
Abstract
Dementia in Parkinson’s disease (PD/PDD) is a common complication with a prevalence of up to 50%, but the specific changes underlying the cognitive decline remain undefined. Neuronal degeneration resulting in the dysfunction of multiple subcortical neurochemical projection systems has been described along with Lewy body-type pathology in cortical and limbic regions. Advanced alpha-synuclein (αSyn) pathology is not necessarily sufficient for producing dementia and concomitant Alzheimer’s disease (AD) change has also been proposed as a possible substrate of PDD. A lack of consensus in the extant literature likely stems from clinical heterogeneity and variable reliability in clinical characterisation as well as other historical and methodological issues. The concurrent presence of abnormally deposited αSyn, amyloid-β and tau proteins in the PDD brain and the interaction of these molecules in a linked pathological cascade of AD and PD-related mechanisms may prove important in determining the underlying pathological process for the development of dementia in PD and this concept of combined pathologies awaits further investigation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In 1817, James Parkinson in his classic treatise “An Essay on the Shaking Palsy” cogently described many of the salient motor features of the disorder but also stated “…the senses and intellects being uninjured”. Today, however, Parkinson’s disease (PD) is viewed as a neurodegenerative condition characterised clinically by a variety of non-motor complications including neuropsychiatric and autonomic/vegetative features in addition to the core motor syndrome. Although the classical motor signs in PD are ascribed in the main to dopaminergic cell loss in the substantia nigra pars compacta [43] resulting in striatal dopamine depletion, the anatomical and pathological substrate underlying dementia has been a matter of controversy. Dementia is highly prevalent in PD and predicts an increased risk for nursing home placement [4], more rapid disease progression [123], greater caregiver burden, reduced patient and caregiver quality of life [3] and increased mortality [90, 92].
In this report, we review the literature concerning the anatomical and pathological substrates underlying dementia and cognitive dysfunction in PD.
Epidemiology
The prevalence of PD is about 1% of those aged 60 years and over and increases with age [36]. The mean age at disease onset is in the early to mid-60s [65]. Life expectancy in Western countries has increased over the last several decades [114] and PD patients share the gain in longevity with the life span of contemporary patients longer than that of 30 or 50 years ago and not significantly lower than the average life expectancy.
The epidemiology of PD with dementia (PDD) is difficult to evaluate accurately due to differences in patient populations, study design and criteria for diagnosing PDD. Amongst all cases of dementia, studies have estimated that 3–4% are due to PDD [35]. Cross-sectional studies in PD cohorts, providing point prevalence estimates of dementia, have yielded frequencies between 10 and 40% [6, 29, 102]. Prospective studies have reported a cumulative incidence of 19–53% [62]. When compared with the age-matched healthy control subjects, the risk for developing dementia in PD patients increases up to sixfold [2]. Because mortality is higher amongst demented subjects, period (or cumulative) prevalence estimates provide a more reliable picture of the presence of dementia in PD [42]. The cumulative prevalence of dementia in PD has been reported to range between 48 and 78% [1, 56] and in patients surviving for 20 years or longer with the illness, as is increasingly common, the vast majority will develop dementia or serious cognitive difficulties [26].
Although cognitive symptoms usually occur at later stages of the disease, a few studies have reported cognitive deficits at the time of diagnosis of PD. Muslimovic et al. [106] found 24% of patients with newly diagnosed PD showing cognitive impairment, whilst in another study by Foltynie et al. [44] 36% of patients amongst 239 newly diagnosed PD patients had similar findings. Thus, even at the time of diagnosis, cognitive decline can be identified in a significant number of PD patients.
There are a number of features that consistently associate with dementia in PD including advanced age at onset, a longer duration of disease, an akinetic rigid syndrome with symmetrical signs, impairment of gait and balance, higher disability and bradykinesia scores [41, 57, 101, 115], major depression or depressive symptoms and other unusual or atypical neurological features such as the early occurrence of autonomic failure, and a poor to moderate response to dopaminergic treatment [6, 8, 42, 58].
Although it is well established that the apolipoprotein E (ApoE) 4 allele confers an increased risk for Alzheimer’s disease (AD), studies examining this gene and susceptibility to dementia in PD have produced contradictory findings, with reports failing to predict a higher risk for dementia [64, 134] contrasting with others implicating a higher prevalence of dementia in PD with the ApoE 4 allele [60, 110]. It is important to note that meta-analysis reviews examining the impact of the ApoE 4 allele on dementia in PD may reflect the inclusion of studies not using uniform diagnostic criteria for dementia in PD along with pathological confirmation.
Clinical features of dementia in PD
Dementia involves the impairment of multiple domains of cognition. Since the mid-1970s, one view has been that dementia can be separated into cortical and subcortical types, a distinction that has found support from both clinical [111] as well as pathological perspectives [34]. Individuals with Parkinsonism are most often elderly and may harbour multiple cerebral pathologies contributing both to motor deficits and cognitive decline yielding a heterogeneous clinical picture. Our discussion relates to the well-recognised disorder of Lewy body PD complicated by progressive cognitive decline culminating in dementia without significant co-morbidities such as AD, significant cerebrovascular disease or other remarkable brain pathology. The dementia syndrome in PD has been extensively studied. Recently, the movement disorder society recruited a task force to define the clinical diagnostic criteria for PDD [42]. According to this diagnostic criteria abnormalities of attention, concentration, memory, word list generation, abstraction and categorisation, judgement, problem resolution, strategy formulation and visuospatial dysfunction (such as problems with visual discrimination, visual organisation, spatial orientation, drawing and angle perception) represent core features of the dementia syndrome in PD [50, 89]. In addition to intellectual/cognitive impairment, hallucinations, delusions, apathy, excessive daytime sleepiness and abnormalities of mood and personality change including, depression, mania and psychosis have also been described in PDD [31, 42]. The neurobehavioural and neuropsychiatric clinical profile of dementia in PD occurs in the absence of prominent apraxia, aphasia and agnosia, clinical features that are more typically characteristic of ‘cortical’ dementias such as AD.
Neuropathology of dementia in PD
α-Synucleinopathy
The classical neuropathological feature of PD is the presence of neuronal intracytoplasmic inclusions known as LBs and inclusions confined to neuronal processes known as Lewy neurites (LN). The discovery that α-synuclein (αSyn) gene mutations can cause PD [88, 113] and that the encoded protein constitutes a major component of LB and LN [122] has provided the basis for a molecular definition of the disease and a better understanding of its pathogenesis. In fact, the term ‘synucleinopathy’ has been used to designate a spectrum of degenerative diseases that share the presence of abnormal αSyn immunoreactive inclusion bodies in neurons and/or glial cells [46, 127] with the term encompassing diverse, but clinically overlapping entities, such as multiple system atrophy, PD, dementia with Lewy bodies (DLB), the LB variant of Alzheimer’s disease (LBVAD) and neurodegeneration with brain iron accumulation type 1 (NBIA I) [9, 13, 38, 45, 107].
