
Abstract
Guam ALS/PDC is a severe tangle forming disorder endemic to Guam with features overlapping such neurodegenerative disorders as Alzheimer disease (AD), Parkinson disease (PD), progressive supranuclear palsy (PSP), ALS, corticobasal degeneration (CBD) and pallido-ponto-nigral degeneration (PPND). Since the prevalence is declining, we examined brain tissue from 35 clinically diagnosed Chamorro patients with ALS/PDC and two Chamorro controls autopsied between 1946 and 2006, to determine if distinct variations in the pathology could be identified up to this time. Although the age at autopsy increased by 4.5–5 years per decade, we identified no qualitative differences in pathological deposits with antibodies against tau, ubiquitin, Aβ, α-synuclein and TDP-43, indicating that these more recently identified proteins have been involved in the neuropathogenesis over the past 6 decades. Tau and TDP-43 positive neuronal, oligodendroglial and astrocytic inclusions involving multiple nerve fiber tracts occurred in both the ALS and PDC types, reinforcing the concept that these forms are part of the same disorder. The results obtained may help to define the commonality of the Guam disease with other tangle forming disorders and may help in monitoring the epidemiological changes that are taking place.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The amyotrophic lateral sclerosis/parkinsonism–dementia complex of Guam (ALS/PDC) is among the most enigmatic of all neurological disorders. It is largely restricted to Guam and occurs with high frequency in certain Chamorro families.
A cluster of cases with ALS was first noted by Zimmerman [46]. This was soon confirmed by several investigators and the incidence was estimated by Kurland and Mulder [22] to be 50–100 times higher than ALS in the rest of the world. The incidence has steadily declined [6, 35, 36, 42] and it has been predicted by some that it will disappear. Mulder and colleagues [29] also noticed that atypical parkinsonism and memory defects were common among Chamorros. This was followed by clinical and pathological studies by Hirano and colleagues in 1961 who introduced the term parkinsonism dementia complex (PDC) [15, 16]. The overlapping pathology with ALS cases led them to conclude that these were variations of the same fundamental disorder, i.e., the ALS/PDC complex of Guam. The cause is unknown. Apo-E genotype is not a factor [1], nor are mutations in the tau gene [28, 34] although certain tau gene polymorphisms were determined to be mild risk factors [41].
ALS/PDC of Guam is a severe tangle forming disorder which affects both neuronal and glial cells [16, 19, 20, 25, 27, 30, 32, 33]. Neurofibrillary tangles (NFTs) found in PDC brains are biochemically and ultrastructurally similar to those in AD [2, 23]. The sarkosyl insoluble tau, as in AD, is composed of all six tau isoforms [3, 24]. However, in Guam ALS/PDC, the type of glial pathology resembles that of PSP, CBD and several sporadic and familial forms of fronto-temporal dementia [21, 43, 45]. Amyloid beta (Aβ) deposits were reported to be rare or nearly absent in Guam ALS/PDC [8], but were later reported to be present by several authors [11, 21, 37, 38]. Focal α-synuclein pathology was also reported to occur in a number of Guam ALS/PDC cases [5, 39, 44] and recent observations showed involvement of TDP-43 as well [9, 14].
The main goal of the present study was to analyze whether the pathology of Guam ALS/PDC has undergone qualitative pathological alterations during the last 6 decades. Such a study may be relevant to defining disease specificity, particularly in view of the similarities of Guam ALS/PDC to other neurodegenerative disorders. The results show that the pathological lesions are qualitatively similar in patients who died recently compared with those who died 4–6 decades ago. In addition to severe glial pathology, we observed Aβ, α-synuclein, and TDP-43 pathology in a significant number of cases. We observed typical tau positive NFTs, oligodendroglial coiled bodies, hazy granules, thorn shaped and tufted astrocytes in both the ALS and PDC forms, further suggesting that these forms are probably variants of the same disease.
Materials and methods
Autopsy cases investigated
The brains of 35 Chamorro patients (average age 63, range 29–88) were analyzed. They died between 1946 and 2006 (see Table 1) with the diagnosis of ALS/PDC. The diagnoses of all 35 cases had previously been neuropathologically confirmed. As it has been suggested by many that the ALS and PDC forms are variations of the same disorder, we have referred to each case as ALS/PDC; however, we indicate in Table 1 whether there was a presence or absence clinically of ALS, parkinsonism or dementia. The brains of two Chamorro residents without symptoms of ALS/PDC (cases 36–37, Table 1) were also analyzed (aged 39 and 47). The causes of death in these patients were cardio-vascular failure and diabetes.
In 22 cases (cases 13, 15–27 and 29–35) whole or half brains were available for analysis. About 7-mm thick transverse tissue blocks from representative levels of the cerebral hemispheres, including hippocampus, frontal, temporal, parietal, primary motor and occipital cortices, the amygdala, basal ganglia, thalamus, mesencephalon, pons, medulla oblongata, cerebellum, and where available cervical spinal cord, were analyzed. In some ALS/PDC cases, the thoracic and lumbar segments of the spinal cord were also analyzed.
These samples were fixed in 4% paraformaldehyde in 0.1 M phosphate buffered saline (pH 7.4) for 2 days, and were maintained in 15% sucrose in 0.01 M phosphate buffered saline (PBS), pH 7.4, until processed. From these tissue samples, 30 μm thick sections were cut on a freezing microtome and were investigated immunohistochemically as free-floating sections.
