
Abstract
Diffuse gliomas are represented in the 2007 WHO classification as astrocytomas, oligoastrocytomas and oligodendrogliomas of grades II and III and glioblastomas WHO grade IV. Molecular data on these tumors have a major impact on prognosis and therapy of the patients. Consequently, the inclusion of molecular parameters in the WHO definition of brain tumors is being planned and has been forwarded as the “ISN-Haarlem” consensus. We, here, analyze markers of special interest including ATRX, IDH and 1p/19q codeletion in a series of 405 adult patients. Among the WHO 2007 classified tumors were 152 astrocytomas, 61 oligodendrogliomas, 63 oligoastrocytomas and 129 glioblastomas. Following the concepts of the “ISN-Haarlem”, we rediagnosed the series to obtain “integrated” diagnoses with 155 tumors being astrocytomas, 100 oligodendrogliomas and 150 glioblastomas. In a subset of 100 diffuse gliomas from the NOA-04 trial with long-term follow-up data available, the “integrated” diagnosis had a significantly greater prognostic power for overall and progression-free survival compared to WHO 2007. Based on the “integrated” diagnoses, loss of ATRX expression was close to being mutually exclusive to 1p/19q codeletion, with only 2 of 167 ATRX-negative tumors exhibiting 1p/19q codeletion. All but 4 of 141 patients with loss of ATRX expression and diffuse glioma carried either IDH1 or IDH2 mutations. Interestingly, the majority of glioblastoma patients with loss of ATRX expression but no IDH mutations exhibited an H3F3A mutation. Further, all patients with 1p/19 codeletion carried a mutation in IDH1 or IDH2. We present an algorithm based on stepwise analysis with initial immunohistochemistry for ATRX and IDH1-R132H followed by 1p/19q analysis followed by IDH sequencing which reduces the number of molecular analyses and which has a far better association with patient outcome than WHO 2007.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Brain tumor classification of diffuse astrocytic and oligodendroglial tumors
The classical way of diagnosing diffuse astrocytoma and oligodendroglial tumors adheres to the World Health Organization (WHO) 2007 guidelines [29]. Along these lines, the calling of oligoastrocytoma poses great problems best seen in the extensive interobserver variance regarding use of this diagnosis [13, 14, 37]. Ever since the association of 1p/19q loss with more favorable prognosis was proposed [9], a dramatic increase of the diagnosis “oligoastrocytoma” has been observed and deserves critical questioning [7]. On the other hand, clinical studies have repeatedly demonstrated the 1p/19q status of oligodendroglial tumors to be of more prognostic or predictive value than the differentiation between oligodendroglioma and oligoastrocytoma [8, 45, 50]. Further, a recent study demonstrated that the majority of IDH-mutated oligoastrocytoma can be resolved as either astrocytoma or oligodendroglioma by histological methods [40]. A novel approach for more stringently diagnosing diffuse astrocytoma and oligodendroglial tumors has been outlined in the recently published “ISN-Haarlem” guidelines.
“ISN-Haarlem” guidelines
To discuss the inclusion of molecular data into the next World Health Organization (WHO) classification of central nervous system tumors, a meeting under sponsorship of the International Society of Neuropathology (ISN) has been held in Haarlem, the Netherlands [30]. Central to the consensus “ISN-Haarlem” guidelines is the proposal to define diagnostic entities as narrowly as possible and to include, where applicable, molecular data to come up with an “integrated diagnosis”. This could lead, for example, to a classification separating astrocytoma and oligodendroglioma into groups based on IDH1/IDH2 mutation and 1p/19q status, leaving aside a probably small group of tumors tentatively called “diffuse glioma, not otherwise specified”.
IDH1 and IDH2 and G-CIMP
IDH1 mutations were first reported in an exome sequencing study on glioblastoma, predominantly in the so-called secondary glioblastoma with a history of a lower grade precursor tumor [36]. A high incidence of IDH1 mutations was subsequently established for diffuse astrocytoma, oligoastrocytoma and oligodendroglioma [2, 6, 19, 48, 55]. IDH2 mutations are much less frequent but occur in the same set of brain tumors [17, 55]. The relevance of IDH1 mutations as a favorable prognostic marker has been demonstrated in several studies [16, 41, 49, 50]. Since IDH1 mutations of the R132H type constitute more than 90 % of all IDH1 and IDH2 mutations in glioma [17], the development of a mutation-specific antibody greatly advanced the determination of IDH1 status in neuropathological diagnosis [12]. Due to the extraordinarily high lineage specificity of IDH1 and IDH2 mutations for diffuse astrocytoma, oligoastrocytoma and oligodendroglioma, IDH1 immunohistochemistry (IHC) is widely applied and has developed into a backbone for differential diagnosis of glioma [10, 11].
The glioma-CpG island methylator phenotype (G-CIMP) was initially identified in glioblastomas, stratifying into a G-CIMP positive cluster with better outcomes and a G-CIMP negative cluster with worse prognosis [34]. All tumors with IDH mutations exhibited a G-CIMP positive phenotype which is due to IDH mutation-induced production of 2-hydroxyglutarate and its subsequent effects on DNA methylation [54]. Thus, a G-CIMP positive phenotype in glioblastoma is highly suggestive for presence of an IDH1 or IDH2 mutation.
