
Abstract
Cancer cells are disordered by nature and thus featured by higher internal redox level than healthy cells. Redox imbalance could trigger programmed cell death if exceeded a certain threshold, rendering therapeutic strategies relying on redox control a possible cancer management solution. Yet, various programmed cell death events have been consecutively discovered, complicating our understandings on their associations with redox imbalance and clinical implications especially therapeutic design. Thus, it is imperative to understand differences and similarities among programmed cell death events regarding their associations with redox imbalance for improved control over these events in malignant cells as well as appropriate design on therapeutic approaches relying on redox control. This review addresses these issues and concludes by bringing affront cold atmospheric plasma as an emerging redox controller with translational potential in clinics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer is a complex disease, the initiation and development of which requires intensive cross talks with its microenvironment and is highly regulated by cellular redox level. Reactive oxygen species (ROS) have been shown to play diverse roles in many critical transition stages of cancer cells such as the life/death transition, tumor angiogenetic switch, and epithelial mesenchymal transition (EMT) [1]. Cancer cells typically have a relatively higher redox level than normal cells due to their chaoticities in organizing cellular functionalities, rendering malignant cells more fragile under redox stress than healthy cells. On the other hand, cancer cells are naturally faced with oxidative stress as a result of imbalanced ROS production and disordered anti-oxidant defense ability [2]. That is, ROS are excessively generated in malignant cells due to increased metabolic rate, accumulated mitochondria dysfunction, elevated cell signaling, enhanced expression of oncogenes, and accelerated peroxisome activities [3], which is a required feature of malignant cells. Therapeutic strategies taking advantages of redox stress may kill cancer cells by triggering programmed cell death (PCD) events. PCD such as apoptosis, paraptosis, mitotic catastrophy, autophagic cell death, ferroptosis, necroptosis, and pyroptosis represents a set of highly ordered and programmed cellular death events that enable the elimination of cells running chaotic or being destined to die. Failed PCD may lead to uncontrolled cell proliferation that is one important cancer hallmark [4]. Thus, it is important to explore differences and commonalities of these varied PCD programs as well as their differential associations with cellular redox imbalance to enable our improved understandings and design on onco-therapeutic strategies relying on redox level control.
Among the various PCD programs identified so far, we selected apoptosis, paraptosis, mitotic catastrophy, autophagic cell death, ferroptosis, necroptosis, and pyroptosis in this review given their representativeness on the stimulating source and their prevalence in literatures. We focus on differences of these diverse PCD events and their associations with redox imbalance in this review, with the aim of differentiating these PCD events by the source of redox imbalance, identifying the link of these programs, and pushing forward possible onco-therapeutic solutions that rely on redox control.
Programmed cell death associated with redox imbalance imposed by DNA damage
Apoptosis
Basics of apoptosis
Apoptosis is the most well-known PCD event that is carefully regulated by many cellular processes to balance cell turnover in proliferating tissues and selectively remove cells that hamper proper organ development and functioning [5]. Apoptosis is accompanied by membrane microvilli loss, cytoplasm condensation, nucleus segmentation, and chromosomal DNA degradation into 180 bp oligomers. During early apoptosis, cells shrink and undergo pyknosis [6] as a result of chromatin condensation [7].Then, budding (i.e., forming a wide range of plasma membrane bubbles) occurs followed by nucleus break and separation of cellular debris corpuscles into the apoptosis body. These small apoptosis bodies are phagocytosed by macrophages, parenchymal cells, or neoplastic cells and degraded within phagolysosomes. Yet, the organelle integrity remains, which is enclosed within the intact plasma membrane.
One important feature of apoptosis is the flipping of phosphatidyl serine groups to the outer membrane surface that enables the common strategy of apoptosis detection through combined use of annexin V and cell-impermeable DNA staining dye such as propidium (PI) or 7-amino-actinomycin (7-AAD) followed by fluorescent microscopy or flow cytometry [8]. Double-stranded breaks could be identified through terminal de-oxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end labeling (TUNEL) technique and comet assay [9]. Alternatively, caspase assay and poly-ADP ribose polymerase (PARP) cleavage assay [10] could be used to evaluate the intermediate modulators of apoptosis.
