidence by age, gender, and race
The incidence of primary brain tumors varies by age, gender, and race. Both in malignant and nonmalignant brain tumors, the overall incidence in adults aged ≥20 years increased with age. In those aged ≥65 years, the incidence of most histological subtypes of brain tumors was highest, which was 1.5–8 times and 2–9 times higher in malignant brain tumors and non-malignant ones, respectively. However, for minors, the incidence rate of malignant brain tumors decreases with age, while it increases with age in non-malignant brain tumors. And the total incidence rate of brain tumors in people aged 0–4 years and 15–19 years is higher than that in children aged 5–14 years. Other gliomas and tumors (incidence rate:0.77/100,000) of the pituitary (incidence rate:0.88/100,000) were the main types.14
In terms of gender, malignant brain tumors (male: 8.28/100,000, female:5.98/100,000) were more easily seen in men than women, and the opposite was true for nonmalignant tumors (male: 13.07/100,000, female:20.97/100,000).14 For malignant brain tumors, gender differences gradually became apparent in adults aged ≥40 years. In adults aged ≥45 years, the gender differences were greatest, with 30% lower rates in females than males (ratio of female to male incidence rate, 0.69; 95% CI: 0.68–0.70).12 The incidences of GBM, diffuse astrocytoma, and other gliomas were higher in males than in females in the same age group. For nonmalignant brain tumors, gender differences gradually became apparent in adults aged ≥20 years. The gender difference was mainly in meningiomas and pituitary tumors. Especially for nonmalignant meningiomas, the incidence rate of females was about twice that of males in adults aged ≥65 years. However, the incidence of pituitary tumors in women decreased with increased age. It was currently believed that this was related to gender differences in lifetime exposure to endogenous hormones.16 In terms of race, the brain tumor total incidence rate in Black was the highest (24.58/100,000), which was the lowest (14.62/100,000) in American Indians/Alaska Natives. Among them, White had the highest morbidity of malignant brain tumors (7.55/100,000) and Black had the highest morbidity of non-malignant ones (20.14/100,000).14
Survival of brain tumors
Between 1975–1977 and 2009–2015, the 5-year survival rate of all malignant brain tumors increased from 23 to 36%, with a greater increase in the younger age group.12 The 5-year relative survival rates for both malignant and nonmalignant brain tumors diagnosed by histology and age were revealed between 2009 and 2015. Overall, the 5-year survival rate was 35.6% for patients with malignant tumors, with GBM having the lowest rate at 6.6%.14 But the survival rate of a pilocytic astrocytoma can reach 94.4%.14 Additionally, the 5-year relative survival rates of people over 40 years old were far lower than that of people aged 0–14 and 15–39 years old.14 In contrast, the overall 5-year survival rate was as high as 91.8% for patients with nonmalignant tumors, with little difference between age groups.14
Risk factors for brain tumors
The research on genetic and environmental risk factors of the brain and other CNS tumors has been continuous but without a breakthrough. To date, some gene loci and rare genetic mutations that may elevate the risk of some brain tumors had been identified.17 Some studies also focused on endogenous factors, such as allergy,18,19 head injury,20 and virus infection.21,22 Ionizing radiation remained the only well-defined environmental risk reason for brain tumors. Many studies had shown that low-dose therapeutic radiation can enhance the risk of many subtypes of brain tumors, including nerve sheath tumors, meningiomas, gliomas, and so on.23,24,25,26,27 Among them, it had the greatest impact on meningiomas.24 For glioma, young people were more vulnerable to ionizing radiation.24 However, the effect of diagnostic radiation exposure on brain tumors was uncertain.23,28,29 The influence of occupational exposure on brain tumors had been tested, but the results were inconsistent because of the small number of brain tumor cases and the difficulty in assessing individual exposure. Common carcinogens in occupational exposure, such as organic solvents, pesticides, and heavy metals (eg. lead, formaldehyde, and sulfur dioxide), had not been observed to link with brain tumors.30,31 Nearly 30–50% of cancers could be defended by appropriate nutrition habits, but their effects on brain tumors had not been fully explained. Foods rich in antioxidants (such as vitamins) and precursors of N-nitroso compounds (such as nitrite) were often considered to be closely related to brain tumors.32,33,34,35,36,37 The latest meta-analysis explored 12 food groups and found that tea and vegetables had a protective effect on glioma, while excessive intake of grains and processed meat significantly increased its risk.38 However, in the large prospective cohort study, the association between the single food group and brain tumors was not observed, while the Mediterranean diet pattern had a more significant impact on it from the perspective of the overall diet.39 Similar to occupational exposure, due to the limitations of dietary survey methods and regional differences in diet, most of the current studies were concentrated in Europe and America, lacking the research results of other populations such as Asia. The relationship between this two still needed to be further explored.
Glioma
Glioma is a frequent primary brain tumor originating from glial cells. Based on the WHO CNS5 in 2021, gliomas are classified into adult-type diffuse gliomas, pediatric-type diffuse low-grade and high-grade gliomas (LGG and HGG), as well as circumscribed astrocytic gliomas.11 The localized gliomas often present benign biological behaviors that could be treated with complete surgical resection. Most diffuse gliomas are malignant and cured only by complete surgical resection. Grading using Arabic numerals is recommended, as highlighted by WHO CNS5.11 LGG comprises CNS WHO grades 1–2, whereas HGG comprises grades 3–4. LGG accounts for 6% of primary adult CNS tumors and usually has a good prognosis,40 but can recur and progress to HGG, especially grade 2 LGG.41 GBM accounts for 57% of all gliomas while 48% of primary CNS malignancies in HGG,42 have a median survival time of fewer than 2 years. Molecular changes and clinical significance in glioma are detailed in Table 1.
Standard treatment of glioma
Although the new version of tumor classification has brought more advantages and significative guidance for clinical practice, it is currently not fully implemented in clinical application. Therefore, this review is based on the grading of gliomas.
For HGG, such as GBM, subtotal gross total resection, concomitant temozolomide (TMZ) radiochemotherapy at a dose, local radiotherapy to the tumor site, and tumor treating fields should be considered as standard treatments.43,44,45,46 All GBM will finally progress or relapse to recurrent GBM (rGBM), while without standard treatment. In addition, bevacizumab, known as an anti-vascular endothelial growth factor (VEGF) antibody, showed improved progression-free survival (PFS) in GBM. Bevacizumab has been applied for rGBM with the approval of the US Food and Drug Administration (FDA).47,48 LGG correlates with a molecular phenotype, and oligodendrogliomas with IDH-mut and 1p19q codeletion possess the best prognosis, and then those with IDH mut and 1p19q intact, while those with IDH wild type have the worst prognosis. Therefore, the patients should be surgically removed as quickly to avoid subsequent malignant tumor progression, while accurate recognition of the molecular subtype of the tumor is very essential for LGG.49 For high-risk LGG, surgical treatment alone is not sufficient, and local postoperative radiotherapy should be administered at 50–54 Gy, accompanied by six cycles of adjuvant procarbazine/lomustine/vincristine (PCV).50 Carboplatin and vincristine are regarded as the standard treatment for some unresectable children with LGG.
Molecular targeted therapy
O6 methylguanine DNA methyltransferase (MGMT) is a repair protein51,52 that is encoded by the MGMT gene, which can reverse DNA alkylation by depleting itself. TMZ, the standard therapy of GBM, is known as an alkylating agent that evokes tumor cell death through DNA alkylation at many sites. In patients with MGMT promoter methylation found in 30–50% of isocitrate dehydrogenase (IDH)-wt GBM,53 gene promoter methylation would repress the expression of this gene. Therefore, with MGMT promoter methylation, glioma patients benefit more from treatment with TMZ.51,54 However, a discordance of MGMT promoter methylation with protein expression was detected in various patient.55,56 This may be related to the regulation of MGMT protein by Wnt signaling in addition to the regulation of MGMT promoter methylation.57 Furthermore, MGMT methylation predicts longer survival at diagnosis, while this was not the case at relapse,58 and presumably, TMZ resistance was also associated with rearrangement mutation or MGMT gene fusion.59 Therefore, it is a reasonable strategy to treat TMZ-resistant glioma patients by developing targeted MGMT-sensitizing TMZ. A phase I trial (NCT01700569) demonstrated that the combination of TMZ, folic acid, as well as radiotherapy was feasible to promote MGMT methylation in patients with unmethylated MGMT.60 In addition, a preclinical study showed that bortezomib can strengthen the GBM’s sensitivity to TMZ by decreasing MGMT levels.61 These suggest that targeting MGMT induces TMZ sensitivity is very promising. According to Kingson Lin et al.,62 mismatch repair (MMR)-independent cell killing can be induced selectively in MGMT-depleted tumors to overcome resistance mechanisms. The agents deposit a kind of dynamic DNA lesion, which can be reversed by MGMT. However, in MGMT-deficient settings, it slowly evolves into an interstrand cross-link, leading to MMR-independent cell death with low toxicity both in vitro and in vivo. This finding may bring new therapies for gliomas and may offer a novel paradigm for the design of chemotherapeutic agents for exploiting specific DNA repair defects.
Mutation of IDH results in altered IDH enzymatic activity, and mutant IDH1 with novel enzymatic activity can generate R-2-hydroxyglutarate (R-2HG).63 The R-2HG alters GBM epigenetics by inhibiting the catalytic activity of tet methylcytosine dioxygenase (TET2), which the a-KG-dependent dioxygenases catalyze the hydroxylation of 5-methylcytosine into 5-hydroxymethylcytosine.64 Accordingly, IDH1R132H mutation triggers the CpG island hyper-methylator phenotype in gliomas.65,66 DNA methylation results in the gliomas’ development by enhancing the number of stem cells and impairing differentiation.67,68 Interestingly, the anti-tumor potencies of R-2HG in impeding proliferation/survival of fat mass and obesity-associated (FTO)-high cancer cells via modulating the FTO/m6A/MYC/CEBPA signaling.69 Moreover, the DNA repair activity of mammalian alkylation protein B homolog 2 (ALKBH2) and alkylation protein B homolog 3 (ALKBH3) could reverse alkylation on 1meA and 3meC,70,71 which is restricted by R-2HG in vitro72 Importantly, the production of R-2HG makes IDH mutant cells sensitive to alkylating agents.72 The clinically significant bifunctional alkylating agents procarbazine and CCNU/lomustine induces highly genotoxic DNA interstrand crosslinks, and are a part of the PCV chemotherapeutic regimen successfully utilized in combination with radiotherapy for the treatment of brain tumors with IDH mutation status.73 In addition, the mutant IDH1/2 and R-2HG exhibit control mechanistic targeted of rapamycin (mTOR) and hypoxia-inducible factor-1 (HIF1) Signaling.74,75,76 Since the mechanisms and clinical implications remain to be clarified and have been discussed in excellent reviews,77 they will not be described here. Although IDH mutation predicted a better clinical prognosis and GBM patients who CNS5 were all IDH-wt. However, there are astrocytomas grade 3–4 IDH mutant and grade 3 IDH mutant oligodendrogliomas. In other tumors, it was found that the efficacy of Ivosidenib targeting IDH was significant (NCT02074839, NCT02677922).78,79,80,81 Ivosidenib showed good tolerability and efficacy in patients with recurrent or progressive IDH-must gliomas (NCT02073994).82 In addition, a vaccine targeting the IDH1 (R132H) mutation showed good tolerability with a high pseudo-progression rate for newly-diagnosed grade 3–4 gliomas (NCT02454634).83 Due to successful attempts in other tumors, many clinical trials targeting IDH mut gliomas are being initiated (NCT02771301, NCT04906473).
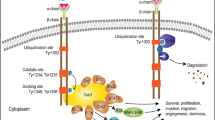
Epidermal growth factor receptor (EGFR) is a common site of oncogenic mutation in IDH-wt GBM84 and has participated in tumor cell proliferation, migration, and escape.85 About 50% of GBM samples have EGFR mutations, of which more than 40% have gene amplification, and the rest consists of gene mutations, rearrangements, etc.84,86,87,88 EGFR variant III (EGFRvIII) (deletion of exons 2–7), as the most significant gene mutation of EGFR, leads to an in-frame deletion variant with a truncated extracellular domain with ligand-independent constitutive activity.84 EGFRvIII induces mTORC2 kinase activity, which is partially restricted by phosphatase and tensin homolog (PTEN). The mTORC2 signaling enhances GBM growth and survival and subsequently activates nuclear transcription factor-kappa B (NF-κB). Moreover, this mTORC2-NF-κB pathway makes cells and tumors of GBM resistant to chemotherapy in a manner independent of V-akt murine thymoma viral oncogene homolog (AKT).89 Furthermore, the EGFRvIII and wild-type EGFR strongly activate the RAS/MEK/ERK signaling, the PI3K/AKT/mTOR signaling, the Notch signaling, and the signal transducer and activator of transcription (STAT) 3/5 signaling.90,91,92 These signalings functions in the regulation of cell activities.91 This is one of the grounds for targeting these signaling pathways to treat GBM, which will be elaborated on later.
There are usually two strategies to target EGFR for GBM treatment: EGFR inhibitors, antibodies, vaccines, chimeric antigen receptor-T (CAR-T) cells, and other therapies to reduce the level of EGFR overexpressing cells. Gefitinib and dacomitinib, as EGFR inhibitors, were not effective in the EGFR-amplified GBM patients (NCT01520870, and NCT02447419),93,94 which may be caused by the low permeability of the blood–brain barrier. However, Osimertinib, a third-generation EGFR inhibitor, has a better blood–brain barrier permeability.95 Preclinical studies have revealed that Osimertinib regulates the mitogen-activated protein kinase (MAPK) pathway and then inhibits the transcription factor EGFR-transcriptional co-activator with PDZ-binding motif (TAZ) to inhibit GBM-patient-derived xenografts (PDX) model.96,97 However, its specific clinical effect remains to be studied.
EGFR antibodies have mostly failed in clinical trials for glioma therapy.98,99 Nevertheless, nimotuzumab is more useful in GBM patients with the activated AKT/mTOR signaling pathway.100 In addition, depatuxizumab mafodotin, an antibody–drug coupling drug, is effective for rGBM that relapses after TMZ standard treatment101,102 but is ineffective in newly diagnosed GBM (NCT02573324).103 In response to rGBM harboring EGFRvIII mutations, the vaccine rindopepimut in combination with TMZ demonstrated efficacy (NCT00458601)104 but failed to exhibit efficacy in phase III clinical trial (NCT01480479),105 see immunotherapy section below. CAR-T regimen is still in phase I trials and has shown the expected effects (NCT02209376).106,107
PI3K/AKT/mTOR is a frequent mutation pathway in IDH-wt GBM patients.108 In particular, mutations in PTEN and PIK3K genes108 lead to abnormal activity of the PI3K/AKT/mTOR pathway, promoting GBM cell viability, stem cell maintenance, and tumor formation.109 This may be linked to the complex and extensive molecular modulation of PI3K/AKT/mTOR. Therefore, the method of improving patients’ tolerance to higher doses to ensure the effect of targeted therapy needs to be proposed urgently.
Like the tumor-suppressor gene tumor protein p53 (TP53) gene, the retinoblastoma tumor suppressor protein (pRB) pathway is very important in the regulation of the cell cycle.110,111 In most of the IDH wild-type GBMs, there are homozygous deletions of cyclin-dependent kinase inhibitor 2 A/B (CDKN2A/B), amplifications of cyclin-dependent kinases 4 and 6 (CDK4/6), and alterations in the RB1 gene in the pRB pathway.84,112 The CDK4/6 form the common functional heterodimeric complexes with cyclin D1-3 (cycD1-3), which can phosphorylate and inactivate the RB protein.113 Inactivation of RB deregulates negative modulation of the E2F transcription factors, thereby inducing the G1/S transition in the cell cycle, thus allowing DNA synthesis and cell growth.113
However, the efficacy of the CDK4/6 inhibitor palbociclib in the treatment of GBM is disappointing (NCT01227434). Clinical trials of another CDK4/6 inhibitor, ribociclib, also showed limited efficacy (NCT02345824).114 In addition, clinical trials of TG02 targeting CDK9 in rGBM therapy and newly diagnosed GBM are ongoing (NCT02942264, NCT03224104).
Telomerase reverse transcriptase (TERT) can maintain the length of telomeres and promote the immortality of tumor cells. The TERT promoter mutations can produce new E-26 transcription factor binding sites, promote transcription, and thus increase activity.115 Interestingly, the combination of the E-26 transcription factor and the mutant TERT promoter is not enough to drive its transcription, but this process requires the non-canonical NF-κB signaling to stimulate a response, and continuous telomerase activity, leading to cancer progression.116 TERT promoter mutations are very common in the GBM of IDH-wt.84 However, in the current clinical trials, it has not been studied as a target for GBM therapy. However, preclinical studies have shown that inhibiting TERT activity can prolong the survival of GBM mice.117 In addition, publications have shown that inhibiting the TERT activity of GBM can sensitize TMZ.118 Therefore, targeting TERT to treat GBM is a worthy strategy for further study.