In the remaining 14 cases (cases 1–12, 14 and 28, Table 1) paraffin blocks were available from the archives of the United States Armed Forces Institute for Pathology. This historical archival material included in all cases samples of the frontal cortex, the mesencephalon and the spinal cord, and, in some cases, the basal ganglia. Sections 20 μm thick were cut and, after paraffin removal in xylene and rehydration in a series of decreasing ethanol solutions, were immunostained as free-floating sections.
Histochemical and immunohistochemical analysis
The paraffin and 4% paraformaldehyde fixed sections were stained with hematoxylin and eosin (H&E), cresyl violet (Nissl), thioflavin S and the Gallyas silver impregnation method for NFTs [7]. The types, dilutions and sources of antibodies used for the immunohistochemical analysis are given in Table 2.
For the detection of Aβ, six different antibodies were used which recognized differing epitopes of the peptide: Aβ 8–17 (6F/3D, DakoCytomation, Carpinteria, CA), Aβ17-24 (4G8, Sigma-Aldrich), Aβ 33–40 (2G3, Elan Pharmaceuticals), Aβ 33–42 (21F12, Elan Pharmaceuticals) and antibodies Aβ40 and Aβ42 (generous gift of Dr. H. Mori) which recognize the C terminal of Aβ40 and Aβ42, respectively. To detect tau, two monoclonal and two polyclonal antibodies were used which recognize non-phosphorylated and phosphorylated tau epitopes (see Table 2). Two monoclonal antibodies to α-synuclein (Synuclein Clone LB509, 1:1,000 and Synuclein VP-A106, Vector Labs, Burlingame, CA; 1:1,000) and two polyclonal antibodies which recognize phosphorylated α-synuclein (#74 and #75, a kind gift of Dr. Kimiko Obi) were utilized. A polyclonal anti-ubiquitin antibody (Z0458, DakoCytomation, Carpinteria, CA, 1:1,000) was used to detect Lewy bodies and tau and α-synuclein negative ubiquitin inclusions.
For the detection of TDP-43, four antibodies were used. A monoclonal (2E2-D3, Abnova Corporation, Taipei, Taiwan), and a polyclonal (10782-1-AP; ProteinTech Group Inc, Chicago, IL), which recognize both non-phosphorylated and phosphorylated TDP-43, and two polyclonal antibodies (pS403/404 and pS409/410; kind gift of Dr. H. Akiyama) which recognize only the pathological phosphorylated forms [14].
Sections being analyzed for Aβ and α-synuclein were pre-treated with 80% formic acid for 20 min before immunostaining. For immunodetection of all sections, the avidin–biotin-peroxidase technique was utilized as previously described in detail [26]. Endogenous peroxidase activity was first removed by incubation in methanol containing 0.3% hydrogen peroxide for 30 min. The sections were then blocked with normal sera of animals in which the secondary antibodies were raised (diluted 1:10 in PBST containing 1% bovine serum albumin, Fluka, 05480), following which they were incubated with a primary antibody overnight at room temperature or for 48–72 h at 4°C. After washing with PBST, the sections were incubated for 30 min at room temperature with the appropriate biotinylated secondary antibody which was diluted at 1:300 in Tris buffer containing 1% BSA. Following a wash in PBST (3× 5 min), the sections were incubated with a mixture of avidin and biotinylated horseradish peroxidase (DAKO, ABComplex/HRP-Kit, K0355) as recommended by the manufacturer. The final reaction solution contained 3,3′-diaminobenzidine (DAB, Sigma, D5637) alone to produce a brown reaction product, or combined with nickel ammonium sulfate to produce a dark-purple reaction product. The sections were rinsed in distilled water, dehydrated in graded ethanol, passed for 2× 5 min in xylene and mounted in Entellan.
Double immunofluorescent staining was used to detect overlapping expression of TDP-43 and tau. For this purpose, sections of the hippocampus, mesencephalon, medulla oblongata and spinal cord of four ALS/PDC cases were incubated overnight at room temperature with a mixture of rabbit anti-TDP-43 (Protein Tech, 1:100) and a monoclonal mouse anti-Tau C3 antibody (dil.1:100). The sections were then incubated with TRITC labeled swine anti-rabbit (R0156, Dako Corporation, Carpinteria, CA, 1:30) for 1 h at room temperature to yield a red fluorescence for TDP-43. Then, following a wash (3× 5 min) with PBST, the sections were incubated for another hour with FITC labeled goat anti-mouse (F0479, Dako Corporation, Carpinteria, CA, 1:30) secondary antibody to reveal tau in green fluorescence. A reverse double immunofluorescence experiment was also performed using the monoclonal anti-TDP-43 antibody (Abnova) and the rabbit anti-tau cocktail antibody (AnaSpec Inc.) for phosphorylated tau. Using the same secondary antibodies, TDP-43 was exhibited in green and tau in red fluorescence. Sudan black (0.3% in 70% ethanol) staining for 10 min was used to quench autofluorescence. After a rinse in PBS the sections were mounted with Fluoromont G (Southern Biotechnology Inc, Birmingham, AL) and examined by confocal microscopy using a Zeiss fluorescent microscope. Capture of the green and red fluorescence, merged images, and 3-D reconstruction of 20–40 serial confocal microscopic images recorded at 0.1–0.3 μm intervals, were performed using the Northern Elite program.
A semiquantitative analysis of NFTs, Aβ plaques, coiled bodies (CBs), Lewy bodies (LBs) and of “hazy” granules (GR) was performed in all cases, based on the density of these pathological changes. Their density was noted in four grades: (absence of lesion −, mild density +, moderate density ++ and severe or high density +++). In each cases the density of NFTs, Aβ plaques, CBs, LBs or GRs of the most affected brain area was entered in Table 3.