1p/19q codeletion
The detection of frequent losses of chromosomal arm 19q in oligodendroglioma [47] was followed by the detection of 1p losses in these same tumors [4], and the recognition that these alterations frequently cooccur [3, 27, 39]. The presence of 1p and 19q deletions was later identified as a decisive predictor for overall survival and response to chemotherapy [9, 43]. However, subsequent studies demonstrated that partial losses of 1p were associated with much poorer survival than loss of the entire arm [20]. Thus, it is now believed, that the combined and entire loss of the chromosomal arms 1p and 19q (1p/19q loss) is the parameter associated with favorable prognosis. The basis of combined 1p/19q loss is believed to be an unbalanced t(1;19) translocation event [15, 21].
ATRX
Somatic mutations in the alpha thalassemia/mental retardation syndrome X-linked (ATRX) gene were first detected in pancreatic neuroendocrine tumors (PanNET) [23], followed by pediatric glioblastomas [42]. Mutation analysis of ATRX in series of adult gliomas revealed a high incidence of this alteration in diffuse astrocytomas opposed by a lower incidence in oligodendrogliomas, oligoastrocytomas and glioblastomas [22, 24, 25]. ATRX status in combination with 1p/19q and IDH1/IDH2 statuses has been used to generate a molecular diagnostic algorithm which turned out to be superior to classical neuropathological techniques in building patient groups with different times to treatment failure [51]. Commonly, ATRX mutation results in a truncated protein and in abrogated protein expression [18, 28]. These findings suggest a considerable potential for ATRX IHC in distinguishing between diffuse astrocytic and oligodendroglial tumors.
Aim of the study
Several studies have identified ATRX IHC as promising tools for classifying brain tumors. However, their conclusions lack the desirable precision for diagnostic use due to heterogeneity of the tumor series examined with problems including underdiagnosed glioblastomas, the wide variation in diagnosing oligoastrocytomas and the shortcoming of precisely differentiating between partial and complete losses on chromosomal arms 1p and 19q. The current series examines ATRX expression on tumors stringently analyzed for molecular alterations and classified according to the Haarlem consensus concepts of the International Society of Neuropathology for nervous system tumor classification [30]. The study has two primary aims: first to establish a technical standard for ATRX IHC and second to establish an algorithm for using ATRX, IDH1-R132H IHC, 1p/19 analyses and IDH sequencing in the diagnosis of diffuse gliomas.
Materials and methods
Tissue selection
Tumor tissues were obtained from the archives of the Departments of Neuropathology at the University Medical Centers of Heidelberg and Tuebingen and the Neurological Institute/Edinger Institute Frankfurt/Main (Germany). Research use of tissues and anonymization of data were in accordance with local ethical approvals. The series contains tumor tissues from 405 patients. All tumors were initially classified and graded according to the current WHO 2007 guidelines [29]. The series consisted of 152 astrocytomas, 61 oligodendrogliomas, 63 oligoastrocytomas and 129 glioblastomas including 93 ordinary glioblastomas, 12 glioblastomas with oligodendroglial component, 7 secondary glioblastomas 3 giant cell glioblastomas and 14 gliosarcomas. Inclusion criteria were the availability of tissue blocks and availability of an Illumina Infinium HumanMethylation450 BeadChip (450k)-based copy number profile. 100 cases from the NOA-04 trial were included [50]. Since only anaplastic gliomas were included in NOA-04, there is an over representation of anaplastic gliomas (WHO grade III) in comparison to the WHO grade II counterparts. All tumors were reevaluated in knowledge of the 1p/19q, 7p/10q and IDH status. This reevaluation was based on the Haarlem consensus concepts of the International Society of Neuropathology for nervous system tumor classification [30]. Final and initial diagnoses are provided in supplementary Table 1.
Determination of 1p/19q codeletion, 7p gain, 10q loss, EGFR amplification, MGMT promoter methylation and G-CIMP phenotype by 450k array analysis
The Illumina Infinium HumanMethylation450 BeadChip (450k) array was used to obtain the DNA methylation status of 482,421 CpG sites (Illumina, San Diego, USA) according to the manufacturer’s instructions at the Genomics and Proteomics Core Facility of the DKFZ. The array data were used to calculate a low-resolution copy number profile as previously described [44]. The probability of MGMT promoter methylation from 450k array data was estimated as previously described [1]. 334 cases were scored as methylated or unmethylated, in 71 cases the methylation status was unsure. Further, the data were analyzed as previously described to allot the tumors to either a G-CIMP or a non-G-CIMP cluster [53].
IDH1/IDH2, H3F3A and TERT promoter mutation analyses
Primer design for sequencing was based on accession numbers NM_005896 for IDH1, NM_002168 for IDH2, NM_002107.4 for H3F3A (http://www.ncbi.nlm.nih.gov). Primers for H3F3A: forward 5′-CATGGCTCGTACAAAGCAGA-3′; reverse 5′-CAAGAGAGACTTTGTCCCATTTTT-3′. PCR and sequencing was performed as previously described [17]. A 163 bp fragment of the TERT promoter region spanning the hotspot mutations at positions 1,295,228 and 1,295,250 on chromosome 5 was amplified using GoTaq G2 Hot Start Polymerase (Promega, Madison, USA) and the primers hTERT-short-for 5′-CAGCGCTGCCTGAAACTC-3′ and hTERT-short-rev, 5′-GTCCTGCCCCTTCACCTT-3′ as previously described [26]. Sequences were determined using a semi-automated sequencer (ABI 3100 Genetic Analyzer, Applied Biosystems, Foster City) and Sequence Pilot version 3.1 (JSI-Medisys, Kippenheim, Germany) software.
