
Abstract
There has been tremendous progress toward understanding the genetic basis of Parkinson’s disease and related movement disorders. We summarize the genetic, clinical and pathological findings of autosomal dominant disease linked to mutations in SNCA, LRRK2, ATXN2, ATXN3, MAPT, GCH1, DCTN1 and VPS35. We then discuss the identification of mutations in PARK2, PARK7, PINK1, ATP13A2, FBXO7, PANK2 and PLA2G6 genes. In particular we discuss the clinical and pathological characterization of these forms of disease, where neuropathology has been important in the likely coalescence of pathways highly relevant to typical PD. In addition to the identification of the causes of monogenic forms of PD, significant progress has been made in defining genetic risk loci for PD; we discuss these here, including both risk variants at LRRK2 and GBA, in addition to discussing the results of recent genome-wide association studies and their implications for PD. Finally, we discuss the likely path of genetic discovery in PD over the coming period and the implications of these findings from a clinical and etiological perspective.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
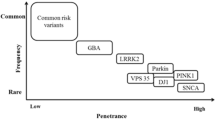
The past 15 years has witnessed a seed change in our concept of the etiologic basis of Parkinson’s disease (PD). This change has been largely driven by genetics, progressing through the early recognition of familial forms of the disease, to include the identification of gene mutations that cause rare familial forms of PD (Table 1), the discovery of more common highly penetrant mutations, finding moderate risk variants, and most recently the mapping of multiple low-risk conferring loci (Fig. 1). Many of the rarer genetic causes have a disease phenotype more consistent with parkinsonism than true idiopathic PD but the considerable overlap in clinical and pathological features, and probably in disease pathways, has prompted us to include them here.

Timeline of genetics in Parkinson’s disease and MSA. Notably some of the risk variants were implicated earlier, but we have shown the date on which the variants role in disease was unequivocally proven
These genetic findings have been central in the field’s attempts to understand the underlying disease process at the molecular level, being used in cell based and animal models in a manner familiar to those working on varied neurodegenerative diseases. The pace of discovery continues at a fast clip, and in large part this is a result of new methods that allow comprehensive and rapid assessment of the genomic landscape of individual subjects. Hand in hand with these genetic discoveries is the clinical and pathological characterization of genetically defined forms of disease. Unfortunately, a number of the PD-like disorders, particularly the recessive parkinsonian conditions have not been neuropathologically characterized. We have still included them in this piece as they provide important insight into disease pathways. This work offers insight into disease processes and, particularly for pathology, points to critical proteins in the disease process.
In this article, we will provide an overview of the genetic discoveries made in PD, not only detailing what these reveal in terms of risk for disease but also discussing the clinical and pathological correlates of the particular genetic flavors of these diseases. We will end this piece with a discussion of what this sum total of discovery has revealed about disease, the likely impact on defining subtypes of disease, and where we think future opportunities for understanding and exploiting the genetic basis of these disorders exist.
Autosomal dominant causes of PD
α-Synuclein (PARK1 and 4)
Nussbaum and colleagues made the discovery of missense mutations in α-synuclein (encoded by SNCA) as a cause of PD in 1997 [102]. Working on a large American–Italian family called the Contursi kindred, this group used a traditional linkage approach to localize the underlying genetic lesion to a region on the long arm of chromosome four [101]. This was the public beginning to the genetic age of PD research. Prior to this finding, there had been much discussion of the relevance of genetic findings in extremely rare and somewhat atypical families with PD, to the sporadic disease. The concern, in its most simple form, was that these were simply unrelated disorders. Therefore, the observations published by Spillantini and colleagues [120] shortly after the report of α-synuclein mutations, were particularly important. In this work, the authors showed that α-synuclein is a major component of Lewy bodies, the pathognomic hallmark of all PD. This work therefore elegantly linked sporadic and familial forms: mutations in α-synuclein could cause a rare and severe familial form of PD, and α-synuclein is a key protein component of all PD cases. Further, this work was extended to show that α-synuclein is a major protein component of glial cytoplasmic inclusions in multiple system atrophy (MSA) [118], and Lewy bodies and Lewy neurites in both PD and dementia with Lewy bodies [119].
To date, there have been three different missense mutations in α-synuclein identified as a cause of PD. The cause of disease in the Contursi kindred was an p.A53T mutation; subsequently p.A30P and p.E46K mutations were identified in German and Spanish PD families, respectively [62, 136]. Six years after the initial discovery of missense mutations in SNCA, triplication of the SNCA locus was identified as the cause of disease in a large family called the Iowa kindred (sometimes also called the Spellman-Muenter or Waters-Miller kindred) [117]. In this family, affected members had three copies of SNCA on the disease-segregating chromosome, meaning that their total genetic complement of SNCA, at four copies, was twice that of individuals in the normal population. Because of this, affected family members produce twice as much of the normal sequence α-synuclein as non-triplication carriers [81], and this increase in α-synuclein protein is believed to be the underlying driver of disease. Subsequent to this finding, additional SNCA triplication and duplication mutations were identified, with a 100 and 50 % increase in genetic load of SNCA, respectively [10, 25, 50, 88].
Notably the onset of disease and co-occurrence of severe dementia and psychiatric problems appear to be associated with the number of copies of SNCA, triplication carriers tending to present with disease a decade earlier than duplication carriers, and with dementia being a much more common feature. This has lead to the simple hypothesis of a dose relationship between α-synuclein levels and disease severity [116], and a more general hypothesis regarding protein levels in disease of abnormal protein deposition [116]. Extending this notion, it is perhaps not difficult to imagine a scenario where small perturbations to α-synuclein levels have a critical role to play in late-onset (and perhaps all) PD.
Clinically, patients with α-synuclein missense mutations present with a severe parkinsonism and an early age at onset; this has been described as a fairly typical levodopa-responsive parkinsonism, however the presence of dementia is quite a common feature [35]. Patients with the most recently described mutation, p.E46K, display a prominent dementia, noted approximately 2 years after disease onset [136]. This is accompanied by hallucinations and fluctuations in consciousness reminiscent of dementia with Lewy bodies. This phenotype is borne out neuropathologically by fulminant Lewy body pathology in subcortical nuclei and in the cortex, albeit with an absence of Alzheimer pathology [136].
Within the Iowa kindred, whose members were followed for many decades, quite a broad array of clinical and pathological features was described. In general, affected family members present with a levodopa-responsive parkinsonism with dementia [84]. The disease onset and duration were earlier and shorter than typical PD, being 33 and 8 years, respectively.
In terms of neuropathology, there is nigral neuronal loss, temporal lobe vacuolation as well as widespread Lewy bodies in both the brainstem and cerebral cortex [25, 40, 84]. In general, other synuclein mutations have significant Lewy body neuropathology usually with brainstem and cortical lesions [35, 136]. The pathology is consistent across the genetic lesions in comparison with the variability observed in LRRK2 and PARK2 cases (Table 2).
Leucine-rich repeat kinase 2 (LRRK2) (PARK8)
Linkage of PD to a pericentromeric region on chromosome 12 was initially reported in 2002 by Funayama and colleagues [27]. The authors described a large Japanese family with apparent autosomal dominant PD, with reduced penetrance. Clinically, the disease resembled a fairly typical PD, with an age of onset in the 50s, and a dopa-responsive disease. In the initial description of this linkage, the authors described neuropathology in a single case, who presented with a striatonigral degeneration with glial cytoplasmic inclusions and was therefore quite distinct from the picture expected for PD. Subsequent clinical and neuropathological assessment of the original Sagamihara kindred revealed quite disparate outcomes, including pure nigral degeneration, Lewy body positive PD, and MSA [42].
