
Abstract
Mutation/loss of alpha-thalassemia/mental retardation syndrome X-linked (ATRX) expression has been described in anaplastic gliomas. The present study explored the role of ATRX status in the molecular classification of anaplastic gliomas and its impact on survival in the biomarker cohort of the NOA-04 anaplastic glioma trial. Patients (n = 133) of the NOA-04 trial were analyzed for ATRX expression using immunohistochemistry. ATRX status was correlated with age, histology, isocitrate dehydrogenase (IDH), 1p/19q, alternative lengthening of telomeres (ALT) and O6-methylguanine-DNA methyltransferase (MGMT) status, and the trial efficacy endpoints. Loss of ATRX expression was detected in 45 % of anaplastic astrocytomas (AA), 27 % of anaplastic oligoastrocytomas (AOA) and 10 % of anaplastic oligodendrogliomas (AO). It was mostly restricted to IDH mutant tumors and almost mutually exclusive with 1p/19q co-deletion. The ALT phenotype was significantly correlated with ATRX loss. ATRX and 1p/19q status were used to re-classify AOA: AOA harboring ATRX loss shared a similar clinical course with AA, whereas AOA carrying 1p/19q co-deletion shared a similar course with AO. Accordingly, in a Cox regression model including ATRX and 1p/19q status, histology was no longer significantly associated with time to treatment failure. Survival analysis showed a marked separation of IDH mutant astrocytic tumors into two groups based on ATRX status: tumors with ATRX loss had a significantly better prognosis (median time to treatment failure 55.6 vs. 31.8 months, p = 0.0168, log rank test). ATRX status helps better define the clinically and morphologically mixed group of AOA, since ATRX loss is a hallmark of astrocytic tumors. Furthermore, ATRX loss defines a subgroup of astrocytic tumors with a favorable prognosis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The WHO classification, based solely on morphological criteria [16], may be increasingly supplemented with defined molecular aberrations [5, 19, 22, 24]. These might help resolve the discrepancy between classification and clinical outcome. A prototypical example for this discrepancy is anaplastic oligoastrocytomas (AOA), which are histologically and molecularly mixed gliomas, displaying both astrocytic and oligodendroglial features [17, 18]. As a result, the diagnosis of oligoastrocytomas is subject to high interobserver variation [12].
The prognostic or predictive impact of mutations of the isocitrate dehydrogenase 1 and 2 genes (IDH1/2), hypermethylation of the O6-methylguanine-DNA methyltransferase (MGMT) promoter and co-deletion of 1p and 19q have been extensively characterized [26]. Recently, mutations and (consequently) loss of expression of alpha-thalassemia/mental retardation syndrome X-linked (ATRX) have been reported in one-third of pediatric glioblastomas [20] and 7 % of adult glioblastomas [7]. Virtually all mutations are inactivating and lead to a loss of protein expression [7, 15]. Mechanistically, loss of ATRX and death-domain associated protein (DAXX) are important factors for a telomere maintenance mechanism not involving telomerases [alternative lengthening of telomeres (ALT)]. A number of studies have analyzed the frequency of ATRX mutation/loss of expression and its correlation with molecular markers in glioma: in pediatric diffuse intrinsic pons gliomas, ATRX mutation occurred in 2 of 22 cases [10]. High-throughput resequencing revealed ATRX mutations to be present in 12 of 32 (37.5 %) grade II and III gliomas examined. In this cohort, ~70 % of IDH mutant, 1p/19q intact tumors carried ATRX mutations [9]. ATRX loss assessed by immunohistochemistry has been reported as 27 % in grade II and 41 % in grade III astrocytomas in adults, compared to a mutation rate of 33 and 46 %, respectively [15]. Another recent study described a significantly higher mutation rate of 73 % in 44 anaplastic astrocytomas [8]. Notably, these studies showed that loss of ATRX expression is more prevalent in astrocytic than in mixed glial tumors and rare in pure oligodendrogliomas. Loss of ATRX expression is highly associated with IDH mutation and almost mutually exclusive with 1p/19q co-deletion, the hallmark of oligodendroglial tumors.