Immunohistochemistry
Immunohistochemistry was conducted on 4-µm-thick formalin-fixed, paraffin-embedded (FFPE) tissue sections mounted on StarFrost Advanced Adhesive slides (Engelbrecht, Kassel, Germany) followed by drying at 80 °C for 15 min. Immunohistochemistry was performed on a BenchMark Ultra immunostainer (Ventana Medical Systems, Tucson, AZ, USA). Sections were stained with anti-IDH1-R132H antibody H09 (Dianova, Hamburg, Germany) as previously described [11].
For ATRX immunohistochemistry, we tested several commercially available antibodies. In our hands the antibody with best specificity for diagnostics is HPA001906 (Sigma-Aldrich, St. Louis, MO; USA). In brief, after deparaffinization, slides were pretreated at 95 °C in Cell Conditioning 1 buffer (Ventana) for 90 min. The sections were incubated with primary antibody (diluted 1:200) for 2 h. Standard Ventana signal amplification was used. The protocol is provided in supplementary file 2. To test for specificity, we used tumors with ATRX mutations confirmed by DNA sequencing. Loss of nuclear ATRX expression was scored as specific if tumor cell nuclei were unstained while nuclei of non-neoplastic cells such as endothelia, microglia, lymphocytes and reactive astrocytes were strongly positive. Of note, moderate staining of tumor cell cytoplasm may occur which most likely is unspecific. However, cytoplasmic staining does not hamper the interpretation of nuclear staining. Parameters to augment staining intensity are increase of antibody concentration, prolongation of pretreatment and of first antibody incubation times. Occasionally, ATRX staining is limited to circumscribed regions of the tissue, most likely due to artificial tissue damage. Evaluation of these partially stained cases seems to be still meaningful because heterogeneous ATRX staining is virtually never observed. In the present study, only a single case of gliosarcoma showed a focal loss of ATRX positivity.
Reagents and staining protocols for IDH1-R132H and ATRX are listed in supplementary file 1.
Statistics
Kaplan–Meier estimator and Cox proportional hazards regression were performed to assess survival data. To compare the performance of Cox regression models, integrated Brier scores displayed as prediction error curves over time were generated using the R package pec [33] (using the “Boot632plus” split method with 1,000 iterations). Briefly, the Brier score represents a weighted (roughly based on the probability of being censored) average of the squared error between estimated survival probability at each time point and observed survival status. All analyses were carried out using R version 3.11 [38]. Fisher’s exact test was used to explore associations between ATRX status and tumor entities or molecular markers.
Results and discussion
Several studies have pointed towards the role of ATRX analysis for the diagnosis of diffuse glioma. Especially, ATRX immunohistochemistry has shown promising results [51]. To test the impact on routine diagnostic neuropathology we here analyzed a series of 154 astrocytomas, 100 oligodendrogliomas and 150 glioblastomas by immunohistochemistry for ATRX expression. Immunohistochemical results were compared with the hallmark markers IDH1/IDH2, 1p/19q codeletion, 7p gain, 10q loss, TERT promoter, EGFR amplification and H3F3A status.
Detection of combined 1p/19q loss and detection of IDH mutations
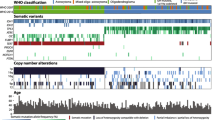
Detection and scoring of combined 1p/19q losses relies on different methods. FISH is probably the most frequently used because this method needs the fewest adaptations to what is available in routine diagnostic laboratories. However, FISH has a major conceptual weakness because reliable information on the integrity of the analyzed chromosome is only obtained on the region the FISH probe is hybridizing to. Thus, it cannot be used to clearly distinguish between partial and complete losses of a chromosomal arm. Classical microsatellite analysis with several probes spanning the targeted chromosome may mostly circumvent this shortcoming; however, this is a tedious approach requiring time, labor and DNA for several rounds of PCR [31]. Multiplex ligation-dependent probe amplification (MLPA) reduces needs for time and material but frequently yields ambiguous results in our hands. The most comprehensive approach allowing data collection on a large set of chromosomal regions is array technology with multiple companies offering solutions. We have adopted an algorithm allowing the generation of a copy number profile (CNP) from hybridizing bisulfite converted DNA to 450K Illumina chips. These CNP provide information on 473 regions on chromosomal arm 1p and 166 regions on chromosomal arm 19q, allowing us to distinguish normal copy number and partial or complete underrepresentation of chromosomal arms with high confidence. Comparison of MLPA and 450k-CNP-based 1p/19q analysis indicated that the latter method more accurately assessed 1p/19q status [52]. Comparable CNP can also be calculated from exome or genome sequencing data sets, but these are more costly and require greater analytical expertise, and are also not so readily applicable to FFPE DNA. Of importance for 1p/19q evaluation is the extent of the deletions on 1p and 19q: There is now general agreement that these losses need to encompass the entire arms, as partial losses have been observed frequently in other gliomas. For example, partial losses of 1p are frequent in glioblastoma and partial losses of 19q are frequently seen in anaplastic astrocytomas WHO grade III [46]. Further, partial losses on 1p have been shown to be associated with poorer survival, in contrast to the favorable prognostic association of entire loss of 1p in oligodendroglioma [20]. Only tumors with loss of both, the entire arms of chromosome 1p and 19q were scored as codeleted for 1p/19q (1p/19q codel) in this study. All other tumors exhibiting balanced profiles, loss of only either 1p or 19q, partial losses of either 1p or 19q, or any combination of these were scored as non-codeleted for 1p/19q (1p/19q noncd). Typical examples for CNP exhibiting 1p/19q codel (tumor 1180) or partial losses on 1p and 19q (tumor 1024) are given in Fig. 1.

