
Abstract
Neurodegeneration of the nigrostriatal dopaminergic system and concurrent dopamine (DA) deficiency in the basal ganglia represent core features of Parkinson’s disease (PD). Despite the central role of DA in the pathogenesis of PD, dopaminergic systems outside of the midbrain have not been systematically investigated for Lewy body pathology or neurodegeneration. Dopaminergic neurons show a surprisingly rich neurobiological diversity, suggesting that there is not one general type of dopaminergic neuron, but rather a spectrum of different dopaminergic phenotypes. This heterogeneity on the cellular level could account for the observed differences in susceptibility of the dopaminergic systems to the PD disease process. In this review, we will summarize the long history from the first description of PD to the rationally derived DA replacement therapy, describe the basal neuroanatomical and neuropathological features of the different dopaminergic systems in health and PD, explore how neuroimaging techniques broadened our view of the dysfunctional dopaminergic systems in PD and discuss how dopaminergic replacement therapy ameliorates the classical motor symptoms but simultaneously induces a new set of hyperdopaminergic symptoms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The basic clinical symptomatology of Parkinson’s disease (PD) has been well known for 200 years and has been expanded ever since. But how can we identify the neuropathological correlates of these symptoms? How can we link symptoms to certain brain circuit dysfunction, or vice versa, how can we predict the clinical manifestation of a dysfunctional system? The comprehension of the association of neuronal systems and physiological functions/dysfunctions is crucial for the rational development of therapy to alleviate disabling symptoms. To investigate the link between symptomatology and neuropathology in humans, we consider the following: (1) post-mortem neuropathological studies to explore neurodegeneration, distribution of Lewy bodies/neurites (LB, LN) and biochemical alterations; (2) neuroimaging studies in combination with radiotracers to examine dysfunctional neurotransmission; and (3) clinical studies investigating the potential of certain medications to alleviate, worsen or even provoke certain symptoms.
In this review, we will briefly summarize the long road to the discovery of a dysfunctional dopaminergic system in PD laying the ground for the still up-to-date gold standard of therapy and then focus on the emerging evidence of the dysfunctional dopaminergic systems of the brain in PD.
A long road to go
The first medical description of PD dates back to 1817 when James Parkinson published his monograph entitled ‘An Essay on the Shaking Palsy’ based on the depiction of the clinical picture of six patients (Fig. 1) (Parkinson 1817). Fifty-five years later, in 1872, Jean-Martin Charcot identified bradykinesia as a defining feature of PD and suggested that tremor is not an obligate symptom of the disease. He therefore proposed the term “Parkinson’s disease”, thereby arguing against the term ‘shaking palsy’. At this time, neuropathological correlate(s) of the diverse symptoms had not been resolved and PD remained a highly debilitating disorder without effective treatment.

Milestones of PD pathogenesis and therapy
Almost 100 years after the first description, neuropathological studies brought a first breakthrough in PD linking degeneration of the substantia nigra (SN) to the characteristic parkinsonian motor symptoms. In 1893, Georges Marinesco and Paul Blocq were the first to suggest that a lesion of the midbrain could contribute to the motor symptoms seen in PD. Their hypothesis was based on an autopsy of a patient with unilateral parkinsonism, which revealed a tuberculous nodule confined to the right cerebral peduncle. Two years later, Edouard Brissaud hypothesized that the SN might be the major pathological site of PD. This hypothesis was validated by the pioneering work of Constantin Trétiakoff in 1919, who demonstrated substantial loss of pigmented nigral cells in post-mortem PD brains and inclusion bodies in the remaining neurons which he called ‘corps de Lewy’ (Lewy body, LB), in honor of their first describer Friederich H. Lewy (Trétiakoff 1919). The neurochemical consequences of SN degeneration, that is, dopamine (DA) deficiency in the basal ganglia of PD patients, however, remained unknown until 1960.
Thus, almost 40 years later, in 1957, Arvid Carlsson demonstrated in a pioneering work that administration of reserpine led to depletion of brain DA levels and onset of motor deficits in animals mimicking the symptomatology of parkinsonism. He also proved that application of l-dopa, a blood–brain barrier-passing precursor of DA, and noradrenaline could alleviate these symptoms by restoring the brain DA to normal levels (Carlsson 1959; Carlsson et al. 1957). This work built the basis for the DA era of PD and was later honored with the Nobel Prize for medicine. Soon after this, Oleh Hornykiewicz and Herbert Ehringer demonstrated that DA is depleted in the putamen, caudate nucleus and SN of post-mortem brains of Parkinson patients (Hornykiewicz 1963; Ehringer and Hornykiewicz 1960). Subsequently, they intravenously administered l-dopa to volunteering patients. The effect of this therapy was the complete abolishment of the akinesia (Birkmayer and Hornykiewicz 1961). Thus, they introduced l-dopa to the field of neurology as the first rationally developed therapy of PD. In 1970, based on the elaborate work of George C. Cotzias (Cotzias et al. 1969), the US Food and Drug Administration (FDA) finally approved l-dopa as the first drug to treat PD.
Although we know for a long time that the dopaminergic system of the brain is neither among the first regions affected in the course of the disease, nor is it solely accountable for the wide spectrum of symptoms, the gold standard of therapy is still based on the restoration of dopaminergic neurotransmission by means of administration of l-dopa or DA receptor agonists (Oertel 2017).
Parkinsonism as the core feature of PD
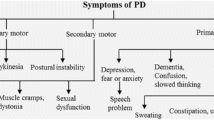
PD is a clinical diagnosis based on the occurrence of the characteristic parkinsonian motor abnormalities plus at least two supportive criteria and the complete absence of absolute exclusion criteria and red flags (Table 1) (Postuma et al. 2015). The three cardinal motor features of PD are bradykinesia/hypokinesia, tremor and rigidity. Typically, PD patients initially present with unilateral motor signs, most commonly with akinesia in combination with resting tremor affecting one of the upper extremities (Pallone 2007). The motor symptoms then gradually spread to the contralateral and lower limbs, but the initial asymmetry remains (Weintraub et al. 2008; Rodriguez-Oroz et al. 2009).
Bradykinesia means slowness of movement, whereas hypo-/akinesia is defined as reduced or diminished amplitude and frequency of spontaneous movements (Rodriguez-Oroz et al. 2009). Patients often describe this as ‘weakening of the limb’, but upon examination, the muscle strength is not altered. Bradykinesia usually presents as a slowness in everyday routine activities and reduced unilaterally arm swing during walking (Lewek et al. 2010; Jankovic 2008). Other signs of this symptom can be a decreased blinking rate (Biousse et al. 2004; Karson 1983), reduced facial expressions (hypomimia) and gesturing, micrographia and a monotone soft speech (hypophonia) (Ho et al. 1999).
Resting tremor (4–6 Hz), representing the most obvious and therefore stigmatizing symptom of PD, is defined as an involuntary rhythmic movement of a body part, which affects usually one of the upper extremities in the early phase of the disease (Jankovic 2008). Tremor is commonly one of the first motor signs to appear and starts generally in the fingers or the thumb, resulting in the typical “pill-rolling tremor” (Kalia and Lang 2015). It becomes apparent during resting state, weakens or even disappears during voluntary movement of the limb and worsens when the patient is stressed or anxious.
Rigidity refers to an increased muscle tone in both the agonist and antagonist muscles resulting in stiffness of the limb. Upon clinical examination, a resistance to passive movement in the extremity can be observed. The resistance may be either smooth (lead-pipe phenomenon) or fluctuating (cogwheel rigidity) (Weintraub et al. 2008), the latter rather representing a mixture of tremor and rigidity.
As the disease advances, postural instability becomes progressively apparent, representing the most common cause of falls and significantly decreasing the quality of life (Williams et al. 2006; Koller et al. 1989; Michalowska et al. 2005). Although postural instability usually develops during the course of the disease, it is mostly not present in early PD and an early occurrence therefore suggests an alternative diagnosis (Jankovic 2008; Postuma et al. 2015). The combination of cardinal motor symptoms, impairment of balance and an anterior shift of the mean center of gravity position finally results in the fully evolved late-stage parkinsonian posture and gait: the patient bends forward into a flexed truncal position and the stride length and walking pace substantially decrease. The patient begins to shuffle and may scrape the foot on the floor with reduced or absent arm swing (Morris et al. 1994; Jankovic 2008; Ebersbach et al. 2013; Błaszczyk et al. 2007).
Apart from these motor symptoms, several non-motor features occur in PD, with a substantial impact on the quality of life of patients (Schrag 2000). The most prevalent non-motor features are the following: reduced gastric and bowel motility resulting in constipation, olfactory dysfunction, sleep disturbances [e.g., REM sleep behavior disorder (RBD)], forgetfulness (cognitive decline), depression, apathy and symptoms of autonomic dysfunction (e.g., urinary urgency, dysfunctional thermoregulation, sweating, orthostatic hypotension, erectile dysfunction) (Martinez-Martin et al. 2007). Importantly, non-motor features often precede the motor symptoms and therefore the diagnosis of PD by decades (Siderowf and Lang 2012).
In contrast to the well-known symptomatology of PD, it has been increasingly difficult to identify the neurobiological correlates underlying the parkinsonian symptoms and integrate them in a pathophysiological model that explains the origin of brady-/akinesia, tremor and rigidity. While striatal DA deficiency could be clearly linked to the onset of motor dysfunction, the wide spectrum of symptoms and compensatory mechanisms in PD cannot be attributed solely to the loss of DA.
The dopaminergic systems of the brain
To understand the link between symptoms and a dysfunctional neuronal brain circuit, it is essential to explore the neurotransmitter system and its physiological functions. Therefore, we will briefly summarize the dopaminergic systems of the brain and their implications in distinct physiological functions.
The dopaminergic neurons of the mammalian central nervous system are distributed along ten distinct neuronal populations located in the ventral mesencephalon (A8–A10), diencephalon (A11–A15), olfactory bulb (A16) and retina (A17) (Fig. 2) (Björklund and Hökfelt 1984; Björklund and Dunnett 2007; Dahlstroem and Fuxe 1964). All of these different subsystems are engaged in several biological functions such as motor, sensory and autonomic control, reward mechanisms and cognition (Smeets and González 2000; Montague et al. 2004).

Localization and PD pathology of the dopaminergic systems in mice (a) and humans (b). The color of the different nuclei refers to the presence or absence of pathology seen in PD. Green—not affected, red—affected, blue—not sufficient data available. a Modified from Björklund and Dunnett (2007)
The neurons of the ventral mesencephalic dopaminergic complex (A8–A10) are morphologically indistinguishable and rather form a continuum without clear anatomical boundaries. The A8 cell group is primarily located in the retrorubral field (RRF), whereas A9 neurons are found in the SN pars compacta, and A10 refers to dopaminergic neurons within the ventral tegmental area (VTA) (Vogt Weisenhorn et al. 2016; Yetnikoff et al. 2014). All of them form one extensive mesotelencephalic dopaminergic projection system comprising three major pathways: (1) a ventral mesostriatal or mesolimbic system which is involved in motivated behaviors predominantly originating in the VTA (A10); (2) a mesolimbocortical or mesocortical system responsible for memory and learning, mainly originating in the VTA (A10); and (3) a dorsal mesostriatal or nigrostriatal pathway, which is engaged in voluntary motor control mainly originating in the SN pars compacta (A9) (Figs. 3, 4) (Björklund and Dunnett 2007; Zeiss 2005; Flückiger et al. 1985).

Mesotelencephalic pathways. The A8–A10 dopaminergic cell groups are found in the ventral midbrain. They form one extensive mesotelencephalic pathway comprising three major pathways: (1) mesolimbic pathway projecting mainly from the VTA to the ventral STR; (2) mesocortical pathway mainly originating in the VTA and projecting to the prefrontal cortex (PFC); and (3) nigrostriatal pathway originating in the substantia nigra and projecting to the dorsal STR. ACB nucleus accumbens, CP caudoputamen, VTA ventral tegmental area, SNr substantia nigra pars reticulata; SNc substantia nigra pars compacta

A traditional dopaminergic neuron. TH tyrosine hydroxylase, AADC aromatic acid decarboxylase, VMAT2 vesicular monoamine transporter 2, DAT dopamine transporter. Modified from (Oertel 2017)
The diencephalic dopaminergic system (A11–A15) contains five distinct cell groups. The neurons of A11 are located in the periventricular gray of the caudal hypothalamus and thalamus and project mainly to the dorsal horn of the spinal cord giving rise to the diencephalospinal pathway (Flückiger et al. 1985; Watson et al. 2012). It was suggested that these neurons contribute to anti-nociception and motor and autonomic reflexes (Clemens and Hochman 2004; Lindvall et al. 1983; Fleetwood-Walker et al. 1988). The tuberoinfundibular dopaminergic neurons of the arcuate nucleus (A12) and the dopaminergic neurons of the preoptic area (A14) are engaged in neuroendocrine functions by secreting DA to the hypophyseal portal blood system, thereby regulating prolactin (PRL) and growth hormone (GH) secretion (Ben-Jonathan and Hnasko 2001; Turiault et al. 2007). The A13 dopaminergic cell group is located within the medial part of the zona incerta and projects locally into the hypothalamus forming the incertohypothalamic pathway (Flückiger et al. 1985). This subsystem is engaged in the regulation of gonadotropin-releasing hormone (GnRH) secretion (Turiault et al. 2007). The neurons of the A15 cell group are located in the rostral hypothalamic periventricular area. Their function is not yet fully understood, but they seem to be involved in the regulation of GnRH as well (Brown et al. 2015; Clarkson and Herbison 2011).
The A16 dopaminergic cells of the olfactory bulb are interneurons found in the periglomerular layer (Halász et al. 1981) and play a pivotal role in odor discrimination and odor processing (Wilson and Sullivan 1995; Tillerson et al. 2006; Taylor et al. 2009). The retinal dopaminergic cells (A17) are neurons of the amacrine subtype found in the inner nuclear and inner plexiform layers of the retina (Archibald et al. 2009). Retinal dopaminergic neurotransmission plays a central role for contrast sensitivity, visual acuity and retinal light adaption by induction of the transition from the rod circuit (dark-adapted state) to the cone circuit (light-adapted state) (Archibald et al. 2009; Jackson et al. 2012; Korshunov et al. 2017; Ribelayga et al. 2008).