Results
A striking finding among these autopsied patients was an apparent linear increase in the age at autopsy and the year of death (Table 1; Fig. 1; P < 0.0005, Fisher’s method). The average increase was 4.5–5 years per decade.

Comparison of age at death with year of death
Table 3 summarizes the results of the immunohistochemical analyses. The types of tau, Aβ, ubiquitin or α-synuclein pathological changes, if present, were identical in the brains of ALS/PDC patients deceased between 1946 and the present. This is illustrated in Figs. 2, 3 and 4 where NFTs, Aβ deposition, and glial pathology in an ALS/PDC patient who died in 1946 are compared to those in a patient who died in 2004. Figure 5 compares α-synuclein positive Lewy bodies in a patient who died in 1954 compared with one who died in 2004.

Neurofibrillary tangles in the frontal cortex (a and b) and substantia nigra (c, d, arrows) of an ALS/PDC patient of Guam who died in 1946 (1st column, a, c) and of another who died in 2003 (2nd column, b, d). In both cases neurofibrillary tangles were immunostained with anti-PHF antibody (clone AT-8, Innogenetics, Belgium, 1:500). e, f Senile plaques in the cerebral cortex of the same ALS/PDC patients, respectively. For immunostaining a monoclonal anti-Aβ 8-17 antibody (M0872, DakoCytomation) was used (g, h). Neurofibrillary tangles exhibiting strong Aβ-immunoreactivity in the frontal cortex of the same Guam ALS/PDC cases. The photomicrographs were taken from the same frontal sections immunostained with the same anti-Aβ antibody as illustrated in e and f (bars 100 μm)

Comparison of oligodendroglial tau positive inclusions in the brains of an ALS/PDC Chamorro patient who died in 1946 (1st column) and one who died in 2003 (2nd column). AT-8 immunoreactive (anti-PHF-tau, Innogenetics, Belgium) coiled bodies along nerve fiber tracts (a–d) and tufted astrocytes (e, f) of the patient who died in 1946 (a, c, e) and in 2003 (b, d, f) (bars 50 μm)

Granular pathology (hazy granules) and thorn-shaped astrocytes in the brains of an ALS/PDC patient who died in 1946 (a, c, e) and one who died in 2003 (b, d, f). Tau positive granules (a, b) and thorn-shaped astrocytes (c–e) are shown in the brainstem. The immunostaining was performed using the anti-PHF antibody clone AT-8, Innogenetics, Belgium (bars 50 μm)

Lewy bodies in substantia nigra (a–d) and cerebral cortex (e, f) neurons in an ALS/PDC Chamorro patient who died in 1954 (a, c, e) and one who died in 2003 (b, d, f). A monoclonal anti-α-synuclein antibody (Zymed Laboratories) was used for immunostaining (bars 100 μm)
NFTs were found in virtually all cases (91.4%, 32/35), three (cases 6, 9 and 11, Table 3), where the hippocampus and entorhinal cortex, the most vulnerable areas for NFT development, were not available for analysis. NFTs were observed in brains of 14 of 15 cases with the ALS form, in 14 of 15 cases with the PDC form and in four of five cases with mixed ALS and PDC forms. NFT accumulation was more severe in cases with PDC and mixed ALS–PDC forms than in cases with the ALS form. The severity and extension of cortical neurofibrillary changes varied from case to case. NFT accumulation was most abundant in the hippocampus and entorhinal cortex, but NFTs also occurred in the frontal, temporal, parietal and primary motor cortical areas. The cortical distribution of NFTs showed a predisposition for layers II and III as previously reported [19, 20]. In cases with more severe NFT accumulation, NFTs were also numerous in cortical layers V and VI. In addition to cortical NFTs, severe neurofibrillary changes were observed in the amygdala, nucleus innominatus and septal nuclei. A number of NFTs were frequently observed in the basal ganglia and thalamus. In the majority of cases, NFTs were present in the substantia nigra, the oculomotor nuclear complex, the tectum, tegmentum and reticular nuclei of the brainstem. In several cases, NFTs were also observed in a few neurons of the spinal cord.
Figure 2a–d illustrates NFTs in two ALS/PDC patients: one deceased in 1946 (a, c) and the other in 2003 (b, d). The NFTs shown in the frontal cortex (a, b) and in the substantia nigra (c, d) were immunostained with a monoclonal anti-PHF antibody (clone AT-8, Innogenetics, Belgium).
Senile plaques were observed in 60% (21/35) of ALS/PDC patients. They were found in 40% of patients autopsied between 1946 and 1973 (4/10) and in 68% in those autopsied between 1982 and 2004 (17/25). Panels e and f of Fig. 2 illustrate cortical Aβ plaques in the frontal cortex of a patient autopsied in 1946 (Fig. 2e) and in another patient autopsied in 2003 (Fig. 2f). In some cases, vascular Aβ deposits were also observed in some leptomeningeal and cortical arteries.
A subset of NFTs strongly immunoreacted with the six antibodies which recognize various epitopes of Aβ in 74.3% (26/35) of the ALS/PDC cases. All cases with Aβ immunoreactive NFTs are labeled with an “a” in the “NFT” column in Table 3. Aβ immunoreactive NFTs were present in high numbers in the hippocampus and entorhinal cortex but were also found in the frontal, temporal and parietal cortices, in the nucleus innominatus, the substantia nigra, and the brainstem. Aβ positive NFTs were also present in five ALS/PDC cases without Aβ plaques. Aβ immunoreactive NFTs in the frontal cortex of two ALS/PDC patients who died in 1946 and 2003 are illustrated in Fig. 2g, h. When Aβ positive senile plaques and Aβ positive NFTs were considered together, 77.1% (27/35) of ALS/PDC cases exhibited Aβ pathology. When comparing the older and more recent groups, Aβ pathology was present in 60% (6/10) of ALS/PDC patients who died between 1946 and 1973 and in 88% (22/25) of those who died between 1982 and 2006.