The underlying genetic cause of chromosome 12-linked PD was shown to be mutation of the gene LRRK2, reported by two groups late in 2004 [95, 139]. In these initial reports a total of six mutations were reported, p.R1441G, p.R1444C, p.Y1699C, p.I1122V, p.I2020T and p.L1114L. Subsequently, an additional LRRK2 mutation, p.G2019S, was described in multiple cases of European descent [18, 33, 47, 86]. This particular mutation is relatively common across several populations, being responsible for PD in ~2 % of sporadic and ~5 % of familial PD cases in Northern European and North American populations; further, certain groups are enriched for the presence of this mutation with reported frequencies of ~10 % in Portuguese PD patients, ~20 % in PD patients of Ashkenazi Jewish ancestry, and ~40 % of North African Berber Arab PD patients [8, 66, 91]. Although it is likely that the p.G2019S mutation has arisen on multiple occasions, for the majority of carriers, this mutation is believed to have been inherited from a common founder [53, 67], dating back 4,500–9,100 years, and it is suggested that the mutation arose in the Near East, then moved throughout the World with the Ashkenazi Diaspora [5].
Although a large number of mutations have been reported and proposed as disease causing, there still remains only a handful with a high degree of proof, based on overwhelming association, or excellent disease segregation data (p.N1437H, p.R1441C, p.R1441G, p.R1441H, p.Y1699C, p.I2012T, p.G2019S, and p.I2020T). The penetrance of LRRK2 mutations has been widely discussed and studied, indeed the original manuscript of Funayama and colleagues commented on the apparent reduced penetrance in their pedigree. The causal mutation in this family was subsequently shown to be p.I2020T, and this discovery confirmed that the mutation was not fully penetrant [27, 42]. Initial estimates of lifetime penetrance centered around 30 % [91] as have kin-cohort based estimates [36], which contrasts sharply with a study in a family-based sample series that produce an estimate of lifetime penetrance of ~90 % [65]. Perhaps the largest study to date addressing this issue presented an age-associated penetrance of the p.G2019S mutation of 28 % at age 59 years, 51 % at 69 years, and 74 % at 79 years [45]. There are likely several factors that influence penetrance and expressivity of LRRK2 mutations, and these probably include genetics, external and environmental factors, and perhaps stochastic events.
On the whole, PD is the most common clinical and pathological diagnosis for LRRK2 mutation positive subjects, this mutation being at most a rare cause of other distinct neurological disorders [46]. In some respects this represents an ascertainment bias, as the majority of samples screened for LRRK2 mutation are typical PD, and when other series have been screened they tend to conform to a distinct diagnostic category. Indeed, some of the first families described to have LRRK2 mutations included affected individuals with quite discordant clinical features, including amyotrophy and dementia [139]. The typical age at onset for LRRK2-linked PD is in the sixth decade of life [45]. When examining metrics of severity of disease such as occurrence of falls, rate of progression, and dyskinesia, LRRK2-linked disease appears to be less rapidly progressing than idiopathic PD, but compared to idiopathic PD LRRK2 patients appear to suffer more from dystonia and tremor tends to be a more frequent presenting symptom [45].
In terms of neuropathology, again, most LRRK2 cases described thus far exhibit a pattern of features consistent with typical PD, namely Lewy bodies in the brainstem and loss of dopaminergic neurons in the substantia nigra. However, additional and sometimes discordant pathological features have been described, including fulminant plaque and tangle pathology in addition to PD pathology, or a pure nigral degeneration without Lewy bodies, and also glial cytoplasmic inclusions reminiscent of MSA [42, 139]. There have been no large series of genetically defined LRRK2 cases that have been neuropathology examined where the brain tissue has been consistently well preserved.
ATXN2 and ATXN3
The spinocerebellar ataxias (SCA) are complex disorders that share a common theme of a progressive deterioration in balance and coordination. While many genetic lesions have been associated with distinct subtypes of SCA, two forms of SCA, SCA2 and SCA3, have been shown to have quite broad presentations that may include a dopa-responsive parkinsonism, sometimes indistinguishable from PD.
The identification of a large Asian–American family with dopa-responsive parkinsonism revealed that the disease in this family was caused by an expansion mutation in ATXN2, the cause of SCA2 [38]. Of the three affected members in this family, two fit the PD Society Brain Bank and National Institute of Neurological Disorders and Stroke criteria for a diagnosis of PD (or probable PD), and the third member, while not fulfilling these criteria, was diagnosed as having PD by their primary care physician. This work was shortly followed by several other reports of similar findings (summarized in [29]). More than 34 CAG repeats within ATXN2 are considered as pathogenic in ataxia, and it appears that the majority of ATXN2 expansion mutations associated with PD tend to be at the lower end of this pathogenic expansion range. Patients may present not only with a pure levodopa-responsive parkinsonism but may also display other features such as ataxia, disease reminiscent of progressive supranuclear palsy, tremor and dementia. To date, no detailed neuropathologic characterization has been presented on any affected patients with ATXN2 expansion mutation and levodopa-responsive parkinsonism.
Triplet repeat expansion mutation in ATXN3 is the cause of SCA3, also called Machado Joseph disease (MJD) [55]. MJD can present with parkinsonism; however, this presentation is usually in combination with other atypical features such as neuropathy and cerebellar and pyramidal signs. In 2001, Gwinn-Hardy and colleagues [39] described an African–American family with members who fulfilled criteria for PD, but with disease caused by ATXN3 expansion mutation. In two of the four affected family members there were no features considered atypical for PD, and three of four were responsive to dopamine replacement therapy. The phenotype of predominant parkinsonism has been subsequently described to be quite common in ATXN3 mutation positive patients of African descent [122], but has also been described in Asian families [71].
The neuropathological features of parkinsonism caused by ATXN2 and ATXN3 expansion mutations remain unclear, however, nigral cell loss has been described as a feature of SCA3 [78].
Vacuolar protein sorting-associated protein 35 (VPS35) (PARK17)
The most recently described cause of monogenic PD is mutation of VPS35 a finding that demonstrates the power of exome sequencing, which was used in both studies [129, 138]. Vilariño–Güell and colleagues described the identification of a p.D620N mutation in VPS35 within affected members of a Swiss kindred with late-onset, autosomal dominant PD. This group then went on to find this mutation in three other families with a similar phenotype and in one sporadic PD case. At the same time, Zimprich and co-workers [138] published the identification of the p.D620N mutation in a large multigenerational Austrian family with PD and in two additional families screened for VPS35 mutations. Both groups also identified additional mutations in VPS35; however, the pathogenicity of these additional variants remains unknown. While the VPS35-linked families are reported to fulfill London Brain Bank criteria for PD, there is somewhat limited clinical and pathological data on these cases.
Rarer causes of autosomal dominant PD or parkinsonism
There are a number of disorders that have PD or dopa-responsive parkinsonism as part of their phenotype. These may occur as a rare phenotypic feature such as in cases with GTP cyclohydrolase 1 (GCH1) mutations [48] or early in the disease process in some kindreds with mutations in the microtubule-associated protein tau (MAPT) gene [49]. Here the PD is only a minor part of the phenotype and only in certain families, such as the very large family with parkinsonism and dementia with pallidopontonigral degeneration (PPND) [135] and the Irish American family with disinhibition–dementia–parkinsonism–amyotrophy complex (DDPAC) [72, 133]. The neuropathology in MAPT families varies considerably, but is dominated by frontotemporal atrophy, with neuronal and/or glial inclusions that stain positively with MAPT antibodies; synuclein and Lewy bodies are not a feature. Perry syndrome [100] is another rare cause of parkinsonism. This neuropsychiatric condition presents in the fifth decade with depression and marked weight loss, later parkinsonism and respiratory failure occur. The disorder is caused by mutations in dynactin 1 (DCTN1) [26] where the neuropathology is dominated by abnormal MAPT deposition but where there are unusual features such as TAR DNA-binding protein 43 (TDP-43) pathology. TDP-43 can also be part of the neuropathology of disease caused by other common autosomal dominant PD genes such as LRRK2 [131].