The prognostic value of ATRX status in different tumor entities has remained controversial; in pancreatic neuroendocrine tumors, altered ATRX expression has been associated with a more aggressive phenotype [25]. In childhood acute myeloid leukemia carrying FLT3 mutations, higher ATRX expression was associated with a superior outcome [13]. In gliomas, ATRX mutation has been associated with a better prognosis in a retrospective cohort of grade II, III and IV tumors [8].
The Neurooncology Working Group of the German Cancer Society (NOA)-04 trial explored the optimal sequence of radiotherapy and alkylating chemotherapy in patients with newly diagnosed, centrally confirmed anaplastic gliomas. NOA-04 revealed initial radiotherapy or chemotherapy to be comparable and IDH mutations as a novel positive prognostic factor in anaplastic gliomas [27]. In the NOA-04 trial cohort, we sought to determine the prognostic value of ATRX status as well as its implications for molecular classification of anaplastic gliomas, especially mixed oligoastrocytomas.
Methods
Patients, evaluations and ethics
The NOA-04 trial (NCT00717210) compared the efficacy and safety of initial radiotherapy, followed by chemotherapy (temozolomide or procarbazine, lomustine and vincristine) at progression or occurrence of unacceptable toxicity with the inverse sequence in patients with newly diagnosed anaplastic gliomas. In this trial, both sequences achieved similar results. Median follow-up time was 54 months [27]. All patients consented to exploratory molecular analyses performed with study data and materials. The original phase III trial was approved by the Ethics Committee (EC) at the University of Tuebingen, Germany, and subsequently all local EC. NOA-04 enrolled patients after written informed consent including future molecular analyses at 39 German sites.
Molecular analyses
IDH1/2 mutations, MGMT promoter methylation and 1p/19q co-deletion were determined as described in the initial trial report [27].
ATRX immunohistochemistry
ATRX immunohistochemistry of polyclonal rabbit antibody (dilution 1:400, product code HPA001906, Sigma-Aldrich, St. Louis, MO, USA) was performed using an automated immunostainer (Benchmark Ultra, Ventana, Tucson, AZ, USA) and standard protocols including pretreatment using Cell Conditioning 1 buffer (Ventana) for 52 min and standard Ventana signal amplification. Evaluation was performed by two observers (BW and DC) simultaneously on a multi-headed microscope and scoring was done in consensus. Only nuclear staining was considered for evaluation. Cases with more than 10 % positive tumor cells were scored positive. Endothelial cells, cortical neurons and infiltrating inflammatory cells were generally positive and served as internal positive controls. Cases with negative tumors cells in which vessel cells and neurons were not stained were not evaluated and not considered for further statistical evaluation (n = 13 cases). In cases with inhomogeneous immunoreaction, areas with highest staining were scored.
ALT fluorescence in situ hybridization
Telomere-specific fluorescence in situ hybridization (FISH) was done using a standard formalin-fixed paraffin-embedded FISH protocol [20], using a FITC peptide nucleic acid telomere probe from Dako. Large, ultra-bright telomere DNA aggregates/clouds are unique to ALT-positive cells and are significantly larger and brighter than the FISH signals emitted from individual telomeres in the same cell population and control CNS tissue samples.
Statistics
The co-occurrence of ATRX loss and reference histology, ALT, IDH1/2 and p53 mutations, MGMT promoter methylation and 1p/19q co-deletion was assessed using Fisher’s exact test. One-way ANOVA was employed to compare mean age between different groups followed by a pairwise t test in comparison with Bonferroni correction. Wilcoxon rank sum test was used for comparing ATRX expression data between ATRX mutated and wild-type groups. Univariate survival analysis was performed using the Kaplan–Meier estimator and log rank test. Multivariate analysis used a Cox proportional hazards model. p < 0.05 was considered statistically significant. All tests were two-sided. Analyses were carried out using Stata IC version 12.1 (StataCorp LP, TX, USA).