Tau positive oligodendroglial coiled bodies were observed in 88.6% (31/35) of the ALS/PDC patients (Table 3). The density of coiled bodies varied from case to case. Coiled bodies were frequently seen in the primary motor cortex, the prefrontal and parietal cortices, the hippocampus and entorhinal cortex, the basal ganglia, particularly in the pallidum, as well as the mesencephalon and brainstem. They were also present in some cases in the cerebellar white matter. The pyramidal tract at levels of the internal capsule, cerebral peduncle and medulla oblongata frequently showed coiled bodies. In the spinal cord, coiled bodies were seen in the crossed and in the uncrossed corticospinal tract. Some coiled bodies were also observed in the anterior horn around motor neurons.
Figure 3a–d illustrates AT-8 (PHF-Tau, Innogenetics, Belgium) immunoreactive coiled bodies in a Guam ALS/PDC patient who died in 1946 (Fig. 3a, c) compared with one who died in 2002 (Fig. 3b, d).
Tau positive granular pathology, named “hazy” granules by Oyanaga et al. [31–33] were frequently associated with thorn-shaped astrocytes, and were observed in all Guam ALS/PDC cases. Again, the severity of granular pathology varied from case to case. Granules were frequently seen in the entorhinal, temporal, frontal, primary motor and parietal cortex. An accentuation of the granular pathology was observed in the amygdala, entorhinal cortex, basal ganglia, brainstem and in the subependymal and subpial areas of the brain. Perivascular accumulations were frequently observed in severely affected subpial and subependymal areas. Similarly to the coiled bodies, “hazy” granules were frequently associated with nerve fiber tracts, which were particularly visible in the basal ganglia and brainstem. The tectum of the mesencephalon, the hilus of the inferior olive, the retro-olivary area of the medulla oblongata and the lateral funiculus of the spinal cord were frequent sites of severe granular pathology. Panels a–f of Fig. 4 illustrate AT-8 tau immunoreactive granules and thorn-shaped astrocytes in the medulla oblongata of an ALS/PDC patients who died in 1946 (Fig. 4 a, c, e) and one who died in 2003 (Fig. 4 b, d, f).
NFTs and glial inclusions all exhibited strong immunostaining with the four different anti-tau antibodies employed (Table 2). Coiled bodies, “hazy” granules, thorn-shaped and tufted astrocytes were all strongly tau positive. Extracellular NFTs showed weaker immunostaining with all of these anti-tau antibodies when compared with the strong tau-immunoreaction of intracellular NFTs.
α-Synuclein positive Lewy bodies were observed in 19 of the 35 (54.3%) cases. Lewy bodies were detected in the brain in 40% of cases deceased between 1946 and 1973 (4/10) and in 60% (15/25) of those deceased between 1982 and 2006. Lewy bodies were frequently observed in the amygdala, and in the nucleus innominatus but they were also seen in the substantia nigra, the raphe and reticular nuclei of the brainstem. In some cases, Lewy bodies were seen in pyramidal neurons of the parahippocampal, frontal, temporal and parietal cortices and in the basal ganglia.
Figure 5a–d illustrates single (Fig. 5a, b) and multiple (Fig. 5c, d) α-synuclein immunoreactive Lewy bodies in dopaminergic neurons of the substantia nigra in a patient who died in 1954 (Fig. 5a, c) compared with one who died in 2003 (Fig. 5b, d). α-Synuclein positive Lewy bodies in pyramidal neurons of the frontal cortex of the same two patients are illustrated in panels e and f.
Ubiquitin and tau positive, α-synuclein negative, intracytoplasmic inclusions were also seen in a few of the older and newer cases. They were observed in the nucleus innominatus, in the substantia nigra, in a few pyramidal neurons of the insular, frontal and temporal cortices and in neurons of the inferior olive. Intranuclear ubiquitin inclusions, which were negative for tau or α-synuclein, were frequently observed in dopaminergic neurons of the substantia nigra of both older and newer cases.
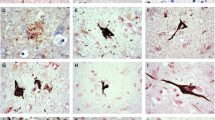
Antibodies which recognize both phosphorylated and non-phosphorylated forms of TDP-43 and those which specifically label the phosphorylated form immunostained a subset of intraneuronal NFTs (Fig. 6a, b) in addition to the normal strong nuclear staining. They also labeled various glial pathologies including coiled bodies (Fig. 6c, d), tufted astrocytes (Fig. 6e, f), granular pathology (Fig. 6g, h), and thorn-shaped astrocytes (Fig. 6i, j). They immunostained some large intracytoplasmic inclusions (Fig. 6k, l). Such immunostaining occurred in both the PDC (left column) and ALS (right column) variants of the disease complex.