Chartier-Harlin et al. [9] recently reported the identification of a new cause of parkinsonism, mutations in eukaryotic translation initiation factor 4 gamma 1 (EIF4G1). A possible founder mutation was identified as p.R1250H in families from France, Ireland and the United States. The phenotype described is of a late-onset PD with a good response to levodopa and neuropathology consistent with α-synuclein deposition and Lewy bodies.
Autosomal recessive causes of PD
Parkin (PARK2)
Kitada [59] first described a homozygous deletion of exons 3–7 of the parkin (PARK2) gene in autosomal recessive juvenile Parkinson’s disease. Since this discovery, a number of mutations and small and large structural changes have been identified in PARK2 that account for up to 10 % of early onset PD cases, depending on the population analysed [11, 54, 80]. Heterozygous PARK2 mutations have frequently been identified and speculated to be associated with PD. The association is not clear and may be due to sequencing bias as variants are also seen in healthy controls at similar frequencies [56, 69]. Asymptomatic heterozygous PARK2 mutation carriers display a small decrease in striatal F-DOPA uptake, suggesting nigral dysfunction but perhaps only relevant in the presence of other genetic factors such as heterozygous mutations in other PD genes or environmental risk factors [57].
The clinical phenotype of homozygous or compound heterozygous PARK2 cases is usually indistinguishable from early onset idiopathic PD, with slowly progressive Levodopa-responsive disease often requiring lower equivalent doses with frequent late motor complications. Atypical and later onset cases are described with prominent dystonia, hyperreflexia and early complications. Interestingly some cases seem responsive to nicotine [70].
The neuropathology of PARK2 cases initially was thought to be deficient in Lewy body and neurofibrillary tangle pathology and was lacking in distinctive pathology at all; however recent reports suggest Lewy bodies in the nigra and locus coeruleus and α-synuclein immunopositive inclusion bodies in the pedunculopontine nucleus exist in some cases [24, 37, 83, 103], although the pathogenicity of at least one of these mutations remains in question [24]. Neurofibrillary tangles have also been reported in PARK2 cases [83]. A recent review of five cases with compound heterozygous PARK2 mutations found the major feature was ventral nigral cell loss, with sparse Lewy bodies present in two cases (Revesz and Lees personnel communication). The group termed the pathology ventral nigropathy. This pathological heterogeneity is not uncommon in other monogenic forms of PD and may be explained by disease duration, mutation type, tissue quality or modifying genes.
DJ1 (PARK7)
In 2003, Bonifati and colleagues [6] carried out homozygosity mapping and positional cloning to identify a homozygous deletion in PARK7 causing early onset PD in a Dutch family and a missense mutation in PARK7 causing disease in an Italian early onset PD family. PARK7 mutations are rare, patients have an age of onset in the 20s or 30s with Levodopa-responsive PD. There are atypical features that occur in reported families such as additional psychiatric features in the Italian family and dystonic features in the Dutch kindred [1, 6, 15]. Structural neuroimaging was unremarkable, but functional imaging of the brain, showed significant evidence for a presynaptic dopamine deficit [15]. No neuropathology has been reported on any PARK7 case to date.
PINK1 (PARK6)
In 2004, Valente and colleagues [127] reported two homozygous mutations in the PTEN-induced kinase 1 gene (PINK1) as a cause of early-onset PD. A homozygous stop mutation in two Italian families and a homozygous missense mutation in a consanguineous Spanish kindred. Mutations in PINK1 are usually private loss of function changes and have been found to be the second most common cause of early onset autosomal recessive PD [76, 89, 130]. As with PARK2 mutations, there has been speculation about the role of single PINK1 heterozygous mutations as a risk factor for PD. Although a subclinical dopaminergic deficit has been identified using 18F-DOPA positron emission tomography (PET), rare PINK1 variants have been identified in PD patients as well as controls [57]. Clinically, patients usually have a later age at onset in the 40s and 50s but otherwise are similar to the PD associated with PARK2 mutations, displaying slowly progressive levodopa-responsive disease. Atypical features are often observed and include prominent dystonia, sleep benefit and pyramidal signs [7, 14, 130].
Recently, neuropathological examination of the brain tissue from a patient with a compound heterozygous PINK1 mutation from a large Spanish kindred was carried out. The phenotype was slowly progressive Levodopa-responsive parkinsonism, initial gait impairment and psychiatric symptoms. This revealed neuronal loss in the substantia nigra pars compacta, Lewy bodies and aberrant neurites in the reticular nuclei of the brainstem, substantia nigra pars compacta and Meynert nucleus; the locus ceruleus and amygdala were spared [107]. These features are comparable with typical idiopathic PD and we speculate that when further brains come to neuropathology there will be heterogeneity as in the case of those with PARK2 mutations.
ATPase (P-type) 13A2 (ATP13A2) (PARK9)
An atypical juvenile form of PD was first reported in a large consanguineous family from Kufor Rakeb, a small homestead in the northern highlands of Jordan [104]. The age of onset was between 12 and 15 years and the PD was Levodopa responsive and slowly progressive. There were atypical features of hyperreflexia, Babinski’s sign, slowed saccades, supranuclear gaze palsy, optokinetic nystagmus, visual hallucinations, mini-myoclonus and oculogyric dystonic spasms. Using homozygosity mapping and positional cloning, Ramirez and colleagues identified mutations in ATP13A2: a homozygous mutation in the Jordanian and compound heterozygous mutation in a Chilean juvenile PD family. The clinical phenotype of this early-onset pallido-pyramidal syndrome varies in severity but only a handful of cases and families have been reported [16, 87, 94, 104, 110]. There has also been a suggestion that heterozygous mutations may be a risk of PD [20]. Mutations in this interesting gene are likely to have a role in lysosome degradation based on the putative gene function and sural nerve pathology [94]. To date, no neuropathology has been described in these cases.
F-box protein 7 (FBXO7) (PARK15)
Shojaee and colleagues [112] reported a large Iranian family with autosomal recessive early onset PD with pyramidal signs, dystonia and equinovarus deformity. The large consanguineous family has pseudo-dominant inheritance with a homozygous missense FBXO7 mutation. Some individuals display both early onset PD and pyramidal signs and others only pyramidal signs. Since the initial report there have been reports of families with homozygous or heterozygous mutations in FBX07 reported from Italy, Holland, Turkey and Pakistan. Most of these mutations are loss of function [17, 94]. The phenotype is similar to the Iranian cases with parkinsonism with pyramidal signs, occasionally other features are present such as psychiatric problems or blephorospasm and all initially respond to levodopa and often develop dyskinesia. Magnetic resonance imaging (MRI) and IBZM-single-photon emission computed tomography (SPECT) in these individuals are normal but FP-CIT SPECT shows severe presynaptic deficits in nigrostriatal dopamine [17]. No neuropathology has been reported in patients with FBXO7 mutations.