V-raf murine viral oncogene homolog B1 (BRAF) is implicated in the MEK/ERK signaling pathway activation and promoting cell proliferation.119 Targeted BRAF mutations, especially BRAFV600E missense mutations, have shown remarkable efficacy in other tumors.120 Although BRAF mutations have been observed in diverse glioma subtypes, they are rare in HGG.121 BRAF’s low mutation rate in HGG limits the therapeutic effect.122
P53 as a tumor suppressor, mouse double minute 2 and 4 (MDM2 and MDM4) as negative modulators of p53 protein, is one of the most frequent mutation sites in glioma.108,123 P53 can block the cell cycle arrest and induce apoptosis in G0/G1 in response to genotoxic stress.124,125 Mutational inactivation of TP53 and censored inactivation of MDM2/4 promote uncontrolled proliferation of glioma cells.84 The interaction between hepatocyte growth factor receptor (MET) and hepatocyte growth factor (HGF) contributes to auto-phosphorylation at diverse tyrosine residues, thereby resulting in the recruitment and activation of many signaling effectors, such as growth factor receptor-bound protein 2 (GRB2), GRB2-associated binding protein 1 (Gab1), Steroid receptor coactivator (SRC), SRC homology collagen (Shc), SRC homology region 2 (SH2)-containing protein tyrosine phosphatase 2 (SHP2), phospholipase C-gamma (PLC-γ), focal adhesion kinase (FAK), and casitas B lineage lymphoma (c-Cbl), along with the subsequent phosphorylation of downstream transducers, including PKCδ/SRC/STAT3, PI3K/Akt, Ras/MAPK/ERK, and Wnt/β-catenin pathway.126,127,128 Approximately 30% of GBM patients are characterized by MET gene fusion and high expression,129,130 which is considered to function in the drug resistance, recurrence, and migratory and invasive capabilities of glioma cells, especially in angiogenesis, radiation resistance, as well as hypoxia.126,131 However, rilotumumab,132 onartuzumab (NCT01632228),133 and cabozantinib (NCT00704288, NCT01870726)134,135,136 targeting MET have limited efficacy in the treatment of GBM.
Trimethylation of lysine 27 on histone H3 (H3K27) alteration occurs in 80% of pediatric diffuse midline gliomas (pDIPGs) and is a driving event leading to tumor initiation and progression.137,138,139,140 The H3K27 alteration elevates the activity of histone deacetylases (HDACs), and HDAC inhibitors are a potent compound for the reduced survival of pDIPG cells.141,142 A clinical trial of the HDAC inhibitor panobinostat for the treatment of pDIPG is ongoing (NCT02717455). In addition, synthetic peptide vaccines directed against the H3.3K27M epitope for the treatment of newly diagnosed DIPG patients and other H3.3K27-positive glioma clinical trials are ongoing (NCT02960230).
The vascular endothelial growth factor receptor (VEGFR) signaling pathway has been considered a key factor in GBM tumor survival.143 Meanwhile, GBM is featured with abnormal vascular proliferation. VEGF is upregulated in GBM and stimulates abnormal proliferation of tumor vessels by activating the essential downstream signaling pathways, such as MAPK/ERK1/2, endothelial nitric oxide synthase (eNOS), as well as mTOR.144 Interestingly, vessel normalization increases tumor blood perfusion and contributes to improved GBM patient survival (NCT00305656).145
Bevacizumab inhibits angiogenesis by acting as a humanized monoclonal antibody against the VEGF-A ligand,.146 A phase III trial of bevacizumab was shown to significantly improve PFS for newly diagnosed GBM and rGBM (NCT00884741) but did not significantly impact overall survival (OS).47,147 Bevacizumab treatment of GBM patients with IDH1-wt showed prolongation of OS (NCT00943826).148 The combination of bevacizumab and TMZshowed excellent efficacy and tolerability in recurrent/progressing GBM.149 In addition, bevacizumab in combination with CCNU and radiotherapy also alleviated PFS in patients with IGS-18 or “classical GBMs”.150
The transforming growth factor beta (TGF-β) protein family has complicated functions in diverse regulatory pathways,151,152 where TGF-β2 is a T cell inhibitor in the GBM tumor microenvironment153 that is found in approximately 90% of GBM tumor cells. Nevertheless, TGF-β1 receptor kinase inhibitor Galunisertib in combination with lomustine has limited efficacy in rGBM therapy (NCT01582269).154 Trabedersen, a TGF-β2-specific antisense oligonucleotide, is helpful for HGG treatment, particularly in patients with Karnofsky Performance Status (KPS) above 80% and under the age of 55 years (NCT00431561),155 but the overall effect is much lower than expected. Recent experiments have noted that TGF-β correlates with TMZ resistance and MGMT expression.156 Thus, TMZ and TGF-β combinations of inhibitors are promising.
Wingless and int-1 (Wnt) signaling modulates the neural progenitor cell (NPC) self-renewal, proliferation, as well as differentiation in the brain at varying stages of CNS development. GBM along with other cancers (e.g., digestive system) is associated with aberrant Wnt pathway activity,157 and in particular, GBM of the mesenchymal type is most active.158 But abnormalities in vital components of the Wnt pathway are not common in GBM.159 Preclinical studies have found that inhibiting the activity of the Wnt pathway inhibits TMZ-induced autophagy, which in turn promotes TMZ re-sensitization.160 However, clinical trials targeting Wnt signaling pathway for glioma treatment are currently lacking. The potency of the Wnt signaling pathway in GBM also still needs more investigation.
The above crucial signaling pathways involved in glioma were demonstrated in Fig. 2.

Signaling pathways, genetic mutations, as well as targeted treatment are implicated in glioma development. Tumorigenesis in glioma is activated through diverse mechanisms. Molecular alteration with clinically significant (such as MGMT, IDH, BRAF) eventually converges to several crucial signaling pathways (EGFR, PTEN, VEGF, MET, PI3K/AKT/mTOR, WNT, and TGF-β), resulting in tumor proliferation, invasion, cell survival, and immune evasion. “P” labeling is attached to the molecules that are phosphorylated due to signaling transduction. Created with BioRender.com (https://biorender.com) and Reactome pathway database (https://reactome.org/)
Immunotherapy
Given the limited effect of standard treatment for gliomas (especially GBM) on survival, immunotherapy may be the future of GBM treatment. However, many difficulties and challenges exist for current immunotherapy of gliomas due to the special immune privileged status of the CNS, the low mutational burden of gliomas themselves, and the presence of a highly immunosuppressive microenvironment, which also implies the great potential of immunotherapy for gliomas.161,162,163 So far, many attempts have been made to develop immunotherapeutic approaches for gliomas, looking for therapeutic potential in immune checkpoint therapy, immune cell therapy, vaccines, oncolytic virotherapy, and other modalities. We will review these potential therapies and the immunological basis underlying glioma.
Immune checkpoint inhibitors (ICIS) have exhibited efficacy in many different clinical trials of malignancies, including in both adjuvant and neoadjuvant settings, accompanied by an overall long-term effect.164,165,166,167 Programmed cell death-1 (PD-1) interacts with programmed cell death ligand 1 (PD-L1). The combination of PD-1/PD-L1 reduces T cell receptor (TCR) and CD28 signaling, suppressing T cell effector activity and driving the immunosuppressive environment development.168,169,170 Although GBMs express elevated levels of PD-L1,171 to date, PD-1/PD-L1 immunotherapy trials using GBM have not been fruitful.48 The possible reason for this is that TMZ used in the trial affects PD-L1 level in GBM172,173 and is associated with suppression of immunity by dexamethasone.174,175
Cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) stimulates negative costimulatory signaling on the activated T cells,176 supporting an immunosuppressive environment by inducing immune tolerance.177 Currently, the CTLA-4 blocker ipilimumab is being assessed in GBM (NCT04323046, NCT04396860, and NCT04817254). A phase I exploratory cohort of the checkmate143 trial (NCT02017717) has demonstrated that ipilimumab plus nivolumab is safe.178 It remains to see whether CTLA-4 inhibitors will bring long-term advantages over the current standard of treatment.
Other immune checkpoints or immune-related molecules implicated in glioma include lymphocyte activation gene 3 (LAG-3), ecto-5’-nucleotidase/cluster of differentiation 73 (CD73), cluster of differentiation 161 (CD161), hepatitis A virus cellular receptor 2 (HAVCR2), indoleamine 2,3-dioxygenase 1 (IDO1), V-domain immunoglobulin suppressor of T cell activation (VISTA), V-set domain containing T cell activation inhibitor 1 (VTCN1), CD27/CD70, B, and T lymphocyte attenuator (BTLA), cluster of differentiation 39 (CD39), CD276, cluster of differentiation 47 (CD47), and many others. These target molecules are in clinical trials or only preclinical studies, which have been elaborated on in excellent reviews.179 Major immune checkpoint molecules were shown in Fig. 3. The clinical trials of immune checkpoint inhibitors in progress were shown in Table 2.

Current immune checkpoint molecules. Given the complex relationship between tumor cells and tumor-infiltrating lymphocytes (TILs), various clinical trials have been implemented based on the immune checkpoint molecules (PD-1/PD-L1, CTLA4, LAG-3, and other classic immune checkpoint molecules). Created with BioRender.com (https://biorender.com) and Reactome pathway database (https://reactome.org/)
Tumor-specific antigen polypeptide vaccines
Tumor-specific antigens refer to antigens that are expressed only by tumor cells, but not by normal tissues. EGFRvIII, introduced in the targeted therapy section, is a tumor-specific antigen. The vaccine peptide rindopepimut was synthesized according to the small amino acid sequence around the fusion site on EGFRvIII.180 This vaccine peptide has shown excellent safety and efficacy in both the phase I and phase II clinical trials.104,181 However, the difference between patients receiving the rindopepimut vaccine and those receiving the placebo vaccine could not be reproduced in phase III clinical trial.105 The possible reason is that rindopepimut selected out GBM cells with unmutated EGFR, leading to tumor recurrence. A phase II clinical trial of rindopepimut plus bevacizumab for recurrent GBM elucidated that patients receiving a peptide vaccine presented a better overall response rate and a longer OS, and could discontinue corticosteroid therapy more frequently than those treated with a placebo.182
Innate immune cell therapies and vaccines
Within the glioma immune microenvironment, innate immune cells are the key component. Glioma-associated innate immune cells mainly include tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and tumor-infiltrating dendritic cells (TIDCs). TAMs mainly consist of a small number of resident microglia and diverse bone marrow-derived macrophages.183,184,185 Microglia comprise only a small fraction of TAMs, which offers essential stimuli for tumors to allow for tumor progression.186 These TAMs were previously regarded as M2 immunosuppressive phenotype, but recent articles have indicated that they are a connection of M1 and M2 phenotype.183,187,188,189 TAMs maintain GBM’s mesenchymal phenotype via multiple mechanisms, which promote tumor growth and increase tumor aggressiveness.184,190 MDSCs can be assigned into 3 categories: CD15+ neutrophils, CD14+ monocytes, as well as CD15− & CD14− immature cells. MDSCs can stimulate T cell dysfunction through multiple mechanisms.191,192 GBM patients have elevated MDSCs in the blood, whose infiltration into the TME strengthens immunosuppressive effects.191,192,193,194 TIDCs are characterized by reduced antigen presentation and elevated expression of regulatory ligands/receptors along with broad immunosuppression. Many preclinical models have unveiled that the dendritic cell (DC) activity can be elevated by supplementation with stimulatory cytokines; this may imply a potent role for DC in GBM treatment. DCs have been recently extensively developed as a cellular platform for delivering antigen-specific vaccines to GBM patients.
Chlorogenic acid can modulate the polarization of TAMs toward the M1 phenotype in GBM.195 A clinical trial of chlorogenic acid for GBM patients is ongoing (NCT03758014). Capecitabine, in a low-dose and time-dependent manner, could attenuate intratumoral MDSCs.196,197 Eleven patients were treated with diverse doses of capecitabine for 5–7 days pre-surgery for recurrent GBM, and low-dose capecitabine and bevacizumab were subsequently used as maintenance therapy. Initial reports suggested that circulating MDSC numbers decreased as time went on in patients treated with higher doses and inflammatory infiltrates (eg. CD8+ T cells and Natural Killer (NK) cells), increased in the TME according to flow cytometry.197 The cytomegalovirus (CMV)-derived antigen pp65 is a novel target for DC therapy. The pp65, together with other CMV antigens, is expressed in approximately 90% of GBM samples.198,199 Reap et al used a vaccine with CMV pp65-specific T cells and CMV pp65 RNA-loaded DCs for treating newly diagnosed CMV seropositive GBM patients and observed that DCs increase T cell polyfunctionality and that this polyfunctionality improves survival.198 Another common DC vaccine strategy is based on tumor antigen profiling, incubating patient-derived DCs with synthetic peptides and, given the heterogeneity of GBM, often including several targets.200 ICT-107 is a hexapeptide DC vaccine for GBM therapy, which consists of gp100, IL13Rα2, peptides human epidermal growth factor receptor 2 (HER2), tyrosinase-related protein 2 (Trp-2), melanoma-associated antigens (MAGE-1), as well as automatic ingestion monitor-2 (AIM-2), all of which are elevated in GBM and glioma stem cells (GSCs). But a phase II placebo-controlled trial focusing on the ICT-107 vaccine in 124 patients showed only an increased PFS by only 2.2 months in vaccinated patients compared with placebo, with no significant difference in overall survival.200 But there were indications that patients’ HLA-A1+ vs HLA-A2+ status and MGMT promoter methylation status had a significant impact on patients’ outcomes. Although these studies have shown mixed outcomes, the ability of DC vaccines to a patient’s tumor cannot be underestimated, and innate immune cell therapies are currently shown to have both the advantages of very low side effects and high specificity.
Adaptive immune cell therapy
Compared to non-GBM controls, GBM and other gliomas can isolate peripheral circulating T cells in the bone marrow, leading to relative lymphopenia.201 In addition, GBM can evoke the invading CD4+ and CD8+ T cell apoptosis via the Fas/FasL signaling.202,203,204 Tregs, by producing TGF-β and IL10, affect tumor immune escape, thereby reducing the capability of CD8+ T cells to respond to their cancer cells.205 GBM cells express chemokine (C–C motif) ligand 2 (CCL2), leukocyte-specific protein-1 (LSP-1), STAT3, HIF-1α, and IDO to enhance the activity and survival of Tregs within the TME.206,207,208,209,210,211
CAR-T is a genetically engineered T cell with an artificial receptor directed against the selected antigen.212,213 These cells can bind tumor-specific antigens, independent of the natural mechanisms of antigen presentation, contributing to full activation of CAR-T cells, with infiltration into the tumor performing effector functions. CAR-T cells have received FDA approval in diffuse large B cell lymphoma (DLBCL) and acute lymphoblastic leukemia (ALL).214,215 The most studied targets of CAR-T in GBM are HER2, EGFRvIII, as well as IL-13αR2, which have already been published in clinical trial results.216,217,218,219,220 O’Rourke, et al stated that EGFRvIII-directed CAR-T cells are effective and safe.106 Brown, et al used CAR-T cells targeting IL-13Rα2 in recurrent GBM patients. The results were dramatic, with complete regression of all lesions and the effect maintained for 7.5 months.217 This clinical trial is still ongoing (NCT02208362).
Similar to CAR-T cells, NK cells have recently been applied in preclinical models of cancer with therapeutic roles for their innate functions to recognize/attack abnormal self-cells and overlook healthy cells by MHC-I recognition.221 Chimeric antigen receptor engineered-Natural Killer (CAR-NK) cells can be generated by chimeric antigen receptor technology and injected like T cells to form a similar type of therapy, though more studies are needed.
