
Abstract
Purpose of Review
Our understanding of the genetic and epigenetic alterations in meningioma and the underlying tumor biology of meningioma has significantly changed over the past decade and resulted in revision of prognostically relevant meningioma subclasses within and beyond the WHO classification of CNS tumors.
Recent Findings
The 2016 WHO classification of CNS tumors recognizes WHO grade I, II, and III based on histopathological features. Recent work has identified genetic alterations with prognostic implications, including mutations of the TERT promoter, loss of function of the DMD gene, and inactivation of the tumor suppressor BAP-1. Studies of DNA methylation patterns in meningiomas have resulted in a novel and prognostically relevant meningioma subclassification schema.
Summary
There have been major advances in our understanding of prognostically relevant genetic and epigenetic changes in meningioma which will hopefully allow for improvement in clinical trial design and the development of more effective therapies for meningioma.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Meningiomas represent 37.6% of primary intracranial and other central nervous system tumors, making them the most common histology of all [1]. They are thought to originate from arachnoid cap cells due to their histologic similarities [2]. In the USA, there are about 30,000 people diagnosed each year, and the incidence is increasing. In 2020, the incidence is projected to increase to 34,210 cases. The median age at diagnosis is 66 years. Females have a higher incidence than males, overall, particularly in non-malignant meningiomas. The 10-year relative survival rate for non-malignant and malignant meningioma is 83.7% and 61.7%, respectively; differences are observed relative to tumor location with higher survival rates for spinal tumors compared to a cerebral location [1]. While the majority (~ 80%) of meningiomas are considered benign and can be observed or cured by surgical resection alone, a subset of up to 20% of meningiomas represent aggressive tumors that recur and continue to grow despite surgical resection and/or radiotherapy [3]. Once patients have failed surgical and radiotherapeutic options, prognosis is typically poor and there are currently no effective medical treatment options.
The development of medical therapies for meningiomas has long been difficult due to multiple and diverse reasons, including incomplete understanding of meningioma pathobiology and therefore lack of meningioma-specific therapeutic targets, the difficulties to develop representative animal models to evaluate novel therapeutic agents, and the significant heterogeneity in clinical trial design [4, 5]. With regard to clinical trials in meningiomas, one major challenge has been the lack of a standardized outcome measure used in these studies. However, a more recent detailed review and meta-analysis of the available clinical studies for progressive meningiomas determined the 6-month progression-free survival (PFS-6) to be the most reliable outcome measure. Based on the studies included in this analysis, the weighted average PFS-6 was 26% (95% CI 19.3–32.7%) for World Health Organization (WHO) grade II/III meningiomas and 29% (95% CI 20.3–37.7%) for WHO I meningiomas. The authors proposed to use these PFS-6 benchmarks for the design of future clinical trials in meningiomas [5]. To further improve and standardize meningioma-specific outcome measures for meningioma studies, the Meningioma Response Assessment in Neuro-Oncology (Meningioma-RANO) working group is working towards standardized radiographic assessment criteria [3, 5].
The past years have brought major advances in our understanding of the molecular underpinnings of meningiomas leading to updated and refined meningioma subclasses based on histopathological, genetic, and epigenetic determinants. This will hopefully spurt the development of more effective and meningioma-specific therapies. This review focuses on the advances in meningioma genetics and epigenetics and will highlight potential therapeutic opportunities.
Histopathological WHO Classification of Meningiomas
Various types of classification systems have been used for meningiomas, dating back to Virchow in the nineteenth century [6]. In 1979, the World Health Organization (WHO) published the “Histologic Typing of Tumours of the Central Nervous System,” known as the “blue book,” in order to establish a standardized grading system of CNS tumors, including meningiomas, based on histological features [7]. The WHO classification of CNS tumors has undergone several updates since then [8,9,10,11]. Based on their malignant potential, the WHO classifies meningiomas as grade I (benign), grade II (intermediate), and grade III (malignant) tumors. Based on the criteria used in the 2007 WHO classification, 80.5% of pathologically confirmed meningiomas were WHO grade I, 17.7% were WHO grade II, and 1.7% were WHO III, respectively [1, 10]. Despite several advances in the recent versions of the WHO classification, the current 2016 WHO classification of CNS tumors remains largely based on histological and cytomorphological criteria as well as mitotic rate. As such, 15 histological meningioma subtypes are recognized and categorized as WHO grade I, II, or III. One change to the previous 2007 WHO classification was that brain invasion was established as an independent histological criterion to justify the diagnosis of a grade II (atypical) meningioma [10, 11]. It is expected that this will result in a proportional increase in WHO grade II meningioma diagnoses.
Overall, a correlation between WHO histopathological grade and patient outcome has been documented in multiple studies [12,13,14,15]. Nevertheless, the current histopathological assessment criteria allow for interrater variability, especially when considering WHO grade II meningiomas. A recent secondary analysis from the NRG Oncology RTOG 0539 trial assessed the concordance rates between histopathological meningioma diagnosis at the enrolling study subsite and at central pathology review. While the concordance rates for histopathological WHO grade I and III diagnoses were 93% and 93.6%, respectively, the concordance rate for WHO grade II meningiomas was only 87.8%. Of the 22 reclassified cases, all but one case involved a WHO grade II correction: 9 were upgraded from a WHO I to WHO II, 8 were upgraded from WHO II to WHO III, two were downgraded from WHO III to II, and two were downgraded from a WHO II to WHO I. Interestingly, the study found the highest concordance rates for the histopathological features of ≥ 20 mitoses/10 HPF (95.3%), anaplasia (93.6%), and brain invasion (92.4%) [16, 17]. Still, the histopathological WHO criteria appear to be insufficient to predict clinical course in some patients; i.e., up to 20% of patients with grade I meningiomas experience tumor recurrence [18,19,20], and some of the patients diagnosed with grade II meningiomas may experience a rather benign clinical course where radiotherapy might confer unnecessary risks [17, 21, 22].