Two typical copy number profiles: tumor 1180 exhibits 1p/19q codel. Note that the entire chromosomal arms are missing. In addition, this tumor also exhibits gain on chromosome 16. Tumor 1024 exhibits partial losses on both, 1p and 19q. These losses do not qualify for the oligodendroglioma defining 1p/19q codel. In addition, this tumor also exhibits gains on 7p, 7q and 19p and EGFR amplification (arrow) as well as losses on 9p, 10p, 10q and 17q
We have generated CNP profiles based on 450k array analysis for all 405 cases included in this study. Combined 1p/19q loss was detected in 100 cases (supplementary Table 1). All 405 cases were subjected to IHC using antibody H09 directed at IDH1-R132H protein. Binding of H09 was taken as proof for an IDH1-R132H mutation. 205/405 tumors carried an IDH1-R132H mutation. All astrocytomas and oligodendrogliomas not binding H09 were subjected to sequencing of exon 4 of both, IDH1 and IDH2. The 16 glioblastomas with a G-CIMP positive phenotype were also subjected to IDH1 and IDH2 sequencing. Altogether, we detected 35 rare IDH1 and 13 IDH2 mutations. 15/16 GBM with G-CIMP tested positive for an IDH mutation (supplementary Table 1).
Establishing the “integrated” diagnosis
All tumors were evaluated by two neuropathologists (DR, AvD) and initially classified according to the current WHO guidelines [29]. We then deviated from the current WHO by not accepting the diagnosis of oligoastrocytoma but reclassifying these tumors either as astrocytomas or oligodendrogliomas. We have previously shown that the vast majority of the so-called oligoastrocytoma can be separated into astrocytoma and oligodendroglioma based on IDH1-R132H, ATRX immunohistochemistry and analysis of 1p/19q status [40]. Our reevaluation was carried out in the knowledge of 1p/19q, 7p/10q and IDH1/IDH2 status.
Cases with 1p/19q codel were defined as oligodendroglioma and 1p/19q noncd gliomas were diagnosed as astrocytoma or glioblastoma depending on the presence of necrosis. Our approach eliminated oligoastrocytomas and glioblastomas with oligodendroglial component (GBMo) from our initially WHO 2007 diagnosed series. Anaplastic astrocytomas without IDH mutation and with 7p gain and 10q loss were defined as glioblastomas. These two lesions have been recognized for a long time as characteristic alterations in GBM [5] and have recently been proposed as the first molecular events in non-IDH-mutated GBM [35].
Our concept adheres to the ideas of the Haarlem consensus guidelines of the International Society of Neuropathology for nervous system tumor classification which will be the basis for discussion on the forthcoming update of the WHO Classification [30]. This revision aims at building up more clearly defined tumor entities and at the inclusion of molecular data where applicable.
Thus, after reevaluation our series contained 42 diffuse astrocytomas WHO grade II, 113 anaplastic astrocytomas WHO grade III, 28 oligodendrogliomas WHO grade II, 72 anaplastic oligodendrogliomas WHO grade III, 150 glioblastomas WHO grade IV including 14 of each gliosarcoma and glioblastoma with IDH mutation and 3 giant cell glioblastomas WHO grade IV (Table 1). The initial and the integrated diagnoses for individual patients are provided in supplementary Table 1. A diagram demonstrating the diagnostic shift is given in Fig. 2.

Changes from initial to integrated diagnosis in 405 adult patients with supratentorial glioma. Width of bars indicates relative proportions of the initial tumor groups. A astrocytoma, OA oligoastrocytoma, O oligodendroglioma, GBM glioblastoma, GBMo glioblastoma with oligodendroglial component, GBMs secondary glioblastoma, gcGBM giant cell glioblastoma, GS gliosarcoma
ATRX expression
We tested several antibodies and staining conditions. In our hands, best results were obtained with “HPA001906” and processing on a Ventana Benchmark. For the evaluation of ATRX immunohistochemistry, only nuclear staining was assessed. Loss of ATRX in tumor cells presents with a typical pattern—all tumor cell nuclei are completely unstained while nuclear positivity is seen in vessels, microglia, reactive astrocytes and entrapped neurons. ATRX immunohistochemistry is significantly affected by the quality of material—tumor portions not sufficiently fixed or thermally altered do not provide satisfactory results. In our experience, however, most tissue blocks contain areas with tumor tissue of sufficient quality for evaluation. In regard to tumor cell content in a sample for ATRX evaluation: We did not systematically assess ATRX loss in the infiltration zone of astrocytoma, however, we advise to perform such analysis preferably in solid tumor tissue. We consider this method not suitable for the analysis of single cells. We detected only one single case with focal ATRX loss that could not be attributed to suboptimal quality of the material. Thus, we assume focal loss of ATRX expression to be diagnostic if internal controls in these regions such as endothelia or microglia demonstrate nuclear staining. In our series, 38/42 (90 %) of WHO II and 101/113 (89 %) of WHO III astrocytomas presented with loss of ATRX expression. In contrast, 0/28 (0 %) oligodendrogliomas and 2/72 (3 %) anaplastic oligodendrogliomas showed loss of ATRX. This difference is significant (p = 1.70 × 10−50; Fisher’s exact test). Further, 25/136 (18 %) of glioblastomas and 1/14 (7 %) gliosarcomas displayed loss of ATRX. Loss of ATRX and mutation of the TERT promoter were almost mutually exclusive in this series (p = 9.88 × 10−47; Fisher’s exact test). Typical examples for ATRX staining are given in Fig. 3.

