
Abstract
We investigated 998 serial Japanese forensic autopsy cases (0–101 years old, mean age 61.7 ± 21.9), with no case selection, using immunohistochemistry to detect cases with progressive supranuclear palsy (PSP). Twenty-nine cases (mean age 82.3 ± 7.2 years, 11 males, 18 females) fulfilled the National Institute of Neuronal Disorders and Stroke (NINDS)-PSP pathological criteria (2.9% of all cases, 4.6% of cases over 60). All had neuronal and glial inclusions in the basal ganglia and brainstem. However, 13 cases had low tau pathology and were categorized as atypical PSP. In addition to PSP pathology, multiple types of astrocytic inclusions and comorbid proteinopathies, particularly a high prevalence of argyrophilic grain disease, were found. All cases had not been diagnosed with PSP and had preserved daily functioning prior to death. However, 14 (48.3%), 11 (37.9%), and 16 (55.2%) cases showed signs of dementia, depressive state, and gait disturbance, respectively. Sixteen accidental death cases (55.2%), including from falls and getting lost, and 11 suicide cases (37.9%) appear to have a relationship with incipient PSP pathology. Cluster analysis using the distribution and amount of 4-repeat-tau pathology classified the cases into three subgroups: Group 1 (10 cases) had typical PSP pathology and seven cases (70.0%) had dementia as the most frequent symptom; Group 2 (7 cases) had significantly higher frequency of gait disorder (6 cases, 85.7%), and less neocortical tau pathology than Group 1; Group 3 (12 cases) had relatively mild PSP pathology and high argyrophilic grain burdens. Granular-shaped astrocytes were the dominant astrocytic inclusion in all cases. We conclude that in forensic cases incipient PSP occurs with a higher prevalence than expected. If these findings can be extrapolated to other population-based cohorts, PSP may be more common than previously thought.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Progressive supranuclear palsy (PSP) is a late-onset neurodegenerative disease with unknown etiology. Supranuclear gaze palsy, progressive axial rigidity, pseudobulbar palsy, and mild dementia were the initial symptoms of the disease described by Steele et al. [65]. Since then, accumulated clinicopathological evidence has shown that the clinical presentation of PSP varies between patients [16, 62]. Recent studies have proposed the existence of clinicopathological variants of PSP: with Parkinsonism (PSP-P) [72], with pure akinesia with gait freezing (PSP-PAGF) [14, 73], with corticobasal syndrome (PSP-CBS) [40, 70], and with cerebellar ataxia (PSP-C) [33].
The PSP-affected brain appears grossly normal, or mildly atrophic. The substantia nigra shows a loss of pigmentation in many cases. The most important microscopic finding of PSP is the accumulation of phosphorylated 4-repeat-tau (4R) as neurofibrillary tangles (NFTs) and neuronal threads (NTs). The globose type NFTs, cortical NFTs with flame-, coiled- or curvilinear-forms, and pretangle neurons are frequently found [16, 17]. The most affected areas are the brainstem and basal ganglia, and in particular the globus pallidus, subthalamic nuclei, substantia nigra, and pontine nucleus. The cerebral cortex, especially the frontal lobe, may also be involved in some cases [13, 16]. The other prominent pathological feature of PSP is tufted astrocytes (TsA) showing 4R- and Gallyas-Braak-positive fine processes [11, 75]. The presence of TsA is important for distinguishing between PSP and corticobasal degeneration (CBD), another 4R-positive neurodegenerative disease [23, 36].
Some studies have revealed that other forms of astrocytes appear in 4-repeat-tauopathies. Granular astrocytes (GsA), which were initially reported as bush-like astrocytes by Botez et al. [6], are frequently found in argyrophilic grain disease (AGD), and and GsA has been considered as one of the distinct morphological form for detecting aging-related tau astrogliopathy (ARTAG) that is a morphological spectrum of astroglial pathology detected by tau immunohistochemistry, especially with phosphorylation-dependent and 4R isoform-specific antibodies [37]. Another form, spiny-shaped astrocytes (SsA), have a TsA-like appearance, but with broader and shorter processes [36]. It is possible that the different pathological appearances of abnormal astrocytes may be useful for differentiating between different 4-repeat-tauopathies.
The prevalence rate of clinical PSP is reported to be 5.8–10 per 100,000 in Japan [34, 59], which may be slightly higher than in Western countries (1.0–6.5 per 100,000) [7, 12, 24, 56, 64, 71]. However, while some incipient PSP cases have been reported [22, 38], the detailed clinicopathological features of such cases have not yet been fully established. We recently detected preclinical, or early clinical, CBD cases by immunohistochemistry, without case selection, in serial forensic autopsy cases [58]. Such analysis is useful for examining the early clinicopathological features and prevalence of disease. Here we examined serial forensic autopsy cases, applying immunohistochemistry to detect incipient PSP and reveal the clinicopathological features of such cases.
Materials and methods
Subjects
We reviewed the archives of all 1239 cases of medicolegal autopsies from our department between 2007 and 2014. Of these cases, brain specimens from 998 people (0–101 years old, mean age 61.7 ± 21.9) without severe injury or severe postmortem degeneration, such as liquefaction preventing histological analysis, were examined. A total of 215 cases had a natural cause of death, and 422 cases suffered accidental traumatic death, such as a fall, traffic accident, burning, drowning, hypothermia, and the like. Suicide or homicide accounted for 332 cases, and there were 29 cases with undetermined causes of death. There were 626 cases (350 male and 276 female) and 121 cases (54 male and 67 female) from patients over 60 and 85 years of age, respectively. The clinical histories of patients were obtained from the family and the records of police examinations. When there was a history of a hospital visit, the medical records were provided by the primary physician.
Tissue sampling and pathological assessment
All brains were fixed in 20% buffered formalin for at least 2 weeks prior to sampling. Specimens that is routinely sampled in our department is shown in Fig. 1. All sections were cut and stained with Luxol fast blue-hematoxylin eosin. Gallyas-Braak and Holzer stainings were also performed [58].