Oncolytic virotherapy
Oncolytic viruses utilize the natural capability of viruses to replicate and lyse cells in combination with the release of neoantigens and damage-associated molecular patterns following tumor cell lysis, thereby invoking a robust immune response in the cancer area that further kills the tumor.222,223,224,225 The most common are herpesviruses, reoviruses, poxviruses, adenoviruses, or Zika virus.223,226,227,228
Markert, et al assessed the impact of G207, which does not function outside of tumor cells by viral ribonucleotide reductase. The trial results exhibited a good safety and efficacy profile; 11/12 had a treatment response, mOS was 12.2 months, and 4/11 were still alive at the 18-month follow-up. A larger phase II clinical trial is in progress (NCT04482933).229 The results of phase I/II, single-arm research assessing the safety of G47∆, in Japanese adults with recurrent/progressive GBM, were reported by Tomoki Todo et al., showing a 1-year survival rate of 38.5% and the mOS of 7.3 months230 (UMIN-CTR Clinical Trial Registry UMIN000002661). Another phase 2 trial assessing the efficacy of G47∆ in residual or recurrent GBM was also reported by Tomoki Todo et al., showing a 1-year survival rate of 84.2% and the mOS of 28.8 months (from initial surgery) and 20.2 months (after G47∆ initiation)231 (UMIN-CTR Clinical Trial Registry UMIN000015995). PVSRIPO, as a recombinant poliovirus that recognizes differentiation cluster (CD155), is upregulated in GBM cells,232,233 and a phase I trial of PVSRIPO in recurrent GBM therapy disclosed that the overall survival of the test group was elevated in contrast to the historical control group (NCT04479241).232 In another important trial, dnx-2401 (an oncolytic adenovirus with tumor selectivity by inactivating the E1A gene), was utilized for GBM treatment, which prevents the virus from replicating in normal cells with a functional RB (retinoblastoma) signaling pathway.222,234 Its results showed a good safety profile and immunoreactivity.222 Excellent reviews exist detailing previous clinical trials.223,226
In addition, the history of glioma clinical trials also suggested that therapeutic strategies targeting single-target, single-pathogenic mechanisms often lead to failure due to GBM being highly plastic with redundant survival mechanisms. Therefore, while developing precision-targeted therapies, it is highly imperative to develop combination therapy strategies, and excellent reviews have detailed the importance of immune combination therapy.225 However, current research to develop optimized combination therapy strategies is challenging due to the unclear tumor mechanism and the existence of a large number of combinable post-permutation therapeutic strategies, and perhaps the development of big data technologies and intelligent experimental platforms can help the field.235 Perhaps the most pressing thing at the moment is when there is a waning of efficacy or an enhancement of toxicity when combining traditional with emerging therapies, as this can lead to the failure of clinical trials. For example, reduced recurrent GBM PD-L1 expression by TMZ may be associated with nivolumab treatment failure in rGBM;173 Immunosuppressive effects of dexamethasone disable immunotherapy (particularly PD-(L) 1 treatment).175 Therefore, an in-depth study of the interaction between traditional and emerging therapies is highly necessary before clinical trials.
Meningiomas
Meningiomas are a primary intracranial tumor in adults, and this disease harbors an annual incidence rate of approximately 8.58 cases per 100,000 population.40,236 Incidence elevates with age, especially in those over 65 years.40,237 The overall proportion of WHO grade 1 was 80%, and about 20% are WHO grade 2 or 3.238,239,240,241 Among WHO grade 1/2 meningiomas, the incidence is 2.3 times higher in women than in men.40 Most patients with meningioma are cured by surgery and radiation therapy. Incomplete resection or aggressive histological features of the tumor may lead to disease recurrence.242,243 Unfortunately, effective drugs have not been observed to date for patients with meningiomas who do not respond to conventional surgery or radiation therapy.243,244,245
At present, the changes in key gene characteristics in meningioma are closely related to tumor recurrence and prognosis and can be used as a promising therapeutic target.246,247,248,249,250,251,252,253 Although the WHO classification and subtypes of meningioma are mainly based on histopathology, 2021 WHO classification is also used for meningioma classification in combination with molecular biomarkers.241,245 Identifying gene mutations and longitudinal heterogeneity of tumor tissues by high-throughput sequencing are helpful for postoperative risk assessment and prognosis guidance, thus achieving personalized treatment of meningioma.246,254,255 Here, we summarize recent advances and ongoing efforts in molecular-driven therapy for meningioma.
Molecular characteristics and signaling pathway in meningioma
2021 WHO classification emphasizes that the criteria for defining atypical or anaplastic (WHO grade 2/3) meningiomas apply to any subtype. Choroid meningioma and clear cell meningioma are re-assigned as CNS WHO grade 2 due to their higher recurrence rate than other CNS WHO grade 1 meningioma. Considering that other invasive features appear in combination with papillary and rhabdoid structures, classification based on rhabdoid cytology or papillary structure alone is not recommended.11,256 Some clinical studies have associated changes in molecular characteristics with histological subtypes of meningioma, and some molecules can be utilized as prognostic biomarkers to guide treatment. The utilization of novel techniques, such as whole genome sequencing (WGS), whole exome sequencing (WES), and transcriptome analysis, can better describe the mutation of these tumors and identify druggable targets.257,258
It is common for atypical meningiomas to show multiple chromosomal gains as well as 1p, 6q, 10q, 14, and 18q chromosomal losses.259 Early studies identified 22q loss, including BAM22, breakpoint cluster region (BCR), and tissue inhibitor of metalloproteinase-1 (TIMP-1) as a common alteration in meningiomas.259,260 The early stages of meningioma tumorigenesis correlate to the inactivation of one or more genes from the 4.1 superfamilies, such as 4.1B (DAL-1) and neurofibromatosis-2 (NF-2).259,261 About 60% of sporadic meningiomas have the inactivation of NF2, which is closely related to disease recurrence.262,263,264 The alteration of NF2 is observed in different histological subtypes. For instance, 70% of fibroblastic and transitional meningiomas have NF2 mutations,265,266,267 but meningothelial, secretory, and microcystic meningioma are rare. In NF2 wild-type meningiomas, other common gene mutations were also associated with the classification and grading of meningiomas, such as AKT1, PIK3CA, tumor necrosis factor receptor-associated factor 7 (TRAF7), Kruppel-like factor 4 (KLF4), and smoothened (SMO).252,268,269,270,271,272 AKT1/TRAF7 and SMO mutations are representative markers of meningothelial meningioma.273,274 Secretory meningioma is often associated with KLF4 and TRAF7 gene changes.275 Nearly 10% of non-NF2 meningiomas harbor mutations in lysine-specific histone demethylase 5C (KDM5C), lysine-specific histone demethylase 6A (KDM6A), or SWI/SNF‐related matrix‐associated actin‐dependent regulator of chromatin subfamily B member 1 (SMARCB1), which encode epigenetic modifiers.269 SMO and AKT1-mTOR mutations are commonly seen in non-NF2, genomically stable meningiomas appearing in the skull base.269 The loss of histone H3K27me3 expression is closely related to meningioma recurrence.276,277 CDKN2A/CDKN2B (tumor suppressor genes on 9p21) loss of function is involved in meningioma progression from WHO grade 2 to grade 3, and TERT promoter mutation have been identified as a diagnostic marker for WHO grade 3 in the new WHO classification.253,278,279,280 In meningiomas, mutations in the Duchenne muscular dystrophy (DMD) gene have also been discovered,258 independently of TERT mutation status, and it was associated with worse clinical outcomes.281 Additionally, different subgroups of WHO grade 3 meningiomas have been identified with novel mutations. Other rarer germline mutations consisted of SWI/SNF Related, Matrix Associated, SMARCE1, (BRCA1-associated protein 1) BAP1, Actin Dependent Regulator of Chromatin, Subfamily B, Member 1 (SMARCB1), as well as a suppressor of fused (SUFU) genes. The BAP1 mutation was first described in rhabdoid meningiomas.282 BAP1 null cells rely on the enhancer of zeste homolog 2 (EZH2) for transformation, which is highly sensitive to EZH2 inhibition, thus opening new therapeutic perspectives.283 However, the work to translate molecular knowledge into clinical management is still ongoing. Consequently, genomics has enhanced our understanding of meningiomas’ molecular underpinnings, directing the way for further research into novel therapeutics. These molecular data from various individual studies were integrated into the activation of several signaling pathways in meningioma, as shown in Fig. 4.

Summary of the activated signaling pathways and drug targets in meningioma. There are many cellular processes involved in meningioma growth, such as the PI3K–AKT–mTORC pathway, MAPK (mitogen-activated protein kinase) pathway, as well as the Hedgehog pathway. The figure shows the current medical therapies for meningioma, which target diverse molecular targets. Created with BioRender.com (https://biorender.com) and Reactome pathway database (https://reactome.org/)
Treatment strategies for meningioma
Drug treatment for meningiomas is an option for patients who cannot undergo surgery or radiation therapy. Most of the better efficacy is still isolated cases and retrospective studies, and there is a lack of a large number of prospective clinical trial data support.245 The main problems in the clinical application of targeted drugs in meningioma include: how to reach the therapeutic target through the blood–brain barrier, how to avoid or reduce the side effects of drug therapy, and how to establish the evaluation criteria of therapeutic effectiveness. Currently, emerging trials of meningioma incorporating genomic information into criteria are expected to improve future clinical outcomes through precision medicine.284 As a common type of intracranial tumor, complete surgical resection is a standard treatment for meningioma. To avoid functional impairment, some patients failed to be successfully treated by surgery alone or completely resected safely. The treatment of refractory diseases is primarily related to WHO grade 2 and 3 meningiomas or inexcision.285 Several drugs are now being studied in clinical trials for meningiomas, focusing on cytotoxic agents,286 hormone agents,287,288 growth factor receptor antagonists,289 angiogenesis inhibitors,290,291,292 and immunotherapy.293 But no studies have revealed marked response, sustainable tumor control, or prolonged survival. Thus, if patients with meningioma fail to benefit from surgery or radiation, the disease becomes more difficult to treat. Recently, considerable achievements have been obtained in molecular gene research of meningioma, and the nature of recurrent refractory meningioma has been further revealed. For example, the expression of fatty acid synthase (FASN) is up-regulation in malignant meningioma, and inhibition of FASN can inhibit the proliferation of meningioma cells.294 Song et al. reported that FASN may be a target for malignant meningioma.295 The future diagnosis and treatment of meningioma based on molecular genes may bring more hope to malignant meningioma patients.
WHO grade 1 meningioma
For asymptomatic and sporadic meningiomas, regular MRI observation is the preferred strategy. For growing, symptomatic tumors, surgery is preferred.296,297 Surgical excision of tumor tissue was performed for histopathological and molecular pathological examination.298 In addition, surgery was evaluated based on Simpson grades of resection, which was used as a prognostic indicator of recurrence risk.299,300 Radiosurgery or fractionated radiotherapy may be used as an alternative to surgery.301 Currently, no useful drugs have been found for routine clinical treatment of WHO grade 1 meningioma. Pay attention to whether the patients have neurological and cognitive dysfunction to avoid affecting their quality of life. MRI evaluation is recommended periodically after observation or treatment.
WHO grade 2 meningioma
Surgical resection of WHO grade 2 meningiomas are preferred. Simpson I resection should be performed as closely as possible.245,302 When meningioma invades complex sites, it is difficult to avoid nerves, large vessels, and functional areas by surgery, and complete tumor resection may not be possible, resulting in an increased risk of recurrence. The follow-up time was shorter than that of WHO grade1 patients, usually 6 months, up to 5 years postoperatively.245 For patients with Simpson IV–V resection, tumor recurrence can be avoided or delayed by combining radiotherapy.245
WHO grade 3 meningioma
WHO grade 3 meningioma has a rapid growth rate, high recurrence tendency, and strong invasiveness, which can lead to systemic metastasis. It is recommended that surgical resection be as complete as possible, combined with fractional radiotherapy with a total dose of not less than 54Gy.303 Follow-up is followed at 3 months after initial treatment and 3 or 6 months thereafter.303 Drug therapy is in clinical trials and lacks data to support it.
Spinal meningiomas
Surgical resection is a preferred approach for spinal meningiomas. On the premise of not damaging the nerve function, the operation should achieve Simpson 1 resection as much as possible. Removal of the dura should not be the target for ventral spinal cord or severely calcified meningiomas.304,305 If surgery is not available or spinal cord decompression is not required, stereotactic radiosurgery or hypofractionated radiotherapy may be used instead.
Targeted therapies for meningioma
Currently, most clinical studies on meningioma are restricted by the small number of patients, the heterogeneity of tumor types and previous treatments, the lack of prospective controlled trials, and underpowered, resulting in the absence of high-grade evidence-based drugs for clinical use. A variety of therapeutic targets have been recognized for targeting the aforesaid genetic biomarkers in meningiomas, including VEGF/VEGFR, platelet-derived growth factors (PDGFs) and their receptors (PDGFR), EGFR, PIK3CA, mTOR pathway, progesterone receptor (PR), somatostatin (SST), PD-1/ PD-L1, etc. Studies have elucidated that VEGF is expressed in 84% of meningiomas, and the expression of VEGF elevates with the increase of meningioma grade.306 The VEGF inhibitor bevacizumab has shown clinical benefit in meningioma patients that are difficult to treat with surgery and radiotherapy.291 Sunitinib is a small-molecule tyrosine kinase inhibitor targeting VEGFR and PDGFR. For the treatment of malignant meningioma, a prospective, multicenter, single-arm Phase II clinical study on sunitinib demonstrated that 42% of patients did not develop tumorigenesis within 6 months.307 Besides, a recent study of the combination of the SST receptor antagonist octreotide and everolimus in recurrent meningiomas found that 6-month and 12-month survival rates were 90 and 75%, respectively. The growth rate of tumor volume reduced in 78% of patients after 3 months of treatment, a decrease of more than 50%. This article showed that the octreotide and everolimus combination had a better anti-meningioma activity.308 PD-L1 and other immune checkpoint inhibitors in phase II clinical trials are being evaluated in high-grade and recurrent meningioma patients. It is hoped that these immunotherapies will elucidate the efficacy of meningioma treatment.
Vismodegib, an inhibitor of the SMO enzyme, has been approved by the FDA for treating advanced basal cell carcinoma.309 Despite active research into TRAF7 and KLF4’s role in meningioma development, neither genetic alteration has been regarded as a potent therapeutic target. There is anecdotal evidence suggesting that AKT inhibitors are effective in meningiomas with AKT1 mutations.251 The tumor suppressor activity of NF-2 is modulated partly by eliminating the interactions with FAK signaling, and NF-2 inactivation or q22 deletion with tumor cells has been revealed to respond to FAK inhibition.310 GSK2256098, a FAK inhibitor, is currently found to function in the treatment of NF-2 mutation-associated meningiomas (NCT02523014). The completed and ongoing studies of meningioma in recent years are summarized in Table 3.
Central nervous system germ cell tumors (CNS GCTs)
CNS GCTs are rare tumors that usually primarily affect in midline location of children and adolescents, and with a different tumor distribution demographically, account for no more than 4% of all primary CNS tumors in Western Europe and reach a high incidence of 11% in Asia, with a male predominance.311,312,313 Radiation therapy status was a vital predictor of death, and chemotherapy was also significant among all histological subtypes, even adjusting for age at diagnosis.311
Currently, there are very few studies on the molecular biology of CNS GCTs. CNS GCTs diagnosis is on account of the combination of clinical features, tumor markers, and neuroimaging features, and is confirmed by cytopathology and histopathology. However, the discovery of human chorionic gonadotropin (HCG) and alpha-fetoprotein (AFP) in CSF and serum is a great step forward for the diagnosis stage, treatment response, detection relapse, and estimated prognosis in intracranial germ cell tumors (ICGCTs).313
The origin of germinomas is still unclear. However, based on immunohistochemical staining and high throughput sequencing, DNA hypomethylation, MAPK, and/or PI3K pathway alterations, as well as chromosomal abnormalities, exhibit a triad implicated in the CNS GCT pathogenesis.
Germinoma cells recapitulate the characteristics of pluripotent human embryonic stem cells (PGCs) by elevating the genes responsible for self-renewal, including pluripotency factor Octamer-binding transcriptional factor 4 (OCT4), NANOG, and KLF4. In contrast, non-germinomatous germ cell tumors (NGGCTs) are featured with the levels of genes related to epithelial–mesenchymal transition, neuronal differentiation, or the Wnt/β-catenin pathway. While chromosomal instability is a characteristic of all CNS GCT, global DNA hypomethylation is only found in germinoma. Somatic tyrosine kinase receptor (KIT)/RAS and PI3K/AKT mutations have been identified in all CNS GCTs, especially germinoma.314,315,316
CNS germinomas treatment
Germinomas are radiosensitive and high cure rate with radiotherapy (RT) alone; in retrospective and prospective series, the 5-year overall survival rates were above 80%.317 Chemotherapy (intensive cisplatin and cyclophosphamide-based chemotherapy) alone could achieve remissions, yet, the long-term outcome was unsatisfactory, including unacceptable morbidity and mortality.318 Thus, the standard germinoma system treatments contain chemotherapy (Carboplatin/Cisplatin and Etoposide ± Ifosfamide) and radiotherapy, to reduce the volume and dose of RT. As for the high radiosensitivity of germinomas, the surgical section is often used for hydrocephalus treatment and obtaining a histological diagnosis and is not necessary for extension tumor resection. Besides, surgical resection is different in the management of pediatric and adult populations. As for pituitary germinomas, the most comment treatment was radiation + chemotherapy in pediatrics, while radiation + gross total resection + chemotherapy in adults.319
For localized CNS germinomas, the treatment may include craniospinal irradiation (CSI) alone, chemotherapy, or reduced-field radiotherapy. The RT treatment is often applied for covering the whole ventricular (WV) system and is followed by primary tumor boost (PTB). However, optimal RT dosage and field inclusion remains controversial. In terms of radiotherapy techniques, passively scattered proton beam therapy (PSPT) provides lower doses of radiation to the healthy tissue around the tumor, and larger temporal lobe and hippocampal volumes were retained when compared to intensity-modulated radiotherapy (IMRT).320 For disseminated germinomas, chemotherapy and CSI combination are almost consistently recommended for patients with disseminated disease at diagnosis.321 For recurrent CNS germinomas, the salvage therapy consists of local or whole-axis RT, surgery, as well as myeloablative high-dose chemotherapy (HDC) with autologous hematopoietic stem cell rescue (ASCR).322
With the development and application of high throughput sequencing technology, the genomic and epigenetic mechanisms of germinomas have been gradually revealed, and molecular targeted therapy is carried out by degree. The KIT mutation and mTOR mutation were confirmed in CNS germinomas and could be the potential target for therapy.316,323 However, no related KIT or mTOR pathway-targeted clinical trials have been recruited or carried out for CNS germinomas. Figure 5 showed potential targeted drugs for CNS germinomas according to the molecular profiles.