What are the prerequisite features a neuron has to possess to be considered dopaminergic? The classical dopaminergic neuron is defined by the presence of: (1) DA, (2) DA-synthetizing enzymes [i.e., tyrosine hydroxylase (TH) and aromatic l-amino acid decarboxylase (AADC)], (3) DA-degrading enzymes (i.e., monoamino oxidases), (4) DA transporters [i.e., vesicular monoamine transporter 2 (VMAT2), DA transporter (DAT)] and (5) autoreceptors (i.e., D2 receptor) (Vernier et al. 2004). Simultaneously, dopaminergic neurons lack dopamine-β-hydoxylase and phenylethyl-N-methyl transferase, the two enzymes required for the conversion of DA into noradrenaline and subsequently adrenaline (Vernier et al. 2004). Importantly, not all of the above-mentioned neuronal populations (A8–A17) contain the complete set of proteins involved in dopaminergic neurotransmission, that is, some cell groups only partially fulfill all of the criteria of a traditional dopaminergic phenotype. For example, in the non-human primate, the A11 neurons contain TH, but at the same time lack detectable levels of AADC or DAT, suggesting that these neurons are l-dopaergic, rather than dopaminergic (Barraud et al. 2010). Among all dopaminergic cell groups, the A8 (RRF), A9 (SN pars compacta) and A10 (VTA) neurons exhibit the most complete dopaminergic phenotype (Vernier et al. 2004). The above introduced traditional map of the brain’s dopaminergic system was generated based on detecting DA complemented with visualizing the distribution of TH immunoreactivity (Björklund and Dunnett 2007, Björklund and Hökfelt 1984; Dahlstroem and Fuxe 1964). As a consequence, the map does not reflect the diversity of the different dopaminergic subsystems. The heterogeneity furthermore suggests that there is not one general type of dopaminergic neurons, but rather a spectrum of different dopaminergic phenotypes.
Neuropathological alterations of the dopaminergic systems in PD
Neuropathological studies, given their cross-sectional nature, allow the investigation of the spatial pattern of pathology, i.e., the aspects of the localization of LB pathology, neurodegeneration and consequent biochemical alterations. The advantages of neuropathological studies are: (1) high resolution in space which enables the detection of pathology at single-cell level and (2) the detection of LB/LN pathology distribution, which is currently not possible with in vivo neuroimaging studies, due to the lack of an α-synuclein radiotracer.
Although it is commonly accepted that a dysfunctional dopaminergic neurotransmission is one of the core features of PD, and that the dopaminergic systems of the brain are heterogeneous and therefore may have different susceptibility to neurodegenerative processes, the dopaminergic neuronal populations outside the midbrain have not been systematically investigated in PD.
Since the discovery of DA depletion in the SN and striatum (STR) of Parkinson patients (Hornykiewicz 1963), several neuropathological studies were conducted to estimate dopaminergic neurodegeneration of the SN pars compacta. The average loss of pigmented nigral neurons compared to age-matched healthy controls ranges between 41 and 79% across studies, on average 67% (Javoy-Agid et al. 1984; Bogerts et al. 1983; Waters et al. 1988; Hirsch et al. 1988; German et al. 1989; Alberico et al. 2015; Zarow et al. 2003; Damier et al. 1999; Gibb and Lees 1991; Kempster et al. 1989; Halliday et al. 1996). Interestingly, the neuropathological process does not homogeneously affect the full extent of the dopaminergic SN. A characteristic topology of neurodegeneration can be observed: neurons of the ventrolateral and caudal subregion, called ventral tier, are primarily affected (around 70–90% cell loss), whereas neurons in the dorsal tier are relatively resistant to the degenerative process (25–70% cell loss) (Damier et al. 1999; Fearnley and Lees 1991; Halliday et al. 1996; Hirsch et al. 1997). In consensus with these findings is the uneven pattern of DA depletion in the STR. The putamen, mostly receiving input from the ventral tier of the SN, shows almost complete DA depletion (< 1% of DA remaining), whereas the caudate nucleus has still substantial levels of DA (~ 40% of DA remaining) (Fig. 6b) (Kish et al. 1988; Waters et al. 1988; Fahn et al. 1971). One study showed that the cell loss of pigmented, neuromelanin-containing SN neurons is less than the loss of TH-positive cells at all studied time points, indicating that prior to cell death, dopaminergic neurons become dysfunctional and decrease their dopaminergic phenotypic expression (ghost cells) (Kordower et al. 2013). This suggests that at the time of the manifestation of the cardinal motor symptoms, symptoms most likely occur due to nigrostriatal dysfunction rather than frank neurodegeneration (Kordower et al. 2013). Besides neurodegeneration, the SN pars compacta also exhibits severe LB and LN pathology (Braak et al. 2003; Gibb and Lees 1989; Seidel et al. 2015). In fact, a combination of nigral Lewy pathology and neurodegeneration of the dopaminergic SN is highly specific for PD and even a prerequisite for the definite neuropathological diagnosis (Gelb et al. 1999; Dickson et al. 2009).

Available radiotracers for in vivo imaging of the dopaminergic systems. Radiotracers can be used to image the presynaptic dopaminergic activity (DA storage, VMAT2 and DAT availability) or the postsynaptic dopaminergic function (D2/D3 receptors) with PET and SPECT approaches
The A8 dopaminergic neurons of the RRF show minor or no degenerative changes in PD (McRitchie et al. 1997), whereas the dopaminergic neurons of the VTA (A10) show abundant LB/LN pathology (Seidel et al. 2015) and substantial neurodegeneration in PD. The reported cell loss of neuromelanin-pigmented VTA neurons in PD ranges between 40 and 77%, on average 53% (Hirsch et al. 1988; German et al. 1989; Alberico et al. 2015; Javoy-Agid et al. 1984; Bogerts et al. 1983; Uhl et al. 1985; Waters et al. 1988; Damier et al. 1999; McRitchie et al. 1997; Javoy-Agid and Agid 1980). A direct comparison of the VTA and SN cell counts is—due to the different samples and statistical methods—difficult. Only a few studies have directly compared nigral and ventral tegmental neuromelanized cell counts by investigating the same midbrain tissue samples. According to their results, the degree of neurodegeneration in the SN usually exceeds that of the VTA by 20% on average (Damier et al. 1999; German et al. 1989; Hirsch et al. 1988). This suggests that, although these two cell populations have a lot of common traits, certain factors partially decrease the susceptibility of VTA (A10) neurons to neurodegeneration and/or increase the vulnerability of SN (A9) neurons.
The diencephalic dopaminergic neuronal populations (A11–A15) have not attracted much attention in PD yet, although their possible involvement in the disease process might contribute to certain autonomic and neuroendocrine dysfunctions seen in PD patients (Politis et al. 2008; Chaudhuri and Schapira 2009). It is reported that virtually all nuclei of the hypothalamus exhibit LB pathology to some extent after a certain disease duration (Langston and Forno 1978). The most severely affected hypothalamic regions are the tuberomammillary nucleus and the lateral and posterior hypothalamic nuclei, regions that do not contain dopaminergic cell groups (Langston and Forno 1978; Braak et al. 2003, 2004). Interestingly, the tuberoinfundibular region which exhibits the highest density of hypothalamic dopaminergic neurons (A12) is relatively spared of LB pathology (Langston and Forno 1978). Nevertheless, to date no study which investigated LB formation specifically in hypothalamic dopaminergic cells exists. In addition, studies on hypothalamic dopaminergic neurodegeneration are also sparse. Only one study aimed to quantify pigmented neuronal cell counts in hypothalamic nuclei of PD patients. Interestingly, no significant cell loss was detected (Matzuk and Saper 1985). Taken together, studies of the hypothalamic dopaminergic system in PD are sparse and their results are controversial.
The olfactory bulb is one of the first brain regions affected during PD (Braak et al. 2003) and hyposmia is present in up to 90% of PD patients (Doty et al. 1988; Haehner et al. 2011), often preceding the classical motor symptoms by more than a decade (Kalia and Lang 2015). Several studies detected dense accumulation of LBs in granule, mitral and tufted cells and the anterior olfactory nucleus (Ubeda-Bañon et al. 2010; Sengoku et al. 2008; Braak et al. 2004). Interestingly, the periglomerular layer, in which the dopaminergic A16 neurons are localized, is relatively spared of the α-synucleinopathy and LBs only occasionally co-localize with TH immunoreactivity (Ubeda-Bañon et al. 2010; Sengoku et al. 2008; Cave et al. 2016). Studies estimating the number of bulbar dopaminergic neurons are contentious. Two independent studies reported that TH-positive neuronal count was doubled in the olfactory bulbs of PD patients compared to healthy controls (Huisman et al. 2004; Mundiñano et al. 2011), while other studies revealed no significant difference between PD and healthy controls (Cave et al. 2016; Ubeda-Bañon et al. 2010; Huisman et al. 2008). Taken together, dopaminergic cells of the olfactory bulb are spared of the α-synucleinopathy. However, whether their neuronal numbers increase during the disease duration and whether this change arises as a consequence of the disease process or DA replacement therapy needs to be further investigated.
Data on the retinal dopaminergic system (A17) in PD are sparse. A very recently published study was the first one to examine and describe phosphorylated α-synuclein-positive, LB- and LN-like inclusions in the retina of PD patients (Ortuño-Lizarán et al. 2018). Morphological changes were exclusively found in the ganglion cell layer and exclusively co-localized with ganglionic cell markers, thereby excluding the possibility of LB formation in dopaminergic amacrine cells. However, despite the lack of α-synucleinopathy in retinal dopaminergic cells, neurochemical evidence of a dysfunctional retinal DA neurotransmission exists. Retinal dopaminergic cells of PD patients show decreased TH immunoreactivity (Nguyen-Legros 1988), and simultaneously significantly lower levels of retinal DA were measured (Harnois and Di Paolo 1990).
It has been hypothesized for a long time that a dysfunctional DA homeostasis might contribute to the selective vulnerability of catecholaminergic neurons in PD (Lotharius 2002; Lohr et al. 2014; Pifl et al. 2014; Uhl 1998; Caudle et al. 2007; Post et al. 2018; Segura-Aguilar et al. 2014; Gandhi et al. 2012; Bayersdorfer et al. 2010; Surmeier 2018; Surmeier et al. 2017). Moreover, DA seems to promote the formation and secretion of SDS-resistant α-synuclein oligomers, thereby eventually contributing to the initiation and progression of the disease (Lee et al. 2011). Despite this central role of DA, extramesencephalic dopaminergic systems have not been systematically investigated for α-synucleinopathy and/or neurodegeneration. Substantial literature exists on the ventral mesencephalic dopaminergic (A8–A10) nuclei considering their involvement in the disease process. These comparative data allow to clearly recognize a spectrum of susceptibility, in which the nigral dopaminergic cells of the ventral tier (A9) are the most vulnerable, followed by the VTA (A10), the dorsal tier of the SN (A9) and the RRF (A8). Identifying the factors which render certain neurons particularly vulnerable or resistant to the disease process remains a key challenge. It has been suggested that the different protein expression patterns and thus the interaction of various proteins influence the susceptibility of these neuronal populations (Double et al. 2010). Specific proteins and protein expression patterns (proteomes) which could account for the observed spectrum of vulnerability within the mesencephalic dopaminergic cell populations have been found. These are of interest for cellular metabolism and also for the electrophysiological firing patterns of these cell groups. Whereas the pacemaking of the less vulnerable VTA neurons relies on voltage-dependent Na+ channels (Puopolo et al. 2007), adult nigral neurons use L-type voltage-gated Ca2+ channels of the Cav1.3 subtype to maintain autonomous pacemaking, leading to sustained Ca2+ influx to the cytosol (Chan et al. 2007; Nedergaard et al. 1993). In the most vulnerable ventral tier of the SN, the latter is combined with a substantially lower intracellular Ca2+ buffering capacity, due to the absence of calbindin and significantly lower expression levels of parvalbumin and calretinin compared to the dorsal tier of the SN or the VTA, respectively (Chung et al. 2005; Yamada et al. 1990; Parent et al. 1996; McRitchie et al. 1996). As a consequence, these neurons have a high intracellular Ca2+ burden leading to high energy demands due to ATP-dependent Ca2+ extrusion mechanisms (Surmeier et al. 2011; Chan et al. 2010). Furthermore, metabolic studies have shown that nigral neurons have an almost threefold higher basal oxidative phosphorylation rate than VTA neurons and thus a substantially elevated basal oxidative stress level and a significantly lower reserve respiratory capacity (Pacelli et al. 2015). This means that nigral neurons are less capable of increasing their ATP production when higher energy demands occur. For thourough reviews on additional potential vulnerability factors, see Double et al. (2010) and Brichta and Greengard (2014).
What we can learn from neuroimaging studies
Neuroimaging studies have become increasingly valuable tools to link pathological alterations with motor and non-motor symptoms, investigate etiology and pathomechanisms, monitor disease progression, support differential diagnosis of parkinsonism and assess the outcome of therapeutic approaches (Politis 2014). In this review, we will focus on three major applications which are relevant regarding dopaminergic dysfunction in PD: (1) the variety of imaging agents allows us to investigate the changes in the dopaminergic systems and metabolic activity caused by PD and thereby broadens our understanding of the molecular and cellular disease pathogenesis and progression (Weingarten et al. 2015; Politis 2014); (2) PET-, SPECT- and MRI-based imaging can be used in the clinical setting to assist in the differential diagnosis of idiopathic PD vs. atypical parkinsonian syndromes or other causes of parkinsonism (Kägi et al. 2010; Scherfler et al. 2007); (3) neuroimaging studies can be used to detect subclinical levels of dopaminergic dysfunction and thus facilitate the identification and risk stratification of prodromal PD patients (Meles et al. 2017; Heller et al. 2017). Apart from these indications, functional neuroimaging has various other applicabilities, such as assessing the therapeutic effect of deep brain stimulation or embryonic cell transplantation (Weingarten et al. 2015; Natale et al. 2018).