Low power view of one histological specimen (Luxol fast blue/hematoxylin eosin). a Frontal lobe. b Nucleus accumbens. c Motor cortex. d Anterior temporal lobe. e Basal ganglia and amygdala. f Anterior hippocampus and thalamus. g Posterior hippocampus and temporal lobe. h, i Posterior lobe. j Cerebellar vermis. k Dentate nucleus. l Mid brain. m Pons. n Medulla oblongata
Immunohistochemistry was routinely performed on the frontal lobe, temporal lobes, basal ganglia, and midbrain of all cases to detect phosphorylated tau (clone AT8, 1:1000; Endogen, Woburn, MA, USA), phosphorylated α-synuclein (clone LB508, 1:500; Zymed, San Francisco, CA, USA), TAR DNA binding protein-43 (TDP-43, 1:5000; Protein Tech group, Chicago, IL, USA), Glial fibrillary acidic protein (GFAP, clone ZCG 29, 1:1000, Nichirei Tokyo, Japan), and β-amyloid (clone 6F/3D, 1:50; Novocastra Vector Labs, Burlingame, CA, USA).
Antibody bindings were detected using a biotin-streptavidin detection system (Nichilei, Tokyo Japan) using 3.3′-diaminobenzidine as the chromogenic substrate. If positive findings were detected in the preliminary immunohistochemistry, an additional staining procedure was performed on subsequent sections. Staining for 3-repeat-tau (3R) and 4R (Merck-Millipore, Billerica, MA, USA) was also performed in cases positive for AT8. In 4-repeat-tauopathy cases, thioflavin-S staining was also performed to differentiate 4R specific NFTs from Alzheimer’s disease (AD) related NFTs [29].
We used the National Institute of Neuronal Disorders and Stroke (NINDS) criteria to define the neuropathological diagnosis of PSP as typical or atypical PSP [25, 43].
The type of astrocytic inclusion was assessed in detail with Gallyas-Braak silver staining, AT8, 3R, and 4R immunostaining. We classified four types of AT8, 4R-positive, and 3R-negative astrocytic inclusions (Fig. 2). TsA was defined by a radial arrangement of thin, long, branching accumulated tau protein from the cytoplasm to the proximal processes of astrocytes that stained well with Gallyas-Braak [75]. The definition of GsA was that its processes stained like beaded fine granules with tau immunostaining, and did not stain with Gallyas-Braak [37]. We evaluated GsA and bush-like astrocytes, which were initially reported by Botez et al., as synonymous because the pathological features of each are considered to be almost identical [6]. Spiny-shaped astrocytes (SsA), which have been reported in atypical PSP cases [36], have broad and spiny stained processes that stain well with Gallyas-Braak, with less staining in the cell body. We also assessed thorn shaped astrocytes (TrA), which along with GsA, are one of the diagnostic hallmarks of ARTAG. TrA have more voluminous perinuclear cytoplasms and their processes are often thicker and shorter [37].

Pathological appearances of three types of astrocytic inclusion. Photomicrograph of AT8 a–c and Gallyas-Braak d–f. a, d Tufted astrocyte. b, e Granular astrocyte. c, f Spiny astrocyte. Scale bar 50 μm
The pathological staging of NFTs was evaluated according to the modified Braak stages of NFT burden using AT8, Gallyas-Braak, and Thioflavin-S [8]. The density of neuritic plaques was evaluated in accordance with the Consortium to Establish a Registry for Alzheimer’s disease (CERAD) criteria using Thioflavin-S and β-amyloid (Aβ) immunostaining [54]. The extent of senile plaques in the brain was evaluated following the criteria of Thal et al. [67]. Based on these results, the level of AD neuropathological change was divided into four categories (Not, Low, Intermediate, High) following the National Institute on Aging-Alzheimer’s Association (NIA-AA) guidelines [27]. The pathology of Lewy body disease (LBD) was assessed according to the Third Consensus Guidelines for Dementia with Lewy body and Braak stages in the development of Parkinson’s disease (PD)-related pathology using α-synuclein immunohistochemistry [9, 15, 52]. The pathological staging of argyrophilic grains was assessed following the AGD system proposed by Saito et al. [63]. The pathological type of TDP-43 proteinopathy was assessed following the stages of AD [32] and the classification system for frontotemporal lobar degeneration (FTLD)-TDP pathology [46]. The pathology of ARTAG and chronic traumatic encephalopathy (CTE), which are both associated with a distinctive pattern of progressive neuronal and glial tau pathology, was assessed following the criteria. The criteria were: ARTAG, the presence of either or both TrA and GSA in the subpial, subependymal, gray matter, white matter, or perivascular region [37]; CTE, the presence of AT8 positive neurons, astrocytes, and cell processes around small vessels in an irregular pattern at the depths of the cortical sulci [49].
Semiquantitative assessment of neuronal loss, and tau-positive neuronal and glial inclusions
The degree of neuronal loss was assessed using a 4-point scale (0, absent; 1, mild; 2, moderate; 3, severe) in the superior frontal gyrus, precentral gyrus, middle temporal gyrus, globus pallidus, putamen, thalamus, subthalamic nucleus, amygdala, CA1, red nucleus, substantia nigra, pontine nuclei, inferior olivary nucleus, and cerebellar dentate nucleus. The degree of neuronal loss in the lesions of PSP cases was compared with 17 age-matched autopsy cases from our department. Control cases were randomly selected from over 70 subjects with both CERAD 0 or A and Braak NFT stage 0–2 and without any of Lewy body and TDP-43 pathology, cerebral hemorrhage, or territorial large cerebral infarction. The density of tau pathology including NFTs, NTs, and glial cell inclusions in PSP cases was also assessed using a 4-point scale (0, absent; 1, sparse; 2, moderate; 3, severe) (Fig. 3).

Grading scale for tau pathology. The occurrence of tau pathology was assessed using a 4-point scale: a Absent (grade 0), b mild (grade 1), c moderate (grade 2), d severe (grade 3). Subthalamic nucleus immunostaining with AT8. Scale bar 200 μm
In same region, the number of TAs, GsA, and SsA were also semiquantitatively evaluated (0, absent; 1, 1 per 250 × field; 2, 2–9 per 250 × field; 3, >10 per 250 × field).
Statistical analysis
Data were analyzed using JMP pro (Version 11.2.0; SAS Institute Inc. Cary, NC, USA) and IBM SPSS Statistics (Version 23; SPSS Inc, Chicago, IL, USA), and the significance level was set at 0.05. Fisher’s exact test was used for categorical variables (sex, symptom presence, and pathological findings). Continuous variables (age at death and brain weight) and ordinal variables (pathological scales and densities) were compared using the Mann-Whitney U test (comparison of two groups) or Kruskal–Wallis test with post hoc test (comparison of three or more groups). Additionally, we performed Ward’s hierarchical cluster analysis using the distribution pattern and amount of 4R pathology in PSP specific regions (frontal cortex, motor cortex, globus pallidus, subthalamic nucleus, red nucleus, substantia nigra, pontine nucleus, inferior olivary nucleus, cerebellar dentate nucleus, and the white matter of the frontal lobe and motor cortex) to classify the pathologically defined PSP cases into subgroups.