Phospholipase A2, group VI (cytosolic, calcium-independent) (PLA2G6) (PARK14) and pantothenate kinase (PANK2)
Mutations in PLA2G6 [82] and PANK2 [44] usually cause an early-onset recessive degenerative disorder with spasticity, ataxia and dystonia; however, later adult onset forms of the disease can present with a dystonia predominant parkinsonism [92]. Imaging has shown neuronal brain iron accumulation (NBIA) and these diseases are mainly classified under this umbrella where the two major types of NBIA are the pantothenate kinase (PANK) associated neurodegeneration (PKAN or NBIA type 1—previously called Hallervorden–Spatz or Martha–Alma disease) caused by mutations in PANK2 and NBIA type 2 or infantile neuroaxonal dystrophy (INAD) (previously Seitelberger’s disease) due to mutations in PLA2G6. The clinical phenotype of NBIA is broad, although there are some characteristic features. Most cases present before the age of 5 years with developmental delay, dystonia, rigidity, dysarthria and ataxia. Optic nerve pallor, spasticity and seizures are also frequently seen. Classical INAD involves onset before 2 years, and has a slow progression. Onset between 2 and 18 years is classified as juvenile disease and after 18 years as adult onset NAD or atypical NAD [64, 92, 94].
Paisan-Ruiz and colleagues [92] carried out homozygosity mapping and positional cloning in three consanguineous Asian families with early onset parkinsonism. Two families were identified with PLA2G6 mutations and the third was later found to have an FBXO7 homozygous mutation. The patients with PLA2G6 homozygous mutations presented in their 20s with slowly progressive gait problems, clumsiness, imbalance, hand tremor, cognitive decline and dysarthria. On examination there was facial hypomimia, eyelid opening apraxia, supranuclear vertical gaze palsy, generalized rigidity and dystonia in all limbs, a left pill rolling rest tremor, mild postural arm tremor, and bilateral bradykinesia. Interestingly using MRI, the brain showed atrophy but no abnormal iron deposition which differs from both the infantile onset cases with PLA2G6 mutations; these typically show abnormal iron accumulation in the globus pallidus and sometimes substantia nigra and also from PANK2 mutation cases that almost always have the “eye of the tiger” sign in the globus pallidus region. Other families have been reported with this phenotype, which forms a rare subgroup of patients with PLA2G6 mutations.
Mutations in PANK2 were first described by Susan Hayflick’s group in 2001 [44, 137]. Patients with homozygous PANK2 mutations and the commoner typical PKAN usually present in the first decade of life with severe extrapyramidal signs, rigidity, ataxia, and dysarthria. The disease progresses rapidly with loss of ambulation and death in early teenage years. In contrast, atypical PKAN usually begins in the second or third decade, and is associated with extrapyramidal signs, slower progression, and patients usually retain ambulatory function. Most patients with parkinsonism are Levodopa responsive at first but this usually lasts only 1–2 years.
Neuropathological examination of brains from patients with genetically proven PLA2G6 and PANK2 disease has only recently been possible and this has shed important light on the pathogenesis of these two disorders. Prior to the genetic dissection, both were considered to be Lewy body disorders but this is only the case for PLA2G6 where there are Lewy bodies and synuclein-positive dystrophic neurites in the substantia nigra and cortex as well as tau immunoreactive cortical neurofibrillary tangles. Identical pathology is seen in PLA2G6 patients with early infantile and late-onset parkinsonian forms of the disease [94]. Patients with PANK2 mutations do not have Lewy bodies or synuclein-positive dystrophic neurites; some cases have no distinctive neuropathology, diffuse tau staining or neurofibrillary tangles [60].
There are a number of additional rare recessive disorders that have PD or parkinsonism within their phenotype. These include rare families with spatacsin (SPG11) [93, 96], fatty acid 2-hydroxylase (FA2H) [19, 61] and alpha chain of type XVIII collagen (COL18A2) [97] mutations, although none of these cases have come to neuropathology as yet.
Risk loci
SNCA
For much of the late 1990s, geneticists spent a lot of time and resources investigating candidate polymorphisms as risk factors for neurodegenerative diseases. For the most part, this was an inherently biased, underpowered, and fruitless endeavor. However, this work provided the initial evidence that two candidate loci were involved in PD risk. Shortly after the identification of SNCA mutations as a rare cause of PD, Kruger and colleagues [63] reported that alleles at a polymorphism in the SNCA promoter region were associated with PD, when combined with APOE genotypes. This polymorphism, called REP1, comprises an imperfect dinucleotide repeat, approximately 10 kb 5′ to the initiation codon of SNCA. Many studies were performed testing the notion that REP1 alleles were associated with risk for disease over the following years, and these were often contradictory in nature. In 2006, a large study that combined previous work and included new genotyping provided compelling evidence of association at the REP1 SNCA locus [74], suggesting this variant, or variant(s) close by were exerting a biological effect on SNCA and modulating risk for disease.
Glucocerebrosidase (GBA)
The keen clinical observation that patients and relatives of patients with Gaucher’s disease seemed to present with PD more often than expected led ultimately to the identification of mutations in GBA as a risk for PD [34, 73, 85, 124, 125]. The initial compelling report of a strong association between single heterozygous GBA mutations and PD came in 2004 when Aharon-Peretz and colleagues [2] showed that within the Ashkenazi Jewish population GBA mutations appeared to be commonly associated with PD, increasing risk ~sevenfold. The generalizability of this finding to PD worldwide was contested for several years, until a large meta-analysis of existing GBA mutation data was performed. This work published in 2009 showed conclusively carriers of a single GBA mutant allele were at ~fivefold greater risk for PD, the most common GBA mutations being present in 15 % of Ashkenazi Jewish patients and 3 % of non-Ashkenazi Jewish patients, compared to 3 % and <1 % of matched controls, respectively. The clinical symptoms associated with possession of a GBA risk allele by a PD patient were quite similar to those observed in typical PD, although there were data that supported a greater rate of cognitive changes, bradykinesia, resting tremor and rigidity in GBA mutation positive patients, in addition to more commonly presenting with symmetric onset [113].
Genome-wide association
The advent of genome-wide association (GWA) studies signaled the end of wholesale candidate gene association studies that were based on function; this method provided a relatively efficient means to identify common genetic risk loci in a genome-wide manner. Two of the first GWA studies were performed in PD [28, 75]. Although these were quite low-powered efforts, they had two consequences; first showing that there were likely no medium to high-risk common risk alleles for PD (i.e. of a similar effect size to APOE ε4 in Alzheimer’s disease), and second placing a large amount of genetic data into the public domain, which could then be mined and augmented by others. The second wave of GWA papers for PD demonstrated clearly that there were risk loci at both the region of the genome encoding α-synuclein (chromosome 4) and MAPT (chromosome 17) [109, 114]. In addition, this work implicated variants close to LRRK2, and at two new loci on chromosome 1 (named PARK16) and chromosome 4, close to the gene bone marrow stromal cell antigen 1 (BST1). This work was shortly followed by an additional GWA study that used new and existing data to search for risk loci; this revealed a consistent association signal at SNCA and MAPT and provided preliminary evidence for an association at a new locus containing the genes cyclin G associated kinase (GAK) and diacylglycerol kinase, theta 110 kDa (DGKQ) [98]. Subsequent to this work, there have been several moderately powered GWA studies across varied populations, in general, the results of each of these have been consistent with the most prominent association signals being at the SNCA and MAPT loci, but have also provided preliminary evidence for other loci [13, 23, 115]. Of note, the major histocompatibility complex (HLA) locus was implicated in risk for PD, work that has been confirmed over several subsequent studies.