PD is the most frequently occurring synucleinopathy. The involvement of the substantia nigra with the destruction of dopaminergic neurons in the pars compacta is the universally acknowledged hallmark of PD [33, 95]. However, PD is a ‘multisystem’ illness with pathology occurring in a variety of cortical and subcortical regions including, but not limited to: the olfactory bulb and related areas, the spinal cord, the dorsal motor nucleus of the vagus nerve, the pedunculopontine nucleus, non-thalamic nuclear nuclei with diffuse projections to cortical and subcortical regions, intralaminar and midline thalamic nuclei, the amygdala, nucleus basalis of Meynert (NBM), transentorhinal cortex, hippocampal formation, temporal and frontal cortices as well as autonomic nerves (e.g. cardiac and abdominopelvic autonomic plexuses) [20, 23, 67, 72, 80, 82, 83, 105, 116, 119].
Controversy over the underlying bases of dementia in PD
The widespread, yet selective, pathology observed in the PD brain accounts for the variety of non-motor symptoms seen in PD including dementia. Nevertheless, there is a debate in the literature regarding the particular changes underlying dementia in PD with no consensus as to whether dementia relates to LB disease-type pathology or rather concomitant AD-type changes or even a combination of both. In addition, the contributions of small vessel subcortical disease along with cortical vascular pathology have likely not received sufficient attention as aggravating factors in reducing cognitive reserve. Reasons for ongoing controversy include clinical heterogeneity in the cases studied with an obvious problem, especially in earlier studies residing in the inclusion of cases with dementia as a presenting feature. The temporal relationship between the clinical onset of parkinsonism and dementia currently defines two distinct clinical entities (PDD and DLB): if extrapyramidal symptoms precede dementia by more than 12 months the patient is classified as having PDD and if dementia and extrapyramidal symptoms develop together within a 12-month period or dementia precedes extrapyramidal symptoms then the patient is classified as having DLB [104]. Exceptions for the temporal relationship between the clinical onset of parkinsonism and dementia have also been described [76].
Difficulty in defining the underlying substrate of dementia in PD (and DLB) also relates to historical methodological/technical issues. LB were initially identified with the use of conventional histological techniques such as haematoxylin and eosin (H&E) staining that underestimates the presence of LB, especially in cortical regions. In the late 1980s immunohistochemistry (IHC) was introduced revealing the presence of a variety of antigenic components within LB. The most consistently recognised proteins were the cytoskeletal proteins neurofilaments (NFs) and ubiquitin (Ub) [51, 93]. Comparison of antibodies raised against NFs and Ub demonstrated that Ub-IHC was a more reliable and sensitive method for the detection of LB because antibodies against NFs only immunolabeled around half of the H&E-stained LB [32]. The use of Ub-IHC was especially helpful in detecting cortical LB that possesses a less conspicuous morphology than their brainstem counterparts. Nevertheless, it soon became apparent that Ub immunoreactivity was not unique to LB, but appears with other inclusions such as neurofibrillary tangles (NFTs) that were difficult to distinguish from LB [93]. αSyn IHC, introduced in 1997 [121, 122], has become the ‘gold standard’ for the detection of LB as it is a much more sensitive and specific technique and enables the distinction of LB from other pathological inclusions. Similarly, early descriptions of the AD histopathological features were based on the silver stains, amyloid stains and modifications thereof such as thioflavin-S fluorescence, Bielschowsky silver stain, Campbell–Switzer silver stain, Bodian stain and the Gallyas silver stain [37]. Although some of these stains (e.g. thioflavin-S fluorescence) are useful for the study of AD, they are not selective for a particular AD neuropathological feature. For instance, Bielschowsky silver stain detects both plaques and NFTs, whereas Campbell–Switzer silver stain and thioflavin-S fluorescence detect plaques, NFTs as well as LB [19, 103]. Immunohistochemical detection of AD-type pathology has proved superior to earlier stains as it is specific and sensitive to particular pathological features (i.e. plaques or NFTs) and provides a biologically meaningful stain that is derived from molecular and biochemical characteristics of the pathological change under study. In view of the methodological limitations of earlier studies, the current lack of consensus in defining the pathological changes producing dementia in PD is not surprising. Also, apart from the presence of classical inclusions, neuronal loss and comorbid vascular disease, the contribution of other less well-studied pathological changes, including glial involvement and impairment of neuronal (and axonal) function in the absence of morphological change on routine staining (e.g. loss of trophic factor and neurotransmitter enzymatic expression), must also be considered [130].
Degeneration of subcortical nuclei/neurochemical deficits and synaptic loss
In the PDD, brain degeneration of subcortical nuclei resulting in multiple neurochemical deficits is a well-established neuropathological finding and one of the earliest studies to report significant subcortical degeneration in demented versus non-demented PD cases was that of Gaspar and Gray [47], with neuronal loss in the locus coeruleus (LC) and in the nucleus basalis of Meynert (NBM) significantly more pronounced in demented cases. In addition, the same study reported LB in the NBM (95% of the cases) and a reduction in choline acetyltransferase activity in both the NBM and cortex. Yoshimura [135] as well as Kosaka et al. [86] and Sudarsky et al. [124] have all reported significant neuronal loss in the LC and NBM in demented PD cases independently of significant concomitant AD pathology. In PD, NBM cell loss averages 30–40% and is much higher in demented than non-demented PD patients [67]. However, severe cell loss in the NBM without overt dementia has also been reported suggesting that the degeneration of the NBM may precede the onset of mental dysfunction, with a critical threshold of neuronal loss along with equivalent cortical cholinergic denervation occurring before dementia becomes apparent [68]. We have also observed that the extent of αSyn pathology in the NBM is high in both demented and cognitively intact cases, highlighting a universal cholinergic deficit in PD with or without clinically evident cognitive decline [80] (Fig. 1b, f, g). The LC, the main source of noradrenergic input to the forebrain and neocortex, demonstrates neuronal loss that ranges from 40 to 50% and cell loss is more severe in demented PD subjects or those with depression than non-demented or non-depressed patients [69]. As a result of LC neuronal depletion, there is a reduced noradrenergic innervation of the forebrain and neocortex and apart from dementia and depression this may also relate to autonomic dysfunction [59]. The LC in PD shows not only cell loss, but also cell shrinkage that is independent of cortical pathological changes suggesting a primary degeneration of this nucleus [59].

Pathology in cortical and subcortical regions. a Cortical LB pathology in the cingulate gyrus of a demented PD case. b LB and LN pathology in the NBM of a demented PD case. c Selective vulnerability of the CA2 sector of the hippocampus to LN-type pathology. Inset higher magnification showing the web of LNs. d Neurofibrillary pathology in the CA2 sector of the hippocampus of a demented PD case. e Multiple ‘classical’ LB in a pigmented neuron of the medial substantia nigra. A single LB in a pigmented neuron is also evident. A single (f) and multiple (g) LB in a neuron of the NBM magnification: a and b ×20, c ×4 and for inset ×10 and d ×20, e, f and g ×100. PD Parkinson’s disease, LB Lewy bodies, LN Lewy neurites, NBM nucleus basalis of Meynert, CA Cornu Ammonis
The dorsal raphe nucleus (DRN) that gives rise to ascending serotonergic pathways also degenerates leading to a reduction in serotonin, its metabolites and receptors in the striatum and medial frontal cortex [7] with cell depletion averaging between 20 and 40% [67]. DRN cell loss is more severe in depressed than non-depressed patients and serotonergic deficiency has also been related to both cognitive dysfunction and dementia [7, 67, 75].