Examples of ATRX staining. Diffuse (a 1313; b 1043) and anaplastic astrocytomas (c 1368; d 1383) with loss of ATRX staining in tumor cell nuclei. The number of non-neoplastic cells, which serve as internal controls, varies markedly between samples but endothelial cells are always positive. Oligodendroglioma (e 1073) and anaplastic oligodendroglioma (f 1308) with strong nuclear ATRX staining. g Glioblastoma (1188) with strong nuclear ATRX expression. Necrotic cells may stain negative. h Glioblastoma (1187) with thermally damaged tissue in which ATRX staining is artificially lost
ATRX association with tumor entity depends on diagnostic approach
Diffuse astrocytoma grade II (AII) diagnosed by WHO 2007 exhibited nuclear ATRX loss in 34/47 (72 %) and anaplastic astrocytoma (AIII) in 72/105 (69 %) of the cases. Applying the “integrated” approach, 36/37 (97 %) AII-IDHmut and 100/103 (97 %) AIII-IDHmut exhibited nuclear ATRX loss. In contrast, only 2/5 of the rare AII-IDHwt and 4/10 (40 %) AIII-IDHwt exhibited nuclear ATRX loss. Oligodendroglioma grade II (OII) diagnosed by WHO 2007 exhibited nuclear ATRX loss in 3/25 (12 %) and anaplastic oligodendroglioma (OIII) in 5/36 (14 %) of the cases. Applying the “integrated” approach, 0/28 OII and 2/72 (3 %) OIII exhibited nuclear ATRX loss. OAII diagnosed by WHO 2007 exhibited nuclear ATRX loss in 5/9 and OAIII in 20/54 (37 %) of the cases. Our “integrated” approach does not recognize the diagnosis of oligoastrocytoma anymore. This distribution clearly shows the potential of ATRX status to differentiate astrocytoma from oligodendroglioma in IDH-mutated tumors and demonstrates the mixed composition of the WHO 2007 oligoastrocytoma groups. Independently of the diagnostic approach, the differences in ATRX association with a tumor entity were remarkably stable across tumor grades. This in fact, holds true also for 1p/19qcodel and IDH mutations.
Association of ATRX expression with 1p/19q and IDH status
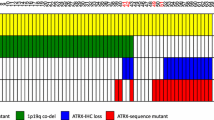
Inclusion of an additional molecular basis for the generation of an integrated diagnosis in our set of 405 tumors resulted in 6 groups. Within these groups, there is strikingly little overlap of the genetic lesions. AII and AIII typically carry IDH mutations and exhibit loss of nuclear ATRX expression. OII and OIII carry 1p/19q codel and IDH mutations and only two of these tumors exhibited loss of nuclear ATRX expression. Interestingly, both of these cases had a wild-type TERT promoter and this is typical for astrocytoma, whereas the vast majority of oligodendrogliomas carry a TERT promoter mutation [25, 26]. GBM and GS typically exhibit the combination of 7p gain and 10q loss accompanied by absence of IDH mutations and maintenance of nuclear ATRX expression. A set of 23 GBM demonstrated nuclear ATRX loss and among these 11 carried an IDH mutation. Interestingly, H3F3A mutations (4 cases with K27M and 4 cases with G34R) in glioblastoma were only seen in combination with nuclear ATRX loss and without IDH mutation—providing evidence for a GBM subset in adult patients with molecular similarities to pediatric GBM [44]. In each of the groups, a few cases did not match the expected pattern: One AII and 6 AIII with IDH mutation did not exhibit ATRX loss. These observations may be explained by hypothesizing that these tumors may carry an ATRX mutation not resulting in the abrogation of protein expression (e.g., a damaging missense mutation). Nine astrocytomas had neither IDH mutation nor ATRX loss. We assume these tumors not to represent a distinct biological entity but to be a mixture of rarer variants of astrocytic tumors lacking some or all known hallmark alterations. Many of these IDH-wt astrocytomas are likely to be variants or early manifestations of glioblastoma which finds support in one exhibiting 7p gain and 10q loss and one an H3F3A mutation. In six astrocytomas with ATRX loss, no IDH mutation could be identified, however, one of these had an H3F3A mutation. A group of 30 GBM did not exhibit any of the mutations tested for in the present study. An overview of these groups and the molecular alterations is given in Fig. 4.

Overview of molecular alterations in 405 patients with “integrated” diagnosis. Each column represents a patient. Red boxes tested positive; dark gray tested balanced for chromosomal status or negative for mutations; light gray IDH1-R132H mutation tested negative, not tested for rare mutations; white not tested. Asterisk indicates 1p/19q status set per definition
Significant differences in “overall survival” in NOA-04 patients depending on diagnostic approach
Comparison of survival plots from 100 NOA-04 study patients demonstrates a significant difference between the WHO 2007 and the “integrated” diagnostic approaches. In regard to both overall survival and time to treatment failure, the “integrated” approach separates groups more stringently. In light of the intermediate position of oligoastrocytoma in the WHO 2007 classification it is of interest that this increase of stringency is achieved although oligoastrocytoma are not diagnosed anymore using the “integrated” diagnosis. The shift in diagnosis in WHO 2007 compared to the “integrated” approach is demonstrated in Fig. 5. With classification aiming at prediction of outcome the “integrated” approach proves much more successful in this series (Table 2; Fig. 6). To compare the performance of Cox regression models based on either the WHO 2007 classification or the “integrated” diagnosis, we calculated the integrated Brier score over time for both models and endpoints (time to treatment failure and overall survival) as well as the R 2 for all models (supplementary Table 2), which showed a markedly improved prediction accuracy (based on the bootstrapped Brier scores) and model fit (R 2) for the “integrated” diagnosis.