Current Clinical Management of Meningiomas
Surgical resection remains the initial step and mainstay of treatment for most symptomatic meningiomas as it leads to decompression of the affected areas of the CNS, symptom improvement, and allows for a histopathological diagnosis.
The extent of initial resection remains a strong predictor of outcome and is traditionally assessed according to the Simpson grade, a grading system which is based on the surgeon’s assessment of the extent of resection, ranging from Simpson grade I (complete removal of tumor, dura, and bone) to grade V (biopsy/decompression only) [23]. More recently, a number of studies have assessed the validity of the Simpson grading system in the modern era and found that maximum safe resection remains a positive predictor of survival, but only in conjunction with other factors such as histopathological grade, mitotic rate, and tumor location [24, 25]. A recent prospective study determined that statistically significant risk factors associated with incomplete meningioma resection were symptomatic presentation, skull-base location, and bone invasion. In contrast, patient gender, preoperative Karnofsky performance status, WHO grade, and patient age were not predictive of incomplete resection [26].
After initial resection, WHO grade I meningiomas are most often followed by clinical and radiographic observation, with re-resection and/or radiation being offered for any subsequent tumor progression. In the case of a WHO grade III diagnosis, radiation is recommended. For WHO grade II meningiomas, however, the role of radiation following initial resection is controversially discussed with no consistent practice across the USA. Few studies investigate this question, including RTOG 0539 (NCT00895622) and NRG-BN003 (NCT03180268). The ongoing RTOG 0539 trial stratifies patients according to WHO Grade and extent of resection. In this study, gross-totally resected WHO grade II and recurrent WHO grade I meningiomas are considered intermediate risk, and these patients are stratified to receive radiotherapy to 54 Gray (Gy). Patients with recurrent grade II meningiomas, newly diagnosed subtotally resected WHO grade II meningioma, and all WHO grade III meningiomas are considered high-risk and receive radiotherapy to 60 Gy. The initial outcome report for the intermediate-risk group documented a favorable 3-year progression-free survival (PFS) of 93.8% and overall survival OS of 98% in 52 evaluable patients [17]. Additionally, the currently ongoing NRG-BN003 trial randomizes patients with gross-totally resected WHO grade II meningiomas to either observation or radiation to address the uncertainty of postsurgical treatment in this meningioma subgroup.
Once surgical and radiotherapeutic options have been exhausted, there are unfortunately no established medical treatment options or preferred regimens despite numerous clinical trials that have evaluated a number of agents including hydroxyurea, temozolomide, irinotecan, imatinib, erlotinib, gefitinib, and antiangiogenic drugs such as bevacizumab and sunitinib, with or without mTORC1 inhibitors such as everolimus. Unfortunately, these agents have been largely ineffective and there remains a dire need for novel treatment targets and improved medical therapy options for meningiomas [4, 5, 27, 28]. Patient enrollment into clinical trials is therefore strongly encouraged whenever possible.
One such clinical study recently evaluated trabectedin, a cytotoxic agent that is frequently used in sarcomas, in the prospective phase II EORTC-1320-BTG study. Patients with progressive WHO grade II and grade III meningiomas were randomized to either receive trabectedin or the investigator’s choice of therapy. However, based on the recently presented preliminary results, there was no significant improvement in PFS or OS for patients receiving trabectedin, and notably, there was significantly higher toxicity in this group [29].
Somatostatin Analogues
Somatostatin receptor type 2 (SSTR2) is overexpressed on the surface of 79–100% of meningiomas, regardless of grade, and represents a specific biomarker that can be applied diagnostically via immunohistochemical staining and radiographically via 68Gallium (Ga)-DOTATATE PET-MR Imaging [30,31,32]. In addition, SSTR2 is a potential therapeutic target [31, 33], although several clinical trials evaluating somatostatin analogues in monotherapy have been disappointing so far [34, 35]. However, more recently, a single-arm phase II study using octreotide, a synthetic somatostatin analog with affinity to SSTR2 and SSTR5, and everolimus in 20 adults with recurrent meningioma showed more encouraging results by reporting PFS-6 of 55%, and a 6- and 12-month OS of 90% and 75%, respectively. Most tumors on study (78%) demonstrated a decrease of over 50% in growth rate at 3 months on treatment. There was no association observed between SSTR2A expression and PFS or growth rate [36]. In contrast to the somatostatin analogues, 177Lutetium (Lu)-DOTATATE (Lutathera) is a radiolabeled antibody that targets SSTR2 with high affinity and is independent from the downstream effects of the somatostatin-receptor pathways. 177Lu-DOTATATE is used as peptide receptor radionuclide therapy (PRRT) and was recently approved for the treatment of advanced, somatostatin-receptor positive gastroenteropancreatic neuroendocrine tumors [37]. Several case series and small prospective clinical studies have evaluated the utility of 177Lu-DOTATATE and other SSTR2-targeting radionuclide agents with encouraging results [38,39,40,41]. Based on these studies, 177Lu-DOTATATE is now investigated for patients with progressive intracranial meningiomas in two ongoing prospective clinical studies (NCT03971461, NCT04082520). In addition to the potential therapeutic value of 177Lu-DOTATATE, both of these studies also investigate 68Ga-DOTATATE, another SSTR2 binding radionuclide, as a possible predictive imaging biomarker.
Hallmark Molecular Alterations in Meningiomas and Potential Therapeutic Opportunities
The past decade has brought an emerging understanding of genetic, epigenetic, and other molecular alterations in meningiomas which will likely further enhance the accuracy of the pathological diagnosis and clinical risk assessment for individual patients (Table 1).