Results
ATRX loss occurs predominantly in astrocytic tumors with IDH mutation and is almost mutually exclusive with 1p/19q co-deletion
The present NOA-04 biomarker cohort comprised 133 of the 274 patients of the NOA-04 intention-to-treat (ITT) population for which unstained paraffin slides were available. Baseline characteristics of the biomarker cohort and the ITT population were similar (Table 1).
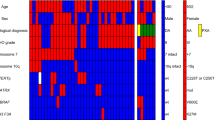
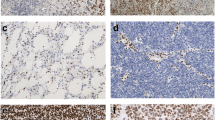
ATRX immunohistochemistry was positive in 89 of 133 cases with a strong nuclear immune reaction in most cases (Fig. 1a). Loss of ATRX was evident in 44 of the 133 cases with the bulk of tumor cells negative for ATRX, whereas endothelia and infiltrating inflammatory cells as well as residual neurons retained ATRX expression (Fig. 1b). In 13 cases, no immunoreaction was observed in the entire tissue, these cases were not scored and consequently not considered for statistical evaluation. As reported earlier, the frequency of ATRX loss was higher in anaplastic astrocytomas (AA, 29/65, 45 %) than in anaplastic oligoastrocytomas (AOA, 13/48, 27 %) and low in anaplastic oligodendrogliomas (AO, 2/20, 10 %, p = 0.009, Fisher’s exact test). ATRX loss occurred almost exclusively in tumors harboring IDH mutations (42/98 IDH mutated tumors, 43 %). Only two patients who were IDH wild type had ATRX loss. Patients with ATRX loss and IDH mutation were younger (mean age 35.7 years, n = 42) than patients with ATRX expression and IDH mutation (mean age 46.8 years, n = 56, p < 0.0001, one-way ANOVA) or IDH wild-type patients with ATRX expression (mean age 54.1 years, n = 30, p < 0.0001, one-way ANOVA).

IHC staining for ATRX. IHC staining of one tumor sample with retained ATRX expression (a) and another tumor sample with ATRX loss (b). Note the ATRX-positive endothelial cells in b
ATRX loss and 1p/19q co-deletion were almost mutually exclusive. Only two AA and one AOA showed both ATRX loss and 1p/19q co-deletion. The two AO with ATRX loss did not have a 1p/19q co-deletion. No association between MGMT promoter methylation and ATRX loss was observed (p = 0.445, Fisher’s exact test; Table 2).
We assessed the association of ATRX status and the ALT phenotype by FISH. In a set of 29 randomly chosen samples, we observed ALT in 11 of 15 samples which harbored ATRX loss, but only in 3 of 14 samples with retained ATRX expression (p = 0.009, Fisher’s exact test).
ATRX loss is a favorable prognostic marker
In a univariate Cox regression model, reference histology was significantly associated with time to treatment failure (TTF), the primary endpoint of the NOA-04 trial (Table 3). Upon incorporation of both ATRX and 1p/19q status into the model (Table 3), the prognostic value of reference histology was no longer significant, while both ATRX loss and 1p/19q co-deletion were associated with TTF. Based on this, we grouped mixed AOA without 1p/19q co-deletion but with ATRX loss and AA (both with and without ATRX loss, but without 1p/19q co-deletion) into “molecular astrocytomas”. Likewise, we combined AOA harboring 1p/19q co-deletion and AO (both with and without 1p/19q co-deletion, but without ATRX loss) into “molecular oligodendrogliomas”. Tumors with IDH wild-type tumors behaved like “molecular glioblastomas” in analogy to [6] (Fig. 2). Kaplan–Meier plots for TTF based on reference histology (Fig. 3a) and “molecular histology” (Fig. 3b–d) demonstrate the highly similar course for AA and AOA (Fig. 3c) as well as AO and AOA (Fig. 3d), respectively, when grouped by ATRX expression and 1p/19q co-deletion status.