Changes from initial to integrated diagnosis in 100 patients with anaplastic astrocytomas, oligoastrocytomas and oligodendrogliomas diagnosed according to WHO 2007 from the NOA04 study. Width of bars indicates relative proportions of the initial tumor groups. A astrocytoma, OA oligoastrocytoma, O oligodendroglioma, GBM glioblastoma

OS (panels on left) and TTF (panels on right) of 100 NOA-04 patients with anaplastic astrocytomas, oligoastrocytomas and oligodendrogliomas diagnosed according to WHO 2007 (upper panels) and receiving an “integrated” diagnosis (middle panels). Lower panels show prediction error curves for OS (left) and TTF (right), depicting the Brier score over time. A higher prediction error indicates a greater difference between observed (known) survival status of patients and the survival probabilities calculated from the respective Cox model, i.e., less prediction accuracy. OS overall survival, TTF time to treatment failure
The combination of ATRX loss and IDH mutation defines the most frequent type of diffuse astrocytoma and progressions thereof
The single finding of most diagnostic potential is the mutual exclusivity of 1p/19q loss and loss of ATRX expression, with the latter found overwhelmingly in IDH-mutated astrocytomas and IDH-mutated glioblastomas. This makes the combination of IDH mutation and ATRX loss the typical molecular fingerprint of diffuse astrocytoma including its manifestation having progressed towards glioblastoma. Likewise, the combination of IDH mutation and ATRX loss virtually excludes the presence of complete 1p/19q loss and thus the diagnosis of oligodendroglioma in its narrower ISN-Haarlem boundaries. The combination of IDH mutation and ATRX loss is, therefore, expected to reduce the need for 1p/19q analysis by a significant fraction of tumors.
Oligoastrocytoma
Oligoastrocytoma is a well-established diagnosis in WHO 2007. Diagnosing tumors on the basis of IDH, ATRX and 1p/19q analysis according to the present suggestion groups these tumors either to astrocytoma or oligodendroglioma. The demonstration of 1p/19q status being of much more relevance to prognosis and treatment response than the morphological differentiation between astrocytoma, oligoastrocytoma and oligodendroglioma clearly warrants such an approach. Further, we recently demonstrated on a series of IDH1-R132H mutated oligoastrocytomas diagnosed at different institutions that these cases could be reliably allotted either to astrocytoma or oligodendroglioma based on molecular data [40], and an epigenome-wide analysis of a large cohort of anaplastic glioma further substantiated that there is no biological basis for the diagnosis of an oligoastrocytoma [53].
Glioblastoma with oligodendroglial differentiation
Four of 12 tumors with initial diagnosis of GBMo exhibited 1p/19q codel, and were therefore classified as anaplastic oligodendroglioma. The remaining 8 tumors were reclassified as GBM, one of which carried an IDH mutation. Separation of these tumors is further supported by the observation that GBMo appears to have a more favorable prognosis, that gliomas with 1p/19q codel nearly inevitably carry an IDH mutation and that IDH mutations are the single most prognostic marker in GBM [49]. Our approach aims at classification, however, it does not solve grading problems. Interestingly, the presence of necrosis appears to be no predictor for poorer overall survival in anaplastic oligodendroglioma [32]. Whether the GBMo reclassified as anaplastic oligodendroglioma should be allotted a WHO grade higher than III needs to be addressed in further studies.
An “integrated” diagnostic approach alters the frequency of astrocytoma and oligodendroglioma diagnoses
Up to WHO 2007, the frequency with which astrocytoma, oligoastrocytoma and oligodendroglioma were diagnosed demonstrated a great inter-institutional variation. Most of this variation was accounted for by the use of the diagnosis oligoastrocytoma. Linking the definition of astrocytoma and oligodendroglioma more strongly to biological parameters and omitting the use of the diagnosis oligoastrocytoma will reduce this inter-institutional variation. In our own experience based on diagnoses from the Department of Neuropathology in Heidelberg, the major part of tumors formerly diagnosed oligoastrocytoma fall into the “integrated” diagnostic group of oligodendroglioma and approximately one-third of all diffuse glioma are diagnosed as oligodendrogliomas, whereas two-thirds of diffuse glioma are diagnosed as astrocytomas.
The integrated approach influences classification, it has less impact on grading of these tumors apart from anaplastic oligoastrocytomas which in our series sometimes were rediagnosed as OII and sometimes as OIII while amongst those rediagnosed as astrocytomas all were anaplastic. This underlines the concept that classification recognizes biological origin while grading reflects to a stage in tumor progression.
Practical approach for diagnosing astrocytomas and oligodendrogliomas using the “integrated” approach
The routine approach to all diffuse astrocytic and oligodendroglial gliomas begins with performing IHC for ATRX and IDH1-R132H expression. Almost all of the tumors exhibiting loss of nuclear ATRX staining are astrocytic. The majority of these tumors will stain positive for IDH1-R132H and nearly all of those which will not are subsequently found to harbor a rare IDH1 or IDH2 or an H3F3A mutation. In contrast, all tumors with nuclear ATRX expression are subjected to 1p/19q analysis. The tumors without a combined deletion of 1p/19q are astrocytic tumors, independent of their IDH status. This group should be sequenced for rare IDH1 and IDH2 mutations based on the important impact of IDH status on prognosis. Astrocytic tumors without ATRX loss, wild type for 1p/19q and without IDH mutation have a strong likelihood of being glioblastoma. Further molecular analyses such as determining 7p/10q status, EGFR or CDK4 amplification are useful.