Results
Clinical profiles and demographics
A total of 29 cases (2.9%, 11 males, 18 females) fulfilled the pathological criteria for PSP. The demographics of these cases are summarized in Table 1, with a summary of their clinicopathological features in Table 2.
The mean age was 82.3 ± 7.2 years (range 64–94). The prevalence rate in cases over 60 years old was 4.6% (29/626) and 9.9% in cases over 85 years old (12/121). No case diagnosed as having clinical PSP was bedridden prior to their death. Prior to their deaths five cases (17.2%) had been diagnosed with Lewy body-related disorders and had responded to levodopa: three had Parkinson’s disease and the other two had Dementia with Lewy bodies. The other cases had not been examined by a neurologist.
Fourteen of the 29 cases (48.3%) showed signs of dementia, 11 cases (37.9%) had signs of depressive states, and 16 cases (55.2%) had gait disturbances. The number of accidental death cases was 16 (55.2%) and 12 these cases died of injuries relating to falls. The number of suicide cases was 11 (37.9%) and two cases (6.9%) had a cause of death of ‘other’.
Neuropathological findings
The mean brain weight was 1263.9 ± 128.5 g (range 1058–1620 g). Case 20, with a previous history of a traffic accident, had a scar from a cerebral contusion on both frontal bases. No case had severe brain atrophy except for Case 19, which had severe atrophy in the inner part of the temporal lobe. Other 28 cases had mild or moderate atrophy in the frontal and temporal lobes. The number of typical PSP cases according to NINDS-PSP criteria was 17 and the other 12 cases had atypical PSP. All of these atypical cases did not fulfill the first criterion of typical PSP, which is “two or more neurons with neurofibrillary tangles or neuropil threads (high density) must be found in the same field in at least three of following areas: pallidum, subthalamic nucleus, substantia nigra, and pons”. However, all the atypical PSP cases in this study had low amounts of exclusively 4R-positive neuronal thread and astrocytic inclusions in these four areas. The distribution of tau pathology in these four areas in atypical PSP cases is shown in Table 3. The globus pallidus and pons were frequent “negative” areas in atypical PSP cases.
In addition to NFTs, NTs, oligodendroglial coiled bodies, and TsA were found in all 29 cases (Fig. 4). GsA and SsA were also found in 27 and 26 cases, respectively. The severity of neuronal loss, amount of tau pathology, and the topographical frequency of the three types of astrocytic inclusions are shown in Supplemental Table 1. Neuronal loss tended to be frequent in the substantia nigra, insula, and amygdala. Besides these regions, mild but significant neuronal loss was found in many other regions, compared with control subjects (Supplemental Table 1). The total amount of tau pathology was higher in the subthalamus, substantia nigra, motor cortex, and amygdala. TsA were frequently observed in the motor cortex (75.9%), thalamus (55.2%), and red nucleus (50.0%). GsA were frequently observed in the limbic cortex, such as in the amygdala (81.5%), and in the temporal cortex (76.9%), putamen and frontal cortex (both 70.4%), and insula (65.4%). SsA were frequently observed in the putamen (80.8%) and globus pallidus (65.4%). Of the three types of astrocytes, 16 cases were TsA dominant and 11 and 2 cases were GsA and SsA dominant, respectively.

PSP and comorbid pathologies. a Neurofibrillary tangle (NFT) of Globose type (4-Repeat-tau immunostaining). b Cortical NFT with curvilinear form (4-Repeat-tau immunostaining). c NFT of pretangle form (4-Repeat-tau immunostaining). d Oligodendroglial coiled body (4-Repeat-tau immunostaining) e Lewy pathology in substantia nigra of Case 8 (α-Synuclein immunostaining). f Argyrophilic grains (Gallyas-Braak staining). Scale bars indicate 50 μm (a–d) and 200 μm (e, f)
Comorbid AD pathology (Fig. 4) was limited, with 14 cases (48.3%) assessed as having Low pathology according to NIA-AA guidelines and only one High case (Case 19, 3.4%) was seen. Case 19 had both 3R and 4R positive NFT and neuronal threads and 4R positive NFT and neuronal threads in the globus pallidus and substantia nigra, AGD was seen in 21 cases (72.4%), and Lewy body pathology was seen in 8 cases (27.6%). Of these latter cases, two (6.9%) were assessed as being in PD Braak stage 6, and both cases had been diagnosed with Lewy body-related disease prior to their death. The stage of the Lewy pathology in these cases could be consistently classified using the criteria recently proposed by Del Tredict and Braak [15]. TDP-43 proteinopathy was seen in 12 cases (41.4%), and the subtype of TDP-43 pathology was type A in all cases. Four cases (13.8%) did not show any obvious comorbid pathology.
Twenty-eight cases fulfilled the criteria for ARTAG, and TrA were found in 21 cases. The case being consistent with CTE was not found in present PSP cases.
Subgroups classified with cluster analysis
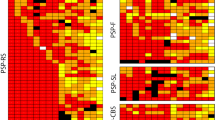
We classified all 29 cases into three groups using cluster analysis of the distribution pattern of tau pathology in PSP specific regions (Fig. 5). The severity and the amount of tau pathology in various areas are shown in Supplemental Table 2, and the pathological features and clinical information of each group are shown in Table 4.

Cluster analysis using the distribution of tau pathology in PSP specific regions. Dendrogram of cluster analysis (upper panel) and the distribution and severity of tau pathology in each area (lower panel)
In Group 1 (10 cases), all cases fulfilled the NINDS criteria for typical PSP. Pathologically, both neuronal loss and tau pathology found in almost all favorite sites. TsA was the dominant astrocytic pathology in all cases. A high prevalence of clinical dementia may be a feature of Group 1. Group 2 (7 cases) had an almost identical burden of tau pathology to Group 1, other than in the neocortical and limbic areas. A higher prevalence of gait disorder (6 cases, 85.7%) and low prevalence of depression (1 case, 14.3%) may be a feature of Group 2 when compared with the other groups. Group 3 (12 cases) had a low prevalence of typical PSP. Significant low intensity tau pathology and a low prevalence of neuronal loss were found in many of the brain areas, other than frontal cortex and limbic area in this group. The number of GsA dominant cases was significantly higher than in Group 1.