Overview of reference histology, molecular aberrations and molecular diagnosis in NOA-04 patients (n = 133). Every column represents a patient. “Molecular diagnosis” is defined as astrocytoma (IDH mutant AOA with ATRX loss and IDH mutant AA with or without ATRX loss, but without 1p/19q co-deletion; n = 49), oligodendroglioma (IDH mutant AOA with 1p/19q co-deletion and IDH mutant AO with or without 1p/19q co-deletion, but without ATRX loss; n = 43), glioblastoma (IDH wild type; n = 32) or non-classifiable (n = 9)

Influence of ATRX on classification and survival. a Time to treatment failure, separated by reference histology. TTF by molecular diagnosis (b) and for “molecular astrocytomas” (c) and “molecular oligodendrogliomas” (d), separated by reference histology. e Time to treatment failure in “molecular astrocytoma” grouped by ATRX status
Survival analysis of the “molecular astrocytomas” demonstrated a marked separation of the clinical course of IDH mutant tumors based on ATRX status. Patients with tumors with ATRX loss had a longer TTF [median TTF 55.6 months (95 % CI 45.1 months–to not reached), n = 40] than patients with IDH mutant tumors with ATRX expression [median TTF 31.8 months (95 % CI 2.8 months–to not reached), n = 9, p = 0.0168, log rank test; Fig. 3e]. For PFS, similar results were obtained (median PFS 37.1 vs. 18.1 months, p = 0.038, log rank test; Supplementary Fig. 1), while for an analysis of OS, the number of events at a median follow-up of 54 months was too small.
To determine predictive properties of the molecular subgrouping, we assessed PFS and TTF for the two treatment arms in “molecular astrocytomas”, “molecular oligodendrogliomas” and “molecular glioblastomas” (Supplementary Fig. 2); we observed a trend toward a longer PFS in “molecular astrocytomas” who initially received radiotherapy compared to chemotherapy (median PFS 50.6 vs. 25.1 months, p = 0.0944; Supplementary Fig. 2a). For TTF, this difference between the groups narrowed (median TTF 55.6 vs. 45.1 months, p = 0.2784, log rank test; Supplementary Fig. 2b), suggesting that patients who initially were treated with chemotherapy still benefited from radiotherapy at recurrence. Treatment efficacy was similar for radiotherapy and chemotherapy in the other two molecular groups (Supplementary Fig. 2c–f).
Discussion
Molecular classification of malignant glioma has been receiving increasing attention lately. ATRX mutation and loss of expression, which have just recently been discovered in gliomas [7], might further refine this classification. Using immunohistochemistry to assess ATRX expression in a subset of the NOA-04 trial patient population, we detected ATRX loss in approximately 40 % of AA and 25 % of mixed AOA. In contrast, ATRX loss was rare in AO. ATRX loss has been reported before to be scarce in oligodendrogliomas [9, 15] and mutually exclusive with 1p/19q co-deletion in non-astrocytic tumors [8].
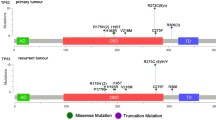
Given the separate clinical course [6] and possibly cell of origin [14] of anaplastic tumors which are IDH wild type, these tumors should be considered a distinct group and seem most closely related to primary glioblastomas. From the distribution of ATRX status and a similar clinical course of IDH mutant AA and AOA (Fig. 3c), we suggest grouping AA and mixed AOA without 1p/19q co-deletion but with ATRX loss as “molecular anaplastic astrocytomas”. 1p/19q co-deletion on the other hand is considered a hallmark of oligodendroglial tumors [21] and was suggested a predictive biomarker for radiochemotherapy with procarbazine, CCNU and vincristine in two independent trials of anaplastic oligodendroglial tumors [2, 4]. Based on the above, AOA which harbor 1p/19q co-deletion and AO can be classified as “molecular anaplastic oligodendroglioma” (Fig. 4). Kaplan–Meier curved indeed demonstrated a very similar course for AA and AOA as well as AO and AOA, when grouped as described above (Fig. 3c, d). Using this approach, only 9 of the 133 tumors (6 %) could not be classified. Of these, two samples carried both 1p/19q co-deletion and ATRX loss and two had a missing IDH status.