All tumors exhibiting 1p/19q loss are oligodendrogliomas. Oligodendrogliomas without IDH1-R132H positivity need not be sequenced for rare IDH mutations, which would almost certainly be detectable in all of them. A diagnostic scheme is provided in Fig. 7.

Diagram for efficient use of IDH1/ATRX-IHC, 1p/19q analysis and IDH sequencing for diagnosing adult diffuse astrocytic and oligodendroglial tumors. − no expression, + expression, wt wild-type status, codel codeletion, i.e., combined loss of the entire chromosomal arms, noncd no codeletion. Single asterisk indicate the series is biased towards diffuse astrocytomas and oligodendrogliomas. Double asterisks indicate two tumors had both, ATRX loss and 1p/19q codel, and received the integrated diagnosis oligodendroglioma. Triple asterisks indicate the small group of IDHwt astrocytomas is likely to contain further underdiagnosed GBM
Conclusions
The present data provide an approach to build an “integrated diagnosis” for adult astrocytic and oligodendroglial glioma based on histology and molecular parameters. ATRX immunohistochemistry constitutes an important parameter for this approach. The “integrated” diagnosis results in stringent separation of astrocytoma from oligodendroglioma, omitting the need for diagnosing oligoastrocytoma or ‘glioblastoma with oligodendroglial component’. Stepwise analysis of the molecular parameters significantly reduces the number of molecular tests required for unequivocal diagnosis.
References
Bady P, Sciuscio D, Diserens AC et al (2012) MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathol 124:547–560
Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116:597–602
Bello MJ, Leone PE, Vaquero J, de Campos JM, Kusak ME, Sarasa JL, Pestana A, Rey JA (1995) Allelic loss at 1p and 19q frequently occurs in association and may represent early oncogenic events in oligodendroglial tumors. Int J Cancer 64:207–210
Bello MJ, Vaquero J, de Campos JM, Kusak ME, Sarasa JL, Saez-Castresana J, Pestana A, Rey JA (1994) Molecular analysis of chromosome 1 abnormalities in human gliomas reveals frequent loss of 1p in oligodendroglial tumors. Int J Cancer 57:172–175
Bigner SH, Mark J, Burger PC, Mahaley SM, Bullard DEJ, Muhlbaier LH, Bigner DD (1988) Specific chromosomal abnormalities in malignant human gliomas. Cancer Res 48:405–411
Bleeker FE, Lamba S, Leenstra S et al (2009) IDH1 mutations at residue p. R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat 30:7–11
Burger PC (2002) What is an oligodendroglioma? Brain Pathol 12:257–259
Cairncross G, Wang M, Shaw E et al (2013) Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol 31:337–343
Cairncross JG, Ueki K, Zlatescu MC et al (1998) Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst 90:1473–1479
Capper D, Sahm S, Hartmann C, Meyermann R, von Deimling A, Schittenhelm J (2010) Application of mutant IDH1 antibody to differentiate diffuse glioma from non-neoplastic central nervous system lesions and therapy induced changes. Am J Surg Pathol 34:1199–1204
Capper D, Weißert S, Balss J et al (2010) Characterization of R132H mutation specific IDH1 antibody binding in brain tumors. Brain Pathol 20:245–254
Capper D, Zentgraf H, Balss J, Hartmann C, von Deimling A (2009) Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol 118:599–601
Coons SW, Johnson PC, Scheithauer BW, Yates AJ, Pearl DK (1997) Improving diagnostic accuracy and interobserver concordance in the classification and grading of primary gliomas. Cancer 79:1381–1393
Fuller CE, Schmidt RE, Roth KA, Burger PC, Scheithauer BW, Banerjee R, Trinkaus K, Lytle R, Perry A (2003) Clinical utility of fluorescence in situ hybridization (FISH) in morphologically ambiguous gliomas with hybrid oligodendroglial/astrocytic features. J Neuropathol Exp Neurol 62:1118–1128
Griffin CA, Burger P, Morsberger L, Yonescu R, Swierczynski S, Weingart JD, Murphy KM (2006) Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J Neuropathol Exp Neurol 65:988–994
Hartmann C, Hentschel B, Wick W et al (2010) Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1 mutated glioblastomas and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol 120:707–718
Hartmann C, Meyer J, Balss J et al (2009) Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 118:469–474
Heaphy CM, de Wilde RF, Jiao Y et al (2011) Altered telomeres in tumors with ATRX and DAXX mutations. Science 333:425
Ichimura K, Pearson DM, Kocialkowski S, Backlund LM, Chan R, Jones DT, Collins VP (2009) IDH1 mutations are present in the majority of common adult gliomas but are rare in primary glioblastomas. Neuro Oncol 11:341–347
Idbaih A, Marie Y, Pierron G et al (2005) Two types of chromosome 1p losses with opposite significance in gliomas. Ann Neurol 58:483–487
Jenkins RB, Blair H, Ballman KV et al (2006) A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 66:9852–9861
Jiao Y, Killela PJ, Reitman ZJ et al (2012) Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 3:709–722
Jiao Y, Shi C, Edil BH et al (2011) DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331:1199–1203
Kannan K, Inagaki A, Silber J et al (2012) Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget 3:1194–1203
Killela PJ, Reitman ZJ, Jiao Y et al (2013) TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci 110:6021–6026
Koelsche C, Sahm F, Capper D et al (2013) Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol 126:907–915