A general advantage of functional neuroimaging studies is their potential to assess in vivo dysfunction of neuronal circuits, i.e., how the affected neurons behave in their neuronal network once they have reached a dysfunctional state. Additionally, they enable the analysis of the spatiotemporal pattern of neuropathology, that is, the progression of cellular and regional dysfunction in space and time. Dopaminergic dysfunction has been one of the major interests over the past 30 years of imaging in PD. The development of different imaging agents and tracers enabled the assessment of presynaptic dopaminergic dysfunction and postsynaptic DA receptor changes (Fig. 5). As expected, the ventral midbrain (A8–A10) of PD patients exhibits a reduction of presynaptic dopaminergic tracer uptake indicating dopaminergic degeneration (Joutsa et al. 2015; Goldstein et al. 2008; Ito et al. 2002; Hsiao et al. 2014). As a consequence, brain regions receiving dopaminergic input from A8–A10, namely the putamen, caudate nucleus and ventral STR (nucleus accumbens and olfactory tubercle), show reduced tracer binding reflecting dopaminergic denervation (Pavese et al. 2011; Joutsa et al. 2015; Lewis et al. 2012; Hsiao et al. 2014). It could be shown that the loss of tracer binding is uneven between the subregions of the STR: the dorsal putamen displays the most severe reduction, followed by the caudate nucleus and the ventral STR (Bohnen et al. 2011; Lewis et al. 2012; Hsiao et al. 2014). This is in accordance with the neuropathological studies describing a stereotypical pattern of ventral mesencephalic dopaminergic neurodegeneration resulting in uneven dopaminergic denervation of the STR (Fig. 6b) (Damier et al. 1999; Fearnley and Lees 1991; Waters et al. 1988; Halliday et al. 1996). The decrease in striatal tracer binding significantly correlates with the degree of locomotor disability, particularly with bradykinesia and rigidity (Vingerhoets et al. 1997; Holthoff-Detto et al. 1997; Rinne et al. 2000; Otsuka et al. 1996). Interestingly, it does not correlate with the degree of rest tremor, suggesting that the neural substrate of this motor symptom might be distinct from the nigrostriatal pathway (Otsuka et al. 1996; Vingerhoets et al. 1997). Longitudinal follow-up PET tracer studies have estimated the progression of mesencephalic dopaminergic dysfunction over time and found that presynaptic dopaminergic function declines exponentially, indicating that the progression of the disease tends to be faster at the early phases (Nandhagopal et al. 2009; Hilker et al. 2005). This finding is of potential importance in therapeutic trials testing compounds with disease-modifying potential, if DAT SPECT is chosen as a surrogate marker for progression of PD. It would mean that such clinical trials should be preferentially performed in de novo PD patients or even in prodromal stages of PD with phenoconversion to manifest motor PD as a clinical end point (see also RBD below).

The relationship between DA levels and physiological functions/dysfunctions. a As a consequence of diverse compensatory mechanisms, mild to moderate changes of DA levels remain asymptomatic (plateau). When the severity of hypo- or hyperdopaminergism increases, compensatory mechanisms fail to retain physiological functions leading to dysfunctional neural circuits manifesting as clinical symptoms. Hypodopaminergic states develop as a consequence of disease progression, whereas hyperdopaminergic states emerge as side effects of DA replacement therapy. b Ventral mesencephalic dopaminergic (A8–A10) neurodegeneration shows a stereotypical pattern resulting in severe hypodopaminergism in the caudoputamen (CP, nigrostriatal pathway) and mild to moderate hypodopaminergism in the ventral STR (ACB—nucleus accumbens, mesolimbic pathway) and prefrontal cortex (PFC, mesocortical pathway). As a consequence, doses of DA replacement therapy which are necessary to remedy the nigrostriatal pathway simultaneously overdose the mesocortical and mesolimbic pathways
Distinct behavioral and pharmacological triggers can be used to detect dysfunctional patterns of brain activity and neurotransmitter release. Several studies have been conducted to measure changes of striatal DA release triggered by l-dopa challenge and correlated this with different symptoms of PD. They have consistently found that l-dopa induces striatal DA release and that the degree of l-dopa-induced DA outflow strongly correlated with disease duration (La Fuente-Fernández et al. 2004), Hoehn–Yahr stage (Pavese et al. 2006) and motor disability measured by the Unified Parkinson’s Disease Rating Scale (UPDRS) (Tedroff et al. 1996). This means that patients who had a longer disease duration, higher Hoehn–Yahr stages or higher UPDRS scores have larger putaminal DA release upon l-dopa administration. Furthermore, the amplitude of striatal DA release positively correlated with dyskinesia scores, indicating that a failure in the regulation of DA release contributes to the development of l-dopa-induced dyskinesias (Pavese et al. 2006, La; Fuente-Fernández et al. 2004), an adverse effect of l-dopa affecting up to 90% of patients after 10 years of DA replacement therapy (Lopez et al. 2010; Hauser et al. 2007).
Reduction of the hypothalamic 18F-dopa uptake indicating monoaminergic dysfunction and reduced AADC activity has been reported in PD patients (Pavese et al. 2010, 2011; Moore et al. 2008). However, since the hypothalamus, apart from its intrinsic dopaminergic neurons (A11–A15), receives dense monoaminergic innervation originating from the serotonergic median and dorsal raphe nuclei and noradrenergic A1 and A6 (locus coeruleus) cell groups (Palkovits et al. 1980; van de Kar and Lorens 1979), changes in 18F-dopa PET reflect the net alterations of all these systems (Pavese et al. 2011). Consequently, direct conclusions on the hypothalamic dopaminergic system (A11–A15) cannot be drawn from 18F-dopa results. Nevertheless, significant reduction of postsynaptic D2 and D3 receptors has been observed in a 11C-raclopride study, indicating dopaminergic dysfunction in the hypothalamus of PD patients (Politis et al. 2008).
Currently, the diagnosis of clinical PD is exclusively based on the presenting symptomatology (Table 1), and neuroimaging techniques such as SPECT, PET or conventional MRI are not recommended as a first-line diagnostic approach. However, under certain conditions (e.g., atypical symptomatology or unclear response to dopaminergic treatment), imaging techniques can be of use to better differentiate idiopathic PD from atypical parkinsonian syndromes such as multiple system atrophy (MSA) or progressive supranuclear palsy (PSP) and from secondary causes of parkinsonism (e.g., vascular parkinsonism, neoplasms or drug-induced parkinsonism). DAT SPECT using the 123I-Ioflupane ligand (DATScan) can be used to measure the strength of dopaminergic innervation of the STR (striatal DAT levels) and is thereby able to differentiate neurodegenerative forms of parkinsonism (e.g., PD, MSA, PSP) which normally show reduced striatal DAT levels, from essential tremor and healthy controls (normal striatal DAT levels) (Kägi et al. 2010; Scherfler et al. 2007). However, this technique does not allow further distinguishing idiopathic PD from the atypical parkinsonian syndromes (MSA, PSP). In this case, one could perform a D2/D3 receptor SPECT with the ligand 123I-iodobenzamide (123I-IBZM). PD patients generally show normal postsynaptic D2/D3 signal intensities, while MSA and PSP patients are commonly characterized by decreased postsynaptic D2/D3 receptor availability. However, a normal D2/D3 signal does not exclude MSA or PSP (Vlaar et al. 2007). Other techniques to differentiate idiopathic PD from atypical parkinsonian syndromes are metabolic PET imaging with 18F-FDG (Hellwig et al. 2012; Juh et al. 2004), which relies on disease-specific alterations of brain glucose metabolism, or MRI-based approaches such as T2-weighted structural MRI, voxel-based morphometry and diffusion tensor imaging (Price et al. 2004; Paviour et al. 2005; Ota et al. 2013).
At the onset of motor symptoms and, consequently, at the time of diagnosis 30% of nigral cells have already been lost and only 50–60% of normal TH immunoreactivity is present in the STR (Fearnley and Lees 1991; Greffard et al. 2006; Cheng et al. 2010; Kordower et al. 2013). The time period, in which the pathological process of PD has already started but the motor symptoms are not yet present, is the so-called premotor or prodromal phase of PD (Kalia and Lang 2015). During this period, functional neuroimaging of the nigrostriatal system is able to detect subclinical levels of dopaminergic dysfunction and thus facilitate the identification and risk stratification of patients with prodromal PD (Bauckneht et al. 2018; Heller et al. 2017; Meles et al. 2017; Iranzo et al. 2010; Stiasny-Kolster et al. 2005; Piccini et al. 1999). Patients suffering from idiopathic RBD, a parasomnia characterized by the absence of atonia during REM sleep in combination with abnormal nocturnal behavior, represent a specific prodromal risk population for developing PD (Iranzo et al. 2013). Several studies found that RBD manifestation precedes the onset of PD, dementia with Lewy bodies and MSA, thereby representing an early and specific symptom of these neurodegenerative α-synucleinopathies (Postuma et al. 2012; Schenck et al. 2013). Applying DAT SPECT imaging with 123I-Ioflupane on RBD patients revealed a progressive decrease of presynaptic striatal DAT availability from “mild” or “subclinical” RBD to manifest RBD to PD (Heller et al. 2017). Moreover, 18F-FDG-PET imaging showed that a subgroup of RBD patients already possessed the same altered brain glucose metabolism pattern which is related to PD patients (Meles et al. 2017). Taken together, neuroimaging techniques can help to differentiate RBD patients from healthy controls, to monitor RBD disease progression, to stratify the risk of phenoconversion to PD and thereby identify and characterize eligible patients for neuroprotective trials (Heller et al. 2017; Bauckneht et al. 2018).
Symptomatology ‘off’/‘on’ dopaminergic medication: conclusions of clinical studies
Clinical drug studies investigating the potential of dopaminergic medication to improve, but also to worsen or even induce some of the PD symptoms, are valuable tools to identify distinct symptoms which are linked to dysfunctional dopaminergic neurotransmission.
Physiological dopaminergic functions, such as voluntary motor control, require optimal DA levels in dopaminergic output regions. Both a hypodopaminergic and hyperdopaminergic state result in neural network dysfunction and eventually in clinical symptoms. At the time of PD motor symptom onset, around 30% of dopaminergic nigral neurons are lost and 50–60% of their axon terminals show a marked decrease of TH immunoreactivity (Fearnley and Lees 1991; Greffard et al. 2006; Cheng et al. 2010; Kordower et al. 2013). This indicates that even when the disease process had started and moderate DA shortage is present in the STR, intrinsic mechanisms compensate the DA deficit retaining normal physiological functions and thereby an asymptomatic state (Fig. 6a) (Perez et al. 2008; Zigmond et al. 1990; Garris et al. 1997; Bergstrom and Garris 2003; Obeso et al. 2004). Interestingly, patients with incidental LB disease, i.e., healthy individuals without apparent parkinsonism or dementia with LB/LN pathology upon autopsy, show a 27% cell loss of pigmented nigral neurons (Fearnley and Lees 1991) and a 33–50% decrease of striatal TH immunoreactivity (DelleDonne et al. 2008; Dickson et al. 2008; Beach et al. 2008). As PD progresses and the severity of hypodopaminergism increases, the compensatory mechanisms fail and symptoms of a hypoactive dopaminergic system manifest. Replenishment of DA neurotransmission in these hypodopaminergic regions via DA replacement therapy (e.g., l-dopa, DA agonists) will ameliorate the manifestation of DA shortage (Fig. 6a, b) (Birkmayer and Hornykiewicz 1961; Cotzias et al. 1969). Concurrently, given the uneven nature of neurodegeneration across the dopaminergic systems of the brain in PD, doses of l-dopa which are necessary to restore dopaminergic neurotransmission in the most severely depleted nigrostriatal system simultaneously ‘overdose’ the better preserved mesolimbic and mesocortical brain networks. Thus, dopaminergic treatment with the focus on the primary clinical aim of ameliorating the motor symptoms leads to overactivation of the mesolimbic and mesocortical systems, thereby resulting in symptoms of hyperdopaminergism (Fig. 6b) (Gotham et al. 1988; Swainson et al. 2000; Voon et al. 2017; Vriend et al. 2014; Joutsa et al. 2015; Vaillancourt et al. 2013). Therefore, symptoms of a dysfunctional dopaminergic neurotransmission represent a continuum in which hypodopaminergic states develop as a consequence of disease progression, whereas hyperdopaminergic states emerge as side effects of DA replacement therapy. In the following section, we will briefly give an insight into the dopaminergic symptoms of PD.
