
Abstract
Extrapyramidal movement disorders include hypokinetic rigid and hyperkinetic or mixed forms, most of them originating from dysfunction of the basal ganglia (BG) and their information circuits. The functional anatomy of the BG, the cortico-BG–thalamocortical, and BG–cerebellar circuit connections are briefly reviewed. Pathophysiologic classification of extrapyramidal movement disorder mechanisms distinguish (1) parkinsonian syndromes, (2) chorea and related syndromes, (3) dystonias, (4) myoclonic syndromes, (5) ballism, (6) tics, and (7) tremor syndromes. Recent genetic and molecular–biologic classifications distinguish (1) synucleinopathies (Parkinson’s disease, dementia with Lewy bodies, Parkinson’s disease–dementia, and multiple system atrophy); (2) tauopathies (progressive supranuclear palsy, corticobasal degeneration, FTLD-17; Guamian Parkinson–dementia; Pick’s disease, and others); (3) polyglutamine disorders (Huntington’s disease and related disorders); (4) pantothenate kinase-associated neurodegeneration; (5) Wilson’s disease; and (6) other hereditary neurodegenerations without hitherto detected genetic or specific markers. The diversity of phenotypes is related to the deposition of pathologic proteins in distinct cell populations, causing neurodegeneration due to genetic and environmental factors, but there is frequent overlap between various disorders. Their etiopathogenesis is still poorly understood, but is suggested to result from an interaction between genetic and environmental factors. Multiple etiologies and noxious factors (protein mishandling, mitochondrial dysfunction, oxidative stress, excitotoxicity, energy failure, and chronic neuroinflammation) are more likely than a single factor. Current clinical consensus criteria have increased the diagnostic accuracy of most neurodegenerative movement disorders, but for their definite diagnosis, histopathological confirmation is required. We present a timely overview of the neuropathology and pathogenesis of the major extrapyramidal movement disorders in two parts, the first one dedicated to hypokinetic-rigid forms and the second to hyperkinetic disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Extrapyramidal movement disorders are divided into hypokinetic rigid, hyperkinetic, and mixed forms, most of which have their origin in dysfunction of the dorsal basal ganglia (BG), which have a multitude of functions associated with cognition and reward, but are primarily involved in motor control. Dysfunction of the cortico-BG–thalamocortical (CBGTC) circuits due to disruption of downstream network activities in cortex, thalamus, and brainstem result in a number of landmark motor disorders such as Parkinson’s and Huntington’s diseases, which disturb motor control in markedly different contexts.
Structure and function of the basal ganglia
The BG are a cluster of subcortical nuclei which include (1) input nuclei [caudate nucleus (CN), putamen (Put), and nucleus accumbens], (2) output nuclei [internal segment of globus pallidus (GPi) and substantia nigra pars reticulata (SNr)], and (3) intrinsic nuclei/external segment of globus pallidus (GPe), subthalamic nucleus (STN), and substantia nigra pars compacta (SNc). According to the current model of the BG circuitry, they are viewed as components of segregated networks that emanate from special cortical areas, traverse the BG and ventral thalamus, and return to the frontal cortex, interacting with internal re-entering circuits engaging motor, associative, and limbic cortical territories in the control of movement, behavior, planning, and emotions, related to a functional interconnection of these areas (Klaus et al. 2019).
The fundamental understanding of the essential anatomical pathways—CBGTC—and the alterations of the neurotransmitter systems located in these circuits are essential for understanding potential pathophysiological mechanisms in the landmark extrapyramidal motor disorders. The functions of these networks are modulated by three main transmitter systems: dopamine (DA), glutamate, and γ-aminobutyric acid (GABA). Normal movement is controlled by the CBGTC circuits. The striatum integrates motor behavior using well-defined circuits, whose individual components are independently affected in various movement disorders. It receives excitatory glutamatergic input from the cerebral cortex, thalamus, and brainstem, mainly from DAergic cells and releases GABAergic output to SNc, SNr, GPi, and GPi, which project to specific nuclei in thalamus and the brainstem tegmentum. The involved thalamic nuclei have an excitatory glutamatergic output to specific regions of the motor cortex. The GABAergic output of SNc and GPi reduces glutamatergic projections from thalamus back to the cortex. Other cortical regions project to subthalamic nucleus (STN), SN, thalamus, ventral tegmental area (VTA) and via pontine nuclei to the cerebellum. GPe, DAergic SNc, and STN modulate the main flow of information through the BG. The classical model of the involved circuits describes a dynamic web of interlinked pathways with inhibitory and excitatory functions providing multiple sites of influence (Young and Sonne 2018) (Fig. 1). Similar to the body regions within the sensory motor cortex, the BG nuclei are somatotopically organized (Simonyan 2019).

Modified from (Haber 2016) with permission from Association La Conférence Hippocrate-Servier. © AICH-Servier
Schematic representation of key structures and pathways of the basal ganglia. Blue arrows: direct pathway; red arrows: indirect pathway; yellow arrow: hyperdirect pathway. Amy amygdala, DS dorsal striatum, GPi globus pallidus, internal segment, GPe globus pallidus, external segment, Hipp hippocampus, PPN pedunculopontine nucleus, SNc substantia nigra compacta, SNr substantia nigra reticulata, STN subthalamic nucleus, Thal thalamus, VA ventral anterior, VM ventral median, VP ventral pallidum, VS ventral striatum, VTA ventral tegmental area, Glu glutamatergic, GABA gabaergic.
Five BG–thalamocortical circuits form a topographically organized functional network: motor and oculomotor circuits, dorsolateral prefrontal, lateral orbitofrontal, and anterior cingulate or limbic circuits involving different parts of the striatum, pallidonigral complex, and medial and ventral thalamus (Simonyan 2019). The functions of these networks are strongly modulated by the release of DA in the striatum. It alters the activity of striatal neurons which, in turn, influences the (inhibitory) BG output.
A nigrostriatal circuit in which SNc gets a GABAergic inhibitory projection from striatum feeds back to striatum as the major source of its DAergic innervation. The medial SN connects with limbic striatal and cortical regions: the ventral SN with associative regions of cortex and striatum and the lateral SN with somatomotor regions of striatum and cortex encoding different functions (Zhang et al. 2017). The retrorubral field (A8) and ventral tegmental area (A10) are integrated in the mesostriatal and mesolimbic DAergic projections. DA induces excitation of striatal neurons that project to GPi and SNr and inhibits thalamic nuclei to maintain normal movements. It inhibits neurons that project to GPe or STN to moderate the normal negative effect on motor speed and tone associated with high output from STN. Its outputs project to GPi, SNr, GPe, striatum, and PPN. DA modulates BG functions, but also acts outside of the striatum, thus contributing to the symptoms of PD and other disorders (Wichmann et al. 2018). GPe receives GABAergic input from striatum and projects to STN, which in turn sends glutamatergic projections to SNr, GPi, and GPe to inhibit glutamatergic excitation of the cortex. Excitatory glutamatergic drive of STN neurons along the cortico-subthalamic pathway triggers GABAergic inhibition of pallidothalamic inputs (Chu et al. 2015). The STN-GPe system is a major input relais station receiving projections from various cortical and subcortical regions, thus modulating the downstream effects of the BG that control both motor function and emotion (Suryanarayana et al. 2019). Many PD symptoms result directly from neurodegeneration; others are driven by aberrant activity patterns in surviving neurons. This latter phenomenon, PD circuit dysfunction, is an area of intense study in view of currently incurable neurodegeneration (McGregor and Nelson 2019).
A commonly presented but overly simplistic model of motor function suggests that BG output structures are controlled by two opposing striatal motor loops, originating from distinct populations of medium-sized spiny projection neurons (MSNs) and projecting to different output structures (Young and Sonne 2018). The direct pathway is a monosynaptic inhibitory projection from the glutamatergic cortex to the GABAergic MSNs, containing DA-D1 receptor neurons projecting to GABAergic neurons in GPi and SNr. Activation of striatal MSNs leads to inhibition of the inhibitory GPi/SNr output and to disinhibition of BG target structures in thalamus and midbrain, thus promoting movement and behavior. The indirect pathway contains disinhibitory projections from the glutamatergic cortex to striatal MSNs (containing GABA and expressing the DA-D2 receptor), with striatal projections to GPe, GABAergic GPe projections to STN, and glutamatergic STN projections to GPi and SNr. The STN as part of the indirect pathway drives pallidal GABAergic output through glutamatergic synapses. The GPi sends inhibitory projections to the ventral anterior and ventral lateral nuclei of the thalamus and will disinhibit motor output by thalamic stimulation of the motor cortex. A signal through the indirect pathway (cortex–striatum–GPe–STN–GPi) ultimately terminates a movement. The SNr, an inhibitory GABAergic nucleus, works together with the GPi as the final output of the BG’s direct and indirect pathways. In turn, both pathways have a reverse effect on spontaneously firing thalamocortical neurons and ultimate motor activity, i.e., activation of the direct pathway facilitates motor activity via disinhibition of thalamocortical neurons, whereas activation of the indirect pathway reduces motor activity by increasing inhibition of the thalamocortical neurons. The thalamus is a neural integrator for the activities of the forebrain, but all the cortico-cerebellocortical loops make relay in the thalamus (Habas et al. 2019).
The parallel circuit model of the BG (Fig. 1) describes how information progresses through the BG in anatomically and functionally distinct channels. Balance between these two pathways at the level of GP and SN is essential for normal functioning of the BG–thalamocortical circuits, the disruption of which is the major locus of PD-related dysfunction (McGregor and Nelson 2019). Increased inhibition of the thalamocortical pathway results in hypokinetic disorders, while decreased inhibition of thalamocortical output induces hyperkinetic disorders (Lanciego et al. 2012). These networks are modulated by the release of DA in the striatum, thus enabling flexible motor and behaviour control (Neumann et al. 2018). In parkinsonism, the loss of striatal DA results in the emergence of oscillatory burst patterns of firing of BG output neurons, increased synchrony of the discharge of neighbouring BG neurons and an overall increase in BG output, thus inhibiting their thalamic and midbrain targets. In PD, DA loss is predicted to cause imbalanced activity between the two pathways.
The reduced activity in the “direct” striato-cortical–nigral–GPi pathway induces akinesia (Beck et al. 2018; Wichmann et al. 2018), which may also be associated with abnormalities outside the DAergic pathways (Spay et al. 2018). The two pathways are not separate parallel systems, but functionally intertwined in- and outside the striatum, collaterals bridging the two pathways (Papa and Wichmann 2015; Simonyan et al. 2017). Other models suggest that they are not alternatively but concomitantly active, and coordinated activity across the two pathways regulates movement initiation and execution (Tecuapetla et al. 2016). While the classical model predicts that increased BG output induces excessive inhibition of thalamus and cortex, leading to a paucity of movement, manipulations of the BG in parkinsonian and healthy animals suggest that other measures of activity such as pattern and synchrony play a role in driving PD motor symptoms. According to the “center-surround” model of the BG, cortical input activates STN neurons that excite GPI neurons, suppressing actions. Concurrently, cortical input to the striatum activates indirect MSNs that shape STN activity through the GPe, as well as direct MSNs that converge and inhibit a subset of GPi neurons to permit selective execution of movement. At the striatal level, inhibitory connections between MSNs may contribute to consensually similar center-surround patterns (McGregor and Nelson 2019).
A different hypothesis of the BG pathways and DA, named the cortico-striatal–temporal-difference (CS–TD) model proposes a new modality that integrates the OpAL and CS–TD models. It suggests that the intratelencephalic (IT–BG pathways represent goodness/badness of current options, while the PT-indirect pathway represents the overall value of the previous option, and both these have influence on the DA neurons, through the BG output. A key assumption is that opposite directions of plasticity are induced upon phasic activation of DA neurons in the IT-indirect pathway and PT-indirect pathway because of different profiles of IT and PT inputs. At PT → indirect MSN synapses, sustained glutamatergic inputs generate rich adenosine, which prevents DA-D2 receptor signaling and instead favors adenosine–A2A receptor signaling. Then, DA-induced phasic adenosine, which reflects TD-RPE, causes long-term synaptic potentiation. In contrast, at IT → indirect MSN synapses, where adenosine is scarce, phasic DA causes long-term synaptic depression via D2 receptor signaling. This new model provides new predictions, part of which is in line with recently reported activity patterns of GPe neurons in the “indirect” pathway (Morita and Kawaguchi 2019).
There are, however, other actions within the BG including communication between DA-D1 and DA-D2 receptor striatal MSNs, with collaterals in both GPi and GPe: GPe projections going back to the striatum, GPi/SNr ones not only to the thalamus, but to pedunculo-pontine tegmental nucleus (PPT), habenula and superior colliculus, as well as a balanced dynamic system regulated by mesolimbic and DAergic neuronal circuits (Cazorla et al. 2015; Hegeman et al. 2016; Schmidt and Berke 2017).
Two “hyperdirect” pathways include a direct cortico–subthalamic–pallidal pathway that increases GPi activity and inhibits thalamocortical targets, thus causing supression of all movements (Wichmann et al. 2018), while three parallel but independent neurotrophic circuits between SN and GABAergic and cholinergic striatal interneurons may exist (Ortega-de San Luis et al. 2018). The hyperdirect and indirect pathways, converging in the STN, are differentially involved in cognitive aspects of motor preparation and gait control during motor performance (Neumann et al. 2018). The thalamostriatal system is a dual system, one originating from midline and intralaminar nuclei, another one from ventral and relais nuclei using glutamate transporters. The source of thalamostriatal projections is highly organized in striatal compartments that are influenced by their cortical and thalamic afferents (Fujiyama et al. 2019). The midbrain locomotor region with the cholinergic PPN that is interconnected with BG, thalamic and brainstem nuclei, spinal effectors, and cerebellum, is crucial for motor and cognitive control (Mori et al. 2016; Vitale et al. 2018). BG and cerebellum are reciprocally interconnected with the neocortex via oligosynaptic loops (Hintzen et al. 2018) as substrate of integrated functional networks between them (Pelzer et al. 2017). They are topographically organized, so that motor, cognitive, and affective territories in the network are interconnected, abnormalities in each node can have network-wide effects (Bostan and Strick 2018). The dorsal motor nucleus of the vagus and SN is connected in a recently discovered monosynaptic nigro–vagal pathway, which is dysfunctional in rodent models of PD (Bove and Travagli 2019).
Classification of major movement disorders
Most extrapyramidal disorders related to BG dysfunction are neurodegenerative diseases featured by neuronal degeneration and astrocytosis in many parts of the nervous system. A classical pathophysiological classification distinguishes: (A) hypokinetic-rigid syndromes: parkinsonian syndromes with rigidity, akinesia/bradykinesia, resting tremor, and postural instability; (B) hyperkinetic syndromes: (1) chorea syndromes with irregular movements; (2) dystonia characterized by involuntary muscle spasms and abnormal posture; (3) ballism with high amplitude movements of the proximal extremities; (4) myoclonus with brief, quick movements; (5) tremor syndromes with rhythmic involuntary movements; and (6) tic disorders with rapid involuntary movements.
Recent genetic and molecular–biologic classification of movement disorders distinguishes (Table 1): (1) Synucleinopathies, a heterogeneous group of neurodegenerative disorders caused by misfolded α-synuclein (α-Syn) protein that forms amyloid-like filamentous inclusions (Alafuzoff and Hartikainen 2017; Goedert et al. 2017b). They include Lewy body (LB) disorders—sporadic and rare familial forms of PD, dementia with Lewy bodies (DLB), pure autonomic failure (PAF), and multiple system atrophy (MSA). Neurodegeneration with brain iron accumulation type I (NBIA-I) or pantothenate kinase-associated neurodegeneration (PKAN) is no longer considered a synucleinopathy (Li et al. 2013). (2) Tauopathies, featured by neurofibrillary tau pathology, include progressive supranuclear palsy (PSP), cortico-basal degeneration (CBD), and frontotemporal lobe degeneration with tau pathology (FTLD/TAU); (3) polyglutamine disorders linked to CAG trinucleotide repeats, such as Huntington’s disease (HD); and (4) paraneoplastic forms (Poplawska-Domaszewicz et al. 2018); those associated with neuronal antibodies (Dash and Pandey 2019) or without hitherto detected genetic or specific disease markers. The various phenotypes are associated with the deposition of pathologic (misfolded) proteins and cytoskeletal abnormalities in distinct neuronal populations, which represent important diagnostic signposts. Recent consensus criteria for their clinical and neuropathologic diagnosis have been established (Ali and Josephs 2018a; Gilman et al. 2008; Hoglinger et al. 2018; Jellinger 2016; McKeith et al. 2017). The first part of this review is dedicated to the hypokinetic-rigid syndromes, the second part to the hyperkinetic disorders.
Synucleinopathies
This heterogeneous group of neurodegenerative disorders caused by misfolded α-synuclein that forms amyloid-like filamentous aggregations in many central nervous system (CNS) areas, include (1) Lewy body diseases (LBD)—Parkinson’s disease (PD) with and without dementia, dementia with Lewy bodies (DLB), and pure autonomic failure (PAF), all morphologically characterized by α-Syn-positive cytoplasmic inclusions in neurons (Lewy bodies/LBs/) and dystrophic neurites (LN) and (2) multiple system atrophy (MSA), the morphological hallmarks of which are α-Syn-positive glial cytoplasmic inclusions (GCI) in oligodendroglia and less frequent neuronal inclusions. Synucleinopathies account for 73–83% of cases of parkinsonism, including 42–63% PD, whereas other degenerative disorders mimicking PD account for 9–33% (Dickson 2018; Horvath et al. 2013a; Savica et al. 2013a).
α-Syn is a 14 kDa intrinsically disordered presynaptic protein with potential for self-oligomerization and fibrillary aggregation under pathologic conditions. Increasing phosphorylation of α-Syn at serine 129 enhances the accumulation and toxicity (Prasad et al. 2019). Pathological α-Syn has the capacity to self-seed and propagate between cells; its intercellular transfer has been implicated in the progression of synucleinopathies (Dehay 2014; Karpowicz et al. 2019; Reyes et al. 2019). For its molecular basis, functions, interaction with DA metabolites, and relevant animal models, see (Alegre-Abarrategui et al. 2019; Benskey et al. 2016; Burre et al. 2018; Das and Eliezer 2019; Dettmer et al. 2016; Ghiglieri et al. 2018; Goedert et al. 2017b; Grozdanov and Danzer 2018; Huang et al. 2019; Jellinger 2013a; Stefanis 2012; Wong and Krainc 2017). α-Syn assembles into oligomers, which lead to impairment of axonal transport (Prots et al. 2018; Volpicelli-Daley 2017), synaptic dysfunction and neuronal death (Calo et al. 2016; McCormack et al. 2019; Mehra et al. 2019; Mor et al. 2017; Snead and Eliezer 2014). Lipid alterations in membranous compartments may have an effect on α-Syn misfolding and neurotoxicity (Canerina-Amaro et al. 2019). Interaction of α-Syn aggregate species with phospholipid membranes causes disruption and cell death (Iyer and Claessens 2019). α-Syn is a multifunctional player in the regulation of exocytosis, endocytosis, and vesicle recycling (Huang et al. 2019), and a major component of LBs, dystrophic Lewy neurites (LNs), and glia in PD and DLB (Spillantini et al. 1998; Wakabayashi et al. 2013), in neuronal and glial inclusions in MSA (Jellinger and Wenning 2016). Elevated levels of soluble α-Syn oligomers were seen in postmortem PD and DLB brains (Tong et al. 2010) with higher intensity in MSA (Sekiya et al. 2019). They mediate early synaptic pathology and cellular dysruption (Bengoa-Vergniory et al. 2017; Roberts and Brown 2015; Rockenstein et al. 2014). Clearance mechanisms of α-Syn are complex and multifaceted in particular related to exosomes (Stefanis et al. 2019).
Co-occurrence of α-Syn, tau, β-amyloid (Aβ) and other proteins, and interaction between their oligomeric forms, promote their mutual aggregation, thereby inducing neuronal damage (Bourdenx et al. 2017; Foguem and Manckoundia 2018; Spires-Jones et al. 2017). Interaction of α-Syn, tau, and Aβ (with metal ions) is responsible for the overlapping pathology of different proteinopathies that are considered a continuum depending upon genetic and environmental factors (Bengoa-Vergniory et al. 2017; Colom-Cadena et al. 2017a; Godini et al. 2019; Spires-Jones et al. 2017; Walker et al. 2015; Yan et al. 2018). Modification of α-Syn may induce both Lewy and tau pathologies, and enhances amyloid and tau accumulation, while tau and Aβ enhance α-Syn aggregation and toxicity (Gerson et al. 2018; Irwin et al. 2013c; Yan et al. 2018). Interaction between Aβ and α-Syn leads to inhibition of Aβ deposition (Bachhuber et al. 2015). In PD and DLB brains, concentrations of soluble pSer129 α-Syn correlated with the levels of Aβ (Swirski et al. 2014). Distinct strains of α-Syn are responsible for propagation and regional distribution of lesions in synucleinopathies (Alegre-Abarrategui et al. 2019; Candelise et al. 2019; Karpowicz et al. 2019), and are involved in their heterogeneity (Peelaerts et al. 2018; Peng et al. 2018b; Tanaka et al. 2019), as observed after the injection of α-Syn aggregates into animal models (Goedert et al. 2017c; Ko and Bezard 2017; Peng et al. 2018a; Polinski et al. 2018; Thakur et al. 2017).
Lewy body disorders
This group of neurodegenerative disorders is morphologically featured by the presence of α-Syn-positive inclusions. Lewy bodies (LBs), α-Syn-positive cytoplasmic inclusions, are the morphological hallmarks of PD and DLB, but are also found in a variety of disorders, e.g., in 7–71% of sporadic and familial forms of AD (Cairns et al. 2015; Savica et al. 2019), in a small proportion of cases of frontotemporal lobar degeneration (FTLD) with parkinsonism (Forrest et al. 2019a), and in 2–61% of aged individuals with or without dementia (Buchman et al. 2018; Jellinger 2004; Markesbery et al. 2009).
LBs occur in two types: the classical brainstem and the cortical type. Classical LBs are spherical cytoplasmic intraneuronal inclusions 8–30 µm in diameter with a hyaline eosinophilic core and a narrow pale-stained halo. Ultrastructurally, classical LBs are non-membrane-bound, granulofilamentous structures composed of radially arranged, 7–20 nm intermediate filaments with electron-dense granule material and vesicular structures: the core shows densely packed filaments and dense granular material, the periphery radially arranged 10-nm filaments (Forno 1996; Tercjak et al. 2014). Cortical LBs, eosinophilic, rounded, angular, or reniform structures without a halo, are poorly organized, granulofibrillary structures with a felt-like arrangement composed of 7–27 nm wide filaments (Ishiyama et al. 2006). They are found in small neurons in lower cortical layers, particularly in insular and entorhinal cortex, amygdala, hippocampal sector CA2/3, and cingulate gyri (Armstrong et al. 2014; Wakabayashi et al. 2013). Similar granular, pale-staining eosinophilic materials displacing neuromelanin (NM) in brainstem neurons—”pale bodies”—are precursors of LBs (Dale et al. 1992).
Both types of LBs share immuno- and biochemical characteristics (Jellinger 2012b; Rocha Cabrero and Morrison 2019). Their major components are α-Syn, ubiquitin (Ub), phosphorylated Ub, and others such as structural fibrillary elements, α-Syn-binding proteins, those implicated in the Ub–proteasome system, synphilin-1, aggresome- and mitochondria-related, cytoskeletal, cytosolic, cellular response proteins, etc. (Kalia and Kalia 2015; Voronkov et al. 2018). LBs have a central Parkin- and Ub-positive domain with peripheral α-Syn. Colocalization of α-Syn, synphilin, and Parkin suggests that Parkin plays a role in ubiquitination and modification of α-Syn, its oligomers inducing Parkin nitrosylation (Wilkaniec et al. 2019). Synapsin III, a key component of α-Syn fibrils, TH, and choline-acetyl transferase (ChAT) are co-localized in cortical LBs (Longhena et al. 2018). Brainstem LBs show TH and ChAT reactivity with peripheral α-Syn (Dugger and Dickson 2010). LBs and pale bodies are reactive for autophagic proteins p62 and NBR1 (Kuusisto et al. 2003; Odagiri et al. 2012), and for TIGAR protein regulating TP53, which is absent in MSA inclusions (Lopez et al. 2019). LBs further contain 14-3-3 proteins that interact with α-Syn and have multiple cellular functions. Leucine-rich repeat kinase 2 (LRRK2) is not a major component of LBs. Purified inclusions contain approximately 50 isoforms of α-Syn (McCormack et al. 2016). Proteomic analysis of cortical LBs revealed 296 proteins related to multiple or unknown functions (Leverenz et al. 2007) and 204 proteins in PD brainstem (Licker et al. 2014). Different conformations of α-Syn fibrils correspond to different stages of maturity of LBs (Covell et al. 2017), but none of the detected α-Syn variants were LB-specific (Bhattacharjee et al. 2019), while phosphorylated NUB1 (an adaptor protein) distinguishes α-Syn in LBs from that in GCIs in MSA (Tanji et al. 2019). Recent studies showed that LBs are rich in protein–lipid structures found in other parts of the brain (Shahmoradian et al. 2018).
The formation of classical LBs begins with intraneuronal dust-like particles related to neuromelanin (NM) or lipofuscin that are cross-linked to α-Syn, with granular or diffuse deposition of α-Syn and Ub in the center, followed by condensation of dense filamentous inclusions, forming “early LBs” later developing to LBs. Extraneuronal LBs after disappearance of the affected neuron are degraded by astroglia (Wakabayashi et al. 2013).
Cortical LBs show diffuse α-Syn and Ub labeling, whereas subcortical LBs have a central Ub-positive domain with peripheral deposition of α-Syn. Initial granular accumulation of α-Syn is followed by accumulation of dense filaments, spreading to dendrites, later deformation of LBs, and final degradation by astrocytes. Coarse, dystrophic neurites (LNs) with α-Syn and Ub inclusions in axonal processes, which may evolve into LBs (Kanazawa et al. 2008). LBs and LNs occur in virtually all brainstem nuclei and fiber tracts, with significant correlations between LBs and LNs, in both PD and DLB (Seidel et al. 2015).
Most toxin animal models of PD, e.g., 6-OHDA and MPTP, lacked LB pathology, although chronic low doses of MPTP occasionally induced α-Syn-positive inclusions (Meredith and Rademacher 2011). However, trichloroethylene caused SN neuron loss, DA depletion in striatum, and accumulation of intraneuronal α-Syn (Liu et al. 2010). On the other hand, most of the α-Syn tg models exhibit key features of human PD including α-Syn-positive inclusions similar to human LBs (Dehay and Fernagut 2016; Feany and Bender 2000). Injection of α-Syn preformed fibrils (PFF), which mimick α-Syn oligomers found in LBs, into the striatum or other brain areas induced PD-like α-Syn pathologies and robust LB and LN formations (Ko and Bezard 2017; Nouraei et al. 2018; Polinski et al. 2018). Intracellular injection of synthetic α-Syn fibrils in marmosets produced robust LB-like inclusions in TH-positive neurons (Shimozawa et al. 2017), whereas no LBs were seen in monkeys with over 10 years of MPTP parkinsonism (Halliday et al. 2009).
Marinesco bodies, intranuclear inclusions in pigmented neurons of SN and locus ceruleus (LC), frequently found in elderly individuals in the presence of AD, are rare in PD and their frequency declines with duration of PD (Abbott et al. 2017). Higher LP has been shown to be associated with lower prevalence of atherosclerotic cardiovascular disease risk factors in PD patients (Driver-Dunckley et al. 2019).
Functional role of Lewy bodies
The pathobiological significance of LBs is poorly understood. As a consequence of α-Syn misfolding, they could represent indicators of toxicity or of neuronal protection or end products or epiphenomena of unknown responses to cellular stress (Chartier and Duyckaerts 2018; Espay et al. 2019; Rocha Cabrero and Morrison 2019; Sian-Hulsmann et al. 2015). LBs interact with DNA to cause nuclear degeneration and cell death (Power et al. 2017). Mitochondrial DNA deletion was highest in LB positive neurons, indicating increased mitochondrial damage (Muller et al. 2013), while accumulation of mitochondrial DNA deletions triggers neuroprotective mechanisms (Ammal Kaidery and Thomas 2018; Michel et al. 2016). Nuclear localization of α-Syn, the effect on gene expression, and its toxicity is modulated by phosphorylation on serine 129 (Prasad et al. 2019), which indicates an interplay between subcellular location, phosphorylation, and toxicity (Pinho et al. 2019). Aggregated forms of Ser129-phosphorylated α-Syn can no longer be degraded by the proteasome and eventually accumulate within LBs (Arawaka et al. 2017). Small α-Syn intermediates termed “soluble oligomers” lead to synaptic dysfunction (Gadad et al. 2011). Their oligomerization in early stages of PD (Kalia and Kalia 2015) induces protein aggregation, disrupts cellular function, and leads to neuronal death due to mitochondrial dysfunction and oxidative stress (OS) (Michel et al. 2016; Mullin and Schapira 2013; Rosborough et al. 2017; Stefanis 2012; Tzoulis et al. 2016; Yasuda et al. 2013; Zeng et al. 2018). The Ub–proteasome system (UPS) and the autophagy–liposome pathway (ALP) that render damaged proteins less toxic than their soluble forms contribute to α-Syn turnover, while alterations in these proteolytic pathways result in the accumulation of pathological proteins due to impaired clearance (Liu et al. 2019c). Ubiquitinated proteins in LBs may be a manifestation of a cytoprotective response to eliminate damaged cellular components and to delay the onset of neuronal degeneration (Grunblatt et al. 2018). LBs could be interpreted as markers of surviving neurons, since they are present in the remaining neurons at post-mortem in PD patients or in tissues of asymptomatic individuals, thus reflecting the inability of cells to clear waste proteins due to dysfunction of clearing mechanisms (e.g., autophagy) with subsequent induction of LP and lysosomal stress (Alegre-Abarrategui et al. 2019). All major brain cell types are able to internalize and degrade extracellular α-Syn, but glial cells appear to be the most efficient scavengers. Impairment of clearance leads to accumulation of toxic α-Syn, and dysfunctions of glia, that is involved in the progression of neurodegeneration (Brück et al. 2016; Chavarria et al. 2018; di Domenico et al. 2019; Filippini et al. 2019).
Sporadic Parkinson’s disease
PD, the second-most frequent neurodegenerative movement disorder (prevalence 100–572/100,000; incidence 4.5–21/100,000 person/year (Marras et al. 2018); proposed twofold rise within the next 20 years (Dorsey et al. 2018)), is clinically featured by bradykinesia, rigidity, resting tremor, postural imbalance, and various nonmotor features. Subtle cognitive dysfunction and depression often occur early in the disease (Lees et al. 2009), dementia being common in later stages (Emre et al. 2007). Progressive degeneration of the DAergic nigrostriatal system and many cortical and subcortical networks are associated with widespread α-Syn pathology. This causes striatal DA deficiency and related biochemical deficits that produce a heterogeneous clinical phenotype (Fereshtehnejad et al. 2017; Lawton et al. 2015, 2018; Selikhova et al. 2009; Thenganatt and Jankovic 2014). Diagnostic accuracy of clinical diagnosis is 73.8–79.6%, according to a recent metaanalysis 82.7% (Rizzo et al. 2016). For the diagnosis of definite PD, histopathological confirmation is required. Although LBs are not specific to PD and occur in a variety of conditions, a positive diagnosis of PD is possible by the demonstration of neuronal loss and the demonstration of LBs in the midportion of the SN. If no LBs are found, two further sections should be examined. Cell loss in SN and LC in the absence of LBs or other α-Syn-positive inclusions suggests an alternative cause of parkinsonism (Dickson et al. 2009).
Neuropathology of Parkinson’s disease
Gross inspection of the brain shows mild cortical atrophy, enlargement of the ventricles, and pallor of SN and LC. Widespread α-Syn-immunoreactive deposits in neurons (LBs) and LNs throughout the nervous system, including the brainstem and many visceral organs are present indicating a multisystem involvement by α-Syn pathology (Beach et al. 2010; Gelpi et al. 2014; Jellinger 2012b; Sulzer and Surmeier 2013; Wakabayashi et al. 2010).
LP is associated with variable neuronal loss in midbrain, other subcortical nuclei and other neuronal systems. Depletion of melanized neurons (45–66%) and DAergic neurons immunoreactive for TH, the key enzyme of DA synthesis (60–85%), affects the ventrolateral part of the A9 group of SNc (91–97% cell loss) projecting to striatum. This corresponds to a somatotopic pattern of DAergic terminal loss being more severe in the dorsal and caudal Put with later involvemen of ventral Put and CN. SN cell degeneration is preceded by loss of neurofilament protein, neuronal TH, and DAT immunoreactivity, indicating functional neuronal damage. Later, extracellular released NM is taken up by macrophages, with rare neuronophagy, and only minor astroglial response. Microglial activation occurs even prior to nigral damage (Duffy et al. 2018). The ventrolateral SNc cell clusters are nearly wiped out, while DAergic neuron loss in the dorsal tier may be as little as 25% (Surmeier et al. 2017), and other DAergic and GABAergic neurons are spared at this time. As the disease processes, the nearby ventral and then dorsal SN cell clusters and their striatal projections are affected (Kordower et al. 2013).
In SN, the proportion of LB-bearing neurons appears to be stable throughout the disease duration, between 3.6 and 15% of surviving SN neurons containing LBs (Greffard et al. 2010). SNc cell loss and the reduction of TH and DAT immunoreactivity in Put followed by CN and NAC correlate with the duration and severity of motor dysfunction (Bernheimer et al. 1973). At 4 year post-diagnosis and thereafter, DAT staining in dorsal Put is almost completely lost with only an occasional DAergic fiber in SNc and a 50–90% loss of TH-positive neurons in striatum (Kordower et al. 2013), whereas in end-stage PD, a stable proportion of LB-bearing SN neurons remains (Greffard et al. 2010). Despite a massive loss of SN neurons with atrophy of the remaining cells (Rudow et al. 2008), degeneration of the striatonigral system is not total, even after many years of illness (Djaldetti et al. 2011). Stereological studies showed no overall loss of neocortical neurons in endstage PD, despite many cortical LBs (Pedersen et al. 2005).
The A10 group of DAergic neurons—VTA, nucleus parabrachialis, and nucleus parabrachialis pigmentosus—projecting to the striatal matrix, thalamus, cortical, and limbic areas (mesocorticolimbic system) show only an average 53% cell loss (Alberico et al. 2015), whereas the periretrorubral A8 region, which contains only a few DAergic but CAB-rich neurons, and the central periventricular gray matter show little or no involvement (Geibl et al. 2019). Cholinergic neurons in the basal forebrain and PNP are lost, but not glutamatergic and GABAergic PPN neurons, while there is a modest loss of glutamatergic neurons in the intralaminar nuclei of the thalamus and basolateral amygdala (Double et al. 2010).
Degeneration of the nigrostriatal system causes denervation in striatum with DA loss ranging from 44 to 98% and progressing from the ventrorostal to posterior Put and CN. In earlier disease stages, an increased number of striatal DAergic neurons, representing a compensatory mechanism, are more efficient in younger PD patients (de la Fuente-Fernandez et al. 2011). More severe nigrostriatal neuron loss occurs in early onset rather than in late-onset PD. At the time of motor symptom onset, the extent of striatal DA marker loss exceeds that of DAergic SN neurons. Neuron loss is more severe in Put (− 98.4%) than in CN (− 89%), whereas in GPi (− 89%) and GPe (− 51%), it is not related to the pattern of Put DA loss (Rajput et al. 2008). The concept that PD motor symptoms first appear when more that 50% of DAergic SN neurons are lost (Bernheimer et al. 1973) has been changed by the notion that at that time only around 30% of DAergic SN neurons, but 50–60% of their axon terminals have been lost (Cheng et al. 2010). This is preceded by loss of DA markers in the nigrostriatal terminals in early PD, while melanin-containing SN neurons may persist for a longer time (Kordower et al. 2013). The duration and severity of motor dysfunction, the corresponding decrease of DA, TH, and vesicular monoamine transporter-2 (VAT2) in striatum are negatively correlated with the total SN α-Syn burden and neuronal loss (Cheng et al. 2010). It shows neither correlation with LB formation (Mori et al. 2006) nor with morphological LB stages, clinical severity of PD, and age at death (Burke et al. 2008), whereas SNc cell loss and α-Syn accumulation are closely related. A significant correlation between the nigral α-Syn burden and DAT immunoreactivity in striatum suggests that the severity of neurodegeneration and local α-Syn burden is closely coupled, whereas nigral TH immunoreactivity did not correlate with α-Syn positivity, which supports the concept of synaptic dysfunction or impairment of axonal transport (Chu et al. 2012). Nigral pigmentation and nigral DAT density show no significant association, wereas pigmentation of the ventral SN tier and DAT binding in related striatal areas are closely related (Martin-Bastida et al. 2019). LP may or may or may not be related to nigral DAergic cell loss (Beach et al. 2009; Colloby et al. 2012; Parkkinen et al. 2011). This suggests that both lesions are not interchangeable hallmarks for disease progression or severity, but could be complementing to each other (Rietdijk et al. 2017). While there are normal levels in the cytosolic fraction of α-Syn without correlation with nigral LB density (Tong et al. 2010), PD brains show a significant increase in soluble and phosphorylated α-Syn (pα-Syn) over the disease course, with progressive decrease of soluble α-Syn (Quinn et al. 2012) and changes of serin 129 pα-Syn (Walker et al. 2013).
Increased pα-Syn precedes its aggregation followed by the formation of LBs and LNs, but it does not necessarily correlate with LP, that shows an inconsistent relationship with clinical disease progression (Lue et al. 2012). Lower neuron densities in SN occur before LB deposition, suggesting that cellular dysfunction precedes LP related to a dying-back mechanism, in which dysfunction is caused by accumulation of small α-Syn aggregates at presynaptic terminals (Schulz-Schaeffer 2015). Accumulation of α-Syn is triggered by presynaptic dysfunction (Nakata et al. 2012), and mediates early synaptic pathology by disrupting synaptic vesicles by retrograde degeneration (Tagliaferro and Burke 2016). α-Syn and synapsin III are suggested to cooperatively regulate DA neuron synaptic function (Zaltieri et al. 2015a), and synapsins have been shown to regulate α-Syn formation (Atias et al. 2019). Early intraaxonal aggregation of α-Syn as “pale neurites” at axon collaterals extending centripetally into proximal segments (Kanazawa et al. 2012) damages the parental neurons by interfering with axonal transport (O’Keeffe and Sullivan 2018; Volpicelli-Daley 2017). Axonopathy in presymptomatic PD is followed by neuronal degeneration (Longhena et al. 2017), suggesting that the loss of DAergic neurons might be a consequence of synaptic loss (Yasuda et al. 2013), defining PD as a “synaptopathy” (Bridi and Hirth 2018; Imbriani et al. 2018; Longhena et al. 2017).
Parkinson disease: a multiorgan disorder
LB/α-Syn pathology in PD is not restricted to DAergic brainstem nuclei, but it is associated with degenerative lesions affecting the central, autonomic, and peripheral system (Beach et al. 2010; Braak and Del Tredici 2009; Wakabayashi and Miki 2018), including the cholinergic basal forebrain, and other neurotransmitter systems (Kalia and Lang 2015; Politis et al. 2010). The extranigral lesions correlate with early premotor symptoms (olfactory, autonomic, sensory symptoms, sleep disturbances, pain, and neuropsychiatric dysfunction), later non-motor fluctuations, and advanced non-DA-responsive nonmotor features (Coon et al. 2018; De Pablo-Fernandez et al. 2017; Jellinger 2015, 2017a, b; Klingelhoefer and Reichmann 2017; Lang 2011; Schapira et al. 2017; Titova et al. 2017). LP involves the spinal cord (Del Tredici and Braak 2012; Nardone et al. 2019), the autonomic and peripheral nervous system, sympathetic and parasympathetic ganglia and plexuses, intramural enteric nervous system, skin, retina, uterus, submandibular gland, bladder, cardiac nervous system, and adrenals (Adler et al. 2016; Braak and Del Tredici 2008; Ma et al. 2019; Orimo et al. 2008; Ortuno-Lizaran et al. 2018; Veys et al. 2019; Wakabayashi and Miki 2018). The musculoskeletal system, and major parts of the sensory nervous system are generally spared (Beach et al. 2009; Cersosimo and Benarroch 2012a, b; Obeso et al. 2017; Oinas et al. 2010), whereas peripheral sympathetic nerves are affected very early (Donadio 2018; Donadio et al. 2019).
Among the earliest involved areas are the olfactory bulb and related olfactory brain nuclei (amygdala and perirhinal cortex), suggesting that olfactory dysfunction in PD is related to the involvement of central pathways rather than peripheral sensory nerve fibers (Attems et al. 2014; Dickson et al. 2009). α-Syn aggregation in the olfactory system and its spreading to the brain may contribute to PD initiation (Cersosimo 2018; Lema Tomé et al. 2013; Rey et al. 2018) by inducing lesions in related brain areas (Niu et al. 2018). Preferential involvement of the olfactory bulb, dmX, and the peripheral autonomic nervous system by LP (Attems et al. 2014; Beach et al. 2010) is related to an increase of pα-Syn in the olfactory bulb and brainstem (Beach et al. 2009; Halliday et al. 2012). Affection of the autonomic nervous system and gastrointestinal tract before involvement of the CNS has suggested a route for spreading α-Syn via the vagus nerve to the brain (Braak and Del Tredici 2008; Holmqvist et al. 2014), confirmed by intragastric rotenone administration or α-Syn inoculation into the mouse gastrointestinal tract (Pan-Montojo et al. 2010). Resection of the vagal nerve interrupted the disease progression to the CNS (Uemura et al. 2018), and appendectomy were associated with reduced risk of PD (Svensson et al. 2015), suggesting a possible role of the gut-brain axis in the pathogenesis of PD (Bove and Travagli 2019; Bu et al. 2019; Perez-Pardo et al. 2017), which has been critically discussed recently (Breen et al. 2019; Kujawska and Jodynis-Liebert 2018; Lionnet et al. 2018). On the other hand, the appearance of α-Syn aggregates in both the submucosal and myenteric plexuses of the enteric nervous system, prior to their appearance in the brain, indicates a possible gut to brain route of α-Syn spread (Felice et al. 2016), and a better understanding of the brain-gut microbiota axis could bring a new insight in the pathophysiology of PD (Fitzgerald et al. 2019; Mulak and Bonaz 2015).
Incidental Lewy body disease (iLBD)
The term iLBD is used when LBs are present in the nervous system in subjects without clinical parkinsonism. Their distribution is similar to that in PD, but often LBs are limited to the limbic cortex, whereas in definite PD cases, LP is present in all regions. A 70% SN cell loss and decreased TH immunoreactivity involve striatum and epicardial nerve fibers, but not to the same extent as in PD (Adler et al. 2010b; Beach et al. 2008; DelleDonne et al. 2008; Dickson et al. 2008), suggesting that it is a preclinical form of PD and that the lack of symptoms is due to subthreshold pathology (Dickson 2018).
Between 5 and 55% of neurologically unremarkable elderly people showed abundant LP with a distribution pattern similar to that seen PD, but relative preservation of pigmented SN neurons (DelleDonne et al. 2008; Jellinger 2004; Markesbery et al. 2009), while LP may be confined to the olfactory bulb. Some had sparse, but widespread LP involving the cortex (Frigerio et al. 2011), which would violate the theory of upward progress from brainstem and would suggest a multicentric disease progress from the onset (Dickson 2012). LP in the spinal cord and dorsal root ganglia in elderly persons was associated with LP in lower brainstem due to retrograde spread (Sumikura et al. 2015).
Staging of Lewy pathology
Three major staging systems currently exist for LB disorders: (1) for PD (Braak et al. 2006; Braak and Del Tredici 2017); (2) for DLB (McKeith et al. 2017); and (3) revised guidelines for LB disease (Zaccai et al. 2008). Based on semiquantitative assessment of LB distribution in a large autopsy series, a staging of the presumed spread of LP was proposed to designate the sequence of lesions in the nervous system (Table 2). LP initially involves the olfactory bulb and related olfactory brain nuclei, the peripheral autonomic system, and adrenal medulla in neurologically unimpaired subjects referred to as iLBD (Beach et al. 2008; DelleDonne et al. 2008; Dickson et al. 2008; Frigerio et al. 2011). In stage 1, the dmX and intermediate reticular zone are involved, while the NBM and midbrain regions are preserved. In stage 2, LNs involve the enteric nervous system, parasympathetic and sympathetic nerves, and medullary nuclei of the level setting system (lower raphe nuclei, gigantocellular reticular nucleus, and ceruleus–subceruleus complex). These a- or presymptomatic stages may explain nonmotor (olfactory and autonomic, e.g., gastrointestinal and urinary) symptoms that precede motor dysfunctions (Cersosimo and Benarroch 2012b; Dickson et al. 2009; Halliday and McCann 2010). In stage 3, LNs and LBs involve PPN, LC, amygdala, upper raphe nuclei, magnocellular nuclei of the basal forebrain, hypothalamic tuberomammillary nucleus, posterolateral/posteromedial SNc, and spinal cord, whereas the allocortex and isocortex are preserved. This stage is associated with disturbed sleep, early motor dysfunction, and several non-motor symptoms. In stage 4, midline and intralaminar thalamic nuclei, anteromedial temporal limbic cortex (transentorhinal and entorhinal region), hippocampus, and the second sector of the Ammon’s horn are affected, associated with severe motor dysfunction. In stage 5, LNs and LBs in cortical areas for regulation of autonomic functions, in higher order sensory association areas and prefrontal fields, are associated with late phase motor disability, and fluctuations. In stage 6, sensory association areas and premotor fields, primary sensory, and motor areas or the entire neocortex are involved (Braak and Del Tredici 2009), causing late motor disability, fluctuations, and cognitive impairment. An increase in the density of α-Syn aggegates and LBs from stages 3–6 correlated negatively with the decrease in neuronal density (Dijkstra et al. 2014).
The validity of the Braak staging scheme, which corresponds roughly to the original classification of LB disorders into three phenotypes—brainstem predominant, limbic/transitional, and diffuse neocortical (Kosaka et al. 1988)—has gained acceptance (Dickson et al. 2010b; Kingsbury et al. 2010), but has been debated (Beach et al. 2010; Burke et al. 2008; Dickson 2012; Jellinger 2009a; Kempster et al. 2010; Parkkinen et al. 2008; Sestini et al. 2019). 51–83% of PD and DLB cases were compatible with this staging (Beach et al. 2009; Jellinger 2009a), but between 6.3% and 47% of autopsy-proven PD cases did not not conform to it (Attems and Jellinger 2008; Beach et al. 2009; Leverenz et al. 2008; Parkkinen et al. 2008). In large autopsy samples, 49–55% of individuals with widespread α-Syn pathology lacked clinical symptoms (Kalaitzakis and Pearce 2009; Leverenz et al. 2008; Zaccai et al. 2008), the determination of cases as atypical being dependent on the staging system applied (Coughlin et al. 2019).
The Braak hypothesis, suggesting predictable caudo-rostral spreading of LP is based exclusively on distribution of LBs but not on neuronal loss, that are not correlated, and it is not identical with α-Syn spreading (Alafuzoff et al. 2009; Rietdijk et al. 2017). While the Braak staging shows only indirect correlations, another scheme based on a limited number of PD cases offered a strong correlation between SN neuronal loss and α-Syn pathology in Braak stages 3–6 (p < 0.001), but no correlation between Hoehn and Yahr and Braak stages (van de Berg et al. 2012). A negative correlation between neuronal density and α-Syn burden was observed in SN, but no relationship with Hoehn and Yahr stage or disease duration (Dijkstra et al. 2014). The Braak staging is valid for PD patients with young onset and long duration with motor symptoms (Halliday et al. 2008), but not for those with late onset and rapid disease course (Jellinger 2019). 10–15% of PD cases associated with genetic mutations show a pattern of LP that is distinct from the Braak scheme (Schneider and Alcalay 2017).
A new unifying system for LB disorders correlates with nigrostriatal degeneration, cognitive impairment, and motor dysfunction (Beach et al. 2009). Whereas the previous systems left 42–50% of elderly individuals unclassified, this new one allowed all cases to be classifiable into one of the following stages: I, olfactory bulb only; IIa, brainstem predominant; IIb, limbic predominant; III, brainstem and limbic; and IV, neocortical. Progression through these stages accompanied by stepwise reduction of striatal TH and SN pigmented cell loss showed significant correlation with clinical and psychometric data (Table 3).
