
Abstract
The role of tumor-induced immune modulation in cancer progression is currently a focus of investigation. The signal transducer and activator of transcription 3 (STAT3) is an established molecular hub of immunosuppression, and its signaling pathways are classically overactivated within malignancies. This article will review STAT3 operational mechanisms within the immune system and the tumor microenvironment, with a focus on therapeutic strategies that may impact outcomes for patients with cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The roles of angiogenesis, invasion, cell proliferation, and survival in tumorigenesis are well established and these elements recognized as hallmarks of cancer. The impact of tumor-induced immune suppression on tumor progression and prognosis has more recently been a subject of intense investigation. The signal transducer and activator of transcription 3 (STAT3) has been demonstrated to be a key mediator in the molecular mechanisms governing tumor escape from immune surveillance, while also driving tumor progression. This article will review the role of STAT3 in the interplay between the tumor microenvironment and the immune system and how this interaction in turn contributes to tumor progression, with a focus on glioma.
STAT3 and tumorigenesis
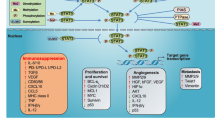
STAT3 belongs to the family of cytoplasmic transcription factors activated by tyrosine kinases. Common STAT3 activators include JAK (Janus family) kinases, SRC kinases, growth factor receptors that have intrinsic tyrosine-kinase activity (i.e., epidermal growth factor receptor [EGFR], platelet-derived growth factor [PDGFR]), and extracellular cytokines such as IL-6 [1]. STAT3 exists in an inactive monomer form. Once activated via phosphorylation, STAT3 (pSTAT3) dimerizes and translocates into the nucleus where it regulates gene transcription [1, 2]. STAT3 influences the expression of a wide array of genes involved in apoptosis, cell migration, cell cycle regulation, angiogenesis, and modulation of immunosuppressive factors [1, 2].
Under normal physiological conditions, STAT3 activation is tightly regulated by a variety of inhibitory molecules. Suppressor of cytokine signaling (SOCS) proteins, specifically SOCS3, disrupt JAK/STAT3 signaling by degradation of JAKs and blocking STAT3 binding to receptor subunits [2]. Alternatively, protein inhibitor of activated STAT (PIAS) functions by blocking STAT3 DNA-binding activity, hence inhibiting gene transcription [3]. Lastly, protein tyrosine phosphatases deactivate STAT3 via dephosphorylation [3].
STAT3 signaling can lead to abnormal overactivation of several pro-oncogenic mechanisms. For example, constitutive activation of STAT3 can confer resistance to apoptosis and enhanced cell proliferation by activation of antiapoptotic molecules such as Bcl-XL and survivin [4]. STAT3 overactivation contributes to tumor invasiveness by upregulation of matrix metallopeptides (MMP-2 and MMP-9) and focal adhesion kinase (FAK) [5]. STAT3 also promotes tumor angiogenesis via interaction with vascular endothelial growth factor (VEGF) [6]. Cumulatively, these discoveries have led to the finding that STAT3 is associated with a spectrum of human cancers [2, 7]. Grandis et al. were among the first to describe the link between constitutive STAT3 activation in malignancy by demonstrating that STAT3 activation is required for growth of human head and neck cancer [8]. Later studies have demonstrated the role of STAT3 in other malignancies [9, 10].
We have previously characterized the effect of STAT3 expression on glioma progression in vivo using the Ntv-A transgenic mouse system. When platelet-derived growth factor B (PDGFB) is expressed in glioneuronal progenitor cells, low-grade gliomas are induced. However, coexpression of STAT3 with PDGFB in mice results in the induction of high-grade gliomas. When mice injected with PDGFB and STAT3 were treated with a STAT3 inhibitor, their median survival time increased, and the incidence of high grade glioma and CD31 (a marker of endothelial proliferation) expression decreased significantly [11]. STAT3 has also been shown to be required for the maintenance of glioblastoma stem cells [12].
Upregulation of STAT3 is also emerging as a mechanism of treatment resistance. For example, anti-PD-1 antibodies have been shown to enhance p-STAT3 expression [13]. BRAF resistance in melanoma has been shown to lead to increased expression of PD-L1, which is mediated by STAT3 [14]. Furthermore, we have shown that the use of bevacizumab in glioblastoma up regulates STAT3 [15].
STAT3 as a biomarker
STAT3 has been shown to be a negative prognostic marker in patients with a variety of malignancies including glioma [16, 17]. Lin et al., examined p-STAT3 expression in 90 newly diagnosed glioblastoma multiforme (GBM) patients and correlated its expression with clinical outcome. Univariate analysis showed a significant correlation between p-STAT3 expression and survival. Specifically low p-STAT3 expression was associated with an overall median survival of 20 months versus 9 months in patients harboring tumors with high p-STAT3 expression. Multivariate analysis confirmed that high p-STAT3 expression was an independent predictor of shorter progression-free and overall survival [16]. Activation of STAT3 via phosphorylation at tyrosine 705 (pTyr705) or serine 727 (pSer727) has been shown to promote tumorgenesis. Lin et al., showed that GBM patients whose tumors expressed both high pTyr705-STAT3 and high pSer727-STAT3 displayed significantly shorter progression free survival compared to patients with phosphorylation at 1 site [17]. Tu et al. investigated the JAK/STAT3 signaling pathway in patients’ glioma samples and found that in glioma patients expressing components of this signaling axis, prognosis was diminished. Once again, multivariate analysis showed that STAT3 was an independent predictor of glioma prognosis [18].
We have analyzed the incidence of activated STAT3 in 129 patient glioma samples and reported an association of STAT3 expression and pathological grade [19]. We did not observe STAT3 expression in the normal brain tissue specimens or in the patients with WHO grade II astrocytomas. In patients with WHO grade III anaplastic astrocytomas (AAs) (n = 17), 9 (53 %) expressed p-STAT3, and in patients with WHO grade IV GBMs or gliosarcomas (n = 60), 32 (53 %) expressed p-STAT3. In our analysis, for patients with GBMs and p-STAT3 expression, the median survival time was 10.7 months, whereas it was 18.1 months for patients without p-STAT3 expression, but this was not statistically significant. However, in patients with AAs expressing p-STAT3, the median survival time was 12.2 months, whereas it was 34.6 months for patients with AAs that lacked p-STAT3 expression (p = 0.02). Interestingly, the association between STAT3 expression and tumor grade did not necessarily hold for the oligodendrogliomas. Specifically, 38 % of the patients with WHO grade II oligodendrogliomas had p-STAT3 expression, and 40 % of the patients with WHO grade II anaplastic oligodendrogliomas, indicating the incidence of p-STAT3 expression did not increase with increasing tumor grade in oligodendrogliomas [19].