The pioneering work of A. Carlsson showed that DA deficiency in the brain of rabbits resulted in parkinsonian symptoms which could be alleviated by administration of l-dopa, a blood–brain barrier crossing precursor of DA (Carlsson 1959; Carlsson et al. 1957). Since then, both neuropathological and neuroimaging studies of PD patients have shown that the degree of DA deficiency in the dorsal STR significantly correlated with the Hoehn–Yahr stage and UPDRS motor disability, especially with bradykinesia and rigidity scores (Hornykiewicz 1963; Morrish et al. 1995; Broussolle et al. 1999; Seibyl et al. 1995; Hsiao et al. 2014; Pavese et al. 2011; Nandhagopal et al. 2009; Price et al. 1978). Consequently, administration of l-dopa significantly improves the two latter motor symptoms of PD (Birkmayer and Hornykiewicz 1961; Cotzias et al. 1969). Although dopaminergic replacement therapy is the most effective symptomatic treatment of PD, long-term l-dopa administration leads to motor side effects, the so-called l-dopa-induced dyskinesias (LIDs). LIDs are among the most common adverse effects of l-dopa therapy and affect up to 80% of patients after 5 years and up to 90% after 10 years of treatment (Hauser et al. 2007; Ahlskog and Muenter 2001; Rajput et al. 1984; Jong et al. 1987). The term LIDs refers to a variety of motor side effects which can be classified based on the clinical movement pattern and the temporal correlation between the occurrence of the dyskinesia and the administration of dopaminergic medication (Luquin et al. 1992; Pandey and Srivanitchapoom 2017; Bastide et al. 2015). Interestingly, severe nigrostriatal damage seems to be a prerequisite of LIDs when l-dopa is administered in pharmacologically relevant doses. Neither healthy controls nor non-human primates with only moderate DA deficiency developed LIDs as a result of long-term l-dopa treatment (Boyce et al. 1990; Schneider 1989; Hagenah et al. 1999; Di Monte et al. 2000; Jenner 2008). This indicates that nigrostriatal hyperdopaminergism per se is not sufficient to induce dyskinesias, but other factors have to be involved additionally. It is hypothesized that a ‘dysregulated DA release’ is responsible for the development of LIDs (see above), which originates in the compensatory mechanisms and changes due to dopaminergic denervation of the STR. It was shown that sprouting of serotonergic axon terminals takes place in the STR of PD patients (Rylander et al. 2010). Serotonergic neurons, due to their partially overlapping protein expression with dopaminergic cells (e.g., AADC, VMAT2), are able to take up l-dopa, convert it to DA and store it in synaptic vesicles (Tison et al. 1991; Arai et al. 1994, 1995; Butcher et al. 1970). As a consequence, these neurons, although do not normally rely on DA as a neurotransmitter, synthesize and release DA upon administration of l-dopa (Carta et al. 2007). However, since they neither express D2 autoreceptors mediating the natural feedback of DA release, nor DAT to clear DA from the synaptic cleft, the dopaminergic neurotransmission becomes dysregulated resulting in swings of synaptic DA levels manifesting as LIDs (Mosharov et al. 2015; Carta and Bezard 2011).
Cognitive deficits ranging from mild cognitive impairment (MCI) not yet qualifying as dementia to manifest dementia are common non-motor symptoms in PD with significant impact on the quality of life (Chaudhuri and Schapira 2009). MCI can be observed in prodromal and manifest PD affecting around 20% of patients at the time of diagnosis (Muslimovic et al. 2005; Aarsland et al. 2009a) and displays a major risk factor for the progression to PD dementia (PDD) (Hoogland et al. 2017; Hobson and Meara 2015; Pedersen et al. 2017). Cognitive impairment most commonly affects executive functions, resulting in a ‘dysexecutive syndrome’ resembling that seen in patients with frontal lobe damage (Owen et al. 1993, 1995; Taylor et al. 1986; Muslimovic et al. 2005; Rowe et al. 2002). The pathophysiology of MCI and dementia in PD is heterogeneous and involves a combination and synergism of distinct pathological changes. These include cortical LB pathology (Mattila et al. 2000; Apaydin et al. 2002; Hurtig et al. 2000; Irwin et al. 2012; Compta et al. 2011), cortical cholinergic deficiency due to the degeneration of the nucleus basalis of Meynert (Mattila et al. 2001; Perry et al. 1991), noradrenergic loss as a consequence of locus coeruleus degeneration, cortical and limbic reduction of DA due to the degeneration of the mesolimbic and mesocortical dopaminergic systems (Rinne et al. 1989; Paulus and Jellinger 1991; Zweig et al. 1993; Ito et al. 2002; Scatton et al. 1983), potential occurrence of Alzheimer’s disease co-pathology (Boller et al. 1980; Paulus and Jellinger 1991; Irwin et al. 2012; Compta et al. 2011) and others—for a thorough review see Halliday et al. (2014). This means that a DA deficit in certain brain regions contributes to the cognitive deficit seen in nondemented PD patients with MCI and in PDD patients (Rinne et al. 1989; Paulus and Jellinger 1991; Zweig et al. 1993; Ito et al. 2002; Scatton et al. 1983). However, DA deficiency per se is not considered to be sufficient for the development of the full range of cognitive deficits (Bosboom et al. 2004; Caballol et al. 2007). Interestingly, studies examining the effects of l-dopa on PD patients with dysexecutive syndrome report beneficial, neutral and detrimental effects (Gotham et al. 1988; Swainson et al. 2000; Kulisevsky 2000; Downes et al. 1989; Bowen et al. 1975; Kulisevsky et al. 1996; Lange et al. 1992; Pillon et al. 1989). This is because executive functions can be split into different components, such as working memory, inhibition, attentional set shifting and planning, whose neurobiological correlates are distinct (Smith 1999; Rabinovici et al. 2015). During an executive task, depending on the subdomain required, different neural networks are active including the prefrontal and parietal cortices, the basal ganglia, the thalamus and the cerebellum (Collette et al. 2005; Monchi et al. 2001, 2006; Wager et al. 2004; Wager and Smith 2003; Cools et al. 2004). As a consequence, cognitive tasks which rely on DA-depleted brain regions (dorsal STR) will be ameliorated by DA replacement therapy, whereas cognitive functions associated with relatively intact or less affected, DA-dependent brain regions (ventral STR and prefrontal cortex) will be impaired due to a relative ‘overactivation’ of these systems (Gotham et al. 1988; Cools et al. 2001). This explains why studies investigating the effect of DA replacement therapy on cognitive function report both detrimental and beneficial effects: depending on the cognitive task, distinct subdomains of executive function are examined, all having a different grade of hypodopaminergism.
Neuropsychiatric syndromes ranging from major depression to psychosis and impulse control disorders (ICDs) are highly prevalent in PD affecting the vast majority of PD patients during the course of the disease (Aarsland et al. 2009b). Major depression occurs in approximately 17% of PD patients (Reijnders et al. 2008) and apathy is present in up to 60% (Yamanishi et al. 2013; Pedersen et al. 2009), whereas the prevalence of anxiety in cross-sectional studies ranges between 20 and 49% (Chen et al. 2010; Dissanayaka et al. 2010; Kulisevsky et al. 2008; Nègre-Pagès et al. 2010; Nuti et al. 2004). All three disorders are suggested to be—even if partly—associated with a deficient mesolimbic dopaminergic neurotransmission, i.e., with a mesolimbic hypodopaminergic state (Remy et al. 2005; Voon et al. 2011; Weintraub et al. 2005). This notion is further supported by clinical trials investigating the efficacy of DA agonists in depressive syndromes showing significant improvement of these symptoms (Reichmann et al. 2002, 2003; Barone et al. 2010; Lemke et al. 2005; Pahwa et al. 2007; Bodkin and Amsterdam 2002; Thobois et al. 2013). A wide range of ICDs, such as pathological gambling, compulsive sexual behavior and binge eating, are associated with dopaminergic treatment affecting around 13.6% of PD patients on DA replacement therapy compared to 1.7% of patients receiving neither DA agonists nor l-dopa (Weintraub et al. 2010). ICDs are suggested to develop as a consequence of a dopaminergic hyperactivity in ventral striatal reward circuitry (mesolimbic system), resulting in an increased drive to perform a certain behavior and to be maintained by an impaired learning from negative consequences due to prefrontal cortical hyperdopaminergism (mesocortical system) (Fig. 6b) (Weintraub 2008; Evans et al. 2006; O’Sullivan et al. 2011; Steeves et al. 2009; Cilia et al. 2008). Interestingly, the largest multicenter study (dominion) investigating the occurrence of ICDs in 3090 PD patients found that the frequency of developing an ICD was twofold higher in patients receiving DA agonists compared to patients taking l-dopa (14.0% vs. 7.2%) (Weintraub et al. 2010). This can be explained by a significantly higher affinity of DA agonists to D3 receptors compared to D1 and D2 receptors (Gerlach et al. 2003). While D1 and D2 receptors are more abundant in the dorsal STR (nigrostriatal pathway) mediating voluntary motor control, D3 receptors are predominantly found in the ventral STR (mesolimbic pathway) playing an important role in reward mechanisms (Sokoloff et al. 1990; Gurevich 1999). As a consequence, doses of DA agonists required to improve motor symptoms may overactivate the mesolimbic system resulting in ICDs (Weintraub 2008; Voon et al. 2017). The association between developing an ICD and a hyperdopaminergic state in the mesocortical and mesolimbic systems is further supported by longitudinal studies showing that DA agonist dose reduction or discontinuation, even in combination with an increased l-dopa dose, significantly improves ICD symptoms (Mamikonyan et al. 2008).
Concluding remarks
Neuropathological analysis, neuroimaging studies and clinical trials have enabled us to better understand dopaminergic dysfunction in PD. It has become evident that, although being one of the core features of PD, nigrostriatal degeneration cannot be solely accountable for the wide range of PD symptoms. Despite the central role of DA in PD, extramesencephalic, i.e., diencephalic, olfactory bulbar and retinal dopaminergic systems have not been systematically investigated yet—not to speak of the dopaminergic system related to the gastrointestinal tract. The distinct dopaminergic systems have a surprisingly high neurobiological diversity, suggesting that there is not one general type of dopaminergic neuron but rather a spectrum of different dopaminergic phenotypes. This heterogeneity on the cellular level could account for the observed differences in susceptibility of the dopaminergic systems to the disease process. To finally understand which factors render neurons particularly vulnerable, we first ought to investigate which neuronal populations are affected in the course of PD, with emphasis on neuronal cell groups sharing common traits, such as the synthetic machinery, the metabolism and the overall reliance on DA as a neurotransmitter.
Arvid Carlsson and his fundamental discovery of DA deficiency in PD paved the way for the still ongoing era of dopaminergic replacement therapy and fueled 50 years of research on the dopaminergic systems in PD. Within the last 2 decades, the research focus slowly shifted toward other important areas such as neuropathological research on other neurotransmitter systems involved in PD, identification of genetic mutations or environmental risk factors. A major focus of the basic research field has been set on unravelling the pathogenesis and progression of PD including research on α-synuclein aggregation and interneuronal trafficking on one side and the identification of cell-autonomous factors rendering certain cell groups more vulnerable to the disease process, such as mitochondrial dysfunction or electrophysiological cell properties on the other side.
Abbreviations
- 123I-IBZM:
-
123I-iodobenzamide
- 18F-FDG:
-
18F-fluorodeoxyglucose
- AADC:
-
Aromatic l-amino acid decarboxylase
- DA:
-
Dopamine
- DAT:
-
Dopamine transporter
- ICD:
-
Impulse control disorder
- LB:
-
Lewy body
- LID:
-
l-dopa-induced dyskinesia
- LN:
-
Lewy neurite
- MCI:
-
Mild cognitive impairment
- MSA:
-
Multiple system atrophy
- PD:
-
Parkinson’s disease
- PDD:
-
Parkinson’s disease dementia
- PSP:
-
Progressive supranuclear palsy
- RBD:
-
REM sleep behavior disorder
- RRF:
-
Retrorubral field
- SN:
-
Substantia nigra
- STR:
-
Striatum
- TH:
-
Tyrosine hydroxylase
- UPDRS:
-
Unified Parkinson’s Disease Rating Scale