Discussion
This study investigated a large autopsy cohort study targeting PSP by examining serial Japanese forensic autopsies. With few exceptions, many forensic autopsies in Japan are performed under the Criminal Code. They are typically performed when the cause of death is suspected to be unnatural or is possibly linked to a crime [58]. We assume that many unusual deaths, as examined in present study, have not been investigated in previous clinical and/or pathological studies relating to PSP. No individual was in a bedridden state, and all had reasonable functioning in daily life, at the time of death. Although our forensic autopsy case series may not accurately represent the general Japanese population, we believe that the results of present study may be useful for exploring the various pathological and clinical features of incipient PSP. This may lead to the prevention of unusual deaths including accidental falls or suicide in older individuals.
Pathological features of the present cases
In this study, immunohistochemistry without case selection detected many incipient PSP cases. A relatively high prevalence of atypical PSP was one of the features of the present study. These cases were clinically heterogeneous and had 3R-enriched tau protein deposits, as found in AD [16]. However, some incipient cases might be identified as typical PSP with a certain level of difficulty, due to the low burden of 4R in the designated area such as pallidum, subthalamic nucleus, substantia nigra, and pons. Immunohistochemistry for 3R and 4R in this study may be sufficient to avoid the inclusion of AD cases. Therefore, we assume that the incomplete distribution and low burden of tau pathology is a feature of incipient PSP rather than atypical PSP in this study. The high prevalence of tau pathology in the substantia nigra and subthalamic nucleus in the atypical PSP cases suggests that these two areas might be the initial sites of tau pathology in the incipient stage of PSP. Our semiquantitative analysis of neuronal loss may have a certain level of unavoidable error. This type of neuronal loss estimate may give inaccurate high values due to the possible shrinkage of neuronal cell bodies or nuclei, making it less likely for cells or nuclei to be included in the plane of the section. Also, the number of cells observed is influenced by variations in section thickness, and, as it is difficult to distinguish small neurons from glia, there may be errors resulting from this. Our comparisons with age-matched healthier controls may show that the present cases, including many incipient ones, already had more mild neuronal loss than “near healthy” cases. A recent quantitative analysis of incidental CBD cases showed that astrocytic rather than neuronal lesions predominate the earliest CBD pathological findings [42]. From that perspective, astrogliopathy proceeding to advanced neuronal loss, as found in this study, may represent early pathology of incipient PSP, like that in early CBD.
Comorbid pathologies were observed in all cases although the type and amount varied. We observed one case with high-staged pathology according to NIA-AA guidelines, and two cases showed advanced Lewy body disease with a clinical diagnosis of PD. These three cases may reflect the clinical appearance of the cases more than the PSP-related pathology. However, a high prevalence of AGD as associated pathology was one of the features of this study. It has been well documented that PSP cases often have various quantities of argyrophilic grains, and a recent study demonstrated that eight out of 30 PSP cases (26.7%) had AGD [66]. The large number of elderly cases in this study may be related to the high frequency of AGD.
GsA and SsA were also found in all cases examined. The distribution of three kind of astrocytic inclusion was different (Supplemental Table 2). The frequent appearance of TsA in the motor cortex was in accordance with previous reports [1, 18, 30]. Conversely, the appearing prevalence of TsA itself in each case varied. We assume that the amount of TsA in the present cases tended to be fewer due to a lower total burden of tau pathology than seen in advanced PSP cases. Caution is required when deciding whether TsA is an absolute requirement for diagnosing incipient PSP, and an additional examination of a larger number of incipient PSP cases is required in the future.
GsA may be a helpful biomarker for the diagnosis of AGD due to its high prevalence in AGD and almost identical topographical distribution with argyrophilic grains [6]. However, as there are some cases with GsA but without argyrophilic grains in both this and a previous study [39], so we cannot use GsA as a specific pathology comorbid with AGD. SsA has a unique distribution with strong pathology in the putamen and motor cortex. This distribution may be consistent with that of TsA in this study. The high prevalence of SsA and its common distribution with TsA in the present incipient cases suggests that SsA and TsA may have a common etiology and diagnostic significance for PSP.
A recent study showed that the prevalence of ARTAG in the brain increases with age, being around 31% in PD, 25.0% in dementia with Lewy bodies, and 41.7% in AD in the degenerating basal forebrain [45]. Whereas the high prevalence of ARTAG in PSP cases in this study may be notable, it is strongly associated with both higher age and a high prevalence of GsA, even though TrA was also found in 21 of the 29 cases. Additional case studies are essential for exploring the significance of the association between PSP, AGD, and ARTAG. On the other hand, CTE is a neurodegenerative sequela of repeated traumatic brain injury [50]. No CTE pathology were found in these asymptomatic and early PSP cases in the present series, while comorbid neurodegenerative disorders have been identified in CTE cases [51] and 12 out of the 50 (24%) end-stage PSP cases had histological evidence of CTE as reported by Ling et al. [41]. One possible explanation is that the early CTE changes in some of the end-stage PSP cases were caused by frequent falls following the onset of clinical symptoms of PSP while frequent falls were not a feature in the asymptomatic and early PSP cases in the present series. Another possibility is that ARTAG with only glial involvement is the earliest feature of CTE, supporting the hypothesis that these two entities share a common etiological pathway [45].
Clinical features of the present PSP cases
A higher PSP prevalence rate (2.9% of all subjects and 4.6% of subjects over 60) than many previous clinical studies targeting Japanese and Western populations is a highlight of this study. The prevalence in subjects over 60 in this study was higher than that observed in a study that prospectively followed 119 normal cognitive and movement disorder participants (mean age 83.5 years, range 67–99) and detected pathologically proven PSP with a prevalence of 3.0% [20]. Due to the certain level of intrinsic bias in this study, we cannot conclude whether the prevalence of PSP in Japan is higher than in other territories. However, some analysis targeting for the MAPT gene in PSP patients may support the possibility of a high prevalence of PSP in the Japanese population. A high frequency of MAPT gene H1 homozygosity, which is strongly associated with increased risk of developing PSP and CBD [2], is common in the Japanese population [21]. Ling et al. recently reported three cases of CBD in the earliest clinical stage and, interestingly, found that the H2 allele may be distinctive protective factor against the progression of CBD [42]. The other possible factor associated with the high prevalence of PSP in the Japanese population is a low frequency of rs8070723-G. rs8070723-A is a major allele but may possibly increase the risk of PSP, whereas individuals with rs8070723-G may have a lower risk of PSP [26]. In our investigation of the Japanese population database of 104 individuals in the 1000 Genomes Browser (https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/) and the Human Genetic Variation Database (HGVD), which includes 1208 Japanese individuals (http://www.hgvd.genome.med.kyoto-u.ac.jp/index.html), the allele frequency of rs8070723-G was much lower in Japanese individuals (0.48% and not detected, respectively).