The most recent phase of GWA studies has involved the combination of existing and new datasets in more extensive meta-analyses; these have provided more persuasive “genome wide significant” results that have been substantiated through replication, both internal and independent. The first of these efforts came from the International Parkinson’s Disease Genomics Consortium in 2011; this work published the discovery and replication of 11 loci that contained genome-wide significant disease-associated variants [52]. Subsequent to this, the same consortium published a secondary analysis in a paper back to back with a study published by the direct to consumer genetic testing company 23andMe. These groups replicated each other’s signals and provided evidence of an additional seven loci [21, 51]. A further meta-analysis of the PD GWAS data from the PDGene database was also recently carried out identifying a 12 risk loci comprehensively tabulated with the risks combined from between 6 and 37 datasets [68]. Taken together these GWA studies identified loci that account for a population attributable risk (PAR) of >60 %, however this is likely an overestimation that reflects the inherent bias of PAR calculations. Perhaps a more pragmatic estimate is reflected in the observation that the 20 % of the population who carry the highest amount of risk based on these variants, are ~3 times more likely to have PD than the 20 % of the population that carry the lowest burden of risk variants. The GWA studies contrast the low heritability of PD demonstrated in twin studies [123, 134]. In dizygotic twin pairs, the hereditability estimates were between 4 % in one study [134] and 11 % in another [123]; in both studies the concordance rates were higher in monozygotic twins (11 and 15.5 %, respectively), although not achieving the concordance rate expected in a simple monogenic disease. Higher concordance was seen in disease with an age at onset less than 50 years. This type of apparent disconnect has many causes, not least of which is the relative inaccuracy inherent in heritability calculations within late-onset disorders [140].
Notably, the identification of risk loci for PD has led to the testing of at least some of these loci in another synucleinopathy, MSA. This work showed clearly that SNCA risk alleles for PD also confer a substantive risk for MSA [111]. The risk conferred by this locus in MSA appeared greater than that for PD, increasing risk ~sixfold under certain models. Larger numbers of clinically and pathologically diagnosed MSA are required to be analyzed to confirm this association. This finding was replicated in a subsequent study [3], and although this was not a wholly independent series, taken together these data argue strongly in favor for variability at SNCA as a genuine biological risk factor for this disease.
Discussion
The past 15 years has seen tremendous progress in our understanding of the genetic basis of PD. The identification of mutations in numerous genes that results in a PD or PD-like phenotype has enabled modeling of disease aimed at understanding the molecular etiology of this complex disorder. An intermediate goal of much of this discovery effort has been to link the protein products of genes associated with PD into a cohesive network or pathway. In part, this premise rests on the notion that all forms of PD share or overlap molecular pathways of pathogenesis. In this regard, the pathology of these monogenic forms of PD has been of critical importance. One of the early arguments against pursuing the genetic cause of rare familial forms of PD centered on the notion that a number of these forms of PD have more parkinsonian phenotypes and are probably not related etiologically to the typical “idiopathic” disease. The identification of α-synuclein mutations was an excellent counterpoint to this argument; showing a genetic link to a protein that when deposited is part of the pathognomic requirement for post-mortem diagnosis of disease. On this basis though, the relevance of other monogenic forms of PD to idiopathic PD, particularly some of the autosomal recessive extreme phenotypes, has been brought into question. The absence of Lewy body pathology was (and is) repeatedly used as an argument that studying these genetic lesions will tell us little about the larger disease. The logic of pathology defining etiology became even foggier with the identification of LRRK2 mutations; as these can cause disease both with or without α-synuclein pathology, even within members of the same family with the same mutation. This remarkable clinical and pathological variability from a single point mutation is unusual in human genetics and has similarities to mitochondrial disorders that have a phenotype dependent on the mutation load [4] or disorders with an element of somatic variability such as neurofibromatiosis [105, 132]. Although LRRK2 has not been shown to vary in this way, a more likely scenario is that genetic forms such as this are more susceptible to other genetic adjuncts or environmental factors. There is compelling data from epidemiology that cigarette smoking and caffeinated-coffee consumption are associated with reduced risk of developing PD and some neurotoxins are associated with increased risk [41]. In truth, while pathology and genetics have both been incredibly informative and intimately related, this argument would not be resolved until we have a clearer picture of the etiologic basis of PD and can understand where each protein fits in the network that leads to disease. This etiologic network seems to be becoming clearer with the identification of new genetic forms of disease, and the functional link between PARK2 and PINK1 has been particularly informative in this regard [12, 31, 79, 99].
Clearly genetics will continue to move forward with the identification of new genetic causes of, and contributors to, PD. It should remain a priority to carefully characterize such cases clinically and pathologically. Knowing the beginning and end of the disease process at least gives us some point of reference to fill in the intervening molecular events.
References
Abou-Sleiman PM, Healy DG, Quinn N, Lees AJ, Wood NW (2003) The role of pathogenic DJ-1 mutations in Parkinson’s disease. Ann Neurol 54:283–286. doi:10.1002/ana.10675
Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R (2004) Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med 351:1972–1977
Al-Chalabi A, Durr A, Wood NW et al (2009) Genetic variants of the alpha-synuclein gene SNCA are associated with multiple system atrophy. PLoS One 4:e7114. doi:10.1371/journal.pone.0007114
Ballana E, Govea N, de Cid R et al (2008) Detection of unrecognized low-level mtDNA heteroplasmy may explain the variable phenotypic expressivity of apparently homoplasmic mtDNA mutations. Hum Mutat 29:248–257. doi:10.1002/humu.20639
Bardien S, Lesage S, Brice A, Carr J (2011) Genetic characteristics of leucine-rich repeat kinase 2 (LRRK2) associated Parkinson’s disease. Parkinsonism Relat Disord 17:501–508. pii: S1353-8020(10)00287-7
Bonifati V, Rizzu P, van Baren MJ et al (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299:256–259. doi:10.1126/science.1077209
Bonifati V, Rohe CF, Breedveld GJ et al (2005) Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology 65:87–95