- VMAT2:
-
Vesicular monoamine transporter 2
- VTA:
-
Ventral tegmental area
References
Aarsland D, Brønnick K, Larsen JP, Tysnes OB, Alves G (2009a) Cognitive impairment in incident, untreated Parkinson disease: the Norwegian ParkWest study. Neurology 72(13):1121–1126
Aarsland D, Marsh L, Schrag A (2009b) Neuropsychiatric symptoms in Parkinson’s disease. Mov Disord 24(15):2175–2186
Ahlskog JE, Muenter MD (2001) Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 16(3):448–458
Alberico SL, Cassell MD, Narayanan NS (2015) The vulnerable ventral tegmental area in Parkinson’s disease. Basal Ganglia 5(2–3):51–55
Apaydin H, Ahlskog JE, Parisi JE, Boeve BF, Dickson DW (2002) Parkinson disease neuropathology: later-developing dementia and loss of the levodopa response. Arch Neurol 59(1):102–112
Arai R, Karasawa N, Geffard M, Nagatsu T, Nagatsu I (1994) Immunohistochemical evidence that central serotonin neurons produce dopamine from exogenous l-dopa in the rat, with reference to the involvement of aromatic l-amino acid decarboxylase. Brain Res 667(2):295–299
Arai R, Karasawa N, Geffard M, Nagatsu I (1995) l-dopa is converted to dopamine in serotonergic fibers of the striatum of the rat: a double-labeling immunofluorescence study. Neurosci Lett 195(3):195–198
Archibald NK, Clarke MP, Mosimann UP, Burn DJ (2009) The retina in Parkinson’s disease. Brain 132(Pt 5):1128–1145
Barone P, Poewe W, Albrecht S, Debieuvre C, Massey D, Rascol O, Tolosa E, Weintraub D (2010) Pramipexole for the treatment of depressive symptoms in patients with Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 9(6):573–580
Barraud Q, Obeid I, Aubert I, Barrière G, Contamin H, McGuire S, Ravenscroft P, Porras G, Tison F, Bezard E, Ghorayeb I (2010) Neuroanatomical study of the A11 diencephalospinal pathway in the non-human primate. PloS One 5(10):e13306
Bastide MF, Meissner WG, Picconi B, Fasano S, Fernagut P-O, Feyder M, Francardo V, Alcacer C, Ding Y, Brambilla R, Fisone G, Jon Stoessl A, Bourdenx M, Engeln M, Navailles S, Deurwaerdère P de, Ko WKD, Simola N, Morelli M, Groc L, Rodriguez M-C, Gurevich EV, Quik M, Morari M, Mellone M, Gardoni F, Tronci E, Guehl D, Tison F, Crossman AR, Kang UJ, Steece-Collier K, Fox S, Carta M, Angela Cenci M, Bézard E (2015) Pathophysiology of l-dopa-induced motor and non-motor complications in Parkinson’s disease. Progr Neurobiol 132:96–168
Bauckneht M, Chincarini A, Carli F de, Terzaghi M, Morbelli S, Nobili F, Arnaldi D (2018) Presynaptic dopaminergic neuroimaging in REM sleep behavior disorder: a systematic review and meta-analysis. Sleep Med Rev 41:266–274
Bayersdorfer F, Voigt A, Schneuwly S, Botella JA (2010) Dopamine-dependent neurodegeneration in Drosophila models of familial and sporadic Parkinson’s disease. Neurobiol Dis 40(1):113–119
Beach TG, Adler CH, Sue LI, Peirce JB, Bachalakuri J, Dalsing-Hernandez JE, Lue LF, Caviness JN, Connor DJ, Sabbagh MN, Walker DG (2008) Reduced striatal tyrosine hydroxylase in incidental Lewy body disease. Acta Neuropathol 115(4):445–451
Ben-Jonathan N, Hnasko R (2001) Dopamine as a prolactin (PRL) inhibitor. Endocr Rev 22(6):724–763
Bergstrom BP, Garris PA (2003) “Passive stabilization” of striatal extracellular dopamine across the lesion spectrum encompassing the presymptomatic phase of Parkinson’s disease: a voltammetric study in the 6-OHDA-lesioned rat. J Neurochem 87(5):1224–1236
Biousse V, Skibell BC, Watts RL, Loupe DN, Drews-Botsch C, Newman NJ (2004) Ophthalmologic features of Parkinson’s disease. Neurology 62(2):177–180
Birkmayer W, Hornykiewicz O (1961) The l-3,4-dioxyphenylalanine (DOPA)-effect in Parkinson-akinesia. Wiener Klin Wochenschr 73:787–788
Björklund A, Dunnett SB (2007) Dopamine neuron systems in the brain: an update. Trends Neurosci 30(5):194–202
Björklund A, Hökfelt T (1984) Distributional of tyrosine hydroxylaseimmunoreactive neurons in the rat brain. In: Handbook of chemical neuroanatomy. (classical transmitters in the CNS, part I), vol 2, pp 277–379
Błaszczyk JW, Orawiec R, Duda-Kłodowska D, Opala G (2007) Assessment of postural instability in patients with Parkinson’s disease. Exp Brain Res 183(1):107–114
Bodkin JA, Amsterdam JD (2002) Transdermal selegiline in major depression: a double-blind, placebo-controlled, parallel-group study in outpatients. Am J Psychiatry 159(11):1869–1875
Bogerts B, Häntsch J, Herzer M (1983) A morphometric study of the dopamine-containing cell groups in the mesencephalon of normals, Parkinson patients, and schizophrenics. Biol Psychiatry 18(9):951–969
Bohnen NI, Müller MLTM, Zarzhevsky N, Koeppe RA, Bogan CW, Kilbourn MR, Frey KA, Albin RL (2011) Leucoaraiosis, nigrostriatal denervation and motor symptoms in Parkinson’s disease. Brain 134(Pt 8):2358–2365
Boller F, Mizutani T, Roessmann U, Gambetti P (1980) Parkinson disease, dementia, and Alzheimer disease: clinicopathological correlations. Ann Neurol 7(4):329–335
Bosboom JLW, Stoffers D, Wolters EC (2004) Cognitive dysfunction and dementia in Parkinson’s disease. J Neural Transm 111(10–11):1303–1315
Bowen FP, Kamienny RS, Burns MM, Yahr M (1975) Parkinsonism: effects of levodopa treatment on concept formation. Neurology 25(8):701–704
Boyce S, Rupniak NM, Steventon MJ, Iversen SD (1990) Nigrostriatal damage is required for induction of dyskinesias by l-dopa in squirrel monkeys. Clin Neuropharmacol 13(5):448–458
Braak H, Del Tredici K, Rüb U, De Vos Rob AI, Steur ENJ, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24(2):197–211
Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K (2004) Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 318(1):121–134
Brichta L, Greengard P (2014) Molecular determinants of selective dopaminergic vulnerability in Parkinson’s disease: an update. Front Neuroanat 8:152
Broussolle E, Dentresangle C, Landais P, Garcia-Larrea L, Pollak P, Croisile B, Hibert O, Bonnefoi F, Galy G, Froment JC, Comar D (1999) The relation of putamen and caudate nucleus 18F-Dopa uptake to motor and cognitive performances in Parkinson’s disease. J Neurol Sci 166(2):141–151
Brown RSE, Herbison AE, Grattan DR (2015) Effects of prolactin and lactation on A15 dopamine neurones in the rostral preoptic area of female mice. J Neuroendocrinol 27(9):708–717
Butcher L, Engel J, Fuxe K (1970) l-dopa induced changes in central monoamine neurons after peripheral decarboxylase inhibition. J Pharm Pharmacol 22(4):313–316
Caballol N, Martí MJ, Tolosa E (2007) Cognitive dysfunction and dementia in Parkinson disease. Mov Disord 22(Suppl 17):S358–S366
Carlsson A (1959) The occurrence, distribution and physiological role of catecholamines in the nervous system. Pharmacol Rev 11(2:part 2):490–493
Carlsson A, Lindqvist M, Magnusson T (1957) 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature 180(4596):1200
Carta M, Bezard E (2011) Contribution of pre-synaptic mechanisms to l-dopa-induced dyskinesia. Neuroscience 198:245–251
Carta M, Carlsson T, Kirik D, Björklund A (2007) Dopamine released from 5-HT terminals is the cause of l-dopa-induced dyskinesia in parkinsonian rats. Brain 130(Pt 7):1819–1833
Caudle WM, Richardson JR, Wang MZ, Taylor TN, Guillot TS, McCormack AL, Colebrooke RE, Di Monte DA, Emson PC, Miller GW (2007) Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci 27(30):8138–8148
Cave JW, Fujiwara N, Weibman AR, Baker H (2016) Cytoarchitectural changes in the olfactory bulb of Parkinson’s disease patients. NPJ Parkinson’s Dis 2:16011
Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ (2007) ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 447:1081
Chan CS, Gertler TS, Surmeier DJ (2010) A molecular basis for the increased vulnerability of substantia nigra dopamine neurons in aging and Parkinson’s disease. Mov Disord 25(Suppl 1):S63–S70
Chaudhuri KR, Schapira AHV (2009) Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol 8(5):464–474
Chen Y-K, Lu J-Y, Chan DML, Mok VCT, Yeung MA, Wong KS, Ungvari GS, Tang WK (2010) Anxiety disorders in Chinese patients with Parkinson’s disease. Int J Psychiatry Med 40(1):97–107
Cheng H-C, Ulane CM, Burke RE (2010) Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 67(6):715–725
Chung CY, Seo H, Sonntag KC, Brooks A, Lin L, Isacson O (2005) Cell type-specific gene expression of midbrain dopaminergic neurons reveals molecules involved in their vulnerability and protection. Hum Mol Genet 14(13):1709–1725
Cilia R, Siri C, Marotta G, Isaias IU, Gaspari D de, Canesi M, Pezzoli G, Antonini A (2008) Functional abnormalities underlying pathological gambling in Parkinson disease. Arch Neurol 65(12):1604–1611
Clarkson J, Herbison AE (2011) Dual phenotype kisspeptin-dopamine neurones of the rostral periventricular area of the third ventricle project to gonadotrophin-releasing hormone neurones. J Neuroendocrinol 23(4):293–301
Clemens S, Hochman S (2004) Conversion of the modulatory actions of dopamine on spinal reflexes from depression to facilitation in D3 receptor knock-out mice. J Neurosci 24(50):11337–11345
Collette F, van der Linden M, Laureys S, Delfiore G, Degueldre C, Luxen A, Salmon E (2005) Exploring the unity and diversity of the neural substrates of executive functioning. Hum Brain Mapp 25(4):409–423
Compta Y, Parkkinen L, O’Sullivan SS, Vandrovcova J, Holton JL, Collins C, Lashley T, Kallis C, Williams DR, Silva R de, Lees AJ, Revesz T (2011) Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: which is more important? Brain 134(Pt 5):1493–1505
Cools R, Barker RA, Sahakian BJ, Robbins TW (2001) Enhanced or impaired cognitive function in Parkinson’s disease as a function of dopaminergic medication and task demands. Cerebral Cortex 11(12):1136–1143 (New York, 1991)
Cools R, Clark L, Robbins TW (2004) Differential responses in human striatum and prefrontal cortex to changes in object and rule relevance. J Neurosci 24(5):1129–1135
Cotzias GC, Papavasiliou PS, Gellene R (1969) Modification of Parkinsonism—chronic treatment with l-dopa. N Engl J Med 280(7):337–345
Dahlstroem A, Fuxe K (1964) Evidence for the existence of monoamine-containing neurons in the central nervous system. I. Demonstration of monoamines in the cell bodies of brain stem neurons. Acta Physiol Scand Suppl 232:1–55
Damier P, Hirsch EC, Agid Y, Graybiel AM (1999) The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 122(Pt 8):1437–1448
DelleDonne A, Klos KJ, Fujishiro H, Ahmed Z, Parisi JE, Josephs KA, Frigerio R, Burnett M, Wszolek ZK, Uitti RJ, Ahlskog JE, Dickson DW (2008) Incidental Lewy body disease and preclinical Parkinson disease. Arch Neurol 65(8):1074–1080
Di Monte DA, McCormack A, Petzinger G, Janson AM, Quik M, Langston WJ (2000) Relationship among nigrostriatal denervation, parkinsonism, and dyskinesias in the MPTP primate model. Mov Disord 15(3):459–466
Dickson DW, Fujishiro H, DelleDonne A, Menke J, Ahmed Z, Klos KJ, Josephs KA, Frigerio R, Burnett M, Parisi JE, Ahlskog JE (2008) Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol 115(4):437–444
Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, Hardy J, Leverenz JB, Del Tredici K, Wszolek ZK, Litvan I (2009) Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol 8(12):1150–1157
Dissanayaka NNW, Sellbach A, Matheson S, O’Sullivan JD, Silburn PA, Byrne GJ, Marsh R, Mellick GD (2010) Anxiety disorders in Parkinson’s disease: prevalence and risk factors. Mov Disord 25(7):838–845
Doty RL, Deems DA, Stellar S (1988) Olfactory dysfunction in parkinsonism: a general deficit unrelated to neurologic signs, disease stage, or disease duration. Neurology 38(8):1237–1244
Double KL, Reyes S, Werry EL, Halliday GM (2010) Selective cell death in neurodegeneration: why are some neurons spared in vulnerable regions? Progr Neurobiol 92(3):316–329
Downes JJ, Roberts AC, Sahakian BJ, Evenden JL, Morris RG, Robbins TW (1989) Impaired extra-dimensional shift performance in medicated and unmedicated Parkinson’s disease: evidence for a specific attentional dysfunction. Neuropsychologia 27(11–12):1329–1343
Ebersbach G, Moreau C, Gandor F, Defebvre L, Devos D (2013) Clinical syndromes: Parkinsonian gait. Mov Disord 28(11):1552–1559
Ehringer H, Hornykiewicz O (1960) Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Klin Wochenschr 38:1236–1239
Evans AH, Pavese N, Lawrence AD, Tai YF, Appel S, Doder M, Brooks DJ, Lees AJ, Piccini P (2006) Compulsive drug use linked to sensitized ventral striatal dopamine transmission. Ann Neurol 59(5):852–858
Fahn S, Libsch LR, Cutler RW (1971) Monoamines in the human neostriatum: topographic distribution in normals and in Parkinson’s disease and their role in akinesia, rigidity, chorea, and tremor. J Neurol Sci 14(4):427–455
Fearnley JM, Lees AJ (1991) Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 114(Pt 5):2283–2301