Other than differences in study populations, discrepancies in the prevalence of PSP between clinical and autopsy studies may be radically underestimated in clinical studies. The clinical identification of PSP depends largely on the presence of the characteristic vertical gaze palsy, but this can be absent in the majority of autopsy-confirmed PSP cases [4, 20]. The high prevalence of PSP in the present study may be the result of the coincidental detection of a considerable number of late-onset incipient cases who died before progressing to the advanced clinical stage. In addition, the mean age in this study (82.3 ± 7.2 years) was higher than the average age at death for PSP samples in the brain bank at the Mayo Clinic and a recent multicenter study of 100 definite cases, which are 75 ± 8 and 65.2 ± 0.9 years, respectively [18, 62]. Conversely, the mean age of the incipient PSP cases in other previous studies tended to be high at 95.1 ± 10.1 and 89.1 ± 5.6 years [20, 22]. Older incipient PSP patients may be considered to have signs of advanced aging or might be overlooked and immunohistochemistry not conducted even if an autopsy was performed.
Litvan et al. reported that patients’ mean time interval between onset and first clinical visit was 3.7 years (range 1–11 years). Most of their patients showed extrapyramidal signs, such as gait disorder and postural instability, bilateral bradykinesia, and history of fall [44]. Although many of our cases did not undergo detailed neurological and psychological examinations before their deaths, many of the present PSP cases already showed some sort of clinical signs possibly associated with PSP lesions. All PSP cases, other than two who died of sudden cardiac death and medical accident, died from unnatural causes such as falls associated with gait disorder, or hypothermia associated with disorientation, before progressing to the advanced stage of PSP. All cases with gait disorder, other than the cases diagnosed with PD, were considered to have locomotor disability when alive, and many cases with mild cognitive impairment were believed to be aging phenomenon. While the pathological PSP cases in which dementia was the only clinical symptom are a few [22, 60], cases in which dementia is an initial clinical symptom of PSP have been reported [19, 61].
A high prevalence of suicide is one of the important findings of this study. Psychiatric symptoms are one of the significant features of PSP, with a prevalence of approximately 20–59.1% [31, 47, 55, 60]. Our recent study showed that post-stroke depression, combined with PSP or AGD, conferred a significant risk of suicide [57]. The present study showed that incipient PSP pathology might be responsible for suicidal attempts related to a depressive state in elderly patients. This is a significant result of this study. Bloise et al. revealed the 8 of 15 PSP cases showed depression that met fourth edition of Diagnostic and Statistical Manual of Mental Disorders (DSM-IV), and they additionally showed the depression occurred more frequently in PSP patients than in controls individual [5]. Also, atypical cases of PSP in which psychiatric symptoms appeared in the early stage have also been reported [5, 35]. Although the mechanism of psychiatric disorders in PSP has been proposed to be dysfunction of the orbitofrontal circuits, with preferential involvement of the mesofrontal targets of striatal projections [53], this circuit is usually associated with symptoms other than depression, which are behavioral disturbance, personality changes, irritability, and apathy [10]. A high prevalence of combined PSP and AGD, which has been considered to be associated with prominent psychiatric symptoms such as aggression, irritability, depression, psychosis, and mild dementia [28, 68, 69], might additionally increase the risk of suicide attempts. We cannot yet conclude whether PSP is an independent risk factor for suicide. Additional examination of suicide cases with other neurodegenerative disorders, such as AD or PD, or a larger number of control cases are essential for exploring the significance of PSP and/or AGD as a risk factor for suicide.
Pathological subclassification of incipient PSP cases
Cluster analysis using the distribution and degree of tau pathology showed that incipient PSP cases can be classified into three groups. However, differences in the pathological or clinical parameters between the groups were not always statistically significant. We propose two reasons for this: one is that the burden of tau pathology was insufficient to reveal differences between the groups, and the other is that comorbid pathology may influence the clinical presentation due to the less advanced PSP pathology.
Group 1 contained the cases with typical and classical PSP pathology, many of which showed dementia (70.0%) as the main symptom. Bigio et al. reported that PSP cases with dementia had more severe pathology in the cortex than PSP cases without dementia [3]. Conversely, Group 1 contained only four cases with a record of gait disturbance, although these cases tended to show tau pathology in the motor cortex, substantia nigra, and dentate nucleus, which are associated with movement disorders. Although we cannot explain the reason for this discrepancy in a reproducible fashion, gait disturbances might be underestimated by family and primary care physicians because of increased attention to the associated dementia.
The severity of the tau pathology in the basal ganglia and substantia nigra in Group 2 were almost identical to Group 1, while the neocortical 4R pathology of Group 2 was milder. In addition to the lower severity of 4R pathology in the limbic system, a higher prevalence of gait disturbance and associated fall-related deaths and lower prevalence of dementia and depressive states were found in Group 2. From the distribution of tau pathology and the clinical appearance, we assume that Group 2 might correspond to PSP-P, pallido–nigro–luysian degeneration and axonal dystrophy as recently reported by Yokoyama et al. [74] or both.
Group 3 had significantly less neuronal loss and tau pathology in some PSP specific regions than Group 1, and a characteristically high prevalence of GsA-predominant astrocytic pathology. Group 3 might consist of additional incipient cases and/or cases associated with advanced AGD pathology. Some cases are consistent with the diffuse form of AGD reported by Maurage et al. [48], and a high prevalence of suicide may be a characteristic of this group. Although not statistically significant, a higher average prevalence and severity of AGD lesions in Group 3 might be related to a higher prevalence of suicide. Further case studies to investigate this are needed.
Limitations
In addition to a certain level of bias in our study population, this study was also limited by the clinical information of some cases was not circumstantiality mainly due to a lack of severe clinical symptoms or low neurologist consultation rates. In particular, the level of education and standard of living of each patient were not fully evaluated. Additionally, ocular motion and other neurological and psychological evaluations were only performed in a very limited number of individuals. In addition, comparison between incipient cases and the cases with advanced PSP may be essential for additionally revealing the clinicopathological features of incipient PSP.