In PD, there is also a considerable loss of neurons in the ventral tegmental area (VTA), providing the major dopaminergic input to meso-limbic and prefrontal areas. Dementia in PD patients has been associated with greater cell depletion in VTA (in the range of 40–60%) than in non-demented patients [137] and the contiguous medial substantia nigra has also been implicated as a substrate for dementia in PD (Fig. 1e) with Rinne et al. [117] reporting that the degree of dementia significantly correlates with the neuronal loss in the medial nigra, independently of concurrent AD pathology, leading the authors to suggest that intact projections to the caudate nucleus, limbic and cortical areas are essential for normal cognitive function. Whilst Jellinger and Paulus [75] have also reported that demented PD subjects demonstrate more cell loss in the medial nigra, this was accompanied by significantly more severe Alzheimer lesions in isocortex and hippocampus than in non-demented subjects.
Overall, these studies indicate that dementia in PD relates to subcortical neuronal degeneration and resultant neurochemical dysfunction of noradrenergic, serotonergic, dopaminergic and cholinergic projection systems and is not always associated with coincidental Alzheimer-type lesions.
In AD, synaptic integrity has been studied using synaptophysin, a 38-kDa integral membrane protein of synaptic vesicles. These studies have demonstrated a strong inverse correlation between levels of synaptophysin and dementia severity as measured by neuropsychological testing [126]. Synaptophysin levels also inversely correlate with indices of NFTs [17, 24]. The loss of synapses in DLB does not correlate as clearly with cognitive status. In studies of DLB, loss of synapses (measured by decreased levels of synaptophysin) has been demonstrated in the entorhinal cortex [133], yet, others using similar methods found no significant decrease in synaptophysin in the frontal cortex [120]. Synaptic loss and its contribution to the development of dementia in PD by and large has not been extensively studied. A study by Zhan et al. [136] found a reduction in synaptophysin in the pyramidal layer of the neocortex in both PD and PDD subjects, with the latter showing a greater loss of synapses compared with control cases. The authors conclude that the loss of synapses in the cortical neuropil may be a significant factor for the development of dementia. Further studies are warranted to investigate the contribution of synaptic loss to the development of dementia in PD as well as its relationship to αSyn, Aβ and tau deposition.
LB and αSyn
Since αSyn was recognised as the main component of LB, many studies have explored the relationship between LB burden and/or αSyn deposition and the presence/or severity of dementia in PD. With some important exceptions, these studies point to limbic and cortical LB pathology as the main determinant of the presence of dementia in PD. Mattila et al. examined the contribution of both PD and AD pathological lesions to dementia in PD and whilst demonstrating a strong association between LB in the frontal cortex and dementia and correlating NFTs in the entorhinal cortex and hippocampal CA1 area with cognitive impairment, using multivariate statistical analysis the authors found that cortical LB alone contribute to the severity of cognitive impairment, independently of concomitant AD pathology [98], results consistent with a previous study [100] by the same research group in which anti-ubiquitin immunostaining was used to identify cortical LB. Similar results have been reported by Hurtig et al., where cortical LB were found to be a more sensitive and specific correlate of dementia than AD-type pathology in 22 demented as compared to 20 non-demented PD subjects [63]. In another study, dementia correlated with LB densities in the entorhinal cortex and anterior cingulate gyrus [87]. In a study by Harding and Halliday [55] cortical LB densities did not separate cases of DLB from those with PDD although semiquantitative thresholds in the parahippocampus could separate demented from non-demented cases with high sensitivity and specificity. We have demonstrated that dementia in PD positively correlates with both αSyn deposition in a variety of cortical and subcortical structures as well as with AD (tau and Aβ) pathology [80]. Specifically, we have reported a significantly positive association of dementia in PD with αSyn in the anterior cingulate gyrus, superior frontal gyrus, temporal cortex, entorhinal cortex, amygdaloid complex and CA2 sector of the hippocampus (Fig. 1a, c). The relationship between αSyn burden in the anterior cingulate gyrus and dementia was so reliable that statistical analysis demonstrated that αSyn deposition in the anterior cingulate gyrus could differentiate cases with dementia from those without with high sensitivity and specificity (75 and 85%, respectively). This finding supports a primary role of αSyn in the pathological processes of cognitive dysfunction in PD as well as pointing to the anterior cingulate gyrus as an important focus of damage in PD. We have also recently demonstrated that αSyn and Aβ pathology in the claustrum, a region of largely obscure physiological function, strongly relates to the presence of dementia in PD and DLB [85].
Although most clinicopathological studies of dementia in PD are retrospective, a recent prospective community-based study conducted by Aarsland et al. [5] also indicated that LB pathology and not AD histopathological changes drive the progression of cognitive impairment in PD. This group showed that the LB score but not Braak and Braak stage, CERAD score, or chronic vascular lesions, was significantly associated with annual decline on Mini-Mental State Examination (MMSE) in a univariate regression analyses. The authors conclude that ‘Lewy body disease’ is the main substrate driving the progression of cognitive impairment in PD.
Others have suggested LN-type pathology in the hippocampus as relating to cognitive decline and dementia in PD [15, 16, 27, 80]. More specifically, Churchyard and Lees [99] found a positive correlation between LN burden in the CA2 sector of the hippocampus and severity of dementia, although this correlation has remained controversial because ubiquitin IHC was used which is a less sensitive method for the visualisation of LB and LN pathology. Nevertheless, using αSyn IHC, we have confirmed numerous LN in the CA2 sector of the hippocampus as well as an association between LN in the CA2 sector of the hippocampus with dementia in PD [80] (Fig. 1c) indicating a role of this anatomical region in PDD.
Contesting these studies, others have found that a severe LB burden does not necessarily predict dementia or cognitive decline in PD. Colosimo et al. [28] demonstrated classic DLB pathology in PD patients without dementia and Parkkinen et al. [108] have also questioned the involvement of LB pathology in cortical and limbic structures in the development of dementia in PD.
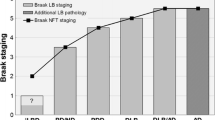
In 2003, Braak et al. [21] devised a staging system according to which αSyn pathology progresses in a systematic fashion and proposed six distinct stages of PD. According to this design, the earliest pathology is to be observed in the DMV and olfactory bulb (stage 1) from where αSyn pathology is thought to proceed in a rostral direction via the pons (stage 2) to the midbrain (stage 3), and from there to the basal forebrain and mesocortex (stage 4) and finally spreading to/involving the neocortex (stages 5–6).