Signaling pathways involved in CNS germinomas pathogenesis and potential molecular targeted therapy. KIT and RAS mutations are the most frequent genes in CNS germinomas. Gain of function mutations of KIT proto-oncogene leads to the KIT protein activation, leading to the upregulation of MAPK or PI3K signaling pathway, which implicated tumor proliferation, migration, and apoptosis resistance. In addition, upregulated PI3K pathway causes a frequent mutation of the mTOR gene, which leads to cell growth via mTOR1 mutation and cell survival via mTOR2 mutation and AKT. Created with BioRender.com (https://biorender.com) and Reactome pathway database (https://reactome.org/)
CNS NGGCTs treatment
CNS NGGCTs are difficult to treat with conventional surgery and RT, and their total cure rate in the era of RT alone was 25%. The current treatment for CNS NGGCT is achieved by combining surgery, chemotherapy, and RT, using a wider field and a higher dose. For malignant NGGCTs, the purposes of treatment are to control the local tumors, with RT covering leptomeningeal tumor spread, as well as chemotherapy eliminating systemic tumor dissemination.324 The combination of surgery, chemotherapy and RT is tailored based on grouping and staging. Among them, the improved survival rate was closely linked to the extent of tumor resection. Metastatic disease is diagnosed by positive CSF cytology and/or distant drops in craniospinal MRI. These malignant GCTs present a high incidence of spinal metastasis or subarachnoid dissemination, which makes CSI with a high-dose local boost essential. Metastatic germinomas may be treated by craniospinal irradiation.325 Chemotherapy is a vital component of multi-modal treatment, while chemotherapy-only strategies are not advised because of the high local treatment failure rate (73.5%). Craniospinal radiotherapy in localized malignant NGGCT could be avoided without enhancing relapses beyond the range of radiotherapy. Chemotherapy and craniospinal radiotherapy are still the gold standards for metastatic disease.326
Central nervous system lymphoma
Central nervous system lymphoma can be classified into two categories: primary and secondary. Primary central nervous system lymphoma (PCNSL) is a rare but aggressive extranodal non-Hodgkin lymphoma (NHL) that impacts the CNS, including the spinal cord, brain, leptomeninges, as well as eyes. About 90% of PCNSL cases are diffuse large B-cell lymphomas (DLBCLs), while the rest are T-cell, Burkitt’s, as well as lymphoblastic and low-grade lymphomas. Currently, PCNSL accounts for approximately 2% of all primary CNS tumors,327 and 4–6% of extranodal lymphomas.328 Secondary central nervous system lymphoma (SCNSL) refers to NHL involving the CNS, which can be manifested as lymphocytic leptomenditis and epidural spinal cord compression signs. SCNSL patients have poor outcomes, and despite dramatic advances in comprehending the mutational landscape of primary diffuse large B-cell lymphoma (DLBCL), there is still a lack of genetic comparison to SCNSL.329
Pathophysiology
At present, the pathogenesis of PCNSL has not been defined.330,331 EB virus has been detected in immunocompromised PCNSL patients, so it is believed that EBV with carcinogenic effects may be related to the pathogenesis of PCNSL, but no EB virus genomic DNA has been detected in patients with normal immune function.332,333 Evidence suggests PCNSL exhibits an overlap of differentiation, expressing germinal center biomarkers such as B cell lymphoma 6 (BCL6) and activation markers such as cyclin D2 and MUM1/Interferon Regulatory Factor 4 (IRF4).334 There is a high frequency of single nucleotide variants and copy number alterations in PCNSL. It has been reported that myeloid differentiation primary response 88 (MYD88) and CD79B are involved in both activated B-cell-like (ABC) and germinal center B-cell-like (GCB) subtypes of PCNSL. The MYD88 and CD79B gene mutation together leads to the B cell receptor signaling pathway activation to promote the development and progression of PCNSL.335 MYD88 missense mutations result in constitutive activation of the TLR pathway,336 while CD79B alteration activates the BCR pathway.337 Caspase activation and recruitment domain 11(CARD11) mutations activate both pathways downstream,337 while tumor necrosis factor alpha-induced protein 3 (TNFAIP3) alterations can cause pathways to lose inhibition.
Many studies indicated that the tumor microenvironment is also an essential factor in PCNSL development. Tumor-associated macrophages (TAMs) have been revealed to be responsible for promoting cancer invasion, proliferation, and immunosuppression in PCNSL cells. The quantification of TAMs may function in prognosis.338 Additionally, TAMs overexpress PD-L1, suggesting that immunotherapy may be effective against them. Activation of the Janus kinase 2 (JAK2)/STAT3 pathway leads to the gene transcription that is implicated in cellular angiogenesis, proliferation, and survival. Meanwhile, the STAT3 gene is found to be expressed in various types of cancer, including PCNSL.339 Amplification of chromosome 9p24.1 leads to elevated expression of PD-L1 and PD-L2, while PD-L1 and PD-L2 can participate in the immune evasion and regulatory mechanisms of PCNSL.340 Additionally, somatic hypermutation (SHM) may lead to PCNSL pathogenesis and may offer a rationale for immunotherapy. Major molecular alterations and related pathways in PCNSL were shown in Fig. 6.

Schematic drawing of the genes, cellular interactions, or signaling pathways targeted by therapeutic strategy for PCNSL. Multi-signaling pathways associated with the malignant B cells’ development and activation are implicated in PCNSLs. Specific targets in B cells and the Toll-like receptor (TLR) pathway, the BCR signaling pathway, and immune microenvironment regulation were exploited for precision therapy. (a) BCR- and TLR-mediated signaling transduction in PCNSL. (b) Immuno-and-targeted therapies in the TME for PCNSL. Created with BioRender.com (https://biorender.com) and Reactome pathway database (https://reactome.org/)
Therapies for PCNSL
Treatment for PCNSL has developed over the last 40 years, while the optimal treatment for PCNSL has not been determined. The advent of high-dose (HD) methotrexate (HD-MTX) therapy has ameliorated PCNSL prognosis.341 At present, HD-MTX-based chemotherapy is recognized as the first-line treatment. Although the initial HD-MTX-based treatment has a high response rate, more than 50% of initial responders relapse.342 PCNSL is sensitive to radiotherapy, and whole-brain radiotherapy (WBRT) can consolidate the response to chemotherapy. Nevertheless, WBRT-related delayed neurotoxicity results in neurocognitive impairment, particularly in elderly patients.343 PCNSL is a DLBCL phenotypic subtype, while the standard DLBCL regimens such as prednisone, doxorubicin, vincristine, cyclophosphamide, and variations, are ineffective in this disease, and the overall effect remains unsatisfactory.344 Other effective approaches consist of rituximab,345 TMZ,346 as well as autologous stem-cell transplantation (ASCT).346 Besides, for novel drugs against PCNSL, including those targeting the B-cell receptor signaling pathway, clinical trials are being conducted. In the mid-to-late 1990s of the 20th century, surgical resection is not regarded as the standard of treatment for PCNSL. There may be a small proportion of patients with large lesions, acute symptoms, as well as signs of brain herniation who will benefit from tumor debulking. Some grow diffusely, and surgical treatment cannot make PCNSL patients benefit, thus enhancing the risk of neurological deficits.347
With the rapid development of precision medicine, targeted therapy is hopeful for further improving the prognosis of PCNSL. It is estimated that 90% of PCNSL is diffuse large B cell lymphoma (DLBCL), which expresses universal B cell markers (CD19, CD20, CD79a). Rituximab is a chimeric monoclonal antibody targeting CD20 and has significant activity in CD20-positive DLBCL. At present, the efficacy of rituximab in PCNSL therapy is still controversial. In phase II randomized controlled clinical trial (IELSG 32), rituximab treatment improved survival in newly diagnosed PCNSL patients.348 However, a recent phase III large randomized controlled clinical study (HOVON 105/ALLG NHL 24) involving 200 PCNSL patients showed that the addition of rituximab to the treatment scheme of newly diagnosed PCNSL patients did not improve the efficacy of PCNSL patients.349 Although the outcomes of the two randomized controlled clinical studies (RCT) were inconsistent, the latest NCCN guidelines recommended rituximab as a first-line combination therapy for PCNSL.
In recent years, targeted therapy and immunotherapy have brought promise for PCNSL treatment. Targeted therapy of PCNSL mainly focuses on Bruton tyrosine kinase (BTK) inhibitors and anti-CD20 monoclonal antibodies. Immunotherapy mainly focuses on immunomodulators, PD-1, and CAR-T. As mentioned earlier, rituximab is a cell–surface protein expressed on mature B cells, while not in neurons or glial cells. The rituximab efficacy in systemic B-cell lymphoma has been well-defined, and regimens containing rituximab have become a better choice for this setting.
Referring to the findings of a phase I study rituximab alone or combined with MTX intraventricular administration was safe and effective in PCNSL.350 At present, HD-MTX combined with rituximab is still the first-line treatment for PCNSL. A clinician can inject rituximab intrathecally if PCNSL has cerebrospinal fluid dissemination and HD-MTX is intolerable but must pay attention to the patient’s health. BTK is a member of the non-receptor tyrosine kinase Tec family, which is mainly expressed in various stages of B cell growth, mediates a series of cellular pathways, including B cell antigen receptor (BCR), and has an important influence on the viability, differentiation, and apoptosis of B cells.351 In a phase I clinical trial, ibutinib, as the first generation of BTK inhibitor, significantly improved the survival of patients.352 In addition, the findings of the Phase Ib clinical trial showed that the combined regimen of Ibutinib and HD-MTX (±rituximab) was well tolerated, with 80% of the total response rate.353 The Phase I/II clinical trial of Tirabrutinib, acting as a second-generation BTK inhibitor, exhibited a certain effect in recurrent/refractory PCNSL.354 Other second-generation BTK inhibitors, such as Zebutenib and Acatinib, have got the approval of the FDA for treating recurrent/refractory mantle cell lymphoma, showing higher efficacy and safety than Ibutinib, and are expected to be developed as the preferred anti-tumor drug to replace Ibutinib. Nowadays, many prospective clinical studies on the treatment of recurrent/refractory PCNSL with second-generation BTK inhibitors are in progress.
Lenalidomide is an immunomodulator that directly or indirectly inhibits tumors through unique immunomodulation. In a phase I clinical trial, lenalidomide resented marked mono-drug activity in patients with recurrent/refractory PCNSL.355 The Phase II REVLRI clinical trial evaluated the clinical efficacy of lenalidomide in recurrent/refractory PCNSL. The total response rate was 39%, with a median PFS of 7.8 months and a median OS of 17.7 months.356 Considering the above encouraging results, NCCN guidelines recommend the use of lenalidomide alone or in conjunction with rituximab for recurrent or refractory PCNSL.
Checkpoints play a part in the human immune system, acting as brakes to prevent excessive activation of T cells from causing inflammatory reactions. PD-1 and its ligand, PD-L1/PD-L2, exert functions in checkpoint pathways. Researchers stated that PD-L1 expression is upregulated in PCNSL.357 In a retrospective study,358 Navuximab was used in patients with relapsed refractory PCNSL/PLT. Four patients obtained complete remission, one patient got partial remission, and the median PFS was 9 months (7–11 months). In light of the above encouraging results, NCCN guidelines recommend lenalidomide alone or combined rituximab as a treatment regimen for recurrent/refractory PCNSL.
Autologous stem cell transplantation is a novel treatment model, and its effectiveness in curing patients with recurrent and high-risk systemic lymphoma has reached clinical recognition and has been applied to the treatment of PCNSL. It is particularly effective for young patients with recurrence but can result in higher treatment-related mortality rates in elderly patients. CAR-T cells targeting CD19 have become the leading engineered T-cell therapy approach for relapsed/refractory B-cell non-Hodgkin lymphoma.343 CAR-T therapy can achieve a complete remission rate of more than 50% in relapsed and refractory DLBCL. Nevertheless, due to the neurotoxicity of CAR-T therapy, patients with CNS involvement were excluded from clinical trials.359 Abramson et al.360 reported that a case of SCNSL treated with CAR-T showed that the lesion disappeared. This research result firstly proves that CAR-T cells can penetrate the blood–brain barrier and achieve the therapeutic response of the CNS, which brings a new dawn for CAR-T to treat PCNSL. A schematic representation of the actions of therapies on these signaling cascades and immune regulation is given in Fig. 6.
Conclusion
Since the implementation of high throughput data analysis, the comprehension of the molecular profile of brain tumors has continued to evolve rapidly. The fifth edition of the WHO classification of CNS tumors in 2021 has incorporated many advanced molecular alterations into the diagnostic standards. These multitudes of cancer-specific genetic alterations, including receptor kinases and their downstream signaling partners, cell cycle regulation, telomere maintenance, and chromatin organization, and the further effects on tumor etiology have reformed the conception of clinical management and prognosis, providing new insights into the transformation of clinical trials. However, the current failure of several targeted agents, especially for GBM, illustrates that CNS tumors do not only rely on a single pathway-driven targeted therapy. Future treatment may be improved in the following ways: 1) the combination strategies of multiple targeted drugs and immunotherapeutic approaches have been proven to be efficacy against brain tumors, especially for recurrent/progressive patients, and could be the trend of treatment management in the future; 2) the limited scale of participation and specific patient groups indicates the necessity of performing more larger and multicenter clinical trials to assess efficacy and safety; 3) developing more effective drug delivery system to overcome the blood–brain barrier, such as nano-drug or extracellular vesicle-based drug delivery system; 4) performing a genetic/precision medical treatments based on the genomics technologies.
References
Louis, D. N. et al. cIMPACT-NOW (the consortium to inform molecular and practical approaches to CNS tumor taxonomy): a new initiative in advancing nervous system tumor classification. Brain Pathol. 27, 851–852 (2017).
Louis, D. N. et al. Announcing cIMPACT-NOW: the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy. Acta Neuropathol. 133, 1–3 (2017).
Louis, D. N. et al. cIMPACT-NOW update 2: diagnostic clarifications for diffuse midline glioma, H3 K27M-mutant and diffuse astrocytoma/anaplastic astrocytoma, IDH-mutant. Acta Neuropathol. 135, 639–642 (2018).
Ellison, D. W. et al. cIMPACT-NOW update 4: diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAF(V600E) mutation. Acta Neuropathol. 137, 683–687 (2019).
Brat, D. J. et al. cIMPACT-NOW update 3: recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol. 136, 805–810 (2018).
Louis, D. N. et al. cIMPACT-NOW update 1: not otherwise specified (NOS) and not elsewhere classified (NEC). Acta Neuropathol. 135, 481–484 (2018).
Louis, D. N. et al. cIMPACT-NOW: a practical summary of diagnostic points from Round 1 updates. Brain Pathol. 29, 469–472 (2019).
Louis, D. N. et al. cIMPACT-NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 30, 844–856 (2020).
Brat, D. J. et al. cIMPACT-NOW update 5: recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol. 139, 603–608 (2020).
Ellison, D. W. et al. cIMPACT-NOW update 7: advancing the molecular classification of ependymal tumors. Brain Pathol. 30, 863–866 (2020).
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 23, 1231–1251 (2021).
Miller, K. D. et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J. Clin. 71, 381–406 (2021).
Li, X. R. et al. Are benign and borderline brain tumors underreported? J. Registry Manag. 43, 187–194 (2016).
Ostrom, Q. T. et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2014-2018. Neuro Oncol. 23, iii1–iii105 (2021).
Fisher, J. L., Schwartzbaum, J. A., Wrensch, M. & Wiemels, J. L. Epidemiology of brain tumors. Neurol. Clin. 25, 867–890, vii (2007).
Ostrom, Q. T. et al. Risk factors for childhood and adult primary brain tumors. Neuro Oncol. 21, 1357–1375 (2019).