Copy Number Alterations
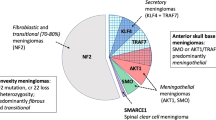
Higher-grade meningiomas have been shown to contain more chromosomal alterations than lower grade tumors. One study showed that while chromosome 22q loss was the most prevalent copy number alteration in both WHO grade II and III meningioma, other variants were also common: 1p loss, 14q loss, and 10q loss. Interestingly, they identified 10q loss in four of the five WHO grade III meningiomas analyzed but in none of the WHO grade II tumors. The authors suggested that this could be one way to differentiate WHO grade II from grade III [42]. In work which we will describe later in this review, classification of meningioma by methylation rather than WHO grading identified patterns of copy alterations in different meningioma subclasses: MC ben-1 nearly always contained deletion 22q (95%), MC ben-2 had no recurrent copy number variations, MC ben-3 often contained chromosome 5 alterations (47%). Intermediate classes commonly had 1p (70% in group A, 89% in group B) and 22q loss (84% in group A and 89% in group B), as well as alterations in chromosome 10 (89% in group B). The malignant class, MC mal, often had CDKN2A deletion (70%) [43].
Neurofibromin 2 and SMARCB1
Alterations in the Neurofibromin 2 (NF2) gene are the most commonly seen aberrations in meningiomas. The NF2 gene is located on Chromosome 22 and encodes for a cell membrane-related protein called merlin or schwannomin that functions as a tumor suppressor [44,45,46]. Alterations in the NF2 gene can be caused by mutation, allelic inactivation, splicing alterations, or Chromosome 22 loss and have been implicated in approximately 40–60% of sporadic meningiomas, making it the single most frequent gene alteration in this tumor [47,48,49,50,51,52]. Additionally, loss of NF2 function is seen in nearly all meningiomas associated with neurofibromatosis type II, an autosomal dominant hereditary condition in which inherited loss of NF2 function leads to development of bilateral vestibular schwannomas, intracranial and spinal meningiomas, and other spinal tumors such as ependymoma. Meningiomas with NF2 mutations and/or chromosome 22 loss are more likely to be atypical and localize to the cerebral and cerebellar hemispheres [51]. In a recent study of intraventricular meningiomas, loss of NF2 function was confirmed to be the most common genetic alteration; loss of Chromosome 22 was seen in 89% of cases, and deleterious NF2 mutations were seen in 44% of cases [53].
The mechanisms by which merlin loss of function leads to meningiomagenesis are insufficiently understood; however, it is speculated that merlin plays a role in contact-dependent inhibition of proliferation, mediated by mTORC1, with effects on the downstream PI3K-Akt-mTOR pathway [54,55,56].
The co-occurrence of NF2 and SMARCB1 mutations, the latter in close proximity to NF2 on Chromosome 22, is seen in some of the meningiomas and schwannomas harboring NF2 alterations [57]. An ongoing Alliance of Clinical Oncology Trial is currently evaluating the efficacy of the focal adhesion kinase (FAK) inhibitor GSK2256098 for meningiomas with NF2 alterations (NCT02523014), which is exciting given that merlin deficiency has been shown to predict for FAK inhibitor sensitivity in tumor xenograft models [58].
Alterations of the PI3K-AKT1-mTOR Pathway
A subset of meningiomas that do not harbor NF2 alterations is characterized by dysregulation of the PI3K-AKT1-mTOR signaling pathway. One study detected the AKT1 p.Glu17Lys mutation in 13% of WHO grade I and II meningiomas which leads to constitutive activation of the AKT1 protein and therefore overactivation of the PI3K-AKT1-mTOR pathway [59]. Other studies confirmed AKT1 mutations as a hallmark alteration in meningiomas of the skull base and higher rate of tumor recurrence [51, 60, 61]. Most recently, the AKT1 p.Glu17Lys mutation was also described as the hallmark mutation in meningiomas located at the foramen magnum [62].
The previously mentioned Alliance in Clinical Oncology trial is evaluating the AKT inhibitor capivasertib (AZD5363) for patients with progressive meningiomas characterized by an AKT1 mutation (NCT02523014). One study evaluated the efficacy of the mTOR inhibitor everolimus with bevacizumab but was unfortunately disappointing [63]. The dual mTORC1 and mTORC2 inhibitor Vistusertib (AZD2014) is currently being explored in an ongoing phase II clinical trial in patients with sporadic grade II and III meningiomas (NCT03071874) and in NF2-associated progressive or symptomatic meningiomas of any grade (NCT02831257).
Other alterations of the PI3K signaling pathway have been described in several studies [64, 65], and the oncogenic PIK3CA mutation is found in up to 7% of non-NF2 altered meningiomas [66]. While PIK3CA and NF2, AKT1, and SMO mutations are mutually exclusive, PIK3CA mutations appeared to co-occur with those in TNF receptor associated factor 7 (TRAF7), which encodes for an E3 ubiquitin ligase that interacts with pro-apoptotic signaling pathways [67]. TRAF7 mutations are seen in up to 24% of WHO grade I and II meningiomas, suggesting a potential cell cycle regulatory function in meningiomas [59].
Alterations of the Hedgehog Signaling Pathway
Activation of the Hedgehog signaling pathway is a characteristic alteration in a subset of meningiomas that is not characterized by alterations in NF2 or AKT1. Hotspot mutations in Smoothened, Frizzled Class Receptor (SMO), are seen in 1–5% of meningiomas and are associated with anterior skull-base/olfactory groove location and higher potential for tumor recurrence [51, 59, 68, 69]. The SMO-inhibitor vismodegib is therefore evaluated in patients with progressive meningiomas characterized by alterations in SMO (NCT02523014).
Genetic Alterations with Potential Prognostic Implications
Telomerase Reverse Transcriptase Promoter Mutations
The presence of either of the two hotspot TERTp mutations C228T and C250T has been associated with more aggressive meningiomas [70, 71]. In a cohort of over 250 meningiomas, these TERTp mutations were shown to occur in 6.4%, most commonly in those with WHO Grade III. Clinical course in patients with TERTp-mutant meningiomas was notable for higher risk of recurrence and shorter time to progression, and this appeared independent from WHO grade. In the study, the median time to progression in those meningiomas possessing a TERTp hotspot mutation was 10.1 months compared with 179 months in TERTp wildtype meningiomas [72]. TERTp mutations were confirmed as poor prognostic indicators in several recent studies [73,74,75,76].