In a later study by the same group [22], an attempt was made to establish a link between cognitive status and the proposed PD stages. A decrease in the median MMSE scores between PD stages 3–6 was found suggesting that the risk of developing dementia increases with the disease progression. Statistical analysis demonstrated a strong positive correlation between the cognitive status of the individual patient and the neuropathological stage and this correlation was characterised by a linear trend.
Aside from several methodological shortcomings of the proposed PD staging system [81, 83, 84], its clinical relevance has also been questioned [25, 70, 71]. Parkkinen et al. [109] examined 226 αSyn-positive individuals and found that 55% of the cases corresponding to Braak’s stages 5–6 lacked clinical signs of dementia or extrapyramidal signs. Given the retrospective nature of the Parkkinen et al. study, it is possible that a proportion of the 55% of cases described had motor and cognitive impairments that were not recorded, but even admitting this caveat, a majority of cases with Braak stages 5–6 would be expected to show obvious clinical signs (i.e. manifest Parkinsonism and cognitive impairment). Parkkinen et al. also demonstrate another weakness of the Braak staging system in that there is no firm requirement for minimum neocortical LB densities to be awarded a neocortical stage.
More recently in an attempt to address the limitations of previous staging systems Beach et al. [14] proposed a new staging system for αSyn with the so-called ‘Unified staging system for Lewy body disorders’. According to Beach et al. subjects with Lewy-type α-synucleinopathy are classified into one of the following stages: I olfactory bulb only; IIa brainstem predominant; IIb limbic predominant; III brainstem and limbic; and IV neocortical. The authors found that progression through these stages is accompanied by a stepwise decline in striatal tyrosine hydroxylase concentration, substantia nigra pigmented neuron loss score, MMSE and Unified Parkinson’s disease Rating Scale (UPDRS) part 3 scores.
AD pathology
AD pathology is commonly found in the PDD brain and clinicopathological studies have also pointed to AD-type pathology as an important determinant of dementia in PD. Hakim and Mathieson [54] examined 34 cases with PD, of which 19 had dementia and demonstrated an increased incidence of AD pathology. Boller et al. [18] and Jellinger and Grisold [66] also suggested AD-type pathology as the main cause of dementia in PD. Hughes et al. [61] found that amongst 100 PD cases examined, 44% had dementia; 29% had AD pathology, 10% had numerous cortical LB and only 6% had a possible vascular cause. Jellinger [68], in a large retrospective pathological study comprising 610 cases, attributed dementia to AD pathology, with only 3.5% of cases with ‘pure’ PD having dementia. Emphasis on AD-type pathology as the main substrate of dementia in PD has also been addressed in studies following the application of αSyn IHC. Jellinger et al. [77] examined the impact of coexisting AD pathology on the natural history of PD. They reported that dementia in PD significantly correlates with coexistent neuritic Alzheimer pathology, particularly when using the CERAD and NIA-R criteria for the diagnosis of AD. Although cerebral amyloid angiopathy (CAA) is not generally thought to relate to dementia or cognitive impairment in PD, a recent study by Jellinger and Attems [74] demonstrated that CAA was more severe in DLB and PDD cases than in subjects with PD without dementia.
Others have not found a strong association between AD pathology and dementia. Mastaglia et al. [97] using Aβ IHC found that dementia in PD is only infrequently due to fully established AD and Jendroska et al. [78] in an earlier study, also reported that dementia cannot be well explained by AD histopathological changes. We have observed a strong positive correlation between the presence of dementia and Aβ deposition in the anterior cingulate gyrus, entorhinal cortex, amygdaloid complex and nucleus basalis of Meynert as well as neurofibrillary changes in the CA2 sector of the hippocampus [80] (Fig. 1d), but αSyn pathology and not AD changes differentiated demented from non-demented PD cases with both high sensitivity and specificity.
Although several studies indicate cortical AD pathology as an important determinant for the development of dementia in PD, the significance of cortical plaques in AD and PD is questionable [10, 11, 87, 98, 128]. For example, there have been inconsistent reports on the correlation between cortical Aβ burden and various measures of clinical deficit in both diseases [132]. In addition, ‘senile’ plaques are found in the neocortex of elderly subjects without overt dementia and are viewed as part of normal ageing [12]. Recently, an increased awareness of striatal pathology in LB diseases has emerged with Duda et al. showing extensive αSyn pathology in the striatum [39]. The greatest density was found in patients with a combination of AD and DLB followed by cases with DLB alone. PD patients also showed mild to moderate changes. Similar findings have been reported by Tsuboi et al. [131]. Furthermore, Liang et al. [91] have shown abundant Aβ pathology in the striatum of cases with DLB that was more pronounced than in cases with PDD. Jellinger and Attems [73] have confirmed these findings in a larger cohort pointing to a morphological distinction between DLB and PDD on the basis of differing striatal pathology. In contrast to a debatable implication of cortical Aβ deposition, in a recent clinicopathological study, we have found Aβ deposition to be significantly greater in the striatum of PDD cases than in non-demented PD cases [82] (Fig. 2). In addition, striatal Aβ deposition was independent of AD changes in the cortex and was minimal in non-demented PD cases. This finding is of particular interest for several reasons. In contrast to cortical Aβ deposition, Aβ deposition in the striatum is universally found in AD brains, but is rarely observed in non-demented elderly individuals [48, 125]. Furthermore, according to phases of β-amyloidosis in the human brain proposed by Thal et al. [129], Aβ deposition in the striatum occurs in phase 3 with associated clinical dementia and, thus, appears to reflect a disease severity-specific pathological change in AD subjects. This evidence points to the striatum as an important anatomical region related to clinically overt dementia in AD. The finding that PDD cases also exhibit a significantly greater Aβ burden in the striatum as opposed to non-demented PD individuals not only makes the striatum a distinctive subcortical region in the development of dementia in PD, but also shows that Aβ striatal pathology strongly associates with clinical dementia. Imaging studies using Aβ ligands also report an increased Aβ burden in the striatum of cases with dementia [118], however, more recent PiB/PET studies have demonstrated minimal cortical or striatal PiB retention in PDD patients, whereas DLB patients show high levels of both cortical and striatal PiB retention, similar to the high levels found in AD patients [40, 52, 94]. Although our finding of high levels of striatal Aβ in PDD individuals contradicts these PiB/PET imaging studies, it is possible that Aβ deposits are present in the striatum in PDD but are not detected by PiB/PET imaging due to PiB having a lower affinity for diffuse Aβ deposits, such as those described in the striatum, compared with the aggregated form of Aβ found in cortical plaques of AD and DLB patients. Because PiB is also able to bind other cross β-structured proteins, such as αSyn more research into PiB–protein interactions is required to draw firm conclusions on the findings of PiB/PET imaging studies in PD and related disorders.