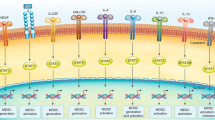
STAT3 and immunosuppression (Fig. 1)
Evidence is accumulating establishing the role of STAT3 in tumor immune evasion. Activated STAT3 induces the production of tumor factors, including VEGF and IL-10, which subsequently activate STAT3 in various immune cell subsets [20]. Activation of STAT3 in these cell types reduces their ability to produce immune stimulatory molecules and conversely, increases the production of immunosuppressive factors such as IL-10, IL-23, and transforming growth factor (TGF-β) [21]. Overall, STAT3 overactivation impacts antitumor immunity by compromising native immune responses via multiple mechanisms [22].

STAT3 mechanisms of tumor-induced immune suppression
Dendritic cell maturation
Dendritic cells (DCs) are differentiated monocytes that function as antigen-presenting cells and are involved in the initiation of T cell responses [3]. They are responsible for activation of tumor-specific T cells through expression of major histocompatibility complex (MHC) class II complexes, costimulatory molecules such as CD80 and CD86 and IL-12, which are critical for T cell activation. A decreased presence of mature DCs impairs the immune system’s ability to induce and maintain an effective antitumor immune response [1, 22].
To confirm the critical role of STAT3 in myeloid cell differentiation, Nefedova et al. isolated hematopoietic progenitor cells (HPCs) from the bone marrow of mice and infected them with a retrovirus construct containing the constitutively active mutant form of STAT3 (STAT3C). STAT3C-transduced HPCs were impaired in their ability to differentiate into DCs. Conversely, to inhibit STAT3 activity, this group used STAT3D, which has mutations in its binding domain rendering it incapable of binding DNA. The authors found that blockade of STAT3 activity in HPCs cultured in the presence of tumor-derived factors restored their ability to differentiate into mature DCs [23]. Kortylewski et al. ablated STAT3 alleles in the hematopoietic compartment of tumor-bearing mice. DCs isolated from STAT3-/-tumor-bearing mice showed enhanced ability to present antigens and to activate antigen-specific CD4+T cells ex vivo. These authors also demonstrated that tumor-infiltrating DCs of STAT3−/− mice displayed increased expression of MHC class II, CD80, and CD86 relative to DCs from STAT3+/+ mice, thus indicating effective antigen presentation and the ability to generate a more robust immune response [20]. Cumulatively, these studies support that STAT3 activation is an important contributor to impaired activation of tumor-infiltrating DCs, making them incapable of activating antigen specific CD8+T cells, and blocking this branch of the antitumor immune response.
Myeloid-derived suppressor cells (MDSCs)
MDSCs are immature cells of the myeloid lineage and are one of the major suppressors of tumor immunity. MDSCs undergo a significant numerical expansion in the presence of malignancy, contributing to tumor immune escape [24]. In normal mice, 2–4 % of nucleated splenocytes are MDSCs, but in murine models of malignancy this number climbs to 20–40 % [24]. The accumulation of MDSCs is associated with worse patient prognosis across a breadth of malignancies [25, 26]. Diaz-Montero et al. reported that levels of circulating MDSCs were significantly increased in patients with solid malignancies of all stages of cancer (multiple pathologies) compared with healthy volunteers. Furthermore, they demonstrated a strong correlation between circulating MDSCs and cancer stage, such that subjects with a significant metastatic tumor burden had the highest numbers of MDSCs [25].
MDSCs suppress T-cell activation via arginase 1, iNOS (which generates nitric oxide, NO) and hyperproduction of reactive oxygen species (ROS) [24, 27, 28]. l-arginine is critical for normal T-cell function, and arginase-1 decreases its levels. In turn, decreased l-arginine reduces T-cell-receptor-chain expression resulting in T-cell dysfunction [28]. Increased ROS production (particularly peroxynitrite) is a key characteristic of MDSCs generated from the tumor microenvironment [24, 29] and induces the post-translational modification of T-cell receptors resulting in antigen-specific T-cell unresponsiveness [24, 29].
MDSCs are a heterogeneous population that consists of myeloid progenitor cells and immature myeloid cells (IMCs). Kusmartsev et al. analyzed the function IMCs isolated from control and tumor-bearing mice. IMCs harvested from tumor-bearing mice had significantly higher levels of ROS than control (tumor-free) mice. Furthermore, in contrast to control mice, IMCs isolated from tumor-bearing mice inhibited antigen-specific responses of CD8+T cells. Subsequent inhibition of ROS abolished the inhibitory effect of IMCs on T cells [30]. In glioblastoma patients, Raychaudhuri et al. examined the peripheral blood mononuclear cells (PBMCs) compared with those in healthy, age-matched controls and found a significant increase in number of circulating MDSCs in patients with GBM, and this was associated with high arginase activity. Furthermore, they demonstrated in vitro that depletion of MDSCs restored normal T-cell function [31].
STAT3 has been proposed to be the main regulator of MDSC expansion [3]. Compared with control mice, MDSCs isolated from tumor-bearing mice were shown to have significantly increased levels of activated STAT3 [23, 32]. Nefedova et al. demonstrated that exposure of myeloid cells to tumor-conditioned medium resulted in up regulation of STAT3 and increased MDSC expansion. Myeloid cell differentiation was subsequently restored after removal of tumor-derived factors [23, 32]. Crozo et al. investigated the mechanisms governing MDSC-mediated immune suppression and reported that increased ROS production is mediated by upregulated activity of NADPH oxidase (NOX2). Moreover, the authors demonstrated that the expression of NOX2 in MDSCs was in fact controlled by STAT3. Treatment with a selective STAT3 inhibitor significantly decreased ROS production in MDSCs. In this state, MDSCs could no longer effectively suppress T-cell immunity [29]. Overall, these data indicate that MDSCs are a major component of the immune suppression network and that STAT3 is a crucial regulator.