Conclusion
We revealed a high prevalence of pathologically proven PSP in Japanese forensic autopsy cases, especially in cases older than a previous report. Although many cases had mild and/or an incomplete distribution of 4R pathology, the high prevalence of fall-related deaths or suicides, which might be associated with PSP, was characteristic. Three types of astrocytic inclusion and comorbid pathologies were also found in many of the cases. Cluster analysis based on the distribution and severity of 4R pathology divided the cases into three groups. Although more study is required, some clinical appearances, comorbid pathologies, and astrocytic pathologies might support the validity of the subclassification of these incipient PSP cases. Early clinical diagnosis of incipient PSP may be a significant factor for preventing some unusual deaths in the elderly. Here we have demonstrated that neuropathological examination, including immunohistochemistry without case selection, is useful in revealing the clinicopathological features of incipient neurodegenerative diseases.
References
Armstrong RA, Lantos PL, Cairns NJ (2007) Progressive supranuclear palsy (PSP): a quantitative study of the pathological changes in cortical and subcortical regions of eight cases. J Neural Transm 114:1569–1577. doi:10.1007/s00702-007-0796-3
Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, Hardy J, Lynch T, Bigio E, Hutton M (1999) Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet 8:711–715. doi:10.1093/hmg/8.4.711
Bigio EH, Brown DF, White CL III (1999) Progressive supranuclear palsy with dementia: cortical pathology. J Neuropathol Exp Neurol 58:359–364. doi:10.1097/00005072-199904000-00006
Birdi S, Rajput AH, Fenton M, Donat JR, Rozdilsky B, Robinson C, Macaulay R, George D (2002) Progressive supranuclear palsy diagnosis and confounding features: report on 16 autopsied cases. Mov Disord 17:1255–1264. doi:10.1002/mds.10211
Bloise MC, Berardelli I, Roselli V, Pasquini M, Stirpe P, Colosimo C, Berardelli A, Fabbrini G (2014) Psychiatric disturbances in patients with progressive supranuclear palsy: a case–control study. Parkinsonism Relat Disord 20:965–968. doi:10.1016/j.parkreldis.2014.05.015
Botez G, Probst A, Ipsen S, Tolnay M (1999) Astrocytes expressing hyperphosphorylated tau protein without glial fibrillary tangles in argyrophilic grain disease. Acta Neuropathol 98:251–256. doi:10.1007/s004010051077
Bower JH, Maraganore DM, McDonnell SK, Rocca WA (1997) Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990. Neurology 49:1284–1288. doi:10.1212/WNL.49.5.1284
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404. doi:10.1007/s00401-006-0127-z
Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K (2004) Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 318:121–134. doi:10.1007/s00441-004-0956-9
Brenneis C, Seppi K, Schocke M, Benke T, Wenning GK, Poewe W (2004) Voxel based morphometry reveals a distinct pattern of frontal atrophy in progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 75:246–249. doi:10.1136/jnnp.2003.015297
Buee L, Delacoute A (1999) Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick’s disease. Brain Pathol 9:681–693. doi:10.1111/j.1750-3639.1999.tb00550.x
Chiò A, Magnani C, Schiffer D (1998) Prevalence of Parkinson’s disease in Northwestern Italy: comparison of tracer methodology and clinical ascertainment of cases. Mov Disord 13:400–405. doi:10.1002/mds.870130305
Colosimo C, Bak TH, Bologna M, Berardelli A (2014) Fifty years of progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 85:938–944. doi:10.1136/jnnp-2013-305740
Compta Y, Valldeoriola F, Tolosa E, Rey MJ, Martí MJ, Valls-Solé J (2007) Long lasting pure freezing of gait preceding progressive supranuclear palsy: a clinicopathological study. Mov Disord 22:1954–1958. doi:10.1002/mds.21612
Del Tredici K, Braak H (2016) Sporadic Parkinson’s disease: development and distribution of a-synuclein pathology. Neuropathol Appl Neurobiol 42:33–50. doi:10.1111/nan.12298
Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA (2010) Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol 23:394–400. doi:10.1097/WCO.0b013e32833be924
Dickson DW, Hauw J, Agid Y, Litvan L (2011) Progressive supranuclear palsy and corticobasal degeneration. In: Dickson DW, Weller RO (eds) Neurodegeneration; the molecular pathology of dementia and movement disorders, 2nd edn. Wiley-Blackwell, Oxford, pp 133–155
Dickson DW, Rademakers R, Hutton ML (2007) Progressive supranuclear palsy: pathology and genetics. Brain Pathol 17:74–82. doi:10.1111/j.1750-3639.2007.00054.x
Donker Kaat L, Boon A, Kamphorst W, Ravid R, Duivenvoorden H, Van Swieten J (2007) Frontal presentation in progressive supranuclear palsy. Neurology 69:723–729. doi:10.1212/01.wnl.0000267643.24870.26
Dugger BN, Hentz JG, Adler CH, Sabbagh MN, Shill HA, Jacobson S, Caviness JN, Belden C, Driver-Dunckley E, Davis KJ (2014) Clinicopathological outcomes of prospectively followed normal elderly brain bank volunteers. J Neuropathol Exp Neurol 73:244–252. doi:10.1097/NEN.0000000000000046
Evans W, Fung HC, Steele J, Eerola J, Tienari P, Pittman A, Silva Rd, Myers A, Vrieze FW, Singleton A, Hardy J (2004) The tau H2 haplotype is almost exclusively Caucasian in origin. Neurosci Lett 369:183–185. doi:10.1016/j.neulet.2004.05.119
Evidente VG, Adler CH, Sabbagh MN, Connor DJ, Hentz JG, Caviness JN, Sue LI, Beach TG (2011) Neuropathological findings of PSP in the elderly without clinical PSP: possible incidental PSP? Parkinsonism Relat Disord 17:365–371. doi:10.1016/j.parkreldis.2011.02.017
Feany MB, Dickson DW (1995) Widespread cytoskeletal pathology characterizes corticobasal degeneration. Am J Pathol 146:1388–1396