Neuronal vulnerability
There is a close relationship between differential expression profiles of α-Syn and selective vulnerability of certain neuronal populations (Taguchi et al. 2019). Degeneration in PD shows a selective vulnerability of neurons located in the caudal and mediolateral region of SNc (area A9), which have an anatomical, physiological, and biochemical phenotype that predisposes them to α-Syn pathology and mitochondrial dysfunction (Surmeier et al. 2017; Surmeier 2018). Some of the factors which determine vulnerability to degeneration in synucleinopathies are best characterized in the DAergic SNc neurons that suffer from an enormous metabolic burden due to this architecture (long unmyelinated axons and large numbers of synapses), Ca2+ handling capacity, and DA itself being potentially toxic (Post et al. 2018). These neurons contain calbindin (CAB) and glycolytic enzymes, but are poor in DAT and arborize profusely in the striatum and extrastriatal components of the BG. NM lipid changes, upregulation of α-Syn, low intrinsic calcium buffering capacity, change in iron levels, long, poorly myelinated, highly branched axons, and various risk factors promote the susceptibility to selective death of these neurons due to disruption of nuclear membrane integrity (Giguere et al. 2018; Jiang et al. 2016; Surmeier et al. 2017; Surmeier 2018). Calcium mediates the localization of α-Syn at the presynaptic terminal and an imbalance in calcium or α-Syn can cause synaptic vesicle clustering (Lautenschlager et al. 2018). Interaction between α-Syn, calcium ions and DA leads to imbalanced protein turnover of these neurons (Post et al. 2018), that show increased iron (Sian-Hulsmann et al. 2011), but much more in microglia obviously originating from phagocytosis of Fe-laden neurons (Horowitz and Greenamyre 2010). An inhibitory effect of α-Syn on proteasomal activities can contribute to the selective vulnerability of DAergic neurons in PD (Zondler et al. 2017). Dysfunctional synaptic vesicle endocytosis may contribute to selective vulnerability of DAergic midbrain neurons (Nguyen et al. 2019). Neurons in STN and GABAergic SNr, that are rich in calcium-binding proteins (calcineurin and parvalbumin), and glycolytic enzymes are either not affected or involved only in the terminal stages (Double et al. 2010). The confluence of disruption of the cellular metabolic state and α-Syn structural equilibrium, and anatomical connectivity as suggested factors to initiate cascades of pathological processes triggered by genetic, environmental, or stochastic events was reviewed recently (Alegre-Abarrategui et al. 2019).
Lesion patterns in clinical subtypes of Parkinson’s disease
Pathological variability of PD contributes to its clinical heterogeneity of the disease. Two major clinical subtypes of PD show specific morphologic patterns of pathophysiologic importance, with different involvements of striatal and cerebello-thalamo-cortical pathways (Figs. 2, 3). The two classical motor subtyping systems of PD poorly overlap, but their temporal instability undermines their prognostic value in the early stage of PD (Erro et al. 2019).

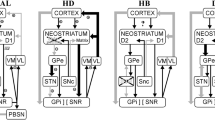
From (Jellinger 2016)
Schematic diagram of the basal ganglia-thalamocortical circuitry under normal conditions and in hypokinetic movement disorders. The width of lines represents the relative change in activity versus normal. Disrupted lines represent altered patterns with an increase or decrease in neuronal activity; dashed arrow, reduced activity; solid arrow, increased activity. D1 and D2 dopamine 1 and 2 receptor subtypes, DYS dystonia, GPe and GPi external and internal segment of the globus pallidus, IP/DP indirect/direct pathway, MSA multiple system atrophy; normal, normal conditions, PD Parkinson’s disease, PPN pedunculopontine nucleus, PSP progressive supranuclear palsy, SNc and SNr substantia nigra pars compacta and reticulata, STN subthalamic nucleus, TH thalamus, VL and VM ventrolateral and ventromedial thalamic nuclei.

Modified from (Helmich et al. 2011)
Model of cerebral mechanisms underlying Parkinson’s disease resting tremor. It emerges from the ventral intermediate nucleus of the thalamus (VIM)–motor cortex (MC)–cerebellum (CBLM) circuit (in blue), when triggered by transient pathological signals from the basal ganglia motor loop (in red). In tremor-dominant PD, the basal ganglia globus pallidus internus, globus pallidus externus and putamen) has increased connectivity with the VIM–MC–CBLM circuit through the MC (thick red line), and the basal ganglia is activated at critical times in the tremor cycle (onset/offset of tremor episodes). These alterations may be caused by loss of dopaminergic projections from retrorubral area 8 in red to the GPi and GPe. These alterations are different from the dopaminergic denervation of the striatum associated with bradykinesia and rigidity. DA dopamine, SNc substantia nigra pars compacta, StN subthalamic nucleus, Vop thalamic ventralis oralis posterior nucleus.
In the akinetic-rigid type (about 50% of PD patients), the ventrolateral SNc projecting to dorsal Put degenerates more severely than the medial parts projecting to CN and anterior Put. Loss of TH- and DAT-reactive fibers and endings progressing from the dorsal to the ventral Put is associated with damage to the met-ENK and SP-rich AChE-poor striosomes projecting to the predominantly affected ventrolateral SNc, that correlates with DA loss in posterior Put and the severity of akinesia/rigidity (Bernheimer et al. 1973). DAergic denervation causes loss of dendrites on type I MSNs, the principal targets of DAergic input from the SN, and decline of nigrostriatal DA. DA modulation of glutamatergic synapses on the striato–pallidal GABA and striato–nigral pathways via heteroreceptor complexes (Borroto-Escuela et al. 2018) is due to the efficacy of inhibitory synaptic plasticity of these BG output nuclei (Milosevic et al. 2019). The beneficial effect of l-dopa on bradykinesia is associated with normalization of the striato–thalamo–cortical motor and STN–cortical motor pathways (Gao et al. 2017).
In early PD stages, overactivation of the BG as a compensation of the DA deficit in the striatal motor circuit (Reetz et al. 2009) and decreased excitation of D1-bearing neurons lead to reduced activity of the “direct” pathway, whereas reduced inhibition of D2-bearing neurons results in decreased activity in striatopallidal GPe projections. In later stages, this filtering mechanism is deranged, and DA depletion shifts the BG toward inhibiting movements by increased activity in the GABAegic “indirect” GPe–STN–GPi network and decreased activity in the “direct” cortico–Put–GPi circuit due to loss of D1 excitation (Calabresi et al. 2009). Excessive glutamatergic drive from GP/SNr leads to an akinetic–rigid syndrome through reduced cortical activation due to inhibition of thalamocortical and brainstem motor systems or due to loss of DA input to prefrontal or motor cortex (Fig. 2).
The tremor-dominant type (about 25% of PD patients) that shows a better prognosis and slower disease progression has less severe depletion of lateral SNc, but damage to the retrorubral A8 field, which is usually preserved in AR PD (Paulus and Jellinger 1991). It projects to the matrix of the dorsolateral striatum and VM thalamus, and influences striatal efflux via the SNc and thalamus-to-prefrontal cortex (Fig. 3). Resting tremor severity is inversely correlated with raphe serotonin transporter availability which, together with Put DA depletion may contribute to it (Pasquini et al. 2018). Resting tremor is associated with increased activity of the ventral intermediate (VIM) thalamus and dysfunction of cerebellar connections (Elias et al. 2008) and is produced by pathological interaction between BG and the cerebello–thalamo–cortical circuit in the presence of striato-pallidal DAergic dysfunction (Dirkx et al. 2017; Helmich 2018). Deficits in cerebellar function with decreased excitability of the cerebello–thalamo–cortical pathway may generate postural tremor, indicating that resting and postural tremor in PD is mediated by different pathways (Ni et al. 2010).
Motor complications, dyskinesia, and freezing
α-Syn pathology in striatum, progressive loss of DAergic neurons and of TH- and DAT-reactive nigrostriatal fibers increase with progression of PD (Sorrentino et al. 2019), and are substrates for motor deficits and decreased efficacy of DAmimetic therapy in late stages of PD (Lane 2019). Prevalence of l-dopa-induced dyskinesia (LID) ranges from 3 to 94% (Rosqvist et al. 2018; Tran et al. 2018). LID can also be present in MSA and PSP, although less frequently, and with varying clinical manifestation (Jost et al. 2019). Dysregulation of striatal projecting neurons in advanced PD (Beck et al. 2018) and degeneration of striatal efferents with transgression to non-DAergic systems cause loss of postsynaptic D2, and muscarinic cholinergic receptors in striatum and of N-methyl-d-aspartate (NMDA) receptors and glutamatergic synapses degenerate, favoring drug resistance and motor complications (Picconi et al. 2008). Impairment of synaptic plasticity of striatal MSNs contributes to the development of motor fluctuations and dyskinesias (Bagetta et al. 2010).
Hyperstimulation of DAergic receptors and impairment of synaptic plasticity of striatal MSNs causing excessive striato-cortical connectivity in response to l-dopa produce aberrant signals that trigger involuntary movements (Herz et al. 2015) and overreaction of the mesocortical and mesolimbic systems results in hyperdopaminergism (Voon et al. 2017) (Fig. 2). Presynaptic dysregulation of DA release after l-dopa, causing stimulation of striatal intraneurons (D1-MSNs), may trigger LIDs (Klietz et al. 2016; Mosharov et al. 2015; Perez et al. 2017). It has become evident that striatal interneurons are major determinants of network activity and behavior in PD and LID (Zhai et al. 2019). Peak-dose dyskinesias are caused by the following mechanisms: (1) marked fluctuation of DA concentrations occur in synaptic clefts of striatal neurons after each l-dopa dose; (2) supersensitive cortico-striatal synapses of direct-pathway spiny neurons; (3) increased production of GABA in the spiny neurons and their axon terminals; (4) each l-dopa dose causes excessive release of GABA into the output nuclei of the BG, resulting in their abnormal firing (Tomiyama 2017); and (5) modifications in perisomatic GABAergic connectivity and neuronal activation of MSN, leading to an imbalance between excitation and inhibition in striatal activity (Gomez et al. 2019). Sprouting of DAergic terminals may contribute to increased DA release/turnover, and increased DA sensitivity of striatal cholinergig neurons, predisposes to motor complications (Bordia and Perez 2019; Perez et al. 2018). Pre- versus postsynaptic mechanisms, changes in DA receptor subtypes, glutamate receptors, striatal spreading depolarization contributing to abnormal BG activity, and non-DAergic transmitter systems including serotonergic and cholinergic mechanisms are also related to LIDs (de Iure et al. 2019; Pagano et al. 2018; Politis et al. 2014). Monoaminergic dysregulation in limbic domains (Engeln et al. 2015) and structures outside the CBGTC circuit, as well as cerebellar dysfunction of the PPN-GB system, may also contribute to LID (Cenci et al. 2018; French and Muthusamy 2018). Since the PPN is densely connected with the BG and the brainstem dysfunctions of this system (Bohnen et al. 2019) or of cerebellar connections (Bhatia et al. 2018) lead to advanced symptomatic progression in PD (French and Muthusamy 2018). The recently described bidirectional connections between BG and cerebellum indicate a key role of the cerebellum in the generation of LID. This model suggests that aberrant neuronal synchrony in PD with LID may propagate from the STN to the cerebellum and “lock” the cerebellar cortex in a hyperactive state. The motor responses are worsened by the lack of normal subcortico-cortical inputs from cerebellum and BG due to of the aberrant plasticity at their own synapses (Kishore and Popa 2014). Animal models of LID in rats and mice with nigrostriatal 6-OHDA lesions treated with l-dopa developed involuntary movements with both hyperkinetic and dystonic components, which enabled insight into the mechanisms of LID (Cenci and Crossman 2018; Keber et al. 2015).
Freezing of gait (FOG), one of the most disabling motor symptoms in PD, reflects a combined motor and cognitive de-automatization deficit, which may be related to structural changes in the PPN network affecting prefrontal cortical areas involved in executive inhibition function (Fling et al. 2013; Snijders et al. 2016), a functional decoupling between the cognitive cortical control network and the BG (Shine et al. 2013), or specific changes in the frontostriatal pathways rather than brainstem lesions (Hall et al. 2014), while others found correlations between the severity of FOG and the density of cortical LB-containing neurons (Virmani et al. 2015).
Pathology of cognitive impairment in Parkinson’s disease
Cognitive impairment (CI), which may precede the onset of dementia up to 10 years, was observed in 19–30% of untreated PD patients (Aarsland and Kurz 2010; Poletti et al. 2012), mild cognitive impairment (MCI), often progressing to dementia in 21–62%, and a mean of 25.8% (Aarsland and Kurz 2010; Jellinger 2013b). The point cumulative prevalence of dementia in PD (48 and 78%), with a mean of 75% after more than 10 years, of 83% after 20 years (Hely et al. 2008) is up to 95% by age 90 years (Rongve and Aarsland 2013). PD dementia (PDD) has a prevalence of 31.3% (95% CI 20.1–42.1) and incidence rates from 42.6 to 112.5/1000 person/years (Marder 2010), indicating that around 10% of a PD population develop dementia per year (Emre et al. 2007). The pathological substrate of CI in PD is heterogenous, related to both LB and AD pathologies, multiple neurotransmitter deficits, and changes in gray and white matter (Hall and Lewis 2019; Wilson et al. 2019). Neuropathology of MCI in PD (PD-MCI) with brainstem–limbic, and rare neocortical LB lesions, amyloid but only rare neuritic plaques in cerebral cortex, mild cerebral amyloid angiopathy (CAA), and lacunar state in BG (Adler et al. 2010a; Jellinger 2013b), or cortical or limbic predominant LB disease, but rare coexisting AD (Molano et al. 2010), is similar to that found in MCI cases without PD (Markesbery 2010; Petersen et al. 2006). Structural brain analyses found unilateral insula involvement in PDD-MCI extending to bilateral insula involvement in PDD indicating both increasing brain atrophy in PD with CI and suggesting the existence of sub-typing in PD-MCI (Mihaescu et al. 2018). In PD-MCI, cholinergic fiber depletion was evident, which was correlated with loss of neurons in hippocampal subfield CA2, whereas only PDD cases had significantly greater LP in CA2 (Liu et al. 2019a).
Cognitive deficits in early PD are associated with impaired striatal and extrastriatal DAergic function (Siepel et al. 2014), due to abnormal processing in the cortico–BG circuit with reduced prefrontal and parietal metabolism (Ekman et al. 2012) and dysfunction of the salience networks of the medial temporal lobe (Christopher et al. 2015). Dysfunction of subcortico–cortical networks is a result of neuronal loss in brainstem and limbic areas, cholinergic deficits in cortex, thalamus, and NBM, striatal DA loss, degeneration of the medial SN, and striatosubfrontal and mesocorticolimbic loops. Cortical cholinergic denervation and early posterior cortical atrophy contribute to CI in PD (Bohnen et al. 2015; Sampedro et al. 2019). Reduction of cholinergic markers is due to early degeneration of the corticopetal basal forebrain projection involving the NBM (70% loss of cholinergic neurons in PDD) (Liu et al. 2018; Ray et al. 2018; Schulz et al. 2018). Muscarinic acetylcholine receptors (mAChRs) are important in the regulation of the striatal network which may have implications in the motor and CI in PD (Ztaou and Amalric 2019). Galanin upregulation in the NBM as a response to loss of cholinergic neurons was higher in the transition between PD and PDD, but failed with increasing AD pathology, thus being uncommon in established AD and DLB (Alexandris et al. 2019). The noradrenergic LC, serotonergic DRN, and VTA are also involved (Del Tredici and Braak 2013; Espay et al. 2014; Halliday et al. 2014; Vermeiren and De Deyn 2017). Aβ pathology is not the primary driver of CI and dementia in PD (Melzer et al. 2019). A systemic review of autopsy studies of PDD was published recently (Smith et al. 2019).
PD patients without dementia may have AD pathology largely restricted to the limbic system (Braak neuritic stages < 4), whereas in 10–50% of PDD cases, it was severe enough to attain the diagnosis of definite AD (Hepp et al. 2016). Neocortical or limbic LP was considered as the most significant correlate of dementia in PD (Horvath et al. 2013b), while recent studies revealed increasing evidence of tau pathology in PD (Whitwell 2018; Zhang et al. 2018b). PD patients with AD co-pathology harbor greater neocortical α-Syn pathology, the latter contributing uniquely to the heterogeneity of CI diseases (Coughlin et al. 2018), while both cognitive and gait disturbances in PD show common underlying pathological mechanisms related to AD pathology (Lim et al. 2018).
Molecular pathology of depression in Parkinson’s disease
Depression is a predominant non-motor symptom involving 30–40% of PD patients (Reijnders et al. 2008). However, the neuropathology of this comorbitity is still unclear. Early neuropathological studies indicated a higher prevalence of lesions in depressed compared to non-depressed PD patients particularly in catecholaminergic brain areas (neuron loss in LC, DVN, and SNc), suggesting that depression in PD is related more to catecholaminergic than serotonergic systems (Frisina et al. 2009). Decreased DAT binding in the CN suggested that depressive symptoms in PD are associated with DA loss in this region related to degeneration of DAergic projections from the VTA (Vriend et al. 2014). Later, imaging studies presented conflicting data about the role of serotonergic degeneration in depression in PD: while some studies suggested that abnormalities in serotonin 1A receptor neurotransmission in the limbic system may be involved in the neural mechanisms underlying depression in PD patients (Ballanger et al. 2012) and emphasized a prominent role of the serotonergic degeneration in apathy, anxiety, and depression in de novo PD (Maillet et al. 2016), others found no association between raphe serotonin transporter availability and depression and other psychiatric symptoms in early drug-naive PD patients (Qamhawi et al. 2015). Other imaging studies demonstrated widespread abnormalities within the limbic circuits notably the orbitofrontal and anterior cingulate cortices, amygdala, thalamus, and ventral striatum involved in the pathophysiology of depression in PD (Thobois et al. 2017). Recent diffusion MRI connectometry studies suggested that the prominent circuits involved in emotion and recognition (fornices, fronto–occipital fasciculus, genu of corpus callosum, etc.) might be impaired in comorbid depressive symptoms in PD (Ansari et al. 2019). Other recent studies indicated that an abnormal mesocorticolimbic system may account for depressive symptoms in PD, suggesting that resting-state functional connectivity of midbrain DAergic nuclei might be useful for understanding the underlying pathology in PD with depression (Wei et al. 2018), while others suggested impaired interhemispheric synchrony as underlying neural mechanism of depression in PD (Zhu et al. 2016). Another study showed significant negative association between depression scores in PD patients and qualitative anisotropy (QA) of left cingulum, genu and splenium of corpus callosum, and anterior and posterior limbs of the right internal capsule (Ghazi Sherbaf et al. 2018). Others suggested a possible role of inflammation and neuromodulation as pathogenic mechanism of depression and cognitive impairment in PD (Pessoa Rocha et al. 2014). The inflammatory hypothesis states that depression in PD is caused by changes in the serotonergic systems induced by neuroinflammation (Santiago et al. 2016), whereas disturbances in monoaminergic transmission and the hypothalamic–pituitary–adrenal axis, increased oxidative and neuroinflammatory events, and impaired trophic transport may be implicated in the relationship between depression and neurodegeneration (Galts et al. 2019). Recent studies failed to verify the vascular depression hypothesis in PD (Ou et al. 2018).
Neuronal basis of drug-induced psychoses in Parkinson’s disease
Psychotic symptoms in PD have a prevalence of 20–40% (Bizzarri et al. 2015) and are associated with high morbidity and mortality (Samudra et al. 2016), but their pathogenesis is unclear. Factors implicated include DAergic medications, neurotransmitter imbalances, neuroanatomic alterations, and genetic disposition (Ffytche et al. 2017). Other factors include LB deposition in the limbic system, cholinergic deficits and impairments of primary visual processing (Williams-Gray et al. 2006), or genetics (e.g., APOE ε4 allele and tau H1H1 genotype) (Zahodne and Fernandez 2008). Current theories on the pathophysiology of PD psychosis implicate pathways involving visual processing and executive function, including temporo-limbic structures and neocortical gray matter with altered neurotransmitter functioning (Chang and Fox 2016), while others described degeneration of specific hippocampal subfields in PD patients with psychosis (Lenka et al. 2018). Unlike patients with PD psychosis who have dementia, those without dementia have no higher LB load in amygdala and hippocampus (Harding et al. 2002; Kalaitzakis et al. 2009a). Definite neuropathological findings for drug-induced psychoses in PD, to the best of our knowledge, are not available.
Genetics of Parkinson’s disease
Familial parkinsonism is rare (5–10%), but the importance of genetic factors is increasingly being recognized (Lill 2016). The heritable estimate of PD is between 23 and 34% (Chang et al. 2017). A minority present a Mendelian form with autosomal-dominant (AutD), e.g., SNCA, LRRK2, and VPS35 genes accounting for 0.1–30% of PD, or with autosomal-recessive (AutR) transmission, e.g., PARK2 (Parkin), PARK7 (DJ-1), or PINK1 genes, depending on family history, age at onset, and population background (Trinh et al. 2018; Volta et al. 2015). Until to date, a total of 23 loci and 19 causative genes have been associated with PD, although some of the PARK loc (PARK3, 10, 12, and 16) have not yet been identified (Del Rey et al. 2018). PD GWAS confirmed 10 candidate genes previously selected and nominated 17 novel candidate gens for sporadic PD (Ferrari et al. 2018). PARK14 (D331Y) PLA2G6 mutation causes degeneration of SNc DAergic neurons by inducing mitochondrial dysfunction, elevated ER stress, mitophagy impairment, and transcriptional abnormality (Chiu et al. 2019). Seven novel candidate genes (VCAM1, BACH1, CALM3, EGR1, IKBKE, MYC, and YWHAG) may play important roles in PD pathogenesis (George et al. 2019). SNCA, the gene encoding α-Syn, is central to the pathogenesis of PD (Lubbe and Morris 2014; Singleton et al. 2013; Verstraeten et al. 2015). It is associated with hereditary AutD forms, and variations of the SNCA gene are associated with increased risk for sporadic PD (Nussbaum 2018). In cases harboring SNCA missense mutations, several mechanisms could lead to a loss of functional mechanisms including haploinsufficiency and epigenetic silencing (Voutsinas et al. 2010). A recently identified SNCA mutation, p.Ala53Glu (A53E), enriches α-Syn oligomers and fibrils dependent on the phosphorylation state (Picillo et al. 2018). Families with SNCA multiplications are rare and globally distributed (Book et al. 2018). A systematic Movement Disorder Society (MDS) gene review identified common variants in SNCA, LRRK2, MAPT and GBA genes contributing to increased PD susceptibility (Lill 2016; Marras et al. 2017). SNCA, TMEM175, SCARB2, BAG3, and GBA have all been shown to be implicated in α-Syn aggregation pathways, while other established risk loci, such as GCH1 and MAPT, show no effect on age at onset of PD (Blauwendraat et al. 2019). Mutations of PARK2 and LRRK2 cause early onset PD with AutR patterns of inheritance. The most common mutation of LRRK2, encoding dardarin, on chromosome 12, a heterozygous G2019S mutation, accounts for approximately 3–10% of familial and 1–8% of sporadic PD (Hernandez et al. 2005). LRRK2 levels are negatively correlated with disease duration; LRRK2 phosphorylation was reduced with clinical PD (Dzamko et al. 2017). Dysfunction or loss of LRRK2 decreased α-Syn aggregation and modifies α-Syn spread in mouse models and human neurons (Bieri et al. 2019), and may influence the accumulation of α-Syn and its pathology to alter cellular functions and signaling pathways (Rui et al. 2018). PRKN, PINK1 and DJ1 cases are associated with early onset PD with slow progression. Mutations in the PRKN gene (encoding parkin) are the most common cause of AutR familial PD, representing up to 50% of all early-onset cases (Schulte and Gasser 2011). PD patients with PARKIN mutation show dystonia at onset and dose-sensitive LID, which is suggested to be caused by other mechanisms than the well-established DA depletion. Since cortical and striatal neurons express PARKIN protein, which modulates the function of ionotropic glutamatergic receptors, PARKIN may have a potential role in controlling the glutamatergic corticostriatal synapse transmission. PARKIN transcript variants 3, 7, and 11 were over-expressed in striatum and cerebellar cortex, together with synphilin-1A and 1C, suggesting that alterations in the regulation of transcription events that may be important for the increased aggregation of α-Syn (Brudek et al. 2016). Patients with PARK2 mutations show increase in the expression of catechol-O-methyltransferase (COMT) and a reduction in DNA methylation in DAergic neurons, which may contribute to the initial neuronal dysfunction in PD (Kuzumaki et al. 2019). Loss of PARKIN function can dysregulate transmission at these synapses where they cause maladaptive changes that co-occur with changes due to DA loss. This suggests an early striatal synaptopathy as the potential cause of early LID in PARKIN mutations (Sassone et al. 2019). PD cases with heterozygous variants in AutR genes suggest that monogenic and idiopathic PD may have shared pathogenic mechanisms (Reed et al. 2019). GBA is a major PD risk factor (Davis et al. 2016; Lunati et al. 2018). GBA mutations influence the age of disease onset, disease severity, motor phenotype, and are associated with a significant risk of dementia (Seto-Salvia et al. 2012). Several α-Syn point mutations associated with familial PD prone to form oligomers tend to form fibrils to a lesser extent (Ruf et al. 2019). In autopsy-proven PD, mutations of the GBA1 gene located on chromosome 1q21 which encodes glucocerebrosidase (GCase) are the most common genetic factor (in 5–20% of PD cases) by interference with α-Syn homoeostasis pathways (Blandini et al. 2018; Mullin et al. 2019; Sidransky and Lopez 2012). GBA mutations induce α-Syn aggregation, lysosomal autophagy changes, and endoplasmic reticulum stress (Balestrino and Schapira 2018; Maor et al. 2019; O’Regan et al. 2017). GCase deficiency, most pronounced in SN, leads to mitochondrial dysfunction, decreased macroautophagy, neuronal ubiquitinopathy and axonal lesions (Gegg and Schapira 2018), and may promote the spread of α-Syn aggregates (Bae et al. 2014; Thomas et al. 2019). The GBA1-substrate glucosylceramide (GluCer) can induce α-Syn aggregation via conversion of physiological α-Syn oligomeric forms into neurotoxic oligomers that are also able to seed amyloid fibril formations (Zunke et al. 2018). No consensus exists regarding the pathogenic mechanism of GBA PD (Mullin et al. 2019). However, the interrelation between GCase, glucosylsphingosine and α-Syn parameters supports the hypothesis that GCase acts as a modulator of PD-DLB (Gundner et al. 2019). Overlap between monogenic and sporadic PD genes is seen for SNCA and LRRK2 loci, LRRK2 and α-Syn showing interaction in PD brains and in cell models (Daher 2017). Mutations of GCH1 that encodes guanosine triphosphate (GTP) cyclohydrolase 1, essential for DA synthesis in nigrostriatal cells, may lead to PD and Dopa-responsive dystonia (Rudakou et al. 2019; Yoshino et al. 2018). Different mutations in a single gene exhibit considerable clinical and neuropathologic variables both within and between kindreds. COQ2 variants, associated with familial MSA, rarely may associate with familial PD (Mikasa et al. 2018). Recent meta-analyses of GWAS data for target genes of brain microRNAs that have been implicated in PD pathogenesis showed significant associations of genetic variants in nine loci (Schulz et al. 2019). PARK1 overexpression was shown to promote PD-like phenotypes by direct phosphorylation of α-Syn at the serine 129 site, inducing DA neuron degeneration in PD (Su et al. 2019). PD risk loci do not lie in specific cell types or brain regions, but rather in global cellular processes detectable across several cell types (Reynolds et al. 2019). DJ-1 (PARK7) can induce activation of transcriptional factors and change redox balance that may protect neurons against α-Syn aggregation and oligomer-induced neurodegeneration (Dolgacheva et al. 2019).
Neuropathology of genetic Parkinson’s disease
Neuropathological features of AutD SNCA (PARK/PARK4) are similar, with LP in all cases. AutR PARKIN (PARK2) mutations usually showed more severe neuronal loss in SNc than in LC, most without LP. Many individuals with A53T mutations (e.g., in the Contursi kindred) had α-Syn neuritic pathology, tau-positive neuritic inclusions, and some had both tau and α-Syn lesions (Kotzbauer et al. 2004; Markopoulou et al. 2008; Polymeropoulos et al. 1997). LRP10 gene defects (at chromosome 14) are implicated in the development of familial PD and DLB, some showing severe LP (Quadri et al. 2018; Sestini et al. 2019). Several forms of PD do not have LBs (Jiang and Dickson 2018). DJ1 (PARK7) AutR cases showed severe SNc and LC neuronal loss with diffuse LP (Taipa et al. 2016) and most GBA PD cases showed LP involving cortical areas (Schneider and Alcalay 2017). LRP10 gene variants showed severe LP (Quadri et al. 2018). PINK1 (PARK6) and PRKN mutations cause AutR early onset PD, with neuronal loss, no LP and inconsistent tau pathology (Schneider and Alcalay 2017). In rare mutations in PLA2G6 (PARK14), cell loss in SN and LC with rare LBs were associated with spheroids and iron deposition in GP (Klein et al. 2016), DJ-1-associated pathology shows damage to SN and LC with diffuse LP (Taipa et al. 2016). TDP-43 pathology is rare in MAPT and SNCA gene mutations (Schneider and Alcalay 2017).
LRRK2 mutations (PARK8), the most common cause of late-onset and AutD PD, are pathologically comparable to sporadic PD (Marras et al. 2016; Pont-Sunyer et al. 2017), with cell loss in SNc and LC but inconsistent LP (Takanashi et al. 2018). LRRK2 is a complex multi-domain protein with kinase and GTPase enzymatic activity. It is associated with mitochondrial functions and autophagy (Gomez-Suaga et al. 2012). Mutations of α-Syn, LRRK2 and tau that have been associated with familial and sporadic forms of PD show a complex interplay (Outeiro et al. 2019a) and a range of tau and TDP-43 pathologies. LRRK2 phosphorylates both tau epitopes and amyloid precursor protein (APP), promotes neurotoxiciy in PD and tauopathy (Bailey et al. 2013; Chen et al. 2017), suggesting an overlap between both AD and PD. Neuropathology in familial PD due to A30P mutant α-Syn was identical to sporadic PD (Seidel et al. 2010). LP was described in heterozygous (R275W) mutations of the PARK2 gene (Ruffmann et al. 2012), and in a family with early-onset PD associated with a heterogenous PARKIN exon 3–4 deletion (Sharp et al. 2014). The MAPT H1 haplotype is related to a higher burden of neocortical LP (Robakis et al. 2016). Other AutD forms pathologically resemble PD with neuronal loss in SN, with or without LBs and NFTs (Tomiyama et al. 2007). G51D SNCA mutations showing neuronal α-Syn and oligodendroglial inclusions may represent a link between PD and MSA (Kiely et al. 2013). The APOE ε4 allele has been considered to be associated also with α-Syn and TDP-43 pathologies (Dickson et al. 2018; Yang et al. 2018).
Kufor-Rakeb disease (KRS/PARK9), a rare AutR young onset disease, the result of mutations of the ATP13A2 gene on chromosome 1p (Park et al. 2015), shows parkinsonism, pyramidal tract signs, supranuclear gaze palsy, dystonic spasms, myoclonus, autonomic dysfunctions, dementia, and good response to l-dopa (Kruer 2013), while other KRS siblings manifested myoclonus and seizure (Rohani et al. 2017). Postmortem studies are so far lacking. Sural nerve biopsy showed reduced myelin fiber density, axonal degeneration, and cytoplasmic inclusion bodies resembling primary lysosomes (Paisan-Ruiz et al. 2010); electron microscopy revealed dense lamellar deposits ca. 1 µm in size (Malandrini et al. 2013). A novel mutation was found in Ashkenazi cases (Inzelberg et al. 2018).
Perry’s syndrome, a rare combination of AutD parkinsonism, depression, hypoventilation, and weight loss, is an early-onset, rapidly progressing disease with neuronal loss in SN without LBs and involvement of putative respiratory neurons in ventral medulla caused by mutations in dynactin p150(Glued) (DCTN1) (Konno et al. 2017; Mishima et al. 2017, 2018). TDP-43-positive, pleomorphic neuronal inclusions, dystrophic neurites, and axonal spheroids were seen in pallidonigral distribution. TDP-43 was neurochemically similar to that found in FTLD-U indicating that Perry’s syndrome is a unique pallidonigral TDP-43 proteinopathy (Mishima et al. 2017; Wider et al. 2009). Three families from distinct parts of the world have been reported (Tacik et al. 2014).
Dementia with Lewy bodies
DLB, accounting for 4.6–10.1% of all dementia cases (Arnaoutoglou et al. 2019; Kane et al. 2018), has an incidence of 3.5/100,000 person/year (Savica et al. 2013b), although it is widely underdiagnosed. A recent meta-analysis reported a pooled sensitivity, specificity, and accuracy of 60.2% (95% CI 30.9–83.7%), 93.8% (83.8–97.6%), and 79.7% (62.6–90.7%) for the diagnostic criteria of DLB, while about 20% of DLB diagnosis are incorrect (Rizzo et al. 2018; Skogseth et al. 2017). Diagnostic accuracy of DAergic imaging in prodromal DLB has a specificity of 89%, but a sensitivity of 61% (Thomas et al. 2019), while occipital hypometabolism has 91% sensitivity and 80% specificity (Hamed et al. 2018). 18FFDG-PET hypometabolism in temporo-parietal and occipital cortex was highly consistent across DLB cases (Caminiti et al. 2019). Clinical features of DLB are spontaneous parkinsonism, recurrent visual hallucinations, fluctuating cognition, RBD, sensitivity to antipsychotic medication, and reduction in striatal DAT (Donaghy and McKeith 2014; Sanford 2018). According to international consensus, DLB is diagnosed when CI precedes parkinsonism, or begins within 1 year of parkinsonism; PDD when parkinsonian symptoms precede CI by more than 1 year (McKeith et al. 2005). Revised criteria for the clinical dagnosis of probable and possible DLB have been reviewed recently (Cousins et al. 2019; Outeiro et al. 2019b; Yousaf et al. 2019), as well as imaging in prodromal DLB (Durcan et al. 2019; Lin and Truong 2019). Complex visual hallucinations are the only symptoms which helped identification of DLB in the context of a mixed AD + DLB dementia (Thomas et al. 2018).
Genetics of Lewy body disease
Our present understanding of the genetic etiology of DLB is limited, although a few families with AutD inheritance and mutations in SNCA and SNCB have been reported (Nervi et al. 2011). The heritable component of DLB was estimated at about 36% (Guerreiro et al. 2018). GBA and APOE ε4 are the most prevalent risk factors for sporadic DLB (Vergouw et al. 2017) and PDD (Brockmann et al. 2015; Sun et al. 2019). PSEN1 and APP are also common (Geiger et al. 2016). GBA mutations are associated with cortical LBs but not with AD pathology (Geiger et al. 2016). The fact that many members of kindreds with mutations in the SNCA gene show some features of DLB, and the frequent occurrence of LBs in familial and sporadic AD, suggested an overlap in their genetic factors, which was not confirmed by GWAS metaanalyses (Moskvina et al. 2013; Orme et al. 2018). MAPT H1 haplotype is associated with enhanced α-Syn deposition, suggesting a genetic association between MAPT haplotype and synucleinopathies (Colom-Cadena et al. 2013b), confirmed by recent GWAS (Outeiro et al. 2019b). Another GWAS reported a wide variety of copy number variations in a large DLB cohort (Kun-Rodrigues et al. 2019). LRP10 (encoding the LDC receptor related protein 10) is a novel disease gene bridging PD and DLB (Quadri et al. 2018). Today, only three genes have been convincingly established to be involved in DLB: APOE, GBA, and SNCA (Orme et al. 2018).
Neuropathology of dementia with Lewy bodies
The DLB brain is macroscopically similar to that in PD. Unlike AD, it shows relative preservation of the medial temporal lobe (hippocampus) (Oppedal et al. 2019). The histologic hallmarks are α-Syn/Lewy pathology or a variable mixture of Lewy and AD pathologies, with three main patterns: (1) widespread LBs associated with cortical diffuse Aβ plaques and low neuritic Braak stages, (2) widespread LBs with neuropathological halmarks of AD, fulfilling the criteria for both diagnoses (mixed AD/DLB), and (3) “pure” LB disease involving widespread cortical areas without significant AD pathology (Irwin and Hurtig 2018). LB density is assessed semiquantitatively, using α-Syn immunohistochemistry, in five cortical regions. According to the density and distribution of LBs, patients are allocated to the brainstem-predominant (PD), limbic (or transitional) type, LBs relatively being restricted to limbic structures, and neocortical type with widespread cortical LBs (Fujishiro et al. 2008a; McKeith 2007). The pattern of LP in brainstem is similar to that of PD (Seidel et al. 2015), but the majority of DLB cases have advanced LB stages. Clinical features are related to the extent of LP and negatively to the severity of tau pathology (Ruffmann et al. 2016; Tiraboschi et al. 2015). Correlates of attentional dysfunction and visual hallucinations are impairment of orbitofrontal, anterior cingulate cortex and secondary visual areas (Heitz et al. 2015), neuronal loss and α-Syn pathology in the superior colliculus (Erskine et al. 2017), less in the pulvinar, which, however, showed decreased protein levels and astrogliosis associated with synaptic changes (Erskine et al. 2018). The striatum exhibits a variable reduction (more than in AD, less than in PD) in TH immunoreactivity, loss of DA markers, reflecting loss of SN neurons and low striatal DAT in DLB compared to AD. Cholinergic denervation is a result of neuronal loss in NBM and cholinergic basal forebrain (Alexandris et al. 2019; Nejad-Davarani et al. 2019). The insular cortex shows severe α-Syn involvement with sparing of insular TH neurons (Fathy et al. 2019). Recent studies showed upregulation of β-synuclein (βSyn) within the frontal cortex and its decrease in occipital cortex of DLB patients, while βSyn-containing neurons were consistently devoid of oligomeric α-Syn in frontal cortex. There was no overall correlation between total βSyn and 5G4 levels (marker of oligomeric α-Syn). The autophagy markers LC3-II and p62 were increased in the areas of βSyn upregulation, which suggests that βSyn changes in DLB may exacerbate neuronal dysfunction caused by accumulation of α-Syn due to influencing protein degradation pathways (Evans et al. 2018).
LP levels were highest in the hippocampal CA2 subregion and entorhinal cortex, while correlation with memory performance was strongest with CA1 burden. This suggests that LP must reach a critical burden across hippocampal circuitry to contribute to memory dysfunction (Adamowicz et al. 2017). Phosphorylated α-Syn at the presynaptic terminals in the form of small aggregates may disrupt structure and function of synapses (Colom-Cadena et al. 2017b; Schulz-Schaeffer 2010). Levels of insoluble pS129-α-Syn are elevated in DLB, whereas increased levels of aqueous-soluble o-α-Syn and detergent-soluble pS129-α-Syn are observed in PD and AD, suggesting different changes across the spectrum of proteinopathies (Vaikath et al. 2018).
All brains in sporadic DLB cases showed important deposits of tau, Aβ and α-Syn that are similar in biochemical quality to those in AD, with less severe tau pathology in DLB and DLB + AD than in “pure” AD (Colom-Cadena et al. 2013a; Deramecourt et al. 2006). Aβ deposition in DLB was associated with more severe hippocampus and subiculum atrophy, reflecting an early process of superimposed AD pathology (Mak et al. 2019). Over 50% of all DLB cases have considerable AD pathology, which is associated with a shorter interval between onset of motor symptoms and development of dementia, and a shorter live span (Irwin and Hurtig 2018). They are classified as AD/DLB or LB variant of AD (LBV/AD) (Hansen et al. 1998). They should be distinguished from cases with more prominent AD pathology and LP limited to the amygdala referred to as AD with amygdala LBs, considered as a distinct form of α-synucleinopathy (Fujishiro et al. 2008b; Uchikado et al. 2006b). “Pure” DLB cases with diffuse Aβ plaques but few neuritic elements or only minimal cerebral Aβ deposition show no significant differences in neocortical synapse density and synaptophysin reactivity, whereas AD/DLB has severe synapse protein loss comparable to AD (Colom-Cadena et al. 2017b). Downregulation of the postsynaptic proteins synaptopodin (SYNPO) and neurogranin indicates defective neurotransmission as a major factor for CI (Bereczki et al. 2016) that is strongly correlated with the DLB hypometabolism pattern (Morbelli et al. 2019). Recent imaging studies suggested a loss of dynamic interactions between the BG-thalamic and large scale cortical networks, which may contribute to fluctuations of cognition in DLB (Schumacher et al. 2019). α-Syn is an important predictor of disease duration, both independently and synergistically with tau and Aβ (Ferman et al. 2018), but concomitant LP and AD involving widespread association cortices contribute to visouspatial dysfunction (Kang et al. 2019). Rapidly progressing DLB cases, clinically resembling Creutzfeldt-Jakob disease, showed higher neocortical α-Syn and Aβ load than those with low or no AD pathology (Geut et al. 2019). CAA in DLB is less severe than in AD, with frontal predominance of cortical microbleeds (De Reuck et al. 2018). Cerebrovascular lesions were lower in DLB than in PD (34.4 vs. 36.7%) (Jellinger 2003). TDP-43 deposition is common in DLB (23.3%) and mixed AD/DLB (52.6%) (McAleese et al. 2017). Grey matter aging-related tau astrogliopathy (ARTAG) has been reported in 36% of LBD (Kovacs et al. 2017).
Dementia with Lewy bodies versus Parkinson’s disease dementia
According to DSM-5, DLB and PDD are major neurocognitive disorders with LP sharing many clinical, genetic, pathophysiological, and morphological features (American P, Association, Force D-T 2013; McKeith et al. 2017). A clear and objective distinction between the two entities other than the arbitrary timing of the appearance of cognitive and motor impairments (1-year rule) has not yet been established (Beach et al. 2009), while others merged the two entities (Berg et al. 2014) or considered them as one disease (Friedman 2018). The clinical features of both phenotypes, DLB (McKeith et al. 2017; Outeiro et al. 2019b) and PDD (Dubois et al. 2007; Emre et al. 2007; Goetz et al. 2008)], despite individual variability, show both overlapping and distinguishing features (Armstrong 2019; Elder et al. 2017; Gomperts 2016; Jellinger and Korczyn 2018; Joki et al. 2018). The latter are that DLB has less severe parkinsonism, and more profound cognitive deficits with higher frequency of visual hallucinations. Moreover, DLB has a unique genetic risk profile in comparison to PD and PDD (Guerreiro et al. 2018; Hansen et al. 2019). The way in which SNCA SNPs modulate risk is complex, and different patterns in PD and DLB show that the effect of each association signal is phenotype-specific: the strongest signal in PD is absent in DLB, while the second signal is significant in both (Pihlstrom et al. 2018). Morphology, molecular isoforms, histochemistry of LBs and the spreading pattern of α-Syn pathology do not significantly differ between both, the late LP stages 5 and 6 suggesting a transition between both phenotypes, although DLB has a higher density of cortical LBs and AD lesions than PDD (Hansen et al. 2019; McKeith et al. 2017).
Morphological differences include higher amyloid load in striatum (Halliday et al. 2011; Jellinger and Attems 2006; Kalaitzakis et al. 2011), in cortex (Fujishiro et al. 2010; Jellinger and Attems 2008; Ruffmann et al. 2016; Walker et al. 2015), entorhinal cortex, amygdala, and putamen in DLB (Hepp et al. 2016), the GP being free of amyloid plaques. Moreover Aβ phases, neuritic plaque scores and CAA severity and frequency are higher in DLB compared to PDD (Halliday et al. 2011; Hepp et al. 2016; Ruffmann et al. 2016; Walker et al. 2015), indicating a hierarchy in both Aβ and tau burden (Bohnen et al. 2017; Kalaitzakis and Pearce 2009; Kalaitzakis et al. 2009b). More severe α-Syn load in hippocampal subareas CA 2/3 and in the entorhinal cortex (EC), implicated a role of the EC-CA 2 circuitry in DLB (Adamowicz et al. 2017). Other deviations are the severity and distribution pattern of SNc lesions (more severe in the ventrolateral cell groups in PDD (Dickson et al. 2009) compared to more severe damage in the dorsolateral SN in DLB). Less nigral neuronal loss causing less severe postsynaptic DAergic upregulation (Jellinger 2006) may be related to the risk for neuroleptic sensibility reaction in DLB. Significantly higher 5-HT1A receptor-binding density in cortex was seen in DLB compared to PDD (Francis and Perry 2007). Thus, DLB and PDD are likely to represent two subtypes of an α-Syn-associated disease spectrum (LBD), beginning with iLBD → PD-nondemented → PDD → DLB → DLB with AD (DLB-AD) at the most severe end, although DLB does not begin with PD and does not always progress to DLB-AD (Jellinger 2018b; Jellinger and Korczyn 2018). The pathology underlying PDD and DLB is heterogeneous, with some differences in the topography and severity of lesions between both phenotypes that need further confirmation. An overlap between FTP and DLB was discussed recently (Gallucci et al. 2019).
Pathogenetic implications
The etiopathogenesis of PD (and other LB diseases) is poorly understood, but the majority is suggested to result from complex interactions between genetic background and environmental factors (Gasser et al. 2011), multiple etiologies being more likely than a single factor (Fig. 4). Genetic susceptibility, e.g., related to mutations in mitochondria-related genes (PARKIN, PINK1) in early onset PD, may be determined through impaired metabolism of free radicals or complex I activity, which, in turn, may be the product of nuclear or mitochondrial genomic deficits (Mullin and Schapira 2013). α-Syn undergoes post-translational modifications that regulate its structure and function, and may be linked to aggregation and/or oligomer formation (Gonzalez et al. 2019). DA modified α-Syn aggregation results in unique DA-induced oligomeric conformations (Mor et al. 2019). α-Syn oligomers induce selective oxidation of the ATP synthase β subunit and mitochondrial lipid peroxidation. These events open the permeability transmission pore (PTP), triggering mitochondrial swelling and ultimately cell death (Ludtmann et al. 2018). Environmental interactions include exogenous compounds with uptake and conversion similar to MPTP, cyanide, 1-trichloromethyl-1,2,3,4-tetrahydro-beta-carboline (TaClo) (Bringmann et al. 1995), or endogenously generated neurotoxins, such as rotenone or tetraisoquinoline, which affect mitochondrial function, produce reactive oxidative species (ROS) (Jiang and Dickson 2018) and cause disruption of calcium homeostasis (Freestone et al. 2009). The neurodegenerative process in PD is thought to be related to a complex cascade of noxious factors (Fig. 4) (Lim and Zhang 2013): imbalanced proteostasis (in particular misfolded α-Syn and its oligomers) (Lehtonen et al. 2019), perturbation of protein degradation systems (Maiti et al. 2017), endolysosomal dysfunctions (Kett and Dauer 2016; Klein and Mazzulli 2018), formation of free radicals, oxidative, nitritive, and proteolytic stress (Janda et al. 2012), production of ROS and advanced glycation products (Guerrero et al. 2013; Munch et al. 2000). There is compelling evidence that the endocytic membrane trafficking pathway plays a relevant role in the etiology of PD (Bandres-Ciga et al. 2019). Mitochondrial dysfunction (Aroso et al. 2016; Chen et al. 2019a; Rocha et al. 2018; Wang et al. 2019; Zaltieri et al. 2015b), impaired bioenergetics (Mullin and Schapira 2013), lipid peroxidation, nuclear RNA deficits, protein-iron and NM-iron interactions (Sian-Hulsmann et al. 2011; Zucca et al. 2017), cause iron-related nigral degeneration (Guan et al. 2019; Sun et al. 2018), and ferroptosis dysregulation inducing cell death (Van Do et al. 2016; Guiney et al. 2017). There is evidence for the existence of a connection between familial mutations of α-Syn, their distinct affinity to lipid membranes and the formation of toxic oligomers of the protein mediated by 3,4-dihydroxyphenylacetaldehyde (DOPAL) (Lima et al. 2019). A number of genetic risk factors of PD, such as PLA2G6 and SCARB2, which are involved in glycerophospholipid and sphingolipid metabolism either directly or indirectly are associated with risk of PD (Alecu and Bennett 2019). ROS-mediated transport impairment occurs early and induces axonal degeneration (Lu et al. 2014), while mitochondrial dysfunction is a common downstream pathogenic mechanism for α-Syn aggregation (Wang et al. 2019) (Fig. 5). Recent studies showed that α-Syn induces a selective loss of the mitochondrial protease ClpP in DA neurons of both α-Syn A53T tg mice and PD patients, which results in an overload of mitochondrial unfolded proteins and increased oxidative damage. Compensation for the loss of ClpP reduced mitochondrial oxidative damage and α-Syn-associated neuropathology. These findings revealed a novel mechanism by which α-Syn induces mitochondrial damage to proceed PD-associated neurodegeneration (Hu et al. 2019). Further mechanisms include transcriptional α-Syn dysregulation (Pinho et al. 2019), defects in intracellular transport pathways (Abeliovich and Gitler 2016), autophagy at presynaptic terminals (Pan et al. 2019), cell membrane disruptions (Iyer and Claessens 2019), disturbed proteasis processes (Zhou et al. 2019), excitotoxicity, neuroinflammation by activation of microglia and production of proinflammatory cytokines (Ferreira and Romero-Ramos 2018; Hirsch et al. 2013; Lema Tomé et al. 2013; Zhang et al. 2018a) and interactions between several noxious factors. Degradation of α-Syn by both UPS and ALP, and the fact that mutated α-Syn inhibits these pathways, support its role as an essential trigger for neurodegeneration (Pan et al. 2008; Puska et al. 2018). Poly(ADP-ribose) accelerates the formation of pathologic α-Syn (Kam et al. 2018), which may contribute to OS inducing microglial activation and antioxidant responses, that could modulate progression of PD (Scudamore and Ciossek 2018) (Fig. 6). Microglia is suggested to play a crucial role in α-Syn transmission via exosome pathways (Xia et al. 2019). Exosomes seem to be a common pathway in the microglia-mediated clearance of toxic aggregated proteins, e.g., Aβ and α-Syn (Paolicelli et al. 2019), which can be affected by lysosomal deficits (Tremblay et al. 2019). In DAergic neurons, increased calcium conductance and greater production of ROS lead to mitochondrial damage. The burden caused by mitochondrial dysfunction will reach a pathological threshold, provoking neuronal dysfunction and death (Callizot et al. 2019). Disruption of the ceramide metabolism may affect endolysosomal and mitochondrial dysfunctions that are important in PD/parkinsonism (Lin et al. 2019). Lowering PARKIN levels by extracellular α-Syn oligomers may contribute to the propagation of neurodegeneration in PD (Wilkaniec et al. 2019). The major components inducing neuronal loss in PD are: (1) presynaptic and/or axonal α-Syn aggregation, synaptic and axon degeneration (2) mitochondrial dysfunction, (3) environmental OS, (4) neuroinflammation and interaction with other noxious factors (Jellinger 2013a; Kouli et al. 2018). Given that a sequence of molecular mechanisms including OS, apoptosis, neuroinflammation, microglia and astrocyte activation and aquaporin 4 (AQP4) are associated with progression of PD (Tamtaji et al. 2019), the role of chemokines in the pathogenesis of PD is of interest, since they may induce microglia activation and apoptosis (Liu et al. 2019b). Microglia activation may contribute to the development of α-Syn pathology, supporting the concept that microglia play an integral role in the propagation and spread of α-Syn pathology (Olanow et al. 2019). Several rodent models of PD showed impairment of major cellular functions (mitochondrial phosphorylation, autophagy-lysosomal changes, protein degradation, and endoplasmic reticulum stress/unfolded protein response) (Jiang and Dickson 2018). Complex cell interactions may induce “prion-like” spreading of α-Syn (Braak and Del Tredici 2017; Duyckaerts et al. 2019; Longhena et al. 2017). Postmortem observation of α-Syn pathology within cell grafts in the striata of PD patients suggested that the spread of α-Syn as a main mechanism underlying disease progression in PD (Angot et al. 2012; Lema Tomé et al. 2013). The three antibodies aSyn-323.1, aSyn-336.1 and aSyn-338.1 that have the highest affinity to recombinant full-length α-Syn were able to neutralize the “seeding” of intracellular α-Syn aggregates in an in vitro assay (Li et al. 2019). α-Syn aggregates form sequence-dependent polymorphic fibrils upon spontaneous aggregation but become seed structure-dependent upon seeding (Tanaka et al. 2019). Transcellular spreading may be responsible for the propagation of neurodegeneration (Brundin et al. 2010; Davis et al. 2018; Freundt et al. 2012; Iljina et al. 2016; Henderson et al. 2019). Misfolded forms of α-Syn and tau can propagate from cell to cell and throughout the brain, thereby templating the misfolding of native forms of the proteins (Vasili et al. 2019). 14-3-3 proteins reduce cell-to-cell transfer and propagation of pathogenic α-Syn (Wang et al. 2018), whereas ‘aggravators’ may induce impaired autophagy and cell-to-cell propagation of α-Syn pathology (Johnson et al. 2019). Rab-GTPase proteins have a fundamental role in the modulation of α-Syn and spreading in PD (Masaracchia et al. 2018). Recent studies provided evidence that the gap junction protein connexin-32 (Cx32) is centrally involved in the uptake and transfer of α-Syn oligomers in neurons and oligodendrocytes. Cx32 levels were significantly decreased in SN of PD and MSA, indicating a potential relationship between human α-Syn and Cx32 in the pathogenesis of both disorders (Reyes et al. 2019). Exocytosis can induce spreading (Emmanouilidou and Vekrellis 2016) and cell-to-cell seeding that explains the formation of LBs and LNs and their wide distribution (Karpowicz et al. 2019). The endocytosis of pathological α-Syn required to facilitate its transmission through synaptically connected neuronal networks has recently be reviewed (Valdinocci et al. 2018). However, recent findings indicate that cellular prion protein PrPC neither binds α-Syn oligomers nor mediates their detrimental actions, suggesting that other pathways may co-exist in PD (La Vitola et al. 2019), whereas the binding between PrPC and α-Syn fibrils prevents the formation and accumulation of PrPSc (De Cecco and Legname 2018). Hence, the prion hypothesis of human synucleinopathies has to be reconsidered (Gelpi and Colom-Cadena 2019; Masaracchia et al. 2018; Tamguney and Korczyn 2018), and there is no evidence that proteins underlying PD (or AD) transmit disease to humans (Irwin et al. 2013a).