Proposed molecular classification of anaplastic gliomas based on histology and molecular markers. Length of the green bars represents the proportion of IDH wild type tumors, whereas the yellow and blue bars represent IDH mutant tumors. Mixed anaplastic oligoastrocytoma harboring IDH mutation are molecularly classified as either oligodendrogliomas (carrying 1p/19q co-deletion) or astrocytomas (carrying ATRX loss)
Prior studies have reported a discrepancy in the frequency of ATRX deficiency: while two studies reported a ~70 % prevalence of ATRX deficiency in IDH mutant, 1p/19q intact tumors [8, 9], another study reported a <50 % prevalence [15]. In the NOA-04 cohort, ATRX loss occurred in 65 % of IDH mutant, 1p/19q intact cases (28 of 43). As expected, ATRX loss was rare both in 1p/19q co-deleted (3 of 43, 7 %) and IDH wild type tumors (2 of 32, 6 %). The high prevalence of ATRX loss in this tumor subset, as reported by others, argues for ATRX deficiency as a class-defining feature.
NOA-04 showed a highly similar clinical course for AO and AOA [27], which others have questioned. In the EORTC 26951 trial, AOA had a worse outcome compared to pure AO [3]. A retrospective panel review of 150 patients from the EORTC 26951 trial, which did not impose such a restrictive central histology, confirmed on consensus only 8 % of the local AOA diagnoses [12]. In NOA-04 on the other hand, a restrictive central histological AOA classification, which required unambiguous oligodendroglial differentiation features, probably enriched the group of AOA for AO. Following the above concept of molecularly assisted subgrouping, of the 48 AOA of the NOA-04 trial, 12 AOA would be grouped with the AA, while 23 tumors would be considered as AO. Eight AOA were IDH wild type, while five AOA could not be re-assigned (for two of these, 1p/19q status is missing; Fig. 2). Due to the retrospective nature of this study, no DNA of sufficient quality was available for these tumors to sequence ATRX and complement 1p/19q testing.
A better prognosis for patients with ATRX mutations has been suggested in a retrospective cohort by Jiao et al. [8]. These authors proposed a classification of anaplastic tumors—irrespective of histology—based on ATRX, CIC, FUBP1 and IDH status and postulated a significantly longer overall survival for patients with either ATRX or CIC and FUBP1 mutations (who all carried IDH mutations as well) compared to patients with IDH mutant tumors which were wild type for ATRX, CIC and FUBP1. We grouped the 133 NOA-04 samples accordingly, resulting in 40 patients with ATRX loss, 40 patients with 1p/19q co-deletion, 17 patients with IDH mutation only and 31 patients who were IDH wild type. Five patients could not be classified using this approach (including 3 patients with both ATRX loss and 1p/19q co-deletion). While median TTF was longer for 1p/19q co-deleted tumors (median TTF not reached), it was similar for tumors with ATRX loss (median TTF 55.6 months) and IDH mutation only (58.3 months, p = 0.3761, log rank test, Supplementary Fig. 3). However, using our approach which incorporates histology (Fig. 4), ATRX status separated “molecular astrocytomas” into two groups, with a significantly better prognosis for patients with tumors with ATRX loss (Fig. 3e). Given the superior clinical course of oligodendroglial tumors, it seems to be important to evaluate the prognostic value of ATRX status only in astrocytic tumors as we did here. To this extent, ATRX status is important as it both helps re-classify mixed oligoastrocytic tumors and defines a subgroup of astrocytic tumors with a superior clinical course.
ATRX plays a role in regulating chromatin remodeling [1] and is tightly associated with IDH mutations, which are linked with a distinct epigenetic signature [23], the glioma CpG island methylator phenotype (G-CIMP). Thus, epigenetic differences between ATRX expressers vs. non-expressers may underlie the distinct clinical course in astrocytic tumors.