TDP-43 positive neuronal and glial abnormalities in a case of the PDC form (left column) and in a case of the ALS form (right column). TDP-43 immunoreactive neurofibrillary tangles (a, b), coiled bodies (c, d), tufted astrocytes (e, f), granular pathology (g, h) thorn-shaped astrocytes (i, j) and large intracytoplasmic inclusions (k, l, asterisks). Note the comparison to the normal cell size (arrow in k) or the TDP-43 staining of nuclei (arrow in l) (bars a, h = 80; b, g = 100 μm; c, d, i, j = 30 μm; f = 40 μm; e, k, l = 50 μm
TDP-43 immunoreactive coiled bodies were associated with several nerve fiber tracts. The corticospinal tract, the frontopontine tract, the accessory striatal circuits, the pallidum, the substantia nigra, the superior colliculus and the olivo-ponto-cerebellar system were the most frequent sites of involvement. In the ALS variant, the involvement was particularly evident in the pyramidal tract, while in the PDC variant, the involvement was dominant in the substantia nigra; however, there was considerable overlap. The majority of patients with the ALS variant also showed TDP-43 positive coiled bodies in the brainstem, mesencephalon, substantia nigra and pallidum, while those with the PDC form frequently had TDP-43 positive coiled bodies in the pyramidal tract. They were particularly visible at the level of the internal capsule, the pedunculus cerebri and the lateral funiculus of the spinal cord.
Only a small subset of NFTs exhibited a TDP-43 immunoreaction. The majority of them were localized in the hippocampus, entorhinal cortex and temporal and frontal cortices. They were also seen in the substantia nigra, in the oculomotor nuclear complex and in the brainstem. TDP-43 immunoreactive NFTs were observed in both the ALS and PDC variants, but their number and their extension was higher in the PDC form. A subset of “hazy” granules, tufted and thorn-shaped astrocytes were also TDP-43 positive. Some large, TDP-43 immunoreactive, well demarcated, round or oval intracytoplasmic inclusions were also seen in a few neurons in both the ALS and PDC forms. They were most frequently seen in the nucleus innominatus, in the substantia nigra, in the inferior olive and in the brainstem reticular formation in both variants. A few of them were also observed in the hippocampus and in pyramidal neurons of the frontal and temporal cortices.
In the ALS variant, TDP-43 positive skein-like inclusions were observed in lower motor neurons. These TDP-43 positive intracytoplasmic skein-like inclusions, together with the TDP-43 positive coiled bodies, in the corticospinal tract and in the anterior horn of the spinal cord (Fig. 7a, b) were identical to those seen in two non-Guam sporadic ALS cases (Fig. 7c, d).

TDP-43 immunoreactive intracytoplasmic skein-like inclusions in motor neurons and coiled bodies in the pyramidal tract at the level of the spinal cord in Guam ALS (a, b) show an identical TDP-43 reaction to those occurring in sporadic non Guam ALS (c, d). e–g Section of the hippocampus of a patient with PDC doubly immunostained with a monoclonal anti-TDP-43 antibody and a polyclonal anti-tau antibody (29582, AnaSpec, San Jose, CA) which specifically recognizes phosphorylated tau. e–g Red fluorescence of tau (e) as revealed by an TRITC tagged secondary antibody, the green fluorescence of TDP-43 (f) as revealed by an FITC tagged secondary antibody and their co-localization in neurofibrillary tangles as shown in orange color (g) on the merged images. Arrows in f and g point to TDP-43 positive nuclei indicating that the TDP-43 positive tangles of these neurons are intracellular (bars a–d = 50 μm; e–g = 150 μm
On doubly immunostained sections, TDP-43 and tau were colocalized in a subset of NFTs, coiled bodies and “hazy” granules. Panels e, f and g of Fig. 7 illustrate the colocalization of phosphorylated tau exhibiting red fluorescence and TDP-43 exhibiting green fluorescence in NFTs. On the merged fluorescent image illustrated in panel g, the overlapping red tau and green TDP-43 fluorescence yields an orange color. Neuronal nuclei show green TDP-43 fluorescence. Reconstruction of serial confocal microscopic images of the same neurons (video, supplementary material online) confirmed the intracellular location of NFTs co-expressing tau and TDP-43.
The brains of the two Chamorro controls (Table 1) did not show any pathological changes.
Discussion
An extensive immunohistochemical study of Guam brains from 35 ALS/PDC Chamorro patients who died during the last 6 decades was performed. The goal was to determine whether the type of histological lesions in historic ALS/PDC cases were identical to those who died more recently. The results establish such an identity.
Guam ALS/PDC is characterized by cortical atrophy and neuronal loss with numerous NFTs widely distributed throughout the hippocampus, entorhinal cortex, neocortex and brain stem. Our results confirm many previous reports of this characteristic finding [3, 17, 18, 24].
The distribution of NFTs in the ALS and PDC variants were similar, although NFTs were more prominent in the PDC form. When compared to AD, the NFT pathology was more severe in the hippocampus, the substantia nigra and brainstem but was less severe in neocortical areas. NFTs were also observed in the spinal cords in several old and new ALS/PDC Chamorro patients, similarly to those seen in PSP [40], but rarely in AD. Despite the high occurrence and the similar topographic distribution of NFTs in both the ALS (14/15) and the PDC (14/15) forms, the NFT pathology was less severe in the ALS form in agreement with previous observations [25, 27, 31].
In the present study, senile plaques were observed in 60% (21/35) of ALS/PDC patients. In addition, a subset of NFTs strongly immunoreacted with the anti-Aβ antibodies employed. Such Aβ immunoreactive NFTs were frequently observed in the hippocampus and entorhinal cortex but they also occurred in the frontal, temporal and parietal cortices, the nucleus innominatus, the substantia nigra, the tegmentum and reticular nuclei of the brainstem. Aβ immunoreactive NFTs were present in 74% (26/35) of ALS/PDC cases. They were also observed in ALS/PDC patients without Aβ plaques. If Aβ deposition in senile plaques and NFTs are both considered together, 77% (27/35) of ALS/PDC cases exhibited Aβ pathology. This indicates that Aβ is involved in the pathogenesis of Guam ALS/PDC. That these Aβ deposits in plaques and NFTs were strongly immunoreactive with the panel of antibodies which detect various epitopes of Aβ 1–42 further indicates that Aβ is implicated in the pathogenesis of ALS/PDC. In some cases, prominent Aβ deposits were also observed in some leptomeningeal and cortical arteries, similarly to those occurring in AD.