From (Jellinger 2012a)
Etiology of PD. Sporadic PD is a complex multifactorial disorder with variable contribution of environmental factors and genetic susceptibility. Mutations of various genes are associated with autosomal-dominant or autosomal-recessive parkinsonism. PARK 16–18: inheritance unknown.

From (Jellinger 2012a)
Common pathways underlying PD pathogenesis. Schematic impairment by α-synuclein and gene mutations enhancing α-synuclein misfolding, fibril formation an Golgi fractionation; impairing proteasome and mitochondrial functions, altering vesicle traffic and translation.

Modified from (Tsang and Chung 2009)
Major pathways underlying Parkinson’s disease pathogenesis related to α-synuclein and dopamine metabolism include proteolytic and mitochondrial dysfunction, oxidative/nitrosative stress, resulting in protein aggregation and reduced energy biosynthesis leading to neuronal degeneration.
Multiple system atrophy
This adult-onset, progressive α-synucleinopathy of presumed sporadic origin, is morphologically characerized by prominent α-Syn inclusions with neuronal multisystem degeneration (Jellinger 2018b). It is clinically characterized by autonomic failure and motor impairment with poorly l-dopa-responsive parkinsonism, cerebellar ataxia, tremor, and corticospinal tract dysfunction (Krismer and Wenning 2017). MSA is an orphan disease (prevalence 1.9–4.9 up to 7.8/100,000, incidence 0.6–0.7/100,000 person-years (Fanciulli and Wenning 2015).
Clinical features of multiple system atrophy
Diagnostic criteria recommend classification into two subtypes: MSA-P (predominant parkinsonism, 70–80% in the Western world) and MSA-C (cerebellar features associated with olivopontocerebellar atrophy/OPCA/, 20–67%), more frequent in Asian populations (67–84%), with rather frequent mixed phenotypes (Ozawa and Onodera 2017; Yabe et al. 2006). Red flag categories—characteristic symptoms including inspiratory stridor, pyramidal signs, postural instability—had a specificity of 98.3% and sensitivity of 84.2% (Köllensperger et al. 2008). Revised criteria differentiate possible, probable, and definite MSA, the latter confirmed by postmortem examination (Gilman et al. 2008). The accuracy of clinical diagnosis of MSA with a positive predictive value even in later stages from 60 to 90% is still unsatisfactory (Joutsa et al. 2014; Koga et al. 2015; Osaki et al. 2009), but the true rate of over- or under-diagnosis is not known. Autonomic dysfunction (urogenital dysfunction, orthostatic hypotension) is common in both variants and reflects degeneration of the central and peripheral autonomic pathways (Ozawa 2007). Motor symptom onset is 56 ± 9 years but 40–75% of MSA cases have a prodromal phase with non-motor and autonomic symptoms (Fanciulli and Wenning 2015). Akinesia and rigidity are prominent in MSA-P but also occur in later stages of MSA-C. Tremor is not rare (Kaindlstorfer et al. 2013). Parkinsonian features are more severe in the MSA-STR group showing DAergic dysfunction than in the MSA-SNc group with predominant pre-synaptic tracer uptake loss in the posterior putamen (Ryu et al. 2019). The goal of a recently established MDS MSA Criteria Revision Task Force is to define principles for a revision of the second consensus criteria for an MSA diagnosis (Stankovic et al. 2019).
Subtypes of multiple system atrophy
MSA diversities are broader than previously considered (Koga and Dickson 2018). Various disorders may mimick MSA (Krismer and Wenning 2017), e.g. PD and PSP (Koga and Dickson 2018); MSA-C may resemble spinocerebellar ataxias (Li et al. 2018). A wide range of subtypes does not fit into the current classifications of MSA (Koga and Dickson 2018; Watanabe et al. 2016): “Minimal-change” MSA-P is a rare aggressive form with CGIs and neurodegeneration restricted to SN and Put (“pure SND”) (Berciano et al. 2002; Ling et al. 2015; Wenning et al. 1994), while a case of “minimal” MSA-C showed widespread GCIs, NCIs and NNIs, and neuronal loss restricted to OPC areas (Wakabayashi et al. 2005). Another with limbic-predominant α-Syn pathology (Koga and Dickson 2019), and those with limbic-predominant α-Syn pathology, dementia and Pick-like inclusions were classified as FTD with α-Syn (FTLD-synuclein) (Aoki et al. 2015; Rohan et al. 2015). Incidental MSA with widespread GCIs without clinical disease is rare (Parkkinen et al. 2007; Wakabayashi et al. 2005; Wenning et al. 1994) as is coexistence of MSA and PSP (Uchikado et al. 2006a). Rare ‘young-onset MSA’ (YOMSA) before age 40 shows more common LID and minimal pathological changes (Batla et al. 2018), while others showed progressed pathological stages of MSA-P or MSA-Mix (Jellinger 2018c). MSA progresses rapidly (Wenning et al. 2013), while MSA-P with slow progression and prolonged survival is an uncommon “benign” subgroup (Petrovic et al. 2012). “Benign” MSA cases show slowly progressing parkinsonism and prolonged survival up to 15 years or more in 2–3% of MSA patients (Kim and Jeon 2012; Petrovic et al. 2012), while others with clinical course of 18 years showed extensive distribution of GCIs (Masui et al. 2012).
Genetics of multiple system atrophy
Familial clustering of MSA is uncommon, but rare familial forms with AutR inheritance have been published (Fujioka et al. 2014a). A recent GWAS found an estimated heretability at 2–7% (Sailer et al. 2016), but unlike PD, no single gene mutation linked to familial forms and no definite risk factor have been identified. Screening for PD causal genes (MAPT, PDYN, Parkin, PINK1) did not reveal any association with MSA (Brooks et al. 2011; Yuan et al. 2015), while LRRK2 exonic variants may contribute to its susceptibility (Heckman et al. 2014). GBA variants were associated with autopsy-proven MSA (Sklerov et al. 2017; Sun et al. 2013), significantly with MSA-C (Mitsui et al. 2015), while others found no association (Srulijes et al. 2013). SNCA polymorphism as a risk factor for MSA (Al-Chalabi et al. 2009; Scholz et al. 2009) was not confirmed (Federoff et al. 2016; Sun et al. 2015). No association of the APOE locus or the prion PRPN with increased risk of MSA was observed (Chelban et al. 2017; Ogaki et al. 2018), and there is no evidence of AutD MSA or of de novo mutations in this disorder (Laurens et al. 2017). A British family with AutD inheritance and G51D SNCA mutation shared neuropathologic features of both PD and MSA (Kiely et al. 2013). They are distinct from PD patients carrying the H50Q or SNCA duplication (Kiely et al. 2015).
The link between V393A mutations in the COQ2 gene, encoding the coenzyme Q10 (COQ10) and familial or sporadic MSA in Japan and other Asian populations (Lin et al. 2015; MSA-Research-Collaboration and Collaboration 2013; Quinzii et al. 2014; Sun et al. 2016; Zhao et al. 2016) was not confirmed in other populations (Ferguson et al. 2014; Ronchi et al. 2016; Sailer et al. 2016; Sharma et al. 2014). Decreased COQ10 levels in cerebellum (Barca et al. 2016; Schottlaender et al. 2016), suggest that its deficiency may contribute to its pathogenesis due to cellular dysfunction (Nakamoto et al. 2018).
Neuropathology and molecular pathology of multiple system atrophy
The brain shows diffuse atrophy, green-gray discoloration of Put in MSA-P and atrophy of the cerebellum, middle cerebellar peduncles, and pons in MSA-C. The pigmented brainstem nuclei are pale. Histopathology shows: (1) selective neuronal loss and axonal degeneration involving multiple NS regions with brunt on the striatonigral and OPC systems; (2) cellular α-Syn-immunoreactive inclusions [glial cytoplasmic inclusions (GCIs) within oligodendrocytes, less frequent glial nuclear inclusions (CNIs), neuronal cytoplasmic inclusions (NCIs), neuronal nuclear inclusions (NNIs)]; (3) astroglial cytoplasmic inclusions and neuronal threads of similar composition; (4) myelin pallor with reduction of myelin basic protein (MBP); and (5) microglial activation. The histologic hallmarks are cytoplasmic α-Syn-immunoreactive GCIs within oligodendroglial cells, the demonstration of which is required for the diagnosis of definite MSA (Trojanowski and Revesz 2007).
GCIs are argyrophilic, triangular, sickle- or half moon-shaped or oval cytoplasmic aggregates composed of fibrillary α-Syn, Ub, and various multifunctional proteins, including 14-3-3 protein, LRRK2, aggresomal proteins (Jellinger and Lantos 2010; Jellinger 2018a). They form a meshwork of loosely packed filaments or tubules 15–30 nm in diameter with a periodicity of 70- to 90-nm and straight filaments, both consisting of polymerized α-Syn, granular material, and variable types of filaments. The central core is formed by phosphorylated (ser129) α-Syn (Gai et al. 2003). The soluble α-Syn in GCIs differs from the insoluble form in LBs (Campbell et al. 2001). Purification of α-Syn containing GCIs revealed 11.9% α-Syn, 2.8% αB-crystallin, and 1.7% 14-3-3 protein compared to 8.5, 2.0, and 1.5% in LBs (McCormack et al. 2016). In MSA brain, α-Syn 140 and 122 isoform levels were significantly increased, whereas α-Syn 126 was decreased in SN, striatum and cerebellum. Early accumulation of p25α (tubulin polymerization-promoting protein/TPPP), a potent stimulator of α-Syn aggregation, may decrease MBP, favoring both the deposition and fibrillation of α-Syn and changing myelin metabolism (Olah et al. 2017). TDP-43 pathology is rare in MSA, but colocalization with α-Syn suggests interaction between the two molecules (Koga et al. 2018b). Widespread accumulation of oligomeric α-Syn in neurons and oligodendrocytes in neocortex and Purkinje cells in MSA is more severe than in PD (Sekiya et al. 2019). α-Syn in brain-derived exosomes distinguishes MSA from PD (Bitan et al. 2019). Cathepsin-D, calpain-1 and kallikrein-6 were elevated in the Put, pontine basis, and cerebellar white matter, indicating that α-Syn accumulation is not due to reduced activity of these proteases, but their upregulation may be compensatory to increased α-Syn (Kiely et al. 2019). Decreased complex II/III activity and increased complex I and IV activity in MSA cerebellar white matter correspond with CoQ10 deficit in MSA and reflect the high regional pathological burden of GCIs, indicating mitochondrial dysfunction in MSA pathogenesis (Foti et al. 2019). Iron levels in BG and SN are higher in MSA than in PD and controls, and resemble that in PSP (Kaindlstorfer et al. 2018; Lee and Lee 2019).
Neuronal cell loss, reactive gliosis, iron deposition, and demyelination involve pons, medulla, Put, cerebellum, SNc, spinal cord, and preganglionic autonomic structures (Ahmed et al. 2012; Holton et al. 2011; Jellinger 2014). The degree and pattern of neurodegeneration and demyelination correlate with the density and distribution of GCIs, and duration of illness, support a direct association, but there is no clear correlation between α-Syn glial burden and neuronal degeneration (Jellinger 2018a). Damage to the striatonigral system is most severe in dorsolateral caudal Put and lateral SN, suggesting transsynaptic degeneration of striatonigral fibers. Based on semiquantitative assessment of neuronal loss, astrocytosis, and GCIs, four degrees of severity were distinguished for both MSA phenotypes (Jellinger et al. 2005). This grading reflects the initial symptoms, disease progression, and clinical key features, but there was a low correlation between involvement of the two major systems and the natural history of the disorder. Postmortem MRI changes in Put (type 1, mild atrophy and isointensity; type 2, atrophy and diffuse hypointensity with a hyperintense putaminal rim [HPR]; type 3, putaminal atrophy and iso- or hypointensity with HPR) reflect various degrees of brain damage (Matsusue et al. 2008). There is an increasing overlap of α-Syn pathology with increasing duration of disease, and with the extent of α-Syn pathology (Brettschneider et al. 2018). Degeneration of GP and STN leads to dysfunction of these inhibitory nuclei projecting to the motor thalamus, a mechanism similar to that in PSP (Fig. 2). In MSA-C, GCIs are most prominent in cerebellum, pons, and medulla (Brettschneider et al. 2017). The cerebellar Purkinje cells are more severely affected in the vermis, with atrophy of olivary nucleus, cerebellopontine fibers, and pontine basis, causing disruption of specific cerebello-cortical circuits (Ren et al. 2019). The motor subnetwork in MSA-C has significant structural alterations in both BG and cerebellar connectivity (Shah et al. 2019). The motor neurons in spinal cord show only mild involvement. Involvement of the autonomic nervous system underlies the multidomain autonomic failure typical of MSA (Iodice et al. 2012; Ozawa 2007). The peripheral nervous system shows α-Syn aggregation in sympathetic ganglia, skin nerve fibres and Schwann cells (Doppler et al. 2015; Mori et al. 2002; Nakamura et al. 2015; Zange et al. 2015). Myelin lesions involve various brain regions, but also affect otherwise apparently normal areas (Matsuo et al. 1998). Demyelination is associated with reduction of myelin proteins by about 50% (Don et al. 2014). GCIs and microglial burden are greatest in mild to moderate white matter lesions and decrease with progression of myelin damage, but showed no correlation with the severity of gray matter damage. Early MSA stages show widespread increase of microglia (about 100%) in the white matter (Kübler et al. 2019) without concomitant astrogliosis or essential oligodendroglial degeneration (Nykjaer et al. 2017). Both microglial activation and α-Syn containing oligodendrocytes trigger neuroinflammation restricted to white matter regions (Hoffmann et al. 2019). MSA-C cases showed increased microglia in cerebellum, not observed in any other region (Kiely et al. 2018). In MSA-C, myoclonus was associated with more α-Syn in the anterior spinal horns and lateral corticospinal tracts (Hwang et al. 2019).
Cognitive impairment (CI) and visual hallucinations, characteristic for DLB, are rare symptoms in MSA (Gilman et al. 2008), although MCI and executive deficits may occur (Fanciulli and Wenning 2015). CI is induced by cortical and subcortical gray matter atrophy and neocortical neuronal loss (Kim et al. 2015; Lee et al. 2015; Salvesen et al. 2015), LB-like inclusions in neocortex (Cykowski et al. 2015), globular inclusion in the medial temporal region (Homma et al. 2016) or corpus callosum involvement (Hara et al. 2018). However, others found no pathological differences between MSA cases with and without cognitive impairment (Asi et al. 2014). Progressive retinal structure abnormalities were seen in visually asymptomatic MSA patients (Mendoza-Santiesteban et al. 2015).
LBs, a classical hallmark of PD, were seen in 10.7–22.7% of autopsy-proven MSA cases (Koga et al. 2017a), while GCI pathology occurred in familial PD cases with rapid disease progression (Houlden and Singleton 2012). α-Syn inclusions in astroglia and oligodendroglia, however, occur in both PD and DLB (Fellner et al. 2011; Ferrer 2018). Limbic TDP-43 pathology is rare in MSA (Koga and Dickson 2018; Koga et al. 2018a, b; Robinson et al. 2018). Grey matter ARTAG has been reported in 17.1% of MSA cases (Kovacs et al. 2017).
Pathogenesis of multiple system atrophy
The role of the neuronal endosomal-lysosomal system in the processing of α-Syn in PD is well established, while lysosomes contribute to the pathogenesis of MSA, enabling oligodendroglial and neuronal uptake of exogenous α-Syn (Puska et al. 2018). α-Syn, primarily generated by neurons, can be toxic once released to the extracellular environment (Guo and Lee 2014), and can spread throughout the brain in a “prion-like” manner like other pathological proteins (Dhillon et al. 2019b; Duyckaerts et al. 2019; Goedert et al. 2017a, c; Karpowicz et al. 2019). Extracellular α-Syn interacting with neuronal and non-neuronal cell types, mediates neuroinflammation, cell-to-cell spread (Davis et al. 2018; Valdinocci et al. 2018). Neuron-to-oligodendrocyte transfer of misfolded α-Syn plays a crucial role in the pathogenesis of MSA (Reyes et al. 2014). MSA and PD show different phosphorylation of α-Syn and distinct α-Syn seed characteristics indicating that distinct strains underlie these diseases (Yamasaki et al. 2019). MSA prions show remarkable stability and resistance to inactivation (Woerman et al. 2018) and their transmission to transgenic mouse lines provided evidence that MSA is a prion disease (Woerman et al. 2019). Human α-Syn forms distinct inclusions and propagates within cultured tg astrocytes exposed to MSA prions, indicating that α-Syn expression determines the tropism of inclusion formation in certain cells (Krejciova et al. 2019). Brain lysates from MSA patients can induce α-Syn pathology similar to what is induced by preformed human α-Syn fibrils in tg mice. This reinforces the idea that the intrinsic traits of the tg mouse model dominates over any prion-like strain properties of MSA α-Syn seeds that can induce pathology (Dhillon et al. 2019a). However, there is currently no evidence of iatrogenic transmission or infectivity of MSA (De Pablo-Fernandez et al. 2018; Wenning et al. 2018).
The pathogenesis of MSA is not fully understood, but the crucial role of oligodendroglial pathology has been confirmed by animal models (Fellner et al. 2015; Stefanova 2014). Decreased expression of glia-derived neurotrophic factor (GDNF) in MSA brains (Ubhi et al. 2010) indicates that α-Syn expression in oligodendrocytes impacts their trophic transport to neurons. Oligodendroglial changes are more widespread than α-Syn-positive GCIs, suggesting that primary oligodendroglial pathology is the main driver of the disease process, inducing degeneration of the oligodendroglia-myelin-axon-neuron complex. Early events are an ectopic appearance of α-Syn in oligodendrocytes, loss of the cAMP-regulated phosphoprotein of 32 kDa (DARPP 32) and calbindin indicating calcium toxicity and disturbance of phosphorylated proteins (Hayakawa et al. 2013). Impaired protein degradation, autophagic and proteasomal dysfunction, alterations of the autophagic pathway (Monzio Compagnoni et al. 2018; Valera et al. 2017), perturbed iron homeostasis (Kaindlstorfer et al. 2018) and lipid dysfunction involved in myelin synthesis by oligodendrocytes (Bleasel et al. 2014; Grigoletto et al. 2017) are other pathogenic factors. Apoptosis and neuroinflammation (Valera et al. 2017) suggest that that multiple mechanisms interact to result in the system-specific pattern of neurodegeneration in MSA (Fellner et al. 2018; Ubhi et al. 2011) (Fig. 7). TNFα-dependent neuroinflammation may play a key role in MSA pathogenesis, and its relevance has been underlined in various models of MSA (Ndayisaba et al. 2019).

From (Jellinger and Wenning 2016)
Putative pathogenic pathways of multiple system atrophy.
Recent animal model studies that only partly replicate the human disorder have provided some progress in our understanding of MSA pathogenesis (Bassil et al. 2017; Bleasel et al. 2016; Fellner et al. 2015; Heras-Garvin et al. 2019; Mandel et al. 2017; Overk et al. 2018; Refolo et al. 2018; Stefanova 2014; Valera et al. 2017). Relocation of p25α from the myelin sheaths to the oligodendroglial soma (due to mitochondrial dysfunction), with formation of cytoplasmic p25α inclusions precedes aggregation of transformed α-Syn in oligodendrocytes. Endogenous α-Syn and p25α orchestrate the formation of pathological α-Syn assemblies in oligodendrocytes and provide in vivo evidence of the contribution of oligodendroglial α-Syn to the pathogenesis of MSA (Mavroeidi et al. 2019). Although large inclusions appear at a late disease state, small, soluble assemblies of α-Syn promoted by p25α are pathogenic (Olah and Ovadi 2019). The source of α-Syn in oligodendroglia is unclear, but it contains α-Syn mRNA expression and α-Syn may be secreted by neurons and taken up by oligodendrocytes, which is facilitated by protein Cx32 (Reyes et al. 2019). 21% of proteins found consistently in GCIs and LBs were synaptic vesicle-related, which suggests that misfolded α-Syn may be targeted via vesicle-mediated transport, which also explains the presence of this neuronal protein within GCIs (McCormack et al. 2019). Secondary events include reduced trophic support to axons and neurons by reduced GDNF. Neuroinflammation, OS, proteasomal dysfunction, proteolytic dysbalance, dysregulation of myelin lipids and energy failure are important factors leading to neurodegeneration in MSA. The disease is currently viewed as a primary synucleinopathy with specific glia-neuronal degeneration developing via the oligo-myelin-axon-neuron complex (Jellinger and Wenning 2016). Thus, MSA represents a specific form of oligodendroglial proteinopathies (Rohan et al. 2016), while others suggest that it is a neuronal disease with secondary accumulation of α-Syn in oligodendrocytes (Cykowski et al. 2015).
Tauopathies
The morphological hallmarks of this heterogeneous group of neurodegenerative diseases are filamentous neuronal and glial tau inclusions associated with the degeneration of affected brain areas showing selective vulnerability (Ferrer et al. 2014; Murray et al. 2014; Spillantini and Goedert 2013). Human tau proteins are encoded by the MAPT gene consisting of 16 exons on chromosome 17q21. The adult human brain has six tau isoforms composed of either three (3R) or four (4R) carboxy-terminal tandem repeat sequences of 31–32 amino acids that are encoded by exons 9–12. The triplets of 3R- and 4R-tau isoforms differ as a result of alternative splicing to generate isoforms with 29 or 58 amino acid inserts (Mietelska-Porowska et al. 2014; Spillantini and Goedert 2013). In tauopathies, the tau protein is hyperphosphorylated, which causes it to lose its affinity for microtubules and becomes resistant to proteolysis; this results in its accumulation and the formation of NFTs (Birdi et al. 2002). Tau filaments comprise two types: straight filaments or tubules with 9–18 nm diameters and “twisted ribbons” composed of two parallel aligned components (Arima 2006). The structures of tau filaments were recently found to differ between distinct tauopathies, e.g., between AD and Pick’s disease (Falcon et al. 2018; Goedert et al. 2019). The patterns of soluble and insoluble tau were the basis for biochemical classification of the major tauopathies (Goedert et al. 2017c; Gotz et al. 2019; Hoglinger et al. 2018; Spillantini and Goedert 2013): AD (as a secondary tauopathy), postencephalitic parkinsonism (PEP), Guamanian amyptrophic lateral sclerosis-Parkinson’s disease complex (ALS/PDC) (3R and 4R triplets), and Pick’s disease (PiD) (predominant 3R), while PSP and CBD contain predominantly 4R-tau doublets with two 68- and 64-kD insoluble tau bands at exon 10. The morphology of the neuronal and glial inclusions and the patterns of cellular vulnerability are specific, but there is frequent overlap between various tauopathies (Irwin et al. 2013b). This has lead to the suggestion that different molecular conformers or strains of aggregated tau exist (Goedert et al. 2017a), which are responsible for the heterogeneity and cell-type specificity of tauopathies. Tau pathology is suggested to spread through “prion-like” propagation (Ayers et al. 2018; Mudher et al. 2017), but does not have a high propensity to spread to peripheral tissues (Dugger et al. 2018). Seeding and spreading of tau occurs in oligodendrocytes, thereby supporting its spreading in the white matter in tauopathies (Ferrer et al. 2019). There are potential barriers in cross-seeding between 3R- and 4R-tau isoforms (Strang et al. 2018; Weismiller et al. 2018). Despite the similarities to prion proteins, there is no evidence that pathological tau is infectious (Gibbons et al. 2019).
Progressive supranuclear palsy
PSP, or Steele-Richardson-Olszewski syndrome, a predominantly sporadic movement disorder, is the most common atypical parkinsonian disease with incidence rates increasing with age from 1.7 to 14/100,000/year and an estimated prevalence of 6.2–74 1.4/100,000. The annual incidence increases with age from 1.7/100,000 at age 50–59 years to 14.7/100,000 at 80–89 years (Coyle-Gilchrist et al. 2016). Mean age at disease onset is 60–65 years, which is older than in IPD, and mean survival is 3–5 years (Ali and Josephs 2018a). PSP is clinically featured by progressive postural instability and falls, supranuclear vertical gaze palsy, frontal cognitive disturbances, and absence of resting tremor (Agarwal and Gilbert 2019). However, atypical cases with a variety of clinical syndromes indicate the heterogeneity of PSP. The new MDS PSP criteria outline 14 core clinical features and 4 clinical clues that combine to diagnose one of eight PSP phenotypes with probable, possible, or suggestive certainty (Armstrong 2018; Hoglinger 2018). The following phenotypes have been established: (1) Richardson’s syndrome (PSP-RS/classic PSP), with early postural instability, falls, vertical gaze palsy and a rapid course (Respondek et al. 2017); (2) PSP-parkinsonism (PSP-P), which often mimics PD; (3) gait freezing form (PSP-PGE), (4) PIGD (postural instability and gait disorder), (5) oculomotor dysfunction (PSP-OM), several others (Ali and Josephs 2018a, b), and overlap with both brFTD (PSP-F) and nfvPPA (PSP-SL) (Hoglinger et al. 2018). The clinical syndrome of PSP may arise through several pathologic processes: PSP-RS, PSP-P, FTLD, CBD, LBD, progressive subcortical gliosis, and MSA (Ali and Josephs 2018a; Respondek et al. 2017). Given these variants, it is not surprising that overall diagnostic accuracy is 70–85.7% (Ali et al. 2019). The proposed four rules for Multiple Allocations eXtinction (MAX) helped to standardize the application of the MDS criteria for PSP (Grimm et al. 2019). Incipient PSP comprises three subgroups with typical or only mild PSP pathology (Yoshida et al. 2017). Divergent brain gene expression patterns associated with distinct cell-specific tau neuropathology, suggest that specific PSP phenotypes may emerge from different tau strains (Narasimhan et al. 2017). Tau strains from human PSP brains showed transneuronal/transsynaptic spreading in a “prion-like” manner via exosomes, nanotubes and endocytosis from the extracellular space (Calafate et al. 2015; Tardivel et al. 2016; Wang et al. 2017).
Genetics of progressive supranuclear palsy
The major genetic risk factor for sporadic PSP, which represents around 85% of all cases, is a variant in the MAP gene, with prevalence of A0/A0 and the H1/H1 genotypes (Houlden et al. 2001). A GWAS study of autopsy-proven PSP identified SNPs mapping to MAPT, STX6, MOBP, STX6, and EIF2AK3 (Hoglinger et al. 2011). Rare monogenetic inheritance of PSP is associated with MAPT mutations (Forrest et al. 2018). Although PSP is defined as a sporadish condition, an increasing number of familial PSP with MAPT mutations have been reported (Donker Kaat et al. 2009; Fujioka et al. 2014b). An atypical PSP-P phenotype with rare variants in FBXO7 and VPS35 genes was associated with LP (Mensikova et al. 2019). PSP variants at MAPT and MOBP loci may confer PSP risk via gene expression and tau pathology (Allen et al. 2016). There is overlap between PSP and CBD in various genes, CXCR4 (chemokine receptor type 4), EGFR (epidermal growth factor), GLDC (glycine dehydrogenase), and MOBP (Yokoyama et al. 2017). Recent studies have identified three novel associations of MAPT H1 subhaplotypes with risk of PSP and their role in susceptibility to and severity of tau pathology (Heckman et al. 2019). A recent GWA analysis of copy number variants found MAPT duplications as a possible genetic cause of PSP, particularly in patients presenting with young age at onset (Chen et al. 2019b). Variation at the TRIM11 locus modifies PSP phenotype (Jabbari et al. 2018).
Neuropathology of progressive supranuclear palsy
Typical PSP cases show pallor of SN, atrophy of STN, midbrain, pontine tegmentum, and superior cerebral peduncle; with mild cortical atrophy (that differentiates PSP-RS from PSP-P) (Schofield et al. 2011). The histological hallmarks of PSP are globose tangles (different from the flame-shaped cortical NFTs), neuronal threads, and tau deposits in glia in BG, diencephalon, many brainstem regions, dentate, and inferior olivary nuclei, and spinal gray matter (Dickson et al. 2011a). Composed of 12- to 15-nm straight tubules/filaments containing 4R-tau with a sequence encoded on exon 10, they differ from those in AD or PEP. Swollen achromatic neurons in cortex and BG contain tau aggregates with straight filaments, which are also present in “tufted” or thorn-shaped astrocytes (with straight, irregular 22-nm filaments, in contrast to the “astrocytic plaques” of CBD) and in oligodendroglia as “coiled bodies” (straight 14-nm filaments) throughout the neuraxis, in particular, the white matter. Tau pathology predominates in precentral gyrus, entorhinal cortex, hippocampus, dentate granule cells, and A10 midbrain cell groups (Dickson et al. 2011a); the distribution of NFTs is similar to that in PEP and Guamanian ALS/PDC. Only few tangle-bearing neurons but many tau-positive oligodendrocytes occur in brainstem and pontine nuclei, not in SNc. Neuropil threads, short, tortouous, cell processes of neuronal and oligodendroglial origin, occur in both cortical and subcortical gray and white matter, the latter predominantly in typical PSP cases (Dickson et al. 2011a). Astrocytic tau pathology, the result of abnormal tau released from damaged axons (Armstrong 2013), and microglial activation correlate with NFT density and neuronal loss (Ito et al. 2008). Severe damage to GPi, GPe, SNr, and STN causes dysfunction of striatal efflux to motor thalamus, accounting for akinesia-rigidity and its resistance to DAergic treatment (Fig. 2).
Neuropathological criteria for clinical variants show significant morphologic and biochemical differences (Respondek et al. 2017): PSP-P and PSP with progressive gait freezing (PGF) have a lower tau pathology score with more restricted involvement of SN, STN, and Gpi and a mean 4R-/3R-tau ratio of 2.8, whereas PSP-RS has more severe and more widely distributed tau pathology in BG, brain stem, and prefrontal cortex, negatively correlated with disease duration (Jellinger 2008b; Williams et al. 2007). PSP-corticobasal syndrome (CBS) brains have greater cortical tau pathology than those with PSP-RS, while PSP-P and PGF have more severe BG degeneration (Dickson et al. 2010a). AD neuropathology is seen in about 26% of PSP cases (Robinson et al. 2018).
Cortical tau pathology in PSP, with the highest density in prefrontal and limbic areas and mainly in deeper cortical layers, differs from its bimodal distribution in AD, while a combination of PSP with AD is rare (Sakamoto et al. 2009). Although tau lesions in central grey matter, SN, and LC are found in both PSP and AD, 4R-selectivity with glial component suggests PSP origin (Ebashi et al. 2019). Hippocampal and amygdala pathology is usually minimal, but 20% of patients show ballooned neurons and argyrophilic grains (AGs) in the limbic region (Togo et al. 2002). TDP-43 pathology in hippocampus, and amygdala may occur in PSP (Koga et al. 2017b). Rare variants are PSP associated with pallidonigro-luysian degeneration and axonal dystrophy (Ahmed et al. 2008; Yokoyama et al. 2016) or presenting with CBS. More tau load in frontal and parietal cortices was seen in a “cortical” variant (PSP-CBS) (Ling et al. 2014). Tau pathology in spinal cord and pyramidal motor system structures is very common in PSP and may supplement the distinction between classical Richardson’s syndrome from other PSP subtypes (Stejskalova et al. 2019).
Nigrostriatal dysfunction in PSP is associated with an 80–90% reduction in DA and 40–50% reduction in homovanillic acid in CN and Put, whereas the mesocortical and mesolimbic DAergic systems remain intact in comparison to PD. The cholinergic systems are severely affected, with a 40–80% reduction in ChAT activity, and 60% loss of neurons in the PPNc. Cognitive decline is related to dysfunction of striatofrontal or prefrontal circuits as a result of degeneration of BG and brainstem tegmental nuclei. Pathogenesis of PSP, in addition to genetic factors, is due to a cascade of events, such as inflammation and oxidative injury leading to tau aggregation in different neuronal populations. Cerebrovascular lesions in PSP are rare, although PSP has been described as a multiinfarct disorder (Josephs et al. 2002). LBs reported in approximately 20% of PSP may represent an independent disease process (Robinson et al. 2018; Uchikado et al. 2006a). Argyrophilic grains are frequent in PSP (18–80%) (Gil et al. 2018a, b) and show overlap with astroglial tau pathology and anatomical vulnerability with PSP (Yokota et al. 2008). NFTs are positively associated with a brain co-expression network enriched for synaptic and PSP candidate risk genes, but are negatively associated with immune system transcripts, indicating diverse molecular mechanisms that underlie cell-specific vulnerability and disease risks in PSP (Allen et al. 2018).
Corticobasal degeneration (CBD)
CBD, previously described as corticodentatonigral degeneration with neuronal achromatism (Rebeiz et al. 1968), is a rare, sporadic, late-onset progressive disorder of unknown etiology. This 4R tauopathy is clinically shows non-l-dopa-responsive rigidity with focal asymmetric cortical signs (apraxia and aphasia; “alien hand syndrome”) and frontal lobe dementia (Wenning et al. 2011). The estimated incidence of CBD is 0.02–0.92/100,000/year and its prevalence 4.9–7.3/100,000 (Ali and Josephs 2018b). The syndrome is not specific, as clinical features of pathologically proven CBD include 4 phenotypes: CBS, frontal behavioral-spatial syndrome (FBS), nonfluent variant of primary progressive aphasia (naPPA), and PSP syndrome (PSP-S) (Shimohata et al. 2015). Current criteria distinguish possible and probable CBD; the diagnosis of probable CBD requires insidious onset, gradual progression for at least 1 year, age at onset > 50 years, no similar family history of known tau mutations, and a clinical phenotype with at least one CBS feature (Armstrong et al. 2013). These criteria, however, did not sufficiently improve the specificity of diagnosis (Alexander et al. 2014; Ali and Josephs 2018b). Their sensitivity for CBS was poor within 2 years of disease onset (Ouchi et al. 2014), which is between 5th and 7th decade; mean duration is 8 ± 2.65 years. Most CBD cases are sporadic, with only rare familial cases of AutD inheritance associated with mutations in tau genes (Kouri et al. 2014). Both CBD and PSP are characterized by accumulation of an isoform of tau containing four tandem repeats in its microtubule-binding domain and both are associated with increased frequency of MAPT H1/H1 genotype (Kouri et al. 2015). A GWAS identified new CBS susceptibility loci and showed that CBD and PSP share a genetic risk factor other than MAPT and MOBR (Kouri et al. 2015). There are shared risk factors between CBD, PSP and FTLD (Yokoyama et al. 2017). Novel GRN mutations were reported in CBS (Taghdiri et al. 2016), while a familial CBS was due to AD pathology and PSEN1 mutations (Lam et al. 2018).
Neuropathology of corticobasal degeneration
Pigment loss in SN and often asymmetric atrophy of the posterior frontal, parietal and perirolandic cortex, are associated with neuronal loss, superficial laminar spongiosis, and gliosis, temporal and occipital lobes being unaffected. Histological hallmarks are neuronal and glial cytoplasmic tau inclusions (ballooned/achromatic neurons) in cortex, BG, brainstem and cerebellum, and extensive accumulation of tau-positive thread-like processes throughout the brain, which are more widespread than in PSP. The ballooned neurons are similar to those seen in PiD and contain phosphorylated neurofilament protein and αB-crystallin. The aggregates composed of predominantly 4R-tau isoforms with exclusively exon 10 isoforms, identical to PSP and certain forms of FTLD-17, do not stain with antibodies to 3R isoforms and Ub (Dickson et al. 2011a). Ultrastructurally, they consist of 10- to 15-nm filaments, with fewer 25- to 30-nm filaments, granular material, and lipofuscin, resembling those in PSP with 20–24 nm. The twisted ribbons and shift from 4R to 3R tau in pretangles in CBD are different from the AD PHFs (Tatsumi et al. 2014b; Uchihara 2014). In the white matter, “astroglial plaques” (APs) and numerous inclusions involve both astrocytes and oligodendroglia (“coiled bodies”). They do not stain for α-Syn or Ub and thus differ from GCIs in MSA. APs, of diagnostic value in CBD, resemble the neuritic plaques in AD, but instead of clustering around amyloid cores, the tau-positive processes surround unstained neuropil. They involve prefrontal and orbital regions and striatum, but are uncommon in brainstem. The presence of tufted astrocytes (TA) and APs differentiates PSP and CBD: proximal-dominant aggregation of TAs and formation of filamentous NFTs in PSP vs. distal-dominant aggregation of APs and less filamentous pretangles in CBD (Yoshida 2014). Typical CBD displays asymmetrical frontoparietal cortical atrophy (with ballooned neurons), nigral degeneration and tau-positive neuronal and glial lesions, especially APs, in the affected cortex, while typical PSP shows loss of subcortical neurons, accumulation of tau-positive inclusions in neurons (NFTs) and in glia (tufted astrocytes). Systemic metaanalysis of the distribution of localized atrophies tried to distinguish between clinical features (CBS) and histopathological findings (CBD) (Albrecht et al. 2017). Brains with CBD or PSP show differences in amino-terminal truncated tau (37 kDa for CBD and 33 kDa for PSP) (Clavaguera et al. 2013), indicating differences in tau proteolytic processing. LBs have been reported in about 20% of CBD cases, comparable to age-matched controls (Robinson et al. 2018). Argyrophilic grains are thought to be a constant feature of CBD (Tatsumi et al. 2014a).
Pathological features of preclinical or early CBD showed significant differences in neuronal loss, cortical atrophy, white matter volume reduction, and asymmetrical cortical tau pathology (Nishida et al. 2015), while early prominence of APs suggests that CBD begins as an astrogliopathy and neuronal lesions occur later (Ling et al. 2016). Unusual variants include CBD with OPCA and TDP-43/CBD-OPCA (Kouri et al. 2013), with accumulation of α-Syn and TDP-43 (Yamashita et al. 2014), and CBD with TDP-43 pathology (45%) presenting with PSP syndrome (Ali and Josephs 2018b), were associated with lower MAPT H1/H1 genotype frequency than TPD-negative CBD (Koga et al. 2018a). AGs, which also have a predominance of 4R-tau, occur in both PSP and CBD more frequently than in controls, but are reliable disease-specific features to CBD (Tatsumi et al. 2014a). CBS with visual hallucinations and probable REM sleep behavior disorders was seen in an autopsy case of CBD with LBD confined to the brain stem (Naasan et al. 2019). Pathogenic factors of CBD are hyperphosphorylated tau, neuroinflammation and oxidative injury, but the role of MAPT-H1 remains unclear. A significant association between tau burden, post-mortem measures of neurodegeneration and in vivo volume loss was found in both PSP and CBD (Spina et al. 2019). Patients with “vascular CBS” have been reported based on MRI findings but none of them was confirmed at autopsy (Kim et al. 2009; Kreisler et al. 2007; Miyaji et al. 2013). Among 217 patients with an antemortem diagnosis of CBS, three were identified as vascular due to chronic cerebrovascular disease, with infarcts or white matter pathology (Koga et al. 2019).
Frontotemporal lobar degeneration-tau (FTLD-tau)
Frontotemporal dementia (FTD) is a heterogeneous clinical syndrome associated with frontotemporal lobar degeneration (FTLD) as a relatively consistent neuropathological hallmark feature. FTD is a diverse condition on the genetic and neuropathological basis. A novel molecular classification of these conditions distinguished three broad molecular subgroups: FTLD with tau, TAR DNA-binding protein 43 (FDP-43) and FET protein accumulation (FTLD-tau, FTLD-TDP and FTLD-FET respectively) (Neumann and Mackenzie 2019).
FTLD-tau, formerly referred to as frontotemporal degeneration and parkinsonism linked to chromosome 17 (FTDP-17), linked the P301L mutation in the MAPT gene, is caused by mutations in either the MAPT or the progranulin (PGRN) gene. The spectrum of sporadic FTLD associated with tau pathology includes PSP, CBD, PiD, and FTDP-17 MAPT (Taniguchi-Watanabe et al. 2016). Depending upon the specific mutation in MAPT, familial FTLD-tau can have 3R, 4R, or a combination of 3R and 4R tau (Dickson et al. 2011b). Positive family history was seen in 25%, 13% with AutD inheritance and 9% MAPT mutations (Forrest et al. 2019b). Mixed FTLD-tau and TDP-43 proteinopathy (FTLD-TDP) is rare (Kim et al. 2018). The tauopathy associated with FTDP-17 MAPT shows a wide range of pathological phenotypes. FTDP-17 mutations contribute to the pathogenesis via increased formation of tau oligomers (Maeda et al. 2018). Neuropathology shows focal symmetrical frontotemporal atrophy with rust-colored appearance of the GP due to increased iron pigment, and depigmented SN (Wszolek et al. 2005). Pretangles in neurons show diffuse tau immunoreactivity, while some cases have also AD-NFTs, globose tangles and astrocytic lesions similar to those in PSP or CBD, or tau-positive glial inclusions resembling those in PSP, CBD, and AG disease. Despite significant pathologic heterogeneity between different mutations, there is broad overlap with other sporadic tauopathies (AD, PSP, CBD, PiD). Ultrastructurally, the filaments vary in structure and appearance, with PHFs, 15- to 27-nm-wide twisted ribbons, and 12- to 15-nm or 15- to 20-nm straight tubules, which are either paired helical, slender or narrow twisted ribbons or straight filaments (Ghetti et al. 2011; van Swieten and Spillantini 2007). They cause cell dysfunction, due to abnormal proteostasis, impaired axonal transport and mitochondrial damage (Irwin et al. 2015). Neuronal loss and astrogliosis affect the frontal, temporal cortical and subcortical gray matter and hippocampus. FTDP-17 with Pick body-like neuronal inclusions and swollen processes in white matter reactive to 3R and 4R tau was associated with a novel tau mutation, p.E372G. FTLD-tau-related pathological lesions in non-diseased individuals suggest that preclinical stages of FTLD-tau exist (Thal et al. 2015). Toxic tau accumulating in neuronal soma and dendrites leads to microtubule depolymerization and synaptic loss (Bodea et al. 2016). However, the hypothesized pathogenic mechanisms by which mutations in the MAPT gene promote tauopathy and the ability of mutant tau protein to support prion-like propagation do not give definite insights into the basis for the reactive vulnerability in FTLD-tau (Strang et al. 2019).
Postencephalitic parkinsonism
This progressive disorder, a sequela of encephalitis lethargica and other viral encephalitides, clinically shows rigid parkinsonism, oculomotor lesions (ocular palsy and oculogyric crises), and cognitive impairment (Jellinger 2011). Depigmentation of SN, marked neuronal loss and astrocytosis in brainstem—particularly in SN (diffuse and more marked than in PD)—, is associated with widespread occurrence of tau-positive globose NFTs, neuropil threads (NTs), glial inclusions in brainstem, BG, NBM, and amygdala, less severe in striatopallidum, thalamus, hypothalamus, and cerebellum. NFTs and NTs, composed of 22-nm twisted tubules with occasional straight filaments showing 3R- plus 4R-tau and Ub immunoreactivity, are identical to those in AD. Tau-immunoreactive astroglia are seen in affected areas, whereas TAs, APs, oligodendroglial inclusions, ballooned neurons or Pick bodies are absent. Perivascular lympho-plasmocytic aggregates and microglial activation can be found in the midbrain for many years after the initial encephalitic illness, but are sparse in long-surviving patients. Cortical pathology is common, with NFTs mainly in hippocampus and less often in cortical layers II and III, differs from that in AD. The distribution of lesions is similar to PSP, although there are subtle deviations: rare involvement of cranial nerves IV and XII, inferior olives, and striatopallidum, different cortical involvement, and less tau pathology in white matter in PEP (Jellinger 2011). Essential differences include PHF and 3 + 4 R tau in PEP, while PSP NFTs have straight filaments and 4R tau. TDP-43 pathology is present in most PEP brains, but there was no correlation between clinical features or hippocampal sclerosis (Ling et al. 2013). Lesions in cholinergic subcortical supranuclear centers of gaze movement cause gaze palsy and lid apraxia similar to that in PSP (Wenning et al. 1997). Amyloid-β deposits are rare. Neither LBs nor α-Syn pathology were detected, thus classifying PEP it as a “pure” tauopathy (Jellinger 2009b). Despite epidemiologic evidence of a viral infection, the etiopathogenesis is unknown, and molecular-biologic studies have failed to identify influenza virus in archival material from PEP brain (Vilensky 2011).
Pick’s disease
This progressive dementia with personality deterioration and signs of frontal disinhibition exhibits rare extrapyramidal symptoms. Age of onset ranges between 40 and 80 years. The prevalence in autopsy series ranged from 8 to 30% of FTD (Munoz et al. 2011). The clinical presentation is often that of the behavioral variant of FTD or progressive non-fluent aphasia, while involvement of the amygdala may mimick Kluver-Bucy syndrome. Most cases are sporadic, but familial cases, usually with AutD inheritance as a result of MAPT mutations (prominently in exon 9) have been reported (Forrest et al. 2019b), while the extended haplotype (H1/H1) of the MAPT gene is not associated with PiD (Russ et al. 2001). Frontotemporal atrophy, often with a “knife blade” appearance of the cortical gyri, is associated with dilated ventricles, and degeneration of striatum and SN. Loss of neurons, astrocytosis, and spongiosis affect the outer cortical layers with swollen neurons (“Pick cells”), indistinguishable from the swollen achromatic (ballooned) neurons in other conditions. Some brains show extensive loss of pigmented SN neurons, in others the SN is preserved. Mature tau pathology is more abundant in frontotemporal mesolimbic regions than in neocortical regions (Irwin et al. 2016). Argyrophilic intraneuronal cytoplasmic inclusions (Pick bodies) are abundant in the granule neurons of dentate fascia and pyramidal neurons of hippocampus. Their major component is 3R-tau (Delacourte et al. 1996), with an occasional mixture of 3R-tau and 4R-tau (Zhukareva et al. 2002), due to the presence of concomitant NFT degeneration or rare AGRs (Kovacs et al. 2013). Ultrastructurally, they consist of narrow protofilaments and of wide filaments (the minority) composed of two narrow filaments packed against each other. The filamentous tau in PiD shows differences in phosphorylation and folding relative to those in AD, indicating the existence of distinct molecular conformers (Falcon et al. 2018; Goedert et al. 2019).