Kraus JA, Koopmann J, Kaskel P, Maintz D, Brandner S, Louis DN, Wiestler OD, von Deimling A (1995) Shared allelic losses on chromosomes 1p and 19q suggest a common origin of oligodendroglioma and oligoastrocytoma. J Neuropathol Exp Neurol 54:91–95
Liu XY, Gerges N, Korshunov A et al (2012) Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol 124:615–625
Louis D, Ohgaki H, Wiestler O, Cavenee W (2007) World Health Organization Classification of Tumours of the Central Nervous System. In: Bosman F, Jaffe E, Lakhani S, Ohgaki H (eds) World Health Organization Classification of Tumours, 4 edn. IARC, Lyon
Louis DN, Perry A, Burger P et al (2014) International Society of Neuropathology-Haarlem Consensus Guidelines, for Nervous System Tumor Classification and Grading. Brain Pathol 24:429–435
Louis DN, von Deimling A, Seizinger BR (1992) A (CA)n dinucleotide repeat assay for evaluating loss of allelic heterozygosity in small and archival human brain tumor specimens. Am J Pathol 141:777–782
Miller CR, Dunham CP, Scheithauer BW, Perry A (2006) Significance of necrosis in grading of oligodendroglial neoplasms: a clinicopathologic and genetic study of newly diagnosed high-grade gliomas. J Clin Oncol 24:5419–5426
Mogensen U, Ishwaran H, Gerds TA (2012) Evaluating random forests for survival analysis using prediction error curves. J Stat Softw 50:1–23
Noushmehr H, Weisenberger DJ, Diefes K et al (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522
Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, Charles N, Michor F, Holland EC (2014) Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell 26:288–300
Parsons DW, Jones S, Zhang X et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812
Perry A (2001) Oligodendroglial neoplasms: current concepts, misconceptions, and folklore. Adv Anat Pathol 8:183–199
R-Core-Team (2014) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.r-project.org/
Reifenberger J, Reifenberger G, Liu L, James CD, Wechsler W, Collins VP (1994) Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol 145:1175–1190
Sahm F, Reuss D, Koelsche C et al (2014) Farewell to oligoastrocytoma: in situ molecular genetics favor classification as either oligodendroglioma or astrocytoma. Acta Neuropathol 128:551–559
Sanson M, Marie Y, Paris S et al (2009) Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 27:4150–4154
Schwartzentruber J, Korshunov A, Liu X et al (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in pediatric glioblastoma. Nature 482:226–231
Smith JS, Perry A, Borell TJ et al (2000) Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J Clin Oncol 18:636–645
Sturm D, Witt H, Hovestadt V et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437
van den Bent MJ, Brandes AA, Taphoorn MJ et al (2013) Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol 31:344–350
von Deimling A, Bender B, Jahnke R et al (1994) Loci associated with malignant progression in astrocytomas: a candidate on chromosome 19q. Cancer Res 54:1397–1401
von Deimling A, Louis DN, von Ammon K, Petersen I, Wiestler OD, Seizinger BR (1992) Evidence for a tumor suppressor gene on chromosome 19q associated with human astrocytomas, oligodendrogliomas and mixed gliomas. Cancer Res 52:4277–4279
Watanabe T, Nobusawa S, Kleihues P, Ohgaki H (2009) IDH1 Mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 174:653–656
Weller M, Felsberg J, Hartmann C et al (2009) Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J Clin Oncol 27:5743–5750
Wick W, Hartmann C, Engel C et al (2009) NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol 27:5874–5880
Wiestler B, Capper D, Holland-Letz T, Korshunov A, von Deimling A, Pfister S, Platten M, Weller M, Wick W (2013) ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol 126:443–451
Wiestler B, Capper D, Hovestadt V et al (2014) Assessing CpG island methylator phenotype, 1p/19q codeletion, and MGMT promoter methylation from epigenome-wide data in the biomarker cohort of the NOA-04 trial. Neuro Oncol 16:1630–1638
Wiestler B, Capper D, Sill M et al (2014) Integrated DNA methylation and copy-number profiling identify three clinically and biologically relevant groups of anaplastic glioma. Acta Neuropathol 128:561–571
Xu W, Yang H, Liu Y et al (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19:17–30
Yan H, Parsons DW, Jin G et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773
Acknowledgments
The authors (WW, MW) conducting this work represent the Neurooncology Working Group (NOA) of the German Cancer Society. We gratefully acknowledge the contributions of Ulrike Ernemann, MD and Christoph Meisner, PhD (Tübingen, Germany), Guido Reifenberger, MD and Michael C. Sabel, MD (Düsseldorf, Germany), Susanne Koeppen, MD (Essen, Germany), Otmar Wiestler, MD and Thorsten Pietsch, MD (Bonn, Germany) and Ralf Ketter, MD to the first publication of the study. The study was supported by the Medical Faculty Heidelberg PostDoc Fellowship and the DKFZ Intramural Funding Program, Priority Topic Intratumoral Heterogeneity, to FS. BW is a scholar of the NCT Heidelberg School of Oncology Postdoc Program. We thank Tanja Göck and Viktoria Zeller for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Reuss, D.E., Sahm, F., Schrimpf, D. et al. ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an “integrated” diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol 129, 133–146 (2015). https://doi.org/10.1007/s00401-014-1370-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-014-1370-3