Bras JM, Guerreiro RJ, Ribeiro MH et al (2005) G2019S dardarin substitution is a common cause of Parkinson’s disease in a Portuguese cohort. Mov Disord : Official J Mov Disord Soc 20:1653–1655. doi:10.1002/mds.20682
Chartier-Harlin MC, Dachsel JC, Vilarino-Guell C et al (2011) Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet 89:398–406. doi:10.1016/j.ajhg.2011.08.009
Chartier-Harlin MC, Kachergus J, Roumier C et al (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364:1167–1169. doi:10.1016/S0140-6736(04)17103-1
Choi JM, Woo MS, Ma HI et al (2008) Analysis of PARK genes in a Korean cohort of early-onset Parkinson disease. Neurogenetics 9:263–269. doi:10.1007/s10048-008-0138-0
Clark IE, Dodson MW, Jiang C et al. (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441: 1162–1166
Consortium UKPsD, Wellcome Trust Case Control Consortium, Spencer CC et al (2011) Dissection of the genetics of Parkinson’s disease identifies an additional association 5′ of SNCA and multiple associated haplotypes at 17q21. Hum Mol Genet 20:345–353. pii: ddq469
Criscuolo C, Volpe G, De Rosa A et al (2006) PINK1 homozygous W437X mutation in a patient with apparent dominant transmission of parkinsonism. Mov Disord: Off J Mov Disord Soc 21:1265–1267. doi:10.1002/mds.20933
Dekker MC, van Swieten JC, Houwing-Duistermaat JJ et al (2003) A clinical-genetic study of Parkinson’s disease in a genetically isolated community. J Neurol 250:1056–1062. doi:10.1007/s00415-003-0151-z
Di Fonzo A, Chien HF, Socal M et al (2007) ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 68:1557–1562. pii: 68/19/1557
Di Fonzo A, Dekker MC, Montagna P et al (2009) FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 72:240–245. doi:10.1212/01.wnl.0000338144.10967.2b
Di Fonzo A, Rohe CF, Ferreira J et al (2005) A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet 365:412–415. doi:S0140-6736(05)17829-5
Dick KJ, Eckhardt M, Paisan-Ruiz C et al (2010) Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35). Hum Mutat 31:E1251–E1260. doi:10.1002/humu.21205
Djarmati A, Hagenah J, Reetz K et al (2009) ATP13A2 variants in early-onset Parkinson’s disease patients and controls. Mov Disord: Off J Mov Disord Soc 24:2104–2111. doi:10.1002/mds.22728
Do CB, Tung JY, Dorfman E et al (2011) Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet 7:e1002141. doi:10.1371/journal.pgen.1002141
Duda JE, Giasson BI, Mabon ME et al (2002) Concurrence of alpha-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol 104:7–11. doi:10.1007/s00401-002-0563-3
Edwards TL, Scott WK, Almonte C et al (2010) Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet 74:97–109
Farrer M, Chan P, Chen R et al (2001) Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol 50:293–300
Farrer M, Kachergus J, Forno L et al (2004) Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol 55:174–179. doi:10.1002/ana.10846
Farrer MJ, Hulihan MM, Kachergus JM et al (2009) DCTN1 mutations in Perry syndrome. Nat Genet 41:163–165. doi:10.1038/ng.293
Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F (2002) A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol 51:296–301. doi:10.1002/ana.10113
Fung HC, Scholz S, Matarin M et al (2006) Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol 5:911–916. pii: S1474-4422(06)70578-6
Furtado S, Payami H, Lockhart PJ et al (2004) Profile of families with parkinsonism-predominant spinocerebellar ataxia type 2 (SCA2). Mov Disord : Off J Mov Disord Soc 19:622–629. doi:10.1002/mds.20074
Gaig C, Marti MJ, Ezquerra M, Rey MJ, Cardozo A, Tolosa E (2007) G2019S LRRK2 mutation causing Parkinson’s disease without Lewy bodies. J Neurol Neurosurg Psychiatry 78:626–628. doi:jnnp.2006.107904
Geisler S, Holmstrom KM, Skujat D et al (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12:119–131. doi:10.1038/ncb2012
Giasson BI, Van Deerlin VM (2008) Mutations in LRRK2 as a cause of Parkinson’s disease. Neurosignals 16:99–105. doi:10.1159/000109764
Gilks WP, Abou-Sleiman PM, Gandhi S et al (2005) A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 365:415–416. doi:10.1016/S0140-6736(05)17830-1
Goker-Alpan O, Lopez G, Vithayathil J, Davis J, Hallett M, Sidransky E (2008) The spectrum of parkinsonian manifestations associated with glucocerebrosidase mutations. Arch Neurol 65:1353–1357
Golbe LI, Di Iorio G, Bonavita V, Miller DC, Duvoisin RC (1990) A large kindred with autosomal dominant Parkinson’s disease. Ann Neurol 27:276–282. doi:10.1002/ana.410270309
Goldwurm S, Tunesi S, Tesei S et al (2011) Kin-cohort analysis of LRRK2-G2019S penetrance in Parkinson’s disease. Mov Disord: Off J Mov Disord Soc. doi:10.1002/mds.23807
Gouider-Khouja N, Larnaout A, Amouri R et al (2003) Autosomal recessive parkinsonism linked to parkin gene in a Tunisian family. Clinical, genetic and pathological study. Parkinsonism Relat Disord 9:247–251 pii: S1353802003000166
Gwinn-Hardy K, Chen JY, Liu HC et al (2000) Spinocerebellar ataxia type 2 with parkinsonism in ethnic Chinese. Neurology 55:800–805
Gwinn-Hardy K, Singleton A, O’Suilleabhain P et al (2001) Spinocerebellar ataxia type 3 phenotypically resembling parkinson disease in a black family. Arch Neurol 58:296–299
Gwinn K, Devine MJ, Jin LW et al (2011) Clinical features, with video documentation, of the original familial Lewy body parkinsonism caused by alpha-synuclein triplication (Iowa kindred). Mov Disord : Off J Mov Disord Soc 26:2134–2136
Hamza TH, Chen H, Hill-Burns EM et al (2011) Genome-wide gene-environment study identifies glutamate receptor gene GRIN2A as a Parkinson’s disease modifier gene via interaction with coffee. PLoS Genet 7:e1002237. doi:10.1371/journal.pgen.1002237
Hasegawa K, Stoessl AJ, Yokoyama T, Kowa H, Wszolek ZK, Yagishita S (2009) Familial parkinsonism: study of original Sagamihara PARK8 (I2020T) kindred with variable clinicopathologic outcomes. Parkinsonism Relat Disord 15:300–306 pii: S1353-8020(08)00233-2
Hayashi S, Wakabayashi K, Ishikawa A et al (2000) An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov Disord: Off J Mov Disord Soc 15:884–888
Hayflick SJ, Westaway SK, Levinson B et al (2003) Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med 348:33–40. doi:10.1056/NEJMoa020817
Healy DG, Falchi M, O’Sullivan SS et al (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case–control study. Lancet Neurol 7:583–590. doi:10.1016/S1474-4422(08)70117-0
Hernandez D, Paisan Ruiz C, Crawley A et al (2005) The dardarin G 2019 S mutation is a common cause of Parkinson’s disease but not other neurodegenerative diseases. Neurosci Lett 389:137–139. pii:S0304-3940(05)00861-X
Hernandez DG, Paisan-Ruiz C, McInerney-Leo A et al (2005) Clinical and positron emission tomography of Parkinson’s disease caused by LRRK2. Ann Neurol 57:453–456. doi:10.1002/ana.20401
Hjermind LE, Johannsen LG, Blau N et al (2006) Dopa-responsive dystonia and early-onset Parkinson’s disease in a patient with GTP cyclohydrolase I deficiency? Mov Disord : Off J Mov Disord Soc 21:679–682. doi:10.1002/mds.20773
Hutton M, Lendon CL, Rizzu P et al (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705. doi:10.1038/31508
Ibanez P, Bonnet AM, Debarges B et al (2004) Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364:1169–1171. doi:10.1016/S0140-6736(04)17104-3
International Parkinson’s Disease Genomics Consortium, Wellcome Trust Case Control Consortium (2011) A two-stage meta-analysis identifies several new loci for Parkinson’s disease. PLoS Genet 7: e1002142. doi:10.1371/journal.pgen.1002142