Guamanian and other forms of western Pacific parkinsonism
A high incidence of ALS and PDC was recognized in three regions of the Western Pacific, the Mariana islands of Guam and Rota, the Muro district on the Kii peninsula in Japan, and Western New Guinea. A doublet of pathological tau at 64 and 69 kDa was observed in brain tissue homogenates. The incidence of ALS/PDC in Guam has declined since the 1960s (Waring et al. 2004). Guamanian PDC and ALS/PDC of the Chamorro population may appear clinically similar to FTLD-U and ALS, with extrapyramidal symptoms, olfactory dysfunction, oculomotor signs, and dementia. Neuropathology shows cerebral and BG atrophy, depigmentation of SN and LC, widespread loss of neurons and gliosis in hippocampus, amygdala, NBM, brainstem tegmentum, and dentate nucleus, with abundant NFTs, granulovacuolar degeneration, and Hirano bodies (Oyanagi et al. 2011). NFTs in cortex involve layers II and III, similar to that in PSP. NTs and tau-positive thread-like structures are sparse or absent. NFTs in both the Japanese and Guamanian forms of PDC show similarities to AD (Oyanagi et al. 2011), and marked deposition of 43-kDa TAR DNA-binding protein, indicating a role for proteostasis imbalance (Verheijen et al. 2018). Glial pathology is prominent in Guam PDC and includes granular astrocytes, coiled inclusions in oligodendroglia, and fine granules in motor cortex, frontal white matter, and amygdala, all composed of 3R + 4R-tau isoforms (Yamazaki et al. 2005). Numerous coiled body-like inclusions occur in cerebral white matter (Hasegawa et al. 2008).
The cortex in PDC is distinguished from that in PSP by the presence of α-Syn and LBs (Miklossy et al. 2008). α-Syn-positive aggregates in the amygdala often colocalize with neurons harboring NFTs, and spherical α-Syn-positive structures in the molecular layer of cerebellar cortex (Yamazaki et al. 2005). Guam PDC is associated with cortical tau-negative, TPD-43-positive dystrophic neurites and neuronoglial inclusions in gray and white matter. Biochemical analysis showed FTLD-U-like insoluble TPD-43, and spinal cord exhibited tau-positive tangles and TDP-43-positive inclusions, suggesting that ALS/PDC is a multiple proteinopathy (Mimuro et al. 2018). The Western Pacific disease clusters show factors similar to that causing AD and other tauopathies, but GWAS failed to identify a single gene locus for Guam PDC, supporting the hypothesis of a mixed genetic/environmental etiology. The cycad hypothesis suggesting that dietary consumption of cycad toxins or sterol glucosides is causative has not been confirmed (Steele and McGeer 2008). The etiopathogenesis remains enigmatic.
In ALS/PDC on the Kii peninsula, ALS and PDC clinically occur separately or in combination, and are considered as different manifestations of a single disease entity. The neuropathological hallmarks are widespread NFTs and NTs, most predominantly in medial temporal and frontal cortices, less in other cortices, subcortical nuclei, brainstem and spinal cord. Tau-positive astrocytes are also present in the white matter (Mimuro et al. 2018). NFTs are ultrastructurally characterized as helical filaments composed of all 6 tau isoforms, similar to those in α-Syn and Guamanian ALS/PDC (Itoh et al. 2003). Kii ALS/PDC differs from AD by differential NFT distribution and the lack of abundant senile plaques. In addition, various types of α-Syn-positive lesions, including NCIs, GCIs and dystrophic neurites are present, mainly in the limbic system and brainstem (Kokubo et al. 2012; Mimuro et al. 2018). A recent study showed phosphorylated tau pathology of various types in dentante nucleus and Purkinje and glia lcells in the cerebellum (Morimoto et al. 2018). Recent PET studies confirmed distinct distribution of tau pathology and lack of β-amyloid in Kii ALS/PDC patients (Shinotoh et al. 2019). Genetic and environmental factors are implicated in the pathogenesis of the disease (Hata et al. 2018).
Secondary parkinsonism
About 10% of all patients with parkinsonism have secondary forms with known specific causes, e.g. certain drugs and toxins, metabolic disorders, viral infections, multiple infarcts, brain tumors, trauma, or hydrocephalus (Table 2).
Vascular parkinsonism (pseudoparkinsonism)
Vascular parkinsonism (VaP) (arteriosclerotic pseudoparkinsonism) (Critchley 1981) is a rare akinetic-rigid syndrome resulting from cerebrovascular disease, with a variety of clinical and pathologic features distinct from those of sporadic PD. Its prevalence is estimated at 2–17% of all parkinsonian syndromes but it is difficult to diagnose with clinical certainty, based on the presence of clinical parkinsonism with variable signs and findings of cerebrovascular disease (Rektor et al. 2018). Three subtypes are considered: (1) acute/subacute post-stroke VaP presenting with acute/subacute onset of parkinsonism responding to DAergic drugs; (2) more frequent insidious onset with progressive parkinsonism and multiple other symptoms, particularly higher-level gait disorder; (3) mixed or overlapping syndrome of VaP with PD and other neurodegenerative parkinsonism (Rektor et al. 2018). Neuropathology shows multiple small ischemic lesions in BG, white matter, and less often the SN that involve the corticostriatopallidal-thalamic and thalamocortical loops (Jellinger 2008a; Zijlmans et al. 2004). Postmortem demonstration of LBs in 13% of patients with multi-infarct encephalopathy, an incidence twice as common as in age-matched controls, suggested subclinical PD, whereas vascular lesions were observed in 44–58% of individuals without dementia and in up to 94% of those with dementia (Jellinger and Attems 2008). Vascular lesions affecting the BG, subcortical and deep white matter changes suggest disruption of the striato-thalamo-cortical circuits leading to motor and cognitive impairment in VaP (Chen et al. 2014; Sibon and Tison 2004).
Drug- and toxin-related parkinsonism
Drug-induced parkinsonism, which can be clinically confused with AR PD, is often associated with neuroleptic drugs, antipsychotics, calcium channel blocking agents, and other substances causing DA depletion, blockage of postsynaptic D1 and D2 receptors, or loss of striatonigral TH immunoreactivity (Wenning et al. 2011). Drug-induced parkinsonism affects 15–60% of patients treated with typical neuroleptics, depending on their type, dose, and the underlying susceptibility of the patients, but neuropathologial data are poor (Shuaib et al. 2016). The mechanisms of DIB and other extrapyramidal effects (dystonia, akathisia, etc.) are thought to be due to antagonistic binding of DAergic receptors in the BG and the mesolimbic and mesocortical pathways (Kamin et al. 2000). Frequent age-related SNc cell loss or iLBD are predisposed to adverse drug effects as a result of relative DA deficiency. Parkinsonism resulting from carbon monoxide, carbon disulfide intoxication or postnarcotic encephalopathy is caused by anoxic lesions with necrosis of GP and SN (Ginsberg 1985). Methamphetamine abuse is linked to injury of SN neurons and increased risk of PD (Rumpf et al. 2018). Methanol intoxication causes bilateral putaminal necrosis and necrosis of subcortical white matter (Franquet et al. 2012). Severe parkinsonism after poisoning with potassium cyanide is due to neuronal loss and gliosis in GP, Put, and SNr, while SNc was spared (Uitti et al. 1985). Chronic lead intoxication causes SN damage, and manganese encephalopathy, e.g., due to welding exposure, is associated with an l-dopa resistent akinetic-rigid syndrome (Racette et al. 2012), related to neuronal loss and gliosis, particularly in GPi, and in striatum with little or no SN damage and absence of LBs (Perl and Olanow 2007). Accumulation of manganese in BG associated with hepatic cirrhosis is a rare disorder with parkinsonism, ataxia, and cognitive impairment (Maffeo et al. 2014). Chronic exposure to trichlorethylene (TCE) and carbondisulfide (CS2) can cause parkinsonism through mitochondrial complex I inhibition (Gash et al. 2008). Severe l-dopa-responsive parkinsonism developed after exposure to MPTP—a synthetic heroin drug that leads to mitochondrial damage and neuronal death—shows diffuse neuronal loss and gliosis in SN, extracellular NM and activated microglia but no typical LBs (Langston et al. 1999). Eosinophilic inclusion bodies resembling LBs have been seen in SN and LC of MPTP-treated aged monkeys, but their ultrastructure differed from that of human LBs (Forno et al. 1996). Other toxins that may cause parkinsonism include paraquat, rotenone, other herbicides and pesticides (Hoglinger et al. 2006; Taba 2017).
Other lesions causing parkinsonism
Parkinsonism has been observed in a wide variety of disorders involving the brainstem or SN, or both, that affect DAergic projections, such as destruction of the SN by bullet injury, after direct traumatic impact, herniation-contusion of the upper brainstem or midbrain damage caused by increased intracranial pressure, with loss of the nigrostriatal pathway (Formisano and Zasler 2014). Chronic traumatic encephalopathy (punch drunk, pugilistic encephalopathy, boxer’s dementia), a consequence of repetitive mild traumatic brain injury, often accompanied by parkinsonian symptoms, is characterized by diffuse cortical atrophy; degeneration of corpus callosum and cerebellum; cell loss in SN, LC, and striatum, deposition of p-tau protein as NFTs, astrocytic tangles in superficial cortical layers, thread-like neurites and astrocytic inclusions around small blood vessels at the sulcal depths of the cortex (McKee et al. 2015, 2018). Parkinsonism has also been observed in rare cases of tuberculoma, brainstem tumors, solid tumors causing brainstem compression, calcification of the BG (Fahr’s disease), viral encephalitis, including HIV infections, subacute sclerosing panencephalitis, multiple sclerosis, paraneoplastic syndromes (Grant and Graus 2009; Yap et al. 2017), normal-pressure hydrocephalus (Wenning et al. 2011), and cerebrovascular diseases (Grabli et al. 2011; Mehanna and Jankovic 2013). Parkinsonism may occur in a variety of inherited metabolic disorders, like Gaucher disease (linking GBA mutations to PD) (Mullin et al. 2019; Sidransky and Lopez 2012), Niemann-Pick disease (parkinsonism with mutated NPC1), lysosomal storage diseases, disorders of amino acid metabolism (phenylketonuria, maple syrup urine disease, methylmalonic acidemia) and inherited mitochondrial disorders, showing genetic evidence of common pathways with PD (Limphaibool et al. 2018). There are links between PD and metal storage disorders (Botsford et al. 2018), in particular iron accumulation causing nigral cell death (Pietracupa et al. 2017). The most frequent metal storage disorders associated with parkinsonism are hereditary hemochromatosis, PKAN and Wilson’s disease (Botsford et al. 2018).
Vision for future research
Despite considerable clinical and pathologic overlapping, most types of movement disorders, particularly those of neurodegenerative origin, show characteristic pathologic pictures. The deposition of pathologic fibrillary proteins or the distribution patterns of CNS lesions may or may not be typical cytoskeletal signposts pointing to the correct diagnosis and to their pathophysiology. Because in vivo markers for most of these disorders (except those with known molecular genetic backgrounds) are still poor, the diagnosis usually depends on clinicomorphologic features. Specific identification and correct diagnosis of some of these disorders may be difficult because they share clinical and morphologic phenotypes with other neurodegenerative diseases or have considerable intrafamilial, interfamilial, and interindividual differences. Therefore, comprehensive morphologic studies using modern methods of neuropathobiology are needed to distinguish the different disease entities. Consensus data on clinical and neuropathologic criteria, together with molecular genetic and biochemical data, will aid in correct classifying and diagnosing neurodegenerative movement disorders and provide further insight into their pathophysiology and pathogenesis as a basis for options of treatment and further directions of research.
Abbreviations
- AGs:
-
Argyrophilic grains
- ALS:
-
Amyptrophic lateral sclerosis
- ALS/PDC:
-
Guamanian ALS–Parkinson’s disease complex
- AP:
-
Astroglial plaque
- APP:
-
Amyloid precursor protein
- AR-PD:
-
Akinesia-and-rigidity type PD
- AS:
-
α-Synuclein
- AutD:
-
Autosomal dominant
- AutR:
-
Autosomal recessive
- βSyn:
-
β-Synuclein
- BG:
-
Basal ganglia
- BHC:
-
Benign hereditary chorea
- BIBD:
-
Basophilic inclusion body disease
- CAA:
-
Cerebral amyloid angiopathy
- CAG:
-
Polyglutamine
- CBD:
-
Corticobasal degeneration
- CBGTC:
-
Cortico-BG-thalamocortical
- CBS:
-
Corticobasal syndrome
- ChAc:
-
Chorea-acanthocytosis
- ChAT:
-
Choline-acetyl transferase
- CI:
-
Cognitive impairment
- CN:
-
Caudate nucleus
- CNS:
-
Central nervous system
- CS–TD:
-
Cortico-striatal–temporal difference
- DA:
-
Dopamine
- DLB:
-
Dementia with Lewy bodies
- DLB-AD:
-
Dementia with Lewy bodies and Alzheimer’s disease
- DRD:
-
Dopa-responsive dystonia
- DRPLA:
-
Dentatorubral-pallidoluysian atrophy
- DS:
-
Dystonia syndrome
- ENK:
-
Enkephalin
- ET:
-
Essential tremor
- FTDP-17:
-
Frontotemporal degeneration and parkinsonism linked to chromosome 17
- FTLD:
-
Frontotemporal lobar degeneration
- GABA:
-
γ-Aminobutyric acid
- GBA:
-
Glucocerebrosidase gene
- GCase:
-
Glucocerebrosidase
- GCIs:
-
Glial cytoplasmic inclusions
- GDNF:
-
Glia-derived neurotrophic factor
- GNIs:
-
Glial nuclear inclusions
- GPe:
-
External segment of globus pallidus
- GPi:
-
Internal segment of globus pallidus
- GTP:
-
Guanosine triphosphate
- HD:
-
Huntington’s disease
- HTT:
-
Huntingtin
- iLBD:
-
Incidental Lewy body disease
- IT:
-
Intratelencephalic
- LB:
-
Lewy body
- LC:
-
Locus ceruleus
- LID:
-
l-Dopa-induced dyskinesia
- LP:
-
Lewy body pathology
- MCI:
-
Mild cognitive impairment
- MD:
-
Menkes’ disease
- MJD:
-
Machado-Joseph disease
- MSA:
-
Multiple system atrophy
- MSA-C:
-
Multiple system atrophy with predominant cerebellar features
- MSA-P:
-
Multiple system atrophy with predominant parkinsonism
- MSN:
-
Medium spiny projection neuron
- NA:
-
Neuroacanthocytosis
- NBIA:
-
Neurodegeneration with brain iron accumulation
- NBM:
-
Nucleus basalis of Meynert
- NCIs:
-
Neuronal cytoplasmic inclusions
- NFTs:
-
Neurofibrillary tangles
- NIID:
-
Neuronal intranuclear inclusion disease
- NM:
-
Neuromelanin
- NNIs:
-
Neuronal nuclear inclusions
- NT:
-
Neuropil threads
- OCD:
-
Obsessive-compulsive disorder
- OPC:
-
Olivopontocerebellar
- OPCA:
-
Olivopontocerebellar atrophy
- OS:
-
Oxidative stress
- pAS:
-
Phosphorylated α-synuclein
- PC:
-
Purkinje cell
- PD:
-
Parkinson’s disease
- PDC:
-
Parkinson’s disease complex
- PDD:
-
Parkinson’s disease dementia
- PEP:
-
Postencephalitic parkinsonism
- PGF:
-
PSP with progressive gait freezing
- PHFs:
-
Paired helical filaments
- PiD:
-
Pick’s disease
- PKAN:
-
Pantothenate-kinase associated neurodegeneration
- PPN:
-
Pedunculopontine nucleus
- PPT:
-
Pedunculo-pontine tegmental
- PSP:
-
Progressive supranuclear palsy
- PSP-CBS:
-
PSP presenting with corticobasal syndrom
- PSP-P:
-
Progressive supranuclear palsy-parkinsonism
- PSP-RS:
-
Richardson’s syndrome
- Put:
-
Putamen
- SCA3:
-
Spinocerebellar ataxia type 3
- SN:
-
Substantia nigra
- SNc:
-
Substantia nigra pars compacta
- SNr:
-
Substantia nigra pars reticulata
- SP:
-
Substance P
- STN:
-
Subthalamic nucleus
- TA:
-
Tufted astrocyte
- TDPD:
-
Tremor-dominant type of PD
- TH:
-
Tyrosine hydoxylase
- TS:
-
Tourette’s syndrome
- VaP:
-
Vascular parkinsonism
- VM:
-
Ventromedial
- VTA:
-
Ventral tegmental area
- WD:
-
Wilson’s disease
- XDP:
-
X-linked dystonia-parkinsonism
References
Aarsland D, Kurz MW (2010) The epidemiology of dementia associated with Parkinson’s disease. Brain Pathol 20:633–639
Abbott RD, Nelson JS, Ross GW, Uyehara-Lock JH, Tanner CM, Masaki KH, Launer LJ, White LR, Petrovitch H (2017) Marinesco bodies and substantia nigra neuron density in Parkinson’s disease. Neuropathol Appl Neurobiol 43:621–630
Abeliovich A, Gitler AD (2016) Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539:207–216
Adamowicz DH, Roy S, Salmon DP, Galasko DR, Hansen LA, Masliah E, Gage FH (2017) Hippocampal alpha-synuclein in dementia with Lewy bodies contributes to memory impairment and is consistent with spread of pathology. J Neurosci 37:1675–1684
Adler CH, Caviness JN, Sabbagh MN, Shill HA, Connor DJ, Sue L, Evidente VG, Driver-Dunckley E, Beach TG (2010a) Heterogeneous neuropathological findings in Parkinson’s disease with mild cognitive impairment. Acta Neuropathol 120:827–828
Adler CH, Connor DJ, Hentz JG, Sabbagh MN, Caviness JN, Shill HA, Noble B, Beach TG (2010b) Incidental Lewy body disease: clinical comparison to a control cohort. Mov Disord 25:642–646
Adler CH, Dugger BN, Hentz JG, Hinni ML, Lott DG, Driver-Dunckley E, Mehta S, Serrano G, Sue LI, Duffy A, Intorcia A, Filon J, Pullen J, Walker DG, Beach TG (2016) Peripheral synucleinopathy in early Parkinson’s disease: submandibular gland needle biopsy findings. Mov Disord 31:250–256
Agarwal S, Gilbert R (2019) Progressive supranuclear palsy, 2018/09/26 edn. StatPearls Publishing, Treasure Island
Ahmed Z, Josephs KA, Gonzalez J, DelleDonne A, Dickson DW (2008) Clinical and neuropathologic features of progressive supranuclear palsy with severe pallido-nigro-luysial degeneration and axonal dystrophy. Brain 131:460–472
Ahmed Z, Asi YT, Sailer A, Lees AJ, Houlden H, Revesz T, Holton JL (2012) The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol Appl Neurobiol 38:4–24
Alafuzoff I, Hartikainen P (2017) Alpha-synucleinopathies. In: Kovacs GG, Alafuzoff I (eds) Handbook of clinical neurology. Neuropathology, vol 145. Elsevier B.V., New York, pp 339–353
Alafuzoff I, Ince PG, Arzberger T, Al-Sarraj S, Bell J, Bodi I, Bogdanovic N, Bugiani O, Ferrer I, Gelpi E, Gentleman S, Giaccone G, Ironside JW, Kavantzas N, King A, Korkolopoulou P, Kovacs GG, Meyronet D, Monoranu C, Parchi P, Parkkinen L, Patsouris E, Roggendorf W, Rozemuller A, Stadelmann-Nessler C, Streichenberger N, Thal DR, Kretzschmar H (2009) Staging/typing of Lewy body related alpha-synuclein pathology: a study of the BrainNet Europe Consortium. Acta Neuropathol 117:635–652
Alberico SL, Cassell MD, Narayanan NS (2015) The vulnerable ventral tegmental area in Parkinson’s disease. Basal Ganglia 5:51–55
Albrecht F, Bisenius S, Morales Schaack R, Neumann J, Schroeter ML (2017) Disentangling the neural correlates of corticobasal syndrome and corticobasal degeneration with systematic and quantitative ALE meta-analyses. NPJ Parkinsons Dis 3:12
Al-Chalabi A, Durr A, Wood NW, Parkinson MH, Camuzat A, Hulot JS, Morrison KE, Renton A, Sussmuth SD, Landwehrmeyer BG, Ludolph A, Agid Y, Brice A, Leigh PN, Bensimon G (2009) Genetic variants of the alpha-synuclein gene SNCA are associated with multiple system atrophy. PLoS One 4:e7114. https://doi.org/10.1371/journal.pone.0007114
Alecu I, Bennett SAL (2019) Dysregulated lipid metabolism and its role in alpha-synucleinopathy in Parkinson’s disease. Front Neurosci 13:328
Alegre-Abarrategui J, Brimblecombe KR, Roberts RF, Velentza-Almpani E, Tilley BS, Bengoa-Vergniory N, Proukakis C (2019) Selective vulnerability in alpha-synucleinopathies. Acta Neuropathol. https://doi.org/10.1007/s00401-00019-02010-00402
Alexander SK, Rittman T, Xuereb JH, Bak TH, Hodges JR, Rowe JB (2014) Validation of the new consensus criteria for the diagnosis of corticobasal degeneration. J Neurol Neurosurg Psychiatry 85:925–929
Alexandris A, Walker L, Liu AKL, McAleese KE, Johnson M, Pearce RK, Gentleman SM, Attems J (2019) Cholinergic deficits and galaninergic hyperinnervation of the nucleus basalis of Meynert in Alzheimer’s disease and Lewy body disorders. Neuropathol Appl Neurobiol (accepted article)
Ali F, Josephs K (2018a) The diagnosis of progressive supranuclear palsy: current opinions and challenges. Expert Rev Neurother 18:603–616
Ali F, Josephs KA (2018b) Corticobasal degeneration: key emerging issues. J Neurol 265:439–445
Ali F, Martin PR, Botha H, Ahlskog JE, Bower JH, Masumoto JY, Maraganore D, Hassan A, Eggers S, Boeve BF, Knopman DS, Drubach D, Petersen RC, Dunkley ED, van Gerpen J, Uitti R, Whitwell JL, Dickson DW, Josephs KA (2019) Sensitivity and specificity of diagnostic criteria for progressive supranuclear palsy. Mov Disord. https://doi.org/10.1002/mds.27619
Allen M, Burgess JD, Ballard T, Serie D, Wang X, Younkin CS, Sun Z, Kouri N, Baheti S, Wang C, Carrasquillo MM, Nguyen T, Lincoln S, Malphrus K, Murray M, Golde TE, Price ND, Younkin SG, Schellenberg GD, Asmann Y, Ordog T, Crook J, Dickson D, Ertekin-Taner N (2016) Gene expression, methylation and neuropathology correlations at progressive supranuclear palsy risk loci. Acta Neuropathol 132:197–211
Allen M, Wang X, Serie DJ, Strickland SL, Burgess JD, Koga S, Younkin CS, Nguyen TT, Malphrus KG, Lincoln SJ, Alamprese M, Zhu K, Chang R, Carrasquillo MM, Kouri N, Murray ME, Reddy JS, Funk C, Price ND, Golde TE, Younkin SG, Asmann YW, Crook JE, Dickson DW, Ertekin-Taner N (2018) Divergent brain gene expression patterns associate with distinct cell-specific tau neuropathology traits in progressive supranuclear palsy. Acta Neuropathol 136:709–727
American P, Association, Force D-T (2013) Diagnostic and statistical manual of mental disorders: DSM–5, 5th edn. American Psychiatric Publishing Inc., Arlington
Ammal Kaidery N, Thomas B (2018) Current perspective of mitochondrial biology in Parkinson’s disease. Neurochem Int 117:91–113
Angot E, Steiner JA, Lema Tome CM, Ekstrom P, Mattsson B, Bjorklund A, Brundin P (2012) Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons In vivo. PLoS One 7:e39465
Ansari M, Adib Moradi S, Ghazi Sherbaf F, Hedayatnia A, Aarabi MH (2019) Comparison of structural connectivity in Parkinson’s disease with depressive symptoms versus non-depressed: a diffusion MRI connectometry study. Int Psychogeriatr 31:5–12
Aoki N, Boyer PJ, Lund C, Lin WL, Koga S, Ross OA, Weiner M, Lipton A, Powers JM, White CL 3rd, Dickson DW (2015) Atypical multiple system atrophy is a new subtype of frontotemporal lobar degeneration: frontotemporal lobar degeneration associated with alpha-synuclein. Acta Neuropathol 130:93–105
Arawaka S, Sato H, Sasaki A, Koyama S, Kato T (2017) Mechanisms underlying extensive Ser129-phosphorylation in alpha-synuclein aggregates. Acta Neuropathol Commun 5:48
Arima K (2006) Ultrastructural characteristics of tau filaments in tauopathies: immuno-electron microscopic demonstration of tau filaments in tauopathies. Neuropathology 26:475–483
Armstrong RA (2013) White matter pathology in progressive supranuclear palsy (PSP): a quantitative study of 8 cases. Clin Neuropathol 32:399–405
Armstrong MJ (2018) Progressive supranuclear palsy: an update. Curr Neurol Neurosci Rep 18:12
Armstrong MJ (2019) Lewy body dementias. Continuum (Minneap Minn) 25:128–146
Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, Boxer AL, Dickson DW, Grossman M, Hallett M, Josephs KA, Kertesz A, Lee SE, Miller BL, Reich SG, Riley DE, Tolosa E, Troster AI, Vidailhet M, Weiner WJ (2013) Criteria for the diagnosis of corticobasal degeneration. Neurology 80:496–503
Armstrong RA, Kotzbauer PT, Perlmutter JS, Campbell MC, Hurth KM, Schmidt RE, Cairns NJ (2014) A quantitative study of alpha-synuclein pathology in fifteen cases of dementia associated with Parkinson disease. J Neural Transm 121:171–181
Arnaoutoglou NA, O’Brien JT, Underwood BR (2019) Dementia with Lewy bodies—from scientific knowledge to clinical insights. Nat Rev Neurol 15:103–112
Aroso M, Ferreira R, Freitas A, Vitorino R, Gomez-Lazaro M (2016) New insights on the mitochondrial proteome plasticity in Parkinson’s disease. Proteom Clin Appl 10:416–429
Asi YT, Ling H, Ahmed Z, Lees AJ, Revesz T, Holton JL (2014) Neuropathological features of multiple system atrophy with cognitive impairment. Mov Disord 29:884–888
Atias M, Tevet Y, Sun J, Stavsky A, Tal S, Kahn J, Roy S, Gitler D (2019) Synapsins regulate alpha-synuclein functions. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1903054116
Attems J, Jellinger KA (2008) The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease. Neuropathol Appl Neurobiol 34:466–467
Attems J, Walker L, Jellinger KA (2014) Olfactory bulb involvement in neurodegenerative diseases. Acta Neuropathol 127:459–475
Ayers JI, Giasson BI, Borchelt DR (2018) Prion-like spreading in tauopathies. Biol Psychiatry 83:337–346
Bachhuber T, Katzmarski N, McCarter JF, Loreth D, Tahirovic S, Kamp F, Abou-Ajram C, Nuscher B, Serrano-Pozo A, Muller A, Prinz M, Steiner H, Hyman BT, Haass C, Meyer-Luehmann M (2015) Inhibition of amyloid-beta plaque formation by alpha-synuclein. Nat Med 21:802–807
Bae EJ, Yang NY, Song M, Lee CS, Lee JS, Jung BC, Lee HJ, Kim S, Masliah E, Sardi SP, Lee SJ (2014) Glucocerebrosidase depletion enhances cell-to-cell transmission of alpha-synuclein. Nat Commun 5:4755
Bagetta V, Ghiglieri V, Sgobio C, Calabresi P, Picconi B (2010) Synaptic dysfunction in Parkinson’s disease. Biochem Soc Trans 38:493–497
Bailey RM, Covy JP, Melrose HL, Rousseau L, Watkinson R, Knight J, Miles S, Farrer MJ, Dickson DW, Giasson BI, Lewis J (2013) LRRK2 phosphorylates novel tau epitopes and promotes tauopathy. Acta Neuropathol 126:809–827
Balestrino R, Schapira AHV (2018) Glucocerebrosidase and Parkinson disease: molecular, clinical, and therapeutic implications. Neuroscientist 24:540–559
Ballanger B, Klinger H, Eche J, Lerond J, Vallet AE, Le Bars D, Tremblay L, Sgambato-Faure V, Broussolle E, Thobois S (2012) Role of serotonergic 1A receptor dysfunction in depression associated with Parkinson’s disease. Mov Disord 27:84–89
Bandres-Ciga S, Saez-Atienzar S, Bonet-Ponce L, Billingsley K, Vitale D, Blauwendraat C, Gibbs JR, Pihlstrom L, Gan-Or Z, Cookson MR, Nalls MA, Singleton AB (2019) The endocytic membrane trafficking pathway plays a major role in the risk of Parkinson’s disease. Mov Disord 34:460–468
Barca E, Kleiner G, Tang G, Ziosi M, Tadesse S, Masliah E, Louis ED, Faust P, Kang UJ, Torres J, Cortes EP, Vonsattel JP, Kuo SH, Quinzii CM (2016) Decreased coenzyme q10 levels in multiple system atrophy cerebellum. J Neuropathol Exp Neurol 75:663–672
Bassil F, Guerin PA, Dutheil N, Li Q, Klugmann M, Meissner WG, Bezard E, Fernagut PO (2017) Viral-mediated oligodendroglial alpha-synuclein expression models multiple system atrophy. Mov Disord 32:1230–1239
Batla A, De Pablo-Fernandez E, Erro R, Reich M, Calandra-Buonaura G, Barbosa P, Balint B, Ling H, Islam S, Cortelli P, Volkmann J, Quinn N, Holton JL, Warner TT, Bhatia KP (2018) Young-onset multiple system atrophy: clinical and pathological features. Mov Disord 33:1099–1107
Beach TG, Adler CH, Sue LI, Peirce JB, Bachalakuri J, Dalsing-Hernandez JE, Lue LF, Caviness JN, Connor DJ, Sabbagh MN, Walker DG (2008) Reduced striatal tyrosine hydroxylase in incidental Lewy body disease. Acta Neuropathol 115:445–451
Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, Sasse J, Boyer S, Shirohi S, Brooks R, Eschbacher J, White CL 3rd, Akiyama H, Caviness J, Shill HA, Connor DJ, Sabbagh MN, Walker DG (2009) Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117:613–634
Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White Iii CL, Akiyama H, Caviness JN, Shill HA, Sabbagh MN, Walker DG (2010) Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol 119:689–702
Beck G, Singh A, Papa SM (2018) Dysregulation of striatal projection neurons in Parkinson’s disease. J Neural Transm (Vienna) 125:449–460
Bengoa-Vergniory N, Roberts RF, Wade-Martins R, Alegre-Abarrategui J (2017) Alpha-Synuclein oligomers: a new hope. Acta Neuropathol 134:819–838
Benskey MJ, Perez RG, Manfredsson FP (2016) The contribution of alpha synuclein to neuronal survival and function—implications for Parkinson’s disease. J Neurochem 137:331–359
Berciano J, Valldeoriola F, Ferrer I, Rumia J, Pascual J, Marin C, Rey MJ, Tolosa E (2002) Presynaptic parkinsonism in multiple system atrophy mimicking Parkinson’s disease: a clinicopathological case study. Mov Disord 17:812–816
Bereczki E, Francis PT, Howlett D, Pereira JB, Hoglund K, Bogstedt A, Cedazo-Minguez A, Baek JH, Hortobagyi T, Attems J, Ballard C, Aarsland D (2016) Synaptic proteins predict cognitive decline in Alzheimer’s disease and Lewy body dementia. Alzheimers Dement 12:1149–1158
Berg D, Postuma RB, Bloem B, Chan P, Dubois B, Gasser T, Goetz CG, Halliday GM, Hardy J, Lang AE, Litvan I, Marek K, Obeso J, Oertel W, Olanow CW, Poewe W, Stern M, Deuschl G (2014) Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson’s disease. Mov Disord 29:454–462
Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F (1973) Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci 20:415–455
Bhatia KP, Bain P, Bajaj N, Elble RJ, Hallett M, Louis ED, Raethjen J, Stamelou M, Testa CM, Deuschl G (2018) Consensus Statement on the classification of tremors from the task force on tremor of the International Parkinson and Movement Disorder Society. Mov Disord 33:75–87
Bhattacharjee P, Ohrfelt A, Lashley T, Blennow K, Brinkmalm A, Zetterberg H (2019) Mass spectrometric analysis of Lewy body-enriched alpha-synuclein in Prkinson’s disease. J Proteome Res 18:2109–2120
Bieri G, Brahic M, Bousset L, Couthouis J, Kramer NJ, Ma R, Nakayama L, Monbureau M, Defensor E, Schule B, Shamloo M, Melki R, Gitler AD (2019) LRRK2 modifies alpha-syn pathology and spread in mouse models and human neurons. Acta Neuropathol 137:961–980
Birdi S, Rajput AH, Fenton M, Donat JR, Rozdilsky B, Robinson C, Macaulay R, George D (2002) Progressive supranuclear palsy diagnosis and confounding features: report on 16 autopsied cases. Mov Disord 17:1255–1264
Bitan G, Dutta S, Del Rosario I, Paul K, Palma JA, Perlman SL, Poon WW, Kaufmann H, Fogel BL, Bronstein JM, Ritz B (2019) Alpha-synuclein in brain-derived exosomes distinguishes Parkinson’s disease from multiple system atrophy (abstr.). AD/PD Conference 2019, Lisbon 14th Int. Conf. on Alzheimer’s & Parkinson’s Disease, March 26–31, 2019, Lisbon. https://cmoffice.kenes.com/cmsearchableprogrammeV15/conferencemanager/programme/personid/anonymous/abstractdetails/0000253720
Bizzarri JV, Giupponi G, Maniscalco I, Schroffenegger P, Conca A, Kapfhammer HP (2015) Parkinson’s disease and psychoses. Neuropsychiatry 29:1–13
Blandini F, Cilia R, Cerri S, Pezzoli G, Schapira AHV, Mullin S, Lanciego JL (2018) Glucocerebrosidase mutations and synucleinopathies: toward a model of precision medicine. Mov Disord 34:9–21
Blauwendraat C, Heilbron K, Vallerga CL, Bandres-Ciga S, von Coelln R, Pihlstrom L, Simon-Sanchez J, Schulte C, Sharma M, Krohn L, Siitonen A, Iwaki H, Leonard H, Noyce AJ, Tan M, Gibbs JR, Hernandez DG, Scholz SW, Jankovic J, Shulman LM, Lesage S, Corvol JC, Brice A, van Hilten JJ, Marinus J, Eerola-Rautio J, Tienari P, Majamaa K, Toft M, Grosset DG, Gasser T, Heutink P, Shulman JM, Wood N, Hardy J, Morris HR, Hinds DA, Gratten J, Visscher PM, Gan-Or Z, Nalls MA, Singleton AB (2019) Parkinson’s disease age at onset genome-wide association study: defining heritability, genetic loci, and alpha-synuclein mechanisms. Mov Disord. https://doi.org/10.1002/mds.27659
Bleasel JM, Wong JH, Halliday GM, Kim WS (2014) Lipid dysfunction and pathogenesis of multiple system atrophy. Acta Neuropathol Commun 2:15
Bleasel JM, Halliday GM, Kim WS (2016) Animal modeling an oligodendrogliopathy–multiple system atrophy. Acta Neuropathol Commun 4:12
Bodea LG, Eckert A, Ittner LM, Piguet O, Gotz J (2016) Tau physiology and pathomechanisms in frontotemporal lobar degeneration. J Neurochem 138(Suppl 1):71–94
Bohnen NI, Albin RL, Muller ML, Petrou M, Kotagal V, Koeppe RA, Scott PJ, Frey KA (2015) Frequency of cholinergic and caudate nucleus dopaminergic deficits across the predemented cognitive spectrum of Parkinson disease and evidence of interaction effects. JAMA Neurol 72:194–200
Bohnen NI, Muller M, Frey KA (2017) Molecular imaging and updated diagnostic criteria in Lewy body dementias. Curr Neurol Neurosci Rep 17:73
Bohnen NI, Kanel P, Zhou Z, Koeppe RA, Frey KA, Dauer WT, Albin RL, Müller MLTM (2019) Cholinergic system changes of falls and freezing of gait in Parkinson disease. Ann Neurol 85:538–549
Book A, Guella I, Candido T, Brice A, Hattori N, Jeon B, Farrer MJ (2018) A meta-analysis of alpha-synuclein multiplication in familial parkinsonism. Front Neurol 9:1021
Bordia T, Perez XA (2019) Cholinergic control of striatal neurons to modulate l-dopa-induced dyskinesias. Eur J Neurosci 49:859–868
Borroto-Escuela DO, Perez De La Mora M, Manger P, Narvaez M, Beggiato S, Crespo-Ramirez M, Navarro G, Wydra K, Diaz-Cabiale Z, Rivera A, Ferraro L, Tanganelli S, Filip M, Franco R, Fuxe K (2018) Brain dopamine transmission in health and Parkinson’s disease: modulation of synaptic transmission and plasticity through volume transmission and dopamine heteroreceptors. Front Synaptic Neurosci 10:20
Bostan AC, Strick PL (2018) The basal ganglia and the cerebellum: nodes in an integrated network. Nat Rev Neurosci 19:338–350
Botsford E, George J, Buckley EE (2018) Parkinson’s disease and metal storage disorders: a systematic review. Brain Sci 8:194. https://doi.org/10.3390/brainsci8110194
Bourdenx M, Koulakiotis NS, Sanoudou D, Bezard E, Dehay B, Tsarbopoulos A (2017) Protein aggregation and neurodegeneration in prototypical neurodegenerative diseases: examples of amyloidopathies, tauopathies and synucleinopathies. Prog Neurobiol 155:171–193
Bove C, Travagli RA (2019) Neurophysiology of the brain stem in Parkinson’s disease. J Neurophysiol 121:1856–1864
Braak H, Del Tredici K (2008) Nervous system pathology in sporadic Parkinson disease. Neurology 70:1916–1925
Braak H, Del Tredici K (2009) Neuroanatomy and pathology of sporadic Parkinson’s disease. Adv Anat Embryol Cell Biol 201:1–119
Braak H, Del Tredici K (2017) Neuropathological staging of brain pathology in sporadic Parkinson’s disease: separating the wheat from the chaff. J Parkinsons Dis 7:S71–S85
Braak H, Bohl JR, Muller CM, Rub U, de Vos RA, Del Tredici K (2006) Stanley Fahn Lecture 2005: the staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov Disord 21:2042–2051
Breen DP, Halliday GM, Lang AE (2019) Gut-brain axis and the spread of alpha-synuclein pathology: Vagal highway or dead end? Mov Disord 34:307–316
Brettschneider J, Irwin DJ, Boluda S, Byrne MD, Fang L, Lee EB, Robinson JL, Suh E, Van Deerlin VM, Toledo JB, Grossman M, Hurtig H, Dengler R, Petri S, Lee VM, Trojanowski JQ (2017) Progression of alpha-synuclein pathology in multiple system atrophy of the cerebellar type. Neuropathol Appl Neurobiol 43:315–329
Brettschneider J, Suh E, Robinson JL, Fang L, Lee EB, Irwin DJ, Grossman M, Van Deerlin VM, Lee VM, Trojanowski JQ (2018) Converging patterns of alpha-synuclein pathology in multiple system atrophy. J Neuropathol Exp Neurol 77:1005–1016
Bridi JC, Hirth F (2018) Mechanisms of alpha-synuclein induced synaptopathy in Parkinson’s disease. Front Neurosci 12:80
Bringmann G, God R, Feineis D, Wesemann W, Riederer P, Rausch WD, Reichmann H, Sontag KH (1995) The TaClo concept: 1-trichloromethyl-1,2,3,4-tetrahydro-beta-carboline (TaClo), a new toxin for dopaminergic neurons. J Neural Transm Suppl 46:235–244
Brockmann K, Srulijes K, Pflederer S, Hauser AK, Schulte C, Maetzler W, Gasser T, Berg D (2015) GBA-associated Parkinson’s disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord 30:407–411
Brooks JA, Houlden H, Melchers A, Islam AJ, Ding J, Li A, Paudel R, Revesz T, Holton JL, Wood N, Lees A, Singleton AB, Scholz SW (2011) Mutational analysis of parkin and PINK1 in multiple system atrophy. Neurobiol Aging 32(548):e545–e547
Brück D, Wenning GK, Stefanova N, Fellner L (2016) Glia and alpha-synuclein in neurodegeneration: A complex interaction. Neurobiol Dis 85:262–274
Brudek T, Winge K, Rasmussen NB, Bahl JM, Tanassi J, Agander TK, Hyde TM, Pakkenberg B (2016) Altered alpha-synuclein, parkin, and synphilin isoform levels in multiple system atrophy brains. J Neurochem 136:172–185
Brundin P, Melki R, Kopito R (2010) Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol 11:301–307
Bu J, Liu J, Liu K, Wang Z (2019) Diagnostic utility of gut alpha-synuclein in Parkinson’s disease: a systematic review and meta-analysis. Behav Brain Res 364:340–347
Buchman AS, Nag S, Leurgans SE, Miller J, VanderHorst V, Bennett DA, Schneider JA (2018) Spinal Lewy body pathology in older adults without an antemortem diagnosis of Parkinson’s disease. Brain Pathol 28:560–568
Burke RE, Dauer WT, Vonsattel JP (2008) A critical evaluation of the Braak staging scheme for Parkinson’s disease. Ann Neurol 64:485–491
Burre J, Sharma M, Sudhof TC (2018) Cell biology and pathophysiology of alpha-synuclein. Cold Spring Harb Perspect Med. https://doi.org/10.1101/cshperspect.a024091
Cairns NJ, Perrin RJ, Franklin EE, Carter D, Vincent B, Xie M, Bateman RJ, Benzinger T, Friedrichsen K, Brooks WS, Halliday GM, McLean C, Ghetti B, Morris JC (2015) Neuropathologic assessment of participants in two multi-center longitudinal observational studies: the Alzheimer Disease Neuroimaging Initiative (ADNI) and the Dominantly Inherited Alzheimer Network (DIAN). Neuropathology 35:390–400
Calabresi P, Mercuri NB, Di Filippo M (2009) Synaptic plasticity, dopamine and Parkinson’s disease: one step ahead. Brain 132:285–287
Calafate S, Buist A, Miskiewicz K, Vijayan V, Daneels G, de Strooper B, de Wit J, Verstreken P, Moechars D (2015) Synaptic contacts enhance cell-to-cell tau pathology propagation. Cell Rep 11:1176–1183
Callizot N, Combes M, Henriques A, Poindron P (2019) Necrosis, apoptosis, necroptosis, three modes of action of dopaminergic neuron neurotoxins. PLoS One 14:e0215277
Calo L, Wegrzynowicz M, Santivanez-Perez J, Grazia Spillantini M (2016) Synaptic failure and alpha-synuclein. Mov Disord 31:169–177
Caminiti SP, Sala A, Iaccarino L, Beretta L, Pilotto A, Gianolli L, Iannaccone S, Magnani G, Padovani A, Ferini-Strambi L, Perani D (2019) Brain glucose metabolism in Lewy body dementia: implications for diagnostic criteria. Alzheimers Res Ther 11:20
Campbell BC, McLean CA, Culvenor JG, Gai WP, Blumbergs PC, Jakala P, Beyreuther K, Masters CL, Li QX (2001) The solubility of alpha-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson’s disease. J Neurochem 76:87–96
Candelise N, Schmitz M, Llorens F, Villar-Pique A, Cramm M, Thom T, da Silva Correia SM, Gomes Eriton, da Cunha J, Mobius W, Outeiro TF, Alvarez VG, Banchelli M, D’Andrea C, de Angelis M, Zafar S, Rabano A, Matteini P, Zerr I (2019) Seeding variability of different alpha synuclein strains in synucleinopathies. Ann Neurol 85:691–703
Canerina-Amaro A, Pereda D, Diaz M, Rodriguez-Barreto D, Casanas-Sanchez V, Heffer M, Garcia-Esparcia P, Ferrer I, Puertas-Avendano R, Marin R (2019) Differential aggregation and phosphorylation of alpha synuclein in membrane compartments associated with Parkinson disease. Front Neurosci 13:382
Cazorla M, Kang UJ, Kellendonk C (2015) Balancing the basal ganglia circuitry: a possible new role for dopamine D2 receptors in health and disease. Mov Disord 30:895–903
Cenci MA, Crossman AR (2018) Animal models of l-dopa-induced dyskinesia in Parkinson’s disease. Mov Disord 33:889–899
Cenci MA, Jorntell H, Petersson P (2018) On the neuronal circuitry mediating l-DOPA-induced dyskinesia. J Neural Transm (Vienna) 125:1157–1169
Cersosimo MG (2018) Propagation of alpha-synuclein pathology from the olfactory bulb: possible role in the pathogenesis of dementia with Lewy bodies. Cell Tissue Res 373:233–243
Cersosimo MG, Benarroch EE (2012a) Autonomic involvement in Parkinson’s disease: pathology, pathophysiology, clinical features and possible peripheral biomarkers. J Neurol Sci 313:57–63
Cersosimo MG, Benarroch EE (2012b) Pathological correlates of gastrointestinal dysfunction in Parkinson’s disease. Neurobiol Dis 46:559–564
Chang A, Fox SH (2016) Psychosis in parkinson’s disease: epidemiology, pathophysiology, and management. Drugs 76:1093–1118
Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, Kerchner GA, Ayalon G, Bingol B, Sheng M, Hinds D, Behrens TW, Singleton AB, Bhangale TR, Graham RR (2017) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 49:1511–1516
Chartier S, Duyckaerts C (2018) Is Lewy pathology in the human nervous system chiefly an indicator of neuronal protection or of toxicity? Cell Tissue Res 373:149–160
Chavarria C, Rodriguez-Bottero S, Quijano C, Cassina P, Souza JM (2018) Impact of monomeric, oligomeric and fibrillar alpha-synuclein on astrocyte reactivity and toxicity to neurons. Biochem J 475:3153–3169
Chelban V, Manole A, Pihlstrom L, Schottlaender L, Efthymiou S, Oconnor E, Meissner WG, Holton JL, Houlden H (2017) Analysis of the prion protein gene in multiple system atrophy. Neurobiol Aging 49:216 e15–216 e18
Chen YF, Tseng YL, Lan MY, Lai SL, Su CS, Liu JS, Chang YY (2014) The relationship of leukoaraiosis and the clinical severity of vascular parkinsonism. J Neurol Sci 346:255–259
Chen ZC, Zhang W, Chua LL, Chai C, Li R, Lin L, Cao Z, Angeles DC, Stanton LW, Peng JH, Zhou ZD, Lim KL, Zeng L, Tan EK (2017) Phosphorylation of amyloid precursor protein by mutant LRRK2 promotes AICD activity and neurotoxicity in Parkinson’s disease. Sci Signal. https://doi.org/10.1126/scisignal.aam6790