Ostrom, Q. T., Francis, S. S. & Barnholtz-Sloan, J. S. Epidemiology of brain and other CNS tumors. Curr. Neurol. Neurosci. Rep. 21, 68 (2021).
Gohar, M. K., Ammar, M. G., Alnagar, A. A. & Abd-ElAziz, H. A. Serum IgE and allergy related genotypes of IL-4R alpha and IL-13 genes: association with glioma susceptibility and glioblastoma prognosis. Egypt J. Immunol. 25, 19–33 (2018).
Turner, M. C. et al. Allergy and brain tumors in the INTERPHONE study: pooled results from Australia, Canada, France, Israel, and New Zealand. Cancer Causes Control 24, 949–960 (2013).
Gurney, J. G. et al. Head injury as a risk factor for brain tumors in children: results from a multicenter case-control study. Epidemiology 7, 485–489 (1996).
Limam, S. et al. Investigation of simian virus 40 (SV40) and human JC, BK, MC, KI, and WU polyomaviruses in glioma. J. Neurovirol. 26, 347–357 (2020).
Rollison, D. E. M. et al. Investigation of human brain tumors for the presence of polyomavirus genome sequences by two independent laboratories. Int J. Cancer 113, 769–774 (2005).
Mathews, J. D. et al. Cancer risk in 680,000 people exposed to computed tomography scans in childhood or adolescence: data linkage study of 11 million Australians. Br. Med. J. 346, f2360 (2013).
Braganza, M. Z. et al. Ionizing radiation and the risk of brain and central nervous system tumors: a systematic review. Neuro Oncol. 14, 1316–1324 (2012).
Taylor, A. J. et al. Population-based risks of CNS tumors in survivors of childhood cancer: the British Childhood Cancer Survivor Study. J. Clin. Oncol. 28, 5287–5293 (2010).
Neglia, J. P. et al. New primary neoplasms of the central nervous system in survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. J. Natl Cancer Inst. 98, 1528–1537 (2006).
Sadetzki, S. et al. Long-term follow-up for brain tumor development after childhood exposure to ionizing radiation for tinea capitis. Radiat. Res. 163, 424–432 (2005).
Pearce, M. S. et al. Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: a retrospective cohort study. Lancet 380, 499–505 (2012).
Davis, F. et al. Medical diagnostic radiation exposures and risk of gliomas. Radiat. Res. 175, 790–796 (2011).
Lacourt, A. et al. INTEROCC case-control study: lack of association between glioma tumors and occupational exposure to selected combustion products, dusts and other chemical agents. BMC Public Health 13, 340 (2013).
Ruder, A. M. et al. The Upper Midwest Health Study: industry and occupation of glioma cases and controls. Am. J. Ind. Med. 55, 747–755 (2012).
Creed, J. H., Smith-Warner, S. A., Gerke, T. A. & Egan, K. M. A prospective study of coffee and tea consumption and the risk of glioma in the UK Biobank. Eur. J. Cancer 129, 123–131 (2020).
Ward, H. A. et al. Meat and haem iron intake in relation to glioma in the European Prospective Investigation into Cancer and Nutrition study. Eur. J. Cancer Prev. 27, 379–383 (2018).
Dubrow, R. et al. Dietary components related to N-nitroso compound formation: a prospective study of adult glioma. Cancer Epidem Biomarker 19, 1709–1722 (2010).
Terry, M. B. et al. An International case control study of adult diet and brain tumor risk: a histology-specific analysis by food group. Ann. Epidemiol. 19, 161–171 (2009).
Holick, C. N. et al. Prospective study of intake of fruit, vegetables, and carotenoids and the risk of adult glioma. Am. J. Clin. Nutr. 85, 877–886 (2007).
Zhang, W. et al. Association between dietary nitrite intake and glioma risk: a systematic review and dose-response meta-analysis of observational studies. Front. Oncol. 12, 910476 (2022).
Zhang, W. et al. Dietary factors and risk of glioma in adults: a systematic review and dose-response meta-analysis of observational studies. Front. Nutr. 9, 834258 (2022).
Kuan, A. S. et al. Diet and risk of glioma: combined analysis of 3 large prospective studies in the UK and USA. Neuro Oncol. 21, 944–952 (2019).
Ostrom, Q. T. et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012-2016. Neuro Oncol. 21, v1–v100 (2019).
Duffau, H. & Taillandier, L. New concepts in the management of diffuse low-grade glioma: proposal of a multistage and individualized therapeutic approach. Neuro Oncol. 17, 332–342 (2015).
Weller, M. & Le Rhun, E. How did lomustine become standard of care in recurrent glioblastoma? Cancer Treat. Rev. 87, 102029 (2020).
Marenco-Hillembrand, L. et al. Trends in glioblastoma: outcomes over time and type of intervention: a systematic evidence based analysis. J. Neurooncol. 147, 297–307 (2020).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005).
Stupp, R. et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. J. Am. Med. Assoc. 318, 2306–2316 (2017).
Weller, M. et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 18, e315–e329 (2017).
Gilbert, M. R. et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 370, 699–708 (2014).
Reardon, D. A. et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the CheckMate 143 Phase 3 randomized clinical trial. JAMA Oncol. 6, 1003–1010 (2020).
Jakola, A. S. et al. Surgical resection versus watchful waiting in low-grade gliomas. Ann. Oncol. 28, 1942–1948 (2017).
Lapointe, S., Perry, A. & Butowski, N. A. Primary brain tumours in adults. Lancet 392, 432–446 (2018).
Hegi, M. E. et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 352, 997–1003 (2005).
Brigliadori, G. et al. Defining the cutoff value of MGMT gene promoter methylation and its predictive capacity in glioblastoma. J. Neurooncol. 128, 333–339 (2016).
Na, K. et al. Targeted next-generation sequencing panel (TruSight Tumor 170) in diffuse glioma: a single institutional experience of 135 cases. J. Neurooncol. 142, 445–454 (2019).
Esteller, M. et al. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 59, 793–797 (1999).
Chen, X. et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat. Commun. 9, 2949 (2018).
Kreth, S. et al. O-methylguanine-DNA methyltransferase (MGMT) mRNA expression predicts outcome in malignant glioma independent of MGMT promoter methylation. PLoS ONE 6, e17156 (2011).
Wickström, M. et al. Wnt/β-catenin pathway regulates MGMT gene expression in cancer and inhibition of Wnt signalling prevents chemoresistance. Nat. Commun. 6, 8904 (2015).
Wang, J. et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 48, 768–776 (2016).
Oldrini, B. et al. MGMT genomic rearrangements contribute to chemotherapy resistance in gliomas. Nat. Commun. 11, 3883 (2020).
Frenel, J. S. et al. 370MO FOLAGLI: A phase I study of folinic acid combined with temozolomide and radiotherapy to modulate MGMT gene promoter methylation in newly diagnosed MGMT non-methytated glioblastoma. 31.
Rahman, M. A. et al. Bortezomib administered prior to temozolomide depletes MGMT, chemosensitizes glioblastoma with unmethylated MGMT promoter and prolongs animal survival. Br. J. Cancer 121, 545–555 (2019).
Lin, K. et al. Mechanism-based design of agents that selectively target drug-resistant glioma. Science 377, 502–511 (2022).
Bady, P. et al. The DNA methylome of DDR genes and benefit from RT or TMZ in IDH mutant low-grade glioma treated in EORTC 22033. Acta Neuropathol. 135, 601–615 (2018).
Xu, W. et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 (2011).
Duncan, C. G. et al. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res. 22, 2339–2355 (2012).
Turcan, S. et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483 (2012).
Bardella, C. et al. Expression of Idh1(R132H) in the murine subventricular zone stem cell niche recapitulates features of early gliomagenesis. Cancer Cell 30, 578–594 (2016).
Lu, C. et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 (2012).
Su, R. et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell 172, 90–105.e123 (2018).
Aas, P. A. et al. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature 421, 859–863 (2003).
Falnes, P., Johansen, R. F. & Seeberg, E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature 419, 178–182 (2002).
Wang, P. et al. Oncometabolite D-2-hydroxyglutarate inhibits ALKBH DNA repair enzymes and sensitizes IDH mutant cells to alkylating agents. Cell Rep. 13, 2353–2361 (2015).
Cairncross, J. G. et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J. Clin. Oncol. 32, 783–790 (2014).
Carbonneau, M. et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 7, 12700 (2016).
Fu, X. et al. 2-Hydroxyglutarate Inhibits ATP Synthase and mTOR Signaling. Cell Metab. 22, 508–515 (2015).
Zhao, S. et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 324, 261–265 (2009).
Laurence, M. G. et al. Oncogenic activities of IDH1/2 mutations: from epigenetics to cellular signaling. Trends Cell Biol. 27, 738–752 (2017).
DiNardo, C. D. et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N. Engl. J. Med. 378, 2386–2398 (2018).
Roboz, G. J. et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 135, 463–471 (2020).
DiNardo, C. D. et al. Mutant isocitrate dehydrogenase 1 inhibitor ivosidenib in combination with azacitidine for newly diagnosed acute myeloid leukemia. J. Clin. Oncol. 39, 57–65 (2021).
Correction to Lancet Oncol 2020; 21: 796–807. Lancet Oncol. 21, e462 (2020).
Mellinghoff, I. K. et al. Ivosidenib in Isocitrate Dehydrogenase 1-Mutated Advanced Glioma. J. Clin. Oncol. 38, 3398–3406 (2020).
Platten, M. et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 592, 463–468 (2021).
Brennan, C. W. et al. The somatic genomic landscape of glioblastoma. Cell 155, 462–477 (2013).
Voldborg, B. R., Damstrup, L., Spang-Thomsen, M. & Poulsen, H. S. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 8, 1197–1206 (1997).
Furnari, F. B., Cloughesy, T. F., Cavenee, W. K. & Mischel, P. S. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer 15, 302–310 (2015).
Libermann, T. A. et al. Expression of epidermal growth factor receptors in human brain tumors. Cancer Res. 44, 753–760 (1984).
Libermann, T. A. et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 313, 144–147 (1985).
Tanaka, K. et al. Oncogenic EGFR signaling activates an mTORC2-NF-κB pathway that promotes chemotherapy resistance. Cancer Discov. 1, 524–538 (2011).
Fan, Q. W. et al. EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell 24, 438–449 (2013).
Huang, P. H., Xu, A. M. & White, F. M. Oncogenic EGFR signaling networks in glioma. Sci. Signal 2, re6 (2009).
Zhao, K. et al. EGFR/c-myc axis regulates TGFβ/Hippo/Notch pathway via epigenetic silencing miR-524 in gliomas. Cancer Lett. 406, 12–21 (2017).
Byeon, S. et al. Use of gefitinib in EGFR-amplified refractory solid tumors: an open-label, single-arm, single-center prospective pilot study. Target Oncol. 15, 185–192 (2020).
Sepulveda-Sanchez, J. M. et al. Phase II trial of dacomitinib, a pan-human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro Oncol. 19, 1522–1531 (2017).
Liu, X. et al. The third-generation EGFR inhibitor AZD9291 overcomes primary resistance by continuously blocking ERK signaling in glioblastoma. J. Exp. Clin. Cancer Res. 38, 219 (2019).
Gao, M. et al. EGFR activates a TAZ-driven oncogenic program in glioblastoma. Cancer Res. 81, 3580–3592 (2021).
Chen, C. et al. Osimertinib successfully combats EGFR-negative glioblastoma cells by inhibiting the MAPK pathway. Acta Pharm. Sin. 42, 108–114 (2021).
Neyns, B. et al. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann. Oncol. 20, 1596–1603 (2009).
Gan, H. K., Burgess, A. W., Clayton, A. H. & Scott, A. M. Targeting of a conformationally exposed, tumor-specific epitope of EGFR as a strategy for cancer therapy. Cancer Res. 72, 2924–2930 (2012).
Ronellenfitsch, M. W. et al. Akt and mTORC1 signaling as predictive biomarkers for the EGFR antibody nimotuzumab in glioblastoma. Acta Neuropathol. Commun. 6, 81 (2018).
Martin, V. et al. ACTR-39. Two-year results of the intellance 2/EORTC Trial 1410 randomized phase II study on depatux–m alone, depatux-m combined with temozolomide (TMZ) and either TMZ or lomustine in recurrent EGFR Amplified glioblastoma (NCT02343406. suppl_6, (2018).
Lassman, A. B. et al. Safety and efficacy of depatuxizumab mafodotin + temozolomide in patients with EGFR-amplified, recurrent glioblastoma: results from an international phase I multicenter trial. Neuro Oncol. 21, 106–114 (2019).
Marin, B. M. et al. Heterogeneous delivery across the blood-brain barrier limits the efficacy of an EGFR-targeting antibody drug conjugate in glioblastoma. Neuro Oncol. 23, 2042–2053 (2021).
Schuster, J. et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol. 17, 854–861 (2015).
Weller, M. et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 18, 1373–1385 (2017).
O’Rourke, D. M. et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 9, eaaa0984 (2017).
Johnson, L. A. et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 7, 275ra222 (2015).
Krauth, J. Comments on the paper by Moller et al. (1989): problems in single-case evaluation. Eur. Arch. Psychiatry Neurol. Sci. 239, 391–394 (1990). discussion 395–397.
Akhavan, D., Cloughesy, T. F. & Mischel, P. S. mTOR signaling in glioblastoma: lessons learned from bench to bedside. Neuro Oncol. 12, 882–889 (2010).
Sellers, W. R. & Kaelin, W. G. Jr Role of the retinoblastoma protein in the pathogenesis of human cancer. J. Clin. Oncol. 15, 3301–3312 (1997).
Santoni, G. et al. Functional in vitro assessment of VEGFA/NOTCH2 signaling pathway and pRB proteasomal degradation and the clinical relevance of mucolipin TRPML2 overexpression in glioblastoma patients. Int. J. Mol. Sci. 23, 688 (2022).
Ferguson, S. D. et al. Targetable gene fusions associate with the IDH wild-type astrocytic lineage in adult gliomas. J. Neuropathol. Exp. Neurol. 77, 437–442 (2018).
Malumbres, M. & Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 30, 630–641 (2005).
Miller, T. W. et al. Tumor pharmacokinetics and pharmacodynamics of the CDK4/6 inhibitor ribociclib in patients with recurrent glioblastoma. J. Neurooncol. 144, 563–572 (2019).
Horn, S. et al. TERT promoter mutations in familial and sporadic melanoma. Science 339, 959–961 (2013).
Li, Y. et al. Non-canonical NF-κB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat. Cell Biol. 17, 1327–1338 (2015).
Takahashi, M. et al. Eribulin penetrates brain tumor tissue and prolongs survival of mice harboring intracerebral glioblastoma xenografts. Cancer Sci. 110, 2247–2257 (2019).
Amen, A. M. et al. Cancer-specific loss of TERT activation sensitizes glioblastoma to DNA damage. Proc. Natl Acad. Sci. USA 118 (2021).
Davies, H. et al. Mutations of the BRAF gene in human cancer. Nature 417, 949–954 (2002).
Robert, C. et al. Five-year outcomes with dabrafenib plus trametinib in metastatic melanoma. N. Engl. J. Med. 381, 626–636 (2019).
Kaley, T. et al. BRAF inhibition in BRAF(V600)-mutant gliomas: results from the VE-BASKET Study. J. Clin. Oncol. 36, 3477–3484 (2018).
Schreck, K. C., Grossman, S. A. & Pratilas, C. A. BRAF mutations and the utility of RAF and MEK inhibitors in primary brain tumors. Cancers 11, 1262 (2019).
Pratt, D. et al. High-grade glioma with pleomorphic and pseudopapillary features (HPAP): a proposed type of circumscribed glioma in adults harboring frequent TP53 mutations and recurrent monosomy 13. Acta Neuropathol. 143, 403–414 (2022).
Levine, A. J. p53, the cellular gatekeeper for growth and division. Cell 88, 323–331 (1997).
Hernandez Borrero, L. J. & El-Deiry, W. S. Tumor suppressor p53: biology, signaling pathways, and therapeutic targeting. Biochim Biophys. Acta Rev. Cancer 1876, 188556 (2021).
Cheng, F. & Guo, D. MET in glioma: signaling pathways and targeted therapies. J. Exp. Clin. Cancer Res. 38, 270 (2019).
Zhang, Y., Du, Z. & Zhang, M. Biomarker development in MET-targeted therapy. Oncotarget 7, 37370–37389 (2016).
Kim, K. H. et al. Wnt/β-catenin signaling is a key downstream mediator of MET signaling in glioblastoma stem cells. Neuro Oncol. 15, 161–171 (2013).
Xie, Q. et al. Hepatocyte growth factor (HGF) autocrine activation predicts sensitivity to MET inhibition in glioblastoma. Proc. Natl Acad. Sci. USA 109, 570–575 (2012).