In pediatric glioblastomas, ATRX mutations have been identified in tumors with H3F3A mutations [20]. However, given that almost all tumors in this NOA-04 biomarker cohort which had ATRX loss were IDH mutated (Table 2), and H3F3A and IDH mutations are mutually exclusive [20, 22], we did not test for H3F3A mutations. We confirmed the association between ATRX loss and the ALT phenotype. Another mechanism for tumor cell telomere maintenance involves the telomerase reverse transcriptase (TERT). Point mutations in the promoter of the TERT gene increase telomerase expression and have been shown to occur frequently in gliomas. In AO and AOA, TERT mutations exclusively occurred in 1p/19 co-deleted tumors. In all anaplastic tumors, TERT mutations and ATRX losses were mutually exclusive [11].
This study is limited by the rather small sample size and the lack of DNA of sufficient quality to verify ATRX mutations. However, the use of immunohistochemistry allows for a broad application of ATRX testing in practice. Furthermore, a good concordance between ATRX mutations and loss of ATRX expression (as assessed through IHC) has been reported [7, 15].
ATRX in conjunction with 1p/19q status may help unequivocally classify mixed gliomas, whose diagnoses are subject to relevant inter-observer variation, as either astrocytic or oligodendroglial. More important than this potential facilitation of the diagnosis is the prognostic value of the molecular classification itself (Fig. 4). In this prospective cohort, patients with astrocytic tumors harboring ATRX loss have a significantly better prognosis than IDH mutant patients with astrocytic tumors who express ATRX.
References
Bassett AR, Cooper SE, Ragab A, Travers AA (2008) The chromatin remodelling factor dATRX is involved in heterochromatin formation. PLoS One 3:e2099
Van den Bent MJ, Brandes AA, Taphoorn MJB, Kros JM, Kouwenhoven MCM, et al. (2012) Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol
Van den Bent MJ, Carpentier AF, Brandes AA, Sanson M, Taphoorn MJB et al (2006) Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer p. J Clin Oncol: Off J Am Soc Clin Oncol 24:2715–2722
Cairncross G, Wang M, Shaw E, Jenkins R, Brachman D, et al. (2012) Phase III Trial of chemo radiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol
Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068
Hartmann C, Hentschel B, Wick W, Capper D, Felsberg J et al (2010) Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol 120:707–718
Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH et al (2011) Altered telomeres in tumors with ATRX and DAXX mutations. Science 333:425
Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM et al (2012) Frequent ATRX, CIC, and FUBP1 mutations refine the classification of malignant gliomas. Oncotarget 3:709–722
Kannan K, Inagaki A, Silber J, Gorovets D, Zhang J et al (2012) Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget 3:1194–2003
Khuong-Quang D-A, Buczkowicz P, Rakopoulos P, Liu X-Y, Fontebasso AM et al (2012) K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124:439–447
Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N et al (2013) TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA 110:6021–6026
Kros JM, Gorlia T, Kouwenhoven MC, Zheng P–P, Collins VP et al (2007) Panel review of anaplastic oligodendroglioma from European Organization for Research and Treatment of Cancer Trial 26951: assessment of consensus in diagnosis, influence of 1p/19q loss, and correlations with outcome. J Neuropathol Exp Neurol 66:545–551
Lacayo NJ, Meshinchi S, Kinnunen P, Yu R, Wang Y et al (2004) Gene expression profiles at diagnosis in de novo childhood AML patients identify FLT3 mutations with good clinical outcomes. Blood 104:2646–2654
Lai A, Kharbanda S, Pope WB, Tran A, Solis OE et al (2011) Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol: Off J Am Soc Clin Oncol 29:4482–4490
Liu X-Y, Gerges N, Korshunov A, Sabha N, Khuong-Quang D-A et al (2012) Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol 124:615–625
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC et al (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109
Maintz D, Fiedler K, Koopmann J, Rollbrocker B, Nechev S et al (1997) Molecular genetic evidence for subtypes of oligoastrocytomas. J Neuropathol Exp Neurol 56:1098–1104
Miller CR, Dunham CP, Scheithauer BW, Perry A (2006) Significance of necrosis in grading of oligodendroglial neoplasms: a clinicopathologic and genetic study of newly diagnosed high-grade gliomas. J Clin Oncol 24:5419–5426