Caspase-dependent apoptosis can be endogenous or exogenous.
Endogenous apoptosis pathway
The endogenous pathway is originated from mitochondria that can be triggered by a variety of environmental and chemical stimuli capable of imposing oxidative stress. When cell redox homeostasis is disrupted, the mitochondrial outer membrane permeability alters that leads to the release of cytochrome C from mitochondria to the cytoplasm; cytochrome C forms complexes with Apaf-1 that further recruits caspase-9 to form the apoptosome in the cytoplasm through the CARD domain; caspase-9 is self-cut followed by caspase-3 activation to initiate apoptosis (Fig. 1).

Graphic illustration on the crosstalk and differences of 3 example DNA damage associated PCD events regarding the mechanism and phenotype
Exogenous apoptosis pathway
The exogenous pathway is mediated by death receptors. Taking Fas as an example, it trimerizes and recruits FADD and procaspase-8 through the cytoplasmic domain to form the death inducing signaling complex (DISC). Through self-cutting, procaspase-8 becomes caspase-8 that cuts procaspase-3 into caspase-3, the executioner of apoptosis (Fig. 1).
Apoptosis in response to DNA damage induced redox imbalance
A variety of factors or chemicals capable of initiating DNA damage signals such as ionizing radiation, ultraviolet radiation and H2O2, etc., can cause redox imbalance and trigger apoptosis [11].
DNA damage is an inciting cause of endoplasmic reticulum (ER) stress that typically occurs when proteins are not or not properly folded. On ER stress, Bax and Bak in the ER membrane allow Ca2+ release from ER to the cytosol where it activates m-Calpain and subsequently caspase-12. This, on one hand, leads to a sequential activation of caspase family members including procaspase-9 and caspase-3; and, on the other hand, causes mitochondrial inner membrane depolarization and the release of cytochrome C to the cytoplasm. Consequently, apoptosome forms, a prerequisite for endogenous caspase-dependent apoptosis [12]. In addition, ER stress can suppress the anti-apoptotic functions of Bcl2 and activate pro-apoptotic proteins such as Bim, Bax and Bak through activating c-Jun N-terminal kinase (JNK) and c/EBP homologous protein (CHOP) [13,14,15,16], linking caspase-independent apoptosis with the ROS/JNK pathway as reported in breast cancer cells [17]. Mammals express at least three different MAPKs, including extracellular signal- regulated kinase (ERK), JNK and p38. These kinases share 60–70% similarity but differ in the signal they sense and the size of the avalanche they trigger. While ERK is stimulated by proliferative signals, JNK and p38 respond to environmental stimuli including ER stress [18].
Reactive oxygen intermediates (ROI) can react with all kinds of unsaturated fatty acids and cholesterols on the cell membrane to generate oxidative damage that can directly initiate cell apoptosis [19]. On the other hand, ROI could cause DNA damage that leads to poly ADP-ribose polymerase (PARP) activation and p53 accumulation [20]. P53 accumulation could activate p21 transcription that arrests cells at the G1 phase until DNA damage is repaired. Otherwise, p53 will continue accumulating to trigger apoptosis through increasing the expression of the pro-apoptotic factor Bax and reducing that of the anti-apoptotic factor Bcl2 [21]; and/or induce apoptosis through activating death receptors such as TNF receptor and Fas [21].
Clinical relevance of apoptosis in cancer treatment
Tumor cells can develop resistance to apoptotic agents and suppress apoptosis by, e.g., up-regulating anti-apoptotic proteins such as Bcl-2 or down-regulating/mutating pro-apoptotic proteins such as Bax [22]. Members of the steroid/retinoid superfamily of ligand-activated transcription factors (SRTFs) could modulate the transcription level of Bcl-2 and Bcl-xL. For example, as Bcl-2 expression is estrogen-dependent in the mammary gland, anti-estrogens such as tamoxifen could inhibit Bcl-2 expression in breast cancer cells and thus promote the sensitivity of tumor cells to anti-cancer drugs [22]. TNF family cytokines, TRAIL and agonistic antibodies against TRAIL receptors have been demonstrated to possess potent antitumor activity [23]. Synthetic triterpenoids such as CDDO and CDDO lm can sensitize solid tumor cells to TRAIL induced apoptosis that functions in both chemo-sensitive and chemo-refractory tumor cells [24,25,26].