Woo, H. Y. et al. Glioblastomas harboring gene fusions detected by next-generation sequencing. Brain Tumor Pathol. 37, 136–144 (2020).
Dean, M. et al. The human met oncogene is related to the tyrosine kinase oncogenes. Nature 318, 385–388 (1985).
Wen, P. Y. et al. A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro Oncol. 13, 437–446 (2011).
Cloughesy, T. et al. Randomized, double-blind, placebo-controlled, multicenter phase II study of onartuzumab plus bevacizumab versus placebo plus bevacizumab in patients with recurrent glioblastoma: efficacy, safety, and hepatocyte growth factor and O(6)-methylguanine-DNA methyltransferase biomarker analyses. J. Clin. Oncol. 35, 343–351 (2017).
Wen, P. Y. et al. Phase II study of cabozantinib in patients with progressive glioblastoma: subset analysis of patients naive to antiangiogenic therapy. Neuro Oncol. 20, 249–258 (2018).
Cloughesy, T. F. et al. Phase II study of cabozantinib in patients with progressive glioblastoma: subset analysis of patients with prior antiangiogenic therapy. Neuro Oncol. 20, 259–267 (2018).
van den Bent, M. et al. A Phase Ib/II, open-label, multicenter study of INC280 (capmatinib) alone and in combination with buparlisib (BKM120) in adult patients with recurrent glioblastoma. J. Neurooncol. 146, 79–89 (2020).
Pathania, M. et al. H3.3(K27M) Cooperates with Trp53 Loss and PDGFRA gain in mouse embryonic neural progenitor cells to induce invasive high-grade gliomas. Cancer Cell 32, 684–700 e689 (2017).
Harutyunyan, A. S. et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 10, 1262 (2019).
Silveira, A. B. et al. H3.3 K27M depletion increases differentiation and extends latency of diffuse intrinsic pontine glioma growth in vivo. Acta Neuropathol. 137, 637–655 (2019).
Larson, J. D. et al. Histone H3.3 K27M accelerates spontaneous brainstem glioma and drives restricted changes in bivalent gene expression. Cancer Cell 35, 140–155 e147 (2019).
Grasso, C. S. et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 21, 555–559 (2015).
Anastas, J. N. et al. Re-programing chromatin with a bifunctional LSD1/HDAC inhibitor induces therapeutic differentiation in DIPG. Cancer Cell 36, 528–544 e510 (2019).
Szabo, E. et al. Autocrine VEGFR1 and VEGFR2 signaling promotes survival in human glioblastoma models in vitro and in vivo. Neuro Oncol. 18, 1242–1252 (2016).
Estrada, C. C., Maldonado, A. & Mallipattu, S. K. Therapeutic inhibition of VEGF signaling and associated nephrotoxicities. J. Am. Soc. Nephrol. 30, 187–200 (2019).
Sorensen, A. G. et al. Increased survival of glioblastoma patients who respond to antiangiogenic therapy with elevated blood perfusion. Cancer Res. 72, 402–407 (2012).
Ferrara, N., Hillan, K. J. & Novotny, W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem. Biophys. Res. Commun. 333, 328–335 (2005).
Chinot, O. L. et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 370, 709–722 (2014).
Sandmann, T. et al. Patients with proneural glioblastoma may derive overall survival benefit from the addition of bevacizumab to first-line radiotherapy and temozolomide: retrospective analysis of the AVAglio trial. J. Clin. Oncol. 33, 2735–2744 (2015).
Gilbert, M. R. et al. NRG oncology RTOG 0625: a randomized phase II trial of bevacizumab with either irinotecan or dose-dense temozolomide in recurrent glioblastoma. J. Neurooncol. 131, 193–199 (2017).
Erdem-Eraslan, L. et al. Identification of patients with recurrent glioblastoma who may benefit from combined bevacizumab and CCNU therapy: a report from the BELOB trial. Cancer Res. 76, 525–534 (2016).
Lin, C. J. et al. Honokiol induces autophagic cell death in malignant glioma through reactive oxygen species-mediated regulation of the p53/PI3K/Akt/mTOR signaling pathway. Toxicol. Appl. Pharm. 304, 59–69 (2016).
Massague, J. TGFbeta in cancer. Cell 134, 215–230 (2008).
Kuppner, M. C. et al. The glioblastoma-derived T-cell suppressor factor/transforming growth factor beta 2 inhibits the generation of lymphokine-activated killer (LAK) cells. Int. J. Cancer 42, 562–567 (1988).
Brandes, A. A. et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol. 18, 1146–1156 (2016).
Bogdahn, U. et al. Targeted therapy for high-grade glioma with the TGF-beta2 inhibitor trabedersen: results of a randomized and controlled phase IIb study. Neuro Oncol. 13, 132–142 (2011).
Nie, E. et al. TGF-beta1 modulates temozolomide resistance in glioblastoma via altered microRNA processing and elevated MGMT. Neuro Oncol. 23, 435–446 (2021).
Mehta, S. & Lo Cascio, C. Developmentally regulated signaling pathways in glioma invasion. Cell Mol. Life Sci. 75, 385–402 (2018).
Lamouille, S., Xu, J. & Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196 (2014).
Lorzadeh, S., Kohan, L., Ghavami, S. & Azarpira, N. Autophagy and the Wnt signaling pathway: a focus on Wnt/beta-catenin signaling. Biochim. Biophys. Acta Mol. Cell Res. 1868, 118926 (2021).
Yun, E. J., Kim, S., Hsieh, J. T. & Baek, S. T. Wnt/beta-catenin signaling pathway induces autophagy-mediated temozolomide-resistance in human glioblastoma. Cell Death Dis. 11, 771 (2020).
Touat, M. et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 580, 517–523 (2020).
Hodges, T. R. et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol. 19, 1047–1057 (2017).
Turajlic, S. et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 18, 1009–1021 (2017).
Weber, J. et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 377, 1824–1835 (2017).
Hellmann, M. D. et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 381, 2020–2031 (2019).
Rizvi, N. A. et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. 16, 257–265 (2015).
Amaria, R. N. et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med. 24, 1649–1654 (2018).
Sun, C., Mezzadra, R. & Schumacher, T. N. Regulation and Function of the PD-L1 Checkpoint. Immunity 48, 434–452 (2018).
Boussiotis, V. A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 375, 1767–1778 (2016).
Hui, E. et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433 (2017).
Nduom, E. K. et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 18, 195–205 (2016).
Heynckes, S. et al. Expression differences of programmed death ligand 1 in de-novo and recurrent glioblastoma multiforme. Oncotarget 8, 74170–74177 (2017).
Heynckes, S. et al. Crosslink between Temozolomide and PD-L1 immune-checkpoint inhibition in glioblastoma multiforme. BMC Cancer 19, 117 (2019).
Iorgulescu, J. B. et al. Concurrent dexamethasone limits the clinical benefit of immune checkpoint blockade in glioblastoma. Clin. Cancer Res. 27, 276–287 (2021).
Giles, A. J. et al. Dexamethasone-induced immunosuppression: mechanisms and implications for immunotherapy. J. Immunother. Cancer 6, 51 (2018).
Wolchok, J. D. & Saenger, Y. The mechanism of anti-CTLA-4 activity and the negative regulation of T-cell activation. Oncologist 13(Suppl 4), 2–9 (2008).
Brown, N. F. et al. A phase II open label, randomised study of ipilimumab with temozolomide versus temozolomide alone after surgery and chemoradiotherapy in patients with recently diagnosed glioblastoma: the Ipi-Glio trial protocol. BMC Cancer 20, 198 (2020).
Omuro, A. et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 20, 674–686 (2018).
Yang, K. et al. Glioma targeted therapy: insight into future of molecular approaches. Mol. Cancer 21, 39 (2022).
Elsamadicy, A. A. et al. Prospect of rindopepimut in the treatment of glioblastoma. Expert Opin. Biol. Ther. 17, 507–513 (2017).
Sampson, J. H. et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 28, 4722–4729 (2010).
Reardon, D. A. et al. Rindopepimut with Bevacizumab for Patients with Relapsed EGFRvIII-Expressing Glioblastoma (ReACT): Results of a Double-Blind Randomized Phase II Trial. Clin. Cancer Res. 26, 1586–1594 (2020).
Hambardzumyan, D., Gutmann, D. H. & Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 19, 20–27 (2016).
Chen, Z. et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. 77, 2266–2278 (2017).
Gutmann, D. H. & Kettenmann, H. Microglia/brain macrophages as central drivers of brain tumor pathobiology. Neuron 104, 442–449 (2019).
Liu, H. et al. Pro-inflammatory and proliferative microglia drive progression of glioblastoma. Cell Rep. 36, 109718 (2021).
Mantovani, A. et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25, 677–686 (2004).
Mantovani, A. et al. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23, 549–555 (2002).
Umemura, N. et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J. Leukoc. Biol. 83, 1136–1144 (2008).
Hara, T. et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell 39, 779–792 e711 (2021).
Rodriguez, P. C. & Ochoa, A. C. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol. Rev. 222, 180–191 (2008).
Raychaudhuri, B. et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol. 13, 591–599 (2011).
Won, W. J. et al. Metabolic and functional reprogramming of myeloid-derived suppressor cells and their therapeutic control in glioblastoma. Cell Stress 3, 47–65 (2019).
Dubinski, D. et al. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro Oncol. 18, 807–818 (2016).
Xue, N. et al. Chlorogenic acid inhibits glioblastoma growth through repolarizating macrophage from M2 to M1 phenotype. Sci. Rep. 7, 39011 (2017).
Peereboom, D. M. et al. Metronomic capecitabine as an immune modulator in glioblastoma patients reduces myeloid-derived suppressor cells. JCI Insight. 4, e130748 (2019).
Otvos, B. et al. Cancer stem cell-secreted macrophage migration inhibitory factor stimulates myeloid derived suppressor cell function and facilitates glioblastoma immune evasion. Stem Cells 34, 2026–2039 (2016).
Reap, E. A. et al. Dendritic cells enhance polyfunctionality of adoptively transferred T cells that target cytomegalovirus in glioblastoma. Cancer Res. 78, 256–264 (2018).
Nair, S. K. et al. Recognition and killing of autologous, primary glioblastoma tumor cells by human cytomegalovirus pp65-specific cytotoxic T cells. Clin. Cancer Res. 20, 2684–2694 (2014).
Wen, P. Y. et al. A randomized double-blind placebo-controlled phase II trial of dendritic cell vaccine ICT-107 in newly diagnosed patients with glioblastoma. Clin. Cancer Res. 25, 5799–5807 (2019).
Chongsathidkiet, P. et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 24, 1459–1468 (2018).
Woroniecka, K. et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin. Cancer Res. 24, 4175–4186 (2018).
Strand, S. et al. Lymphocyte apoptosis induced by CD95 (APO-1/Fas) ligand-expressing tumor cells—a mechanism of immune evasion? Nat. Med. 2, 1361–1366 (1996).
Walker, D. G., Chuah, T., Rist, M. J. & Pender, M. P. T-cell apoptosis in human glioblastoma multiforme: implications for immunotherapy. J. Neuroimmunol. 175, 59–68 (2006).
Fecci, P. E. et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 66, 3294–3302 (2006).
Chang, A. L. et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. 76, 5671–5682 (2016).
Cao, J. Y. et al. Elevated lymphocyte specific protein 1 expression is involved in the regulation of leukocyte migration and immunosuppressive microenvironment in glioblastoma. Aging 12, 1656–1684 (2020).
Ferguson, S. D., Srinivasan, V. M. & Heimberger, A. B. The role of STAT3 in tumor-mediated immune suppression. J. Neurooncol. 123, 385–394 (2015).
Piperi, C., Papavassiliou, K. A. & Papavassiliou, A. G. Pivotal role of STAT3 in shaping glioblastoma immune microenvironment. Cells 8, 1398 (2019).
Miska, J. et al. HIF-1alpha is a metabolic switch between glycolytic-driven migration and oxidative phosphorylation-driven immunosuppression of Tregs in glioblastoma. Cell Rep. 27, 226–237 e224 (2019).
Wainwright, D. A. et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin. Cancer Res. 18, 6110–6121 (2012).
Bagley, S. J. et al. CAR T-cell therapy for glioblastoma: recent clinical advances and future challenges. Neuro Oncol. 20, 1429–1438 (2018).
Stock, S., Schmitt, M. & Sellner, L. Optimizing manufacturing protocols of chimeric antigen receptor T cells for improved anticancer immunotherapy. Int. J. Mol. Sci. 20, 6223 (2019).
Maude, S. L. et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 378, 439–448 (2018).
Schuster, S. J. et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 380, 45–56 (2019).
Brown, C. E. et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res. 21, 4062–4072 (2015).
Brown, C. E. et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 375, 2561–2569 (2016).
Goff, S. L. et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J. Immunother. 42, 126–135 (2019).
Zhang, C. et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J. Natl Cancer Inst. 108, 375 (2016).
Ahmed, N. et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 3, 1094–1101 (2017).
Wu, S. Y., Fu, T., Jiang, Y. Z. & Shao, Z. M. Natural killer cells in cancer biology and therapy. Mol. Cancer 19, 120 (2020).
Lang, F. F. et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J. Clin. Oncol. 36, 1419–1427 (2018).
Martikainen, M. & Essand, M. Virus-based immunotherapy of glioblastoma. Cancers 11, 186 (2019).
Bartee, E. & Li, Z. In vivo and in situ programming of tumor immunity by combining oncolytics and PD-1 immune checkpoint blockade. Exp. Hematol. Oncol. 6, 15 (2017).
Zhu, S. et al. Combination strategies to maximize the benefits of cancer immunotherapy. J. Hematol. Oncol. 14, 156 (2021).
Farrera-Sal, M., Moya-Borrego, L., Bazan-Peregrino, M. & Alemany, R. Evolving status of clinical immunotherapy with oncolytic adenovirus. Clin. Cancer Res. 27, 2979–2988 (2021).
Zhu, Z. et al. Zika Virus Targets Glioblastoma Stem Cells through a SOX2-Integrin alphavbeta5 Axis. Cell Stem Cell 26, 187–204 e110 (2020).
Nair, S. et al. Zika virus oncolytic activity requires CD8+ T cells and is boosted by immune checkpoint blockade. JCI Insight 6, e144619 (2021).
Friedman, G. K. et al. Oncolytic HSV-1 G207 immunovirotherapy for pediatric high-grade gliomas. N. Engl. J. Med. 384, 1613–1622 (2021).
Todo, T. et al. A phase I/II study of triple-mutated oncolytic herpes virus G47∆ in patients with progressive glioblastoma. Nat. Commun. 13, 4119 (2022).
Todo, T. et al. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: a phase 2 trial. Nat. Med. 28, 1630–1639 (2022).
Desjardins, A. et al. Recurrent glioblastoma treated with recombinant poliovirus. N. Engl. J. Med. 379, 150–161 (2018).
Merriam, B. Back pain. Practitioner 233, 649–650 (1989).
Fueyo, J. et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 19, 2–12 (2000).
Mottini, C. et al. Computer-aided drug repurposing for cancer therapy: Approaches and opportunities to challenge anticancer targets. Semin. Cancer Biol. 68, 59–74 (2021).
Preusser, M., Brastianos, P. K. & Mawrin, C. Advances in meningioma genetics: novel therapeutic opportunities. Nat. Rev. Neurol. 14, 106–115 (2018).
Ostrom, Q. T. et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008-2012. Neuro Oncol. 17(Suppl 4), iv1–iv62 (2015).
Louis, D. N. et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 131, 803–820 (2016).
Riemenschneider, M. J., Perry, A. & Reifenberger, G. Histological classification and molecular genetics of meningiomas. Lancet Neurol. 5, 1045–1054 (2006).
Harter, P. N., Braun, Y. & Plate, K. H. Classification of meningiomas-advances and controversies. Chin. Clin. Oncol. 6, S2 (2017).
Gritsch, S., Batchelor, T. T. & Gonzalez Castro, L. N. Diagnostic, therapeutic, and prognostic implications of the 2021 World Health Organization classification of tumors of the central nervous system. Cancer 128, 47–58 (2022).
Gousias, K., Schramm, J. & Simon, M. The Simpson grading revisited: aggressive surgery and its place in modern meningioma management. J. Neurosurg. 125, 551–560 (2016).
Kaley, T. et al. Historical benchmarks for medical therapy trials in surgery- and radiation-refractory meningioma: a RANO review. Neuro Oncol. 16, 829–840 (2014).
Norden, A. D. et al. Phase II study of monthly pasireotide LAR (SOM230C) for recurrent or progressive meningioma. Neurology 84, 280–286 (2015).
Goldbrunner, R. et al. EANO guideline on the diagnosis and management of meningiomas. Neuro Oncol. 23, 1821–1834 (2021).