Fleetwood-Walker SM, Hope PJ, Mitchell R (1988) Antinociceptive actions of descending dopaminergic tracts on cat and rat dorsal horn somatosensory neurones. J Physiol 399(1):335–348
Flückiger E, Müller EE, Thorner MO, Halász B, Fuxe K, Agnati LF, Kalia M, Goldstein M, Andersson K, Härfstrand A, Clark B (1985) The dopaminergic system, vol 1. Springer, Berlin
Gandhi S, Vaarmann A, Yao Z, Duchen MR, Wood NW, Abramov AY (2012) Dopamine induced neurodegeneration in a PINK1 model of Parkinson’s disease. PLoS One 7(5):e37564
Garris PA, Walker QD, Wightman RM (1997) Dopamine release and uptake rates both decrease in the partially denervated striatum in proportion to the loss of dopamine terminals. Brain Res 753(2):225–234
Gelb DJ, Oliver E, Gilman S (1999) Diagnostic criteria for Parkinson disease. Arch Neurol 56(1):33–39
Gerlach M, Double K, Arzberger T, Leblhuber F, Tatschner T, Riederer P (2003) Dopamine receptor agonists in current clinical use: comparative dopamine receptor binding profiles defined in the human striatum. J Neural Transm 110(10):1119–1127
German DC, Manaye K, Smith WK, Woodward DJ, Saper CB (1989) Midbrain dopaminergic cell loss in Parkinson’s disease: computer visualization. Ann Neurol 26(4):507–514
Gibb WRG, Lees AJ (1989) The significance of the lewy body in the diagnosis of idiopathic Parkinson’s disease. Neuropathol Appl Neurobiol 15(1):27–44
Gibb WR, Lees AJ (1991) Anatomy, pigmentation, ventral and dorsal subpopulations of the substantia nigra, and differential cell death in Parkinson’s disease. J Neurol Neurosurg Psychiatry 54(5):388–396
Goldstein DS, Holmes C, Bentho O, Sato T, Moak J, Sharabi Y, Imrich R, Conant S, Eldadah BA (2008) Biomarkers to detect central dopamine deficiency and distinguish Parkinson disease from multiple system atrophy. Parkinsonism Relat Disord 14(8):600–607
Gotham AM, Brown RG, Marsden CD (1988) ‘Frontal’ cognitive function in patients with Parkinson’s disease ‘on’and ‘off’ levodopa. Brain 111(2):299–321
Greffard S, Verny M, Bonnet A-M, Beinis J-Y, Gallinari C, Meaume S, Piette F, Hauw J-J, Duyckaerts C (2006) Motor score of the Unified Parkinson Disease Rating Scale as a good predictor of Lewy body-associated neuronal loss in the substantia nigra. Arch Neurol 63(4):584–588
Gurevich E (1999) Distribution of dopamine D3 receptor expressing neurons in the human forebrain comparison with D2 receptor expressing neurons. Neuropsychopharmacology 20(1):60–80
Haehner A, Hummel T, Reichmann H (2011) Olfactory loss in Parkinson’s disease. Parkinsons Dis 2011:1–6
Hagenah J, Klein C, Sieberer M, Vieregge P (1999) Exogenous levodopa is not toxic to elderly subjects with non-parkinsonian movement disorders: further clinical evidence. J Neural Transm 106(3–4):301–307
Halász N, Johansson O, Hökfelt T, Ljungdahl Å, Goldstein M (1981) Immunohistochemical identification of two types of dopamine neuron in the rat olfactory bulb as seen by serial sectioning. J Neurocytol 10(2):251–259
Halliday GM, McRitchie DA, Cartwright H, Pamphlett R, Hely MA, Morris JGL (1996) Midbrain neuropathology in idiopathic Parkinson’s disease and diffuse Lewy body disease. J Clin Neurosci 3(1):52–60
Halliday GM, Leverenz JB, Schneider JS, Adler CH (2014) The neurobiological basis of cognitive impairment in Parkinson’s disease. Mov Disord 29(5):634–650
Harnois C, Di Paolo T (1990) Decreased dopamine in the retinas of patients with Parkinson’s disease. Investig Ophthalmol Vis Sci 31(11):2473–2475
Hauser RA, Rascol O, Korczyn AD, Jon Stoessl A, Watts RL, Poewe W, Deyn PP de, Lang AE (2007) Ten-year follow-up of Parkinson’s disease patients randomized to initial therapy with ropinirole or levodopa. Mov Disord 22(16):2409–2417
Heller J, Brcina N, Dogan I, Holtbernd F, Romanzetti S, Schulz JB, Schiefer J, Reetz K (2017) Brain imaging findings in idiopathic REM sleep behavior disorder (RBD)—a systematic review on potential biomarkers for neurodegeneration. Sleep Med Rev 34:23–33
Hellwig S, Amtage F, Kreft A, Buchert R, Winz OH, Vach W, Spehl TS, Rijntjes M, Hellwig B, Weiller C, Winkler C, Weber WA, Tüscher O, Meyer PT (2012) 18FFDG-PET is superior to 123IIBZM-SPECT for the differential diagnosis of parkinsonism. Neurology 79(13):1314–1322
Hilker R, Schweitzer K, Coburger S, Ghaemi M, Weisenbach S, Jacobs AH, Rudolf J, Herholz K, Heiss W-D (2005) Nonlinear progression of Parkinson disease as determined by serial positron emission tomographic imaging of striatal fluorodopa F 18 activity. Arch Neurol 62(3):378–382
Hirsch E, Graybiel AM, Agid YA (1988) Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature 334:345
Hirsch EC, Faucheux B, Damier P, Mouatt-Prigent A, Agid Y (1997) Neuronal vulnerability in Parkinson’s disease. J Neural Transm Suppl 50:79–88
Ho AK, Iansek R, Marigliani C, Bradshaw JL, Gates S (1999) Speech impairment in a large sample of patients with Parkinson’s disease. Behav Neurol 11(3):131–137
Hobson P, Meara J (2015) Mild cognitive impairment in Parkinson’s disease and its progression onto dementia: a 16-year outcome evaluation of the Denbighshire cohort. Int J Geriatr Psychiatry 30(10):1048–1055
Holthoff-Detto VA, Kessler J, Herholz K, Bonner H, Pietrzyk U, Wurker M, Ghaemi M, Wienhard K, Wagner R, Heiss W-D (1997) Functional effects of striatal dysfunction in Parkinson disease. Arch Neurol 54(2):145–150
Hoogland J, Boel JA, Bie RMA de, Geskus RB, Schmand BA, Dalrymple-Alford JC, Marras C, Adler CH, Goldman JG, Tröster AI, Burn DJ, Litvan I, Geurtsen GJ (2017) Mild cognitive impairment as a risk factor for Parkinson’s disease dementia. Mov Disord 32(7):1056–1065
Hornykiewicz O (1963) The tropical localization and content of noradrenalin and dopamine (3-hydroxytyramine) in the substantia nigra of normal persons and patients with Parkinson’s disease. Wiener Klin Wochenschr 75:309–312
Hsiao I-T, Weng Y-H, Hsieh C-J, Lin W-Y, Wey S-P, Kung M-P, Yen T-C, Lu C-S, Lin K-J (2014) Correlation of Parkinson disease severity and 18F-DTBZ positron emission tomography. JAMA Neurol 71(6):758–766
Huisman E, Uylings HBM, Hoogland PV (2004) A 100% increase of dopaminergic cells in the olfactory bulb may explain hyposmia in Parkinson’s disease. Mov Disord 19(6):687–692
Huisman E, Uylings HBM, Hoogland PV (2008) Gender-related changes in increase of dopaminergic neurons in the olfactory bulb of Parkinson’s disease patients. Mov Disord 23(10):1407–1413
Hurtig HI, Trojanowski JQ, Galvin J, Ewbank D, Schmidt ML, Lee VM-Y, Clark CM, Glosser G, Stern MB, Gollomp SM, Arnold SE (2000) Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology 54(10):1916–1921
Iranzo A, Lomeña F, Stockner H, Valldeoriola F, Vilaseca I, Salamero M, Molinuevo JL, Serradell M, Duch J, Pavía J, Gallego J, Seppi K, Högl B, Tolosa E, Poewe W, Santamaria J (2010) Decreased striatal dopamine transporter uptake and substantia nigra hyperechogenicity as risk markers of synucleinopathy in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a prospective study. Lancet Neurol 9(11):1070–1077
Iranzo A, Tolosa E, Gelpi E, Molinuevo JL, Valldeoriola F, Serradell M, Sanchez-Valle R, Vilaseca I, Lomeña F, Vilas D, LLadó A, Gaig C, Santamaria J (2013) Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder. An observational cohort study. Lancet Neurol 12(5):443–453
Irwin DJ, White MT, Toledo JB, Xie SX, Robinson JL, van Deerlin V, Lee VM-Y, Leverenz JB, Montine TJ, Duda JE, Hurtig HI, Trojanowski JQ (2012) Neuropathologic substrates of Parkinson disease dementia. Ann Neurol 72(4):587–598
Ito K, Nagano-Saito A, Kato T, Arahata Y, Nakamura A, Kawasumi Y, Hatano K, Abe Y, Yamada T, Kachi T, Brooks DJ (2002) Striatal and extrastriatal dysfunction in Parkinson’s disease with dementia: a 6-18Ffluoro-l-dopa PET study. Brain 125(Pt 6):1358–1365
Jackson CR, Ruan G-X, Aseem F, Abey J, Gamble K, Stanwood G, Palmiter RD, Iuvone PM, McMahon DG (2012) Retinal dopamine mediates multiple dimensions of light-adapted vision. J Neurosci 32(27):9359–9368
Jankovic J (2008) Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry 79(4):368–376
Javoy-Agid F, Agid Y (1980) Is the mesocortical dopaminergic system involved in Parkinson disease? Neurology 30(12):1326
Javoy-Agid F, Ruberg M, Taquet H, Bokobza B, Agid Y, Gaspar P, Berger B, N’Guyen-Legros J, Alvarez C, Gray F (1984) Biochemical neuropathology of Parkinson’s disease. Adv Neurol 40:189–198
Jenner P (2008) Preventing and controlling dyskinesia in Parkinson’s disease—a view of current knowledge and future opportunities. Mov Disord 23(Suppl 3):S585–S598
Jong GJ de, Meerwaldt JD, Schmitz PI (1987) Factors that influence the occurrence of response variations in Parkinson’s disease. Ann Neurol 22(1):4–7
Joutsa J, Johansson J, Seppanen M, Noponen T, Kaasinen V (2015) Dorsal-to-ventral shift in midbrain dopaminergic projections and increased thalamic/raphe serotonergic function in early Parkinson disease. J Nucl Med 56(7):1036–1041
Juh R, Kim J, Moon D, Choe B, Suh T (2004) Different metabolic patterns analysis of parkinsonism on the 18F-FDG PET. Eur J Radiol 51(3):223–233
Kägi G, Bhatia KP, Tolosa E (2010) The role of DAT-SPECT in movement disorders. J Neurol Neurosurg Psychiatry 81(1):5–12
Kalia LV, Lang AE (2015) Parkinson’s disease. Lancet 386(9996):896–912
Karson CN (1983) Spontaneous eye-blink rates and dopaminergic systems. Brain 106(3):643–653
Kempster PA, Gibb WR, Stern GM, Lees AJ (1989) Asymmetry of substantia nigra neuronal loss in Parkinson’s disease and its relevance to the mechanism of levodopa related motor fluctuations. J Neurol Neurosurg Psychiatry 52(1):72–76
Kish SJ, Shannak K, Hornykiewicz O (1988) Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. Pathophysiologic and clinical implications. N Engl J Med 318(14):876–880
Koller WC, Glatt S, Vetere-Overfield B, Hassanein R (1989) Falls and Parkinson’s disease. Clin Neuropharmacol 12(2):98–105
Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM, Bartus RT (2013) Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136(Pt 8):2419–2431
Korshunov KS, Blakemore LJ, Trombley PQ (2017) Dopamine: a modulator of circadian rhythms in the central nervous system. Front Cell Neurosci 11:91
Kulisevsky J (2000) Role of dopamine in learning and memory: implications for the treatment of cognitive dysfunction in patients with Parkinson’s disease. Drugs Aging 16(5):365–379
Kulisevsky J, Avila A, Barbanoj M, Antonijoan R, Berthier ML, Gironell A (1996) Acute effects of levodopa on neuropsychological performance in stable and fluctuating Parkinson’s disease patients at different levodopa plasma levels. Brain 119(Pt 6):2121–2132
Kulisevsky J, Pagonabarraga J, Pascual-Sedano B, García-Sánchez C, Gironell A (2008) Prevalence and correlates of neuropsychiatric symptoms in Parkinson’s disease without dementia. Mov Disord 23(13):1889–1896
La Fuente-Fernández R de, Sossi V, Huang Z, Furtado S, Lu J-Q, Calne DB, Ruth TJ, Stoessl AJ (2004) Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain 127(Pt 12):2747–2754
Lange KW, Robbins TW, Marsden CD, James M, Owen AM, Paul GM (1992) L-dopa withdrawal in Parkinson’s disease selectively impairs cognitive performance in tests sensitive to frontal lobe dysfunction. Psychopharmacology 107(2–3):394–404
Langston JW, Forno LS (1978) The hypothalamus in Parkinson disease. Ann Neurol 3(2):129–133
Lee H-J, Baek SM, Ho D-H, Suk J-E, Cho E-D, Lee S-J (2011) Dopamine promotes formation and secretion of non-fibrillar alpha-synuclein oligomers. Exp Mol Med 43(4):216–222
Lemke MR, Brecht HM, Koester J, Kraus PH, Reichmann H (2005) Anhedonia, depression, and motor functioning in Parkinson’s disease during treatment with pramipexole. J Neuropsychiatry Clin Neurosci 17(2):214–220
Lewek MD, Poole R, Johnson J, Halawa O, Huang X (2010) Arm swing magnitude and asymmetry during gait in the early stages of Parkinson’s disease. Gait Posture 31(2):256–260
Lewis SJ, Pavese N, Rivero-Bosch M, Eggert K, Oertel W, Mathias CJ, Brooks DJ, Gerhard A (2012) Brain monoamine systems in multiple system atrophy: a positron emission tomography study. Neurobiol Dis 46(1):130–136
Lindvall O, Björklund A, Skagerberg G (1983) Dopamine-containing neurons in the spinal cord: anatomy and some functional aspects. Ann Neurol 14(3):255–260
Lohr KM, Bernstein AI, Stout KA, Dunn AR, Lazo CR, Alter SP, Wang M, Li Y, Fan X, Hess EJ, Yi H, Vecchio LM, Goldstein DS, Guillot TS, Salahpour A, Miller GW (2014) Increased vesicular monoamine transporter enhances dopamine release and opposes Parkinson disease-related neurodegeneration in vivo. Proc Natl Acad Sci USA 111(27):9977–9982
Lopez IC, Ruiz PJG, Del Pozo SVF, Bernardos VS (2010) Motor complications in Parkinson’s disease: ten year follow-up study. Mov Disord 25(16):2735–2739