Commercialized onco-therapeutic products triggering apoptosis in cancer cells include CDDO for solid tumor treatment [24,25,26], and Venetoclax for treating acute myeloid leukemia and small lymphocytic lymphoma [27] (Table 1). Many drugs have been under clinical trials, including ABBV-621 (NCT: 03082209) [28], GEN1029 (NCT: 3576131) [29], ALRN-6924 (NCT: 03725436) [30], BI907828 (NCT: 03449381) [31]for the treatment of malignant solid tumors, ABBV-155 (NCT: 03595059) for treating refractory solid tumors [32], ABT-737 (NCT: 00902018) for treating small cell lung cancer and hematological tumors [33], APG-1252 (NCT: 03080311) for the treatment of solid tumors as represented by small cell lung cancer [34], Siremadlin (NCT: 04097821) for treating uveal melanoma [35], SMACmimetic (NCT: 02890069) for the treatment of breast cancer [36], Lexatumumab (NCT: 00428272) for treating osteosarcoma, neuroblastoma and pancreatic cancer [37], Mapatumumab (NCT: 00315757) for the treatment of multiple myeloma, renal carcinoma, bladder cancer, etc. [38], MIK665 (NCT: 02992483) for the treatment of multiple myeloma and lymphoma [39], Navitoclax (NCT: 01557777) [40] and APG-2575 (NCT: 03913949) [41] for the treatment of chronic lymphocytic leukemia, AMG176 (NCT: 02675452) for treating chronic lymphocytic leukemia and acute myeloid leukemia [42], APR-246 (NCT: 04214860) for treating myeloid malignancy [43], and AZD5991 (NCT: 03218683) for the treatment of blood tumors [44] (Table 1).
Paraptosis
Basics of paraptosis
Paraptosis is a form of PCD displaying mitochondria swelling and/or ER and cytoplasmic vacuolization [45, 46]. It differs from apoptosis in that it is not affected by caspase inhibitors or anti-apoptotic proteins such as Bcl2 [47]. Paraptosis is induced by insulin-like growth factor I receptor (IGF-IR) and suppressed by ALG-2-interacting protein (AIP1) (Fig. 1). IGF-IR induced paraptosis is primarily mediated via mitogen-activated protein kinase (MAPK) family members.
No assay is so far available for paraptosis detection except for electron microscopy where the appearance of multiple single-membraned cytoplasmic vacuoles could be considered as a symbol of paraptosis [48].
Paraptosis in response to DNA damage imposed redox imbalance
Paraptosis could be induced through ER stress as a result of DNA damage signaling. Lots of studies have suggested the association of paraptosis with redox imbalance and accumulation of misfolded proteins in ER [49, 50]. It was reported that ginger extract triggers cytoplasmic vacuolation, ER dilation, mitochondrial dysfunction, and DNA fragmentation in response to DNA damage induced ER stress that ultimately leads to paraptosis as a result of excess ROS generation [51]. As a DNA damage response sensor, p53 was reported to suppress paraptosis through inhibiting IGF-IR and transactivating IGF-BP3 expression, whereas the binding of IGF-BP3 to IGFs suppresses IGF-IR signaling [52, 53].
Clinical relevance of paraptosis in cancer treatment
Many natural compounds such as taxol, cyclosporine A, tunicamycin, procyanidins, curcumin, honokiol, ginsenosides, tocotrienols, celastrol, ophobiolin A, hesperidin, morusin, 6-shogaol, chalcomoracin, gambogic acid, plumbagin, 8-p-hdroxybenzoyl tovarol, cis-nerolidol, manumycin A, DL-selenocystine, 15-deoxy-Δ12,14-prostaglandin J2, yessotoxin and 1-desulfoyessotoxin have shown great promise and translational potential in triggering paraptosis in a variety of human cancer cell lines [54]. Among them, taxol [55, 56] (for treating ovarian, breast, and lung cancers) and curcumin (for treating colorectal cancer) [54] have been commercialized for clinical use (Table 1).