Loss of Function of the Dystrophin-encoding and Muscular Dystrophy-associated Gene
In a recent study of 169 meningiomas of 53 patients, DMD inactivation by genomic deletion or loss of protein expression was detected in 32% of patients with progressive meningiomas and associated with shorter overall survival. While TERTp alterations co-occurred in 18.8% of these patients, multivariate analysis revealed DMD inactivation and TERTp mutations as independent predictors of poor prognosis [74].
Loss of BRCA1 associated protein-1, Tumor Protein p53 (TP53) Mutations, and Loss of CDKN2A/B
BAP-1 is a tumor suppressor gene encoding for a deubiquitylating enzyme, the ubiquitin carboxyl-terminal hydrolase BAP1. Inactivating BAP-1 mutations have been associated with rhabdoid meningioma (WHO III) histology, heralding early meningioma recurrence and aggressive clinical course [77, 78]. Similar to other cancers and CNS tumors, mutations in the tumor suppressor gene TP53 and loss of function of CDKN2A/B are found in higher-grade meningiomas (WHO II/III) and are associated with cell anaplasia and high risk of recurrence [79, 80]. In a study evaluating a total of 30 recurrent and non-recurrent meningiomas, identification of a new single nucleotide variant in CDKN2A was significantly associated with risk of recurrence. Additionally, the majority of recurrent tumors included in this study were noted to have one of three well-known CDKN2A alterations [71, 81].
Forkhead Box M1
Another gene that appears to signal more aggressive behavior in meningioma is Forkhead box M1 (FOXM1), which encodes for the FOXM1 transcription factor, a regulator of the cell cycle and therefore integral to tumor proliferation. Two studies have shown that FOXM1 mRNA expression levels were highest in grade III, followed by grade II, and were lowest in grade I meningiomas [69, 82]. A FOXM1 inhibitor, siomycin A, has shown activity against cell proliferation in cell lines [82].
Kruppel-like factor 4
In meningioma, the KLF4 p.K409Q hotspot mutation nearly always co-occurs with TRAF7 mutations and was in approximately 11% of non-NF2 meningiomas [59, 83, 84]. Like the other members of the large family of Kruppel-like factors, KLF4 is involved in regulation of gene transcription, inhibiting cell cycle progression by binding to the cell cycle inhibitor p21Cip1/Waf1 promoter and recruiting p53 [85]. In addition, the Kruppel-like factors may play an important role in the induction and maintenance of pluripotent stem cells in cell cultures [86]. Interestingly, KLF4 mutations have been primarily associated with WHO grade 1 secretory meningiomas—a meningioma subtype that is characterized by low risk of recurrence [59, 61, 83].
RNA Polymerase II Subunit A
One study found recurrent somatic mutations in POLR2A in 6% of meningiomas and appeared mutually exclusive to any of the other known meningioma driver mutations. Presence of a POLR2A mutation was associated with location in the tuberculum sellae and meningothelial (WHO I) histology, i.e., low risk of recurrence [87].
Methylation Classification of Meningiomas
In a recent multicenter study, 497 meningiomas were analyzed for their genome-wide DNA methylation patterns and additionally characterized by DNA copy number analysis, mutational profiling, and RNA sequencing. The DNA methylation profiling led to segregation of six meningioma subclasses with distinct clinical behavior, i.e., methylation class (MC) benign 1–3, MC intermediate A and B, and MC malignant. Although WHO grade I tumors were enriched in MC benign 1–3, and grade III tumors were enriched in MC malignant, WHO grade II tumors were scattered throughout the six classes. In this analysis, the methylation subclasses were more accurately predictive of clinical course and risk of recurrence compared with WHO grade. In addition, the known hallmark molecular aberrations demonstrated enrichment in certain methylation classes (Fig. 1) [43]. Another study established two distinct methylation subclasses based on analysis of 140 meningioma samples [88].

Schematic overview of the six identified methylation classes and their molecular and clinical characteristics [43]
Most recently, a model of 5-year recurrence free survival (RFS) was developed to assess individual patient risk of meningioma recurrence, generating a probability score based on methylation results, extent of resection (Simpson grade), and WHO grade. The model was developed based on a discovery cohort of 282 meningiomas and validated in two independent validation cohorts comprising of 140 and 46 samples, respectively [89]. It is expected that these and other methylation classification schemata will be adopted to risk-stratify meningioma patients in future clinical trials.
H3K27 Trimethylation
Beyond the epigenetic patterns of DNA methylation in the characterized meningioma subclasses [43], epigenetic modification on the level of histones, in particular trimethylation at the 27th lysine residue of Histone H3 (H3K27me3), was recently established to represent a poor prognostic biomarker in meningioma. In a study of 232 meningiomas, reduced staining for H3K27me3 by immunohistochemistry (IHC) was significantly associated with increased risk for more rapid progression and association with the MC-malignant methylation subclass mentioned previously [81]. In addition, H3K27me3 negative cases were enriched for tumors containing NF2 and SUFU mutations. IHC staining for H3K27me3 may represent a useful adjunct diagnostic marker to refine the histopathological diagnosis of meningiomas; however, this needs to be validated in future prognostic studies [90].
Meningiomas and Their Immunological Tumor Microenvironment
The immunological tumor microenvironment of meningiomas is insufficiently understood. In one study, 51 frozen meningioma samples were evaluated for tumor and non-tumor cell composition; this demonstrated that the majority (76 ± 20%) of cells represented CD45(−) neoplastic cells, and the majority of tumor infiltrating CD45(+) immune cells were tissue macrophages (22 ± 18%) with low numbers of tumor infiltrating lymphocytes (2 ± 2%) [91]. Various studies have evaluated the degree of programmed death–ligand 1 (PD-L1) expression in meningiomas with highly variable expression levels ranging from anywhere between 0.9 and 40.9% in grade I, 2.6 and 60.5% in grade II, and 8.8 and 88.9% in grade III meningiomas [92,93,94]. This may, at least in part, be due to the various assays being used in clinical practice to test for PD-L1 expression [95]. In another study, immunogenic neoantigens were detected in the majority of the meningiomas tested [96]. However, tumor mutational burden (TMB) of meningiomas was generally low; one study sequenced 228 meningioma samples and reported a mean TMB of 4.2 mutations/Mb only. In a pooled data set of 843 samples, only 21 (2.5%) had a total of TMB > 10 mutations/Mb which would be conventionally considered as high [97]. Nevertheless, of these 21 patients, the authors detailed one patient whose tumor had a documented MSH2/MSH6 inactivation and TMB of 38 mutations/Mb and had a durable response to nivolumab. Another report documented treatment response of a presumed sphenoid wing meningioma in a patient receiving nivolumab for concomitant metastatic lung cancer although no biopsy was obtained [98].