Sahm, F. et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol. 18, 682–694 (2017).
Clark, V. E. et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat. Genet. 48, 1253–1259 (2016).
Petrilli, A. M. & Fernandez-Valle, C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene 35, 537–548 (2016).
James, M. F. et al. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Mol. Cell Biol. 29, 4250–4261 (2009).
Youngblood, M. W. et al. Correlations between genomic subgroup and clinical features in a cohort of more than 3000 meningiomas. J. Neurosurg. 133, 1345–1354 (2019).
Weller, M. et al. Durable control of metastatic AKT1-mutant WHO grade 1 meningothelial meningioma by the AKT inhibitor, AZD5363. J. Natl Cancer Inst. 109, 1–4 (2017).
Youngblood, M. W. et al. Associations of meningioma molecular subgroup and tumor recurrence. Neuro Oncol. 23, 783–794 (2021).
Sahm, F. et al. TERT promoter mutations and risk of recurrence in meningioma. J. Natl Cancer Inst. 108, 377 (2016).
Nassiri, F. et al. DNA methylation profiling to predict recurrence risk in meningioma: development and validation of a nomogram to optimize clinical management. Neuro Oncol. 21, 901–910 (2019).
Olar, A. et al. Global epigenetic profiling identifies methylation subgroups associated with recurrence-free survival in meningioma. Acta Neuropathol. 133, 431–444 (2017).
Vaubel, R. A. et al. Meningiomas with rhabdoid features lacking other histologic features of malignancy: a study of 44 cases and review of the literature. J. Neuropathol. Exp. Neurol. 75, 44–52 (2016).
Prager, B. C. et al. The meningioma enhancer landscape delineates novel subgroups and drives druggable dependencies. Cancer Discov. 10, 1722–1741 (2020).
Paramasivam, N. et al. Mutational patterns and regulatory networks in epigenetic subgroups of meningioma. Acta Neuropathol. 138, 295–308 (2019).
Mawrin, C. & Perry, A. Pathological classification and molecular genetics of meningiomas. J. Neurooncol. 99, 379–391 (2010).
Zang, K. D. & Singer, H. Chromosomal consitution of meningiomas. Nature 216, 84–85 (1967).
Trofatter, J. A. et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 72, 791–800 (1993).
Lekanne Deprez, R. H. et al. Frequent NF2 gene transcript mutations in sporadic meningiomas and vestibular schwannomas. Am. J. Hum. Genet. 54, 1022–1029 (1994).
Ruttledge, M. H. et al. Evidence for the complete inactivation of the NF2 gene in the majority of sporadic meningiomas. Nat. Genet. 6, 180–184 (1994).
Zang, K. D. Meningioma: a cytogenetic model of a complex benign human tumor, including data on 394 karyotyped cases. Cytogenet. Cell Genet. 93, 207–220 (2001).
Kros, J. et al. NF2 status of meningiomas is associated with tumour localization and histology. J. Pathol. 194, 367–372 (2001).
Wellenreuther, R. et al. Analysis of the neurofibromatosis 2 gene reveals molecular variants of meningioma. Am. J. Pathol. 146, 827–832 (1995).
Lekanne Deprez, R. H. et al. Cytogenetic, molecular genetic and pathological analyses in 126 meningiomas. J. Neuropathol. Exp. Neurol. 54, 224–235 (1995).
Clark, V. E. et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 339, 1077–1080 (2013).
Brastianos, P. K. et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat. Genet. 45, 285–289 (2013).
Williams, S. R. et al. Genomic analysis of posterior fossa meningioma demonstrates frequent AKT1 E17K mutations in foramen magnum meningiomas. J. Neurol. Surg. B Skull Base 80, 562–567 (2019).
Williams, E. A. et al. Distinct genomic subclasses of high-grade/progressive meningiomas: NF2-associated, NF2-exclusive, and NF2-agnostic. Acta Neuropathol. Commun. 8, 171 (2020).
Abedalthagafi, M. et al. Oncogenic PI3K mutations are as common as AKT1 and SMO mutations in meningioma. Neuro Oncol. 18, 649–655 (2016).
Birzu, C., Peyre, M. & Sahm, F. Molecular alterations in meningioma: prognostic and therapeutic perspectives. Curr. Opin. Oncol. 32, 613–622 (2020).
Cordova, C. & Kurz, S. C. Advances in molecular classification and therapeutic opportunities in meningiomas. Curr. Oncol. Rep. 22, 84 (2020).
Reuss, D. E. et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta Neuropathol. 125, 351–358 (2013).
Nassiri, F. et al. Loss of H3K27me3 in meningiomas. Neuro Oncol. 23, 1282–1291 (2021).
Katz, L. M. et al. Loss of histone H3K27me3 identifies a subset of meningiomas with increased risk of recurrence. Acta Neuropathol. 135, 955–963 (2018).
Sievers, P. et al. CDKN2A/B homozygous deletion is associated with early recurrence in meningiomas. Acta Neuropathol. 140, 409–413 (2020).
Mirian, C. et al. Poor prognosis associated with TERT gene alterations in meningioma is independent of the WHO classification: an individual patient data meta-analysis. J. Neurol. Neurosurg. Psychiatry 91, 378–387 (2020).
Goutagny, S. et al. High incidence of activating TERT promoter mutations in meningiomas undergoing malignant progression. Brain Pathol. 24, 184–189 (2014).
Juratli, T. A. et al. DMD genomic deletions characterize a subset of progressive/higher-grade meningiomas with poor outcome. Acta Neuropathol. 136, 779–792 (2018).
Shankar, G. M. et al. Germline and somatic BAP1 mutations in high-grade rhabdoid meningiomas. Neuro Oncol. 19, 535–545 (2017).
Shankar, G. M. & Santagata, S. BAP1 mutations in high-grade meningioma: implications for patient care. Neuro Oncol. 19, 1447–1456 (2017).
Gupta, S., Bi, W. L. & Dunn, I. F. Medical management of meningioma in the era of precision medicine. Neurosurg. Focus 44, E3 (2018).
Gelerstein, E. et al. Regression of intracranial meningioma following treatment with nivolumab: case report and review of the literature. J. Clin. Neurosci. 37, 51–53 (2017).
Chamberlain, M. C. & Johnston, S. K. Hydroxyurea for recurrent surgery and radiation refractory meningioma: a retrospective case series. J. Neurooncol. 104, 765–771 (2011).
Grunberg, S. M. et al. Long-term administration of mifepristone (RU486): clinical tolerance during extended treatment of meningioma. Cancer Invest. 24, 727–733 (2006).
Ji, Y. et al. Double-blind phase III randomized trial of the antiprogestin agent mifepristone in the treatment of unresectable meningioma: SWOG S9005. J. Clin. Oncol. 33, 4093–4098 (2015).
Norden, A. D. et al. Phase II trials of erlotinib or gefitinib in patients with recurrent meningioma. J. Neurooncol. 96, 211–217 (2010).
Nayak, L. et al. Atypical and anaplastic meningiomas treated with bevacizumab. J. Neurooncol. 109, 187–193 (2012).
Dasanu, C. A., Alvarez-Argote, J., Limonadi, F. M. & Codreanu, I. Bevacizumab in refractory higher-grade and atypical meningioma: the current state of affairs. Expert Opin. Biol. Ther. 19, 99–104 (2019).
Scerrati, A. et al. The controversial role of Bevacizumab in the treatment of patients with intracranial meningioma: a comprehensive literature review. Expert Rev. Anticancer Ther. 20, 197–203 (2020).
Bi, W. L. et al. Activity of PD-1 blockade with nivolumab among patients with recurrent atypical/anaplastic meningioma: phase II trial results. Neuro Oncol. 24, 101–113 (2022).
Haase, D. et al. Fatty acid synthase as a novel target for meningioma therapy. Neuro Oncol. 12, 844–854 (2010).
Song, L. R. et al. MicroRNA-195 functions as a tumor suppressor by directly targeting fatty acid synthase in malignant meningioma. World Neurosurg. 136, e355–e364 (2020).
Nowak-Choi, K. et al. Resected WHO grade I meningioma and predictors of local control. J. Neurooncol. 152, 145–151 (2021).
Paldor, I. et al. Review of controversies in management of non-benign meningioma. J. Clin. Neurosci. 31, 37–46 (2016).
Huntoon, K., Toland, A. M. S. & Dahiya, S. Meningioma: a review of clinicopathological and molecular aspects. Front Oncol. 10, 579599 (2020).
Slot, K. M. et al. Agreement between extent of meningioma resection based on surgical simpson grade and based on postoperative magnetic resonance imaging findings. World Neurosurg. 111, e856–e862 (2018).
Przybylowski, C. J. et al. Prognostic value of the Simpson grading scale in modern meningioma surgery: Barrow Neurological Institute experience. J. Neurosurg. 135, 515–523 (2020).
Biau, J., Khalil, T., Verrelle, P. & Lemaire, J. J. Fractionated radiotherapy and radiosurgery of intracranial meningiomas. Neurochirurgie 64, 29–36 (2018).
Simpson, D. The recurrence of intracranial meningiomas after surgical treatment. J. Neurol. Neurosurg. Psychiatry 20, 22–39 (1957).
Goldbrunner, R. et al. EANO guidelines for the diagnosis and treatment of meningiomas. Lancet Oncol. 17, e383–e391 (2016).
Klekamp, J. & Samii, M. Surgical results for spinal meningiomas. Surg. Neurol. 52, 552–562 (1999).
Tsuda, K. et al. Is Simpson grade I removal necessary in all cases of spinal meningioma? Assessment of postoperative recurrence during long-term follow-up. Neurol. Med Chir. 54, 907–913 (2014).
Ragel, B. T. & Jensen, R. L. Aberrant signaling pathways in meningiomas. J. Neurooncol. 99, 315–324 (2010).
Kaley, T. J. et al. Phase II trial of sunitinib for recurrent and progressive atypical and anaplastic meningioma. Neuro Oncol. 17, 116–121 (2015).
Graillon, T. et al. Everolimus and octreotide for patients with recurrent meningioma: results from the phase II CEVOREM trial. Clin. Cancer Res. 26, 552–557 (2020).
Leavitt, E., Lask, G. & Martin, S. Sonic Hedgehog pathway inhibition in the treatment of advanced basal cell carcinoma. Curr. Treat. Options Oncol. 20, 84 (2019).
Shapiro, I. M. et al. Merlin deficiency predicts FAK inhibitor sensitivity: a synthetic lethal relationship. Sci. Transl. Med. 6, 237ra268 (2014).
Gittleman, H. et al. Descriptive epidemiology of germ cell tumors of the central nervous system diagnosed in the United States from 2006 to 2015. J. Neurooncol. 143, 251–260 (2019).
Kurucu, N. et al. Primary intracranial germ cell tumors in children 36-year experience of a single center. J. Cancer Res. Therap. 16, 1459–1465 (2020).
Udaka, Y. T. & Packer, R. J. Pediatric brain tumors. Neurol. Clin. 36, 533–556 (2018).
Fukushima, S. et al. Mutually exclusive mutations of KIT and RAS are associated with KIT mRNA expression and chromosomal instability in primary intracranial pure germinomas. Acta Neuropathol. 127, 911–925 (2014).
Schulte, S. L. et al. CNS germinomas are characterized by global demethylation, chromosomal instability and mutational activation of the Kit-, Ras/Raf/Erk- and Akt-pathways. Oncotarget 7, 55026–55042 (2016).
Ichimura, K. et al. Recurrent neomorphic mutations of MTOR in central nervous system and testicular germ cell tumors may be targeted for therapy. Acta Neuropathol. 131, 889–901 (2016).
Alapetite, C. et al. Pattern of relapse and outcome of non-metastatic germinoma patients treated with chemotherapy and limited field radiation: the SFOP experience. Neuro Oncol. 12, 1318–1325 (2010).
Kellie, S. J. et al. Intensive cisplatin and cyclophosphamide-based chemotherapy without radiotherapy for intracranial germinomas: failure of a primary chemotherapy approach. Pediatr. Blood Cancer 43, 126–133 (2004).
Bhimani, A. D. et al. Pituitary germinomas: a multi-institutional study analyzing patient demographics and management patterns. Pituitary 23, 381–388 (2020).
Park, J. et al. Differential dosimetric benefit of proton beam therapy over intensity modulated radiotherapy for a variety of targets in patients with intracranial germ cell tumors. Radiat. Oncol. 10, 135 (2015).
Chen, Y. W. et al. Treatment strategies for initially disseminated intracranial germinomas: experiences at a single institute. Child’s Nerv. Syst. 28, 557–563 (2012).
Hu, Y. W. et al. Salvage treatment for recurrent intracranial germinoma after reduced-volume radiotherapy: a single-institution experience and review of the literature. Int. J. Radiat. Oncol. Biol. Phys. 84, 639–647 (2012).
Takami, H. Advances in molecular profiling and developing clinical trials of CNS germ cell tumors: present and future directions. Curr. Oncol. Rep. 24, 105–112 (2022).
Calaminus, G. et al. Impact of surgery, chemotherapy and irradiation on long term outcome of intracranial malignant non-germinomatous germ cell tumors: results of the German Cooperative Trial MAKEI 89. Klin. Padiatrie 216, 141–149 (2004).
Frappaz, D. et al. EANO, SNO and Euracan consensus review on the current management and future development of intracranial germ cell tumors in adolescents and young adults. Neuro Oncol. 24, 516–527 (2022).
Calaminus, G. et al. Outcome of patients with intracranial non-germinomatous germ cell tumors-lessons from the SIOP-CNS-GCT-96 trial. Neuro Oncol. 19, 1661–1672 (2017).
Ostrom, Q. T. et al. CBTRUS Statistical Report: primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro Oncol. Supplement_1 (2020).
Villano, J. L. et al. Age, gender, and racial differences in incidence and survival in primary CNS lymphoma. Br. J. Cancer 105, 1414–1418 (2011).
Magnes, T. et al. Clonal evolution in diffuse large B-cell lymphoma with central nervous system recurrence. ESMO Open 6, 100012 (2021).
Batchelor & T., T. Primary central nervous system lymphoma. Hematol. Am Soc Hematol Educ Program. 2016, 379–385 (2016).
Ponzoni, M., Issa, S., Batchelor, T. T. & Rubenstein, J. L. Beyond high-dose methotrexate and brain radiotherapy: novel targets and agents for primary CNS lymphoma. Ann. Oncol. 25, 316–322 (2013).
Mccann, K. J. et al. Primary central nervous system lymphoma: tumor-related clones exist in the blood and bone marrow with evidence for separate development. Blood 113, 4677 (2009).
Casamayor-Palleja‘, M. et al. Expression of macrophage inflammatory protein-3alpha, stromal cell-derived factor-1, and B-cell-attracting chemokine-1 identifies the tonsil crypt as an attractive site for B cells. Blood 97, 3992–3994 (2001).
Rubenstein, J. L. et al. Gene expression and angiotropism in primary CNS lymphoma. Blood 107, 3716–3723 (2006).
Montesinos-Rongen, M. et al. Activating L265P mutations of the MYD88 gene are common in primary central nervous system lymphoma. Acta Neuropathol. 122, 791–792 (2011).
Ngo, V. N. et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 470, 115–119 (2011).
Davis, R. E. et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463, 88–92 (2010).
You, H., Wei, L. & Kaminska, B. Emerging insights into origin and pathobiology of primary central nervous system lymphoma. Cancer Lett. 509, 121–129 (2021).
Mizowaki, T. et al. STAT3 activation is associated with cerebrospinal fluid interleukin-10 (IL-10) in primary central nervous system diffuse large B cell lymphoma. J. Neurooncol. 124, 165–174 (2015).
Gonzalez-Aguilar, A. et al. Recurrent Mutations of MYD88 and TBL1XR1 in primary central nervous system lymphomas. Clin. Cancer Res. 18, 5203 (2012).
Calimeri, T. et al. How we treat primary central nervous system lymphoma. ESMO Open. 6, 100213 (2021).
Batchelor & T. Primary CNS lymphoma. J. Clin. Oncol. 24, 1281–1288 (2006).
Grommes, C. et al. Comprehensive approach to diagnosis and treatment of newly diagnosed primary CNS lymphoma. Neuro Oncol. 21, 296–305 (2018).
Omuro, A. et al. R-MPV followed by high-dose chemotherapy with TBC and autologous stem-cell transplant for newly diagnosed primary CNS lymphoma. Blood. 125, 1403–1410 (2015).
Cher, L., Glass, J., Harsh, G. R. & Hochberg, F. H. Therapy of primary CNS lymphoma with methotrexate-based chemotherapy and deferred radiotherapy: preliminary results. Neurology 46, 1757–1759 (1996).
Frigault, M. J. et al. Tisagenlecleucel CAR T-cell therapy in secondary CNS lymphoma. Blood 134, blood.2019001694 (2019).