Macrophages/microglia
Macrophages that infiltrate tumors are referred to as tumor-associated macrophages (TAMs). They arise from circulating monocytes and are thought to be recruited by the tumor through a variety of signals such as monocyte chemoattractant-protein (MCP-1) and CCL2 (chemokine, C–C motif ligand). In general, macrophages can differentiate into 2 phenotypes: M1 and M2. M1 is capable of inducing an antitumor response via the release of inflammatory cytokines, antigen presentation, and phagocytosis of tumor cells [33, 34]. However, after attraction to tumor, macrophages may polarize into the M2 phenotype and contribute to the tumor immunosuppressive environment. M2 macrophages express and secrete tumor supportive factors, which promote angiogenesis (e.g., VEGF, CCL-2), cell proliferation (e.g., EGF), extracellular matrix remodeling (e.g., MMPs, plasmin), and immunosuppression (IL-10, TGF-β) [35].
Elevated trafficking of M2 TAMs has been associated with poor patient prognosis [35–37]. Komohara et al. analyzed microglia/macrophage polarization in patients’ glioma samples and found that the ratio of M2 macrophages was associated with histological grade [38]. Ding et al. examined 50 glioma samples and also reported a significant correlation between WHO grade and M2 phenotype. Furthermore, progression-free survival and overall survival were lower in patients with elevated M2 macrophage expression [37].
Growing data indicate that STAT3 may play a role in this aspect of immunosuppression. Using a murine glioma model, Zhang et al. examined STAT3 activity in microglia/macrophages and reported that tumor-associated microglia/macrophages demonstrated higher STAT3 activity [39]. Furthermore inhibition of STAT3 in tumor-associated microglia/macrophages resulted in delayed tumor growth and improved survival in glioma bearing mice [39]. After screening multiple compounds Fujiwara et al., reported that oleanolic acid (OA) inhibited macrophage polarization to the M2 phenotype by suppressing STAT3 signaling in macrophages and subsequently inhibited proliferation of U373 human glioblastoma cells [40]. We have shown that human glioblastoma cancer stem cells (gCSCs) induce M2 polarization, inhibit phagocytosis, induced the secretion of the immunosuppressive cytokines IL-10 and TGF-β1 by the microglia/macrophages, and enhanced their capacity to inhibit T-cell proliferation. This was a STAT3-mediated process that could be reversed by inhibiting phosphorylated-STAT3 [41]. These results indicate that STAT3 activation may play a role in M2 macrophage polarization, directly contributing to local immunosuppression.
T regulatory cells (Tregs)
T regulatory cells (Tregs) are a population of CD4+T cells that express the transcription factor forkhead box P3 (FoxP3) and high levels of the IL-2-α receptor chain (CD25), which develop in the thymus (natural Tregs) or from mature CD4+T cells under specific conditions. Tregs modulate the immune system to maintain tolerance to self antigens and to prevent autoimmune reactions. In the presence of tumors, Tregs can accumulate within the tumor microenvironment, releasing immunosuppressive mediators (e.g., TGF-β and IL-10) and ultimately suppressing immune responses mediated by CD8+T cells [3, 22]. These cells have been demonstrated in multiple tumor models and tumor samples from patients including those with glioma [42–44]. El Andaloussi and Lesniak found that Tregs were common within GBMs and that FoxP3 expression correlated with glioma WHO grade, with grade IV having the highest frequency, followed by grade III and grade II [44]. Heimberger et al. also reported a strong positive correlation between FoxP3+Treg numbers and pathological grade, with high grade lesions (gliosarcomas, GBMs, and AAs) displaying the most Treg infiltration [45].
The increased trafficking of Tregs has been reported as a marker of poor patient outcome. Curiel et al., examined tumor specimens from patients with untreated ovarian cancer and reported a significant accumulation of Tregs, particularly at the later stages of disease. The frequency of Tregs was a significant predictor of death after controlling for stage, surgical debulking, and other factors known to affect survival [46]. Other studies have reported a striking association between Treg accumulation and cancer patient survival, and this is thought to be due to the suppression of native antitumor immunity [42, 43, 47]. Yue et al. examined the prognostic significance of Tregs in GBM samples from 62 patients and found that FoxP3 density in tumor-infiltrating lymphocytes was a predictor of patient progression-free survival [48]; however, this conflicts with prior studies that did not identify this as a prognosticator and also examined Treg to immune effector ratios [45]. The mechanisms and pathways of glioblastoma-mediated immune suppression have been shown to be markedly heterogeneous, including within tumor subtypes (classical, proneural, mesenchymal, neural) [49], and this may partially account for discrepancies in results, if the analysis has enrichment for a particular subtype.
STAT3 has been shown to be an inducer of FoxP3 [50]. Ablation of STAT3 in the haematopoietic system of tumor-bearing mice has been associated with a significant reduction the in number of tumor-infiltrating Tregs, proliferation of CD8+T cells, and subsequent increased antitumor responses [20]. Kinjyo et al. generated SOCS3 knockout mice [21]; SOCS3 is a known negative regulator of STAT3 activity. They found that these mice had reduced immune responses due to increased STAT3 signaling, and they reported further that in vitro, SOCS3-deficient T cells produced more TGF-β and IL-10, which are known immunosuppressive factors. Overall, these reports support the role of STAT3 in Treg cell accumulation in the tumor stroma, contributing to the suppression of antitumor immunity.
Th17 cells
Th17 cells develop from naïve CD4+T cells in the presence of TGF-β and IL-6 and are maintained by IL-21 and IL-23. They are characterized by their ability to secrete several cytokines, most importantly IL-17 [3, 51]. The role of Th17 in malignancy is controversial, with some reports showing that Th17 cells eradicate tumors while other report promotion of tumor progression [51]. Although this matter is not resolved, it is suggested that these cells adopt a pro- or antitumor function depending on the type of stimulation encountered. Th17 cells also exhibit plasticity and may convert to Tregs, hence promoting immunosuppression [51, 52]. Th17 cells can express CD39 and CD73 on their cell surface, which cleave ATP into adenosine–a molecule that causes cytotoxic T cell suppression [51, 53]. STAT3 has been shown to play a role in this process by specifically binding to the promoter regions of CD39 and CD73 [51, 53].