In spite of these studies, which suggest that only a small subset of meningiomas may respond to immunotherapies, several clinical trials are underway that evaluate the efficacy of various checkpoint inhibitors for patients with progressive meningiomas (e.g., NCT02648997, NCT03279692, NCT03604978, NCT032667836).
Conclusions
Meningiomas are the most common primary intracranial and spinal tumors, the majority of which are benign and will not recur. However, a subset of them represent aggressive brain tumors for which there are no effective medical therapies once surgery and radiation have failed. Currently, ongoing studies evaluate the efficacy of agents targeting NF2, AKT1, SMO, and SSTR2, as well as immunotherapies. There have been significant advances in the understanding of the genetic and epigenetic alterations in meningiomas, particularly in their characterization into prognostically relevant methylation subclasses. As with other intracranial tumors, histology and WHO grade may not be sufficient for accurate prediction of recurrence risk. Therefore, these advances will hopefully result in improved patient risk stratification for enrollment in future clinical trials and help to advance the field in the quest to find effective medical therapies for meningioma.
References
Ostrom QT, Cioffi G, Gittleman H, Patil N, Waite K, Kruchko C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro Oncol. 2019;21(Supplement_5):v1–v100.
Wiemels J, Wrensch M, Claus EB. Epidemiology and etiology of meningioma. J Neuro-Oncol. 2010;99(3):307–14.
Rogers L, Barani I, Chamberlain M, Kaley TJ, McDermott M, Raizer J, et al. Meningiomas: knowledge base, treatment outcomes, and uncertainties. A RANO review. 2015;122(1):4.
Wen PY, Quant E, Drappatz J, Beroukhim R, Norden AD. Medical therapies for meningiomas. J Neuro-Oncol. 2010;99(3):365–78. https://doi.org/10.1007/s11060-010-0349-8.
Kaley T, Barani I, Chamberlain M, McDermott M, Panageas K, Raizer J, et al. Historical benchmarks for medical therapy trials in surgery- and radiation-refractory meningioma: a RANO review. Neuro Oncol. 2014;16(6):829–40.
Patil CG, Laws ER. Chapter 1: Meningioma: history of the tumor and its management. In: Pamir M, Black PM, Fahlbusch R, editors. Meningiomas: a comprehensive text; 2010. p. 3–9.
Zülch KJ. Histological typing of tumours of the central nervous system. Geneva: World Health Organization; 1979.
Kleihues P, Burger PC, Scheithauer BW. The new WHO classification of brain tumours. Brain Pathol. 1993;3(3):255–68. https://doi.org/10.1111/j.1750-3639.1993.tb00752.x.
Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, Burger PC, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61(3):215–25.
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous. System. 2007;114(2):97–109.
Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–20. https://doi.org/10.1007/s00401-016-1545-1.
Ho DM, Hsu CY, Ting LT, Chiang H. Histopathology and MIB-1 labeling index predicted recurrence of meningiomas: a proposal of diagnostic criteria for patients with atypical meningioma. Cancer. 2002;94(5):1538–47. https://doi.org/10.1002/cncr.10351.
Combs SE, Schulz-Ertner D, Debus J, von Deimling A, Hartmann C. Improved correlation of the neuropathologic classification according to adapted World Health Organization classification and outcome after radiotherapy in patients with atypical and anaplastic meningiomas. Int J Radiat Oncol Biol Phys. 2011;81(5):1415–21.
Domingues PH, Sousa P, Otero Á, Gonçalves JM, Ruiz L, de Oliveira C, et al. Proposal for a new risk stratification classification for meningioma based on patient age, WHO tumor grade, size, localization, and karyotype. Neuro Oncol. 2014;16(5):735–47.
Olar A, Wani KM, Sulman EP, Mansouri A, Zadeh G, Wilson CD, et al. Mitotic index is an independent predictor of recurrence-free survival in meningioma. Brain Pathol. 2015;25(3):266–75.
Rogers CL, Perry A, Pugh S, Vogelbaum MA, Brachman D, McMillan W, et al. Pathology concordance levels for meningioma classification and grading in NRG Oncology RTOG Trial 0539. Neuro Oncol. 2016;18(4):565–74.
Rogers L, Zhang P, Vogelbaum MA, et al. Intermediate-risk meningioma: initial outcomes from NRG Oncology RTOG 0539 [published correction appears in J Neurosurg. 2018 Dec 1;129(6):1650]. J Neurosurg. 2018;129(1):35–47. https://doi.org/10.3171/2016.11.JNS161170.
Jääskeläinen J, Haltia M, Servo A. Atypical and anaplastic meningiomas: radiology, surgery, radiotherapy, and outcome. Surg Neurol. 1986;25(3):233–42.
van Alkemade H, de Leau M, Dieleman EMT, Kardaun JWPF, van Os R, Vandertop WP, et al. Impaired survival and long-term neurological problems in benign meningioma. Neuro Oncol. 2012;14(5):658–66.
Gallagher MJ, Jenkinson MD, Brodbelt AR, Mills SJ, Chavredakis E. WHO grade 1 meningioma recurrence: are location and Simpson grade still relevant? Clin Neurol Neurosurg. 2016;141:117–21.
Talacchi A, Muggiolu F, De Carlo A, Nicolato A, Locatelli F, Meglio M. Recurrent atypical meningiomas: combining surgery and radiosurgery in one effective multimodal treatment. World Neurosurg. 2016;87:565–72.