Golbe LI, Davis PH, Schoenberg BS, Duvoisin RC (1988) Prevalence and natural history of progressive supranuclear palsy. Neurology 38:1031–1034. doi:10.1212/WNL.38.7.1031
Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, McKee A, Tabaton M, Litvan I (1994) Preliminary NINDS neuropathologic criteria for Steele–Richardson–Olszewski syndrome (progressive supranuclear palsy). Neurology 44:2015–2019. doi:10.1212/WNL.44.11.2015
Höglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L et al (2011) Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet 43:699–705. doi:10.1038/ng.859
Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8:1–13. doi:10.1016/j.jalz.2011.10.007
Ikeda K, Akiyama H, Arai T, Matsushita M, Tsuchiya K, Miyazaki H (2000) Clinical aspects of argyrophilic grain disease. Clin Neuropathol 19:278–284
Irwin DJ, Cohen TJ, Grossman M, Arnold SE, McCarty-Wood E, Van Deerlin VM, Lee VM, Trojanowski JQ (2013) Acetylated tau neuropathology in sporadic and hereditary tauopathies. Am J Pathol 183:344–351. doi:10.1016/j.ajpath.2013.04.025
Iwasaki Y, Yoshida M, Hattori M, Goto A, Aiba I, Hashizume Y, Sobue G (2004) Distribution of tuft-shaped astrocytes in the cerebral cortex in progressive supranuclear palsy. Acta Neuropathol 108:399–405. doi:10.1007/s00401-004-0904-5
Jackson JA, Jankovic J, Ford J (1983) Progressive supranuclear palsy: clinical features and response to treatment in 16 patients. Ann Neurol 13:273–278. doi:10.1002/ana.410130308
Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L, Liesinger AM, Petersen RC, Parisi JE, Dickson DW (2016) Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol 131:571–585. doi:10.1007/s00401-016-1537-1
Kanazawa M, Shimohata T, Toyoshima Y, Tada M, Kakita A, Morita T, Ozawa T, Takahashi H, Nishizawa M (2009) Cerebellar involvement in progressive supranuclear palsy: a clinicopathological study. Mov Disord 24:1312–1318. doi:10.1002/mds.22583
Kawashima M, Miyake M, Kusumi M, Adachi Y, Nakashima K (2004) Prevalence of progressive supranuclear palsy in Yonago, Japan. Mov Disord 19:1239–1240. doi:10.1002/mds.20149
Kim WH, Lee YS, Jung SH, Choi HJ, Lee MJ, Kang MH, Kim CE, Lee JS, Bae JN (2009) Major depressive disorder preceding the onset of progressive supranuclear palsy. Psychiatry Investig 6:112–114. doi:10.4306/pi.2009.6.2.112
Komori T, Arai N, Oda M, Nakayama H, Mori H, Yagishita S, Takahashi T, Amano N, Murayama S, Murakami S (1998) Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol 96:401–408. doi:10.1007/s004010050911
Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H, Cairns NJ, Crary JF, Duyckaerts C, Ghetti B et al (2016) Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol 131:87–102. doi:10.1007/s00401-015-1509-x
Kovacs GG, Milenkovic I, Wöhrer A, Höftberger R, Gelpi E, Haberler C, Hönigschnabl S, Reiner-Concin A, Heinzl H, Jungwirth S (2013) Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol 126:365–384. doi:10.1007/s00401-013-1157-y
Kovacs GG, Molnár K, László L, Ströbel T, Botond G, Hönigschnabl S, Reiner-Concin A, Palkovits M, Fischer P, Budka H (2011) A peculiar constellation of tau pathology defines a subset of dementia in the elderly. Acta Neuropathol 122:205–222. doi:10.1007/s00401-011-0819-x
Ling H, de Silva R, Massey LA, Courtney R, Hondhamuni G, Bajaj N, Lowe J, Holton JL, Lees A, Revesz T (2014) Characteristics of progressive supranuclear palsy presenting with corticobasal syndrome: a cortical variant. Neuropathol Appl Neurobiol 40:149–163. doi:10.1111/nan.12037
Ling H, Holton JL, Shaw K, Davey K, Lashley T, Revesz T (2015) Histological evidence of chronic traumatic encephalopathy in a large series of neurodegenerative diseases. Acta Neuropathol 130:891–893. doi:10.1007/s00401-015-1496-y
Ling H, Kovacs GG, Vonsattel JP, Davey K, Mok KY, Hardy J, Morris HR, Warner TT, Holton JL, Revesz T (2016) Astrogliopathy predominates the earliest stage of corticobasal degeneration. Brain 126:3237–3252. doi:10.1093/brain/aww256
Litvan I, Mangone CA, McKee A, Verny M, Parsa A, Jellinger K, D’Olhaberriague L, Chaudhuri KR, Pearce RK (1996) Natural history of progressive supranuclear palsy (Steele–Richardson–Olszewski syndrome) and clinical predictors of survival: a clinicopathological study. J Neurol Neurosurg Psychiatry 61:615–620. doi:10.1136/jnnp.60.6.615
Litvan I, Hauw JJ, Bartko JJ, Lantos PL, Daniel SE, Horoupian DS, McKee A, Dickson D, Bancher C, Tabaton M, Jellinger K, Anderson DW (1996) Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol 55:97–105. doi:10.1097/00005072-199601000-00010
Liu AK, Goldfinger MH, Questali HE, Pearce RKB, Gentleman SM (2016) ARTAG in the basal forebrain: widening the constellation of astrocytic tau pathology. Acta Neuropathol Commun 4:59. doi:10.1186/s40478-016-0330-7
Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DM, Lee VM (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122:111–113. doi:10.1007/s00401-011-0845-8
Maher E, Lees A (1986) The clinical features and natural history of the Steele–Richardson–Olszewski syndrome (progressive supranuclear palsy). Neurology 36:1005–1008. doi:10.1212/WNL.36.7.1005
Maurage C-A, Sergeant N, Schraen-Maschke S, Lebert F, Ruchoux M-M, Sablonnière B, Pasquier F, Delacourte A (2003) Diffuse form of argyrophilic grain disease: a new variant of four-repeat tauopathy different from limbic argyrophilic grain disease. Acta Neuropathol 106:575–583. doi:10.1007/S00401-003-0762-6
Mckee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, Perl DP, Stein TD, Vonsattel JP, Stewart W, Tripodis Y, Crary JF, Bieniek KF, Dams-O’Connor K, Alvarez VE, Gordon WA (2016) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131:75–86. doi:10.1007/s00401-015-1515-z
McKee AC, Stein TD, Kiernan PT, Alvarez VE (2015) The neuropathology of chronic traumatic encephalopathy. Brain Pathol 25:350–364. doi:10.1111/bpa.12248
McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH et al (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136:43–64. doi:10.1093/brain/aws307
McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, et al, Consortium on DLB (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65:1863–1872. doi:10.1212/01.wnl.0000187889.17253.b1
Millar D, Griffiths P, Zermansky AJ, Burn DJ (2006) Characterizing behavioral and cognitive dysexecutive changes in progressive supranuclear palsy. Mov Disord 21:199–207. doi:10.1002/mds.20707
Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L (1991) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41:479–486. doi:10.1212/WNL.41.4.479
Nath U, Ben-Shlomo Y, Thomson RG, Lees AJ, Burn DJ (2003) Clinical features and natural history of progressive supranuclear palsy: a clinical cohort study. Neurology 60:910–916. doi:10.1212/01.WNL.0000052991.70149.68
Nath U, Ben-Shlomo Y, Thomson RG, Morris HR, Wood NW, Lees AJ, Burn DJ (2001) The prevalence of progressive supranuclear palsy (Steele–Richardson–Olszewski syndrome) in the UK. Brain 124:1438–1449. doi:10.1093/brain/124.7.1438
Nishida N, Hata Y, Yoshida K, Kinoshita K (2015) Neuropathologic features of suicide victims who presented with acute poststroke depression: significance of association with neurodegenerative disorders. J Neuropathol Exp Neurol 74:401–410. doi:10.1097/NEN.0000000000000184
Nishida N, Yoshida K, Hata Y, Arai Y, Kinoshita K (2015) Pathological features of preclinical or early clinical stages of corticobasal degeneration: a comparison with advanced cases. Neuropathol Appl Neurobiol 41:893–905. doi:10.1111/nan.12229
Osaki Y, Morita Y, Kuwahara T, Miyano I, Doi Y (2011) Prevalence of Parkinson’s disease and atypical parkinsonian syndromes in a rural Japanese district. Acta Neurol Scand 124:182–187. doi:10.1111/j.1600-0404.2010.01442.x
Papapetropoulos S, Gonzalez J, Mash DC (2005) Natural history of progressive supranuclear palsy: a clinicopathologic study from a population of brain donors. Eur Neurol 54:1–9. doi:10.1159/000086754
Quante A, Jakob F, Wolf J (2008) Depression preceding the onset of progressive supranuclear paralysis: a case report. J Neuropsychiatry Clin Neurosci 20:247–248. doi:10.1176/jnp.2008.20.2.247a
Respondek G, Stamelou M, Kurz C, Ferguson LW, Rajput A, Chiu WZ, van Swieten JC, Troakes C, Al Sarraj S, Gelpi E (2014) The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord 29:1758–1766. doi:10.1002/mds.26054
Saito Y, Ruberu NN, Sawabe M, Arai T, Tanaka N, Kakuta Y, Yamanouchi H, Murayama S (2004) Staging of argyrophilic grains: an age-associated tauopathy. J Neuropathol Exp Neurol 63:911–918. doi:10.1093/jnen/63.9.911
Schrag A, Ben-Shlomo Y, Quinn NP (1999) Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet 354:1771–1775. doi:10.1016/S0140-6736(99)04137-9
Steele JC, Richardson JC, Olszewski J (1964) Progressive Supranuclear Palsy. A Heterogeneous Degeneration Involving the Brain Stem, Basal Ganglia and Cerebellum with Vertical Gaze and Pseudobulbar Palsy. Nuchal Dystonia and Dementia. Arch Neurol 10:333–359. doi:10.1001/archneur.1964.00460160003001
Tatsumi S, Mimuro M, Iwasaki Y, Takahashi R, Kakita A, Takahashi H, Yoshida M (2014) Argyrophilic grains are reliable disease-specific features of corticobasal degeneration. J Neuropathol Exp Neurol 73:30–38. doi:10.1097/NEN.0000000000000022
Thal DR, Rub U, Orantes M, Braak H (2002) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800. doi:10.1212/WNL.58.12.1791
Togo T, Isojima D, Akatsu H, Suzuki K, Uchikado H, Katsuse O, Iseki E, Kosaka K, Hirayasu Y (2005) Clinical features of argyrophilic grain disease: a retrospective survey of cases with neuropsychiatric symptoms. Am J Geriatr Psychiatry 13:1083–1091. doi:10.1176/appi.ajgp.13.12.1083
Tolney M, Monsch AU, Probst A (2001) Argyrophilic grain disease. A frequent dementing disorder in aged patients. Adv Exp Med Biol 487:39–58
Tsuboi Y, Josephs KA, Boeve BF, Litvan I, Caselli RJ, Caviness JN, Uitti RJ, Bott AD, Dickson DW (2005) Increased tau burden in the cortices of progressive supranuclear palsy presenting with corticobasal syndrome. Mov Disord 20:982–988. doi:10.1002/mds.20478
Wermuth L, Joensen P, Bünger N, Jeune B (1997) High prevalence of Parkinson’s disease in the Faroe Islands. Neurology 49:426–432. doi:10.1212/WNL.49.2.426
Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L, Holton JL, Revesz T, Lees AJ (2005) Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain 128:1247–1258. doi:10.1093/brain/awh488
Williams DR, Holton JL, Strand K, Revesz T, Lees AJ (2007) Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Mov Disord 22:2235–2241. doi:10.1002/mds.21698
Yokoyama Y, Toyoshima Y, Shiga A, Tada M, Kitamura H, Hasegawa K, Onodera O, Ikeuchi T, Someya T, Nishizawa M, Kakita A, Takahashi H (2015) Pathological and clinical spectrum of progressive supranuclear palsy: with special reference to astrocytic tau pathology. Brain Pathol 26:155–166. doi:10.1111/bpa.12265
Yoshida M (2014) Astrocytic inclusions in progressive supranuclear palsy and corticobasal degeneration. Neuropathology 34:555–570. doi:10.1111/neup.12143
Acknowledgements
The authors thank Ms. Syuko Matsumori, Ms. Tamae Sasakura, Mr. Noboru Onozuka, and Mr. Osamu Yamamoto for their technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
All procedures performed in studies involving human participants in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yoshida, K., Hata, Y., Kinoshita, K. et al. Incipient progressive supranuclear palsy is more common than expected and may comprise clinicopathological subtypes: a forensic autopsy series. Acta Neuropathol 133, 809–823 (2017). https://doi.org/10.1007/s00401-016-1665-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-016-1665-7