International Parkinson Disease Genomics Consortium, Nalls MA, Plagnol V et al (2011) Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet 377:641–649. [pii:S0140-6736(10)62345-8]
Kachergus J, Mata IF, Hulihan M et al (2005) Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet 76:672–680. pii:S0002-9297(07)62878-X
Kann M, Jacobs H, Mohrmann K et al (2002) Role of parkin mutations in 111 community-based patients with early-onset parkinsonism. Ann Neurol 51:621–625. doi:10.1002/ana.10179
Kawaguchi Y, Okamoto T, Taniwaki M et al (1994) CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 8:221–228. doi:10.1038/ng1194-221
Kay DM, Moran D, Moses L et al (2007) Heterozygous parkin point mutations are as common in control subjects as in Parkinson’s patients. Ann Neurol 61:47–54. doi:10.1002/ana.21039
Khan NL, Brooks DJ, Pavese N et al (2002) Progression of nigrostriatal dysfunction in a parkin kindred: an [18F]dopa PET and clinical study. Brain 125:2248–2256
Khan NL, Jain S, Lynch JM et al (2005) Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain 128:2786–2796. doi:awh667
Kitada T, Asakawa S, Hattori N et al (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608. doi:10.1038/33416
Kruer MC, Hiken M, Gregory A et al (2011) Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration. Brain 134:947–958
Kruer MC, Paisan-Ruiz C, Boddaert N et al (2010) Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA). Ann Neurol 68:611–618. doi:10.1002/ana.22122
Kruger R, Kuhn W, Muller T et al (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 18:106–108. doi:10.1038/ng0298-106
Kruger R, Vieira-Saecker AM, Kuhn W et al (1999) Increased susceptibility to sporadic Parkinson’s disease by a certain combined alpha-synuclein/apolipoprotein E genotype. Ann Neurol 45:611–617
Kurian MA, Morgan NV, MacPherson L et al (2008) Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (PLAN). Neurology 70:1623–1629
Latourelle JC, Sun M, Lew MF et al (2008) The Gly2019Ser mutation in LRRK2 is not fully penetrant in familial Parkinson’s disease: the GenePD study. BMC Med 6:32. doi:10.1186/1741-7015-6-32
Lesage S, Durr A, Tazir M et al (2006) LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N Engl J Med 354:422–423
Lesage S, Leutenegger AL, Ibanez P et al (2005) LRRK2 haplotype analyses in European and North African families with Parkinson disease: a common founder for the G2019S mutation dating from the 13th century. Am J Hum Genet 77:330–332 pii: S0002-9297(07)62924-3
Lill CM, Roehr JT, McQueen MB et al (2012) Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: the PDGene database. PLoS Genet 8:e1002548. doi:10.1371/journal.pgen.1002548
Lincoln SJ, Maraganore DM, Lesnick TG et al (2003) Parkin variants in North American Parkinson’s disease: cases and controls. Mov Disord : Off J Mov Disord Soc 18:1306–1311. doi:10.1002/mds.10601
Lohmann E, Periquet M, Bonifati V et al (2003) How much phenotypic variation can be attributed to parkin genotype? Ann Neurol 54:176–185. doi:10.1002/ana.10613
Lu CS, Chang HC, Kuo PC et al (2004) The parkinsonian phenotype of spinocerebellar ataxia type 3 in a Taiwanese family. Parkinsonism Relat Disord 10:369–373. doi:10.1016/j.parkreldis.2004.03.009
Lynch T, Sano M, Marder KS et al (1994) Clinical characteristics of a family with chromosome 17-linked disinhibition-dementia-parkinsonism-amyotrophy complex. Neurology 44:1878–1884
Machaczka M, Rucinska M, Skotnicki AB, Jurczak W (1999) Parkinson’s syndrome preceding clinical manifestation of Gaucher’s disease. Am J Hematol 61:216–217. doi:10.1002/(SICI)1096-8652(199907)61:3<216:AID-AJH11>3.0.CO;2-E
Maraganore DM, de Andrade M, Elbaz A et al (2006) Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA 296:661–670 pii: 296/6/661
Maraganore DM, de Andrade M, Lesnick TG et al (2005) High-resolution whole-genome association study of Parkinson disease. Am J Hum Genet 77:685–693 pii: S0002-9297(07)63354-0
Marongiu R, Brancati F, Antonini A et al (2007) Whole gene deletion and splicing mutations expand the PINK1 genotypic spectrum. Hum Mutat 28:98. doi:10.1002/humu.9472
Marti-Masso JF, Ruiz-Martinez J, Bolano MJ et al (2009) Neuropathology of Parkinson’s disease with the R1441G mutation in LRRK2. Mov Disord: Off J Mov Disord Soc 24:1998–2001. doi:10.1002/mds.22677
Matilla T, McCall A, Subramony SH, Zoghbi HY (1995) Molecular and clinical correlations in spinocerebellar ataxia type 3 and Machado-Joseph disease. Ann Neurol 38:68–72. doi:10.1002/ana.410380113
Matsuda N, Sato S, Shiba K et al (2010) PINK1 stabilized by mitochondrial depolarization recruits parkin to damaged mitochondria and activates latent parkin for mitophagy. J Cell Biol 189:211–221. doi:10.1083/jcb.200910140
Mellick GD, Siebert GA, Funayama M et al (2009) Screening PARK genes for mutations in early-onset Parkinson’s disease patients from Queensland, Australia. Parkinsonism Relat Disord 15:105–109. [pii:S1353-8020(08)00108-9]
Miller DW, Hague SM, Clarimon J et al (2004) Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 62:1835–1838
Morgan NV, Westaway SK, Morton JE et al (2006) PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet 38:752–754
Mori H, Kondo T, Yokochi M et al (1998) Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology 51:890–892
Muenter MD, Forno LS, Hornykiewicz O et al (1998) Hereditary form of parkinsonism–dementia. Ann Neurol 43:768–781. doi:10.1002/ana.410430612
Neudorfer O, Giladi N, Elstein D et al (1996) Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 89:691–694
Nichols WC, Pankratz N, Hernandez D et al (2005) Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet 365:410–412. doi:10.1016/S0140-6736(05)17828-3
Ning YP, Kanai K, Tomiyama H et al (2008) PARK9-linked parkinsonism in eastern Asia: mutation detection in ATP13A2 and clinical phenotype. Neurology 70:1491–1493
Nishioka K, Ross OA, Ishii K et al (2009) Expanding the clinical phenotype of SNCA duplication carriers. Mov Disord : Off J Mov Disord Soc 24:1811–1819. doi:10.1002/mds.22682
Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C (2010) Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat 31:763–780. doi:10.1002/humu.21277
Obi T, Nishioka K, Ross OA et al (2008) Clinicopathologic study of a SNCA gene duplication patient with Parkinson disease and dementia. Neurology 70:238–241
Ozelius LJ, Senthil G, Saunders-Pullman R et al (2006) LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med 354:424–425
Paisan-Ruiz C, Bhatia KP, Li A et al (2009) Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol 65:19–23. doi:10.1002/ana.21415
Paisan-Ruiz C, Dogu O, Yilmaz A, Houlden H, Singleton A (2008) SPG11 mutations are common in familial cases of complicated hereditary spastic paraplegia. Neurology 70:1384–1389. doi:10.1212/01.wnl.0000294327.66106.3d
Paisan-Ruiz C, Guevara R, Federoff M et al (2010) Early-onset l-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov Disord: Off J Mov Disord Soc 25:1791–1800. doi:10.1002/mds.23221
Paisan-Ruiz C, Jain S, Evans EW et al (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44:595–600. doi:10.1016/S0896627304006890
Paisan-Ruiz C, Nath P, Wood NW, Singleton A, Houlden H (2008) Clinical heterogeneity and genotype-phenotype correlations in hereditary spastic paraplegia because of Spatacsin mutations (SPG11). Eur J Neurol: Off J Eur Fed Neurol Soc 15:1065–1070. doi:10.1111/j.1468-1331.2008.02247.x