Tau positive oligodendroglial inclusions were observed in more than 88% (31/35) of ALS/PDC patients and were frequently associated with tufted astrocytes. Tau positive granular pathology, known as hazy granules [33], was observed in all ALS/PDC cases, and was frequently associated with thorn-shaped astrocytes. The density of coiled bodies and hazy granules varied from case to case. They were frequently associated with nerve fiber tracts and were similar to those occurring in PSP, CBD, and PPND.
α-Synuclein is the major constituent of Lewy bodies as well as the glial cytoplasmic inclusions of multiple system atrophy [4]. Lewy bodies were identified in the substantia nigra in Guamanian PDC patients as early as 1966 by Hirano et al. [17]. More recently, α-synuclein pathology has also been detected in the amygdala and cerebellum in a subset of ALS/PDC cases [5, 39, 43, 44]. Our results showed that α-synuclein positive Lewy bodies occurred in more than 50% of cases in old and new cohorts and in both forms of the complex.
Tau and ubiquitin positive, but α-synuclein negative, neuronal intracellular inclusions in the substantia nigra, amygdala and cortical pyramidal neurons were also observed in some recent and old ALS/PDC cases. Ubiquitin positive intranuclear inclusions were also frequently seen.
In harmony with the recent findings of Hasegawa et al. [14] and Geser et al. [9], we observed TDP-43 in a subset of pathological neuronal and glial inclusions in both variants. In addition to the ubiquitin positive, tau negative skein-like inclusions of the motor neurons in the ALS form, a subset of NFTs, coiled bodies, ‘hazy” granules, and a few tufted and thorn-shaped astrocytes showed a distinct TDP-43 immunoreaction. In a subset of NFTs and coiled bodies, TDP-43 was colocalized with phosphorylated tau. TDP-43 immunoreactive coiled bodies were intimately associated with various nerve fiber tracts. The presence of TDP-43 positive oligodendroglial inclusions, and their association with nerve fiber tracts of the motor system in both forms further suggests that they may be variants of the same disease. We suggest that the severity of involvement of particular fiber tracts may determine the variant observed clinically.
Although the present study shows the persistence of tau, Aβ, α-synuclein, ubiquitin, TDP-43 immunoreactive aggregated proteins in ALD/PDC, it cannot judge whether the severity and distribution of these pathological lesions have changed during the past 6 decades. Careful analysis of a much broader sample would be required, with particular attention being paid to the actual causes of death since this terminates lesion development.
These results, taken together with previous publications, strongly indicate that, in addition to tau, Aβ, α-synuclein and TDP-43 are involved in the degenerative process of Guam ALS/PDC. With respect to Aβ, Gouras et al. [12] suggested that intraneuronal Aβ42 accumulation is an early pathological step in AD. It has also been shown that soluble Aβ and tau interact with each other and may promote tau phosphorylation and Aβ aggregation in AD [13]. Since Aβ pathology was present in 77.1% (27/35) cases in our series, the question is raised as to whether a similar mechanism may play a role in the Guam ALS/PDC complex. In addition, coincubation of tau with α-synuclein promotes fibrilization of both tau and α-synuclein [10]. This may also contribute to the very severe tau pathology in this disorder.
In conclusion, the types of histological changes in Guam ALS/PDC have not changed during the last 6 decades even though the ALS form has largely disappeared and the PDC form may be declining. Severe tau pathology dominates the pathological picture but Aβ, α-synuclein and TDP-43 pathology are also implicated in the pathogenesis of the disease. Despite the fact that the NFT pathology shares similarities with AD and the presence of Lewy bodies are identical to those occurring in Parkinson disease, the tau positive glial pathology frequently associated with nerve fiber tracts shows strong similarities to PSP, CBD and several FTD tauopathies. Tau positive NFTs, coiled bodies, hazy granules, tufted astrocytes and thorn-shaped astrocytes and the association of coiled bodies and hazy granules with nerve fiber tracts, may occur in both the ALS and PDC forms. This further suggests that these forms are variants of the same disease.
The reason why the age at autopsy is significantly increasing with each passing decade, coupled with a declining incidence, suggests that the causative factors may be diminishing or may even have disappeared. Future trends cannot be predicted from the present data and continued careful research will be required.