Immunostaining for β-amyloid deposits in the caudate nucleus. a A large number of Aβ deposits in the caudate nucleus of a PD case with dementia. b Caudate nucleus without Aβ deposits from a non-demented PD case. Magnification ×20. PD Parkinson’s disease, Aβ amyloid β peptide
Molecular interaction of αSyn, tau and Aβ proteins
At least 70% of dementias in the elderly are related to abnormalities of αSyn, tau and Aβ proteins and this figure increases to more than 90% for dementias of neurodegenerative origin [30]. It has become apparent that there is a strong interaction between αSyn, tau and Aβ at a molecular level. In vitro studies have shown αSyn binding to tau and inducing its phosphorylation [79]. αSyn induces fibrillisation of tau and coincubation of tau and αSyn synergistically promote mutual fibrillisation [49]. In vivo evidence of an interaction between αSyn and tau has also been reported with mice overexpressing human A53T SNCA demonstrating inclusions positive for αSyn as well as for tau [49]. Similarly, a link between Aβ and αSyn has been described with in vitro experiments showing Aβ42 promoting the formation of αSyn oligomers and polymers [96]. Cells transfected with SNCA and treated with Aβ42 form more inclusions than in the absence of Aβ42 [96]. Pletnikova et al. found that Aβ deposits in the cerebral cortex in PDD were associated with extensive αSyn lesions and higher levels of insoluble αSyn [112]. Therefore, Aβ peptides may contribute to an aggravation of LB pathology by promoting an aggregation of αSyn and exacerbating αSyn-dependent neuronal pathologies. An interaction between Aβ and tau has also been demonstrated with injection of β-amyloid Aβ42 fibrils into the brains of P301L mutant tau transgenic mice causing a fivefold increase in the number of NFTs in cell bodies [53] supporting the view that Aβ42 fibrils can accelerate NFTs formation in vivo. Given that in the PDD brain these proteins will co-exist to a varying degree, the identification for a specific primary pathological substrate for dementia in PD remains difficult. Further studies are needed to examine the interactions and impact of the coexistence of these pathogenic proteins in producing dementia in PD.
Coda
Dementia has a major impact on the natural history and prognosis of PD. The putative brain changes underlying dementia in PD have not yet been explored in sufficient detail to permit a consensus definition. Although some studies indicate LB-type pathology in cortical and limbic structures as the main histological substrate of PDD, it now appears that even widespread αSyn lesions often cannot reliably account for the presence of dementia with frequently encountered concomitant AD-type changes suggesting a synergism of multiple pathogenic molecules that requires future investigation. Further work from valid retrospective as well as prospective clinicopathological studies embracing the multiple implied pathologies underlying dementia in PD will provide novel mechanism-based treatments, with the potential to prevent, delay or arrest the development of cognitive impairment in PD.
References
Aarsland D, Andersen K, Larsen JP, Lolk A, Kragh-Sorensen P (2003) Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol 60:387–392
Aarsland D, Andersen K, Larsen JP, Lolk A, Nielsen H, Kragh-Sorensen P (2001) Risk of dementia in Parkinson’s disease: a community-based, prospective study. Neurology 56:730–736
Aarsland D, Larsen JP, Karlsen K, Lim NG, Tandberg E (1999) Mental symptoms in Parkinson’s disease are important contributors to caregiver distress. Int J Geriatr Psychiatry 14:866–874
Aarsland D, Larsen JP, Tandberg E, Laake K (2000) Predictors of nursing home placement in Parkinson’s disease: a population-based, prospective study. J Am Geriatr Soc 48:938–942
Aarsland D, Perry R, Brown A, Larsen JP, Ballard C (2005) Neuropathology of dementia in Parkinson’s disease: a prospective, community-based study. Ann Neurol 58:773–776
Aarsland D, Tandberg E, Larsen JP, Cummings JL (1996) Frequency of dementia in Parkinson disease. Arch Neurol 53:538–542
Agid Y, Graybiel AM, Ruberg M, Hirsch E, Blin J, Dubois B, Javoy-Agid F (1990) The efficacy of levodopa treatment declines in the course of Parkinson’s disease: do nondopaminergic lesions play a role? Adv Neurol 53:83–100
Apaydin H, Ahlskog JE, Parisi JE, Boeve BF, Dickson DW (2002) Parkinson disease neuropathology: later-developing dementia and loss of the levodopa response. Arch Neurol 59:102–112
Arawaka S, Saito Y, Murayama S, Mori H (1998) Lewy body in neurodegeneration with brain iron accumulation type 1 is immunoreactive for alpha-synuclein. Neurology 51:887–889
Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW (1991) The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex 1:103–116
Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42:631–639
Arriagada PV, Marzloff K, Hyman BT (1992) Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology 42:1681–1688
Bayer TA, Jakala P, Hartmann T, Havas L, McLean C, Culvenor JG, Li QX, Masters CL, Falkai P, Beyreuther K (1999) Alpha-synuclein accumulates in Lewy bodies in Parkinson’s disease and dementia with Lewy bodies but not in Alzheimer’s disease beta-amyloid plaque cores. Neurosci Lett 266:213–216
Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, Sasse J, Boyer S, Shirohi S, Brooks R, Eschbacher J, White CL 3rd, Akiyama H, Caviness J, Shill HA, Connor DJ, Sabbagh MN, Walker DG (2009) Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117:613–634
Bertrand E, Lechowicz W, Lewandowska E, Szpak GM, Dymecki J, Kosno-Kruszewska E, Wierzba-Bobrowicz T (2003) Degenerative axonal changes in the hippocampus and amygdala in Parkinson’s disease. Folia Neuropathol 41:197–207
Bertrand E, Lechowicz W, Szpak GM, Lewandowska E, Dymecki J, Wierzba-Bobrowicz T (2004) Limbic neuropathology in idiopathic Parkinson’s disease with concomitant dementia. Folia Neuropathol 42:141–150
Bierer LM, Hof PR, Purohit DP, Carlin L, Schmeidler J, Davis KL, Perl DP (1995) Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch Neurol 52:81–88
Boller F, Mizutani T, Roessmann U, Gambetti P (1980) Parkinson disease, dementia, and Alzheimer disease: clinicopathological correlations. Ann Neurol 7:329–335
Braak E, Braak H (1999) Silver staining method for demonstrating Lewy bodies in Parkinson’s disease and argyrophilic oligodendrocytes in multiple system atrophy. J Neurosci Methods 87:111–115
Braak H, Braak E (2000) Pathoanatomy of Parkinson’s disease. J Neurol 247(Suppl 2):II3–II10