Chen C, Turnbull DM, Reeve AK (2019a) Mitochondrial dysfunction in Parkinson’s disease-cause or consequence? Biology (Basel) 8:38
Chen Z, Chen JA, Shatunov A, Jones AR, Kravitz SN, Huang AY, Lawrence L, Lowe JK, Lewis CM, Payan CAM, Lieb W, Franke A, Deloukas P, Amouyel P, Tzourio C, Dartigues JF, Ludolph A, Bensimon G, Leigh PN, Bronstein JM, Coppola G, Geschwind DH, Al-Chalabi A (2019b) Genome-wide survey of copy number variants finds MAPT duplications in progressive supranuclear palsy. Mov Disord
Cheng HC, Ulane CM, Burke RE (2010) Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 67:715–725
Chiu CC, Lu CS, Weng YH, Chen YL, Huang YZ, Chen RS, Cheng YC, Huang YC, Liu YC, Lai SC, Lin KJ, Lin YW, Chen YJ, Chen CL, Yeh TH, Wang HL (2019) PARK14 (D331Y) PLA2G6 causes early-onset degeneration of substantia nigra dopaminergic neurons by inducing mitochondrial dysfunction, er stress, mitophagy impairment and transcriptional dysregulation in a knockin mouse model. Mol Neurobiol 56:3835–3853
Christopher L, Duff-Canning S, Koshimori Y, Segura B, Boileau I, Chen R, Lang AE, Houle S, Rusjan P, Strafella AP (2015) Salience network and parahippocampal dopamine dysfunction in memory-impaired Parkinson disease. Ann Neurol 77:269–280
Chu Y, Morfini GA, Langhamer LB, He Y, Brady ST, Kordower JH (2012) Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain 135:2058–2073
Chu HY, Atherton JF, Wokosin D, Surmeier DJ, Bevan MD (2015) Heterosynaptic regulation of external globus pallidus inputs to the subthalamic nucleus by the motor cortex. Neuron 85:364–376
Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, Probst A, Winkler DT, Reichwald J, Staufenbiel M, Ghetti B, Goedert M, Tolnay M (2013) Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA 110:9535–9540
Colloby SJ, McParland S, O’Brien JT, Attems J (2012) Neuropathological correlates of dopaminergic imaging in Alzheimer’s disease and Lewy body dementias. Brain 135:2798–2808
Colom-Cadena M, Gelpi E, Charif S, Belbin O, Blesa R, Marti MJ, Clarimon J, Lleo A (2013a) Confluence of alpha-synuclein, tau, and beta-amyloid pathologies in dementia with Lewy bodies. J Neuropathol Exp Neurol 72:1203–1212
Colom-Cadena M, Gelpi E, Marti MJ, Charif S, Dols-Icardo O, Blesa R, Clarimon J, Lleo A (2013b) MAPT H1 haplotype is associated with enhanced alpha-synuclein deposition in dementia with Lewy bodies. Neurobiol Aging 34:936–942
Colom-Cadena M, Grau-Rivera O, Planellas L, Cerquera C, Morenas E, Helgueta S, Munoz L, Kulisevsky J, Marti MJ, Tolosa E, Clarimon J, Lleo A, Gelpi E (2017a) Regional overlap of pathologies in Lewy body disorders. J Neuropathol Exp Neurol 76:216–224
Colom-Cadena M, Pegueroles J, Herrmann AG, Henstridge CM, Munoz L, Querol-Vilaseca M, Martin-Paniello CS, Luque-Cabecerans J, Clarimon J, Belbin O, Nunez-Llaves R, Blesa R, Smith C, McKenzie CA, Frosch MP, Roe A, Fortea J, Andilla J, Loza-Alvarez P, Gelpi E, Hyman BT, Spires-Jones TL, Lleo A (2017b) Synaptic phosphorylated alpha-synuclein in dementia with Lewy bodies. Brain 140:3204–3214
Coon EA, Cutsforth-Gregory JK, Benarroch EE (2018) Neuropathology of autonomic dysfunction in synucleinopathies. Mov Disord 33:349–358
Coughlin D, Xie SX, Liang M, Williams A, Peterson C, Weintraub D, McMillan CT, Wolk DA, Akhtar RS, Hurtig HI, Branch Coslett H, Hamilton RH, Siderowf AD, Duda JE, Rascovsky K, Lee EB, Lee VM, Grossman M, Trojanowski JQ, Irwin DJ (2018) Cognitive and pathological influences of tau pathology in Lewy body disorders. Ann Neurol 85:259–271
Coughlin DG, Petrovitch H, White LR, Noorigian JV, Masaki K, Webster RG, Duda JE (2019) Most cases with Lewy pathology in a population-based cohort adhere to the Braak progression pattern but ‘failure to fit’ is highly dependent on staging system applied. Parkinsonism Relat Disord. https://doi.org/10.1016/j.parkreldis.2019.1003.1023
Cousins O, Yousaf T, Wilson H, Pagano G, Politis M (2019) Molecular imaging of dementia with Lewy bodies. Int Rev Neurobiol 144:59–93
Covell DJ, Robinson JL, Akhtar RS, Grossman M, Weintraub D, Bucklin HM, Pitkin RM, Riddle D, Yousef A, Trojanowski JQ, Lee VM (2017) Novel conformation-selective alpha-synuclein antibodies raised against different in vitro fibril forms show distinct patterns of Lewy pathology in Parkinson’s disease. Neuropathol Appl Neurobiol 43:604–620
Coyle-Gilchrist IT, Dick KM, Patterson K, Vazquez Rodriquez P, Wehmann E, Wilcox A, Lansdall CJ, Dawson KE, Wiggins J, Mead S, Brayne C, Rowe JB (2016) Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology 86:1736–1743
Critchley M (1981) Arteriosclerotic pseudoparkinsonism. In: Rose FC, Capildeo R (eds) Research Progress in Parkinson’s disease. Pitman, London
Cykowski MD, Coon EA, Powell SZ, Jenkins SM, Benarroch EE, Low PA, Schmeichel AM, Parisi JE (2015) Expanding the spectrum of neuronal pathology in multiple system atrophy. Brain 138:2293–2309
Daher JP (2017) Interaction of LRRK2 and alpha-synuclein in Parkinson’s disease. Adv Neurobiol 14:209–226
Dale GE, Probst A, Luthert P, Martin J, Anderton BH, Leigh PN (1992) Relationships between Lewy bodies and pale bodies in Parkinson’s disease. Acta Neuropathol (Berl) 83:525–529
Das T, Eliezer D (2019) Membrane interactions of intrinsically disordered proteins: the example of alpha-synuclein. Biochim Biophys Acta Proteins Proteomics. https://doi.org/10.1016/j.bbapap.2019.1005.1001
Dash D, Pandey S (2019) Movement disorders associated with neuronal antibodies. Acta Neurol Scand 139:106–117
Davis MY, Johnson CO, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, Van Deerlin VM, Quinn JF, Chung KA, Peterson-Hiller AL, Rosenthal LS, Dawson TM, Albert MS, Goldman JG, Stebbins GT, Bernard B, Wszolek ZK, Ross OA, Dickson DW, Eidelberg D, Mattis PJ, Niethammer M, Yearout D, Hu SC, Cholerton BA, Smith M, Mata IF, Montine TJ, Edwards KL, Zabetian CP (2016) Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol 73:1217–1224
Davis AA, Leyns CEG, Holtzman DM (2018) Intercellular spread of protein aggregates in neurodegenerative disease. Annu Rev Cell Dev Biol 34:545–568
De Cecco E, Legname G (2018) The role of the prion protein in the internalization of alpha-synuclein amyloids. Prion 12:23–27
de Iure A, Napolitano F, Beck G, Quiroga Varela A, Durante V, Sciaccaluga M, Mazzocchetti P, Megaro A, Tantucci M, Cardinale A, Punzo D, Mancini A, Costa C, Ghiglieri V, Tozzi A, Picconi B, Papa SM, Usiello A, Calabresi P (2019) Striatal spreading depolarization: Possible implication in levodopa-induced dyskinetic-like behavior. Mov Disord. https://doi.org/10.1002/mds.27632
de la Fuente-Fernandez R, Schulzer M, Kuramoto L, Cragg J, Ramachandiran N, Au WL, Mak E, McKenzie J, McCormick S, Sossi V, Ruth TJ, Lee CS, Calne DB, Stoessl AJ (2011) Age-specific progression of nigrostriatal dysfunction in Parkinson’s disease. Ann Neurol 69:803–810
De Pablo-Fernandez E, Breen DP, Bouloux PM, Barker RA, Foltynie T, Warner TT (2017) Neuroendocrine abnormalities in Parkinson’s disease. J Neurol Neurosurg Psychiatry 88:176–185
De Pablo-Fernandez E, Cerdan Santacruz D, Warner TT, Holton JL (2018) No evidence of iatrogenic human transmission in autopsy confirmed multiple system atrophy. Mov Disord 33:1183–1184
De Reuck J, Auger F, Durieux N, Cordonnier C, Maurage CA, Deramecourt V, Pasquier F, Leys D, Bordet R (2018) The impact of cerebral amyloid angiopathy in Lewy body dementia: a neuropathological study with magnetic resonance imaging correlations. J Neurodegener Dis Res 1:101
Dehay B (2014) Evidence piles up for prion-like propagation mechanisms in synucleinopathies. Mov Disord 29:187
Dehay B, Fernagut PO (2016) Alpha-synuclein-based models of Parkinson’s disease. Rev Neurol (Paris) 172:371–378
Del Rey NL, Quiroga-Varela A, Garbayo E, Carballo-Carbajal I, Fernandez-Santiago R, Monje MHG, Trigo-Damas I, Blanco-Prieto MJ, Blesa J (2018) Advances in Parkinson’s disease: 200 years later. Front Neuroanat 12:113
Del Tredici K, Braak H (2012) Spinal cord lesions in sporadic Parkinson’s disease. Acta Neuropathol 124:643–664
Del Tredici K, Braak H (2013) Dysfunction of the locus coeruleus-norepinephrine system and related circuitry in Parkinson’s disease-related dementia. J Neurol Neurosurg Psychiatry 84:774–783
Delacourte A, Robitaille Y, Sergeant N, Buee L, Hof PR, Wattez A, Laroche-Cholette A, Mathieu J, Chagnon P, Gauvreau D (1996) Specific pathological Tau protein variants characterize Pick’s disease. J Neuropathol Exp Neurol 55:159–168
DelleDonne A, Klos KJ, Fujishiro H, Ahmed Z, Parisi JE, Josephs KA, Frigerio R, Burnett M, Wszolek ZK, Uitti RJ, Ahlskog JE, Dickson DW (2008) Incidental Lewy body disease and preclinical Parkinson disease. Arch Neurol 65:1074–1080
Deramecourt V, Bombois S, Maurage CA, Ghestem A, Drobecq H, Vanmechelen E, Lebert F, Pasquier F, Delacourte A (2006) Biochemical staging of synucleinopathy and amyloid deposition in dementia with Lewy bodies. J Neuropathol Exp Neurol 65:278–288
Dettmer U, Selkoe D, Bartels T (2016) New insights into cellular alpha-synuclein homeostasis in health and disease. Curr Opin Neurobiol 36:15–22
Dhillon JS, Trejo-Lopez JA, Riffe C, Levites Y, Sacino AN, Borchelt DR, Yachnis AY, Giasson BI (2019a) Comparative analyses of the in vivo induction and transmission of alpha-synuclein pathology in transgenic mice by MSA brain lysate and recombinant alpha-synuclein fibrils. Acta Neuropathol Commun 7:80
Dhillon JS, Trejo-Lopez JA, Riffe C, McFarland NR, Hiser WM, Giasson BI, Yachnis AT (2019b) Dissecting alpha-synuclein inclusion pathology diversity in multiple system atrophy: implications for the prion-like transmission hypothesis. Lab Invest. https://doi.org/10.1038/s41374-41019-40198-41379
di Domenico A, Carola G, Calatayud C, Pons-Espinal M, Munoz JP, Richaud-Patin Y, Fernandez-Carasa I, Gut M, Faella A, Parameswaran J, Soriano J, Ferrer I, Tolosa E, Zorzano A, Cuervo AM, Raya A, Consiglio A (2019) Patient-specific IPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson’s disease. Stem Cell Rep 12:213–229
Dickson DW (2012) Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med 2:a009258
Dickson DW (2018) Neuropathology of Parkinson disease. Parkinsonism Relat Disord 46(Suppl 1):S30–S33
Dickson DW, Fujishiro H, Delledonne A, Menke J, Ahmed Z, Klos KJ, Josephs KA, Frigerio R, Burnett M, Parisi JE, Ahlskog JE (2008) Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol 115:437–444
Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, Hardy J, Leverenz JB, Del Tredici K, Wszolek ZK, Litvan I (2009) Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol 8:1150–1157
Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA (2010a) Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol 23:394–400
Dickson DW, Uchikado H, Fujishiro H, Tsuboi Y (2010b) Evidence in favor of Braak staging of Parkinson’s disease. Mov Disord 25(Suppl 1):S78–S82
Dickson DW, Hauw JJ, Agid Y, Litvan I (2011a) Progressive supranuclear palsy and corticobasal degeneration. In: Dickson DW, Weller RO (eds) Neurodegeneration: the molecular pathology of dementia and movement disorders, 2nd edn. Blackwell Publishing Ltd., Oxford, pp 135–155
Dickson DW, Kouri N, Murray ME, Josephs KA (2011b) Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J Mol Neurosci 45:384–389
Dickson DW, Heckman MG, Murray ME, Soto AI, Walton RL, Diehl NN, van Gerpen JA, Uitti RJ, Wszolek ZK, Ertekin-Taner N, Knopman DS, Petersen RC, Graff-Radford NR, Boeve BF, Bu G, Ferman TJ, Ross OA (2018) APOE epsilon4 is associated with severity of Lewy body pathology independent of Alzheimer pathology. Neurology 91:e1182–e1195
Dijkstra AA, Voorn P, Berendse HW, Groenewegen HJ, Rozemuller AJ, van de Berg WD (2014) Stage-dependent nigral neuronal loss in incidental Lewy body and Parkinson’s disease. Mov Disord 29:1244–1251
Dirkx MF, den Ouden HE, Aarts E, Timmer MH, Bloem BR, Toni I, Helmich RC (2017) Dopamine controls Parkinson’s tremor by inhibiting the cerebellar thalamus. Brain 140:721–734
Djaldetti R, Lorberboym M, Karmon Y, Treves TA, Ziv I, Melamed E (2011) Residual striatal dopaminergic nerve terminals in very long-standing Parkinson’s disease: a single photon emission computed tomography imaging study. Mov Disord 26:327–330
Dolgacheva LP, Berezhnov AV, Fedotova EI, Zinchenko VP, Abramov AY (2019) Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J Bioenerg Biomembr 51:175–188
Don AS, Hsiao JH, Bleasel JM, Couttas TA, Halliday GM, Kim W (2014) Altered lipid levels provide evidence for myelin dysfunction in multiple system atrophy. Acta Neuropathol Commun 2:150
Donadio V (2018) Skin nerve alpha-synuclein deposits in Parkinson’s disease and other synucleinopathies: a review. Clin Auton Res. https://doi.org/10.1007/s10286-10018-10581-10284
Donadio V, Doppler K, Incensi A, Kuzkina A, Janzen A, Mayer G, Volkmann J, Rizzo G, Antelmi E, Plazzi G, Sommer C, Liguori R, Oertel WH (2019) Abnormal alpha-synuclein deposits in skin nerves: intra- and inter-laboratory reproducibility. Eur J Neurol. https://doi.org/10.1111/ene.13939
Donaghy PC, McKeith I (2014) The clinical characteristics of dementia with Lewy bodies and a consideration of prodromal diagnosis. Alzheimers Res Ther 6:46
Donker Kaat L, Boon AJ, Azmani A, Kamphorst W, Breteler MM, Anar B, Heutink P, van Swieten JC (2009) Familial aggregation of parkinsonism in progressive supranuclear palsy. Neurology 73:98–105
Doppler K, Weis J, Karl K, Ebert S, Ebentheuer J, Trenkwalder C, Klebe S, Volkmann J, Sommer C (2015) Distinctive distribution of phospho-alpha-synuclein in dermal nerves in multiple system atrophy. Mov Disord 30:1688–1692
Dorsey ER, Sherer T, Okun MS, Bloem BR (2018) The emerging evidence of the Parkinson pandemic. J Parkinsons Dis 8:S3–S8
Double KL, Reyes S, Werry EL, Halliday GM (2010) Selective cell death in neurodegeneration: why are some neurons spared in vulnerable regions? Prog Neurobiol 92:316–329
Driver-Dunckley ED, Zhang N, Adler CH, Serrano GE, Sue LI, Shill H, Mehta SH, Belden CM, Zamrini E, Davis K, Beach T (2019) Brain Lewy-type synucleinopathy density is associated with a lower prevalence of atherosclerotic cardiovascular disease risk factors in patients with Parkinson’s disease. J Parkinsons Dis (in print)
Dubois B, Burn D, Goetz C, Aarsland D, Brown RG, Broe GA, Dickson D, Duyckaerts C, Cummings J, Gauthier S, Korczyn A, Lees A, Levy R, Litvan I, Mizuno Y, McKeith IG, Olanow CW, Poewe W, Sampaio C, Tolosa E, Emre M (2007) Diagnostic procedures for Parkinson’s disease dementia: recommendations from the movement disorder society task force. Mov Disord 22:2314–2324
Duffy MF, Collier TJ, Patterson JR, Kemp CJ, Luk KC, Tansey MG, Paumier KL, Kanaan NM, Fischer DL, Polinski NK, Barth OL, Howe JW, Vaikath NN, Majbour NK, El-Agnaf OMA, Sortwell CE (2018) Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. J Neuroinflamm 15:129
Dugger BN, Dickson DW (2010) Cell type specific sequestration of choline acetyltransferase and tyrosine hydroxylase within Lewy bodies. Acta Neuropathol 120:633–639
Dugger BN, Hoffman BR, Scroggins A, Serrano GE, Adler CH, Shill HA, Belden CM, Sabbagh MN, Caviness JN, Driver Dunckley E, Beach TG (2018) Tau immunoreactivity in peripheral tissues of human aging and select tauopathies. Neurosci Lett 696:132–139
Durcan R, Donaghy P, Osborne C, Taylor JP, Thomas AJ (2019) Imaging in prodromal dementia with Lewy bodies: where do we stand? Int J Geriatr Psychiatry 34:635–646
Duyckaerts C, Clavaguera F, Potier MC (2019) The prion-like propagation hypothesis in Alzheimer’s and Parkinson’s disease. Curr Opin Neurol 32:266–271
Dzamko N, Gysbers AM, Bandopadhyay R, Bolliger MF, Uchino A, Zhao Y, Takao M, Wauters S, van de Berg WD, Takahashi-Fujigasaki J, Nichols RJ, Holton JL, Murayama S, Halliday GM (2017) LRRK2 levels and phosphorylation in Parkinson’s disease brain and cases with restricted Lewy bodies. Mov Disord 32:423–432
Ebashi M, Ito Y, Uematsu M, Nakamura A, Hirokawa K, Kamei S, Uchihara T (2019) How to demix Alzheimer-type and PSP-type tau lesions out of their mixture-hybrid approach to dissect comorbidity. Acta Neuropathol Commun 7:71
Ekman U, Eriksson J, Forsgren L, Mo SJ, Riklund K, Nyberg L (2012) Functional brain activity and presynaptic dopamine uptake in patients with Parkinson’s disease and mild cognitive impairment: a cross-sectional study. Lancet Neurol 11:679–687
Elder GJ, Mactier K, Colloby SJ, Watson R, Blamire AM, O’Brien JT, Taylor JP (2017) The influence of hippocampal atrophy on the cognitive phenotype of dementia with Lewy bodies. Int J Geriatr Psychiatry 32:1182–1189
Elias S, Israel Z, Bergman H (2008) Physiology of Parkinson’s disease. In: Hallett M, Poewe W (eds) Therapeutics of Parkinson’s disease and other movement disorders. Wiley-Blackwell, New York, pp 25–36
Emmanouilidou E, Vekrellis K (2016) Exocytosis and spreading of normal and aberrant alpha-synuclein. Brain Pathol 26:398–403
Emre M, Aarsland D, Brown R, Burn DJ, Duyckaerts C, Mizuno Y, Broe GA, Cummings J, Dickson DW, Gauthier S, Goldman J, Goetz C, Korczyn A, Lees A, Levy R, Litvan I, McKeith I, Olanow W, Poewe W, Quinn N, Sampaio C, Tolosa E, Dubois B (2007) Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord 22:1689–1707
Engeln M, De Deurwaerdere P, Li Q, Bezard E, Fernagut PO (2015) Widespread monoaminergic dysregulation of both motor and non-motor circuits in parkinsonism and dyskinesia. Cereb Cortex 25:2783–2792
Erro R, Picillo M, Amboni M, Savastano R, Scannapieco S, Cuoco S, Santangelo G, Vitale C, Pellecchia MT, Barone P (2019) Comparing postural instability and gait disorder and akinetic-rigid subtyping of Parkinson disease and their stability over time. Eur J Neurol 1:5. https://doi.org/10.1111/ene.13968
Erskine D, Thomas AJ, Taylor JP, Savage MA, Attems J, McKeith IG, Morris CM, Khundakar AA (2017) Neuronal loss and alpha-synuclein pathology in the superior colliculus and its relationship to visual hallucinations in dementia with Lewy bodies. Am J Geriatr Psychiatry 25:595–604
Erskine D, Ding J, Thomas AJ, Kaganovich A, Khundakar AA, Hanson PS, Taylor JP, McKeith IG, Attems J, Cookson MR, Morris CM (2018) Molecular changes in the absence of severe pathology in the pulvinar in dementia with Lewy bodies. Mov Disord 33:982–991
Espay AJ, LeWitt PA, Kaufmann H (2014) Norepinephrine deficiency in Parkinson’s disease: the case for noradrenergic enhancement. Mov Disord 29:1710–1719
Espay AJ, Vizcarra JA, Marsili L, Lang AE, Simon DK, Merola A, Josephs KA, Fasano A, Morgante F, Savica R, Greenamyre JT, Cambi F, Yamasaki TR, Tanner CM, Gan-Or Z, Litvan I, Mata IF, Zabetian CP, Brundin P, Fernandez HH, Standaert DG, Kauffman MA, Schwarzschild MA, Sardi SP, Sherer T, Perry G, Leverenz JB (2019) Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases. Neurology 92:329–337
Evans T, Kok WL, Cowan K, Hefford M, Anichtchik O (2018) Accumulation of beta-synuclein in cortical neurons is associated with autophagy attenuation in the brains of dementia with Lewy body patients. Brain Res 1681:1–13
Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, Crowther RA, Ghetti B, Scheres SHW, Goedert M (2018) Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 561:137–140
Fanciulli A, Wenning GK (2015) Multiple-system atrophy. N Engl J Med 372:249–263
Fathy YY, Jonker AJ, Oudejans E, de Jong FJJ, van Dam AW, Rozemuller AJM, van de Berg WDJ (2019) Differential insular cortex subregional vulnerability to alpha-synuclein pathology in Parkinson’s disease and dementia with Lewy bodies. Neuropathol Appl Neurobiol 45:262–277
Feany MB, Bender WW (2000) A Drosophila model of Parkinson’s disease. Nature 404:394–398
Federoff M, Price TR, Sailer A, Scholz S, Hernandez D, Nicolas A, Singleton AB, Nalls M, Houlden H (2016) Genome-wide estimate of the heritability of multiple system atrophy. Parkinsonism Relat Disord 22:35–41
Felice VD, Quigley EM, Sullivan AM, O’Keeffe GW, O’Mahony SM (2016) Microbiota-gut-brain signalling in Parkinson’s disease: Implications for non-motor symptoms. Parkinsonism Relat Disord 27:1–8
Fellner L, Jellinger KA, Wenning GK, Stefanova N (2011) Glial dysfunction in the pathogenesis of alpha-synucleinopathies: emerging concepts. Acta Neuropathol 121:675–693
Fellner L, Wenning GK, Stefanova N (2015) Models of multiple system atrophy. Curr Top Behav Neurosci 22:369–393
Fellner L, Buchinger E, Brueck D, Irschick R, Wenning GK, Stefanova N (2018) Limited effects of dysfunctional macroautophagy on the accumulation of extracellularly derived alpha-synuclein in oligodendroglia: implications for MSA pathogenesis. BMC Neurosci 19:32
Fereshtehnejad SM, Zeighami Y, Dagher A, Postuma RB (2017) Clinical criteria for subtyping Parkinson’s disease: biomarkers and longitudinal progression. Brain 140:1959–1976
Ferguson MC, Garland EM, Hedges L, Womack-Nunley B, Hamid R, Phillips JA 3rd, Shibao CA, Raj SR, Biaggioni I, Robertson D (2014) SHC2 gene copy number in multiple system atrophy (MSA). Clin Auton Res 24:25–30
Ferman TJ, Aoki N, Crook JE, Murray ME, Graff-Radford NR, van Gerpen JA, Uitti RJ, Wszolek ZK, Graff-Radford J, Pedraza O, Kantarci K, Boeve BF, Dickson DW (2018) The limbic and neocortical contribution of alpha-synuclein, tau, and amyloid beta to disease duration in dementia with Lewy bodies. Alzheimers Dement 14:330–339
Ferrari R, Kia DA, Tomkins JE, Hardy J, Wood NW, Lovering RC, Lewis PA, Manzoni C (2018) Stratification of candidate genes for Parkinson’s disease using weighted protein-protein interaction network analysis. BMC Genom 19:452
Ferreira SA, Romero-Ramos M (2018) Microglia response during Parkinson’s disease: alpha-synuclein intervention. Front Cell Neurosci 12:247
Ferrer I (2018) Oligodendrogliopathy in neurodegenerative diseases with abnormal protein aggregates: the forgotten partner. Prog Neurobiol 169:24–54
Ferrer I, Lopez-Gonzalez I, Carmona M, Arregui L, Dalfo E, Torrejon-Escribano B, Diehl R, Kovacs GG (2014) Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol 73:81–97
Ferrer I, Aguiló García M, Carmona M, Andrés-Benito P, Torrejón-Escribano B, Garcia-Esparcia P, del Rio JA (2019) Involvement of oligodendrocytes in tau seeding and spreading in tauopathies. Front Aging Neurosci 11:112
Ffytche DH, Creese B, Politis M, Chaudhuri KR, Weintraub D, Ballard C, Aarsland D (2017) The psychosis spectrum in Parkinson disease. Nat Rev Neurol 13:81–95
Filippini A, Gennarelli M, Russo I (2019) Alpha-synuclein and glia in Parkinson’s disease: a beneficial or a detrimental duet for the endo-lysosomal system? Cell Mol Neurobiol 39:161–168
Fitzgerald E, Murphy S, Martinson HA (2019) Alpha-synuclein pathology and the role of the microbiota in Parkinson’s disease. Front Neurosci 13:369
Fling BW, Cohen RG, Mancini M, Nutt JG, Fair DA, Horak FB (2013) Asymmetric pedunculopontine network connectivity in parkinsonian patients with freezing of gait. Brain 136:2405–2418
Foguem C, Manckoundia P (2018) Lewy body disease: clinical and pathological “overlap syndrome” between synucleinopathies (Parkinson disease) and tauopathies (Alzheimer disease). Curr Neurol Neurosci Rep 18:24
Formisano R, Zasler ND (2014) Posttraumatic parkinsonism. J Head Trauma Rehabil 29:387–390
Forno LS (1996) Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol 55:259–272
Forno LS, DeLanney LE, Irwin I, Langston JW (1996) Electron microscopy of Lewy bodies in the amygdala-parahippocampal region. Comparison with inclusion bodies in the MPTP-treated squirrel monkey. Adv Neurol 69:217–228
Forrest SL, Kril JJ, Stevens CH, Kwok JB, Hallupp M, Kim WS, Huang Y, McGinley CV, Werka H, Kiernan MC, Gotz J, Spillantini MG, Hodges JR, Ittner LM, Halliday GM (2018) Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain 141:521–534
Forrest SL, Crockford DR, Sizemova A, McCann H, Shepherd CE, McGeachie AB, Affleck AJ, Carew-Jones F, Bartley L, Kwok JB, Kim WS, Jary E, Tan RH, McGinley CV, Piguet O, Hodges JR, Kril JJ, Halliday GM (2019a) Coexisting Lewy body disease and clinical parkinsonism in frontotemporal lobar degeneration. Neurology 92:e2472–e2482
Forrest SL, Halliday GM, McCann H, McGeachie AB, McGinley CV, Hodges JR, Piguet O, Kwok JB, Spillantini MG, Kril JJ (2019b) Heritability in frontotemporal tauopathies. Alzheimers Dement (Amst) 11:115–124
Foti SC, Hargreaves I, Carrington S, Kiely AP, Houlden H, Holton JL (2019) Cerebral mitochondrial electron transport chain dysfunction in multiple system atrophy and Parkinson’s disease. Sci Rep 9:6559
Francis PT, Perry EK (2007) Cholinergic and other neurotransmitter mechanisms in Parkinson’s disease, Parkinson’s disease dementia, and dementia with Lewy bodies. Mov Disord 22(Suppl 17):S351–S357
Franquet E, Salvado-Figueres M, Lorenzo-Bosquet C, Cuberas-Borros G, Rovira A, Castell-Conesa J, Hernandez-Vara J (2012) Nigrostriatal pathway dysfunction in a methanol-induced delayed dystonia-parkinsonism. Mov Disord 27:1220–1221
Freestone PS, Chung KK, Guatteo E, Mercuri NB, Nicholson LF, Lipski J (2009) Acute action of rotenone on nigral dopaminergic neurons—involvement of reactive oxygen species and disruption of Ca homeostasis. Eur J Neurosci 30:1849–1859
French IT, Muthusamy KA (2018) A review of the pedunculopontine nucleus in Parkinson’s disease. Front Aging Neurosci 10:99
Freundt EC, Maynard N, Clancy EK, Roy S, Bousset L, Sourigues Y, Covert M, Melki R, Kirkegaard K, Brahic M (2012) Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann Neurol 72:517–524
Friedman JH (2018) Dementia with Lewy bodies and Parkinson’s disease dementia become the same disease. Parkinsonism Relat Disord 46(Suppl 1):S6–S9
Frigerio R, Fujishiro H, Ahn TB, Josephs KA, Maraganore DM, Delledonne A, Parisi JE, Klos KJ, Boeve BF, Dickson DW, Ahlskog JE (2011) Incidental Lewy body disease: Do some cases represent a preclinical stage of dementia with Lewy bodies? Neurobiol Aging 32:857–863
Frisina PG, Haroutunian V, Libow LS (2009) The neuropathological basis for depression in Parkinson’s disease. Parkinsonism Relat Disord 15:144–148
Fujioka S, Ogaki K, Tacik PM, Uitti RJ, Ross OA, Wszolek ZK (2014a) Update on novel familial forms of Parkinson’s disease and multiple system atrophy. Parkinsonism Relat Disord 20(Suppl 1):S29–S34
Fujioka S, Van Gerpen JA, Uitti RJ, Dickson DW, Wszolek ZK (2014b) Familial progressive supranuclear palsy: a literature review. Neurodegener Dis 13:180–182
Fujishiro H, Ferman TJ, Boeve BF, Smith GE, Graff-Radford NR, Uitti RJ, Wszolek ZK, Knopman DS, Petersen RC, Parisi JE, Dickson DW (2008a) Validation of the neuropathologic criteria of the third consortium for dementia with Lewy bodies for prospectively diagnosed cases. J Neuropathol Exp Neurol 67:649–656
Fujishiro H, Tsuboi Y, Lin WL, Uchikado H, Dickson DW (2008b) Co-localization of tau and alpha-synuclein in the olfactory bulb in Alzheimer’s disease with amygdala Lewy bodies. Acta Neuropathol 116:17–24
Fujishiro H, Iseki E, Higashi S, Kasanuki K, Murayama N, Togo T, Katsuse O, Uchikado H, Aoki N, Kosaka K, Arai H, Sato K (2010) Distribution of cerebral amyloid deposition and its relevance to clinical phenotype in Lewy body dementia. Neurosci Lett 486:19–23
Fujiyama F, Unzai T, Karube F (2019) Thalamostriatal projections and striosome-matrix compartments. Neurochem Int 125:67–73
Gadad BS, Britton GB, Rao KS (2011) Targeting oligomers in neurodegenerative disorders: lessons from alpha-synuclein, tau, and amyloid-beta peptide. J Alzheimers Dis 24(Suppl 2):223–232
Gai WP, Pountney DL, Power JH, Li QX, Culvenor JG, McLean CA, Jensen PH, Blumbergs PC (2003) Alpha-synuclein fibrils constitute the central core of oligodendroglial inclusion filaments in multiple system atrophy. Exp Neurol 181:68–78
Galts CPC, Bettio LEB, Jewett DC, Yang CC, Brocardo PS, Rodrigues ALS, Thacker JS, Gil-Mohapel J (2019) Depression in neurodegenerative diseases: common mechanisms and current treatment options. Neurosci Biobehav Rev 102:56–84
Gallucci M, Dell’Acqua C, Boccaletto F, Fenoglio C, Galimberti D, Di Battista ME (2019) Overlap between frontotemporal dementia and dementia with Lewy bodies: a Treviso Dementia (TREDEM) registry case report. J Alzheimers Dis 69:839–847
Gao LL, Zhang JR, Chan P, Wu T (2017) Levodopa effect on basal ganglia motor circuit in Parkinson’s disease. CNS Neurosci Ther 23:76–86
Gash DM, Rutland K, Hudson NL, Sullivan PG, Bing G, Cass WA, Pandya JD, Liu M, Choi DY, Hunter RL, Gerhardt GA, Smith CD, Slevin JT, Prince TS (2008) Trichloroethylene: parkinsonism and complex 1 mitochondrial neurotoxicity. Ann Neurol 63:184–192
Gasser T, Hardy J, Mizuno Y (2011) Milestones in PD genetics. Mov Disord 26:1042–1048
Gegg ME, Schapira AHV (2018) The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J 285:3591–3603
Geibl FF, Henrich MT, Oertel WH (2019) Mesencephalic and extramesencephalic dopaminergic systems in Parkinson’s disease. J Neural Transm (Vienna) 126:377–396
Geiger JT, Ding J, Crain B, Pletnikova O, Letson C, Dawson TM, Rosenthal LS, Pantelyat A, Gibbs JR, Albert MS, Hernandez DG, Hillis AE, Stone DJ, Singleton AB, Hardy JA, Troncoso JC, Scholz SW (2016) Next-generation sequencing reveals substantial genetic contribution to dementia with Lewy bodies. Neurobiol Dis 94:55–62
Gelpi E, Colom-Cadena M (2019) Oligomers: a hot topic for neurodegeneration and a note of caution for experimental models. Brain 142:228–230
Gelpi E, Navarro-Otano J, Tolosa E, Gaig C, Compta Y, Rey MJ, Marti MJ, Hernandez I, Valldeoriola F, Rene R, Ribalta T (2014) Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov Disord 29:1010–1018
George G, Valiya Parambath S, Lokappa SB, Varkey J (2019) Construction of Parkinson’s disease marker-based weighted protein-protein interaction network for prioritization of co-expressed genes. Gene 697:67–77
Gerson JE, Farmer KM, Henson N, Castillo-Carranza DL, Carretero Murillo M, Sengupta U, Barrett A, Kayed R (2018) Tau oligomers mediate alpha-synuclein toxicity and can be targeted by immunotherapy. Mol Neurodegener 13:13
Geut H, Vergouw LJM, Galis Y, Ingrassia A, de Jong FJ, Quadri M, Bonifati V, Lemstra AW, Rozemuller AJM, van de Berg WDJ (2019) Neuropathological and genetic characteristics of a post-mortem series of cases with dementia with Lewy bodies clinically suspected of Creutzfeldt-Jakob’s disease. Parkinsonism Relat Disord. https://doi.org/10.1016/j.parkreldis.2019.1002.1011
Ghazi Sherbaf F, Same K, Aarabi MH (2018) High angular resolution diffusion imaging correlates of depression in Parkinson’s disease: a connectometry study. Acta Neurol Belg 118:573–579
Ghetti B, Wszolek ZK, Boeve BF, Spina S, Goedert M (2011) Frontotemporal dementia and parkinsonism linked to chromosome 17. In: Dickson DW, Weller RO (eds) Neurodegeneration: the molecular pathology of dementia and movement disorders, 2nd edn. Blackwell Publishing Ltd., Oxford, pp 110–134
Ghiglieri V, Calabrese V, Calabresi P (2018) Alpha-synuclein: from early synaptic dysfunction to neurodegeneration. Front Neurol 9:295
Gibbons GS, Lee VMY, Trojanowski JQ (2019) Mechanisms of cell-to-cell transmission of pathological tau: a review. JAMA Neurol 76:101–108
Giguere N, Burke Nanni S, Trudeau LE (2018) On cell loss and selective vulnerability of neuronal populations in Parkinson’s disease. Front Neurol 9:455
Gil MJ, Manzano MS, Cuadrado ML, Fernandez C, Gomez E, Matesanz C, Calero M, Rabano A (2018a) Frontotemporal lobar degeneration: Study of a clinicopathological cohort. J Clin Neurosci 58:172–180
Gil MJ, Manzano MS, Cuadrado ML, Fernandez C, Gomez E, Matesanz C, Calero M, Rabano A (2018b) Argyrophilic grain pathology in frontotemporal lobar degeneration: demographic, clinical, neuropathological, and genetic features. J Alzheimers Dis 63:1109–1117
Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, Wood NW, Colosimo C, Durr A, Fowler CJ, Kaufmann H, Klockgether T, Lees A, Poewe W, Quinn N, Revesz T, Robertson D, Sandroni P, Seppi K, Vidailhet M (2008) Second consensus statement on the diagnosis of multiple system atrophy. Neurology 71:670–676
Ginsberg MD (1985) Carbon monoxide intoxication: clinical features, neuropathology and mechanisms of injury. J Toxicol Clin Toxicol 23:281–288
Godini R, Fallahi H, Ebrahimie E (2019) A comparative system-level analysis of the neurodegenerative diseases. J Cell Physiol 234:5215–5229
Goedert M, Eisenberg DS, Crowther RA (2017a) Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci 40:189–210
Goedert M, Jakes R, Spillantini MG (2017b) The synucleinopathies: twenty years on. J Parkinsons Dis 7:S51–S69
Goedert M, Masuda-Suzukake M, Falcon B (2017c) Like prions: the propagation of aggregated tau and alpha-synuclein in neurodegeneration. Brain 140:266–278
Goedert M, Falcon B, Zhang W, Ghetti B, Scheres SHW (2019) Distinct conformers of assembled tau in Alzheimer’s and Pick’s diseases. Cold Spring Harb Symp Quant Biol. https://doi.org/10.1101/sqb.2018.1183.037580
Goetz CG, Emre M, Dubois B (2008) Parkinson’s disease dementia: definitions, guidelines, and research perspectives in diagnosis. Ann Neurol 64(Suppl 2):S81–S92
Gomez G, Escande MV, Suarez LM, Rela L, Belforte JE, Moratalla R, Murer MG, Gershanik OS, Taravini IRE (2019) Changes in dendritic spine density and inhibitory perisomatic connectivity onto medium spiny neurons in l-dopa-induced dyskinesia. Mol Neurobiol. https://doi.org/10.1007/s12035-12019-11515-12034
Gomez-Suaga P, Fdez E, Blanca Ramirez M, Hilfiker S (2012) A link between autophagy and the pathophysiology of LRRK2 in Parkinson’s disease. Parkinsons Dis 2012:324521
Gomperts SN (2016) Lewy body dementias: dementia with Lewy bodies and Parkinson disease dementia. Continuum (Minneap Minn) 22:435–463
Gonzalez N, Arcos-Lopez T, Konig A, Quintanar L, Menacho Marquez M, Outeiro TF, Fernandez CO (2019) Effects of alpha-synuclein posttranslational modifications on metal binding. J Neurochem. https://doi.org/10.1111/jnc.14721
Gotz J, Halliday G, Nisbet RM (2019) Molecular pathogenesis of the tauopathies. Annu Rev Pathol 14:239–261
Grabli D, Auré K, Vidailhet M, Roze E (2011) Movement disorders in neurometabolic diseases. In: Gálvez-Jiménez N, Tuite P (eds) Uncommon causes of movement disorders. Cambridge University Press, Cambridge, pp 245–257
Grant R, Graus F (2009) Paraneoplastic movement disorders. Mov Disord 24:1715–1724
Greffard S, Verny M, Bonnet AM, Seilhean D, Hauw JJ, Duyckaerts C (2010) A stable proportion of Lewy body bearing neurons in the substantia nigra suggests a model in which the Lewy body causes neuronal death. Neurobiol Aging 31:99–103
Grigoletto J, Pukass K, Gamliel A, Davidi D, Katz-Brull R, Richter-Landsberg C, Sharon R (2017) Higher levels of myelin phospholipids in brains of neuronal alpha-synuclein transgenic mice precede myelin loss. Acta Neuropathol Commun 5:37
Grimm MJ, Respondek G, Stamelou M, Arzberger T, Ferguson L, Gelpi E, Giese A, Grossman M, Irwin DJ, Pantelyat A, Rajput A, Roeber S, van Swieten JC, Troakes C, Antonini A, Bhatia KP, Colosimo C, van Eimeren T, Kassubek J, Levin J, Meissner WG, Nilsson C, Oertel WH, Piot I, Poewe W, Wenning GK, Boxer A, Golbe LI, Josephs KA, Litvan I, Morris HR, Whitwell JL, Compta Y, Corvol JC, Lang AE, Rowe JB, Hoglinger GU (2019) How to apply the Movement Disorder Society criteria for diagnosis of progressive supranuclear palsy. Mov Disord. https://doi.org/10.1002/mds.27666
Grozdanov V, Danzer KM (2018) Release and uptake of pathologic alpha-synuclein. Cell Tissue Res 373:175–182
Grunblatt E, Ruder J, Monoranu CM, Riederer P, Youdim MB, Mandel SA (2018) Differential alterations in metabolism and proteolysis-related proteins in human Parkinson’s disease substantia nigra. Neurotox Res 33:560–568
Guan X, Zhang Y, Wei H, Guo T, Zeng Q, Zhou C, Wang J, Gao T, Xuan M, Gu Q, Xu X, Huang P, Pu J, Zhang B, Liu C, Zhang M (2019) Iron-related nigral degeneration influences functional topology mediated by striatal dysfunction in Parkinson’s disease. Neurobiol Aging 75:83–97
Guerreiro R, Ross OA, Kun-Rodrigues C, Hernandez DG, Orme T, Eicher JD, Shepherd CE, Parkkinen L, Darwent L, Heckman MG, Scholz SW, Troncoso JC, Pletnikova O, Ansorge O, Clarimon J, Lleo A, Morenas-Rodriguez E, Clark L, Honig LS, Marder K, Lemstra A, Rogaeva E, St George-Hyslop P, Londos E, Zetterberg H, Barber I, Braae A, Brown K, Morgan K, Troakes C, Al-Sarraj S, Lashley T, Holton J, Compta Y, Van Deerlin V, Serrano GE, Beach TG, Lesage S, Galasko D, Masliah E, Santana I, Pastor P, Diez-Fairen M, Aguilar M, Tienari PJ, Myllykangas L, Oinas M, Revesz T, Lees A, Boeve BF, Petersen RC, Ferman TJ, Escott-Price V, Graff-Radford N, Cairns NJ, Morris JC, Pickering-Brown S, Mann D, Halliday GM, Hardy J, Trojanowski JQ, Dickson DW, Singleton A, Stone DJ, Bras J (2018) Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet Neurol 17:64–74
Guerrero E, Vasudevaraju P, Hegde ML, Britton GB, Rao KS (2013) Recent advances in alpha-synuclein functions, advanced glycation, and toxicity: implications for Parkinson’s disease. Mol Neurobiol 47:525–536
Guiney SJ, Adlard PA, Bush AI, Finkelstein DI, Ayton S (2017) Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem Int 104:34–48
Gundner AL, Duran-Pacheco G, Zimmermann S, Ruf I, Moors T, Baumann K, Jagasia R, van de Berg WDJ, Kremer T (2019) Path mediation analysis reveals GBA impacts Lewy body disease status by increasing alpha-synuclein levels. Neurobiol Dis 121:205–213
Guo JL, Lee VM (2014) Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 20:130–138
Habas C, Manto M, Cabaraux P (2019) The cerebellar thalamus. Cerebellum 18:635–648
Haber SN (2016) Corticostriatal circuitry. Dialogues Clin Neurosci 18:7–21
Hall JM, Lewis SJG (2019) Neural correlates of cognitive impairment in Parkinson’s disease: a review of structural MRI findings. Int Rev Neurobiol 144:1–28
Hall JM, Shine JM, Walton CC, Gilat M, Kamsma YP, Naismith SL, Lewis SJ (2014) Early phenotypic differences between Parkinson’s disease patients with and without freezing of gait. Parkinsonism Relat Disord 20:604–607
Halliday GM, McCann H (2010) The progression of pathology in Parkinson’s disease. Ann N Y Acad Sci 1184:188–195
Halliday G, Hely M, Reid W, Morris J (2008) The progression of pathology in longitudinally followed patients with Parkinson’s disease. Acta Neuropathol 115:409–415
Halliday G, Herrero MT, Murphy K, McCann H, Ros-Bernal F, Barcia C, Mori H, Blesa FJ, Obeso JA (2009) No Lewy pathology in monkeys with over 10 years of severe MPTP parkinsonism. Mov Disord 24:1519–1523
Halliday GM, Song YJ, Harding AJ (2011) Striatal beta-amyloid in dementia with Lewy bodies but not Parkinson’s disease. J Neural Transm 118:713–719
Halliday G, McCann H, Shepherd C (2012) Evaluation of the Braak hypothesis: how far can it explain the pathogenesis of Parkinson’s disease? Expert Rev Neurother 12:673–686
Halliday GM, Leverenz JB, Schneider JS, Adler CH (2014) The neurobiological basis of cognitive impairment in Parkinson’s disease. Mov Disord 29:634–650
Hamed M, Schraml F, Wilson J, Galvin J, Sabbagh MN (2018) Occipital and cingulate hypometabolism are significantly under-reported on 18-fluorodeoxyglucose positron emission tomography scans of patients with Lewy body dementia. J Alzheimers Dis Parkinsonism 8:428
Hansen LA, Daniel SE, Wilcock GK, Love S (1998) Frontal cortical synaptophysin in Lewy body diseases: relation to Alzheimer’s disease and dementia. J Neurol Neurosurg Psychiatry 64:653–656
Hansen D, Ling H, Lashley T, Holton JL, Warner TT (2019) Clinical, neuropathological and genetic features of Lewy body dementias. Neuropathol Appl Neurobiol 11:78. https://doi.org/10.1111/nan.12554
Hara K, Watanabe H, Bagarinao E, Kawabata K, Yoneyama N, Ohdake R, Imai K, Masuda M, Yokoi T, Ogura A, Tsuboi T, Ito M, Atsuta N, Niwa H, Taoka T, Maesawa S, Naganawa S, Katsuno M, Sobue G (2018) Corpus callosal involvement is correlated with cognitive impairment in multiple system atrophy. J Neurol 265:2079–2087
Harding AJ, Stimson E, Henderson JM, Halliday GM (2002) Clinical correlates of selective pathology in the amygdala of patients with Parkinson’s disease. Brain 125:2431–2445
Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, Beach TG, Buratti E, Baralle F, Morita M, Nakano I, Oda T, Tsuchiya K, Akiyama H (2008) Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 64:60–70
Hata Y, Ma N, Yoneda M, Morimoto S, Okano H, Murayama S, Kawanishi S, Kuzuhara S, Kokubo Y (2018) Nitrative stress and tau accumulation in amyotrophic lateral sclerosis/parkinsonism-dementia complex (ALS/PDC) in the Kii Peninsula, Japan. Front Neurosci 11:751
Hayakawa H, Nagai M, Kawanami A, Nakata Y, Nihira T, Ogino M, Takada M, Saido T, Takano J, Saegusa M, Mikami T, Hamada J, Nishiyama K, Mochizuki H, Mizuno Y (2013) Loss of DARPP-32 and calbindin in multiple system atrophy. J Neural Transm 120:1689–1698
Heckman MG, Schottlaender L, Soto-Ortolaza AI, Diehl NN, Rayaprolu S, Ogaki K, Fujioka S, Murray ME, Cheshire WP, Uitti RJ, Wszolek ZK, Farrer MJ, Sailer A, Singleton AB, Chinnery PF, Keogh MJ, Gentleman SM, Holton JL, Aoife K, Mann DM, Al-Sarraj S, Troakes C, Dickson DW, Houlden H, Ross OA (2014) LRRK2 exonic variants and risk of multiple system atrophy. Neurology 83:2256–2261
Heckman MG, Brennan RR, Labbe C, Soto AI, Koga S, DeTure MA, Murray ME, Petersen RC, Boeve BF, van Gerpen JA, Uitti RJ, Wszolek ZK, Rademakers R, Dickson DW, Ross OA (2019) Association of MAPT subhaplotypes with risk of progressive supranuclear palsy and severity of tau pathology. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2019.0250
Hegeman DJ, Hong ES, Hernandez VM, Chan CS (2016) The external globus pallidus: progress and perspectives. Eur J Neurosci 43:1239–1265