Lotharius J (2002) Impaired dopamine storage resulting from alpha-synuclein mutations may contribute to the pathogenesis of Parkinson’s disease. Hum Mol Genet 11(20):2395–2407
Luquin MR, Scipioni O, Vaamonde J, Gershanik O, Obeso JA (1992) Levodopa-induced dyskinesias in Parkinson’s disease: clinical and pharmacological classification. Mov Disord 7(2):117–124
Mamikonyan E, Siderowf AD, Duda JE, Potenza MN, Horn S, Stern MB, Weintraub D (2008) Long-term follow-up of impulse control disorders in Parkinson’s disease. Mov Disord 23(1):75–80
Martinez-Martin P, Schapira AHV, Stocchi F, Sethi K, Odin P, MacPhee G, Brown RG, Naidu Y, Clayton L, Abe K, Tsuboi Y, MacMahon D, Barone P, Rabey M, Bonuccelli U, Forbes A, Breen K, Tluk S, Olanow CW, Thomas S, Rye D, Hand A, Williams AJ, Ondo W, Chaudhuri KR (2007) Prevalence of nonmotor symptoms in Parkinson’s disease in an international setting; study using nonmotor symptoms questionnaire in 545 patients. Mov Disord 22(11):1623–1629
Mattila PM, Rinne JO, Helenius H, Dickson DW, Röyttä M (2000) Alpha-synuclein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson’s disease. Acta Neuropathol 100(3):285–290
Mattila PM, Röyttä M, Lönnberg P, Marjamäki P, Helenius H, Rinne JO (2001) Choline acetyltransferase activity and striatal dopamine receptors in Parkinson’s disease in relation to cognitive impairment. Acta Neuropathol 102(2):160–166
Matzuk MM, Saper CB (1985) Preservation of hypothalamic dopaminergic neurons in Parkinson’s disease. Ann Neurol 18(5):552–555
McRitchie DA, Hardman CD, Halliday GM (1996) Cytoarchitectural distribution of calcium binding proteins in midbrain dopaminergic regions of rats and humans. J Comp Neurol 364(1):121–150
McRitchie DA, Cartwright HR, Halliday GM (1997) Specific A10 dopaminergic nuclei in the midbrain degenerate in Parkinson’s disease. Exp Neurol 144(1):202–213
Meles SK, Vadasz D, Renken RJ, Sittig-Wiegand E, Mayer G, Depboylu C, Reetz K, Overeem S, Pijpers A, Reesink FE, van Laar T, Heinen L, Teune LK, Hoffken H, Luster M, Kesper K, Adriaanse SM, Booij J, Leenders KL, Oertel WH (2017) FDG PET, dopamine transporter SPECT, and olfaction: combining biomarkers in REM sleep behavior disorder. Mov Disord 32(10):1482–1486
Michalowska M, Fiszer U, Krygowska-Wajs A, Owczarek K (2005) Falls in Parkinson’s disease. Causes and impact on patients’ quality of life. Funct Neurol 20(4):163–168
Monchi O, Petrides M, Petre V, Worsley K, Dagher A (2001) Wisconsin card sorting revisited: distinct neural circuits participating in different stages of the task identified by event-related functional magnetic resonance imaging. J Neurosci 21(19):7733–7741
Monchi O, Petrides M, Strafella AP, Worsley KJ, Doyon J (2006) Functional role of the basal ganglia in the planning and execution of actions. Ann Neurol 59(2):257–264
Montague PR, Hyman SE, Cohen JD (2004) Computational roles for dopamine in behavioural control. Nature 431:760
Moore RY, Whone AL, Brooks DJ (2008) Extrastriatal monoamine neuron function in Parkinson’s disease: an 18F-dopa PET study. Neurobiol Dis 29(3):381–390
Morris ME, Iansek R, Matyas TA, Summers JJ (1994) The pathogenesis of gait hypokinesia in Parkinson’s disease. Brain 117(5):1169–1181
Morrish PK, Sawle GV, Brooks DJ (1995) Clinical and 18F dopa PET findings in early Parkinson’s disease. J Neurol Neurosurg Psychiatry 59(6):597–600
Mosharov EV, Borgkvist A, Sulzer D (2015) Presynaptic effects of levodopa and their possible role in dyskinesia. Mov Disord 30(1):45–53
Mundiñano I-C, Caballero M-C, Ordóñez C, Hernandez M, DiCaudo C, Marcilla I, Erro M-E, Tuñon M-T, Luquin M-R (2011) Increased dopaminergic cells and protein aggregates in the olfactory bulb of patients with neurodegenerative disorders. Acta Neuropathol 122(1):61–74
Muslimovic D, Post B, Speelman JD, Schmand B (2005) Cognitive profile of patients with newly diagnosed Parkinson disease. Neurology 65(8):1239–1245
Nandhagopal R, Kuramoto L, Schulzer M, Mak E, Cragg J, Lee CS, McKenzie J, McCormick S, Samii A, Troiano A, Ruth TJ, Sossi V, La Fuente-Fernandez R de, Calne DB, Stoessl AJ (2009) Longitudinal progression of sporadic Parkinson’s disease: a multi-tracer positron emission tomography study. Brain 132(Pt 11):2970–2979
Natale ER de, Wilson H, Pagano G, Politis M (2018) Imaging transplantation in movement disorders. Int Rev Neurobiol 143:213–263
Nedergaard S, Flatman JA, Engberg I (1993) Nifedipine- and omega-conotoxin-sensitive Ca2+ conductances in guinea-pig substantia nigra pars compacta neurones. J Physiol 466:727–747
Nègre-Pagès L, Grandjean H, Lapeyre-Mestre M, Montastruc JL, Fourrier A, Lépine JP, Rascol O (2010) Anxious and depressive symptoms in Parkinson’s disease: the French cross-sectionnal DoPaMiP study. Mov Disord 25(2):157–166
Nguyen-Legros J (1988) Functional neuroarchitecture of the retina: hypothesis on the dysfunction of retinal dopaminergic circuitry in Parkinson’s disease. Surg Radiol Anat 10(2):137–144
Nuti A, Ceravolo R, Piccinni A, Dell’Agnello G, Bellini G, Gambaccini G, Rossi C, Logi C, Dell’Osso L, Bonuccelli U (2004) Psychiatric comorbidity in a population of Parkinson’s disease patients. Eur J Neurol 11(5):315–320
O’Sullivan SS, Wu K, Politis M, Lawrence AD, Evans AH, Bose SK, Djamshidian A, Lees AJ, Piccini P (2011) Cue-induced striatal dopamine release in Parkinson’s disease-associated impulsive-compulsive behaviours. Brain 134(Pt 4):969–978
Obeso JA, Rodriguez-Oroz MC, Lanciego JL, Rodriguez Diaz M (2004) How does Parkinson’s disease begin? The role of compensatory mechanisms. Trends Neurosci 27(3):125–127 (author reply 127–8)
Oertel WH (2017) Recent advances in treating Parkinson’s disease. F1000Research 6:260
Ortuño-Lizarán I, Beach TG, Serrano GE, Walker DG, Adler CH, Cuenca N (2018) Phosphorylated α-synuclein in the retina is a biomarker of Parkinson’s disease pathology severity. Mov Disord 33(8):1315–1324
Ota M, Nakata Y, Ito K, Kamiya K, Ogawa M, Murata M, Obu S, Kunugi H, Sato N (2013) Differential diagnosis tool for parkinsonian syndrome using multiple structural brain measures. Comput Math Methods Med 2013:1–10
Otsuka M, Ichiya Y, Kuwabara Y, Hosokawa S, Sasaki M, Yoshida T, Fukumura T, Masuda K, Kato M (1996) Differences in the reduced 18F-Dopa uptakes of the caudate and the putamen in Parkinson’s disease: correlations with the three main symptoms. J Neurol Sci 136(1–2):169–173
Owen AM, Roberts AC, Hodges JR, Summers BA, Polkey CE, Robbins TW (1993) Contrasting mechanisms of impaired attentional set-shifting in patients with frontal lobe damage or Parkinson’s disease. Brain 116(Pt 5):1159–1175
Owen AM, Sahakian BJ, Hodges JR, Summers BA, Polkey CE, Robbins TW (1995) Dopamine-dependent frontostriatal planning deficits in early Parkinson’s disease. Neuropsychology 9(1):126–140
Pacelli C, Giguère N, Bourque M-J, Lévesque M, Slack RS, Trudeau L-É (2015) Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr Biol 25(18):2349–2360
Pahwa R, Stacy MA, Factor SA, Lyons KE, Stocchi F, Hersh BP, Elmer LW, Truong DD, Earl NL (2007) Ropinirole 24-hour prolonged release: randomized, controlled study in advanced Parkinson disease. Neurology 68(14):1108–1115
Palkovits M, Záborszky L, Feminger A, Mezey É, Fekete MIK, Herman JP, Kanyicska B, Szabó D (1980) Noradrenergic innervation of the rat hypothalamus: experimental biochemical and electron microscopic studies. Brain Res 191(1):161–171
Pallone JA (2007) Introduction to Parkinson’s disease. Dis Month 53(4):195–199
Pandey S, Srivanitchapoom P (2017) Levodopa-induced dyskinesia: clinical features, pathophysiology, and medical management. Ann Indian Acad Neurol 20(3):190–198
Parent A, Fortin M, Côté PY, Cicchetti F (1996) Calcium-binding proteins in primate basal ganglia. Neurosci Res 25(4):309–334
Parkinson J (1817) An essay on the shaking palsy. Whittingham and Rowland for Sherwood, London
Paulus W, Jellinger K (1991) The neuropathologic basis of different clinical subgroups of Parkinson’s disease. J Neuropathol Exp Neurol 50(6):743–755
Pavese N, Evans AH, Tai YF, Hotton G, Brooks DJ, Lees AJ, Piccini P (2006) Clinical correlates of levodopa-induced dopamine release in Parkinson disease: a PET study. Neurology 67(9):1612–1617
Pavese N, Moore RY, Scherfler C, Khan NL, Hotton G, Quinn NP, Bhatia KP, Wood NW, Brooks DJ, Lees AJ, Piccini P (2010) In vivo assessment of brain monoamine systems in parkin gene carriers: a PET study. Exp Neurol 222(1):120–124
Pavese N, Rivero-Bosch M, Lewis SJ, Whone AL, Brooks DJ (2011) Progression of monoaminergic dysfunction in Parkinson’s disease: a longitudinal 18F-dopa PET study. NeuroImage 56(3):1463–1468
Paviour DC, Price SL, Stevens JM, Lees AJ, Fox NC (2005) Quantitative MRI measurement of superior cerebellar peduncle in progressive supranuclear palsy. Neurology 64(4):675–679
Pedersen KF, Alves G, Aarsland D, Larsen JP (2009) Occurrence and risk factors for apathy in Parkinson disease: a 4-year prospective longitudinal study. J Neurol Neurosurg Psychiatry 80(11):1279–1282
Pedersen KF, Larsen JP, Tysnes O-B, Alves G (2017) Natural course of mild cognitive impairment in Parkinson disease: a 5-year population-based study. Neurology 88(8):767–774
Perez XA, Parameswaran N, Huang LZ, O’Leary KT, Quik M (2008) Pre-synaptic dopaminergic compensation after moderate nigrostriatal damage in non-human primates. J Neurochem 105(5):1861–1872
Perry EK, McKeith I, Thompson P, Marshall E, Kerwin J, Jabeen S, Edwardson JA, Ince P, Blessed G, Irving D, Perry RH (1991) Topography, extent, and clinical relevance of neurochemical deficits in dementia of Lewy body type, Parkinson’s disease, and Alzheimer’s disease. Ann N Y Acad Sci 640(1):197–202
Piccini P, Burn DJ, Ceravolo R, Maraganore D, Brooks DJ (1999) The role of inheritance in sporadic Parkinson’s disease: evidence from a longitudinal study of dopaminergic function in twins. Ann Neurol 45(5):577–582
Pifl C, Rajput A, Reither H, Blesa J, Cavada C, Obeso JA, Rajput AH, Hornykiewicz O (2014) Is Parkinson’s disease a vesicular dopamine storage disorder? Evidence from a study in isolated synaptic vesicles of human and nonhuman primate striatum. J Neurosci 34(24):8210–8218
Pillon B, Dubois B, Bonnet AM, Esteguy M, Guimaraes J, Vigouret JM, Lhermitte F, Agid Y (1989) Cognitive slowing in Parkinson’s disease fails to respond to levodopa treatment: the 15-objects test. Neurology 39(6):762–768
Politis M (2014) Neuroimaging in Parkinson disease: from research setting to clinical practice. Nat Rev Neurol 10(12):708–722
Politis M, Piccini P, Pavese N, Koh S-B, Brooks DJ (2008) Evidence of dopamine dysfunction in the hypothalamus of patients with Parkinson’s disease: an in vivo 11C-raclopride PET study. Exp Neurol 214(1):112–116
Post MR, Lieberman OJ, Mosharov EV (2018) Can interactions between α-synuclein, dopamine and calcium explain selective neurodegeneration in Parkinson’s disease? Front Neurosci 12:161
Postuma RB, Lang AE, Gagnon JF, Pelletier A, Montplaisir JY (2012) How does parkinsonism start? Prodromal parkinsonism motor changes in idiopathic REM sleep behaviour disorder. Brain 135(Pt 6):1860–1870
Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, Obeso J, Marek K, Litvan I, Lang AE, Halliday G, Goetz CG, Gasser T, Dubois B, Chan P, Bloem BR, Adler CH, Deuschl G (2015) MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 30(12):1591–1601
Price KS, Farley IJ, Hornykiewicz O (1978) Neurochemistry of Parkinson’s disease: relation between striatal and limbic dopamine. Adv Biochem Psychopharmacol 19:293–300
Price S, Paviour D, Scahill R, Stevens J, Rossor M, Lees A, Fox N (2004) Voxel-based morphometry detects patterns of atrophy that help differentiate progressive supranuclear palsy and Parkinson’s disease. NeuroImage 23(2):663–669
Puopolo M, Raviola E, Bean BP (2007) Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci 27(3):645–656
Rabinovici GD, Stephens ML, Possin KL (2015) Executive dysfunction. Continuum: lifelong learning in neurology. Behav Neurol Neuropsychiatry 21(3):646–659
Rajput AH, Stern W, Laverty WH (1984) Chronic low-dose levodopa therapy in Parkinson’s disease: an argument for delaying levodopa therapy. Neurology 34(8):991–996
Reichmann H, Brecht HM, Kraus PH, Lemke MR (2002) Pramipexol bei der Parkinson-Krankheit. Ergebnisse einer Anwendungsbeobachtung (Pramipexole in Parkinson disease. Results of a treatment observation). Der Nervenarzt 73(8):745–750
Reichmann H, Brecht MH, Köster J, Kraus PH, Lemke MR (2003) Pramipexole in routine clinical practice: a prospective observational trial in Parkinson’s disease. CNS Drugs 17(13):965–973
Reijnders JSAM, Ehrt U, Weber WEJ, Aarsland D, Leentjens AFG (2008) A systematic review of prevalence studies of depression in Parkinson’s disease. Mov Disord 23(2):183–189 (quiz 313)