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Braak H, Rub U, Jansen Steur EN, Del Tredici K, de Vos RA (2005) Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology 64:1404–1410
Braak H, Sastre M, Bohl JR, de Vos RA, Del Tredici K (2007) Parkinson’s disease: lesions in dorsal horn layer I, involvement of parasympathetic and sympathetic pre- and postganglionic neurons. Acta Neuropathol 113:421–429
Brown DF, Risser RC, Bigio EH, Tripp P, Stiegler A, Welch E, Eagan KP, Hladik CL, White CL 3rd (1998) Neocortical synapse density and Braak stage in the Lewy body variant of Alzheimer disease: a comparison with classic Alzheimer disease and normal aging. J Neuropathol Exp Neurol 57:955–960
Burke RE, Dauer WT, Vonsattel JP (2008) A critical evaluation of the Braak staging scheme for Parkinson’s disease. Ann Neurol 64:485–491
Caballol N, Marti MJ, Tolosa E (2007) Cognitive dysfunction and dementia in Parkinson disease. Mov Disord 22(Suppl 17):S358–S366
Churchyard A, Lees AJ (1997) The relationship between dementia and direct involvement of the hippocampus and amygdala in Parkinson’s disease. Neurology 49:1570–1576
Colosimo C, Hughes AJ, Kilford L, Lees AJ (2003) Lewy body cortical involvement may not always predict dementia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 74:852–856
Cummings JL (1988) Intellectual impairment in Parkinson’s disease: clinical, pathologic, and biochemical correlates. J Geriatr Psychiatry Neurol 1:24–36
Cummings JL (2003) Toward a molecular neuropsychiatry of neurodegenerative diseases. Ann Neurol 54:147–154
Cummings JL, Benson DF (1984) Subcortical dementia: review of an emerging concept. Arch Neurol 41:874–879
Dale GE, Probst A, Luthert P, Martin J, Anderton BH, Leigh PN (1992) Relationships between Lewy bodies and pale bodies in Parkinson’s disease. Acta Neuropathol 83:525–529
Damier P, Hirsch EC, Agid Y, Graybiel AM (1999) The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 122(Pt 8):1437–1448
Darvesh S, Freedman M (1996) Subcortical dementia: a neurobehavioral approach. Brain Cogn 31:230–249
de Lau LM, Schipper CM, Hofman A, Koudstaal PJ, Breteler MM (2005) Prognosis of Parkinson disease: risk of dementia and mortality: the Rotterdam Study. Arch Neurol 62:1265–1269
de Rijk MC, Launer LJ, Berger K, Breteler MM, Dartigues JF, Baldereschi M, Fratiglioni L, Lobo A, Martinez-Lage J, Trenkwalder C, Hofman A (2000) Prevalence of Parkinson’s disease in Europe: a collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 54:S21–S23
Dickson DW (2005) Required techniques and useful molecular markers in the neuropathologic diagnosis of neurodegenerative diseases. Acta Neuropathol 109:14–24
Dickson DW, Liu W, Hardy J, Farrer M, Mehta N, Uitti R, Mark M, Zimmerman T, Golbe L, Sage J, Sima A, D’Amato C, Albin R, Gilman S, Yen SH (1999) Widespread alterations of alpha-synuclein in multiple system atrophy. Am J Pathol 155:1241–1251
Duda JE, Giasson BI, Mabon ME, Lee VM, Trojanowski JQ (2002) Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann Neurol 52:205–210
Edison P, Rowe CC, Rinne JO, Ng S, Ahmed I, Kemppainen N, Villemagne VL, O’Keefe G, Nagren K, Chaudhury KR, Masters CL, Brooks DJ (2008) Amyloid load in Parkinson’s disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry 79:1331–1338
Emre M (2003) What causes mental dysfunction in Parkinson’s disease? Mov Disord 18(Suppl 6):S63–S71
Emre M, Aarsland D, Brown R, Burn DJ, Duyckaerts C, Mizuno Y, Broe GA, Cummings J, Dickson DW, Gauthier S, Goldman J, Goetz C, Korczyn A, Lees A, Levy R, Litvan I, McKeith I, Olanow W, Poewe W, Quinn N, Sampaio C, Tolosa E, Dubois B (2007) Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord 22:1689–1707 (quiz 1837)
Fearnley JM, Lees AJ (1991) Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 114(Pt 5):2283–2301
Foltynie T, Brayne CE, Robbins TW, Barker RA (2004) The cognitive ability of an incident cohort of Parkinson’s patients in the UK: the CamPaIGN study. Brain 127:550–560
Gai WP, Power JH, Blumbergs PC, Blessing WW (1998) Multiple-system atrophy: a new alpha-synuclein disease? Lancet 352:547–548
Galvin JE, Lee VM, Trojanowski JQ (2001) Synucleinopathies: clinical and pathological implications. Arch Neurol 58:186–190
Gaspar P, Gray F (1984) Dementia in idiopathic Parkinson’s disease: a neuropathological study of 32 cases. Acta Neuropathol 64:43–52
Gearing M, Wilson RW, Unger ER, Shelton ER, Chan HW, Masters CL, Beyreuther K, Mirra SS (1993) Amyloid precursor protein (APP) in the striatum in Alzheimer’s disease: an immunohistochemical study. J Neuropathol Exp Neurol 52:22–30
Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM (2003) Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300:636–640
Goetz CG, Emre M, Dubois B (2008) Parkinson’s disease dementia: definitions, guidelines, and research perspectives in diagnosis. Ann Neurol 64(Suppl 2):S81–S92
Goldman JE, Yen SH, Chiu FC, Peress NS (1983) Lewy bodies of Parkinson’s disease contain neurofilament antigens. Science 221:1082–1084
Gomperts SN, Rentz DM, Moran E, Becker JA, Locascio JJ, Klunk WE, Mathis CA, Elmaleh DR, Shoup T, Fischman AJ, Hyman BT, Growdon JH, Johnson KA (2008) Imaging amyloid deposition in Lewy body diseases. Neurology 71:903–910
Gotz J, Chen F, van Dorpe J, Nitsch RM (2001) Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 293:1491–1495
Hakim AM, Mathieson G (1979) Dementia in Parkinson disease: a neuropathologic study. Neurology 29:1209–1214
Harding AJ, Halliday GM (2001) Cortical Lewy body pathology in the diagnosis of dementia. Acta Neuropathol 102:355–363
Hely MA, Morris JG, Reid WG, Trafficante R (2005) Sydney multicenter study of Parkinson’s disease: non-l-dopa-responsive problems dominate at 15 years. Mov Disord 20:190–199
Hietanen M, Teravainen H (1988) The effect of age of disease onset on neuropsychological performance in Parkinson’s disease. J Neurol Neurosurg Psychiatry 51:244–249
Hobson P, Meara J (1999) The detection of dementia and cognitive impairment in a community population of elderly people with Parkinson’s disease by use of the CAMCOG neuropsychological test. Age Ageing 28:39–43
Hoogendijk WJ, Pool CW, Troost D, van Zwieten E, Swaab DF (1995) Image analyser-assisted morphometry of the locus coeruleus in Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis. Brain 118(Pt 1):131–143
Huang X, Chen P, Kaufer DI, Troster AI, Poole C (2006) Apolipoprotein E and dementia in Parkinson disease: a meta-analysis. Arch Neurol 63:189–193
Hughes AJ, Daniel SE, Blankson S, Lees AJ (1993) A clinicopathologic study of 100 cases of Parkinson’s disease. Arch Neurol 50:140–148