Heitz C, Noblet V, Cretin B, Philippi N, Kremer L, Stackfleth M, Hubele F, Armspach JP, Namer I, Blanc F (2015) Neural correlates of visual hallucinations in dementia with Lewy bodies. Alzheimers Res Ther 7:6
Helmich RC (2018) The cerebral basis of Parkinsonian tremor: a network perspective. Mov Disord 33:219–231
Helmich RC, Janssen MJ, Oyen WJ, Bloem BR, Toni I (2011) Pallidal dysfunction drives a cerebellothalamic circuit into Parkinson tremor. Ann Neurol 69:269–281
Hely MA, Reid WG, Adena MA, Halliday GM, Morris JG (2008) The Sydney multicenter study of Parkinson’s disease: the inevitability of dementia at 20 years. Mov Disord 23:837–844
Henderson MX, Trojanowski JQ, Lee VM (2019) α-Synuclein pathology in Parkinson's disease and related alpha-synucleinopathies. Neurosci Lett. https://doi.org/10.1016/j.neulet.2019.134316
Hepp DH, Vergoossen DL, Huisman E, Lemstra AW, Berendse HW, Rozemuller AJ, Foncke EM, van de Berg WD (2016) Distribution and load of amyloid-beta pathology in Parkinson disease and dementia with Lewy bodies. J Neuropathol Exp Neurol 75:936–945
Heras-Garvin A, Weckbecker D, Ryazanov S, Leonov A, Griesinger C, Giese A, Wenning GK, Stefanova N (2019) Anle138b modulates alpha-synuclein oligomerization and prevents motor decline and neurodegeneration in a mouse model of multiple system atrophy. Mov Disord 34:255–263
Hernandez D, Paisan Ruiz C, Crawley A, Malkani R, Werner J, Gwinn-Hardy K, Dickson D, Wavrant Devrieze F, Hardy J, Singleton A (2005) The dardarin G 2019 S mutation is a common cause of Parkinson’s disease but not other neurodegenerative diseases. Neurosci Lett 389:137–139
Herz DM, Haagensen BN, Christensen MS, Madsen KH, Rowe JB, Lokkegaard A, Siebner HR (2015) Abnormal dopaminergic modulation of striato-cortical networks underlies levodopa-induced dyskinesias in humans. Brain 138:1658–1666
Hintzen A, Pelzer EA, Tittgemeyer M (2018) Thalamic interactions of cerebellum and basal ganglia. Brain Struct Funct 223:569–587
Hirsch EC, Jenner P, Przedborski S (2013) Pathogenesis of Parkinson’s disease. Mov Disord 28:24–30
Hoffmann A, Ettle B, Battis K, Reiprich S, Schlachetzki JCM, Masliah E, Wegner M, Kuhlmann T, Riemenschneider MJ, Winkler J (2019) Oligodendroglial a-synucleinopathy-driven neuroinflammation in multiple system atrophy. Brain Pathol 29:380–396
Hoglinger GU (2018) Is it useful to classify progressive supranuclear palsy and corticobasal degeneration as different disorders? No. Mov Disord Clin Pract 5:141–144
Hoglinger GU, Oertel WH, Hirsch EC (2006) The rotenone model of parkinsonism—the 5 years inspection. J Neural Transm Suppl (70):269–272
Hoglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, van Swieten JC, Heutink P, Wszolek ZK, Uitti RJ, Vandrovcova J, Hurtig HI, Gross RG, Maetzler W, Goldwurm S, Tolosa E, Borroni B, Pastor P, Cantwell LB, Han MR, Dillman A, van der Brug MP, Gibbs JR, Cookson MR, Hernandez DG, Singleton AB, Farrer MJ, Yu CE, Golbe LI, Revesz T, Hardy J, Lees AJ, Devlin B, Hakonarson H, Muller U, Schellenberg GD (2011) Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet 43:699–705
Hoglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, Mollenhauer B, Muller U, Nilsson C, Whitwell JL, Arzberger T, Englund E, Gelpi E, Giese A, Irwin DJ, Meissner WG, Pantelyat A, Rajput A, van Swieten JC, Troakes C, Antonini A, Bhatia KP, Bordelon Y, Compta Y, Corvol JC, Colosimo C, Dickson DW, Dodel R, Ferguson L, Grossman M, Kassubek J, Krismer F, Levin J, Lorenzl S, Morris HR, Nestor P, Oertel WH, Poewe W, Rabinovici G, Rowe JB, Schellenberg GD, Seppi K, van Eimeren T, Wenning GK, Boxer AL, Golbe LI, Litvan I (2018) Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord 32:853–864
Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Bjorklund T, Wang ZY, Roybon L, Melki R, Li JY (2014) Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol 128:805–820
Holton JL, Lees AL, Revesz T (2011) Multiple system atrophy. In: Dickson DW, Weller RO (eds) Neurodegeneration: the molecular pathology of dementia and movement disorders, 2nd edn. Blackwell Publishing Ltd., Oxford, pp 242–252
Homma T, Mochizuki Y, Komori T, Isozaki E (2016) Frequent globular neuronal cytoplasmic inclusions in the medial temporal region as a possible characteristic feature in multiple system atrophy with dementia. Neuropathology 36:421–431
Horowitz MP, Greenamyre JT (2010) Mitochondrial iron metabolism and its role in neurodegeneration. J Alzheimers Dis 20(Suppl 2):S551–S568
Horvath J, Burkhard PR, Bouras C, Kovari E (2013a) Etiologies of parkinsonism in a century-long autopsy-based cohort. Brain Pathol 23:28–33
Horvath J, Herrmann FR, Burkhard PR, Bouras C, Kovari E (2013b) Neuropathology of dementia in a large cohort of patients with Parkinson’s disease. Parkinsonism Relat Disord 19:864–868 (discussion 864)
Houlden H, Singleton AB (2012) The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol 124:325–338
Houlden H, Baker M, Morris HR, MacDonald N, Pickering-Brown S, Adamson J, Lees AJ, Rossor MN, Quinn NP, Kertesz A, Khan MN, Hardy J, Lantos PL, St George-Hyslop P, Munoz DG, Mann D, Lang AE, Bergeron C, Bigio EH, Litvan I, Bhatia KP, Dickson D, Wood NW, Hutton M (2001) Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology 56:1702–1706
Hu D, Sun X, Liao X, Zhang X, Zarabi S, Schimmer A, Hong Y, Ford C, Luo Y, Qi X (2019) Alpha-synuclein suppresses mitochondrial protease ClpP to trigger mitochondrial oxidative damage and neurotoxicity. Acta Neuropathol 137:939–960
Huang M, Wang B, Li X, Fu C, Wang C, Kang X (2019) Alpha-synuclein: a multifunctional player in exocytosis, endocytosis, and vesicle recycling. Front Neurosci 13:28
Hwang J, Bank AM, Mortazavi F, Oakley DH, Frosch MP, Schmahmann JD (2019) Spinal cord a-synuclein deposition associated with myoclonus in patients with MSA-C. Neurology (accepted paper)
Iljina M, Garcia GA, Horrocks MH, Tosatto L, Choi ML, Ganzinger KA, Abramov AY, Gandhi S, Wood NW, Cremades N, Dobson CM, Knowles TP, Klenerman D (2016) Kinetic model of the aggregation of alpha-synuclein provides insights into prion-like spreading. Proc Natl Acad Sci USA 113:E1206–E1215
Imbriani P, Schirinzi T, Meringolo M, Mercuri NB, Pisani A (2018) Centrality of early synaptopathy in Parkinson’s disease. Front Neurol 9:103
Inzelberg R, Estrada-Cuzcano A, Laitman Y, De Vriendt E, Friedman E, Jordanova A (2018) Kufor–Rakeb syndrome/PARK9: one novel and one possible recurring Ashkenazi ATP13A2 mutation. J Parkinsons Dis 8:399–403
Iodice V, Lipp A, Ahlskog JE, Sandroni P, Fealey RD, Parisi JE, Matsumoto JY, Benarroch EE, Kimpinski K, Singer W, Gehrking TL, Gehrking JA, Sletten DM, Schmeichel AM, Bower JH, Gilman S, Figueroa J, Low PA (2012) Autopsy confirmed multiple system atrophy cases: Mayo experience and role of autonomic function tests. J Neurol Neurosurg Psychiatry 83:453–459
Irwin DJ, Hurtig HI (2018) The contribution of tau, amyloid-beta and alpha-synuclein pathology to dementia in Lewy body disorders. J Alzheimers Dis Parkinsonism. https://doi.org/10.4172/2161-0460.1000444
Irwin DJ, Abrams JY, Schonberger LB, Leschek EW, Mills JL, Lee VM, Trojanowski JQ (2013a) Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurol 70:462–468
Irwin DJ, Cohen TJ, Grossman M, Arnold SE, McCarty-Wood E, Van Deerlin VM, Lee VM, Trojanowski JQ (2013b) Acetylated tau neuropathology in sporadic and hereditary tauopathies. Am J Pathol 183:344–351
Irwin DJ, Lee VM, Trojanowski JQ (2013c) Parkinson’s disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci 14:626–636
Irwin DJ, Cairns NJ, Grossman M, McMillan CT, Lee EB, Van Deerlin VM, Lee VM, Trojanowski JQ (2015) Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol 129:469–491
Irwin DJ, Brettschneider J, McMillan CT, Cooper F, Olm C, Arnold SE, Van Deerlin VM, Seeley WW, Miller BL, Lee EB, Lee VM, Grossman M, Trojanowski JQ (2016) Deep clinical and neuropathological phenotyping of Pick disease. Ann Neurol 79:272–287
Ishiyama M, Yagishita S, Hasegawa K, Yokoyama T (2006) Ultrastructural study (tEM and sEM) of cortical Lewy bodies (abstract). Neuropathol 26(2):A58
Ito K, Arai K, Yoshiyama Y, Kashiwado K, Sakakibara Y, Hattori T (2008) Astrocytic tau pathology positively correlates with neurofibrillary tangle density in progressive supranuclear palsy. Acta Neuropathol 115:623–628
Itoh N, Ishiguro K, Arai H, Kokubo Y, Sasaki R, Narita Y, Kuzuhara S (2003) Biochemical and ultrastructural study of neurofibrillary tangles in amyotrophic lateral sclerosis/parkinsonism-dementia complex in the Kii peninsula of Japan. J Neuropathol Exp Neurol 62:791–798
Iyer A, Claessens M (2019) Disruptive membrane interactions of alpha-synuclein aggregates. Biochim Biophys Acta Proteins Proteom 1867:468–482
Jabbari E, Woodside J, Tan MMX, Shoai M, Pittman A, Ferrari R, Mok KY, Zhang D, Reynolds RH, de Silva R, Grimm MJ, Respondek G, Muller U, Al-Sarraj S, Gentleman SM, Lees AJ, Warner TT, Hardy J, Revesz T, Hoglinger GU, Holton JL, Ryten M, Morris HR (2018) Variation at the TRIM11 locus modifies progressive supranuclear palsy phenotype. Ann Neurol 84:485–496
Janda E, Isidoro C, Carresi C, Mollace V (2012) Defective autophagy in Parkinson’s disease: role of oxidative stress. Mol Neurobiol 46:639–661
Jellinger KA (2003) Prevalence of vascular lesions in dementia with Lewy bodies. A postmortem study. J Neural Transm (Vienna) 110:771–778
Jellinger KA (2004) Lewy body-related alpha-synucleinopathy in the aged human brain. J Neural Transm 111:1219–1235
Jellinger KA (2006) Pathological substrate of dementia in Parkinson’s disease–its relation to DLB and DLBD. Parkinsonism Relat Disord 12:119–120
Jellinger KA (2008a) Vascular parkinsonism. Therapy 5:237–255
Jellinger KA (2008b) Different tau pathology pattern in two clinical phenotypes of progressive supranuclear palsy. Neurodegener Dis 5:339–346
Jellinger KA (2009a) A critical evaluation of current staging of alpha-synuclein pathology in Lewy body disorders. Biochim Biophys Acta 1792:730–740
Jellinger KA (2009b) Significance of brain lesions in Parkinson disease dementia and Lewy body dementia. Front Neurol Neurosci 24:114–125
Jellinger KA (2011) Postencephalitic Parkinsonism. In: Dickson DW, Weller RO (eds) Neurodegeneration: the molecular pathology of dementia and movement disorders, 2nd edn. Blackwell Publishing Ltd., Oxford, pp 179–187
Jellinger KA (2012a) The role of alpha-synuclein in neurodegeneration—an update. Transl Neurosci 3:75–122
Jellinger KA (2012b) Neuropathology of sporadic Parkinson’s disease: evaluation and changes of concepts. Mov Disord 27:8–30
Jellinger KA (2013a) Synuclein and Parkinson’s disease: an update. In: Martinez A, Gil C (eds) Emerging drugs and targets for Parkinson’s disease. The Royal Society of Chemistry, London, pp 175–214
Jellinger KA (2013b) Mild cognitive impairment in Parkinson disease: heterogenous mechanisms. J Neural Transm (Vienna) 120:157–167
Jellinger KA (2014) Neuropathology. In: Wenning GK, Fanciulli A (eds) Multiple system atrophy. Springer, Vienna, pp 17–55
Jellinger KA (2015) Neuropathobiology of non-motor symptoms in Parkinson disease. J Neural Transm (Vienna) 122:1429–1440
Jellinger KA (2016) Neuropathology of movement disorders, vol 1. In: Winn HR (ed) Youmans Neurological surgery, 7th edn. Elsevier-Saunders, Philadelphia, pp e840–e880
Jellinger KA (2017a) Neuropathology of movement disorders. In: Falup-Pecurariu C, Ferreira J, Martinez-Martin P, Chaudhuri KR (eds) Movement disorders curricula. Springer, Wien, pp 43–48
Jellinger KA (2017b) Neuropathology of nonmotor symptoms of Parkinson’s disease. Int Rev Neurobiol 133:13–62
Jellinger KA (2018a) Multiple system atrophy: an oligodendroglioneural synucleinopathy. J Alzheimers Dis 62:1141–1179
Jellinger KA (2018b) Dementia with Lewy bodies and Parkinson’s disease-dementia: current concepts and controversies. J Neural Transm (Vienna) 125:615–650
Jellinger KA (2018c) Young-onset multiple system atrophy. Mov Disord 33:1974–1975
Jellinger KA (2019) Is Braak staging valid for all types of Parkinson’s disease? J Neural Transm (Vienna) 126:423–431
Jellinger KA, Attems J (2006) Does striatal pathology distinguish Parkinson disease with dementia and dementia with Lewy bodies? Acta Neuropathol 112:253–260
Jellinger KA, Attems J (2008) Prevalence and impact of vascular and Alzheimer pathologies in Lewy body disease. Acta Neuropathol 115:127–136
Jellinger KA, Korczyn AD (2018) Are dementia with Lewy bodies and Parkinson’s disease dementia the same disease? BMC Med 16:34
Jellinger KA, Lantos PL (2010) Papp-Lantos inclusions and the pathogenesis of multiple system atrophy: an update. Acta Neuropathol 119:657–667
Jellinger KA, Wenning GK (2016) Multiple system atrophy: pathogenic mechanisms and biomarkers. J Neural Transm (Vienna) 123:555–572
Jellinger KA, Seppi K, Wenning GK (2005) Grading of neuropathology in multiple system atrophy: proposal for a novel scale. Mov Disord 20(Suppl 12):S29–S36
Jiang P, Dickson DW (2018) Parkinson’s disease: experimental models and reality. Acta Neuropathol 135:13–32
Jiang P, Gan M, Yen SH, Moussaud S, McLean PJ, Dickson DW (2016) Proaggregant nuclear factor(s) trigger rapid formation of alpha-synuclein aggregates in apoptotic neurons. Acta Neuropathol 132:77–91
Johnson ME, Stecher B, Labrie V, Brundin L, Brundin P (2019) Triggers, facilitators, and aggravators: redefining Parkinson’s disease pathogenesis. Trends Neurosci 42:4–13
Joki H, Higashiyama Y, Nakae Y, Kugimoto C, Doi H, Kimura K, Kishida H, Ueda N, Nakano T, Takahashi T, Koyano S, Takeuchi H, Tanaka F (2018) White matter hyperintensities on MRI in dementia with Lewy bodies, Parkinson’s disease with dementia, and Alzheimer’s disease. J Neurol Sci 385:99–104
Josephs KA, Ishizawa T, Tsuboi Y, Cookson N, Dickson DW (2002) A clinicopathological study of vascular progressive supranuclear palsy: a multi-infarct disorder presenting as progressive supranuclear palsy. Arch Neurol 59:1597–1601
Jost WH, Lingor P, Tonges L, Schwarz J, Buhmann C, Kassubek J, Schrag A (2019) Dyskinesia in multiple system atrophy and progressive supranuclear palsy. J Neural Transm (Vienna). https://doi.org/10.1007/s00702-00019-02012-00700
Joutsa J, Gardberg M, Roytta M, Kaasinen V (2014) Diagnostic accuracy of parkinsonism syndromes by general neurologists. Parkinsonism Relat Disord 20:840–844
Kaindlstorfer C, Granata R, Wenning GK (2013) Tremor in multiple system atrophy—a review. Tremor Other Hyperkinet Mov (N Y). https://doi.org/10.7916/d7918nv7919gz7919
Kaindlstorfer C, Jellinger KA, Eschlbock S, Stefanova N, Weiss G, Wenning GK (2018) The relevance of iron in the pathogenesis of multiple system atrophy: a viewpoint. J Alzheimers Dis 61:1253–1273
Kalaitzakis ME, Pearce RK (2009) The morbid anatomy of dementia in Parkinson’s disease. Acta Neuropathol 118:587–598
Kalaitzakis ME, Christian LM, Moran LB, Graeber MB, Pearce RK, Gentleman SM (2009a) Dementia and visual hallucinations associated with limbic pathology in Parkinson’s disease. Parkinsonism Relat Disord 15:196–204
Kalaitzakis ME, Pearce RK, Gentleman SM (2009b) Clinical correlates of pathology in the claustrum in Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 461:12–15
Kalaitzakis ME, Walls AJ, Pearce RK, Gentleman SM (2011) Striatal Abeta peptide deposition mirrors dementia and differentiates DLB and PDD from other Parkinsonian syndromes. Neurobiol Dis 41:377–384
Kalia LV, Kalia SK (2015) Alpha-synuclein and Lewy pathology in Parkinson’s disease. Curr Opin Neurol 28:375–381
Kalia LV, Lang AE (2015) Parkinson’s disease. Lancet 386:896–912
Kam TI, Mao X, Park H, Chou SC, Karuppagounder SS, Umanah GE, Yun SP, Brahmachari S, Panicker N, Chen R, Andrabi SA, Qi C, Poirier GG, Pletnikova O, Troncoso JC, Bekris LM, Leverenz JB, Pantelyat A, Ko HS, Rosenthal LS, Dawson TM, Dawson VL (2018) Poly(ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson’s disease. Science. https://doi.org/10.1126/science.aat8407
Kamin J, Manwani S, Hughes D (2000) Emergency psychiatry: extrapyramidal side effects in the psychiatric emergency service. Psychiatr Serv 51:287–289
Kanazawa T, Uchihara T, Takahashi A, Nakamura A, Orimo S, Mizusawa H (2008) Three-layered structure shared between Lewy bodies and Lewy neurites—three-dimensional reconstruction of triple-labeled sections. Brain Pathol 18:415–422
Kanazawa T, Adachi E, Orimo S, Nakamura A, Mizusawa H, Uchihara T (2012) Pale neurites, premature alpha-synuclein aggregates with centripetal extension from axon collaterals. Brain Pathol 22:67–78
Kane JPM, Surendranathan A, Bentley A, Barker SAH, Taylor JP, Thomas AJ, Allan LM, McNally RJ, James PW, McKeith IG, Burn DJ, O’Brien JT (2018) Clinical prevalence of Lewy body dementia. Alzheimers Res Ther 10:19
Kang SW, Jeon S, Yoo HS, Chung SJ, Lee PH, Sohn YH, Yun M, Evans AC, Ye BS (2019) Effects of Lewy body disease and Alzheimer disease on brain atrophy and cognitive dysfunction. Neurology 92:e2015–e2026
Karpowicz RJ Jr, Trojanowski JQ, Lee VM (2019) Transmission of alpha-synuclein seeds in neurodegenerative disease: recent developments. Lab Investig. https://doi.org/10.1038/s41374-41019-40195-z
Kempster PA, O’Sullivan SS, Holton JL, Revesz T, Lees AJ (2010) Relationships between age and late progression of Parkinson’s disease: a clinico-pathological study. Brain 133:1755–1762
Kett LR, Dauer WT (2016) Endolysosomal dysfunction in Parkinson’s disease: recent developments and future challenges. Mov Disord 31:1433–1443
Kiely AP, Asi YT, Kara E, Limousin P, Ling H, Lewis P, Proukakis C, Quinn N, Lees AJ, Hardy J, Revesz T, Houlden H, Holton JL (2013) Alpha-synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol 125:753–769
Kiely AP, Ling H, Asi YT, Kara E, Proukakis C, Schapira AH, Morris HR, Roberts HC, Lubbe S, Limousin P, Lewis PA, Lees AJ, Quinn N, Hardy J, Love S, Revesz T, Houlden H, Holton JL (2015) Distinct clinical and neuropathological features of G51D SNCA mutation cases compared with SNCA duplication and H50Q mutation. Mol Neurodegener 10:41
Kiely AP, Murray CE, Foti SC, Benson BC, Courtney R, Strand C, Lashley T, Holton JL (2018) Immunohistochemical and molecular investigations show alteration in the inflammatory profile of multiple system atrophy brain. J Neuropathol Exp Neurol 77:598–607
Kiely AP, Miners JS, Courtney R, Strand C, Love S, Holton JL (2019) Exploring the putative role of kallikrein-6, calpain-1 and cathepsin-D in the proteolytic degradation of alpha-synuclein in multiple system atrophy. Neuropathol Appl Neurobiol 45:347–360
Kim HJ, Jeon BS (2012) Multiple system atrophy with prolonged survival. Mov Disord 27:1834
Ryu H-S, Oh M, Oh JS, Moon H, Park KW, Lee CS, You S, Kim M-J, Kim Y, J., Kim J, Kim KM, Kim JS, Chung SJ (2019) Distinct clinical features of predominant pre-synaptic and transsynaptic nigrostriatal dysfunction in multiple system atrophy. J Neurol Sci (accepted article)
Keber U, Klietz M, Carlsson T, Oertel WH, Weihe E, Schafer MK, Hoglinger GU, Depboylu C (2015) Striatal tyrosine hydroxylase-positive neurons are associated with L-DOPA-induced dyskinesia in hemiparkinsonian mice. Neuroscience 298:302–317
Kim YD, Kim JS, Lee ES, Yang DW, Lee KS, Kim YI (2009) Progressive “vascular” corticobasal syndrome due to bilateral ischemic hemispheric lesions. Intern Med 48:1699–1702
Kim JS, Yang JJ, Lee DK, Lee JM, Youn J, Cho JW (2015) Cognitive impairment and its structural correlates in the parkinsonian subtype of multiple system atrophy. Neurodegener Dis 15:294–300
Kim EJ, Brown JA, Deng J, Hwang JL, Spina S, Miller ZA, DeMay MG, Valcour V, Karydas A, Ramos EM, Coppola G, Miller BL, Rosen HJ, Seeley WW, Grinberg LT (2018) Mixed TDP-43 proteinopathy and tauopathy in frontotemporal lobar degeneration: nine case series. J Neurol 265:2960–2971
Kingsbury AE, Bandopadhyay R, Silveira-Moriyama L, Ayling H, Kallis C, Sterlacci W, Maeir H, Poewe W, Lees AJ (2010) Brain stem pathology in Parkinson’s disease: an evaluation of the Braak staging model. Mov Disord 25:2508–2515
Kishore A, Popa T (2014) Cerebellum in levodopa-induced dyskinesias: the unusual suspect in the motor network. Front Neurol 5:157
Klaus A, da Silva JA, Costa RM (2019) What, if, and when to move: basal ganglia circuits and self-paced action initiation. Annu Rev Neurosci. https://doi.org/10.1146/annurev-neuro-072116-031033
Klein AD, Mazzulli JR (2018) Is Parkinson’s disease a lysosomal disorder? Brain 141:2255–2262
Klein C, Lochte T, Delamonte SM, Braenne I, Hicks AA, Zschiedrich-Jansen K, Simon DK, Friedman JH, Lohmann K (2016) PLA2G6 mutations and parkinsonism: long-term follow-up of clinical features and neuropathology. Mov Disord 31:1927–1929
Klietz M, Keber U, Carlsson T, Chiu WH, Hoglinger GU, Weihe E, Schafer MK, Depboylu C (2016) l-DOPA-induced dyskinesia is associated with a deficient numerical downregulation of striatal tyrosine hydroxylase mRNA-expressing neurons. Neuroscience 331:120–133
Klingelhoefer L, Reichmann H (2017) Parkinson’s disease as a multisystem disorder. J Neural Transm (Vienna) 124:709–713
Ko WKD, Bezard E (2017) Experimental animal models of Parkinson’s disease: a transition from assessing symptomatology to alpha-synuclein targeted disease modification. Exp Neurol 298:172–179
Koga S, Dickson DW (2018) Recent advances in neuropathology, biomarkers and therapeutic approach of multiple system atrophy. J Neurol Neurosurg Psychiatry 89:175–184
Koga S, Dickson DW (2019) “Minimal change” multiple system atrophy with limbic-predominant alpha-synuclein pathology. Acta Neuropathol 137:167–169
Koga S, Aoki N, Uitti RJ, van Gerpen JA, Cheshire WP, Josephs KA, Wszolek ZK, Langston JW, Dickson DW (2015) When DLB, PD, and PSP masquerade as MSA: an autopsy study of 134 patients. Neurology 85:404–412
Koga S, Parks A, Uitti RJ, van Gerpen JA, Cheshire WP, Wszolek ZK, Dickson DW (2017a) Profile of cognitive impairment and underlying pathology in multiple system atrophy. Mov Disord 32:405–413
Koga S, Roemer SF, Kasanuki K, Dickson DW (2019) Cerebrovascular pathology presenting as corticobasal syndrome: an autopsy case series of “vascular CBS”. Parkinsonism Relat Disord (in press)
Koga S, Sanchez-Contreras M, Josephs KA, Uitti RJ, Graff-Radford N, van Gerpen JA, Cheshire WP, Wszolek ZK, Rademakers R, Dickson DW (2017b) Distribution and characteristics of transactive response DNA binding protein 43 kDa pathology in progressive supranuclear palsy. Mov Disord 32:246–255
Koga S, Kouri N, Walton RL, Ebbert MTW, Josephs KA, Litvan I, Graff-Radford N, Ahlskog JE, Uitti RJ, van Gerpen JA, Boeve BF, Parks A, Ross OA, Dickson DW (2018a) Corticobasal degeneration with TDP-43 pathology presenting with progressive supranuclear palsy syndrome: a distinct clinicopathologic subtype. Acta Neuropathol 136:389–404
Koga S, Lin WL, Walton RL, Ross OA, Dickson DW (2018b) TDP-43 pathology in multiple system atrophy: colocalization of TDP-43 and alpha-synuclein in glial cytoplasmic inclusions. Neuropathol Appl Neurobiol 44:707–721
Kokubo Y, Taniguchi A, Hasegawa M, Hayakawa Y, Morimoto S, Yoneda M, Hirokawa Y, Shiraishi T, Saito Y, Murayama S, Kuzuhara S (2012) Alpha-synuclein pathology in the amyotrophic lateral sclerosis/parkinsonism dementia complex in the Kii Peninsula, Japan. J Neuropathol Exp Neurol 71:625–630
Köllensperger M, Geser F, Seppi K, Stampfer-Kountchev M, Sawires M, Scherfler C, Boesch S, Mueller J, Koukouni V, Quinn N, Pellecchia MT, Barone P, Schimke N, Dodel R, Oertel W, Dupont E, Ostergaard K, Daniels C, Deuschl G, Gurevich T, Giladi N, Coelho M, Sampaio C, Nilsson C, Widner H, Sorbo FD, Albanese A, Cardozo A, Tolosa E, Abele M, Klockgether T, Kamm C, Gasser T, Djaldetti R, Colosimo C, Meco G, Schrag A, Poewe W, Wenning GK (2008) Red flags for multiple system atrophy. Mov Disord 23:1093–1099
Konno T, Ross OA, Teive HAG, Slawek J, Dickson DW, Wszolek ZK (2017) DCTN1-related neurodegeneration: Perry syndrome and beyond. Parkinsonism Relat Disord 41:14–24
Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM, Bartus RT (2013) Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136:2419–2431
Kosaka K, Tsuchiya K, Yoshimura M (1988) Lewy body disease with and without dementia: a clinicopathological study of 35 cases. Clin Neuropathol 7:299–305
Kotzbauer PT, Giasson BI, Kravitz AV, Golbe LI, Mark MH, Trojanowski JQ, Lee VM (2004) Fibrillization of alpha-synuclein and tau in familial Parkinson’s disease caused by the A53T alpha-synuclein mutation. Exp Neurol 187:279–288
Kouli A, Torsney KM, Kuan WL (2018) Parkinson’s disease: etiology, neuropathology, and pathogenesis. In: Stoker TB, Greenland JC (eds) Parkinson’s disease: pathogenesis and clinical aspects, 2019/02/01 edn. Codon Publications, Brisbane, pp 3–26. https://doi.org/10.15586/codonpublications.parkinsonsdisease.12018
Kouri N, Oshima K, Takahashi M, Murray ME, Ahmed Z, Parisi JE, Yen SH, Dickson DW (2013) Corticobasal degeneration with olivopontocerebellar atrophy and TDP-43 pathology: an unusual clinicopathologic variant of CBD. Acta Neuropathol 125:741–752
Kouri N, Carlomagno Y, Baker M, Liesinger AM, Caselli RJ, Wszolek ZK, Petrucelli L, Boeve BF, Parisi JE, Josephs KA, Uitti RJ, Ross OA, Graff-Radford NR, DeTure MA, Dickson DW, Rademakers R (2014) Novel mutation in MAPT exon 13 (p. N410H) causes corticobasal degeneration. Acta Neuropathol 127:271–282
Kouri N, Ross OA, Dombroski B, Younkin CS, Serie DJ, Soto-Ortolaza A, Baker M, Finch NC, Yoon H, Kim J, Fujioka S, McLean CA, Ghetti B, Spina S, Cantwell LB, Farlow MR, Grafman J, Huey ED, Ryung Han M, Beecher S, Geller ET, Kretzschmar HA, Roeber S, Gearing M, Juncos JL, Vonsattel JP, Van Deerlin VM, Grossman M, Hurtig HI, Gross RG, Arnold SE, Trojanowski JQ, Lee VM, Wenning GK, White CL, Hoglinger GU, Muller U, Devlin B, Golbe LI, Crook J, Parisi JE, Boeve BF, Josephs KA, Wszolek ZK, Uitti RJ, Graff-Radford NR, Litvan I, Younkin SG, Wang LS, Ertekin-Taner N, Rademakers R, Hakonarsen H, Schellenberg GD, Dickson DW (2015) Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun 6:7247
Kovacs GG, Rozemuller AJ, van Swieten JC, Gelpi E, Majtenyi K, Al-Sarraj S, Troakes C, Bodi I, King A, Hortobagyi T, Esiri MM, Ansorge O, Giaccone G, Ferrer I, Arzberger T, Bogdanovic N, Nilsson T, Leisser I, Alafuzoff I, Ironside JW, Kretzschmar H, Budka H (2013) Neuropathology of the hippocampus in FTLD-Tau with Pick bodies: a study of the BrainNet Europe Consortium. Neuropathol Appl Neurobiol 39:166–178
Kovacs GG, Robinson JL, Xie SX, Lee EB, Grossman M, Wolk DA, Irwin DJ, Weintraub D, Kim CF, Schuck T, Yousef A, Wagner ST, Suh E, Van Deerlin VM, Lee VM, Trojanowski JQ (2017) Evaluating the patterns of aging-related tau astrogliopathy unravels novel insights into brain aging and neurodegenerative diseases. J Neuropathol Exp Neurol 76:270–288
Kreisler A, Mastain B, Tison F, Fenelon G, Destee A (2007) Multi-infarct disorder presenting as corticobasal degeneration (DCB): vascular pseudo-corticobasal degeneration? Rev Neurol (Paris) 163:1191–1199
Krejciova Z, Carlson GA, Giles K, Prusiner SB (2019) Replication of multiple system atrophy prions in primary astrocyte cultures from transgenic mice expressing human alpha-synuclein. Acta Neuropathol Commun 7:81
Krismer F, Wenning GK (2017) Multiple system atrophy: insights into a rare and debilitating movement disorder. Nat Rev Neurol 13:232–243
Kruer MC (2013) The neuropathology of neurodegeneration with brain iron accumulation. Int Rev Neurobiol 110:165–194
Kübler D, Wachter T, Cabanel N, Su Z, Turkheimer FE, Dodel R, Brooks DJ, Oertel WH, Gerhard A (2019) Widespread microglial activation in multiple system atrophy. Mov Disord 34:564–568
Kujawska M, Jodynis-Liebert J (2018) What is the evidence that Parkinson’s disease is a prion disorder, which originates in the gut? Int J Mol Sci 19:3573
Kun-Rodrigues C, Orme T, Carmona S, Hernandez DG, Ross OA, Eicher JD, Shepherd C, Parkkinen L, Darwent L, Heckman MG, Scholz SW, Troncoso JC, Pletnikova O, Dawson T, Rosenthal L, Ansorge O, Clarimon J, Lleo A, Morenas-Rodriguez E, Clark L, Honig LS, Marder K, Lemstra A, Rogaeva E, St George-Hyslop P, Londos E, Zetterberg H, Barber I, Braae A, Brown K, Morgan K, Troakes C, Al-Sarraj S, Lashley T, Holton J, Compta Y, Van Deerlin V, Serrano GE, Beach TG, Lesage S, Galasko D, Masliah E, Santana I, Pastor P, Diez-Fairen M, Aguilar M, Tienari PJ, Myllykangas L, Oinas M, Revesz T, Lees A, Boeve BF, Petersen RC, Ferman TJ, Escott-Price V, Graff-Radford N, Cairns NJ, Morris JC, Pickering-Brown S, Mann D, Halliday GM, Hardy J, Trojanowski JQ, Dickson DW, Singleton A, Stone DJ, Guerreiro R, Bras J (2019) A comprehensive screening of copy number variability in dementia with Lewy bodies. Neurobiol Aging 75:223e1–223e10
Kuusisto E, Parkkinen L, Alafuzoff I (2003) Morphogenesis of Lewy bodies: dissimilar incorporation of alpha-synuclein, ubiquitin, and p62. J Neuropathol Exp Neurol 62:1241–1253
Kuzumaki N, Suda Y, Iwasawa C, Narita M, Sone T, Watanabe M, Maekawa A, Matsumoto T, Akamatsu W, Igarashi K, Tamura H, Takeshima H, Tawfik VL, Ushijima T, Hattori N, Okano H (2019) Cell-specific overexpression of COMT in dopaminergic neurons of Parkinson’s disease. Brain 142:1675–1689
La Vitola P, Beeg M, Balducci C, Santamaria G, Restelli E, Colombo L, Caldinelli L, Pollegioni L, Gobbi M, Chiesa R, Forloni G (2019) Cellular prion protein neither binds to alpha-synuclein oligomers nor mediates their detrimental effects. Brain 142:249–254
Lam B, Khan A, Keith J, Rogaeva E, Bilbao J, St George-Hyslop P, Ghani M, Freedman M, Stuss DT, Chow T, Black SE, Masellis M (2018) Characterizing familial corticobasal syndrome due to Alzheimer’s disease pathology and PSEN1 mutations. Alzheimers Dement 13:520–530
Lanciego JL, Luquin N, Obeso JA (2012) Functional neuroanatomy of the basal ganglia. Cold Spring Harb Perspect Med 2:a009621
Lane EL (2019) l-DOPA for Parkinson’s disease-a bittersweet pill. Eur J Neurosci 49:384–398
Lang AE (2011) A critical appraisal of the premotor symptoms of Parkinson’s disease: potential usefulness in early diagnosis and design of neuroprotective trials. Mov Disord 26:775–783
Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D (1999) Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol 46:598–605
Laurens B, Vergnet S, Lopez MC, Foubert-Samier A, Tison F, Fernagut PO, Meissner WG (2017) Multiple system atrophy—state of the art. Curr Neurol Neurosci Rep 17:41
Lautenschlager J, Stephens AD, Fusco G, Strohl F, Curry N, Zacharopoulou M, Michel CH, Laine R, Nespovitaya N, Fantham M, Pinotsi D, Zago W, Fraser P, Tandon A, St George-Hyslop P, Rees E, Phillips JJ, De Simone A, Kaminski CF, Schierle GSK (2018) C-terminal calcium binding of alpha-synuclein modulates synaptic vesicle interaction. Nat Commun 9:712
Lawton M, Baig F, Rolinski M, Ruffman C, Nithi K, May MT, Ben-Shlomo Y, Hu MT (2015) Parkinson’s disease subtypes in the Oxford Parkinson Disease Centre (OPDC) discovery cohort. J Parkinsons Dis 5:269–279
Lawton M, Ben-Shlomo Y, May MT, Baig F, Barber TR, Klein JC, Swallow DMA, Malek N, Grosset KA, Bajaj N, Barker RA, Williams N, Burn DJ, Foltynie T, Morris HR, Wood NW, Grosset DG, Hu MTM (2018) Developing and validating Parkinson’s disease subtypes and their motor and cognitive progression. J Neurol Neurosurg Psychiatry 89:1279–1287
Lee J-H, Lee M-S (2019) Iron accumulation in atypical parkinsonian syndromes: in vivo MRI evidences for distinctive patterns. Front Neurol 10:74. https://doi.org/10.3389/fneur.2019.00074
Lee MJ, Shin JH, Seoung JK, Lee JH, Yoon U, Oh JH, Jung DS, Kim EJ (2015) Cognitive impairments associated with morphological changes in cortical and subcortical structures in multiple system atrophy of the cerebellar type. Eur J Neurol 23:92–100
Lees AJ, Hardy J, Revesz T (2009) Parkinson’s disease. Lancet 373:2055–2066
Lehtonen S, Sonninen TM, Wojciechowski S, Goldsteins G, Koistinaho J (2019) Dysfunction of cellular proteostasis in Parkinson’s disease. Front Neurosci 13:457
Lema Tomé CM, Tyson T, Rey NL, Grathwohl S, Britschgi M, Brundin P (2013) Inflammation and alpha-synuclein’s prion-like behavior in Parkinson’s disease—is there a link? Mol Neurobiol 47:561–574
Lenka A, Ingalhalikar M, Shah A, Saini J, Arumugham SS, Hegde S, George L, Reddy V, Reddy YCJ, Yadav R, Pal PK (2018) Hippocampal subfield atrophy in patients with Parkinson’s disease and psychosis. J Neural Transm (Vienna) 125:1361–1372
Leverenz JB, Umar I, Wang Q, Montine TJ, McMillan PJ, Tsuang DW, Jin J, Pan C, Shin J, Zhu D, Zhang J (2007) Proteomic identification of novel proteins in cortical lewy bodies. Brain Pathol 17:139–145
Leverenz JB, Hamilton R, Tsuang DW, Schantz A, Vavrek D, Larson EB, Kukull WA, Lopez O, Galasko D, Masliah E, Kaye J, Woltjer R, Clark C, Trojanowski JQ, Montine TJ (2008) Empiric refinement of the pathologic assessment of Lewy-related pathology in the dementia patient. Brain Pathol 18:220–224
Li A, Paudel R, Johnson R, Courtney R, Lees AJ, Holton JL, Hardy J, Revesz T, Houlden H (2013) Pantothenate kinase-associated neurodegeneration is not a synucleinopathy. Neuropathol Appl Neurobiol 39:121–131
Li M, Ma Q, Zhao X, Wang C, Wu H, Li J, Yang W (2018) Dilemma of multiple system atrophy and spinocerebellar ataxias. J Neurol 265:2764–2772
Li X, Koudstaal W, Fletcher L, Costa M, van Winsen M, Siregar B, Inganas H, Kim J, Keogh E, Macedo J, Holland T, Perry S, Bard F, Hoozemans JJ, Goudsmit J, Apetri A, Pascual G (2019) Naturally occurring antibodies isolated from PD patients inhibit synuclein seeding in vitro and recognize Lewy pathology. Acta Neuropathol 137:825–836
Licker V, Turck N, Kovari E, Burkhardt K, Cote M, Surini-Demiri M, Lobrinus JA, Sanchez JC, Burkhard PR (2014) Proteomic analysis of human substantia nigra identifies novel candidates involved in Parkinson’s disease pathogenesis. Proteomics 14:784–794
Lill CM (2016) Genetics of Parkinson’s disease. Mol Cell Probes 30:386–396
Lim KL, Zhang CW (2013) Molecular events underlying Parkinson’s disease—an interwoven tapestry. Front Neurol 4:33
Lim EW, Aarsland D, Ffytche D, Taddei RN, van Wamelen DJ, Wan YM, Tan EK, Ray Chaudhuri K (2018) Amyloid-beta and Parkinson’s disease. J Neurol. https://doi.org/10.1007/s00415-00018-09100-00418
Lima VA, do Nascimento LA, Eliezer D, Follmer C (2019) Role of Parkinson’s disease-linked mutations and n-terminal acetylation on the oligomerization of alpha-synuclein induced by 3,4-dihydroxyphenylacetaldehyde. ACS Chem Neurosci 10:690–703
Limphaibool N, Iwanowski P, Holstad MJV, Perkowska K (2018) Parkinsonism in inherited metabolic disorders: key considerations and major features. Front Neurol 9:857
Lin YW, Truong D (2019) Diffuse Lewy body disease. J Neurol Sci 399:144–150
Lin CH, Tan EK, Yang CC, Yi Z, Wu RM (2015) COQ2 gene variants associate with cerebellar subtype of multiple system atrophy in Chinese. Mov Disord 30:436–437
Lin G, Wang L, Marcogliese PC, Bellen HJ (2019) Sphingolipids in the pathogenesis of Parkinson’s disease and parkinsonism. Trends Endocrinol Metab 30:106–117
Ling H, Holton JL, Lees AJ, Revesz T (2013) TDP-43 pathology is present in most post-encephalitic parkinsonism brains (Scientific correspondence). Neuropathol Appl Neurobiol 40:654–657
Ling H, de Silva R, Massey LA, Courtney R, Hondhamuni G, Bajaj N, Lowe J, Holton JL, Lees A, Revesz T (2014) Characteristics of progressive supranuclear palsy presenting with corticobasal syndrome: a cortical variant. Neuropathol Appl Neurobiol 40:149–163
Ling H, Asi YT, Petrovic IN, Ahmed Z, Prashanth LK, Hazrati LN, Nishizawa M, Ozawa T, Lang A, Lees AJ, Revesz T, Holton JL (2015) Minimal change multiple system atrophy: an aggressive variant? Mov Disord 30:960–967
Ling H, Kovacs GG, Vonsattel JP, Davey K, Mok KY, Hardy J, Morris HR, Warner TT, Holton JL, Revesz T (2016) Astrogliopathy predominates the earliest stage of corticobasal degeneration pathology. Brain 139:3237–3252
Lionnet A, Leclair-Visonneau L, Neunlist M, Murayama S, Takao M, Adler CH, Derkinderen P, Beach TG (2018) Does Parkinson’s disease start in the gut? Acta Neuropathol 135:1–12
Liu M, Choi DY, Hunter RL, Pandya JD, Cass WA, Sullivan PG, Kim HC, Gash DM, Bing G (2010) Trichloroethylene induces dopaminergic neurodegeneration in Fisher 344 rats. J Neurochem 112:773–783
Liu AKL, Lim EJ, Ahmed I, Chang RC, Pearce RKB, Gentleman SM (2018) Review: revisiting the human cholinergic nucleus of the diagonal band of Broca. Neuropathol Appl Neurobiol 44:647–662
Liu AKL, Chau TW, Lim EJ, Ahmed I, Chang RC, Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RKB (2019a) Hippocampal CA2 Lewy pathology is associated with cholinergic degeneration in Parkinson’s disease with cognitive decline. Acta Neuropathol Commun 7:61
Liu X, Hebron M, Shi W, Lonskaya I, Moussa CE (2019b) Ubiquitin specific protease-13 independently regulates parkin ubiquitination and alpha-synuclein clearance in alpha-synucleinopathies. Hum Mol Genet 28:548–560
Liu JQ, Chu SF, Zhou X, Zhang DY, Chen NH (2019c) Role of chemokines in Parkinson’s disease. Brain Res Bull. https://doi.org/10.1016/j.brainresbull.2019.1005.1020
Longhena F, Faustini G, Missale C, Pizzi M, Spano P, Bellucci A (2017) The contribution of alpha-synuclein spreading to Parkinson’s disease synaptopathy. Neural Plast 2017:5012129
Longhena F, Faustini G, Varanita T, Zaltieri M, Porrini V, Tessari I, Poliani PL, Missale C, Borroni B, Padovani A, Bubacco L, Pizzi M, Spano P, Bellucci A (2018) Synapsin III is a key component of alpha-synuclein fibrils in Lewy bodies of PD brains. Brain Pathol 28:875–888
Lopez KLR, Simpson JE, Watson LC, Mortiboys H, Hautbergue GM, Bandmann O, Highley JR (2019) TIGAR inclusion pathology is specific for Lewy body diseases. Brain Res 1706:218–223
Lu X, Kim-Han JS, Harmon S, Sakiyama-Elbert SE, O’Malley KL (2014) The Parkinsonian mimetic, 6-OHDA, impairs axonal transport in dopaminergic axons. Mol Neurodegener 9:17
Lubbe S, Morris HR (2014) Recent advances in Parkinson’s disease genetics. J Neurol 261:259–266
Ludtmann MHR, Angelova PR, Horrocks MH, Choi ML, Rodrigues M, Baev AY, Berezhnov AV, Yao Z, Little D, Banushi B, Al-Menhali AS, Ranasinghe RT, Whiten DR, Yapom R, Dolt KS, Devine MJ, Gissen P, Kunath T, Jaganjac M, Pavlov EV, Klenerman D, Abramov AY, Gandhi S (2018) Alpha-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat Commun 9:2293
Lue L-F, Walker DG, Adler CH, Shill H, Tran H, Akiyama H, Sue LI, Caviness J, Sabbagh MN, Beach TG (2012) Biochemical increase in phosphorylated a-synuclein precedes histopathology of Lewy-type synucleinopathies. Brain Pathol 22:745–756
Lunati A, Lesage S, Brice A (2018) The genetic landscape of Parkinson’s disease. Rev Neurol (Paris) 174:628–643
Ma LY, Liu GL, Wang DX, Zhang MM, Kou WY, Feng T (2019) Alpha-synuclein in peripheral tissues in Parkinson’s disease. ACS Chem Neurosci 10:812–823
Maeda S, Sato Y, Takashima A (2018) Frontotemporal dementia with parkinsonism linked to chromosome-17 mutations enhance tau oligomer formation. Neurobiol Aging 69:26–32
Maffeo E, Montuschi A, Stura G, Giordana MT (2014) Chronic acquired hepatocerebral degeneration, pallidal T1 MRI hyperintensity and manganese in a series of cirrhotic patients. Neurol Sci 35:523–530
Maillet A, Krack P, Lhommee E, Metereau E, Klinger H, Favre E, Le Bars D, Schmitt E, Bichon A, Pelissier P, Fraix V, Castrioto A, Sgambato-Faure V, Broussolle E, Tremblay L, Thobois S (2016) The prominent role of serotonergic degeneration in apathy, anxiety and depression in de novo Parkinson’s disease. Brain 139:2486–2502
Maiti P, Manna J, Dunbar GL (2017) Current understanding of the molecular mechanisms in Parkinson’s disease: targets for potential treatments. Transl Neurodegener 6:28
Mak E, Donaghy PC, McKiernan E, Firbank MJ, Lloyd J, Petrides GS, Thomas AJ, O’Brien JT (2019) Beta amyloid deposition maps onto hippocampal and subiculum atrophy in dementia with Lewy bodies. Neurobiol Aging 73:74–81
Malandrini A, Rubegni A, Battisti C, Berti G, Federico A (2013) Electron-dense lamellated inclusions in 2 siblings with Kufor-Rakeb syndrome. Mov Disord 28:1751–1752
Mandel RJ, Marmion DJ, Kirik D, Chu Y, Heindel C, McCown T, Gray SJ, Kordower JH (2017) Novel oligodendroglial alpha synuclein viral vector models of multiple system atrophy: studies in rodents and nonhuman primates. Acta Neuropathol Commun 5:47
Maor G, Rapaport D, Horowitz M (2019) The effect of mutant GBA1 on accumulation and aggregation of alpha-synuclein. Hum Mol Genet 28:1768–1781
Marder K (2010) Cognitive impairment and dementia in Parkinson’s disease. Mov Disord 25(Suppl 1):S110–S116
Markesbery WR (2010) Neuropathologic alterations in mild cognitive impairment: a review. J Alzheimers Dis 19:221–228
Markesbery WR, Jicha GA, Liu H, Schmitt FA (2009) Lewy body pathology in normal elderly subjects. J Neuropathol Exp Neurol 68:816–822
Markopoulou K, Dickson DW, McComb RD, Wszolek ZK, Katechalidou L, Avery L, Stansbury MS, Chase BA (2008) Clinical, neuropathological and genotypic variability in SNCA A53T familial Parkinson’s disease. Variability in familial Parkinson’s disease. Acta Neuropathol 116:25–35
Marras C, Alcalay RN, Caspell-Garcia C, Coffey C, Chan P, Duda JE, Facheris MF, Fernandez-Santiago R, Ruiz-Martinez J, Mestre T, Saunders-Pullman R, Pont-Sunyer C, Tolosa E, Waro B (2016) Motor and nonmotor heterogeneity of LRRK2-related and idiopathic Parkinson’s disease. Mov Disord 31:1192–1202