Kshettry VR, Ostrom QT, Kruchko C, Al-Mefty O, Barnett GH, Barnholtz-Sloan JS. Descriptive epidemiology of World Health Organization grades II and III intracranial meningiomas in the United States. Neuro Oncol. 2015;17(8):1166–73.
Simpson D. The recurrence of intracranial meningiomas after surgical treatment. J Neurol Neurosurg Psychiatry. 1957;20(1):22–39.
Ehresman JS, Garzon-Muvdi T, Rogers D, Lim M, Gallia GL, Weingart J, et al. The relevance of Simpson grade resections in modern neurosurgical treatment of World Health Organization Grade I, II, and III meningiomas. World Neurosurg. 2018;109:e588–e93.
Gousias K, Schramm J, Simon M. The Simpson grading revisited: aggressive surgery and its place in modern meningioma management. J Neurosurg. 2016;125(3):551–60. https://doi.org/10.3171/2015.9.JNS15754.
Lemée J-M, Corniola MV, Da Broi M, Joswig H, Scheie D, Schaller K, et al. Extent of resection in meningioma: predictive factors and clinical implications. Sci Rep. 2019;9(1):5944.
Kaley TJ, Wen P, Schiff D, Ligon K, Haidar S, Karimi S, et al. Phase II trial of sunitinib for recurrent and progressive atypical and anaplastic meningioma. Neuro-Oncology. 2015;17(1):116–21.
Network NCC. Central nervous systems Cancer (Version 1.2020).62.
Preusser M, Silvani A, Le Rhun E, Soffietti R, Lombardi G, Sepulveda J, et al. PL3.2 Trabectedin for recurrent WHO grade II or III meningioma: a randomized phase II study of the EORTC Brain Tumor Group (EORTC-1320-BTG). Neuro Oncol. 2019;21(Supplement_3):iii2–3.
Barresi V, Alafaci C, Salpietro F, Tuccari G. Sstr2A immunohistochemical expression in human meningiomas: is there a correlation with the histological grade, proliferation or microvessel density? Oncol Rep. 2008;20(3):485–92.
de Oliveira Silva CB, Ongaratti BR, Trott G, Haag T, Ferreira NP, Leães CGS, et al. Expression of somatostatin receptors (SSTR1-SSTR5) in meningiomas and its clinicopathological significance. Int J Clin Exp Pathol. 2015;8(10):13185–92.
Arena S, Barbieri F, Thellung S, Pirani P, Corsaro A, Villa V, et al. Expression of somatostatin receptor mRNA in human meningiomas and their implication in in vitro antiproliferative activity. J Neuro Oncol. 2004;66(1–2):155–66.
Sharpe C, Duong J, Law WP. 68Ga-DOTATATE PET/MRI in radiation therapy planning for meningioma – the benefits of hybrid imaging. J Nucl Med. 2017;58(supplement 1):1111.
Nguyen E, Fu B, Dandekar M, Carrillo J, Kong X-T, Cadena G, et al. MNGI-12. A retrospective interventional cohort study to assess the efficacy and safety of Sandostatin LAR (octreotide acetate) for the treatment of meningiomas in adult patients. Neuro Oncol. 2017;19(Suppl 6):vi134–vi.
Ortola Buigues A, Crespo Hernandez I, Jorquera Moya M, Diaz Perez JA. Unresectable recurrent multiple meningioma: a case report with radiological response to Somatostatin analogues. Case Rep Oncol. 2016;9(2):520–5.
Graillon T, Sanson M, Campello C, et al. Everolimus and octreotide for patients with recurrent meningioma: results from the Phase II CEVOREM Trial. Clin Cancer Res. 2020;26(3):552–7. https://doi.org/10.1158/1078-0432.CCR-19-2109.
Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B, et al. Phase 3 trial of (177)Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125–35.
Seystahl K, Stoecklein V, Schuller U, Rushing E, Nicolas G, Schafer N, et al. Somatostatin receptor-targeted radionuclide therapy for progressive meningioma: benefit linked to 68Ga-DOTATATE/-TOC uptake. Neuro Oncol. 2016;18(11):1538–47.
Gerster-Gillieron K, Forrer F, Maecke H, Mueller-Brand J, Merlo A, Cordier D. 90Y-DOTATOC as a therapeutic option for complex recurrent or progressive Meningiomas. J Nucl Med. 2015;56(11):1748–51.
Marincek N, Radojewski P, Dumont RA, Brunner P, Muller-Brand J, Maecke HR, et al. Somatostatin receptor-targeted radiopeptide therapy with 90Y-DOTATOC and 177Lu-DOTATOC in progressive meningioma: long-term results of a phase II clinical trial. J Nucl Med. 2015;56(2):171–6.
Makis W, McCann K, McEwan AJ. Rhabdoid papillary meningioma treated with 177Lu DOTATATE PRRT. Clin Nucl Med. 2015;40(3):237–40.
McNulty SN, Schwetye K, Goldstein M, Carter J, Schmidt RE, Ansstas G, et al. Analysis of point mutations and copy number variation in Grade II and III meningioma. Exp Mol Pathol. 2018;105(3):328–33.
Sahm F, Schrimpf D, Stichel D, Jones DTW, Hielscher T, Schefzyk S, et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol. 2017;18(5):682–94.
Trofatter JA, MacCollin MM, Rutter JL, Murrell JR, Duyao MP, Parry DM, et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;75(4):826.
Rouleau GA, Merel P, Lutchman M, Sanson M, Zucman J, Marineau C, et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993;363(6429):515–21.
Evans DGR. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis. 2009;4:16.
Seizinger BR, de la Monte S, Atkins L, Gusella JF, Martuza RL. Molecular genetic approach to human meningioma: loss of genes on chromosome 22. Proc Natl Acad Sci U S A. 1987;84(15):5419–23.
Ruttledge MH, Sarrazin J, Rangaratnam S, Phelan CM, Twist E, Merel P, et al. Evidence for the complete inactivation of the NF2 gene in the majority of sporadic meningiomas. Nat Genet. 1994;6(2):180–4.