Targeting STAT3
Immune suppression is a well-recognized feature of glioblastomas, which have multiple operational mechanisms that include: (1) secretion of immunosuppressive factors; (2) down regulation of costimulatory molecules; (3) induction of T-cell apoptosis; (4) inhibition of natural killer (NK) cells; (5) aberrant antigen recognition through reduction of MHC molecules; and (6) recruitment of suppressive immune cells, to name a few [54, 55]. Many of these mechanisms are tied to STAT3 signaling and therapeutically targeting a key hub with dual functions in immune suppression and gliomagenesis is compelling. There are multiple ways of targeting STAT3 including: indirect signaling inhibition, RNA interference, and direct targeting of the STAT3 protein itself.
Blockade of upstream signaling
One of the most investigated methods of inhibiting STAT3 is the use of tyrosine kinase inhibitors, which can block STAT3 signaling by disrupting upstream tyrosine kinases responsible for STAT3 phosphorylation, hence blocking STAT3 activation [2, 9]. To this end, JAK/STAT3 inhibitors have demonstrated some success in a variety of tumor models, including glioma. Fujita et al. tested the JAK/STAT3 inhibitor, JSI-124 (cucurbitacin I), in a murine GBM model (GL261), with the specific goal of evaluating its impact on glioma-induced immunomodulation. By exposing immune cells (splenocytes) to GL261-conditioned medium, they found that soluble factors from glioma cells induced the phosphorylation of STAT3, but this was inhibited by treatment with JSI-124. Additionally, JSI-124 enhanced DC maturation, as demonstrated by up regulation and surface expression of MHC class II and costimulatory molecules [56]. However, this agent has not been advanced into clinical trials.
WP1066 is a caffeic acid analogue that blocks the nuclear translocation of p-STAT3 into the nucleus. Therapeutic efficacy with WP1066 has been demonstrated in multiple tumor models [57–59]. For example, we have shown that WP1066 has therapeutic efficacy against metastatic [60] and established central nervous system (CNS) melanoma in murine models [59]. The therapeutic effects of WP1066 can be partially ablated in C57BL/6 J mice with B16 melanoma by using in vivo depletions of the CD4 and CD8 population or by implanting B16 or GL261 gliomas in nude (athymic) mice, indicating that part of the therapeutic effect of WP1066 is immunologically mediated [59]. WP1066 has also demonstrated therapeutic efficacy against subcutaneously implanted U-87 cells [61] and in two distinct Ntv-A transgenic murine models of glioma [11, 62].
We have also demonstrated that STAT3 blockade with WP1066 can significantly modulate tumor-mediated immune suppression. WP1066 can induce the expression of costimulatory molecules on peripheral macrophages and tumor-infiltrating microglia ex vivo in tumor samples from GBM patients. Treatment of the peripheral blood from GBM patients who are immunologically anergic with WP1066 resulted in marked production of proinflammatory cytokines (e.g. IL-2, IL-4, IL-12, IL-15). STAT3 blockade with WP1066 induced proliferation of effector T cells from GBM patients, mechanistically, this was found to be secondary to the activation of ZAP-70 in the T cells and inhibition of Tregs [63]. Furthermore, we found that the immunosuppressive properties of glioma cancer stem cells (gCSCs) were markedly diminished following treatment with WP1066 (or siRNA targeting STAT3) including Treg induction, macrophage/microglia polarization to the immune suppressive M2 phenotype and their secretion of immunosuppressive cytokines (IL-10, TGF-β1). Combined, these data indicate that WP1066 can reverse both innate and adaptive tumor-mediated immune suppression [41, 64].
Nucleic acid-based strategies
An alternative method of STAT3 inhibition is disruption of STAT3 mRNA translation by coding RNA interference. Several methods of RNA interference have been developed including: dominant-negative mutants, decoy oligonucleotides, and antisense approaches. Dominant negative strategies entail mutating the STAT3 functional domain to generate a gene product that disrupts STAT3 function. For example, STAT3D has an amino acid substitution in the DNA binding domain inhibiting STAT3 signaling [23, 65]. This method of STAT3 inhibition has been shown to hinder tumorigenesis in a variety of tumor models but use is limited preclinical studies involving gliomas [66].
STAT3 decoy oligonucleotides (ODNs) function via competitive inhibition. The transfected decoy ODN interacts with activated STAT3, occupying the DNA binding domain and subsequently inhibiting STAT3 from binding to its DNA-binding site [65, 67]. Gu et al. demonstrated that STAT3 decoy OGN reduced STAT3 transcriptional activity and subsequently reduced glioma cell proliferation in 2 different glioma cell lines [67]. This technique may be limited due to ODN degradation in the presence of nucleases [7, 65]. Investigation of methods to increase stability of these molecules is under way.
Antisense strategies have been successfully used to inhibit STAT3 in preclinical models. This approach is particularly advantageous owing to its specificity for targeting gene expression. Small inferring RNAs (siRNAs) are a class of double-stranded RNA molecules that can cause RNA interference and suppression of gene expression. Herrmann et al., used an aptamer (apt) based system to deliver siRNA to tumor associated T-cells. The authors fused STAT3 siRNA to an apt that binds cytotoxic T-lymphocyte-associated antigen 4 (CTLA4). They found that tumor- bearing mice treated with CTLA4apt–STAT3 siRNA had a reduction in tumor-associated Tregs. Furthermore, mice with B16 melanoma experimental lung metastases treated systemically with CTLA4apt–STAT3 siRNA, had significant reduction in lung metastases [68]. See et al. evaluated the role of STAT3 activation on the immunological microenvironment of gliomas using siRNA inhibition. Inhibition of STAT3 by siRNA significantly reduced the level of DNA binding of pSTAT3, returned expression of proinflammatory cytokines back to normal levels (IL-6, IL-8) and enhanced the maturation of dendritic cells boosting antitumor activity in a T-cell-independent manner. Thus, the authors showed that by targeting STAT3, the antitumor immune response could be restored [69].