Marras C, Lang A, van de Warrenburg BP, Sue CM, Tabrizi SJ, Bertram L, Mercimek-Mahmutoglu S, Ebrahimi-Fakhari D, Warner TT, Durr A, Assmann B, Lohmann K, Kostic V, Klein C (2017) Nomenclature of genetic movement disorders: recommendations of the International Parkinson and Movement Disorder Society task force. Mov Disord 32:724–725
Marras C, Beck JC, Bower JH, Roberts E, Ritz B, Ross GW, Abbott RD, Savica R, Van Den Eeden SK, Willis AW, Tanner CM (2018) Prevalence of Parkinson’s disease across North America. NPJ Parkinsons Dis 4:21
Martin-Bastida A, Lao-Kaim NP, Roussakis AA, Searle GE, Xing Y, Gunn RN, Schwarz ST, Barker RA, Auer DP, Piccini P (2019) Relationship between neuromelanin and dopamine terminals within the Parkinson's nigrostriatal system. Brain. https://doi.org/10.1093/brain/awz1120
Masaracchia C, Hnida M, Gerhardt E, Lopes da Fonseca T, Villar-Pique A, Branco T, Stahlberg MA, Dean C, Fernandez CO, Milosevic I, Outeiro TF (2018) Membrane binding, internalization, and sorting of alpha-synuclein in the cell. Acta Neuropathol Commun 6:79
Masui K, Nakata Y, Fujii N, Iwaki T (2012) Extensive distribution of glial cytoplasmic inclusions in an autopsied case of multiple system atrophy with a prolonged 18-year clinical course. Neuropathology 32:69–76
Matsuo A, Akiguchi I, Lee GC, McGeer EG, McGeer PL, Kimura J (1998) Myelin degeneration in multiple system atrophy detected by unique antibodies. Am J Pathol 153:735–744
Matsusue E, Fujii S, Kanasaki Y, Sugihara S, Miyata H, Ohama E, Ogawa T (2008) Putaminal lesion in multiple system atrophy: postmortem MR-pathological correlations. Neuroradiology 50:559–567
Mavroeidi P, Arvanitaki F, Karakitsou AK, Vetsi M, Kloukina I, Zweckstetter M, Giller K, Becker S, Sorrentino ZA, Giasson BI, Jensen PH, Stefanis L, Xilouri M (2019) Endogenous oligodendroglial alpha-synuclein and TPPP/p25alpha orchestrate alpha-synuclein pathology in experimental multiple system atrophy models. Acta Neuropathol. https://doi.org/10.1007/s00401-00019-02014-y
McAleese KE, Walker L, Erskine D, Thomas AJ, McKeith IG, Attems J (2017) TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing. Brain Pathol 27:472–479
McCormack A, Chegeni N, Chegini F, Colella A, Power J, Keating D, Chataway T (2016) Purification of alpha-synuclein containing inclusions from human post mortem brain tissue. J Neurosci Methods 266:141–150
McCormack A, Keating DJ, Chegeni N, Colella A, Wang JJ, Chataway T (2019) Abundance of synaptic vesicle-related proteins in alpha-synuclein-containing protein inclusions suggests a targeted formation mechanism. Neurotox Res 35:883–897
McGregor MM, Nelson AB (2019) Circuit mechanisms of Parkinson’s disease. Neuron 101:1042–1056
McKee AC, Stein TD, Kiernan PT, Alvarez VE (2015) The neuropathology of chronic traumatic encephalopathy. Brain Pathol 25:350–364
McKee AC, Abdolmohammadi B, Stein TD (2018) The neuropathology of chronic traumatic encephalopathy. Handb Clin Neurol 158:297–307
McKeith I (2007) Dementia with Lewy bodies and Parkinson’s disease with dementia: where two worlds collide. Pract Neurol 7:374–382
McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65:1863–1872
McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D, Aarsland D, Galvin J, Attems J, Ballard CG, Bayston A, Beach TG, Blanc F, Bohnen N, Bonanni L, Bras J, Brundin P, Burn D, Chen-Plotkin A, Duda JE, El-Agnaf O, Feldman H, Ferman TJ, Ffytche D, Fujishiro H, Galasko D, Goldman JG, Gomperts SN, Graff-Radford NR, Honig LS, Iranzo A, Kantarci K, Kaufer D, Kukull W, Lee VMY, Leverenz JB, Lewis S, Lippa C, Lunde A, Masellis M, Masliah E, McLean P, Mollenhauer B, Montine TJ, Moreno E, Mori E, Murray M, O’Brien JT, Orimo S, Postuma RB, Ramaswamy S, Ross OA, Salmon DP, Singleton A, Taylor A, Thomas A, Tiraboschi P, Toledo JB, Trojanowski JQ, Tsuang D, Walker Z, Yamada M, Kosaka K (2017) Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89:88–100
Mehanna R, Jankovic J (2013) Movement disorders in cerebrovascular disease. Lancet Neurol 12:597–608
Mehra S, Sahay S, Maji SK (2019) Alpha-synuclein misfolding and aggregation: implications in Parkinson’s disease pathogenesis. Biochim Biophys Acta Proteins Proteomics. https://doi.org/10.1016/j.bbapap.2019.1003.1001
Melzer TR, Stark MR, Keenan RJ, Myall DJ, MacAskill MR, Pitcher TL, Livingston L, Grenfell S, Horne KL, Young BN, Pascoe MJ, Almuqbel MM, Wang J, Marsh SH, Miller DH, Dalrymple-Alford JC, Anderson TJ (2019) Beta amyloid deposition is not associated with cognitive impairment in Parkinson’s disease. Front Neurol 10:391
Mendoza-Santiesteban CE, Palma JA, Martinez J, Norcliffe-Kaufmann L, Hedges TR 3rd, Kaufmann H (2015) Progressive retinal structure abnormalities in multiple system atrophy. Mov Disord 30:1944–1953
Mensikova K, Tuckova L, Kolarikova K, Bartonikova T, Vodicka R, Ehrmann J, Vrtel R, Prochazka M, Kanovsky P, Kovacs GG (2019) Atypical parkinsonism of progressive supranuclear palsy-parkinsonism (PSP-P) phenotype with rare variants in FBXO7 and VPS35 genes associated with Lewy body pathology. Acta Neuropathol 137:171–173
Meredith GE, Rademacher DJ (2011) MPTP mouse models of Parkinson’s disease: an update. J Parkinsons Dis 1:19–33
Michel PP, Hirsch EC, Hunot S (2016) Understanding dopaminergic cell death pathways in Parkinson disease. Neuron 90:675–691
Mietelska-Porowska A, Wasik U, Goras M, Filipek A, Niewiadomska G (2014) Tau protein modifications and interactions: their role in function and dysfunction. Int J Mol Sci 15:4671–4713
Mihaescu AS, Masellis M, Graff-Guerrero A, Kim J, Criaud M, Cho SS, Ghadery C, Valli M, Strafella AP (2018) Brain degeneration in Parkinson’s disease patients with cognitive decline: a coordinate-based meta-analysis. Brain Imaging Behav. https://doi.org/10.1007/s11682-11018-19922-11680
Mikasa M, Kanai K, Li Y, Yoshino H, Mogushi K, Hayashida A, Ikeda A, Kawajiri S, Okuma Y, Kashihara K, Sato T, Kondo H, Funayama M, Nishioka K, Hattori N (2018) COQ2 variants in Parkinson’s disease and multiple system atrophy. J Neural Transm (Vienna) 125:937–944
Miklossy J, Steele JC, Yu S, McCall S, Sandberg G, McGeer EG, McGeer PL (2008) Enduring involvement of tau, beta-amyloid, alpha-synuclein, ubiquitin and TDP-43 pathology in the amyotrophic lateral sclerosis/parkinsonism-dementia complex of Guam (ALS/PDC). Acta Neuropathol 116:625–637
Milosevic L, Gramer R, Kim TH, Algarni M, Fasano A, Kalia SK, Hodaie M, Lozano AM, Popovic MR, Hutchison WD (2019) Modulation of inhibitory plasticity in basal ganglia output nuclei of patients with Parkinson’s disease. Neurobiol Dis 124:46–56
Mimuro M, Yoshida M, Kuzuhara S, Kokubo Y (2018) Amyotrophic lateral sclerosis and parkinsonism-dementia complex of the Hohara focus of the Kii Peninsula: a multiple proteinopathy? Neuropathology 38:98–107
Mishima T, Koga S, Lin WL, Kasanuki K, Castanedes-Casey M, Wszolek ZK, Oh SJ, Tsuboi Y, Dickson DW (2017) Perry syndrome: a distinctive type of TDP-43 proteinopathy. J Neuropathol Exp Neurol 76:676–682
Mishima T, Fujioka S, Tomiyama H, Yabe I, Kurisaki R, Fujii N, Neshige R, Ross OA, Farrer MJ, Dickson DW, Wszolek ZK, Hattori N, Tsuboi Y (2018) Establishing diagnostic criteria for Perry syndrome. J Neurol Neurosurg Psychiatry 89:482–487
Mitsui J, Matsukawa T, Sasaki H, Yabe I, Matsushima M, Durr A, Brice A, Takashima H, Kikuchi A, Aoki M, Ishiura H, Yasuda T, Date H, Ahsan B, Iwata A, Goto J, Ichikawa Y, Nakahara Y, Momose Y, Takahashi Y, Hara K, Kakita A, Yamada M, Takahashi H, Onodera O, Nishizawa M, Watanabe H, Ito M, Sobue G, Ishikawa K, Mizusawa H, Kanai K, Hattori T, Kuwabara S, Arai K, Koyano S, Kuroiwa Y, Hasegawa K, Yuasa T, Yasui K, Nakashima K, Ito H, Izumi Y, Kaji R, Kato T, Kusunoki S, Osaki Y, Horiuchi M, Kondo T, Murayama S, Hattori N, Yamamoto M, Murata M, Satake W, Toda T, Filla A, Klockgether T, Wullner U, Nicholson G, Gilman S, Tanner CM, Kukull WA, Stern MB, Lee VM, Trojanowski JQ, Masliah E, Low PA, Sandroni P, Ozelius LJ, Foroud T, Tsuji S (2015) Variants associated with Gaucher disease in multiple system atrophy. Ann Clin Transl Neurol 2:417–426
Miyaji Y, Koyama K, Kurokawa T, Mitomi M, Suzuki Y, Kuroiwa Y (2013) Vascular corticobasal syndrome caused by unilateral internal carotid artery occlusion. J Stroke Cerebrovasc Dis 22:1193–1195
Molano J, Boeve B, Ferman T, Smith G, Parisi J, Dickson D, Knopman D, Graff-Radford N, Geda Y, Lucas J, Kantarci K, Shiung M, Jack C, Silber M, Pankratz VS, Petersen R (2010) Mild cognitive impairment associated with limbic and neocortical Lewy body disease: a clinicopathological study. Brain 133:540–556
Monzio Compagnoni G, Kleiner G, Samarani M, Aureli M, Faustini G, Bellucci A, Ronchi D, Bordoni A, Garbellini M, Salani S, Fortunato F, Frattini E, Abati E, Bergamini C, Fato R, Tabano S, Miozzo M, Serratto G, Passafaro M, Deleidi M, Silipigni R, Nizzardo M, Bresolin N, Comi GP, Corti S, Quinzii CM, Di Fonzo A (2018) Mitochondrial dysregulation and impaired autophagy in iPSC-derived dopaminergic neurons of multiple system atrophy. Stem Cell Rep 11:1185–1198
Mor DE, Tsika E, Mazzulli JR, Gould NS, Kim H, Daniels MJ, Doshi S, Gupta P, Grossman JL, Tan VX, Kalb RG, Caldwell KA, Caldwell GA, Wolfe JH, Ischiropoulos H (2017) Dopamine induces soluble alpha-synuclein oligomers and nigrostriatal degeneration. Nat Neurosci 20:1560–1568
Mor DE, Daniels MJ, Ischiropoulos H (2019) The usual suspects, dopamine and alpha-synuclein, conspire to cause neurodegeneration. Mov Disord 34:167–179
Morbelli S, Chincarini A, Brendel M, Rominger A, Bruffaerts R, Vandenberghe R, Kramberger MG, Trost M, Garibotto V, Nicastro N, Frisoni GB, Lemstra AW, van der Zande J, Pilotto A, Padovani A, Garcia-Ptacek S, Savitcheva I, Ochoa-Figueroa MA, Davidsson A, Camacho V, Peira E, Arnaldi D, Bauckneht M, Pardini M, Sambuceti G, Aarsland D, Nobili F (2019) Metabolic patterns across core features in dementia with Lewy bodies. Ann Neurol 85:715–725
Mori F, Inenaga C, Yoshimoto M, Umezu H, Tanaka R, Takahashi H, Wakabayashi K (2002) Alpha-synuclein immunoreactivity in normal and neoplastic Schwann cells. Acta Neuropathol 103:145–151
Mori F, Nishie M, Kakita A, Yoshimoto M, Takahashi H, Wakabayashi K (2006) Relationship among alpha-synuclein accumulation, dopamine synthesis, and neurodegeneration in Parkinson disease substantia nigra. J Neuropathol Exp Neurol 65:808–815
Mori F, Okada KI, Nomura T, Kobayashi Y (2016) The pedunculopontine tegmental nucleus as a motor and cognitive interface between the cerebellum and basal ganglia. Front Neuroanat 10:109
Morimoto S, Hatsuta H, Kokubo Y, Nakano Y, Hasegawa M, Yoneda M, Hirokawa Y, Kuzuhara S, Shiraishi T, Murayama S (2018) Unusual tau pathology of the cerebellum in patients with amyotrophic lateral sclerosis/parkinsonism-dementia complex from the Kii Peninsula, Japan. Brain Pathol 28:287–291
Morita K, Kawaguchi Y (2019) A dual role hypothesis of the cortico-basal-ganglia pathways: opponency and temporal difference through dopamine and adenosine. Front Neural Circuits 12:111
Mosharov EV, Borgkvist A, Sulzer D (2015) Presynaptic effects of levodopa and their possible role in dyskinesia. Mov Disord 30:45–53
Moskvina V, Harold D, Russo G, Vedernikov A, Sharma M, Saad M, Holmans P, Bras JM, Bettella F, Keller MF, Nicolaou N, Simon-Sanchez J, Gibbs JR, Schulte C, Durr A, Guerreiro R, Hernandez D, Brice A, Stefansson H, Majamaa K, Gasser T, Heutink P, Wood N, Martinez M, Singleton AB, Nalls MA, Hardy J, Owen MJ, O’Donovan MC, Williams J, Morris HR, Williams NM (2013) Analysis of genome-wide association studies of Alzheimer disease and of Parkinson disease to determine if these 2 diseases share a common genetic risk. JAMA Neurol 70:1268–1276
MSA-Research-Collaboration, Collaboration M-SAR (2013) Mutations in COQ2 in familial and sporadic multiple-system atrophy. N Engl J Med 369:233–244
Mudher A, Colin M, Dujardin S, Medina M, Dewachter I, Alavi Naini SM, Mandelkow EM, Mandelkow E, Buee L, Goedert M, Brion JP (2017) What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol Commun 5:99
Mulak A, Bonaz B (2015) Brain-gut-microbiota axis in Parkinson’s disease. World J Gastroenterol 21:10609–10620
Muller SK, Bender A, Laub C, Hogen T, Schlaudraff F, Liss B, Klopstock T, Elstner M (2013) Lewy body pathology is associated with mitochondrial DNA damage in Parkinson’s disease. Neurobiol Aging 34:2231–2233
Mullin S, Schapira A (2013) Alpha-synuclein and mitochondrial dysfunction in Parkinson’s disease. Mol Neurobiol 47:587–597
Mullin S, Hughes D, Mehta A, Schapira AHV (2019) Neurological effects of glucocerebrosidase gene mutations. Eur J Neurol 26:e329–e388
Munch G, Luth HJ, Wong A, Arendt T, Hirsch E, Ravid R, Riederer P (2000) Crosslinking of alpha-synuclein by advanced glycation endproducts—an early pathophysiological step in Lewy body formation? J Chem Neuroanat 20:253–257
Munoz DG, Morris HR, Rossor M (2011) Pick’s disease. In: Dickson DW, Weller RO (eds) Neurodegeneration: the molecular pathology of dementia and movement disorders, 2nd edn. Blackwell Publishing Ltd., Oxford, pp 156–164
Murray ME, Kouri N, Lin WL, Jack CR Jr, Dickson DW, Vemuri P (2014) Clinicopathologic assessment and imaging of tauopathies in neurodegenerative dementias. Alzheimers Res Ther 6:1
Naasan G, Shany-Ur T, Sidhu M, Barton C, Ketelle R, Shdo SM, Kramer JH, Miller BL, Seeley WW (2019) Corticobasal syndrome with visual hallucinations and probable REM-sleep behavior disorder: an autopsied case report of a patient with CBD and LBD pathology. Neurocase 25:26–33
Nakamoto FK, Okamoto S, Mitsui J, Sone T, Ishikawa M, Yamamoto Y, Kanegae Y, Nakatake Y, Imaizumi K, Ishiura H, Tsuji S, Okano H (2018) The pathogenesis linked to coenzyme Q10 insufficiency in iPSC-derived neurons from patients with multiple-system atrophy. Sci Rep 8:14215
Nakamura K, Mori F, Kon T, Tanji K, Miki Y, Tomiyama M, Kurotaki H, Toyoshima Y, Kakita A, Takahashi H, Yamada M, Wakabayashi K (2015) Filamentous aggregations of phosphorylated alpha-synuclein in Schwann cells (Schwann cell cytoplasmic inclusions) in multiple system atrophy. Acta Neuropathol Commun 3:29
Nakata Y, Yasuda T, Fukaya M, Yamamori S, Itakura M, Nihira T, Hayakawa H, Kawanami A, Kataoka M, Nagai M, Sakagami H, Takahashi M, Mizuno Y, Mochizuki H (2012) Accumulation of alpha-synuclein triggered by presynaptic dysfunction. J Neurosci 32:17186–17196
Narasimhan S, Guo JL, Changolkar L, Stieber A, McBride JD, Silva LV, He Z, Zhang B, Gathagan RJ, Trojanowski JQ, Lee VMY (2017) Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J Neurosci 37:11406–11423
Nardone R, Holler Y, Brigo F, Versace V, Sebastianelli L, Florea C, Schwenker K, Golaszewski S, Saltuari L, Trinka E (2019) Spinal cord involvement in Lewy body-related alpha-synucleinopathies. J Spinal Cord Med. https://doi.org/10.1080/10790268.10792018.11557863
Ndayisaba A, Jellinger K, Berger T, Wenning GK (2019) TNFalpha inhibitors as targets for protective therapies in MSA: a viewpoint. J Neuroinflamm 16:80
Nejad-Davarani S, Koeppe RA, Albin RL, Frey KA, Muller M, Bohnen NI (2019) Quantification of brain cholinergic denervation in dementia with Lewy bodies using PET imaging with [(18)F]-FEOBV. Mol Psychiatry 24:322–327
Nervi A, Reitz C, Tang MX, Santana V, Piriz A, Reyes D, Lantigua R, Medrano M, Jimenez-Velazquez IZ, Lee JH, Mayeux R (2011) Familial aggregation of dementia with Lewy bodies. Arch Neurol 68:90–93
Neumann M, Mackenzie IRA (2019) Review: neuropathology of non-tau frontotemporal lobar degeneration. Neuropathol Appl Neurobiol 45:19–40
Neumann WJ, Schroll H, de Almeida Marcelino AL, Horn A, Ewert S, Irmen F, Krause P, Schneider GH, Hamker F, Kuhn AA (2018) Functional segregation of basal ganglia pathways in Parkinson’s disease. Brain 141:2655–2669
Nguyen M, Wong YC, Ysselstein D, Severino A, Krainc D (2019) Synaptic, mitochondrial, and lysosomal dysfunction in Parkinson’s disease. Trends Neurosci 42:140–149
Ni Z, Pinto AD, Lang AE, Chen R (2010) Involvement of the cerebellothalamocortical pathway in Parkinson disease. Ann Neurol 68:816–824
Nishida N, Yoshida K, Hata Y, Arai Y, Kinoshita K (2015) Pathological features of preclinical or early clinical stages of corticobasal degeneration: a comparison with advanced cases. Neuropathol Appl Neurobiol 41:893–905
Niu H, Shen L, Li T, Ren C, Ding S, Wang L, Zhang Z, Liu X, Zhang Q, Geng D, Wu X, Li H (2018) Alpha-synuclein overexpression in the olfactory bulb initiates prodromal symptoms and pathology of Parkinson’s disease. Transl Neurodegener 7:25
Nouraei N, Mason DM, Miner KM, Carcella MA, Bhatia TN, Dumm BK, Soni D, Johnson DA, Luk KC, Leak RK (2018) Critical appraisal of pathology transmission in the alpha-synuclein fibril model of Lewy body disorders. Exp Neurol 299:172–196
Nussbaum RL (2018) Genetics of synucleinopathies. Cold Spring Harb Perspect Med 8:78. https://doi.org/10.1101/cshperspect.a024109
Nykjaer C, Brudek T, Salvesen L, Pakkenberg B (2017) Changes in the cell population in brain white matter in multiple system atrophy. Mov Disord 32:1074–1082
Obeso JA, Stamelou M, Goetz CG, Poewe W, Lang AE, Weintraub D, Burn D, Halliday GM, Bezard E, Przedborski S, Lehericy S, Brooks DJ, Rothwell JC, Hallett M, DeLong MR, Marras C, Tanner CM, Ross GW, Langston JW, Klein C, Bonifati V, Jankovic J, Lozano AM, Deuschl G, Bergman H, Tolosa E, Rodriguez-Violante M, Fahn S, Postuma RB, Berg D, Marek K, Standaert DG, Surmeier DJ, Olanow CW, Kordower JH, Calabresi P, Schapira AHV, Stoessl AJ (2017) Past, present, and future of Parkinson’s disease: a special essay on the 200th Anniversary of the Shaking Palsy. Mov Disord 32:1264–1310
Odagiri S, Tanji K, Mori F, Kakita A, Takahashi H, Wakabayashi K (2012) Autophagic adapter protein NBR1 is localized in Lewy bodies and glial cytoplasmic inclusions and is involved in aggregate formation in alpha-synucleinopathy. Acta Neuropathol 124:173–186
Ogaki K, Martens YA, Heckman MG, Koga S, Labbe C, Lorenzo-Betancor O, Wernick AI, Walton RL, Soto AI, Vargas ER, Nielsen HM, Fujioka S, Kanekiyo T, Uitti RJ, van Gerpen JA, Cheshire WP, Wszolek ZK, Low PA, Singer W, Dickson DW, Bu G, Ross OA (2018) Multiple system atrophy and apolipoprotein E. Mov Disord 33:647–650
Oinas M, Paetau A, Myllykangas L, Notkola IL, Kalimo H, Polvikoski T (2010) Alpha-synuclein pathology in the spinal cord autonomic nuclei associates with alpha-synuclein pathology in the brain: a population-based Vantaa 85+ study. Acta Neuropathol 119:715–722
O’Keeffe GW, Sullivan AM (2018) Evidence for dopaminergic axonal degeneration as an early pathological process in Parkinson’s disease. Parkinsonism Relat Disord 56:9–15
Olah J, Ovadi J (2019) Pharmacological targeting of alpha-synuclein and TPPP/p25 in Parkinson’s disease: challenges and opportunities in a Nutshell. FEBS Lett. https://doi.org/10.1002/1873-3468.13464
Olah J, Bertrand P, Ovadi J (2017) Role of the microtubule-associated TPPP/p25 in Parkinson’s and related diseases and its therapeutic potential. Expert Rev Proteom 14:301–309
Olanow CW, Savolainen M, Chu Y, Halliday GM, Kordower JH (2019) Temporal evolution of microglia and alpha-synuclein accumulation following foetal grafting in Parkinson’s disease. Brain 142:1690–1700
Oppedal K, Ferreira D, Cavallin L, Lemstra AW, Ten Kate M, Padovani A, Rektorova I, Bonanni L, Wahlund LO, Engedal K, Nobili F, Kramberger M, Taylor JP, Hort J, Snaedal J, Blanc F, Walker Z, Antonini A, Westman E, Aarsland D (2019) A signature pattern of cortical atrophy in dementia with Lewy bodies: a study on 333 patients from the European DLB consortium. Alzheimers Dement 15:400–409
O’Regan G, deSouza RM, Balestrino R, Schapira AH (2017) Glucocerebrosidase mutations in Parkinson disease. J Parkinsons Dis 7:411–422
Orimo S, Uchihara T, Nakamura A, Mori F, Kakita A, Wakabayashi K, Takahashi H (2008) Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 131:642–650
Orme T, Guerreiro R, Bras J (2018) The genetics of dementia with Lewy bodies: current understanding and future directions. Curr Neurol Neurosci Rep 18:67
Ortega-de San Luis C, Sanchez-Garcia MA, Nieto-Gonzalez JL, Garcia-Junco-Clemente P, Montero-Sanchez A, Fernandez-Chacon R, Pascual A (2018) Substantia nigra dopaminergic neurons and striatal interneurons are engaged in three parallel but interdependent postnatal neurotrophic circuits. Aging Cell 17:e12821
Ortuno-Lizaran I, Esquiva G, Beach TG, Serrano GE, Adler CH, Lax P, Cuenca N (2018) Degeneration of human photosensitive retinal ganglion cells may explain sleep and circadian rhythms disorders in Parkinson’s disease. Acta Neuropathol Commun 6:90
Osaki Y, Ben-Shlomo Y, Lees AJ, Wenning GK, Quinn NP (2009) A validation exercise on the new consensus criteria for multiple system atrophy. Mov Disord 24:2272–2276
Ou R, Wei Q, Hou Y, Yuan X, Song W, Cao B, Liu H, Zhang L, Chen Y, Shang H (2018) Vascular risk factors and depression in Parkinson’s disease. Eur J Neurol 25:637–643
Ouchi H, Toyoshima Y, Tada M, Oyake M, Aida I, Tomita I, Satoh A, Tsujihata M, Takahashi H, Nishizawa M, Shimohata T (2014) Pathology and sensitivity of current clinical criteria in corticobasal syndrome. Mov Disord 29:238–244
Outeiro TF, Harvey K, Dominguez-Meijide A, Gerhardt E (2019) LRRK2, alpha-synuclein, and tau: partners in crime or unfortunate bystanders? Biochem Soc Trans. https://doi.org/10.1042/BST20180466
Outeiro TF, Koss DJ, Erskine D, Walker L, Kurzawa-Akanbi M, Burn D, Donaghy P, Morris C, Taylor JP, Thomas A, Attems J, McKeith I (2019b) Dementia with Lewy bodies: an update and outlook. Mol Neurodegener 14:5
Overk C, Rockenstein E, Valera E, Stefanova N, Wenning G, Masliah E (2018) Multiple system atrophy: experimental models and reality. Acta Neuropathol 135:33–47
Oyanagi K, Hashimoto T, Yamazaki M (2011) Parkinsonism-dementia complex of Guam. In: Dickson DW, Weller RO (eds) Neurodegeneration: the molecular pathology of dementia and movement disorders, 2nd edn. Blackwell Publishing Ltd., Oxford, pp 171–178
Ozawa T (2007) Morphological substrate of autonomic failure and neurohormonal dysfunction in multiple system atrophy: impact on determining phenotype spectrum. Acta Neuropathol (Berl) 114:201–211
Ozawa T, Onodera O (2017) Multiple system atrophy: clinicopathological characteristics in Japanese patients. Proc Jpn Acad Ser B Phys Biol Sci 93:251–258
Pagano G, Niccolini F, Politis M (2018) The serotonergic system in Parkinson’s patients with dyskinesia: evidence from imaging studies. J Neural Transm (Vienna) 125:1217–1223
Paisan-Ruiz C, Guevara R, Federoff M, Hanagasi H, Sina F, Elahi E, Schneider SA, Schwingenschuh P, Bajaj N, Emre M, Singleton AB, Hardy J, Bhatia KP, Brandner S, Lees AJ, Houlden H (2010) Early-onset l-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov Disord 25:1791–1800
Pan T, Kondo S, Le W, Jankovic J (2008) The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain 131:1969–1978
Pan PY, Zhu Y, Shen Y, Yue Z (2019) Crosstalk between presynaptic trafficking and autophagy in Parkinson’s disease. Neurobiol Dis 122:64–71
Pan-Montojo F, Anichtchik O, Dening Y, Knels L, Pursche S, Jung R, Jackson S, Gille G, Spillantini MG, Reichmann H, Funk RH (2010) Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS One 5:e8762
Paolicelli RC, Bergamini G, Rajendran L (2019) Cell-to-cell communication by extracellular vesicles: focus on microglia. Neuroscience 405:148–157
Papa SM, Wichmann T (2015) Interaction between hyperdirect and indirect basal ganglia pathways. Mov Disord 30:909
Park JS, Blair NF, Sue CM (2015) The role of ATP13A2 in Parkinson’s disease: clinical phenotypes and molecular mechanisms. Mov Disord 30:770–779
Parkkinen L, Hartikainen P, Alafuzoff I (2007) Abundant glial alpha-synuclein pathology in a case without overt clinical symptoms. Clin Neuropathol 26:276–283
Parkkinen L, Pirttila T, Alafuzoff I (2008) Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. Acta Neuropathol 115:399–407
Parkkinen L, O’Sullivan SS, Collins C, Petrie A, Holton JL, Revesz T, Lees AJ (2011) Disentangling the relationship between Lewy bodies and nigral neuronal loss in Parkinson’s disease. J Parkinsons Dis 1:277–286
Pasquini J, Ceravolo R, Qamhawi Z, Lee JY, Deuschl G, Brooks DJ, Bonuccelli U, Pavese N (2018) Progression of tremor in early stages of Parkinson’s disease: a clinical and neuroimaging study. Brain 141:811–821
Paulus W, Jellinger K (1991) The neuropathologic basis of different clinical subgroups of Parkinson’s disease. J Neuropathol Exp Neurol 50:743–755
Pedersen KM, Marner L, Pakkenberg H, Pakkenberg B (2005) No global loss of neocortical neurons in parkinson’s disease: a quantitative stereological study. Mov Disord 20:164–171
Peelaerts W, Bousset L, Baekelandt V, Melki R (2018) a-Synuclein strains and seeding in Parkinson’s disease, incidental Lewy body disease, dementia with Lewy bodies and multiple system atrophy: similarities and differences. Cell Tissue Res 373:195–212
Pelzer EA, Melzer C, Timmermann L, von Cramon DY, Tittgemeyer M (2017) Basal ganglia and cerebellar interconnectivity within the human thalamus. Brain Struct Funct 222:381–392
Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, Zhang B, Pitkin RM, Olufemi MF, Luk KC, Trojanowski JQ, Lee VM (2018a) Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 557:558–563
Peng C, Gathagan RJ, Lee VM (2018b) Distinct alpha-synuclein strains and implications for heterogeneity among alpha-synucleinopathies. Neurobiol Dis 109:209–218
Perez XA, Zhang D, Bordia T, Quik M (2017) Striatal D1 medium spiny neuron activation induces dyskinesias in parkinsonian mice. Mov Disord 32:538–548
Perez XA, Bordia T, Quik M (2018) The striatal cholinergic system in l-dopa-induced dyskinesias. J Neural Transm (Vienna) 125:1251–1262
Perez-Pardo P, Kliest T, Dodiya HB, Broersen LM, Garssen J, Keshavarzian A, Kraneveld AD (2017) The gut-brain axis in Parkinson’s disease: possibilities for food-based therapies. Eur J Pharmacol 817:86–95
Perl DP, Olanow CW (2007) The neuropathology of manganese-induced parkinsonism. J Neuropathol Exp Neurol 66:675–682
Pessoa Rocha N, Reis HJ, Vanden Berghe P, Cirillo C (2014) Depression and cognitive impairment in Parkinson’s disease: a role for inflammation and immunomodulation? Neuroimmunomodulation 21:88–94
Petersen RC, Parisi JE, Dickson DW, Johnson KA, Knopman DS, Boeve BF, Jicha GA, Ivnik RJ, Smith GE, Tangalos EG, Braak H, Kokmen E (2006) Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol 63:665–672
Petrovic IN, Ling H, Asi Y, Ahmed Z, Kukkle PL, Hazrati LN, Lang AE, Revesz T, Holton JL, Lees AJ (2012) Multiple system atrophy-parkinsonism with slow progression and prolonged survival: a diagnostic catch. Mov Disord 27:1186–1190
Picconi B, Paille V, Ghiglieri V, Bagetta V, Barone I, Lindgren HS, Bernardi G, Angela Cenci M, Calabresi P (2008) l-DOPA dosage is critically involved in dyskinesia via loss of synaptic depotentiation. Neurobiol Dis 29:327–335
Picillo M, Lizarraga KJ, Friesen EL, Chau H, Zhang M, Sato C, Rooke G, Munhoz RP, Rogaeva E, Fraser PE, Kalia SK, Kalia LV (2018) Parkinsonism due to A53E alpha-synuclein gene mutation: clinical, genetic, epigenetic, and biochemical features. Mov Disord 33:1950–1955
Pietracupa S, Martin-Bastida A, Piccini P (2017) Iron metabolism and its detection through MRI in parkinsonian disorders: a systematic review. Neurol Sci 38:2095–2101
Pihlstrom L, Blauwendraat C, Cappelletti C, Berge-Seidl V, Langmyhr M, Henriksen SP, van de Berg WDJ, Gibbs JR, Cookson MR, Singleton AB, Nalls MA, Toft M (2018) A comprehensive analysis of SNCA-related genetic risk in sporadic Parkinson disease. Ann Neurol 84:117–129
Pinho R, Paiva I, Jercic KG, Fonseca-Ornelas L, Gerhardt E, Fahlbusch C, Garcia-Esparcia P, Kerimoglu C, Pavlou MAS, Villar-Pique A, Szego E, Lopes da Fonseca T, Odoardi F, Soeroes S, Rego AC, Fischle W, Schwamborn JC, Meyer T, Kugler S, Ferrer I, Attems J, Fischer A, Becker S, Zweckstetter M, Borovecki F, Outeiro TF (2019) Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum Mol Genet 28:31–50
Poletti M, Frosini D, Pagni C, Baldacci F, Nicoletti V, Tognoni G, Lucetti C, Del Dotto P, Ceravolo R, Bonuccelli U (2012) Mild cognitive impairment and cognitive-motor relationships in newly diagnosed drug-naive patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry 83:601–606
Polinski NK, Volpicelli-Daley LA, Sortwell CE, Luk KC, Cremades N, Gottler LM, Froula J, Duffy MF, Lee VMY, Martinez TN, Dave KD (2018) Best practices for generating and using alpha-synuclein pre-formed fibrils to model Parkinson’s disease in rodents. J Parkinsons Dis 8:303–322
Politis M, Wu K, Loane C, Kiferle L, Molloy S, Brooks DJ, Piccini P (2010) Staging of serotonergic dysfunction in Parkinson’s disease: an in vivo 11C-DASB PET study. Neurobiol Dis 40:216–221
Politis M, Wu K, Loane C, Brooks DJ, Kiferle L, Turkheimer FE, Bain P, Molloy S, Piccini P (2014) Serotonergic mechanisms responsible for levodopa-induced dyskinesias in Parkinson’s disease patients. J Clin Investig 124:1340–1349
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047
Pont-Sunyer C, Tolosa E, Caspell-Garcia C, Coffey C, Alcalay RN, Chan P, Duda JE, Facheris M, Fernandez-Santiago R, Marek K, Lomena F, Marras C, Mondragon E, Saunders-Pullman R, Waro B (2017) The prodromal phase of leucine-rich repeat kinase 2-associated Parkinson disease: clinical and imaging Studies. Mov Disord 32:726–738
Poplawska-Domaszewicz K, Florczak-Wyspianska J, Kozubski W, Michalak S (2018) Paraneoplastic movement disorders. Rev Neurosci 29:745–755
Post MR, Lieberman OJ, Mosharov EV (2018) Can interactions between alpha-synuclein, dopamine and calcium explain selective neurodegeneration in Parkinson’s disease? Front Neurosci 12:161
Power JH, Barnes OL, Chegini F (2017) Lewy bodies and the mechanisms of neuronal cell death in Parkinson’s disease and dementia with Lewy bodies. Brain Pathol 27:3–12
Prasad V, Wasser Y, Hans F, Goswami A, Katona I, Outeiro TF, Kahle PJ, Schulz JB, Voigt A (2019) Monitoring alpha-synuclein multimerization in vivo. FASEB J 33:2116–2131
Prots I, Grosch J, Brazdis RM, Simmnacher K, Veber V, Havlicek S, Hannappel C, Krach F, Krumbiegel M, Schutz O, Reis A, Wrasidlo W, Galasko DR, Groemer TW, Masliah E, Schlotzer-Schrehardt U, Xiang W, Winkler J, Winner B (2018) Alpha-synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc Natl Acad Sci USA 115:7813–7818
Puska G, Lutz MI, Molnar K, Regelsberger G, Ricken G, Pirker W, Laszlo L, Kovacs GG (2018) Lysosomal response in relation to alpha-synuclein pathology differs between Parkinson’s disease and multiple system atrophy. Neurobiol Dis 114:140–152
Qamhawi Z, Towey D, Shah B, Pagano G, Seibyl J, Marek K, Borghammer P, Brooks DJ, Pavese N (2015) Clinical correlates of raphe serotonergic dysfunction in early Parkinson’s disease. Brain 138:2964–2973
Quadri M, Mandemakers W, Grochowska MM, Masius R, Geut H, Fabrizio E, Breedveld GJ, Kuipers D, Minneboo M, Vergouw LJM, Carreras Mascaro A, Yonova-Doing E, Simons E, Zhao T, Di Fonzo AB, Chang HC, Parchi P, Melis M, Correia Guedes L, Criscuolo C, Thomas A, Brouwer RWW, Heijsman D, Ingrassia AMT, Calandra Buonaura G, Rood JP, Capellari S, Rozemuller AJ, Sarchioto M, Fen Chien H, Vanacore N, Olgiati S, Wu-Chou YH, Yeh TH, Boon AJW, Hoogers SE, Ghazvini M, Ijpma AS, van Ijcken WFJ, Onofrj M, Barone P, Nicholl DJ, Puschmann A, De Mari M, Kievit AJ, Barbosa E, De Michele G, Majoor-Krakauer D, van Swieten JC, de Jong FJ, Ferreira JJ, Cossu G, Lu CS, Meco G, Cortelli P, van de Berg WDJ, Bonifati V (2018) LRP10 genetic variants in familial Parkinson’s disease and dementia with Lewy bodies: a genome-wide linkage and sequencing study. Lancet Neurol 17:597–608
Quinn JG, Coulson DT, Brockbank S, Beyer N, Ravid R, Hellemans J, Irvine GB, Johnston JA (2012) Alpha-synuclein mRNA and soluble alpha-synuclein protein levels in post-mortem brain from patients with Parkinson’s disease, dementia with Lewy bodies, and Alzheimer’s disease. Brain Res 1459:71–80
Quinzii CM, Hirano M, DiMauro S (2014) Mutant COQ2 in multiple-system atrophy. N Engl J Med 371:81–82
Racette BA, Criswell SR, Lundin JI, Hobson A, Seixas N, Kotzbauer PT, Evanoff BA, Perlmutter JS, Zhang J, Sheppard L, Checkoway H (2012) Increased risk of parkinsonism associated with welding exposure. Neurotoxicology 33:1356–1361
Rajput AH, Sitte HH, Rajput A, Fenton ME, Pifl C, Hornykiewicz O (2008) Globus pallidus dopamine and Parkinson motor subtypes: clinical and brain biochemical correlation. Neurology 70:1403–1410
Ray NJ, Bradburn S, Murgatroyd C, Toseeb U, Mir P, Kountouriotis GK, Teipel SJ, Grothe MJ (2018) In vivo cholinergic basal forebrain atrophy predicts cognitive decline in de novo Parkinson’s disease. Brain 141:165–176
Rebeiz JJ, Kolodny EH, Richardson EP Jr (1968) Corticodentatonigral degeneration with neuronal achromasia. Arch Neurol 18:20–33
Reed X, Bandres-Ciga S, Blauwendraat C, Cookson MR (2019) The role of monogenic genes in idiopathic Parkinson’s disease. Neurobiol Dis 124:230–239
Reetz K, Gaser C, Klein C, Hagenah J, Buchel C, Gottschalk S, Pramstaller PP, Siebner HR, Binkofski F (2009) Structural findings in the basal ganglia in genetically determined and idiopathic Parkinson’s disease. Mov Disord 24:99–103
Refolo V, Bez F, Polissidis A, Kuzdas-Wood D, Sturm E, Kamaratou M, Poewe W, Stefanis L, Angela Cenci M, Romero-Ramos M, Wenning GK, Stefanova N (2018) Progressive striatonigral degeneration in a transgenic mouse model of multiple system atrophy: translational implications for interventional therapies. Acta Neuropathol Commun 6:2
Reijnders JS, Ehrt U, Weber WE, Aarsland D, Leentjens AF (2008) A systematic review of prevalence studies of depression in Parkinson’s disease. Mov Disord 23:183–189 (quiz 313)
Rektor I, Bohnen NI, Korczyn AD, Gryb V, Kumar H, Kramberger MG, de Leeuw FE, Pirtosek Z, Rektorova I, Schlesinger I, Slawek J, Valkovic P, Vesely B (2018) An updated diagnostic approach to subtype definition of vascular parkinsonism—recommendations from an expert working group. Parkinsonism Relat Disord 49:9–16
Ren S, Zhang H, Zheng W, Liu M, Gao F, Wang Z, Chen Z (2019) Altered functional connectivity of cerebello-cortical circuit in multiple system atrophy (cerebellar-type). Front Neurosci 12:996
Respondek G, Kurz C, Arzberger T, Compta Y, Englund E, Ferguson LW, Gelpi E, Giese A, Irwin DJ, Meissner WG, Nilsson C, Pantelyat A, Rajput A, van Swieten JC, Troakes C, Josephs KA, Lang AE, Mollenhauer B, Muller U, Whitwell JL, Antonini A, Bhatia KP, Bordelon Y, Corvol JC, Colosimo C, Dodel R, Grossman M, Kassubek J, Krismer F, Levin J, Lorenzl S, Morris H, Nestor P, Oertel WH, Rabinovici GD, Rowe JB, van Eimeren T, Wenning GK, Boxer A, Golbe LI, Litvan I, Stamelou M, Hoglinger GU (2017) Which ante mortem clinical features predict progressive supranuclear palsy pathology? Mov Disord 32:995–1005
Rey NL, Wesson DW, Brundin P (2018) The olfactory bulb as the entry site for prion-like propagation in neurodegenerative diseases. Neurobiol Dis 109:226–248
Reyes JF, Rey NL, Bousset L, Melki R, Brundin P, Angot E (2014) Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 62:387–398
Reyes JF, Sackmann C, Hoffmann A, Svenningsson P, Winkler J, Ingelsson M, Hallbeck M (2019) Binding of alpha-synuclein oligomers to Cx32 facilitates protein uptake and transfer in neurons and oligodendrocytes. Acta Neuropathol. https://doi.org/10.1007/s00401-00019-02007-x
Reynolds RH, Botia J, Nalls MA, Hardy J, Gagliano Taliun SA, Ryten M (2019) Moving beyond neurons: the role of cell type-specific gene regulation in Parkinson’s disease heritability. NPJ Parkinsons Dis 5:6
Rietdijk CD, Perez-Pardo P, Garssen J, van Wezel RJ, Kraneveld AD (2017) Exploring Braak’s hypothesis of Parkinson’s disease. Front Neurol 8:37
Rizzo G, Copetti M, Arcuti S, Martino D, Fontana A, Logroscino G (2016) Accuracy of clinical diagnosis of Parkinson disease: a systematic review and meta-analysis. Neurology 86:566–576
Rizzo G, Arcuti S, Copetti M, Alessandria M, Savica R, Fontana A, Liguori R, Logroscino G (2018) Accuracy of clinical diagnosis of dementia with Lewy bodies: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 89:358–366
Robakis D, Cortes E, Clark LN, Vonsattel JP, Virmani T, Alcalay RN, Crary JF, Levy OA (2016) The effect of MAPT haplotype on neocortical Lewy body pathology in Parkinson disease. J Neural Transm (Vienna) 123:583–588
Roberts HL, Brown DR (2015) Seeking a mechanism for the toxicity of oligomeric alpha-synuclein. Biomolecules 5:282–305
Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, Caswell C, Van Deerlin VM, Yan N, Yousef A, Hurtig HI, Siderowf A, Grossman M, McMillan CT, Miller B, Duda JE, Irwin DJ, Wolk D, Elman L, McCluskey L, Chen-Plotkin A, Weintraub D, Arnold SE, Brettschneider J, Lee VM, Trojanowski JQ (2018) Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 141:2181–2193
Rocha Cabrero F, Morrison EH (2019) Lewy Bodies, 2019/02/07 edn. StatPearls Publishing, Treasure Island
Rocha EM, De Miranda B, Sanders LH (2018) Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol Dis 109:249–257
Rockenstein E, Nuber S, Overk CR, Ubhi K, Mante M, Patrick C, Adame A, Trejo-Morales M, Gerez J, Picotti P, Jensen PH, Campioni S, Riek R, Winkler J, Gage HH, Winner B, Masliah E (2014) Accumulation of oligomer-prone a-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain 137:1496–1513
Rohan Z, Rahimi J, Weis S, Kapas I, Auff E, Mitrovic N, Liberski PP, Sikorska B, Matej R, Kovacs GG (2015) Screening for alpha-synuclein immunoreactive neuronal inclusions in the hippocampus allows identification of atypical MSA (FTLD-synuclein). Acta Neuropathol 130:299–301
Rohan Z, Milenkovic I, Lutz MI, Matej R, Kovacs GG (2016) Shared and distinct patterns of oligodendroglial response in alpha-synucleinopathies and tauopathies. J Neuropathol Exp Neurol 75:1100–1109
Rohani M, Lang AE, Sina F, Elahi E, Fasano A, Hardy J, Bras J, Alavi A (2017) Action myoclonus and seizure in Kufor–Rakeb syndrome. Mov Disord Clin Pract 5:195–199
Ronchi D, Di Biase E, Franco G, Melzi V, Del Sorbo F, Elia A, Barzaghi C, Garavaglia B, Bergamini C, Fato R, Mora G, Del Bo R, Fortunato F, Borellini L, Trezzi I, Compagnoni GM, Monfrini E, Frattini E, Bonato S, Cogiamanian F, Ardolino G, Priori A, Bresolin N, Corti S, Comi GP, Di Fonzo A (2016) Mutational analysis of COQ2 in patients with MSA in Italy. Neurobiol Aging 45:213e1–213e2
Rongve A, Aarsland D (2013) Dementia in Parkinson’s disease and dementia with Lewy bodies. In: Dening T, Thomas A (eds) Oxford textbook of old age psychiatry 2e. Oxford University Press, Oxford, pp 469–478
Rosborough K, Patel N, Kalia LV (2017) Alpha-synuclein and parkinsonism: updates and future perspectives. Curr Neurol Neurosci Rep 17:31
Rosqvist K, Horne M, Hagell P, Iwarsson S, Nilsson MH, Odin P (2018) Levodopa effect and motor function in late stage Parkinson’s disease. J Parkinsons Dis 8:59–70
Rudakou U, Ouled Amar Bencheikh B, Ruskey JA, Krohn L, Laurent SB, Spiegelman D, Liong C, Fahn S, Waters C, Monchi O, Fon EA, Dauvilliers Y, Alcalay RN, Dupre N, Gan-Or Z (2019) Common and rare GCH1 variants are associated with Parkinson’s disease. Neurobiol Aging 73:231
Rudow G, O’Brien R, Savonenko AV, Resnick SM, Zonderman AB, Pletnikova O, Marsh L, Dawson TM, Crain BJ, West MJ, Troncoso JC (2008) Morphometry of the human substantia nigra in ageing and Parkinson’s disease. Acta Neuropathol 115:461–470
Ruf VC, Nubling GS, Willikens S, Shi S, Schmidt F, Levin J, Botzel K, Kamp F, Giese A (2019) Different effects of alpha-synuclein mutants on lipid binding and aggregation detected by single molecule fluorescence spectroscopy and tht fluorescence-based measurements. ACS Chem Neurosci 10:1649–1659
Ruffmann C, Zini M, Goldwurm S, Bramerio M, Spinello S, Rusconi D, Gambacorta M, Tagliavini F, Pezzoli G, Giaccone G (2012) Lewy body pathology and typical Parkinson disease in a patient with a heterozygous (R275 W) mutation in the Parkin gene (PARK2). Acta Neuropathol 123:901–903
Ruffmann C, Calboli FC, Bravi I, Gveric D, Curry LK, de Smith A, Pavlou S, Buxton JL, Blakemore AI, Takousis P, Molloy S, Piccini P, Dexter DT, Roncaroli F, Gentleman SM, Middleton LT (2016) Cortical Lewy bodies and Abeta burden are associated with prevalence and timing of dementia in Lewy body diseases. Neuropathol Appl Neurobiol 42:436–450
Rui Q, Ni H, Li D, Gao R, Chen G (2018) The role of LRRK2 in neurodegeneration of Parkinson disease. Curr Neuropharmacol 16:1348–1357
Rumpf JJ, Albers J, Fricke C, Mueller W, Classen J (2018) Structural abnormality of substantia nigra induced by methamphetamine abuse. Mov Disord 32:1784–1788
Russ C, Lovestone S, Baker M, Pickering-Brown SM, Andersen PM, Furlong R, Mann D, Powell JF (2001) The extended haplotype of the microtubule associated protein tau gene is not associated with Pick’s disease. Neurosci Lett 299:156–158
Sailer A, Scholz SW, Nalls MA, Schulte C, Federoff M, Price TR, Lees A, Ross OA, Dickson DW, Mok K, Mencacci NE, Schottlaender L, Chelban V, Ling H, O’Sullivan SS, Wood NW, Traynor BJ, Ferrucci L, Federoff HJ, Mhyre TR, Morris HR, Deuschl G, Quinn N, Widner H, Albanese A, Infante J, Bhatia KP, Poewe W, Oertel W, Hoglinger GU, Wullner U, Goldwurm S, Pellecchia MT, Ferreira J, Tolosa E, Bloem BR, Rascol O, Meissner WG, Hardy JA, Revesz T, Holton JL, Gasser T, Wenning GK, Singleton AB, Houlden H (2016) A genome-wide association study in multiple system atrophy. Neurology 87:1591–1598
Sakamoto R, Tsuchiya K, Yoshida R, Itoh Y, Furuta N, Kosuga A, Sugai Y, Mimura M (2009) Progressive supranuclear palsy combined with Alzheimer’s disease: a clinicopathological study of two autopsy cases. Neuropathology 29:219–229