Weller, M. et al. Surgery for primary CNS lymphoma? Challenging a paradigm. Neuro Oncol. 14, 1481–1484 (2012).
Ferreri, A. J. et al. Chemoimmunotherapy with methotrexate, cytarabine, thiotepa, and rituximab (MATRix regimen) in patients with primary CNS lymphoma: results of the first randomisation of the International Extranodal Lymphoma Study Group-32 (IELSG32) phase 2 trial. Lancet Haematol. 3, e217–e227 (2016).
Bromberg, J. E. C. et al. Rituximab in patients with primary CNS lymphoma (HOVON 105/ALLG NHL 24): a randomised, open-label, phase 3 intergroup study. Lancet Oncol. 20, 216–228 (2019).
Rubenstein, J. L. et al. Multicenter phase 1 trial of intraventricular immunochemotherapy in recurrent CNS lymphoma. Blood 121, 745–751 (2013).
Dal Porto, J. M. et al. B cell antigen receptor signaling 101. Mol. Immunol. 41, 599–613 (2004).
Grommes, C. et al. Ibrutinib Unmasks Critical Role of Bruton Tyrosine Kinase in Primary CNS Lymphoma. Cancer Discov. 7, 1018–1029 (2017).
Grommes, C. et al. Phase 1b trial of an ibrutinib-based combination therapy in recurrent/refractory CNS lymphoma. Blood 133, 436–445 (2019).
Narita, Y. et al. Phase I/II study of tirabrutinib, a second-generation Bruton’s tyrosine kinase inhibitor, in relapsed/refractory primary central nervous system lymphoma. Neuro Oncol. 23, 122–133 (2021).
Rubenstein, J. L. et al. Phase 1 investigation of lenalidomide/rituximab plus outcomes of lenalidomide maintenance in relapsed CNS lymphoma. Blood Adv. 2, 1595–1607 (2018).
Ghesquieres, H. et al. Lenalidomide in combination with intravenous rituximab (REVRI) in relapsed/refractory primary CNS lymphoma or primary intraocular lymphoma: a multicenter prospective ‘proof of concept’ phase II study of the French Oculo-Cerebral lymphoma (LOC) Network and the Lymphoma Study Association (LYSA)†. Ann. Oncol. 30, 621–628 (2019).
Chapuy, B. et al. Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood 127, 869–881 (2016).
Nayak, L. et al. PD-1 blockade with nivolumab in relapsed/refractory primary central nervous system and testicular lymphoma. Blood 129, 3071–3073 (2017).
Turtle, C. J. et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci. Transl. Med. 8, 355ra116 (2016).
Abramson, J. S. et al. Anti-CD19 CAR T cells in CNS diffuse large-B-cell lymphoma. N. Engl. J. Med. 377, 783–784 (2017).
Youssef, G. & Miller, J. J. Lower grade gliomas. Curr. Neurol. Neurosci. Rep. 20, 21 (2020).
Wickstrom, M. et al. Wnt/beta-catenin pathway regulates MGMT gene expression in cancer and inhibition of Wnt signalling prevents chemoresistance. Nat. Commun. 6, 8904 (2015).
Butler, M. et al. MGMT status as a clinical biomarker in glioblastoma. Trends Cancer 6, 380–391 (2020).
Hegi, M. E. et al. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J. Clin. Oncol. 26, 4189–4199 (2008).
Chen, R. et al. Molecular features assisting in diagnosis, surgery, and treatment decision making in low-grade gliomas. Neurosurg. Focus 38, E2 (2015).
Watanabe, T. et al. Phenotype versus genotype correlation in oligodendrogliomas and low-grade diffuse astrocytomas. Acta Neuropathol. 103, 267–275 (2002).
Aldape, K., Burger, P. C. & Perry, A. Clinicopathologic aspects of 1p/19q loss and the diagnosis of oligodendroglioma. Arch. Pathol. Lab. Med. 131, 242–251 (2007).
Ruda, R. et al. Efficacy of initial temozolomide for high-risk low grade gliomas in a phase II AINO (Italian Association for Neuro-Oncology) study: a post-hoc analysis within molecular subgroups of WHO 2016. J. Neurooncol. 145, 115–123 (2019).
Tanaka, K. et al. Oncogenic EGFR signaling activates an mTORC2-NF-kappaB pathway that promotes chemotherapy resistance. Cancer Discov. 1, 524–538 (2011).
Al-Nedawi, K. et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 10, 619–624 (2008).
Cancer Genome Atlas Research, N. et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N. Engl. J. Med. 372, 2481–2498 (2015).
Zheng, S. et al. Prospective clinical sequencing of adult glioma. Mol. Cancer Ther. 18, 991–1000 (2019).
Yalon, M. et al. A feasibility and efficacy study of rapamycin and erlotinib for recurrent pediatric low-grade glioma (LGG). Pediatr. Blood Cancer 60, 71–76 (2013).
Brennan, C. et al. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS ONE 4, e7752 (2009).
Filbin, M. G. et al. Coordinate activation of Shh and PI3K signaling in PTEN-deficient glioblastoma: new therapeutic opportunities. Nat. Med. 19, 1518–1523 (2013).
Ermoian, R. P. et al. Dysregulation of PTEN and protein kinase B is associated with glioma histology and patient survival. Clin. Cancer Res. 8, 1100–1106 (2002).
Kita, D., Yonekawa, Y., Weller, M. & Ohgaki, H. PIK3CA alterations in primary (de novo) and secondary glioblastomas. Acta Neuropathol. 113, 295–302 (2007).
Kaley, T. J. et al. Phase I clinical trial of temsirolimus and perifosine for recurrent glioblastoma. Ann. Clin. Transl. Neurol. 7, 429–436 (2020).
Wakimoto, H. et al. Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin. Cancer Res. 20, 2898–2909 (2014).
Parsons, D. W. et al. An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 (2008).
Reuss, D. E. et al. ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an “integrated” diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol. 129, 133–146 (2015).
Killela, P. J. et al. Mutations in IDH1, IDH2, and in the TERT promoter define clinically distinct subgroups of adult malignant gliomas. Oncotarget 5, 1515–1525 (2014).
Koelsche, C. et al. Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol. 126, 907–915 (2013).
Li, Y. et al. Non-canonical NF-kappaB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat. Cell Biol. 17, 1327–1338 (2015).
Asby, D. J. et al. Combined use of CDK4/6 and mTOR inhibitors induce synergistic growth arrest of diffuse intrinsic pontine glioma cells via mutual downregulation of mTORC1 activity. Cancer Manag. Res. 10, 3483–3500 (2018).
Hoeman, C., Shen, C. & Becher, O. J. CDK4/6 and PDGFRA signaling as therapeutic targets in diffuse intrinsic pontine glioma. Front. Oncol. 8, 191 (2018).
Liu, S. et al. Inhibition of Rb and mTOR signaling associates with synergistic anticancer effect of palbociclib and erlotinib in glioblastoma cells. Invest. N. Drugs 36, 961–969 (2018).
Korshunov, A. et al. Epithelioid glioblastomas stratify into established diagnostic subsets upon integrated molecular analysis. Brain Pathol. 28, 656–662 (2018).
Schindler, G. et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 121, 397–405 (2011).
Banerjee, A. et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro Oncol. 19, 1135–1144 (2017).
Hargrave, D. R. et al. Efficacy and safety of dabrafenib in pediatric patients with BRAF V600 mutation-positive relapsed or refractory low-grade glioma: results from a phase I/IIa study. Clin. Cancer Res. 25, 7303–7311 (2019).
von Deimling, A., Korshunov, A. & Hartmann, C. The next generation of glioma biomarkers: MGMT methylation, BRAF fusions and IDH1 mutations. Brain Pathol. 21, 74–87 (2011).
Wick, W. et al. N2M2 (NOA-20) phase I/II trial of molecularly matched targeted therapies plus radiotherapy in patients with newly diagnosed non-MGMT hypermethylated glioblastoma. Neuro Oncol. 21, 95–105 (2019).
Miles, X., Vandevoorde, C., Hunter, A. & Bolcaen, J. MDM2/X inhibitors as radiosensitizers for glioblastoma targeted therapy. Front. Oncol. 11, 703442 (2021).
Gluck, W. L. et al. Phase 1 study of the MDM2 inhibitor AMG 232 in patients with advanced P53 wild-type solid tumors or multiple myeloma. Invest. N. Drugs 38, 831–843 (2020).
Pierscianek, D. et al. MET gain in diffuse astrocytomas is associated with poorer outcome. Brain Pathol. 23, 13–18 (2013).
Zarghooni, M. et al. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J. Clin. Oncol. 28, 1337–1344 (2010).
Koschmann, C. et al. Characterizing and targeting PDGFRA alterations in pediatric high-grade glioma. Oncotarget 7, 65696–65706 (2016).
Verhaak, R. G. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010).
Lapin, D. H., Tsoli, M. & Ziegler, D. S. Genomic insights into diffuse intrinsic pontine glioma. Front. Oncol. 7, 57 (2017).
Di Stefano, A. L. et al. Detection, characterization, and inhibition of FGFR-TACC fusions in IDH wild-type glioma. Clin. Cancer Res. 21, 3307–3317 (2015).
Tabernero, J. et al. Phase I dose-escalation study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 33, 3401–3408 (2015).
Sharma, M. et al. Phase II study of Dovitinib in recurrent glioblastoma. J. Neurooncol. 144, 359–368 (2019).
Ohashi, R., Matsuda, Y., Ishiwata, T. & Naito, Z. Downregulation of fibroblast growth factor receptor 2 and its isoforms correlates with a high proliferation rate and poor prognosis in high-grade glioma. Oncol. Rep. 32, 1163–1169 (2014).
Torre, M. et al. Molecular and clinicopathologic features of gliomas harboring NTRK fusions. Acta Neuropathol. Commun. 8, 107 (2020).
Okamura, R. et al. Analysis of NTRK alterations in pan-cancer adult and pediatric malignancies: implications for NTRK-targeted therapeutics. JCO Precis. Oncol. 1–20 (2018).
Wu, G. et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 46, 444–450 (2014).
Solomon, J. P. et al. NTRK fusion detection across multiple assays and 33,997 cases: diagnostic implications and pitfalls. Mod. Pathol. 33, 38–46 (2020).
Shepherd, D. J. et al. Mosaicism for receptor tyrosine kinase activation in a glioblastoma involving both PDGFRA amplification and NTRK2 fusion. Oncologist 26, 919–924 (2021).
Alharbi, M. et al. Regression of ETV6-NTRK3 infantile glioblastoma after first-line treatment with larotrectinib. JCO Precis. Oncol. 4, 796–800 (2020).
Castel, D. et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 130, 815–827 (2015).
Hu, X. M. et al. H3K27M Mutation Doesn’t Mean Worse Prognosis in Old Patients. Front. Oncol. 12, 912166 (2022).
Schwartzentruber, J. et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 (2012).
Venneti, S. et al. Histone 3 lysine 9 trimethylation is differentially associated with isocitrate dehydrogenase mutations in oligodendrogliomas and high-grade astrocytomas. J. Neuropathol. Exp. Neurol. 72, 298–306 (2013).
Li, J. et al. Notch1 is an independent prognostic factor for patients with glioma. J. Surg. Oncol. 103, 813–817 (2011).
Fan, X. et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 28, 5–16 (2010).
Gilbert, C. A., Daou, M. C., Moser, R. P. & Ross, A. H. Gamma-secretase inhibitors enhance temozolomide treatment of human gliomas by inhibiting neurosphere repopulation and xenograft recurrence. Cancer Res. 70, 6870–6879 (2010).
Hai, L. et al. Notch1 is a prognostic factor that is distinctly activated in the classical and proneural subtype of glioblastoma and that promotes glioma cell survival via the NF-kappaB(p65) pathway. Cell Death Dis. 9, 158 (2018).
Zhang, X. et al. Notch1 promotes glioma cell migration and invasion by stimulating beta-catenin and NF-kappaB signaling via AKT activation. Cancer Sci. 103, 181–190 (2012).
Bai, M. et al. Dissecting and analyzing the subclonal mutations associated with poor prognosis in diffuse glioma. Biomed. Res. Int. 2022, 4919111 (2022).
Wang, D., Liu, S. & Wang, G. Establishment of an endocytosis-related prognostic signature for patients with low-grade glioma. Front. Genet. 12, 709666 (2021).
Ullrich, N. J. et al. A phase II study of continuous oral mTOR inhibitor everolimus for recurrent, radiographic-progressive neurofibromatosis type 1-associated pediatric low-grade glioma: a Neurofibromatosis Clinical Trials Consortium study. Neuro Oncol. 22, 1527–1535 (2020).
Wahl, M. et al. Probing the phosphatidylinositol 3-kinase/mammalian target of rapamycin pathway in gliomas: A phase 2 study of everolimus for recurrent adult low-grade gliomas. Cancer 123, 4631–4639 (2017).
Wiestler, B. et al. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol. 126, 443–451 (2013).
Haase, S. et al. Mutant ATRX: uncovering a new therapeutic target for glioma. Expert Opin. Ther. Targets 22, 599–613 (2018).
Nagaishi, M. et al. Alpha-internexin and altered CIC expression as a supportive diagnostic marker for oligodendroglial tumors with the 1p/19q co-deletion. Brain Tumor Pathol. 31, 257–264 (2014).
Baumgarten, P. et al. Loss of FUBP1 expression in gliomas predicts FUBP1 mutation and is associated with oligodendroglial differentiation, IDH1 mutation and 1p/19q loss of heterozygosity. Neuropathol. Appl. Neurobiol. 40, 205–216 (2014).
Chamberlain, M. C. Hydroxyurea for recurrent surgery and radiation refractory high-grade meningioma. J. Neurooncol. 107, 315–321 (2012).
Chamberlain, M. C. & Glantz, M. J. Interferon-alpha for recurrent World Health Organization grade 1 intracranial meningiomas. Cancer 113, 2146–2151 (2008).
Chamberlain, M. C. IFN-alpha for recurrent surgery- and radiation-refractory high-grade meningioma: a retrospective case series. CNS Oncol. 2, 227–235 (2013).
Lou, E. et al. Bevacizumab therapy for adults with recurrent/progressive meningioma: a retrospective series. J. Neurooncol. 109, 63–70 (2012).
Simo, M. et al. Recurrent high-grade meningioma: a phase II trial with somatostatin analogue therapy. Cancer Chemother. Pharm. 73, 919–923 (2014).
Chamberlain, M. C., Glantz, M. J. & Fadul, C. E. Recurrent meningioma: salvage therapy with long-acting somatostatin analogue. Neurology 69, 969–973 (2007).
Chamberlain, M. C., Tsao-Wei, D. D. & Groshen, S. Temozolomide for treatment-resistant recurrent meningioma. Neurology 62, 1210–1212 (2004).
Preusser, M. et al. Trabectedin for recurrent WHO grade 2 or 3 meningioma: a randomized phase II study of the EORTC Brain Tumor Group (EORTC-1320-BTG). Neuro Oncol. 24, 755–767 (2022).
Shih, K. C. et al. A phase II trial of bevacizumab and everolimus as treatment for patients with refractory, progressive intracranial meningioma. J. Neurooncol. 129, 281–288 (2016).
Acknowledgements
This study was sponsored by The National Natural Science Foundation of China, grant number 81972338. This work was also supported by the National Science and Technology Major Project of China (No.2016ZX09101017) and Clinical Major Specialty Projects of Beijing. This research was also supported by Beijing Advanced Innovation Center for Big Data-based Precision Medicine (No.2021YFF0901404).
Author information
Authors and Affiliations
Contributions
W.B.L. and S.L.L. designed and wrote the manuscript. C.W., W.C.B.Z., Z.K., Y.Z., J.Y.Y., and R.Z. did a literature search and wrote the paper, and drafted figures. J.Y.C. and Y.J.L. revised the paper. All authors listed have made a substantial contribution to the work. All authors listed have made a substantial contribution to this work. All authors have read and approved the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests. Wenbin Li is a member of the editorial board of Signal Transduction and Targeted Therapy, but he does not participate in the paper processing.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, S., Wang, C., Chen, J. et al. Signaling pathways in brain tumors and therapeutic interventions. Sig Transduct Target Ther 8, 8 (2023). https://doi.org/10.1038/s41392-022-01260-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-01260-z
- Springer Nature Limited
This article is cited by
-
Prognostic values and immune infiltration of KLF15, AQP7, AGPAT9 in glioma and glioblastoma
Future Journal of Pharmaceutical Sciences (2024)
-
Design and experimental validation of a metamaterial-based sensor for microwave imaging in breast, lung, and brain cancer detection
Scientific Reports (2024)
-
Gliomas: a reflection of temporal gliogenic principles
Communications Biology (2024)
-
A viral attack on brain tumors: the potential of oncolytic virus therapy
Journal of NeuroVirology (2024)