Paisan-Ruiz C, Scopes G, Lee P, Houlden H (2009) Homozygosity mapping through whole genome analysis identifies a COL18A1 mutation in an Indian family presenting with an autosomal recessive neurological disorder. Am J Med Genet Part B, Neuropsychiatric Genet: Off Publ Int Soc Psychiatric Genet 150B:993–997. doi:10.1002/ajmg.b.30929
Pankratz N, Wilk JB, Latourelle JC et al (2009) Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet 124:593–605. doi:10.1007/s00439-008-0582-9
Park J, Lee SB, Lee S et al (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441:1157–1161
Perry TL, Bratty PJ, Hansen S, Kennedy J, Urquhart N, Dolman CL (1975) Hereditary mental depression and Parkinsonism with taurine deficiency. Arch Neurol 32:108–113
Polymeropoulos MH, Higgins JJ, Golbe LI et al (1996) Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science 274:1197–1199
Polymeropoulos MH, Lavedan C, Leroy E et al (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047
Pramstaller PP, Schlossmacher MG, Jacques TS et al (2005) Lewy body Parkinson’s disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol 58:411–422. doi:10.1002/ana.20587
Ramirez A, Heimbach A, Grundemann J et al (2006) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 38:1184–1191
Rieley MB, Stevenson DA, Viskochil DH, Tinkle BT, Martin LJ, Schorry EK (2011) Variable expression of neurofibromatosis 1 in monozygotic twins. Am J Med Genet Part A 155A:478–485. doi:10.1002/ajmg.a.33851
Ross OA, Toft M, Whittle AJ et al (2006) Lrrk2 and Lewy body disease. Ann Neurol 59:388–393. doi:10.1002/ana.20731
Samaranch L, Lorenzo-Betancor O, Arbelo JM et al (2010) PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 133:1128–1142
Sasaki S, Shirata A, Yamane K, Iwata M (2004) Parkin-positive autosomal recessive juvenile Parkinsonism with alpha-synuclein-positive inclusions. Neurology 63:678–682
Satake W, Nakabayashi Y, Mizuta I et al (2009) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 41:1303–1307
Schneider SA, Paisan-Ruiz C, Quinn NP et al (2010) ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov Disord : Off J Mov Disord Soc 25:979–984. doi:10.1002/mds.22947
Scholz SW, Houlden H, Schulte C et al (2009) SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol 65:610–614. doi:10.1002/ana.21685
Shojaee S, Sina F, Banihosseini SS et al (2008) Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am J Hum Genet 82:1375–1384. [pii:S0002-9297(08)00314-5]
Sidransky E, Nalls MA, Aasly JO et al (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361:1651–1661
Simon-Sanchez J, Schulte C, Bras JM et al (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41:1308–1312
Simon-Sanchez J, van Hilten JJ, van de Warrenburg B et al (2011) Genome-wide association study confirms extant PD risk loci among the Dutch. Eur J Hum Genet 19:655–661
Singleton A, Gwinn-Hardy K (2004) Parkinson’s disease and dementia with Lewy bodies: a difference in dose? Lancet 364:1105–1107. doi:10.1016/S0140-6736(04)17117-1
Singleton AB, Farrer M, Johnson J et al (2003) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302:841. doi:10.1126/science.1090278
Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M (1998) Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 251:205–208
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 95:6469–6473
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840. doi:10.1038/42166
Spira PJ, Sharpe DM, Halliday G, Cavanagh J, Nicholson GA (2001) Clinical and pathological features of a Parkinsonian syndrome in a family with an Ala53Thr alpha-synuclein mutation. Ann Neurol 49:313–319
Subramony SH, Hernandez D, Adam A et al (2002) Ethnic differences in the expression of neurodegenerative disease: Machado-Joseph disease in Africans and Caucasians. Mov Disord: Off J Mov Disord Soc 17:1068–1071. doi:10.1002/mds.10241
Tanner CM, Ottman R, Goldman SM et al (1999) Parkinson disease in twins: an etiologic study. JAMA 281:341–346
Tayebi N, Callahan M, Madike V et al (2001) Gaucher disease and parkinsonism: a phenotypic and genotypic characterization. Mol Genet Metab 73:313–321. doi:10.1006/mgme.2001.3201
Tayebi N, Walker J, Stubblefield B et al (2003) Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol Genet Metab 79:104–109. doi:10.1016/S1096719203000714
Thomas M, Hayflick SJ, Jankovic J (2004) Clinical heterogeneity of neurodegeneration with brain iron accumulation (Hallervorden-Spatz syndrome) and pantothenate kinase-associated neurodegeneration. Mov Disord: Off J Mov Disord Soc 19:36–42. doi:10.1002/mds.10650
Valente EM, Abou-Sleiman PM, Caputo V et al (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304:1158–1160. doi:10.1126/science.1096284
van de Warrenburg BP, Lammens M, Lucking CB et al (2001) Clinical and pathologic abnormalities in a family with parkinsonism and parkin gene mutations. Neurology 56:555–557
Vilarino-Guell C, Wider C, Ross OA et al (2011) VPS35 mutations in Parkinson disease. Am J Hum Genet 89:162–167 pii:S0002-9297(11)00242-4
Weng YH, Chou YH, Wu WS et al (2007) PINK1 mutation in Taiwanese early-onset parkinsonism: clinical, genetic, and dopamine transporter studies. J Neurol 254:1347–1355. doi:10.1007/s00415-007-0534-7
Wider C, Dickson DW, Wszolek ZK (2010) Leucine-rich repeat kinase 2 gene-associated disease: redefining genotype-phenotype correlation. Neuro-degenerative diseases 7:175–179. doi:10.1159/000289232
Wiest V, Eisenbarth I, Schmegner C, Krone W, Assum G (2003) Somatic NF1 mutation spectra in a family with neurofibromatosis type 1: toward a theory of genetic modifiers. Hum Mutat 22:423–427. doi:10.1002/humu.10272
Wilhelmsen KC, Lynch T, Pavlou E, Higgins M, Nygaard TG (1994) Localization of disinhibition–dementia–parkinsonism–amyotrophy complex to 17q21-22. Am J Hum Genet 55:1159–1165
Wirdefeldt K, Gatz M, Reynolds CA, Prescott CA, Pedersen NL (2011) Heritability of Parkinson disease in Swedish twins: a longitudinal study. Neurobiol Aging 32(1923):e1921–e1928. doi:10.1016/j.neurobiolaging.2011.02.017
Wszolek ZK, Pfeiffer RF, Bhatt MH et al (1992) Rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Ann Neurol 32:312–320. doi:10.1002/ana.410320303
Zarranz JJ, Alegre J, Gomez-Esteban JC et al (2004) The new mutation, E46 K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55:164–173. doi:10.1002/ana.10795
Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ (2001) A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet 28:345–349. doi:10.1038/ng572
Zimprich A, Benet-Pages A, Struhal W et al (2011) A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 89:168–175. [pii:S0002-9297(11)00261-8]
Zimprich A, Biskup S, Leitner P et al (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44:601–607. [pii:S0896627304007202]
Zuk O, Hechter E, Sunyaev SR, Lander ES (2012) The mystery of missing heritability: genetic interactions create phantom heritability. Proc Natl Acad Sci USA. doi:10.1073/pnas.1119675109
Acknowledgments
This work was supported in part by the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Department of Health and Human Services; projects number Z01 AG000957-08 and AG000958-08. We are also grateful to the Medical Research Council (MRC), NORD, the Parkinson’s Disease Foundation (PDF), the DMRF and The Wellcome Trust.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Houlden, H., Singleton, A.B. The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol 124, 325–338 (2012). https://doi.org/10.1007/s00401-012-1013-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-012-1013-5