Salvesen L, Ullerup BH, Sunay FB, Brudek T, Lokkegaard A, Agander TK, Winge K, Pakkenberg B (2015) Changes in total cell numbers of the basal ganglia in patients with multiple system atrophy—a stereological study. Neurobiol Dis 74:104–113
Sampedro F, Marin-Lahoz J, Martinez-Horta S, Pagonabarraga J, Kulisevsky J (2019) Dopaminergic degeneration induces early posterior cortical thinning in Parkinson’s disease. Neurobiol Dis 124:29–35
Samudra N, Patel N, Womack KB, Khemani P, Chitnis S (2016) Psychosis in Parkinson disease: a review of etiology, phenomenology, and management. Drugs Aging 33:855–863
Sanford AM (2018) Lewy body dementia. Clin Geriatr Med 34:603–615
Santiago RM, Vital MABF, Sato MDO, Adam GP (2016) Depression in Parkinson’s disease is associated with a serotoninergic system change secondary to neuroinflammation. Int J Neurol Neurother 3:061
Sassone J, Valtorta F, Ciammola A (2019) Early dyskinesias in Parkinson’s disease patients with parkin mutation: a primary corticostriatal synaptopathy? Front Neurosci 13:273
Savica R, Grossardt BR, Bower JH, Ahlskog JE, Rocca WA (2013a) Incidence and pathology of synucleinopathies and tauopathies related to parkinsonism. JAMA Neurol 70:859–866
Savica R, Grossardt BR, Bower JH, Boeve BF, Ahlskog JE, Rocca WA (2013b) Incidence of dementia with Lewy bodies and Parkinson disease dementia. JAMA Neurol 70:1396–1402
Savica R, Beach TG, Hentz JG, Sabbagh MN, Serrano GE, Sue LI, Dugger BN, Shill HA, Driver-Dunckley E, Caviness JN, Mehta SH, Jacobson SA, Belden CM, Davis KJ, Zamrini E, Shprecher DR, Adler CH (2019) Lewy body pathology in Alzheimer’s disease: a clinicopathological prospective study. Acta Neurol Scand 139:76–81
Schapira AHV, Chaudhuri KR, Jenner P (2017) Non-motor features of Parkinson disease. Nat Rev Neurosci 18:435–450
Schmidt R, Berke JD (2017) A pause-then-cancel model of stopping: evidence from basal ganglia neurophysiology. Philos Trans R Soc Lond B Biol Sci. https://doi.org/10.1098/rstb.2016.0202
Schneider SA, Alcalay RN (2017) Neuropathology of genetic synucleinopathies with parkinsonism: review of the literature. Mov Disord 32:1504–1523
Schofield EC, Hodges JR, Macdonald V, Cordato NJ, Kril JJ, Halliday GM (2011) Cortical atrophy differentiates Richardson’s syndrome from the parkinsonian form of progressive supranuclear palsy. Mov Disord 26:256–263
Scholz SW, Houlden H, Schulte C, Sharma M, Li A, Berg D, Melchers A, Paudel R, Gibbs JR, Simon-Sanchez J, Paisan-Ruiz C, Bras J, Ding J, Chen H, Traynor BJ, Arepalli S, Zonozi RR, Revesz T, Holton J, Wood N, Lees A, Oertel W, Wullner U, Goldwurm S, Pellecchia MT, Illig T, Riess O, Fernandez HH, Rodriguez RL, Okun MS, Poewe W, Wenning GK, Hardy JA, Singleton AB, Del Sorbo F, Schneider S, Bhatia KP, Gasser T (2009) SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol 65:610–614
Schottlaender LV, Bettencourt C, Kiely AP, Chalasani A, Neergheen V, Holton JL, Hargreaves I, Houlden H (2016) Coenzyme Q10 levels are decreased in the cerebellum of multiple-system atrophy patients. PLoS One 11:e0149557
Schulte C, Gasser T (2011) Genetic basis of Parkinson’s disease: inheritance, penetrance, and expression. Appl Clin Genet 4:67–80
Schulz J, Pagano G, Fernandez Bonfante JA, Wilson H, Politis M (2018) Nucleus basalis of Meynert degeneration precedes and predicts cognitive impairment in Parkinson’s disease. Brain 141:1501–1516
Schulz J, Takousis P, Wohlers I, Itua IOG, Dobricic V, Rucker G, Binder H, Middleton L, Ioannidis JPA, Perneczky R, Bertram L, Lill CM (2019) Meta-analyses identify differentially expressed microRNAs in Parkinson’s disease. Ann Neurol 85:835–851
Schulz-Schaeffer WJ (2010) The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol 120:131–143
Schulz-Schaeffer WJ (2015) Is cell death primary or secondary in the pathophysiology of idiopathic Parkinson’s disease? Biomolecules 5:1467–1479
Schumacher J, Peraza LR, Firbank M, Thomas AJ, Kaiser M, Gallagher P, O’Brien JT, Blamire AM, Taylor JP (2019) Dysfunctional brain dynamics and their origin in Lewy body dementia. Brain 142:1767–1782
Scudamore O, Ciossek T (2018) Increased oxidative stress exacerbates alpha-synuclein aggregation in vivo. J Neuropathol Exp Neurol 77:443–453
Seidel K, Schols L, Nuber S, Petrasch-Parwez E, Gierga K, Wszolek Z, Dickson D, Gai WP, Bornemann A, Riess O, Rami A, Den Dunnen WF, Deller T, Rub U, Kruger R (2010) First appraisal of brain pathology owing to A30P mutant alpha-synuclein. Ann Neurol 67:684–689
Seidel K, Mahlke J, Sonny S, Krüger R, Heinsen H, Auburger G, Bouzrou M, Grinberg LT, Wicht H, Korf H-W, den Dunnen W, Rüb U (2015) The brainstem pathologies of Parkinson’s disease and dementia with Lewy bodies. Brain Pathol 25:121–135
Sekiya H, Kowa H, Koga H, Takata M, Satake W, Futamura N, Funakawa I, Jinnai K, Takahashi M, Kondo T, Ueno Y, Kanagawa M, Kobayashi K, Toda T (2019) Wide distribution of alpha-synuclein oligomers in multiple system atrophy brain detected by proximity ligation. Acta Neuropathol 137:455–466
Selikhova M, Williams DR, Kempster PA, Holton JL, Revesz T, Lees AJ (2009) A clinico-pathological study of subtypes in Parkinson’s disease. Brain 132:2947–2957
Sestini S, Alongi P, Berti V, Calcagni ML, Cecchin D, Chiaravalloti A, Chincarini A, Cistaro A, Guerra UP, Pappatà S, Tiraboschi P, Nobili F (2019) The role of molecular imaging in the frame of the revised dementia with Lewy body criteria. Clin Transl Imaging 7:83–98
Seto-Salvia N, Pagonabarraga J, Houlden H, Pascual-Sedano B, Dols-Icardo O, Tucci A, Paisan-Ruiz C, Campolongo A, Anton-Aguirre S, Martin I, Munoz L, Bufill E, Vilageliu L, Grinberg D, Cozar M, Blesa R, Lleo A, Hardy J, Kulisevsky J, Clarimon J (2012) Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson’s disease course. Mov Disord 27:393–399
Shah A, Prasad S, Rastogi B, Dash S, Saini J, Pal PK, Ingalhalikar M (2019) Altered structural connectivity of the motor subnetwork in multiple system atrophy with cerebellar features. Eur Radiol 29:2783–2791
Shahmoradian SH, Lewis AJ, Genoud C, Graff-Meyer A, Hench J, Moors T, Schweighauser G, Wang J, Goldie KN, Suetterlin R, Castano-Diez D, Perez-Navarro P, Huisman E, Ipsen S, Ingrassia A, de Gier Y, Rozemuller AJM, Da Paepe A, Erny J, Staempfli A, Hoernschemeyer J, Grosserueschkamp F, Niedieker D, El-Mashtoly SF, Quadri M, van Ijcken WFJ, Bonifati V, Gerwert K, Bohrmann B, Frank S, Britschgi M, Stahlberg H, van de Berg W, Lauer ME (2018) Lewy pathology in Parkinson’s disease consists of a crowded organellar, membranous medley (This article is a preprint and has not been peer-reviewed). bioRxiv. https://doi.org/10.1101/137976
Sharma M, Wenning G, Krüger R (2014) Mutant COQ2 in multiple-system atrophy. N Engl J Med 371:80–83
Sharp ME, Marder KS, Cote L, Clark LN, Nichols WC, Vonsattel JP, Alcalay RN (2014) Parkinson’s disease with Lewy bodies associated with a heterozygous PARKIN dosage mutation. Mov Disord 29:566–568
Shimohata T, Aiba I, Nishizawa M (2015) Criteria for the diagnosis of corticobasal degeneration. Brain Nerve 67:513–523
Shimozawa A, Ono M, Takahara D, Tarutani A, Imura S, Masuda-Suzukake M, Higuchi M, Yanai K, Hisanaga SI, Hasegawa M (2017) Propagation of pathological alpha-synuclein in marmoset brain. Acta Neuropathol Commun 5:12
Shine JM, Matar E, Ward PB, Frank MJ, Moustafa AA, Pearson M, Naismith SL, Lewis SJ (2013) Freezing of gait in Parkinson’s disease is associated with functional decoupling between the cognitive control network and the basal ganglia. Brain 136:3671–3681
Shinotoh H, Shimada H, Kokubo Y, Tagai K, Niwa F, Kitamura S, Endo H, Ono M, Kimura Y, Hirano S, Mimuro M, Ichise M, Sahara N, Zhang MR, Suhara T, Higuchi M (2019) Tau imaging detects distinctive distribution of tau pathology in ALS/PDC on the Kii Peninsula. Neurology 92:e136–e147
Shuaib UA, Rajput AH, Robinson CA, Rajput A (2016) Neuroleptic-induced parkinsonism: clinicopathological study. Mov Disord 31:360–365
Sian-Hulsmann J, Mandel S, Youdim MB, Riederer P (2011) The relevance of iron in the pathogenesis of Parkinson’s disease. J Neurochem 118:939–957
Sian-Hulsmann J, Monoranu C, Strobel S, Riederer P (2015) Lewy bodies: a spectator or salient killer? CNS Neurol Disord Drug Targets 14:947–955
Sibon I, Tison F (2004) Vascular Parkinsonism. Curr Opin Neurol 17:49–54
Sidransky E, Lopez G (2012) The link between the GBA gene and parkinsonism. Lancet Neurol 11:986–998
Siepel FJ, Bronnick KS, Booij J, Ravina BM, Lebedev AV, Pereira JB, Gruner R, Aarsland D (2014) Cognitive executive impairment and dopaminergic deficits in de novo Parkinson’s disease. Mov Disord 29:1802–1808
Simonyan K (2019) Recent advances in understanding the role of the basal ganglia [version 1; peer review: 2 approved]. F1000 Res 8:122. https://doi.org/10.12688/f11000research.16524.12681
Simonyan K, Cho H, Hamzehei Sichani A, Rubien-Thomas E, Hallett M (2017) The direct basal ganglia pathway is hyperfunctional in focal dystonia. Brain 140:3179–3190
Singleton AB, Farrer MJ, Bonifati V (2013) The genetics of Parkinson’s disease: progress and therapeutic implications. Mov Disord 28:14–23
Sklerov M, Kang UJ, Liong C, Clark L, Marder K, Pauciulo M, Nichols WC, Chung WK, Honig LS, Cortes E, Vonsattel JP, Alcalay RN (2017) Frequency of GBA variants in autopsy-proven multiple system atrophy. Mov Disord Clin Pract 4:574–581
Skogseth RE, Hortobagyi T, Soennesyn H, Chwiszczuk L, Ffytche D, Rongve A, Ballard C, Aarsland D (2017) Accuracy of clinical diagnosis of dementia with Lewy bodies versus neuropathology. J Alzheimers Dis 59:1139–1152
Smith C, Malek N, Grosset KA, Cullen B, Gentleman SM, Grosset DG (2019) The neuropathology of dementia in patients with Parkinson’s disease: a systematic review of autopsy studies. J Neurol Neurosurg Psychiatry (in press)
Snead D, Eliezer D (2014) Alpha-synuclein function and dysfunction on cellular membranes. Exp Neurobiol 23:292–313
Snijders AH, Takakusaki K, Debu B, Lozano AM, Krishna V, Fasano A, Aziz TZ, Papa SM, Factor SA, Hallett M (2016) Physiology of freezing of gait. Ann Neurol 80:644–659
Sorrentino ZA, Giasson BI, Chakrabarty P (2019) Alpha-synuclein and astrocytes: tracing the pathways from homeostasis to neurodegeneration in Lewy body disease. Acta Neuropathol. https://doi.org/10.1007/s00401-00019-01977-00402
Spay C, Meyer G, Welter ML, Lau B, Boulinguez P, Ballanger B (2018) Functional imaging correlates of akinesia in Parkinson’s disease: still open issues. Neuroimage Clin 21:101644
Spillantini MG, Goedert M (2013) Tau pathology and neurodegeneration. Lancet Neurol 12:609–622
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 95:6469–6473
Spina S, Brown JA, Deng J, Gardner RC, Nana AL, Hwang JL, Gaus SE, Huang EJ, Kramer JH, Rosen HJ, Kornak J, Neuhaus J, Miller BL, Grinberg LT, Boxer AL, Seeley WW (2019) Neuropathological correlates of structural and functional imaging biomarkers in 4-repeat tauopathies. Brain. https://doi.org/10.1093/brain/awz1122
Spires-Jones TL, Attems J, Thal DR (2017) Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol 134:187–205
Srulijes K, Hauser AK, Guella I, Asselta R, Brockmann K, Schulte C, Solda G, Cilia R, Maetzler W, Schols L, Wenning GK, Poewe W, Barone P, Wullner U, Oertel W, Berg D, Goldwurm S, Gasser T (2013) No association of GBA mutations and multiple system atrophy. Eur J Neurol 20:e61–e62
Stankovic I, Quinn N, Vignatelli L, Antonini A, Berg D, Coon E, Cortelli P, Fanciulli A, Ferreira JJ, Freeman R, Halliday G, Hoglinger GU, Iodice V, Kaufmann H, Klockgether T, Kostic V, Krismer F, Lang A, Levin J, Low P, Mathias C, Meissner WG, Kaufmann LN, Palma JA, Panicker JN, Pellecchia MT, Sakakibara R, Schmahmann J, Scholz SW, Singer W, Stamelou M, Tolosa E, Tsuji S, Seppi K, Poewe W, Wenning GK (2019) A critique of the second consensus criteria for multiple system atrophy. Mov Disord. https://doi.org/10.1002/mds.27701
Steele JC, McGeer PL (2008) The ALS/PDC syndrome of Guam and the cycad hypothesis. Neurology 70:1984–1990
Stefanis L (2012) Alpha-synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med 2:a009399
Stefanis L, Emmanouilidou E, Pantazopoulou M, Kirik D, Vekrellis K, Tofaris GK (2019) How is alpha-synuclein cleared from the cell? J Neurochem. https://doi.org/10.1111/jnc.14704
Stefanova N (2014) Animal models. In: Wenning GK, Fanciulli A (eds) Multiple system atrophy. Springer, Vienna, pp 83–96
Stejskalova Z, Rohan Z, Rusina R, Tesar A, Kukal J, Kovacs GG, Bartos A, Matej R (2019) Pyramidal system involvement in progressive supranuclear palsy—a clinicopathological correlation. BMC Neurol 19:42
Strang KH, Croft CL, Sorrentino ZA, Chakrabarty P, Golde TE, Giasson BI (2018) Distinct differences in prion-like seeding and aggregation between tau protein variants provide mechanistic insights into tauopathies. J Biol Chem 293:2408–2421
Strang KH, Golde TE, Giasson BI (2019) MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab Invest. https://doi.org/10.1038/s41374-41019-40197-x
Su Y, Deng MF, Xiong W, Xie AJ, Guo J, Liang ZH, Hu B, Chen JG, Zhu X, Man HY, Lu Y, Liu D, Tang B, Zhu LQ (2019) MicroRNA-26a/death-associated protein kinase 1 signaling induces synucleinopathy and dopaminergic neuron degeneration in Parkinson’s disease. Biol Psychiatry 85:769–781
Sulzer D, Surmeier DJ (2013) Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov Disord 28:41–50
Sumikura H, Takao M, Hatsuta H, Ito S, Nakano Y, Uchino A, Nogami A, Saito Y, Mochizuki H, Murayama S (2015) Distribution of alpha-synuclein in the spinal cord and dorsal root ganglia in an autopsy cohort of elderly persons. Acta Neuropathol Commun 3:57
Sun QY, Guo JF, Han WW, Zuo X, Wang L, Yao LY, Pan Q, Xia K, Yan XX, Tang BS (2013) Genetic association study of glucocerebrosidase gene L444P mutation in essential tremor and multiple system atrophy in mainland China. J Clin Neurosci 20:217–219
Sun Z, Xiang X, Tang B, Chen Z, Peng H, Xia K, Jiang H (2015) SNP rs11931074 of the SNCA gene may not be associated with multiple system atrophy in Chinese population. Int J Neurosci 125:612–615
Sun Z, Ohta Y, Yamashita T, Sato K, Takemoto M, Hishikawa N, Abe K (2016) New susceptible variant of COQ2 gene in Japanese patients with sporadic multiple system atrophy. Neurol Genet 2:e54
Sun Y, Pham AN, Hare DJ, Waite TD (2018) Kinetic modeling of pH-dependent oxidation of dopamine by iron and its relevance to Parkinson’s disease. Front Neurosci 12:859
Sun R, Yang S, Zheng B, Liu J, Ma X (2019) Apolipoprotein E polymorphisms and Parkinson disease with or without dementia: a meta-analysis including 6453 participants. J Geriatr Psychiatry Neurol 32:3–15
Surmeier DJ (2018) Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J 285:3657–3668
Surmeier DJ, Obeso JA, Halliday GM (2017) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18:101–113
Suryanarayana SM, Hellgren Kotaleski J, Grillner S, Gurney KN (2019) Roles for globus pallidus externa revealed in a computational model of action selection in the basal ganglia. Neural Netw 109:113–136
Svensson E, Horvath-Puho E, Thomsen RW, Djurhuus JC, Pedersen L, Borghammer P, Sorensen HT (2015) Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol 78:522–529
Swirski M, Miners JS, de Silva R, Lashley T, Ling H, Holton J, Revesz T, Love S (2014) Evaluating the relationship between amyloid-ß and a-synuclein phosphorylated at Ser129 in dementia with Lewy bodies and Parkinson’s disease. Alzheimers Res Ther 6:77
Taba P (2017) Toxic-induced parkinsonism. In: Falup-Pecurariu C, Ferreira J, Martinez-Martin P, Chaudhuri KR (eds) Movement disorders curricula. Springer, Wien, pp 225–232
Tacik P, Fiesel FC, Fujioka S, Ross OA, Pretelt F, Castaneda Cardona C, Kidd A, Hlavac M, Raizis A, Okun MS, Traynor S, Strongosky AJ, Springer W, Wszolek ZK (2014) Three families with Perry syndrome from distinct parts of the world. Parkinsonism Relat Disord 20:884–888
Taghdiri F, Sato C, Ghani M, Moreno D, Rogaeva E, Tartaglia MC (2016) Novel GRN mutations in patients with corticobasal syndrome. Sci Rep 6:22913
Tagliaferro P, Burke RE (2016) Retrograde axonal degeneration in Parkinson disease. J Parkinsons Dis 6:1–15
Taguchi K, Watanabe Y, Tsujimura A, Tanaka M (2019) Expression of alpha-synuclein is regulated in a neuronal cell type-dependent manner. Anat Sci Int 94:11–22
Taipa R, Pereira C, Reis I, Alonso I, Bastos-Lima A, Melo-Pires M, Magalhaes M (2016) DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain 139:1680–1687
Takanashi M, Funayama M, Matsuura E, Yoshino H, Li Y, Tsuyama S, Takashima H, Nishioka K, Hattori N (2018) Isolated nigral degeneration without pathological protein aggregation in autopsied brains with LRRK2 p.R1441H homozygous and heterozygous mutations. Acta Neuropathol Commun 6:105
Tamguney G, Korczyn AD (2018) A critical review of the prion hypothesis of human synucleinopathies. Cell Tissue Res 373:213–220
Tamtaji OR, Behnam M, Pourattar MA, Jafarpour H, Asemi Z (2019) Aquaporin 4: a key player in Parkinson’s disease. J Cell Physiol. https://doi.org/10.1002/jcp.28871
Tanaka G, Yamanaka T, Furukawa Y, Kajimura N, Mitsuoka K, Nukina N (2019) Sequence- and seed-structure-dependent polymorphic fibrils of alpha-synuclein. Biochim Biophys Acta Mol Basis Dis 1865:1410–1420
Taniguchi-Watanabe S, Arai T, Kametani F, Nonaka T, Masuda-Suzukake M, Tarutani A, Murayama S, Saito Y, Arima K, Yoshida M, Akiyama H, Robinson A, Mann DMA, Iwatsubo T, Hasegawa M (2016) Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau. Acta Neuropathol 131:267–280
Tanji K, Miki Y, Mori F, Kon T, Kakita A, Takahashi H, Wakabayashi K (2019) Phosphorylated NUB1 distinguishes alpha-synuclein in Lewy bodies from that in glial cytoplasmic inclusions in multiple system atrophy. Brain Pathol. https://doi.org/10.1111/bpa.12728
Tardivel M, Begard S, Bousset L, Dujardin S, Coens A, Melki R, Buee L, Colin M (2016) Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological tau protein assemblies. Acta Neuropathol Commun 4:117
Tatsumi S, Mimuro M, Iwasaki Y, Takahashi R, Kakita A, Takahashi H, Yoshida M (2014a) Argyrophilic grains are reliable disease-specific features of corticobasal degeneration. J Neuropathol Exp Neurol 73:30–38
Tatsumi S, Uchihara T, Aiba I, Iwasaki Y, Mimuro M, Takahashi R, Yoshida M (2014b) Ultrastructural differences in pretangles between Alzheimer disease and corticobasal degeneration revealed by comparative light and electron microscopy. Acta Neuropathol Commun 2:161
Tecuapetla F, Jin X, Lima SQ, Costa RM (2016) Complementary contributions of striatal projection pathways to action initiation and execution. Cell 166:703–715
Tercjak A, Bergareche A, Caballero C, Tunon T, Linazasoro G (2014) Lewy bodies under atomic force microscope. Ultrastruct Pathol 38:1–5
Thakur P, Breger LS, Lundblad M, Wan OW, Mattsson B, Luk KC, Lee VMY, Trojanowski JQ, Bjorklund A (2017) Modeling Parkinson’s disease pathology by combination of fibril seeds and alpha-synuclein overexpression in the rat brain. Proc Natl Acad Sci USA 114:E8284–E8293
Thal DR, von Arnim CA, Griffin WS, Mrak RE, Walker L, Attems J, Arzberger T (2015) Frontotemporal lobar degeneration FTLD-tau: preclinical lesions, vascular, and Alzheimer-related co-pathologies. J Neural Transm (Vienna) 122:1007–1018
Thenganatt MA, Jankovic J (2014) Parkinson disease subtypes. JAMA Neurol 71:499–504
Thobois S, Prange S, Sgambato-Faure V, Tremblay L, Broussolle E (2017) Imaging the etiology of apathy, anxiety, and depression in Parkinson’s disease: implication for treatment. Curr Neurol Neurosci Rep 17:76
Thomas AJ, Mahin-Babaei F, Saidi M, Lett D, Taylor JP, Walker L, Attems J (2018) Improving the identification of dementia with Lewy bodies in the context of an Alzheimer’s-type dementia. Alzheimers Res Ther 10:27
Thomas AJ, Donaghy P, Roberts G, Colloby SJ, Barnett NA, Petrides G, Lloyd J, Olsen K, Taylor JP, McKeith I, O’Brien JT (2019) Diagnostic accuracy of dopaminergic imaging in prodromal dementia with Lewy bodies. Psychol Med 49:396–402
Tiraboschi P, Attems J, Thomas A, Brown A, Jaros E, Lett DJ, Ossola M, Perry RH, Ramsay L, Walker L, McKeith IG (2015) Clinicians’ ability to diagnose dementia with Lewy bodies is not affected by beta-amyloid load. Neurology 84:496–499
Titova N, Qamar MA, Chaudhuri KR (2017) The nonmotor features of Parkinson’s disease. Int Rev Neurobiol 132:33–54
Togo T, Cookson N, Dickson DW (2002) Argyrophilic grain disease: neuropathology, frequency in a dementia brain bank and lack of relationship with apolipoprotein E. Brain Pathol 12:45–52
Tomiyama M (2017) Symptoms and pathophysiology of dyskinesias. Brain Nerve 69:1409–1416
Tomiyama H, Hatano T, Hattori N (2007) Clinical molecular genetics for PARK8 (LRRK2). Brain Nerve 59:839–850
Tong J, Wong H, Guttman M, Ang LC, Forno LS, Shimadzu M, Rajput AH, Muenter MD, Kish SJ, Hornykiewicz O, Furukawa Y (2010) Brain alpha-synuclein accumulation in multiple system atrophy, Parkinson’s disease and progressive supranuclear palsy: a comparative investigation. Brain 133:172–188
Tran TN, Vo TNN, Frei K, Truong DD (2018) Levodopa-induced dyskinesia: clinical features, incidence, and risk factors. J Neural Transm (Vienna) 125:1109–1117
Tremblay ME, Cookson MR, Civiero L (2019) Glial phagocytic clearance in Parkinson’s disease. Mol Neurodegener 14:16
Trinh J, Zeldenrust FMJ, Huang J, Kasten M, Schaake S, Petkovic S, Madoev H, Grunewald A, Almuammar S, Konig IR, Lill CM, Lohmann K, Klein C, Marras C (2018) Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov Disord 33:1857–1870
Trojanowski JQ, Revesz T (2007) Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol 33:615–620
Tsang AH, Chung KK (2009) Oxidative and nitrosative stress in Parkinson’s disease. Biochim Biophys Acta 1792:643–650
Tzoulis C, Schwarzlmuller T, Biermann M, Haugarvoll K, Bindoff LA (2016) Mitochondrial DNA homeostasis is essential for nigrostriatal integrity. Mitochondrion 28:33–37
Ubhi K, Rockenstein E, Mante M, Inglis C, Adame A, Patrick C, Whitney K, Masliah E (2010) Neurodegeneration in a transgenic mouse model of multiple system atrophy is associated with altered expression of oligodendroglial-derived neurotrophic factors. J Neurosci 30:6236–6246
Ubhi K, Low P, Masliah E (2011) Multiple system atrophy: a clinical and neuropathological perspective. Trends Neurosci 34:581–590
Uchihara T (2014) Pretangles and neurofibrillary changes: Similarities and differences between AD and CBD based on molecular and morphological evolution. Neuropathology 34:571–577
Uchikado H, DelleDonne A, Ahmed Z, Dickson DW (2006a) Lewy bodies in progressive supranuclear palsy represent an independent disease process. J Neuropathol Exp Neurol 65:387–395
Uchikado H, Lin WL, DeLucia MW, Dickson DW (2006b) Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol 65:685–697
Uemura N, Yagi H, Uemura MT, Hatanaka Y, Yamakado H, Takahashi R (2018) Inoculation of alpha-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol Neurodegener 13:21
Uitti RJ, Rajput AH, Ashenhurst EM, Rozdilsky B (1985) Cyanide-induced parkinsonism: a clinicopathologic report. Neurology 35:921–925
Vaikath NN, Erskine D, Morris CM, Majbour NK, Vekrellis K, Li JY, El-Agnaf OMA (2018) Heterogeneity in alpha-synuclein subtypes and their expression in cortical brain tissue lysates from Lewy body diseases and Alzheimer’s disease. Neuropathol Appl Neurobiol. https://doi.org/10.1111/nan.12531
Valdinocci D, Radford RAW, Goulding M, Hayashi J, Chung RS, Pountney DL (2018) Extracellular Interactions of alpha-synuclein in multiple system atrophy. Int J Mol Sci 19:4129
Valera E, Spencer B, Mott J, Trejo M, Adame A, Mante M, Rockenstein E, Troncoso JC, Beach TG, Masliah E, Desplats P (2017) MicroRNA-101 modulates autophagy and oligodendroglial alpha-synuclein accumulation in multiple system atrophy. Front Mol Neurosci 10:329
van de Berg WD, Hepp DH, Dijkstra AA, Rozemuller JA, Berendse HW, Foncke E (2012) Patterns of alpha-synuclein pathology in incidental cases and clinical subtypes of Parkinson’s disease. Parkinsonism Relat Disord 18(Suppl 1):S28–S30
Van Do B, Gouel F, Jonneaux A, Timmerman K, Gele P, Petrault M, Bastide M, Laloux C, Moreau C, Bordet R, Devos D, Devedjian JC (2016) Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol Dis 94:169–178
van Swieten J, Spillantini MG (2007) Hereditary frontotemporal dementia caused by Tau gene mutations. Brain Pathol 17:63–73
Vasili E, Dominguez-Meijide A, Outeiro TF (2019) Spreading of alpha-synuclein and tau: a systematic comparison of the mechanisms involved. Front Mol Neurosci 12:107
Vergouw LJM, van Steenoven I, van de Berg WDJ, Teunissen CE, van Swieten JC, Bonifati V, Lemstra AW, de Jong FJ (2017) An update on the genetics of dementia with Lewy bodies. Parkinsonism Relat Disord 43:1–8
Verheijen BM, Oyanagi K, van Leeuwen FW (2018) Dysfunction of protein quality control in parkinsonism–dementia complex of Guam. Front Neurol 9:173
Vermeiren Y, De Deyn PP (2017) Targeting the norepinephrinergic system in Parkinson’s disease and related disorders: the locus coeruleus story. Neurochem Int 102:22–32
Verstraeten A, Theuns J, Van Broeckhoven C (2015) Progress in unraveling the genetic etiology of Parkinson disease in a genomic era. Trends Genet 31:140–149
Veys L, Vandenabeele M, Ortuno-Lizaran I, Baekelandt V, Cuenca N, Moons L, De Groef L (2019) Retinal alpha-synuclein deposits in Parkinson’s disease patients and animal models. Acta Neuropathol 137:379–395
Vilensky JA (ed) (2011) Encephalitis lethargica: during and after the epidemic. Oxford University Press, Oxford
Virmani T, Moskowitz CB, Vonsattel JP, Fahn S (2015) Clinicopathological characteristics of freezing of gait in autopsy-confirmed Parkinson’s disease. Mov Disord 30:1874–1884
Vitale F, Capozzo A, Mazzone P, Scarnati E (2018) Neurophysiology of the pedunculopontine tegmental nucleus. Neurobiol Dis. https://doi.org/10.1016/j.nbd.2018.1003.1004
Volpicelli-Daley LA (2017) Effects of alpha-synuclein on axonal transport. Neurobiol Dis 105:321–327
Volta M, Milnerwood AJ, Farrer MJ (2015) Insights from late-onset familial parkinsonism on the pathogenesis of idiopathic Parkinson’s disease. Lancet Neurol 14:1054–1064
Voon V, Napier TC, Frank MJ, Sgambato-Faure V, Grace AA, Rodriguez-Oroz M, Obeso J, Bezard E, Fernagut PO (2017) Impulse control disorders and levodopa-induced dyskinesias in Parkinson’s disease: an update. Lancet Neurol 16:238–250
Voronkov DN, Salkov VN, Anufriev PL, Khudoerkov RM (2018) Lewy bodies in Parkinson’s disease: histological, immunohistochemical, and interferometric examinations. Arkhiv Patologii 80:9–13
Voutsinas GE, Stavrou EF, Karousos G, Dasoula A, Papachatzopoulou A, Syrrou M, Verkerk AJ, van der Spek P, Patrinos GP, Stoger R, Athanassiadou A (2010) Allelic imbalance of expression and epigenetic regulation within the alpha-synuclein wild-type and p.Ala53Thr alleles in Parkinson disease. Hum Mutat 31:685–691
Vriend C, Raijmakers P, Veltman DJ, van Dijk KD, van der Werf YD, Foncke EM, Smit JH, Berendse HW, van den Heuvel OA (2014) Depressive symptoms in Parkinson’s disease are related to reduced [123I]FP-CIT binding in the caudate nucleus. J Neurol Neurosurg Psychiatry 85:159–164
Wakabayashi K, Miki Y (2018) Multi-organ distribution of alpha-synuclein pathology in dementia with Lewy bodies. Brain Nerve 70:489–500
Wakabayashi K, Mori F, Nishie M, Oyama Y, Kurihara A, Yoshimoto M, Kuroda N (2005) An autopsy case of early (“minimal change”) olivopontocerebellar atrophy (multiple system atrophy-cerebellar). Acta Neuropathol 110:185–190
Wakabayashi K, Mori F, Tanji K, Orimo S, Takahashi H (2010) Involvement of the peripheral nervous system in synucleinopathies, tauopathies and other neurodegenerative proteinopathies of the brain. Acta Neuropathol 120:1–12
Wakabayashi K, Tanji K, Odagiri S, Miki Y, Mori F, Takahashi H (2013) The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol Neurobiol 47:495–508
Walker DG, Lue LF, Adler CH, Shill HA, Caviness JN, Sabbagh MN, Akiyama H, Serrano GE, Sue LI, Beach TG (2013) Changes in properties of serine 129 phosphorylated alpha-synuclein with progression of Lewy-type histopathology in human brains. Exp Neurol 240:190–204
Walker L, McAleese KE, Thomas AJ, Johnson M, Martin-Ruiz C, Parker C, Colloby SJ, Jellinger K, Attems J (2015) Neuropathologically mixed Alzheimer’s and Lewy body disease: burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol 129:729–748
Wang Y, Balaji V, Kaniyappan S, Kruger L, Irsen S, Tepper K, Chandupatla R, Maetzler W, Schneider A, Mandelkow E, Mandelkow EM (2017) The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener 12:5
Wang B, Underwood R, Kamath A, Britain C, McFerrin MB, McLean PJ, Volpicelli-Daley LA, Whitaker RH, Placzek WJ, Becker K, Ma J, Yacoubian TA (2018) 14-3-3 proteins reduce cell-to-cell transfer and propagation of pathogenic alpha-synuclein. J Neurosci 38:8211–8232
Wang X, Becker K, Levine N, Zhang M, Lieberman AP, Moore DJ, Ma J (2019) Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol Commun 7:41
Waring SC, Esteban-Santillan C, Reed DM, Craig UK, Labarthe DR, Petersen RC, Kurland LT (2004) Incidence of amyotrophic lateral sclerosis and of the parkinsonism-dementia complex of Guam, 1950-1989. Neuroepidemiology 23:192–200
Watanabe H, Riku Y, Nakamura T, Hara K, Ito M, Hirayama M, Yoshida M, Katsuno M, Sobue G (2016) Expanding concept of clinical conditions and symptoms in multiple system atrophy. Rinsho Shinkeigaku 56:457–464
Wei L, Hu X, Yuan Y, Liu W, Chen H (2018) Abnormal ventral tegmental area-anterior cingulate cortex connectivity in Parkinson’s disease with depression. Behav Brain Res 347:132–139
Weismiller HA, Murphy R, Wei G, Ma B, Nussinov R, Margittai M (2018) Structural disorder in four-repeat Tau fibrils reveals a new mechanism for barriers to cross-seeding of Tau isoforms. J Biol Chem 293:17336–17348
Wenning GK, Quinn N, Magalhaes M, Mathias C, Daniel SE (1994) “Minimal change” multiple system atrophy. Mov Disord 9:161–166
Wenning GK, Jellinger K, Litvan I (1997) Supranuclear gaze palsy and eyelid apraxia in postencephalitic parkinsonism. J Neural Transm 104:845–865
Wenning GK, Litvan I, Tolosa E (2011) Milestones in atypical and secondary parkinsonisms. Mov Disord 26:1083–1095
Wenning GK, Geser F, Krismer F, Seppi K, Duerr S, Boesch S, Kollensperger M, Goebel G, Pfeiffer KP, Barone P, Pellecchia MT, Quinn NP, Koukouni V, Fowler CJ, Schrag A, Mathias CJ, Giladi N, Gurevich T, Dupont E, Ostergaard K, Nilsson CF, Widner H, Oertel W, Eggert KM, Albanese A, del Sorbo F, Tolosa E, Cardozo A, Deuschl G, Hellriegel H, Klockgether T, Dodel R, Sampaio C, Coelho M, Djaldetti R, Melamed E, Gasser T, Kamm C, Meco G, Colosimo C, Rascol O, Meissner WG, Tison F, Poewe W (2013) The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol 12:264–274
Wenning G, Trojanowski JQ, Kaufmann H, Wisniewski T, Rocca WA, Low PA (2018) Is multiple system atrophy an infectious disease? Ann Neurol 83:10–12
Whitwell JL (2018) Tau imaging in parkinsonism: what have we learned so far? Mov Disord Clin Pract 5:118–130
Wichmann T, Bergman H, DeLong MR (2018) Basal ganglia, movement disorders and deep brain stimulation: advances made through non-human primate research. J Neural Transm (Vienna) 125:419–430
Wider C, Dickson DW, Stoessl AJ, Tsuboi Y, Chapon F, Gutmann L, Lechevalier B, Calne DB, Personett DA, Hulihan M, Kachergus J, Rademakers R, Baker MC, Grantier LL, Sujith OK, Brown L, Calne S, Farrer MJ, Wszolek ZK (2009) Pallidonigral TDP-43 pathology in Perry syndrome. Parkinsonism Relat Disord 15:281–286
Wilkaniec A, Lenkiewicz AM, Czapski GA, Jesko HM, Hilgier W, Brodzik R, Gassowska-Dobrowolska M, Culmsee C, Adamczyk A (2019) Extracellular alpha-synuclein oligomers induce parkin s-nitrosylation: relevance to sporadic Parkinson’s disease etiopathology. Mol Neurobiol 56:125–140
Williams DR, Holton JL, Strand K, de Silva R, Lees AJ, Revesz T (2007) Differences in tau load are associated with different clinical phenotypes in progressive supranuclear palsy observations (abstr.). Neuropathol Appl Neurobiol 33:248–275
Williams-Gray CH, Foltynie T, Lewis SJ, Barker RA (2006) Cognitive deficits and psychosis in Parkinson’s disease: a review of pathophysiology and therapeutic options. CNS Drugs 20:477–505
Wilson H, Niccolini F, Pellicano C, Politis M (2019) Cortical thinning across Parkinson’s disease stages and clinical correlates. J Neurol Sci 398:31–38
Woerman AL, Kazmi SA, Patel S, Freyman Y, Oehler A, Aoyagi A, Mordes DA, Halliday GM, Middleton LT, Gentleman SM, Olson SH, Prusiner SB (2018) MSA prions exhibit remarkable stability and resistance to inactivation. Acta Neuropathol 135:49–63
Woerman AL, Oehler A, Kazmi SA, Lee J, Halliday GM, Middleton LT, Gentleman SM, Mordes DA, Spina S, Grinberg LT, Olson SH, Prusiner SB (2019) Multiple system atrophy prions retain strain specificity after serial propagation in two different Tg(SNCA*A53T) mouse lines. Acta Neuropathol 137:437–454
Wong YC, Krainc D (2017) Alpha-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med 23:1–13
Wszolek ZK, Slowinski J, Golan M, Dickson DW (2005) Frontotemporal dementia and parkinsonism linked to chromosome 17. Folia Neuropathol 43:258–270
Xia Y, Zhang G, Han C, Ma K, Guo X, Wan F, Kou L, Yin S, Liu L, Huang J, Xiong N, Wang T (2019) Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis 10:174
Yabe I, Soma H, Takei A, Fujiki N, Yanagihara T, Sasaki H (2006) MSA-C is the predominant clinical phenotype of MSA in Japan: analysis of 142 patients with probable MSA. J Neurol Sci 249:115–121
Yamasaki TR, Holmes BB, Furman JL, Dhavale DD, Su BW, Song ES, Cairns NJ, Kotzbauer PT, Diamond MI (2019) Parkinson’s disease and multiple system atrophy have distinct alpha-synuclein seed characteristics. J Biol Chem 294:1045–1058
Yamashita S, Sakashita N, Yamashita T, Tawara N, Tasaki M, Kawakami K, Komohara Y, Fujiwara Y, Kamikawa M, Nakagawa T, Hirano T, Maeda Y, Hasegawa M, Takeya M, Ando Y (2014) Concomitant accumulation of alpha-synuclein and TDP-43 in a patient with corticobasal degeneration. J Neurol 261:2209–2217
Yamazaki M, Hasegawa M, Mori O, Murayama S, Tsuchiya K, Ikeda K, Chen KM, Katayama Y, Oyanagi K (2005) Tau-positive fine granules in the cerebral white matter: a novel finding among the tauopathies exclusive to parkinsonism-dementia complex of Guam. J Neuropathol Exp Neurol 64:839–846
Yan X, Uronen RL, Huttunen HJ (2018) The interaction of alpha-synuclein and Tau: a molecular conspiracy in neurodegeneration? Semin Cell Dev Biol. https://doi.org/10.1016/j.semcdb.2018.1005.1005
Yang HS, Yu L, White CC, Chibnik LB, Chhatwal JP, Sperling RA, Bennett DA, Schneider JA, De Jager PL (2018) Evaluation of TDP-43 proteinopathy and hippocampal sclerosis in relation to APOE epsilon4 haplotype status: a community-based cohort study. Lancet Neurol 17:773–781
Yap SM, Lynch T, MacMahon P, Murray B (2017) Paraneoplastic atypical parkinsonism with anti-crmp5 antibodies and severe caudate and putaminal hypometabolism on 18-fluorodeoxyglucose positron emission tomography of the brain. Mov Disord Clin Pract 4:263–265. https://doi.org/10.1002/mdc1003.12370
Yasuda T, Nakata Y, Mochizuki H (2013) Alpha-synuclein and neuronal cell death. Mol Neurobiol 47:466–483
Yokota O, Tsuchiya K, Terada S, Ishizu H, Uchikado H, Ikeda M, Oyanagi K, Nakano I, Murayama S, Kuroda S, Akiyama H (2008) Basophilic inclusion body disease and neuronal intermediate filament inclusion disease: a comparative clinicopathological study. Acta Neuropathol 115:561–575
Yokoyama Y, Toyoshima Y, Shiga A, Tada M, Kitamura H, Hasegawa K, Onodera O, Ikeuchi T, Someya T, Nishizawa M, Kakita A, Takahashi H (2016) Pathological and clinical spectrum of progressive supranuclear palsy: with special reference to astrocytic tau pathology. Brain Pathol 26:155–166
Yokoyama JS, Karch CM, Fan CC, Bonham LW, Kouri N, Ross OA, Rademakers R, Kim J, Wang Y, Hoglinger GU, Muller U, Ferrari R, Hardy J, Momeni P, Sugrue LP, Hess CP, James Barkovich A, Boxer AL, Seeley WW, Rabinovici GD, Rosen HJ, Miller BL, Schmansky NJ, Fischl B, Hyman BT, Dickson DW, Schellenberg GD, Andreassen OA, Dale AM, Desikan RS (2017) Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta Neuropathol 133:825–837
Yoshida M (2014) Astrocytic inclusions in progressive supranuclear palsy and corticobasal degeneration. Neuropathology 34:555–570
Yoshida K, Hata Y, Kinoshita K, Takashima S, Tanaka K, Nishida N (2017) Incipient progressive supranuclear palsy is more common than expected and may comprise clinicopathological subtypes: a forensic autopsy series. Acta Neuropathol 133:809–823
Yoshino H, Nishioka K, Li Y, Oji Y, Oyama G, Hatano T, Machida Y, Shimo Y, Hayashida A, Ikeda A, Mogushi K, Shibagaki Y, Hosaka A, Iwanaga H, Fujitake J, Ohi T, Miyazaki D, Sekijima Y, Oki M, Kusaka H, Fujimoto KI, Ugawa Y, Funayama M, Hattori N (2018) GCH1 mutations in dopa-responsive dystonia and Parkinson’s disease. J Neurol 265:1860–1870
Young CB, Sonne J (2018) Neuroanatomy, Basal Ganglia. StatPearls Publishing, Treasure Island
Yousaf T, Dervenoulas G, Valkimadi PE, Politis M (2019) Neuroimaging in Lewy body dementia. J Neurol 266:1–26
Yuan X, Chen Y, Cao B, Zhao B, Wei Q, Guo X, Yang Y, Yuan L, Shang H (2015) An association analysis of the R1628P and G2385R polymorphisms of the LRRK2 gene in multiple system atrophy in a Chinese population. Parkinsonism Relat Disord 21:147–149
Zaccai J, Brayne C, McKeith I, Matthews F, Ince PG (2008) Patterns and stages of alpha-synucleinopathy: relevance in a population-based cohort. Neurology 70:1042–1048
Zahodne LB, Fernandez HH (2008) Pathophysiology and treatment of psychosis in Parkinson’s disease: a review. Drugs Aging 25:665–682
Zaltieri M, Grigoletto J, Longhena F, Navarria L, Favero G, Castrezzati S, Colivicchi MA, Della Corte L, Rezzani R, Pizzi M, Benfenati F, Spillantini MG, Missale C, Spano P, Bellucci A (2015a) Alpha-synuclein and synapsin III cooperatively regulate synaptic function in dopamine neurons. J Cell Sci 128:2231–2243
Zaltieri M, Longhena F, Pizzi M, Missale C, Spano P, Bellucci A (2015b) Mitochondrial dysfunction and alpha-synuclein synaptic pathology in Parkinson’s disease: who’s on first? Parkinsons Dis 2015:108029
Zange L, Noack C, Hahn K, Stenzel W, Lipp A (2015) Phosphorylated alpha-synuclein in skin nerve fibres differentiates Parkinson’s disease from multiple system atrophy. Brain 138:2310–2321
Zeng XS, Geng WS, Jia JJ, Chen L, Zhang PP (2018) Cellular and molecular basis of neurodegeneration in Parkinson disease. Front Aging Neurosci 10:109
Zhai S, Shen W, Graves SM, Surmeier DJ (2019) Dopaminergic modulation of striatal function and Parkinson’s disease. J Neural Transm (Vienna) 126:411–422
Zhang Y, Larcher KM, Misic B, Dagher A (2017) Anatomical and functional organization of the human substantia nigra and its connections. Elife 6:e26653
Zhang G, Xia Y, Wan F, Ma K, Guo X, Kou L, Yin S, Han C, Liu L, Huang J, Xiong N, Wang T (2018a) New perspectives on roles of alpha-synuclein in Parkinson’s disease. Front Aging Neurosci 10:370
Zhang X, Gao F, Wang D, Li C, Fu Y, He W, Zhang J (2018b) Tau pathology in Parkinson’s disease. Front Neurol 9:809
Zhao Q, Yang X, Tian S, An R, Zheng J, Xu Y (2016) Association of the COQ2 V393A variant with risk of multiple system atrophy in East Asians: a case-control study and meta-analysis of the literature. Neurol Sci 37:423–430
Zhou ZD, Selvaratnam T, Lee JCT, Chao YX, Tan EK (2019) Molecular targets for modulating the protein translation vital to proteostasis and neuron degeneration in Parkinson’s disease. Transl Neurodegener 8:6
Zhu Y, Song X, Xu M, Hu X, Li E, Liu J, Yuan Y, Gao JH, Liu W (2016) Impaired interhemispheric synchrony in Parkinson’s disease with depression. Sci Rep 6:27477
Zhukareva V, Shah K, Uryu K, Braak H, Del Tredici K, Sundarraj S, Clark C, Trojanowski JQ, Lee VM (2002) Biochemical analysis of tau proteins in argyrophilic grain disease, Alzheimer’s disease, and Pick’s disease : a comparative study. Am J Pathol 161:1135–1141
Zijlmans JC, Daniel SE, Hughes AJ, Revesz T, Lees AJ (2004) Clinicopathological investigation of vascular parkinsonism, including clinical criteria for diagnosis. Mov Disord 19:630–640
Zondler L, Kostka M, Garidel P, Heinzelmann U, Hengerer B, Mayer B, Weishaupt JH, Gillardon F, Danzer KM (2017) Proteasome impairment by alpha-synuclein. PLoS One 12:e0184040
Ztaou S, Amalric M (2019) Contribution of cholinergic interneurons to striatal pathophysiology in Parkinson’s disease. Neurochem Int 126:1–10
Zucca FA, Segura-Aguilar J, Ferrari E, Munoz P, Paris I, Sulzer D, Sarna T, Casella L, Zecca L (2017) Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog Neurobiol 155:96–119
Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, Toker NJ, Jeon S, Fredriksen K, Mazzulli JR (2018) Reversible conformational conversion of alpha-synuclein into toxic assemblies by glucosylceramide. Neuron 97(92–107):e110
Acknowledgements
The author thanks Mr. E. Mitter-Ferstl, PhD, for secretarial and graphical work. The study was partially funded by the Society for the Promotion of Research in Experimental Neurology, Vienna, Austria.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that he has no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jellinger, K.A. Neuropathology and pathogenesis of extrapyramidal movement disorders: a critical update—I. Hypokinetic-rigid movement disorders. J Neural Transm 126, 933–995 (2019). https://doi.org/10.1007/s00702-019-02028-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-019-02028-6