MicroRNAs (miRs) can modulate critical gene transcripts involved in tumorigenesis and can target signaling networks. On the basis of miRNA gene expression arrays of glioblastoma and molecular modeling, miR-124 was identified as being able to modulate STAT3. miR-124 is absent in all grades and pathological types of gliomas. We have demonstrated that by upregulating miR-124 in gCSCs, multiple components of the STAT3 pathway were inhibited. miR-124 reversed gCSC-mediated immune suppression of T-cell proliferation and induction of Tregs. Treatment of T cells with miR-124 induced marked effector response, including up regulation of IL-2, IFN-γ, and tumor necrosis factor (TNF)-α. Both systemic administration of miR-124 and adoptive miR-124-transfected T-cell transfers were able to exert potent anti-glioma therapeutic effects in murine models of glioblastoma. These therapeutic effects were ablated in both CD4+ and CD8+ depleted mice and nude mouse systems, indicating that the therapeutic effect of miR-124 depends on the presence of a T-cell-mediated antitumor immune response [70].
Targeting STAT3 protein
Finally, another possible therapeutic strategy is to target STAT3 protein directly. STAT3 has 3 domains, and the one most commonly targeted is the Src Homology-2 (SH2) domain. After activation, STAT3 forms a dimer through its SH2 domain, allowing for subsequent nuclear translocation and gene expression. See et al. utilized two small molecule inhibitors (NSC 74859 and Stattic) that block STAT3 dimerization by inhibiting the SH2 domain. The authors reported that both of these compounds reduce pSTAT3 expression in glioma cells [69]. Fu et al. investigated effects of the inhibitor LLL-3 on the viability of human GBM cells in a mouse xenograft model. They reported that LLL-3 inhibited STAT3 DNA-binding activity in GBM cell lines. Moreover, tumor-bearing mice treated with LLL-3 had decreased tumor size, decreased contralateral tumor invasion, and significantly increased survival times [71].
Combination immunotherapy
STAT3 inhibitors have been shown to potentiate a wide variety of immunotherapeutic modalities including adoptive transfer of cytotoxic T cells [56], dendritic cell activation [72, 73], cytokines [74] and Treg inhibitors [60]. Using a GL261 intracranial glioma model, Fujita et al. demonstrated that STAT3 inhibition (with JSI-124) combined with adoptive transfer of cytotoxic T-cells resulted in prolonged survival in glioma-bearing mice compared to JSI-124 or adoptive transfer therapy alone. Specifically, the authors reported that treatment with STAT3 inhibitor improved the tumor-homing capability and persistence of transferred T-cells [56].
Multiple murine melanoma models have demonstrated the utility of combination immunotherapy involving dendritic cell activation and STAT3 inhibition [72, 73]. For example, Molavi et al. examined the synergistic anti-tumor effect of JSI-124 and a known dendritic cell activator, CpG oligodeoxynucleotide. These oligodeoxynucleotides have an unmethylated CpG motif and stimulate dendritic cells through their Toll-like receptors. The authors found that intratumoral injection of CpG plus JSI-124 inhibited tumor growth and improved survival in tumor-bearing mice compared to treatment with monotherapy. Furthermore, compared to the control group (PBS), monotherapy with JSI-124 or CpG resulted in a 2-5-fold percentage increase in intra-tumoral NK cells, CD8+ and CD4+ T-cells. On the other hand, combination therapy (JSI-124 plus CpG) resulted in a 125-fold, 75-fold and 50-fold percent increase in tumor infiltrating CD4+ T cells, NK cells, and CD8+ T cells respectively. Combination therapy also resulted in enhanced dendritic cell activation, increased levels of pro-inflammatory cytokines (i.e. IL-12, IL-2, TNF-α) and reduced levels of immunosuppressive factors (i.e. TGF-β). This landmark study was one of the earliest to provide evidence of the utility of dendritic cell activation in combination of STAT3 inhibition as an immunotherapeutic approach [72].
STAT3 inhibition has also been combined with immunomodulatory cytokines and agents in order to produce anti-tumor effects. Kong et al. used the STAT3 inhibitor WP1193 in combination with IFN-α (known to have multiple anti-tumor immune effects) to enhance therapeutic efficacy in a mouse metastatic melanoma model. They reported that mice with established intracranial melanoma treated with IFN-α plus WP1193 had superior median survival times compared to those treated with IFN-α or WP1193 monotherapy [74]. Cyclophosphamide (CTX) is a chemotherapeutic agent with known immunomodulatory properties including decreasing Tregs. Using a murine intracranial melanoma model, Hatiboglu et al. examined the synergistic effect of CTX combined with STAT3 inhibitor WP1066. Tumor bearing mice treated with combination therapy were found to have significantly longer survival times compared with monotherapy with WP1066 or CTX [60].
Collectively, these data speak to the promising immunomodulatory and subsequent anti-tumor effects of combination therapy involving STAT3 inhibition. Further clinical testing of combination therapy is needed to fully elucidate clinical applicability.
Summary
STAT3 is persistently activated in several different cancers and promotes pro-oncogenic mechanisms including immune escape. STAT3 activation within immune cells governs multiple immunosuppressive mechanisms including macrophage polarization to the M2 phenotype, inhibition of DC development, and accumulation of immunosuppressive cells such as Tregs, Th17 cells, and MDSCs. In light of its multifaceted role in tumorigenesis, the STAT3 signaling pathway has become a promising therapeutic target. Refinement of the available targeting strategies and their combination with other immunotherapeutics will be a powerful tool in anticancer therapy.