Hughes TA, Ross HF, Musa S, Bhattacherjee S, Nathan RN, Mindham RH, Spokes EG (2000) A 10-year study of the incidence of and factors predicting dementia in Parkinson’s disease. Neurology 54:1596–1602
Hurtig HI, Trojanowski JQ, Galvin J, Ewbank D, Schmidt ML, Lee VM, Clark CM, Glosser G, Stern MB, Gollomp SM, Arnold SE (2000) Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology 54:1916–1921
Inzelberg R, Chapman J, Treves TA, Asherov A, Kipervasser S, Hilkewicz O, Verchovsky R, Klimowitzky S, Korczyn AD (1998) Apolipoprotein E4 in Parkinson disease and dementia: new data and meta-analysis of published studies. Alzheimer Dis Assoc Disord 12:45–48
Inzelberg R, Schechtman E, Paleacu D (2002) Onset age of Parkinson disease. Am J Med Genet 111:459–460 (author reply 461)
Jellinger K, Grisold W (1982) Cerebral atrophy in Parkinson syndrome. Exp Brain Res Suppl 5:26–35
Jellinger KA (1991) Pathology of Parkinson’s disease: changes other than the nigrostriatal pathway. Mol Chem Neuropathol 14:153–197
Jellinger KA (1997) Morphological substrates of dementia in parkinsonism: a critical update. J Neural Transm Suppl 51:57–82
Jellinger KA (2000) Morphological substrates of mental dysfunction in Lewy body disease: an update. J Neural Transm Suppl 59:185–212
Jellinger KA (2008) A critical reappraisal of current staging of Lewy-related pathology in human brain. Acta Neuropathol 116:1–16
Jellinger KA (2009) A critical evaluation of current staging of alpha-synuclein pathology in Lewy body disorders. Biochim Biophys Acta 1792:730–740
Jellinger KA (2009) Significance of brain lesions in Parkinson disease dementia and Lewy body dementia. Front Neurol Neurosci 24:114–125
Jellinger KA, Attems J (2006) Does striatal pathology distinguish Parkinson disease with dementia and dementia with Lewy bodies? Acta Neuropathol 112:253–260
Jellinger KA, Attems J (2008) Prevalence and impact of vascular and Alzheimer pathologies in Lewy body disease. Acta Neuropathol 115:427–436
Jellinger KA, Paulus W (1992) Clinico-pathological correlations in Parkinson’s disease. Clin Neurol Neurosurg 94(Suppl):S86–S88
Jellinger KA, Seppi K, Wenning GK (2003) Clinical and neuropathological correlates of Lewy body disease. Acta Neuropathol 106:188–189 author reply 190
Jellinger KA, Seppi K, Wenning GK, Poewe W (2002) Impact of coexistent Alzheimer pathology on the natural history of Parkinson’s disease. J Neural Transm 109:329–339
Jendroska K, Lees AJ, Poewe W, Daniel SE (1996) Amyloid beta-peptide and the dementia of Parkinson’s disease. Mov Disord 11:647–653
Jensen PH, Hager H, Nielsen MS, Hojrup P, Gliemann J, Jakes R (1999) alpha-synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J Biol Chem 274:25481–25489
Kalaitzakis ME, Christian LM, Moran LB, Graeber MB, Pearce RK, Gentleman SM (2008) Dementia and visual hallucinations associate with limbic pathology in Parkinson’s disease. Parkinsonism Relat Disord 15:196–204
Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK (2008) Controversies over the staging of alpha-synuclein pathology in Parkinson’s disease. Acta Neuropathol 116:125–128 (author reply 129–131)
Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK (2008) Striatal beta-amyloid deposition in Parkinson disease with dementia. J Neuropathol Exp Neurol 67:155–161
Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK (2008) The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease: a critical analysis of alpha-synuclein staging. Neuropathol Appl Neurobiol 34:284–295
Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK (2009) Evidence against a reliable staging system of alpha-synuclein pathology in Parkinson’s disease. Neuropathol Appl Neurobiol 35:125–126
Kalaitzakis ME, Pearce RK, Gentleman SM (2009) Clinical correlates of pathology in the claustrum in Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 461:12–15
Kosaka K, Tsuchiya K, Yoshimura M (1988) Lewy body disease with and without dementia: a clinicopathological study of 35 cases. Clin Neuropathol 7:299–305
Kovari E, Gold G, Herrmann FR, Canuto A, Hof PR, Bouras C, Giannakopoulos P (2003) Lewy body densities in the entorhinal and anterior cingulate cortex predict cognitive deficits in Parkinson’s disease. Acta Neuropathol 106:83–88
Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 18:106–108
Kulisevsky J, Pagonabarraga J (2009) Cognitive impairment in Parkinson’s disease: tools for diagnosis and assessment. Mov Disord 24:1103–1110
Levy G, Tang MX, Louis ED, Cote LJ, Alfaro B, Mejia H, Stern Y, Marder K (2002) The association of incident dementia with mortality in PD. Neurology 59:1708–1713
NJ LiangT, Duda JE (2006) Does striatal pathology distinguish DLB from PDD? Mov Disord 21:S69–S70
Louis ED, Marder K, Cote L, Tang M, Mayeux R (1997) Mortality from Parkinson disease. Arch Neurol 54:260–264
Lowe J, Blanchard A, Morrell K, Lennox G, Reynolds L, Billett M, Landon M, Mayer RJ (1988) Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson’s disease, Pick’s disease, and Alzheimer’s disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and Mallory bodies in alcoholic liver disease. J Pathol 155:9–15
Maetzler W, Reimold M, Liepelt I, Solbach C, Leyhe T, Schweitzer K, Eschweiler GW, Mittelbronn M, Gaenslen A, Uebele M, Reischl G, Gasser T, Machulla HJ, Bares R, Berg D (2008) [11C]PIB binding in Parkinson’s disease dementia. Neuroimage 39:1027–1033
Mann DM, Yates PO (1983) Possible role of neuromelanin in the pathogenesis of Parkinson’s disease. Mech Ageing Dev 21:193–203
Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, Mucke L (2001) beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc Natl Acad Sci USA 98:12245–12250
Mastaglia FL, Johnsen RD, Byrnes ML, Kakulas BA (2003) Prevalence of amyloid-beta deposition in the cerebral cortex in Parkinson’s disease. Mov Disord 18:81–86
Mattila PM, Rinne JO, Helenius H, Dickson DW, Roytta M (2000) Alpha-synuclein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson’s disease. Acta Neuropathol 100:285–290
Mattila PM, Rinne JO, Helenius H, Roytta M (1999) Neuritic degeneration in the hippocampus and amygdala in Parkinson’s disease in relation to Alzheimer pathology. Acta Neuropathol 98:157–164
Mattila PM, Roytta M, Torikka H, Dickson DW, Rinne JO (1998) Cortical Lewy bodies and Alzheimer-type changes in patients with Parkinson’s disease. Acta Neuropathol 95:576–582
Mayeux R, Denaro J, Hemenegildo N, Marder K, Tang MX, Cote LJ, Stern Y (1992) A population-based investigation of Parkinson’s disease with and without dementia: relationship to age and gender. Arch Neurol 49:492–497