References
Buée L, Pérez-Tur J, Leveugle B, Buée-Scherrer V, Mufson EJ, Loerzel AJ, Chartier-Harlin MC, Perl DP, Delacourte A, Hof PR (1996) Apolipoprotein E in Guamanian amyotrophic lateral sclerosis/parkinsonism–dementia complex: genotype analysis and relationships to neuropathological changes. Acta Neuropathol 91:247–253
Buée L, Bussiere T, Buée-Scherrer V, Delacourte A, Hof PR (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 33:95–130
Buée-Scherrer V, Buée L, Hof PR, Leveugle B, Gilles C, Loerzel AJ, Perl DP, Delacourte A (1995) Neurofibrillary degeneration in amyotrophic lateral sclerosis/parkinsonism–dementia complex of Guam. Immunochemical characterization of tau proteins. Am J Pathol 146:924–932
Duda JE, Lee VM-Y, Trojanowski JQ (2000) Neuropathology of synuclein aggregates. J Neurosci Res 61:121–127
Forman MS, Schmidt ML, Kasturi S, Perl DP, Lee VM-Y, Trojanowski JQ (2002) Tau and alpha-synuclein pathology in amygdala of parkinsonism–dementia complex patients of Guam. Am J Pathol 160:1725–1731
Galasko D, Salmon DP, Craig UK, Thal LJ, Schellenberg G, Wiederholt W (2002) Clinical features and changing patterns of neurodegenerative disorders on Guam, 1997–2000. Neurology 58:90–97
Gallyas F (1971) Silver staining of Alzheimer’s neurofibrillary changes by means of physical development. Acta Morphol Acad Sci Hung 19:1–8
Gentleman SM, Perl D, Allsop D, Clinton J, Royston MC, Roberts GW (1991) Beta (A4)-amyloid protein and parkinsonian–dementia complex of Guam. Lancet 337:55–56
Geser F, Winton MJ, Kwong LK, Xu Y, Xie SX, Igaz LM, Garruto RM, Perl DP, Galasko D, Lee VM, Trojanowski JQ (2008) Pathological TDP-43 in parkinsonism–dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol 115(1):133–145
Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM (2003) Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300:636–640
Guiroy DC, Mellini M, Miyazaki M, Hilbich C, Safar J, Garruto RM, Yanagihara R, Beyreuther K, Gajdusek DC (1993) Neurofibrillary tangles of the Guamanian amyotrophic lateral sclerosis, parkinsonism–dementia and neurologically normal Guamanians contain a 4-to 4.5 kilodalton protein which is immunoreactive to anti-amyloid beta/A4-protein antibodies. Acta Neuropathol 86(3):265–274
Gouras GK, Almeida CG, Takahashi RH (2005) Intraneuronal Abeta accumulation and origin of plaques in Alzheimer’s disease. Neurobiol Aging 26:1235–1244
Guo J, Arai T, Miklossy J, McGeer PL (2006) A-beta and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer disease. Proc Natl Acad Sci USA 103:1953–1958
Hasegawa M, Arai T, Akiyama H, Nonaka T, Mori H, Hashimoto T, Yamazaki M, Oyanagi K (2007) TDP-43 is deposited in the Guam parkinsonism–dementia complex brains. Brain 130(Pt 5):1386–1394
Hirano A, Kurland LT, Krooth RS, Lessel S (1961) Parkinsonism–dementia complex, an endemic disease on the island of Guam. I. Clin Features Brain 84:642–661
Hirano A, Malamud N, Kurland LT (1961) Parkinsonism–dementia complex, an endemic disease on the island of Guam. II. Pathological features. Brain 84:662–679
Hirano A, Malamud N, Elizan TS, Kurland LT (1966) Amyotrophic lateral sclerosis and parkinsonism–dementia complex on Guam: further pathologic studies. Arch Neurol 15:35–51
Hirano A, Dembitzer HM, Kurland LT, Zimmerman HM (1968) The fine structure of some intraganglionic alterations: neurofibrillary tangles, granulovacuolar bodies, and “rod-like” structures as seen in Guam amyotrophic lateral sclerosis and parkinsonism–dementia complex. J Neuropathol Exp Neurol 27:167–182
Hof PR, Perl DP, Loerzel AJ, Morrison JH (1991) Neurofibrillary tangle distribution in the cerebral cortex of parkinsonism–dementia cases from Guam: differences with Alzheimer’s disease. Brain Res 564:306–313
Hof PR, Nimchinsky EA, Buée-Scherrer V, Buée L, Nasrallah J, Hottinger AF, Purohit DP, Loerzel AJ, Steele JC, Delacourte A et al (1994) Amyotrophic lateral sclerosis/parkinsonism–dementia complex of Guam: quantitative neuropathology, immunohistochemical analysis of neuronal vulnerability, and comparison with related neurodegenerative disorders. Acta Neuropathol 88:397–404
Ito H, Hirano H, Yen SH, Kato S (1991) Demonstration of beta amyloid protein-containing neurofibrillary tangles in parkinsosm–dementia complex on Guam. Neuropathol Appl Neurobiol 17:365–373
Kurland LT, Mulder DW (1954) Epidemiological investigations of amyotrophic lateral sclerosis: 1. Preliminary report on geographic distribution with special reference to the Mariana Islands, including clinical and pathological observations. Neurology 4:355–378
Lee VM-Y, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159
Mawal-Dewan M, Schmidt L, Balin B, Perl DP, Lee VMY, Trojanowski JQ (1996) Identification of phosphorylation sites in PHF-tau from patients with Guam amyotrophic lateral sclerosis/parkinsonism–dementia complex. J Neuropathol Exp Neurol 55:1051–1059
McGeer PL, Schwab C, McGeer EG, Haddock RL, Steele JC (1997) Familial nature and continuing morbidity of the amyotrophic lateral sclerosis-parkinsonism–dementia complex of Guam. Neurology 49:400–409