References
Kortylewski M, Yu H (2007) Stat3 as a potential target for cancer immunotherapy. J Immunother 30(2):131–139. doi:10.1097/01.cji.0000211327.76266.65.00002371-200702000-00001
Kim JE, Patel M, Ruzevick J, Jackson CM, Lim M (2014) STAT3 activation in glioblastoma: biochemical and therapeutic implications. Cancers (Basel) 6(1):376–395. doi:10.3390/cancers6010376
Rebe C, Vegran F, Berger H, Ghiringhelli F (2013) STAT3 activation: a key factor in tumor immunoescape. JAKSTAT 2(1):e23010. doi:10.4161/jkst.23010.2012JAKS0054R
Chen F, Xu Y, Luo Y, Zheng D, Song Y, Yu K, Li H, Zhang L, Zhong W, Ji Y (2010) Down-regulation of Stat3 decreases invasion activity and induces apoptosis of human glioma cells. J Mol Neurosci 40(3):353–359. doi:10.1007/s12031-009-9323-3
Wei Z, Jiang X, Qiao H, Zhai B, Zhang L, Zhang Q, Wu Y, Jiang H, Sun X (2013) STAT3 interacts with Skp2/p27/p21 pathway to regulate the motility and invasion of gastric cancer cells. Cell Signal 25(4):931–938. doi:10.1016/j.cellsig.2013.01.011
Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, Zhang S, Wang T, Sinibaldi D, Coppola D, Heller R, Ellis LM, Karras J, Bromberg J, Pardoll D, Jove R, Yu H (2002) Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 21(13):2000–2008
Yu H, Jove R (2004) The STATs of cancer–new molecular targets come of age. Nat Rev Cancer 4(2):97–105
Grandis JR, Drenning SD, Chakraborty A, Zhou MY, Zeng Q, Pitt AS, Tweardy DJ (1998) Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor- mediated cell growth In vitro. J Clin Invest 102(7):1385–1392. doi:10.1172/JCI3785
Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernandez-Luna JL, Nunez G, Dalton WS, Jove R (1999) Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 10(1):105–115. doi:10.1016/S1074-7613(00)80011-4
Ding BB, Yu JJ, Yu RY, Mendez LM, Shaknovich R, Zhang Y, Cattoretti G, Ye BH (2008) Constitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood 111(3):1515–1523. doi:10.1182/blood-2007-04-087734
Doucette TA, Kong LY, Yang Y, Ferguson SD, Yang J, Wei J, Qiao W, Fuller GN, Bhat KP, Aldape K, Priebe W, Bogler O, Heimberger AB, Rao G (2012) Signal transducer and activator of transcription 3 promotes angiogenesis and drives malignant progression in glioma. Neuro Oncol 14(9):1136–1145. doi:10.1093/neuonc/nos139/nos139
Sherry MM, Reeves A, Wu JK, Cochran BH (2009) STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 27(10):2383–2392. doi:10.1002/stem.185
Bandaru A, Devalraju KP, Paidipally P, Dhiman R, Venkatasubramanian S, Barnes PF, Vankayalapati R, Valluri V (2014) Phosphorylated STAT3 and PD-1 regulate IL-17 production and IL-23 receptor expression in Mycobacterium tuberculosis infection. Eur J Immunol 44(7):2013–2024. doi:10.1002/eji.201343680
Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS (2013) The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3 K inhibition. Clin Cancer Res 19(3):598–609. doi:10.1158/1078-0432.CCR-12-2731
de Groot J, Liang J, Kong LY, Wei J, Piao Y, Fuller G, Qiao W, Heimberger AB (2012) Modulating antiangiogenic resistance by inhibiting the signal transducer and activator of transcription 3 pathway in glioblastoma. Oncotarget 3(9):1036–1048
Lin GS, Yang LJ, Wang XF, Chen YP, Tang WL, Chen L, Lin ZX (2014) STAT3 Tyr705 phosphorylation affects clinical outcome in patients with newly diagnosed supratentorial glioblastoma. Med Oncol 31(4):924. doi:10.1007/s12032-014-0924-5
Lin GS, Chen YP, Lin ZX, Wang XF, Zheng ZQ, Chen L (2014) STAT3 serine 727 phosphorylation influences clinical outcome in glioblastoma. Int J Clin Exp Pathol 7(6):3141–3149
Tu Y, Zhong Y, Fu J, Cao Y, Fu G, Tian X, Wang B (2011) Activation of JAK/STAT signal pathway predicts poor prognosis of patients with gliomas. Med Oncol 28(1):15–23. doi:10.1007/s12032-010-9435-1
Abou-Ghazal M, Yang DS, Qiao W, Reina-Ortiz C, Wei J, Kong LY, Fuller GN, Hiraoka N, Priebe W, Sawaya R, Heimberger AB (2008) The incidence, correlation with tumor-infiltrating inflammation, and prognosis of phosphorylated STAT3 expression in human gliomas. Clin Cancer Res 14(24):8228–8235. doi:10.1158/1078-0432.CCR-08-1329
Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mule J, Kerr WG, Jove R, Pardoll D, Yu H (2005) Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 11(12):1314–1321. doi:10.1038/nm1325
Kinjyo I, Inoue H, Hamano S, Fukuyama S, Yoshimura T, Koga K, Takaki H, Himeno K, Takaesu G, Kobayashi T, Yoshimura A (2006) Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-β1. J Exp Med 203(4):1021–1031
Yu H, Kortylewski M, Pardoll D (2007) Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 7(1):41–51. doi:10.1038/nri1995
Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, Jove R, Gabrilovich D (2004) Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol 172(1):464–474
Gabrilovich DI, Nagaraj S (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9(3):162–174. doi:10.1038/nri2506
Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ (2009) Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother 58(1):49–59. doi:10.1007/s00262-008-0523-4
Gorgun GT, Whitehill G, Anderson JL, Hideshima T, Maguire C, Laubach J, Raje N, Munshi NC, Richardson PG, Anderson KC (2013) Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood 121(15):2975–2987. doi:10.1182/blood-2012-08-448548
Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI (2007) Altered recognition of antigen is a mechanism of CD8 + T cell tolerance in cancer. Nat Med 13(7):828–835. doi:10.1038/nm1609