Wellenreuther R, Kraus JA, Lenartz D, Menon AG, Schramm J, Louis DN, et al. Analysis of the neurofibromatosis 2 gene reveals molecular variants of meningioma. Am J Pathol. 1995;146(4):827–32.
Hartmann C, Sieberns J, Gehlhaar C, Simon M, Paulus W, von Deimling A. NF2 mutations in secretory and other rare variants of meningiomas. Brain Pathol. 2006;16(1):15–9.
Brastianos PK, Horowitz PM, Santagata S, Jones RT, McKenna A, Getz G, et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet. 2013;45(3):285–9.
Bi WL, Mei Y, Agarwalla PK, Beroukhim R, Dunn IF. Genomic and epigenomic landscape in meningioma. Neurosurg Clin N Am. 2016;27(2):167–79.
Jungwirth G, Warta R, Beynon C, Sahm F, von Deimling A, Unterberg A, et al. Intraventricular meningiomas frequently harbor NF2 mutations but lack common genetic alterations in TRAF7, AKT1, SMO, KLF4, PIK3CA, and TERT. Acta Neuropathol Commun. 2019;7(1):140.
James MF, Han S, Polizzano C, Plotkin SR, Manning BD, Stemmer-Rachamimov AO, et al. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Mol Cell Biol. 2009;29(15):4250–61.
James MF, Stivison E, Beauchamp R, Han S, Li H, Wallace MR, et al. Regulation of mTOR complex 2 signaling in neurofibromatosis 2-deficient target cell types. Mol Cancer Res. 2012;10(5):649–59.
Curto M, McClatchey AI. Nf2/Merlin: a coordinator of receptor signalling and intercellular contact. Br J Cancer. 2008;98(2):256–62.
Hadfield KD, Newman WG, Bowers NL, Wallace A, Bolger C, Colley A, et al. Molecular characterisation of SMARCB1 and NF2 in familial and sporadic schwannomatosis. J Med Genet. 2008;45(6):332–9.
Shapiro IM, Kolev VN, Vidal CM, Kadariya Y, Ring JE, Wright Q, et al. Merlin deficiency predicts FAK inhibitor sensitivity: a synthetic lethal relationship. Sci Transl Med. 2014;6(237):237ra68.
Clark VE, Erson-Omay EZ, Serin A, Yin J, Cotney J, Ozduman K, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. 2013;339(6123):1077–80.
Keppler-Noreuil KM, Baker EH, Sapp JC, Lindhurst MJ, Biesecker LG. Somatic AKT1 mutations cause meningiomas colocalizing with a characteristic pattern of cranial hyperostosis. Am J Med Genet A. 2016;170(10):2605–10.
Yesiloz U, Kirches E, Hartmann C, Scholz J, Kropf S, Sahm F, et al. Frequent AKT1E17K mutations in skull base meningiomas are associated with mTOR and ERK1/2 activation and reduced time to tumor recurrence. Neuro Oncol. 2017;19(8):1088–96.
Williams SR, Juratli TA, Castro BA, Lazaro TT, Gill CM, Nayyar N, et al. Genomic analysis of posterior Fossa meningioma demonstrates frequent AKT1 E17K mutations in foramen magnum meningiomas. J Neurol Surg B Skull Base. 2019;80(6):562–7.
Shih KC, Chowdhary S, Rosenblatt P, Weir AB 3rd, Shepard GC, Williams JT, et al. A phase II trial of bevacizumab and everolimus as treatment for patients with refractory, progressive intracranial meningioma. J Neuro Oncol. 2016;129(2):281–8.
Mawrin C, Sasse T, Kirches E, Kropf S, Schneider T, Grimm C, et al. Different activation of mitogen-activated protein kinase and Akt signaling is associated with aggressive phenotype of human meningiomas. Clin Cancer Res. 2005;11(11):4074–82.
El-Habr EA, Levidou G, Trigka EA, Sakalidou J, Piperi C, Chatziandreou I, et al. Complex interactions between the components of the PI3K/AKT/mTOR pathway, and with components of MAPK, JAK/STAT and Notch-1 pathways, indicate their involvement in meningioma development. Virchows Archiv. 2014;465(4):473–85.
Abedalthagafi M, Bi WL, Aizer AA, Merrill PH, Brewster R, Agarwalla PK, et al. Oncogenic PI3K mutations are as common as AKT1 and SMO mutations in meningioma. Neuro Oncol. 2016;18(5):649–55.
Xu LG, Li LY, Shu HB. TRAF7 potentiates MEKK3-induced AP1 and CHOP activation and induces apoptosis. J Biol Chem. 2004;279(17):17278–82.
Boetto J, Bielle F, Sanson M, Peyre M, Kalamarides M. SMO mutation status defines a distinct and frequent molecular subgroup in olfactory groove meningiomas. Neuro Oncol. 2017;19(3):345–51.
Laurendeau I, Ferrer M, Garrido D, D’Haene N, Ciavarelli P, Basso A, et al. Gene expression profiling of the hedgehog signaling pathway in human meningiomas. Mol Med. 2010;16(7–8):262–70.
Kalala JP, Maes L, Vandenbroecke C, de Ridder L. The hTERT protein as a marker for malignancy in meningiomas. Oncol Rep. 2005;13(2):273–7.
Goutagny S, Nault JC, Mallet M, Henin D, Rossi JZ, Kalamarides M. High incidence of activating TERT promoter mutations in meningiomas undergoing malignant progression. Brain Pathol. 2014;24(2):184–9.
Sahm F, Schrimpf D, Olar A, Koelsche C, Reuss D, Bissel J, et al. TERT promoter mutations and risk of recurrence in meningioma. J Natl Cancer Inst. 2015;108(5):djv377.
Juratli TA, Thiede C, Koerner MVA, Tummala SS, Daubner D, Shankar GM, et al. Intratumoral heterogeneity and TERT promoter mutations in progressive/higher-grade meningiomas. Oncotarget. 2017;8(65):109228–37.