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K et al (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522
Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E et al (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482:226–231
Smith JS, Perry A, Borell TJ, Lee HK, O’Fallon J et al (2000) Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J Clin Oncol 18:636–645
Sturm D, Witt H, Hovestadt V, Khuong-Quang D-A, Jones DTW et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437
Turcan S, Rohle D, Goenka A, Walsh LA, Fang F et al (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483:479–483
Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y et al (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110
Weisbrod AB, Zhang L, Jain M, Barak S, Quezado MM, Kebebew E (2013) Altered PTEN, ATRX, CHGA, CHGB, and TP53 expression are associated with aggressive VHL-associated pancreatic neuroendocrine tumors. Horm cancer 4:165–175
Weller M, Stupp R, Hegi ME, van den Bent M, Tonn JC et al (2012) Personalized care in neurooncology coming of age: why we need MGMT and 1p/19q testing for malignant glioma patients in clinical practice. Neurooncology 14(Suppl 4):iv100–iv108
Wick W, Hartmann C, Engel C, Stoffels M, Felsberg J et al (2009) NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol 27:5874–5880
Acknowledgments
The authors (WW, MW) conducting this work represent the Neurooncology Working Group (NOA) of the German Cancer Society. BW is a scholar of the NCT Heidelberg School of Oncology Postdoc Program. We gratefully acknowledge the contributions of Ulrike Ernemann, MD and Christoph Meisner, PhD (Tübingen, Germany), Guido Reifenberger, MD and Michael C. Sabel, MD (Düsseldorf, Germany), Susanne Koeppen, MD (Essen, Germany), Otmar Wiestler, MD and Thorsten Pietsch, MD (Bonn, Germany) and Ralf Ketter, MD to the first publication of the study. We are indebted to Diana Rieker and Tanja Göck for excellent technical assistance with the ATRX immunohistochemistry. The Charitable Hertie Foundation and National Genome Network of the BMBF provided funding.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
401_2013_1156_MOESM1_ESM.jpg
{kind=link}
Supplementary material 1 (JPG 798 kb) PFS of “molecular astrocytomas” by ATRX status. Tumors with ATRX loss had a significantly longer PFS than ATRX expressing tumors (median PFS 37.1 vs. 18.1 months, p = 0.038, log rank test)
Correlation coefficients of growth indices with precipitation and temperature data corresponding to August (Year prior to growth) to December (Year of growth) period, for Prades (a) and Arcalís (b). Climate and growth indices data are from the period 1952 to 2008. Asterisks indicate significant relationships (p < 0.05) (JPEG 1.15 MB)
401_2013_1156_MOESM2_ESM.jpg
{kind=link}
Supplementary material 2 (JPG 1459 kb) PFS and TTF by treatment arm and molecular diagnosis. In “molecular astrocytomas”, initial radiotherapy was associated with a trend towards longer PFS (log rank p = 0.0944; a) and TTF (log rank p = 0.2784; b), while in “molecular oligodendrogliomas” and “molecular glioblastomas”, efficacy of both treatment arms was similar
Correlation coefficients of growth indices with precipitation and temperature data corresponding to August (Year prior to growth) to December (Year of growth) period, for Prades (a) and Arcalís (b). Climate and growth indices data are from the period 1952 to 2008. Asterisks indicate significant relationships (p < 0.05) (JPEG 1.15 MB)
401_2013_1156_MOESM3_ESM.jpg
{kind=link}
Supplementary material 3 (JPG 813 kb) TTF by genetic signature. NOA-04 samples were grouped as described previously into tumors harboring ATRX loss (and IDH mutation), 1p/19q co-deletion (and IDH mutation), IDH mutation only or IDH wild type
Correlation coefficients of growth indices with precipitation and temperature data corresponding to August (Year prior to growth) to December (Year of growth) period, for Prades (a) and Arcalís (b). Climate and growth indices data are from the period 1952 to 2008. Asterisks indicate significant relationships (p < 0.05) (JPEG 1.15 MB)
Rights and permissions
About this article
Cite this article
Wiestler, B., Capper, D., Holland-Letz, T. et al. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol 126, 443–451 (2013). https://doi.org/10.1007/s00401-013-1156-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-013-1156-z