Rodriguez PC, Ochoa AC (2008) Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev 222:180–191. doi:10.1111/j.1600-065X.2008.00608
Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI (2009) Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol 182(9):5693–5701. doi:10.4049/jimmunol.0900092
Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI (2004) Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol 172(2):989–999
Raychaudhuri B, Rayman P, Ireland J, Ko J, Rini B, Borden EC, Garcia J, Vogelbaum MA, Finke J (2011) Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol 13(6):591–599. doi:10.1093/neuonc/nor042
Nefedova Y, Nagaraj S, Rosenbauer A, Muro-Cacho C, Sebti SM, Gabrilovich DI (2005) Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res 65(20):9525–9535
Sica A, Schioppa T, Mantovani A, Allavena P (2006) Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer 42(6):717–727. doi:10.1016/j.ejca.2006.01.003
Yang I, Han SJ, Kaur G, Crane C, Parsa AT (2010) The role of microglia in central nervous system immunity and glioma immunology. J Clin Neurosci 17(1):6–10. doi:10.1016/j.jocn.2009.05.006
Mantovani A, Schioppa T, Porta C, Allavena P, Sica A (2006) Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev 25(3):315–322. doi:10.1007/s10555-006-9001-7
Lievense LA, Bezemer K, Aerts JG, Hegmans JP (2013) Tumor-associated macrophages in thoracic malignancies. Lung Cancer 80(3):256–262. doi:10.1016/j.lungcan.2013.02.017
Ding P, Wang W, Wang J, Yang Z, Xue L (2014) Expression of Tumor-Associated Macrophage in Progression of Human Glioma. Cell Biochem Biophys. doi:10.1007/s12013-014-0105-3
Komohara Y, Ohnishi K, Kuratsu J, Takeya M (2008) Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol 216(1):15–24
Zhang L, Alizadeh D, Van Handel M, Kortylewski M, Yu H, Badie B (2009) Stat3 inhibition activates tumor macrophages and abrogates glioma growth in mice. Glia 57(13):1458–1467. doi:10.1002/glia.20863
Fujiwara Y, Komohara Y, Kudo R, Tsurushima K, Ohnishi K, Ikeda T, Takeya M (2011) Oleanolic acid inhibits macrophage differentiation into the M2 phenotype activation of STAT3. Oncol Rep 26(6):1533–1537. doi:10.3892/or.2011.1454
Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, Sawaya R, Heimberger AB (2010) Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol 12(11):1113–1125. doi:10.1093/neuonc/noq082
Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, Xu Y, Li YW, Tang ZY (2007) Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol 25(18):2586–2593
Hiraoka N, Onozato K, Kosuge T, Hirohashi S (2006) Prevalence of FOXP3 + regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res 12(18):5423–5434
El Andaloussi A, Lesniak MS (2007) CD4 + CD25 + FoxP3 + T-cell infiltration and heme oxygenase-1 expression correlate with tumor grade in human gliomas. J Neurooncol 83(2):145–152
Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, Yang DS, Sun W, Qiao W, Hiraoka N, Fuller GN (2008) Incidence and prognostic impact of FoxP3 + regulatory T cells in human gliomas. Clin Cancer Res 14(16):5166–5172. doi:10.1158/1078-0432.CCR-08-0320
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W (2004) Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10(9):942–949. doi:10.1038/nm1093
Shen Z, Zhou S, Wang Y, Li RL, Zhong C, Liang C, Sun Y (2010) Higher intratumoral infiltrated Foxp3+ Treg numbers and Foxp3+/CD8+ ratio are associated with adverse prognosis in resectable gastric cancer. J Cancer Res Clin Oncol 136(10):1585–1595. doi:10.1007/s00432-010-0816-9
Yue Q, Zhang X, Ye HX, Wang Y, Du ZG, Yao Y, Mao Y (2014) The prognostic value of Foxp3 + tumor-infiltrating lymphocytes in patients with glioblastoma. J Neurooncol 116(2):251–259. doi:10.1007/s11060-013-1314-0
Doucette TA, Rao G, Rao A, Shen L, Aldape K, Wei J, Dziurzynski K, Gilbert M, Heimberger AB (2013) Immune heterogeneity of glioblastoma subtypes: extrapolation from the cancer genome atlas. Cancer Immunol Res 1(2):112–122
Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, Bellucci R, Raderschall E, Canning C, Soiffer RJ, Frank DA, Ritz J (2006) IL-2 regulates FOXP3 expression in human CD4+ CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 108(5):1571–1579. doi:blood-2006-02-004747
Bailey SR, Nelson MH, Himes RA, Li Z, Mehrotra S, Paulos CM (2014) Th17 cells in cancer: the ultimate identity crisis. Front Immunol 5:276. doi:10.3389/fimmu.2014.00276
Gomez-Rodriguez J, Wohlfert EA, Handon R, Meylan F, Wu JZ, Anderson SM, Kirby MR, Belkaid Y, Schwartzberg PL (2014) Itk-mediated integration of T cell receptor and cytokine signaling regulates the balance between Th17 and regulatory T cells. J Exp Med 211(3):529–543. doi:10.1084/jem.20131459
Chalmin F, Mignot G, Bruchard M, Chevriaux A, Vegran F, Hichami A, Ladoire S, Derangere V, Vincent J, Masson D, Robson SC, Eberl G, Pallandre JR, Borg C, Ryffel B, Apetoh L, Rebe C, Ghiringhelli F (2012) Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity 36(3):362–373. doi:10.1016/j.immuni.2011.12.019
Albesiano E, Han JE, Lim M (2010) Mechanisms of local immunoresistance in glioma. Neurosurg Clin N Am 21(1):17–29. doi:10.1016/j.nec.2009.08.008
Rolle CE, Sengupta S, Lesniak MS (2012) Mechanisms of immune evasion by gliomas. Adv Exp Med Biol 746:53–76. doi:10.1007/978-1-4614-3146-6_5