Mayeux R, Stern Y, Rosenstein R, Marder K, Hauser A, Cote L, Fahn S (1988) An estimate of the prevalence of dementia in idiopathic Parkinson’s disease. Arch Neurol 45:260–262
McKee AC, Kosik KS, Kowall NW (1991) Neuritic pathology and dementia in Alzheimer’s disease. Ann Neurol 30:156–165
McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology 65:1863–1872
Minguez-Castellanos A, Chamorro CE, Escamilla-Sevilla F, Ortega-Moreno A, Rebollo AC, Gomez-Rio M, Concha A, Munoz DG (2007) Do alpha-synuclein aggregates in autonomic plexuses predate Lewy body disorders? A cohort study. Neurology 68:2012–2018
Muslimovic D, Post B, Speelman JD, Schmand B (2005) Cognitive profile of patients with newly diagnosed Parkinson disease. Neurology 65:1239–1245
Papp MI, Lantos PL (1994) The distribution of oligodendroglial inclusions in multiple system atrophy and its relevance to clinical symptomatology. Brain 117(Pt 2):235–243
Parkkinen L, Kauppinen T, Pirttila T, Autere JM, Alafuzoff I (2005) Alpha-synuclein pathology does not predict extrapyramidal symptoms or dementia. Ann Neurol 57:82–91
Parkkinen L, Pirttila T, Alafuzoff I (2008) Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. Acta Neuropathol 115:399–407
Parsian A, Racette B, Goldsmith LJ, Perlmutter JS (2002) Parkinson’s disease and apolipoprotein E: possible association with dementia but not age at onset. Genomics 79:458–461
Pillon B, Deweer B, Agid Y, Dubois B (1993) Explicit memory in Alzheimer’s, Huntington’s, and Parkinson’s diseases. Arch Neurol 50:374–379
Pletnikova O, West N, Lee MK, Rudow GL, Skolasky RL, Dawson TM, Marsh L, Troncoso JC (2005) Abeta deposition is associated with enhanced cortical alpha-synuclein lesions in Lewy body diseases. Neurobiol Aging 26:1183–1192
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047
Rajput AH, Rozdilsky B, Rajput A, Ang L (1990) Levodopa efficacy and pathological basis of Parkinson syndrome. Clin Neuropharmacol 13:553–558
Reid WG, Hely MA, Morris JG, Broe GA, Adena M, Sullivan DJ, Williamson PM (1996) A longitudinal of Parkinson’s disease: clinical and neuropsychological correlates of dementia. J Clin Neurosci 3:327–333
Richard IH, Papka M, Rubio A, Kurlan R (2002) Parkinson’s disease and dementia with Lewy bodies: one disease or two? Mov Disord 17:1161–1165
Rinne JO, Rummukainen J, Paljarvi L, Rinne UK (1989) Dementia in Parkinson’s disease is related to neuronal loss in the medial substantia nigra. Ann Neurol 26:47–50
Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, Cowie TF, Dickinson KL, Maruff P, Darby D, Smith C, Woodward M, Merory J, Tochon-Danguy H, O’Keefe G, Klunk WE, Mathis CA, Price JC, Masters CL, Villemagne VL (2007) Imaging beta-amyloid burden in aging and dementia. Neurology 68:1718–1725
Rub U, Del Tredici K, Schultz C, Ghebremedhin E, de Vos RA, Jansen Steur E, Braak H (2002) Parkinson’s disease: the thalamic components of the limbic loop are severely impaired by alpha-synuclein immunopositive inclusion body pathology. Neurobiol Aging 23:245–254
Samuel W, Alford M, Hofstetter CR, Hansen L (1997) Dementia with Lewy bodies versus pure Alzheimer disease: differences in cognition, neuropathology, cholinergic dysfunction, and synapse density. J Neuropathol Exp Neurol 56:499–508
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 95:6469–6473
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840
Starkstein SE, Bolduc PL, Mayberg HS, Preziosi TJ, Robinson RG (1990) Cognitive impairments and depression in Parkinson’s disease: a follow up study. J Neurol Neurosurg Psychiatry 53:597–602
Sudarsky L, Morris J, Romero J, Walshe TM (1989) Dementia in Parkinson’s disease: the problem of clinicopathological correlation. J Neuropsychiatry Clin Neurosci 1:159–166
Suenaga T, Hirano A, Llena JF, Yen SH, Dickson DW (1990) Modified Bielschowsky stain and immunohistochemical studies on striatal plaques in Alzheimer’s disease. Acta Neuropathol 80:280–286
Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ (1997) Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol 56:933–944
Takeda A, Mallory M, Sundsmo M, Honer W, Hansen L, Masliah E (1998) Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders. Am J Pathol 152:367–372
Terry RD (1996) The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J Neuropathol Exp Neurol 55:1023–1025
Thal DR, Rub U, Orantes M, Braak H (2002) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800
Tooyama I, Kawamata T, Walker D, Yamada T, Hanai K, Kimura H, Iwane M, Igarashi K, McGeer EG, McGeer PL (1993) Loss of basic fibroblast growth factor in substantia nigra neurons in Parkinson’s disease. Neurology 43:372–376
Tsuboi Y, Uchikado H, Dickson DW (2007) Neuropathology of Parkinson’s disease dementia and dementia with Lewy bodies with reference to striatal pathology. Parkinsonism Relat Disord 13(Suppl 3):S221–S224
Vickers JC, Dickson TC, Adlard PA, Saunders HL, King CE, McCormack G (2000) The cause of neuronal degeneration in Alzheimer’s disease. Prog Neurobiol 60:139–165
Wakabayashi K, Honer WG, Masliah E (1994) Synapse alterations in the hippocampal-entorhinal formation in Alzheimer’s disease with and without Lewy body disease. Brain Res 667:24–32
Williams-Gray CH, Goris A, Saiki M, Foltynie T, Compston DA, Sawcer SJ, Barker RA (2009) Apolipoprotein E genotype as a risk factor for susceptibility to and dementia in Parkinson’s disease. J Neurol 256:493–498
Yoshimura M (1988) Pathological basis for dementia in elderly patients with idiopathic Parkinson’s disease. Eur Neurol 28(Suppl 1):29–35
Zhan SS, Beyreuther K, Schmitt HP (1993) Quantitative assessment of the synaptophysin immuno-reactivity of the cortical neuropil in various neurodegenerative disorders with dementia. Dementia 4:66–74
Zweig RM, Cardillo JE, Cohen M, Giere S, Hedreen JC (1993) The locus coeruleus and dementia in Parkinson’s disease. Neurology 43:986–991
Acknowledgments
The authors would like to thank the UK Parkinson’s Disease Society, registered charity 948776, for their continual support. The valuable comments of Professor Manuel Graeber and Dr Stephen Gentleman are also greatly appreciated. The present work was funded through a grant from the UK Parkinson’s Disease Society (G-0709).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kalaitzakis, M.E., Pearce, R.K.B. The morbid anatomy of dementia in Parkinson’s disease. Acta Neuropathol 118, 587–598 (2009). https://doi.org/10.1007/s00401-009-0597-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-009-0597-x