Juratli TA, McCabe D, Nayyar N, Williams EA, Silverman IM, Tummala SS, et al. DMD genomic deletions characterize a subset of progressive/higher-grade meningiomas with poor outcome. Acta Neuropathol. 2018;136(5):779–92.
Biczok A, Kraus T, Suchorska B, Terpolilli NA, Thorsteinsdottir J, Giese A, et al. TERT promoter mutation is associated with worse prognosis in WHO grade II and III meningiomas. J Neuro-Oncol. 2018;139(3):671–8.
Spiegl-Kreinecker S, Lotsch D, Neumayer K, Kastler L, Gojo J, Pirker C, et al. TERT promoter mutations are associated with poor prognosis and cell immortalization in meningioma. Neuro Oncol. 2018;20(12):1584–93.
Shankar GM, Santagata S. BAP1 mutations in high-grade meningioma: implications for patient care. Neuro Oncol. 2017;19(11):1447–56.
Shankar GM, Abedalthagafi M, Vaubel RA, Merrill PH, Nayyar N, Gill CM, et al. Germline and somatic BAP1 mutations in high-grade rhabdoid meningiomas. Neuro Oncol. 2017;19(4):535–45.
Phillips JJ, Gong H, Chen K, et al. The genetic landscape of anaplastic pleomorphic xanthoastrocytoma. Brain Pathol. 2019;29(1):85–96. https://doi.org/10.1111/bpa.12639.
Aoki K, Nakamura H, Suzuki H, Matsuo K, Kataoka K, Shimamura T, et al. Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro Oncol. 2018;20(1):66–77.
Guyot A, Duchesne M, Robert S, et al. Analysis of CDKN2A gene alterations in recurrent and non-recurrent meningioma. J Neuro-Oncol. 2019;145(3):449–59. https://doi.org/10.1007/s11060-019-03333-6.
Kim H, Park K-J, Ryu B-K, Park D-H, Kong D-S, Chong K, et al. Forkhead box M1 (FOXM1) transcription factor is a key oncogenic driver of aggressive human meningioma progression.n/a(n/a).
Reuss DE, Piro RM, Jones DT, Simon M, Ketter R, Kool M, et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta Neuropathol. 2013;125(3):351–8.
Yuzawa S, Nishihara H, Yamaguchi S, Mohri H, Wang L, Kimura T, et al. Clinical impact of targeted amplicon sequencing for meningioma as a practical clinical-sequencing system. Mod Pathol. 2016;29(7):708–16.
McConnell BB, Yang VW. Mammalian Kruppel-like factors in health and diseases. Physiol Rev. 2010;90(4):1337–81.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76.
Clark VE, Harmanci AS, Bai H, Youngblood MW, Lee TI, Baranoski JF, et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat Genet. 2016;48(10):1253–9.
Olar A, Wani KM, Wilson CD, Zadeh G, DeMonte F, Jones DT, et al. Global epigenetic profiling identifies methylation subgroups associated with recurrence-free survival in meningioma. Acta Neuropathol. 2017;133(3):431–44.
Nassiri F, Mamatjan Y, Suppiah S, Badhiwala JH, Mansouri S, Karimi S, et al. DNA methylation profiling to predict recurrence risk in meningioma: development and validation of a nomogram to optimize clinical management. Neuro Oncol. 2019;21(7):901–10.
Katz LM, Hielscher T, Liechty B, Silverman J, Zagzag D, Sen R, et al. Loss of histone H3K27me3 identifies a subset of meningiomas with increased risk of recurrence. Acta Neuropathol. 2018;135(6):955–63.
Domingues PH, Teodosio C, Ortiz J, Sousa P, Otero A, Maillo A, et al. Immunophenotypic identification and characterization of tumor cells and infiltrating cell populations in meningiomas. Am J Pathol. 2012;181(5):1749–61.
Johnson MD. PD-L1 expression in meningiomas. J Clin Neurosci. 2018;57:149–51.
Han SJ, Reis G, Kohanbash G, Shrivastav S, Magill ST, Molinaro AM, et al. Expression and prognostic impact of immune modulatory molecule PD-L1 in meningioma. J Neuro-Oncol. 2016;130(3):543–52.
Du Z, Abedalthagafi M, Aizer AA, McHenry AR, Sun HH, Bray M-A, et al. Increased expression of the immune modulatory molecule PD-L1 (CD274) in anaplastic meningioma. Oncotarget. 2015;6(7):4704–16.
Kintsler S, Cassataro MA, Drosch M, Holenya P, Knuechel R, Braunschweig T. Expression of programmed death ligand (PD-L1) in different tumors. Comparison of several current available antibody clones and antibody profiling. Ann Diagn Pathol. 2019;41:24–37.
Bi WL, Greenwald NF, Abedalthagafi M, Wala J, Gibson WJ, Agarwalla PK, et al. Genomic landscape of high-grade meningiomas. NPJ Genom Med. 2017;2:15.
Dunn IF, Du Z, Touat M, Sisti MB, Wen PY, Umeton R, et al. Mismatch repair deficiency in high-grade meningioma: a rare but recurrent event associated with dramatic immune activation and clinical response to PD-1 blockade. JCO Precis Oncol. 2018. https://doi.org/10.1200/PO.18.00190.
Gelerstein E, Berger A, Jonas-Kimchi T, Strauss I, Kanner AA, Blumenthal DT, et al. Regression of intracranial meningioma following treatment with nivolumab: case report and review of the literature. J Clin Neurosci. 2017;37:51–3.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Christine Cordova is a co-investigator of NCT03971461. She did not receive research funding NCT03971461. Sylvia C. Kurz is study chair of NCT03971461, a study funded by Advanced Accelerator Applications evaluating Lutathera for advanced meningiomas.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Neuro-oncology
Rights and permissions
About this article

Cite this article
Cordova, C., Kurz, S.C. Advances in Molecular Classification and Therapeutic Opportunities in Meningiomas. Curr Oncol Rep 22, 84 (2020). https://doi.org/10.1007/s11912-020-00937-4
Published:
DOI: https://doi.org/10.1007/s11912-020-00937-4