Remy P, Doder M, Lees A, Turjanski N, Brooks D (2005) Depression in Parkinson’s disease: loss of dopamine and noradrenaline innervation in the limbic system. Brain 128(Pt 6):1314–1322
Ribelayga C, Cao Y, Mangel SC (2008) The circadian clock in the retina controls rod-cone coupling. Neuron 59(5):790–801
Rinne JO, Rummukainen J, Paljärvi L, Rinne UK (1989) Dementia in Parkinson’s disease is related to neuronal loss in the medial substantia nigra. Ann Neurol 26(1):47–50
Rinne JO, Portin R, Ruottinen H, Nurmi E, Bergman J, Haaparanta M, Solin O (2000) Cognitive impairment and the brain dopaminergic system in Parkinson disease. Arch Neurol 57(4):470
Rodriguez-Oroz MC, Jahanshahi M, Krack P, Litvan I, Macias R, Bezard E, Obeso JA (2009) Initial clinical manifestations of Parkinson’s disease: features and pathophysiological mechanisms. Lancet Neurol 8(12):1128–1139
Rowe J, Stephan KE, Friston K, Frackowiak R, Lees A, Passingham R (2002) Attention to action in Parkinson’s disease: impaired effective connectivity among frontal cortical regions. Brain 125(Pt 2):276–289
Rylander D, Parent M, O’Sullivan SS, Dovero S, Lees AJ, Bezard E, Descarries L, Cenci MA (2010) Maladaptive plasticity of serotonin axon terminals in levodopa-induced dyskinesia. Ann Neurol 68(5):619–628
Scatton B, Javoy-Agid F, Rouquier L, Dubois B, Agid Y (1983) Reduction of cortical dopamine, noradrenaline, serotonin and their metabolites in Parkinson’s disease. Brain Res 275(2):321–328
Schenck CH, Boeve BF, Mahowald MW (2013) Delayed emergence of a parkinsonian disorder or dementia in 81% of older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder: a 16-year update on a previously reported series. Sleep Med 14(8):744–748
Scherfler C, Schwarz J, Antonini A, Grosset D, Valldeoriola F, Marek K, Oertel W, Tolosa E, Lees AJ, Poewe W (2007) Role of DAT-SPECT in the diagnostic work up of parkinsonism. Mov Disord 22(9):1229–1238
Schneider JS (1989) Levodopa-induced dyskinesias in parkinsonian monkeys: relationship to extent of nigrostriatal damage. Pharmacol Biochem Behav 34(1):193–196
Schrag A (2000) What contributes to quality of life in patients with Parkinson’s disease? J Neurol Neurosurg Psychiatry 69(3):308–312
Segura-Aguilar J, Paris I, Muñoz P, Ferrari E, Zecca L, Zucca FA (2014) Protective and toxic roles of dopamine in Parkinson’s disease. J Neurochem 129(6):898–915
Seibyl JP, Marek KL, Quinlan D, Sheff K, Zoghbi S, Zea-Ponce Y, Baldwin RM, Fussell B, Smith EO, Charney DS, van Dyck C (1995) Decreased single-photon emission computed tomographic 123Ibeta-CIT striatal uptake correlates with symptom severity in Parkinson’s disease. Ann Neurol 38(4):589–598
Seidel K, Mahlke J, Siswanto S, Krüger R, Heinsen H, Auburger G, Bouzrou M, Grinberg LT, Wicht H, Korf H-W, den Dunnen W, Rüb U (2015) The brainstem pathologies of Parkinson’s disease and dementia with Lewy bodies. Brain Pathol (Zurich Switzerland) 25(2):121–135
Sengoku R, Saito Y, Ikemura M, Hatsuta H, Sakiyama Y, Kanemaru K, Arai T, Sawabe M, Tanaka N, Mochizuki H, Inoue K, Murayama S (2008) Incidence and extent of Lewy body-related alpha-synucleinopathy in aging human olfactory bulb. J Neuropathol Exp Neurol 67(11):1072–1083
Siderowf A, Lang AE (2012) Premotor Parkinson’s disease. Concepts and definitions. Mov Disord 27(5):608–616
Smeets WJAJ, González A (2000) Catecholamine systems in the brain of vertebrates: new perspectives through a comparative approach. Brain Res Rev 33(2–3):308–379
Smith EE (1999) Storage and executive processes in the frontal lobes. Science 283(5408):1657–1661
Sokoloff P, Giros B, Martres M-P, Bouthenet M-L, Schwartz J-C (1990) Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 347:146
Steeves TDL, Miyasaki J, Zurowski M, Lang AE, Pellecchia G, van Eimeren T, Rusjan P, Houle S, Strafella AP (2009) Increased striatal dopamine release in Parkinsonian patients with pathological gambling: a 11C raclopride PET study. Brain 132(Pt 5):1376–1385
Stiasny-Kolster K, Doerr Y, Moller JC, Hoffken H, Behr TM, Oertel WH, Mayer G (2005) Combination of ‘idiopathic’ REM sleep behaviour disorder and olfactory dysfunction as possible indicator for alpha-synucleinopathy demonstrated by dopamine transporter FP-CIT-SPECT. Brain 128(Pt 1):126–137
Surmeier DJ (2018) Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J 285:3657–3668
Surmeier DJ, Guzman JN, Sanchez-Padilla J, Goldberg JA (2011) The origins of oxidant stress in Parkinson’s disease and therapeutic strategies. Antioxid Redox Signal 14(7):1289–1301
Surmeier DJ, Obeso JA, Halliday GM (2017) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18(2):101–113
Swainson R, Rogers RD, Sahakian BJ, Summers BA, Polkey CE, Robbins TW (2000) Probabilistic learning and reversal deficits in patients with Parkinson’s disease or frontal or temporal lobe lesions: possible adverse effects of dopaminergic medication. Neuropsychologia 38(5):596–612
Taylor AE, Saint-Cyr JA, Lang AE (1986) Frontal lobe dysfunction in Parkinson’s disease. The cortical focus of neostriatal outflow. Brain 109(Pt 5):845–883
Taylor TN, Caudle WM, Shepherd KR, Noorian A, Jackson CR, Iuvone PM, Weinshenker D, Greene JG, Miller GW (2009) Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci 29(25):8103–8113
Tedroff J, Pedersen M, Aquilonius S-M, Hartvig P, Jacobsson G, Langstrom B (1996) Levodopa-induced changes in synaptic dopamine in patients with Parkinson’s disease as measured by [11C]raclopride displacement and PET. Neurology 46(5):1430
Thobois S, Lhommée E, Klinger H, Ardouin C, Schmitt E, Bichon A, Kistner A, Castrioto A, Xie J, Fraix V, Pelissier P, Chabardes S, Mertens P, Quesada J-L, Bosson J-L, Pollak P, Broussolle E, Krack P (2013) Parkinsonian apathy responds to dopaminergic stimulation of D2/D3 receptors with piribedil. Brain 136(Pt 5):1568–1577
Tillerson JL, Caudle WM, Parent JM, Gong C, Schallert T, Miller GW (2006) Olfactory discrimination deficits in mice lacking the dopamine transporter or the D2 dopamine receptor. Behav Brain Res 172(1):97–105
Tison F, Mons N, Geffard M, Henry P (1991) The metabolism of exogenous l-dopa in the brain: an immunohistochemical study of its conversion to dopamine in non-catecholaminergic cells of the rat brain. J Neural Transm 3(1):27–39
Trétiakoff C (1919) Contribution a l’etude de l’anatomie pathologique du locus niger de Soemmering avec quelques deductions relatives a la pathogenie des troubles du tonus musculaire et de la maladie de Parkinson
Turiault M, Parnaudeau S, Milet A, Parlato R, Rouzeau J-D, Lazar M, Tronche F (2007) Analysis of dopamine transporter gene expression pattern—generation of DAT-iCre transgenic mice. FEBS J 274(14):3568–3577
Ubeda-Bañon I, Saiz-Sanchez D, La Rosa-Prieto C de, Argandoña-Palacios L, Garcia-Muñozguren S, Martinez-Marcos A (2010) alpha-Synucleinopathy in the human olfactory system in Parkinson’s disease: involvement of calcium-binding protein- and substance P-positive cells. Acta Neuropathol 119(6):723–735
Uhl GR (1998) Hypothesis: the role of dopaminergic transporters in selective vulnerability of cells in Parkinson’s disease. Ann Neurol 43(5):555–560
Uhl GR, Hedreen JC, Price DL (1985) Parkinson’s disease: loss of neurons from the ventral tegmental area contralateral to therapeutic surgical lesions. Neurology 35(8):1215–1218
Vaillancourt DE, Schonfeld D, Kwak Y, Bohnen NI, Seidler R (2013) Dopamine overdose hypothesis: evidence and clinical implications. Mov Disord 28(14):1920–1929
van de Kar LD, Lorens SA (1979) Differential serotonergic innervation of individual hypothalamic nuclei and other forebrain regions by the dorsal and median midbrain raphe nuclei. Brain Res 162(1):45–54
Vernier P, Moret F, Callier S, Snapyan M, Wersinger C, Sidhu A (2004) The degeneration of dopamine neurons in Parkinson’s disease: insights from embryology and evolution of the mesostriatocortical system. Ann N Y Acad Sci 1035:231–249
Vingerhoets FJ, Schulzer M, Calne DB, Snow BJ (1997) Which clinical sign of Parkinson’s disease best reflects the nigrostriatal lesion? Ann Neurol 41(1):58–64
Vlaar AMM, van Kroonenburgh MJPG, Kessels AGH, Weber WEJ (2007) Meta-analysis of the literature on diagnostic accuracy of SPECT in parkinsonian syndromes. BMC Neurol 7:27
Vogt Weisenhorn DM, Giesert F, Wurst W (2016) Diversity matters—heterogeneity of dopaminergic neurons in the ventral mesencephalon and its relation to Parkinson’s disease. J Neurochem 139(Suppl 1):8–26
Voon V, Mehta AR, Hallett M (2011) Impulse control disorders in Parkinson’s disease: recent advances. Curr Opin Neurol 24(4):324–330
Voon V, Napier TC, Frank MJ, Sgambato-Faure V, Grace AA, Rodriguez-Oroz M, Obeso J, Bezard E, Fernagut P-O (2017) Impulse control disorders and levodopa-induced dyskinesias in Parkinson’s disease: an update. Lancet Neurol 16(3):238–250
Vriend C, Pattij T, van der Werf YD, Voorn P, Booij J, Rutten S, Berendse HW, van den Heuvel OA (2014) Depression and impulse control disorders in Parkinson’s disease: two sides of the same coin? Neurosci Biobehav Rev 38:60–71
Wager TD, Smith EE (2003) Neuroimaging studies of working memory: a meta-analysis. Cogn Affect Behav Neurosci 3(4):255–274
Wager TD, Jonides J, Reading S (2004) Neuroimaging studies of shifting attention: a meta-analysis. NeuroImage 22(4):1679–1693
Waters CM, Peck R, Rossor M, Reynolds GP, Hunt SP (1988) Immunocytochemical studies on the basal ganglia and substantia nigra in Parkinson’s disease and Huntington’s chorea. Neuroscience 25(2):419–438
Watson C, Paxinos G, Puelles L (eds) (2012) The mouse nervous system, 1st edn. Elsevier/Academic Press, London
Weingarten CP, Sundman MH, Hickey P, Chen N-k (2015) Neuroimaging of Parkinson’s disease: expanding views. Neurosci Biobehav Rev 59:16–52
Weintraub D (2008) Dopamine and impulse control disorders in Parkinson’s disease. Ann Neurol 64(Suppl 2):S93–S100
Weintraub D, Newberg AB, Cary MS, Siderowf AD, Moberg PJ, Kleiner-Fisman G, Duda JE, Stern MB, Mozley D, Katz IR (2005) Striatal dopamine transporter imaging correlates with anxiety and depression symptoms in Parkinson’s disease. J Nucl Med 46(2):227–232
Weintraub D, Comella CL, Horn S (2008) Parkinson’s disease—Part 1: Pathophysiology, symptoms, burden, diagnosis, and assessment. Am J Manag Care 14(2 Suppl):S40–S48
Weintraub D, Koester J, Potenza MN, Siderowf AD, Stacy M, Voon V, Whetteckey J, Wunderlich GR, Lang AE (2010) Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol 67(5):589–595
Williams DR, Watt HC, Lees AJ (2006) Predictors of falls and fractures in bradykinetic rigid syndromes: a retrospective study. J Neurol Neurosurg Psychiatry 77(4):468–473
Wilson DA, Sullivan RM (1995) The D2 antagonist spiperone mimics the effects of olfactory deprivation on mitral/tufted cell odor response patterns. J Neurosci 15(8):5574–5581
Yamada T, McGeer PL, Baimbridge KG, McGeer EG (1990) Relative sparing in Parkinson’s disease of substantia nigra dopamine neurons containing calbindin-D28K. Brain Res 526(2):303–307
Yamanishi T, Tachibana H, Oguru M, Matsui K, Toda K, Okuda B, Oka N (2013) Anxiety and depression in patients with Parkinson’s disease. Intern Med (Tokyo Japan) 52(5):539–545
Yetnikoff L, Lavezzi HN, Reichard RA, Zahm DS (2014) An update on the connections of the ventral mesencephalic dopaminergic complex. Neuroscience 282:23–48
Zarow C, Lyness SA, Mortimer JA, Chui HC (2003) Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol 60(3):337–341
Zeiss CJ (2005) Neuroanatomical phenotyping in the mouse: the dopaminergic system. Vet Pathol 42(6):753–773
Zigmond MJ, Abercrombie ED, Berger TW, Grace AA, Stricker EM (1990) Compensations after lesions of central dopaminergic neurons: some clinical and basic implications. Trends Neurosci 13(7):290–296
Zweig RM, Cardillo JE, Cohen M, Giere S, Hedreen JC (1993) The locus ceruleus and dementia in Parkinson’s disease. Neurology 43(5):986
Acknowledgements
WHO is supported by the Charitable Hertie Foundation, Frankfurt/Main, Germany. WHO received personal fees for educational talks and/or consultancy, outside of the submitted work, from Abbvie, Adamas, Bristol-Myer-Squibb, Desitin, Mundipharma, Neuropore, Novartis, Roche and UCB Pharma, and grants from the Deutscher Akademischer Austauschdienst, the Deutsche Forschungsgemeinschaft, the International Parkinson-Fonds The Netherlands, the ParkinsonFonds Deutschland, the Michael J. Fox Foundation, USA, the National Research Fond Luxembourg, from Roche International, Switzerland and Novartis Pharma, Germany.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Geibl, F.F., Henrich, M.T. & Oertel, W.H. Mesencephalic and extramesencephalic dopaminergic systems in Parkinson’s disease. J Neural Transm 126, 377–396 (2019). https://doi.org/10.1007/s00702-019-01970-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-019-01970-9