Fujita M, Zhu X, Sasaki K, Ueda R, Low KL, Pollack IF, Okada H (2008) Inhibition of STAT3 promotes the efficacy of adoptive transfer therapy using type-1 CTLs by modulation of the immunological microenvironment in a murine intracranial glioma. J Immunol 180(4):2089–2098. doi:10.4049/jimmunol.180.4.2089
Bao JJ, Fokt I, Szymanski S, Priebe W (2005) Inhibition of constitutively active STAT3 by WP1066 suppresses proliferation and induces apoptosis in pancreatic cancer cells. Clin Cancer Res 11(24):9026S–9027S
Kupferman ME, Zhou G, Zhao M, Jasser S, Dakak-Yazici Y, Priebe W, Myers JN (2006) A novel inhibitor of STAT3 signaling in head and neck squamous cell carcinoma. In: 97th American Association of Cancer Research Annual Meeting, Washington, DC
Kong LY, Abou-Ghazal MK, Wei J, Chakraborty A, Sun W, Qiao W, Fuller GN, Fokt I, Grimm EA, Schmittling RJ, Archer GE Jr, Sampson JH, Priebe W, Heimberger AB (2008) A novel inhibitor of signal transducers and activators of transcription 3 activation is efficacious against established central nervous system melanoma and inhibits regulatory T cells. Clin Cancer Res 14(18):5759–5768
Hatiboglu MA, Kong LY, Wei J, Wang Y, McEnery KA, Fuller GN, Qiao W, Davies MA, Priebe W, Heimberger AB (2012) The tumor microenvironment expression of p-STAT3 influences the efficacy of cyclophosphamide with WP1066 in murine melanoma models. Int J Cancer 131(1):8–17. doi:10.1002/ijc.26307
Iwamaru A, Szymanski S, Iwado E, Aoki H, Yokoyama T, Fokt I, Hess K, Conrad C, Madden T, Sawaya R, Kondo S, Priebe W, Kondo Y (2007) A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene 26(17):2435–2444. doi:10.1038/sj.onc.1210031
Kong LY, Wu AS, Doucette T, Wei J, Priebe W, Fuller GN, Qiao W, Sawaya R, Rao G, Heimberger AB (2010) Intratumoral mediated immunosuppression is prognostic in genetically engineered murine models of glioma and correlates to immunotherapeutic responses. Clin Cancer Res 16(23):5722–5733. doi:10.1158/1078-0432.CCR-10-1693
Hussain SF, Kong LY, Jordan J, Conrad C, Madden T, Fokt I, Priebe W, Heimberger AB (2007) A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res 67(20):9630–9636. doi:10.1158/0008-5472.CAN-07-1243
Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, Gumin J, Henry V, Colman H, Priebe W, Sawaya R, Lang FF, Heimberger AB (2010) Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther 9(1):67–78. doi:10.1158/1535-7163.MCT-09-0734
Sen M, Grandis JR (2012) Nucleic acid-based approaches to STAT inhibition. JAKSTAT 1(4):285–291. doi:10.4161/jkst.22312
Wang X, Crowe PJ, Goldstein D, Yang JL (2012) STAT3 inhibition, a novel approach to enhancing targeted therapy in human cancers (review). Int J Oncol 41(4):1181–1191. doi:10.3892/ijo.2012.1568
Gu J, Li G, Sun T, Su Y, Zhang X, Shen J, Tian Z, Zhang J (2008) Blockage of the STAT3 signaling pathway with a decoy oligonucleotide suppresses growth of human malignant glioma cells. J Neurooncol 89(1):9–17. doi:10.1007/s11060-008-9590-9
Herrmann A, Priceman SJ, Kujawski M, Xin H, Cherryholmes GA, Zhang W, Zhang C, Lahtz C, Kowolik C, Forman SJ, Kortylewski M, Yu H (2014) CTLA4 aptamer delivers STAT3 siRNA to tumor-associated and malignant T cells. J Clin Invest 124(7):2977–2987. doi:10.1172/JCI73174
See AP, Han JE, Phallen J, Binder Z, Gallia G, Pan F, Jinasena D, Jackson C, Belcaid Z, Jeong SJ, Gottschalk C, Zeng J, Ruzevick J, Nicholas S, Kim Y, Albesiano E, Pardoll DM, Lim M (2012) The role of STAT3 activation in modulating the immune microenvironment of GBM. J Neurooncol 110(3):359–368. doi:10.1007/s11060-012-0981-6
Wei J, Wang F, Kong LY, Xu S, Doucette T, Ferguson SD, Yang Y, McEnery K, Jethwa K, Gjyshi O, Qiao W, Levine NB, Lang FF, Rao G, Fuller GN, Calin GA, Heimberger AB (2013) miR-124 inhibits STAT3 signaling to enhance T cell-mediated immune clearance of glioma. Casncer Res 73(13):3913–3926. doi:10.1158/0008-5472.CAN-12-4318
Fuh B, Sobo M, Cen L, Josiah D, Hutzen B, Cisek K, Bhasin D, Regan N, Lin L, Chan C, Caldas H, DeAngelis S, Li C, Li PK, Lin J (2009) LLL-3 inhibits STAT3 activity, suppresses glioblastoma cell growth and prolongs survival in a mouse glioblastoma model. Br J Cancer 100(1):106–112. doi:10.1038/sj.bjc.6604793.6604793
Molavi O, Ma Z, Hamdy S, Lai R, Lavasanifar A, Samuel J (2008) Synergistic antitumor effects of CpG oligodeoxynucleotide and STAT3 inhibitory agent JSI-124 in a mouse melanoma tumor model. Immunol Cell Biol 86(6):506–514. doi:10.1038/icb.2008.27
Molavi O, Ma Z, Hamdy S, Lavasanifar A, Samuel J (2009) Immunomodulatory and anticancer effects of intra-tumoral co-delivery of synthetic lipid A adjuvant and STAT3 inhibitor, JSI-124. Immunopharmacol Immunotoxicol 31(2):214–221. doi:10.1080/08923970802380452
Kong LY, Gelbard A, Wei J, Reina-Ortiz C, Wang Y, Yang EC, Hailemichael Y, Fokt I, Jayakumar A, Qiao W, Fuller GN, Overwijk WW, Priebe W, Heimberger AB (2010) Inhibition of p-STAT3 enhances IFN-alpha efficacy against metastatic melanoma in a murine model. Clin Cancer Res 6(9):2550–2561. doi:10.1158/1078-0432.CCR-10-0279
Acknowledgments
We thank Audria Patrick for assistance with manuscript preparation and David M. Wildrick, Ph.D. for editorial assistance.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ferguson, S.D., Srinivasan, V.M. & Heimberger, A.B. The role of STAT3 in tumor-mediated immune suppression. J Neurooncol 123, 385–394 (2015). https://doi.org/10.1007/s11060-015-1731-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-015-1731-3