Miklossy J, Arai T, Guo JP, Klegeris A, Yu S, McGeer EG, McGeer PL (2006) LRRK2 expression in normal and pathologic human brain and in human cell lines. J Neuropathol Exp Neurol 65:953–963
Morris HR, Al-Sarraj S, Schwab C, Gwinn-Hardy K, Pérez-Tur J, Wood NW, Hardy J, Lees AJ, McGeer PL, Daniel SE, Steele JC (2001) A clinical and pathological study of motor neurone disease on Guam. Brain 124:2215–2222
Morris HR, Steele JC, Crook R, Wavrant-De Vrièze F, Onstead-Cardinale L, Gwinn-Hardy K, Wood NW, Farrer M, Lees AJ, McGeer PL, Siddique T, Hardy J, Perez-Tur J (2004) Genome-wide analysis of the parkinsonism–dementia complex of Guam. Arch Neurol 61:1889–1897
Mulder DW, Kurland LT, Iriarte LLG (1954) Neurological diseases on the island of Guam. US Armed Forces Med J 5:1724–1739
Nakano I, Hirano A (1983) Neuron loss in the nucleus basalis of Meynert in parkinsonism–dementia complex of Guam. Ann Neurol 13:87–91
Oyanagi K, Makifuchi T, Ohtoh T, Chen KM, van der Schaaf T, Gajdusek DC, Chase TN, Ikuta F (1994) Amyotrophic lateral sclerosis of Guam: the nature of the neuropathological findings. Acta Neuropathol 88:405–412
Oyanagi K, Makifuchi T, Ohtoh T, Chen KM, Gajdusek DC, Chase TN, Ikuta F (1994) The neostriatum and nucleus accumbens in parkinsonism–dementia complex of Guam: a pathological comparison with Alzheimer’s disease and progressive supranuclear palsy. Acta Neuropathol 88:122–128
Oyanagi K, Makifuchi T, Ohtoh T, Chen KM, Gajdusek DC, Chase TN (1997) Distinct pathological features of the Gallyas- and tau-positive glia in the Parkinsonism–dementia complex and amyotrophic lateral sclerosis of Guam. J Neuropathol Exp Neurol 56:308–316
Pérez-Tur J, Buée L, Morris HR, Waring SC, Onstead L, Wavrant-De Vrièze F, Crook R, Buée-Scherrer V, Hof PR, Petersen RC, McGeer PL, Delacourte A, Hutton M, Siddique T, Ahlskog JE, Hardy J, Steele JC (1999) Neurodegenerative diseases of Guam: analysis of TAU. Neurology 53:411–413
Plato CC, Galasko D, Garruto RM, Plato M, Gamst A, Craig UK, Torres JM, Wiederholt W (2002) ALS and PDC of Guam: forty-year follow-up. Neurology 58:765–773
Plato CC, Garruto RM, Galasko D, Craig UK, Plato M, Gamst A, Torres JM, Wiederholt W (2003) Amyotrophic lateral sclerosis and parkinsonism–dementia complex of Guam: changing incidence rates during the past 60 years. Am J Epidemiol 157:149–157
Schmidt ML, Lee VM, Saido T, Perl D, Schuck T, Iwatsubo T, Trojanowski JQ (1998) Amyloid plaques in Guam amyotrophic lateral sclerosis/parkinsonism–dementia complex contain species of A beta similar to those found in the amyloid plaques of Alzheimer’s disease and pathological aging. Acta Neuropathol 95:117–122
Schwab C, Steele JC, Akiyama H, McGeer EG, McGeer PL (1995) Relationship of amyloid β/A4 protein to the neurofibrillary tangles in Guamanian parkinsonism–dementia. Acta Neuropathol 90:287–298
Sebeo J, Hof PR, Perl DP (2004) Occurrence of alpha-synuclein pathology in the cerebellum of Guamanian patients with parkinsonism–dementia complex. Acta Neuropathol 107:497–503
Steele JC, Richardson JC, Olszewski J (1964) Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 10:333–359
Sundar PD, Yu C-E, Sieh W et al (2007) Two sites in the MAPT region confer genetic risk for Guam ALS/PDC and dementia. Human Mol Gen 16:295–306
Wiederholt WC (1999) Neuroepidemiologic research initiatives on Guam: past and present. Neuroepidemiology 18:279–291
Winton MJ, Joyce S, Zhukareva V, Practico D, Perl DP, Galasko D, Craig U, Trojanowski JQ, Lee VM (2006) Characterization of tau pathologies in gray and white matter of Guam parkinsonism–dementia complex. Acta Neuropathol 111:401–412
Yamazaki M, Arai Y, Baba M, Iwatsubo T, Mori O, Katayama Y, Oyanagi K (2000) Alpha-synuclein inclusions in amygdala in the brains of patients with the parkinsonism–dementia complex of Guam. J Neuropathol Exp Neurol 59:585–591
Yoshiyama Y, Lee VM-Y, Trojanowski JQ (2001) Frontotemporal dementia and tauopathy. Curr Neurol Neurosci Rep 1:413–421
Zimmerman H (1945) Progress report of work in the laboratory of pathology during May 1945. Guam. US Naval Medical Research Unit Number 2, 1 June. Washington, DC
Acknowledgments
This paper was supported by a Grant from the Pacific Alzheimer Research Foundation. We thank H. Akiyama, L. Binder, B. Giasson, H. Mori and K. Obi for their generous gifts of key antibodies and H. Martin and J. P. Guo for technical assistance in preparing figures.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Electronic supplementary material (MPG 264 kb)
Rights and permissions
About this article
Cite this article
Miklossy, J., Steele, J.C., Yu, S. et al. Enduring involvement of tau, β-amyloid, α-synuclein, ubiquitin and TDP-43 pathology in the amyotrophic lateral sclerosis/parkinsonism–dementia complex of Guam (ALS/PDC). Acta Neuropathol 116, 625–637 (2008). https://doi.org/10.1007/s00401-008-0439-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-008-0439-2