Mitotic catastrophe
Basics of mitotic catastrophe
Mitotic catastrophe is a form of PCD due to failed or incomplete mitosis, which is featured by chromosome breaks and poor karyokinesis [57]. During mitosis, the CDK1/cyclin B1 complex promotes the G2/M cell cycle transition and plays essential roles in microtubule reorganization, chromatin condensation, and nuclear membrane breakdown [58] (Fig. 1). Over-activated CDK1 can lead to mitotic catastrophe [59]. Prolonged suppression of anaphase-promoting complex (APC) can result in mitotic catastrophe [59] as APC could degrade cyclin B to promote cells transit from the metaphase to anaphase [58].
Dis-functionalities of cell cycle checkpoint regulators are vital to initiate mitotic catastrophe. Inhibition of G2/M checkpoint regulators such as checkpoint kinase 1/2 (Chk1/2), ataxia telangiectasia mutated (ATM), ataxia telangiectasia mutated and Rad3 related (ATR) are known to induce mitotic catastrophe [60, 61]. Chemotherapies such as 5-fluorouracil and doxorubicin were reported to trigger mitotic catastrophe through increasing cyclin B1 [62]. CDK1 promotes mitotic catastrophe through inhibiting the phosphorylation of survivin, a protein that promotes mitotic progression and inhibits apoptosis [63].
Mitotic catastrophe has been conventionally detected via continuous observation under microscopy. An automated fluorescence videomicroscopy assay was developed for the real-time detection of mitotic catastrophe in a high-throughput fashion[64].
Mitotic catastrophe in response to DNA damage imposed redox imbalance
Mitotic catastrophe is triggered by DNA damage signals that can be sensed by p53. P53 is a gatekeeper of genome integrity and inhibits mitotic catastrophe through various pathways including, e.g., transcriptionally inhibiting CDK1 and cyclin B1 [65, 66], and upregulating the expression of CDK1 inhibitors such as p21 [67]. Thus, mitotic catastrophe occurs predominantly in p53-deficient cells as a result of genomic instability [68]. On the other hand, p53 may also play a promotive role in mitotic catastrophe as it could transcriptionally repress surviving [69].
Clinical relevance of mitotic catastrophe in cancer treatment
Induction of mitotic catastrophe such as anti-mitotic agents has been implicated as an efficient strategy for cancer management. Microtubule destabilizers such as vinblastine [70] and vincristine [71] have been used in the treatment of hematological malignancies; microtubule stabilizers such as taxanes drugs paclitaxel [72] and docetaxel [73] have been applied in clinics for treating ovarian, breast cancer, peritoneal malignancy, and for treating breast cancer, ovarian cancer, and non-small cell lung cancer, respectively (Table 1). Drugs taking advantages of mitotic catastrophe and are being under clinical trials include BI2536) for treating non-small cell lung cancer [74], ON01910) for treating chronic lymphocytic leukemia [75], and MK-1775) for the treatment of acute lymphoblastic leukemia and nasopharynx cancer [76] (Table 1).
Programmed cell death associated with redox imbalance imposed by metabolic stress
Autophagic cell death
Basics of autophagic cell death
Autophagic cell death is a type of PCD as a result of autophagy [45]. Autophagy is a regulated catabolic lysosomal-dependent process that facilitates cells to eliminate misfolded proteins, damaged or non-functional cellular components, etc. to maintain cellular homeostasis [77]. When autophagy occurs, cytoplasmic contents are sequestered by phagophores to form autophagosomes that merge with lysosomes and form autophagolysomes to degrade engulfed materials, where ATG proteins and beclin-1 are well-known regulators of autophagy [78] (Fig. 2). Mammalian target of rapamycin (mTOR) can inhibit autophagy through suppressing ATG13 and ULK1 [79], where the complex formed by ATG13, ULK1 and FIP200 plays essential roles in phagophore formation and autophagy [79]. Beclin-1 was shown to induce autophagic cell death by promoting autophagosome formation [80].

Graphic illustration on the crosstalk and differences of 2 example metabolic disorder associated PCD events regarding the mechanism and phenotype
Autophagy is a self-protective phenomenon in response to cellular stress such as metabolic stress, whereas cells are committed to death if the cellular stress is irreversible. Autophagic cell death is featured by the presence of large intracellular vesicles, membrane blebbing, enlarged organelles, and depletion of cytoplasmic organelles without chromatin condensation [81].
Autophagic cell death could be detected through direct autophagic activity measurement or indirect antibody (autophagy specific) based analyses such as western blot, fluorescence microscopy and flow cytometry [82].
Autophagic cell death in response to metabolic stress associated redox imbalance
Metabolic stress is characterized by nutrient, oxygen and growth factor deprivation [83]. Autophagic cell death is regulated by the endocrine system on nutrient starvation that constitutes a primary source of metabolic stress in cancer cells. Specifically, glucose concentration in malignant cells is estimated to be 3-to-10 folds lower than that in normal tissues [84, 85]. This nutrient deficiency directly reduces ATP production and results in excess ROS generation [86].
In response to metabolic stress imposed redox imbalance, p53 plays dual roles in autophagic cell death depending on its cellular localization. While nuclear p53 promotes autophagy, cytoplasmic p53 suppresses it [87]. Nuclear p53 triggers autophagy by transactivating its target genes [87, 88]. For example, nuclear p53 promotes autophagy through transactivating TSC2 and AMPK, both of which are upstream regulators of mTOR [88, 89]. P53 can also upregulate the AMPK activators sestrins ½ [90] and the autophagy activator DRAM [91]. Besides, apoptosis-associated genes under p53 regulation such as PUMA, BAX, Bnip3, and BAD are also involved in autophagy [92,93,94]. On the other hand, cytoplasmic p53 suppresses autophagy through binding Beclin-1 that promotes its ubiquitination and degradation [95].
Clinical relevance of autophagic cell death in cancer treatment
Impaired autophagy has been associated with many diseases including human cancers [96]. Inhibitors of autophagy such as chloroquine and hydroxychloroquine have been proposed for the treatment of multiple malignancies in clinics despite the questionable unresolved safety issues [97].
Clinical therapeutic strategies using autophagic cell death include everolimus [98] and CCI-779 [99] for the treatment of renal cell carcinoma, as well as vorinostat for treating cutaneous T-cell lymphoma [100] (Table 1).
Ferroptosis
Basics of ferroptosis
Ferroptosis, an iron-dependent oxidative PCD, was firstly discovered in 2012 by Brent R. Stockwell et al. [101]. Different from apoptosis, cells undergoing ferroptosis do not show cell shrinkage and chromatin agglutination, but exhibit increased lipid peroxidation, elevated ROS, shrinked mitochondria, increased mitochondria membrane density and decreased ridge amount. While inhibitors of cell apoptosis, autophagy, pyroptosis do not inhibit ferroptosis, this process can be suppressed by iron chelators, lipophilic antioxidants, lipid peroxidation inhibitors, and polyunsaturated fatty acid depletion [102]. Ferroptosis inducers such as erastin decreases cellular antioxidant capacity through acting on the glutathione peroxidase (GPXs) [103].
Essential ferroptosis suppressor genes have been identified that can be used in ferroptosis detection. Glutathione peroxidase 4 (GPX4), an antioxidant defense enzyme, functions to repair lipid oxidative damage and plays a primary suppressive role in ferroptosis [104, 105]. Ferroptosis suppressor protein 1 (FSP1) is a GSH-independent ferroptosis suppressor [106] that co-operates with coenzyme Q10 [107] and acts in parallel with GPX4 to inhibit ferroptosis [108]. Several ferroptosis-promotive markers have also been reported. For example, acyl-CoA synthetase long-chain family member 4 (ACSL4) expression was remarkably lower in ferroptosis-resistant than -sensitive cells which, once knocked down, inhibited erastin-induced ferroptosis [109]. Other pro-ferroptosis factors include, e.g., cox-2, PTGS2, NOX1 [110], and transferrin receptor 1 (TfR1) [111]. The lethal accumulation of lipid peroxides in plasma membranes as characterized by ferroptosis makes it possible to detect ferroptosis through determining the amount of lipid peroxides in cellular membranes using BODIPY-C11 probe and flow cytometry [112].
Ferroptosis has two primary pathways, i.e., the canonical GPX4-meidated GSH-dependent pathway and the newly identified FSP1-meidated GSH-independent pathway.
GPX4-mediated GSH-dependent pathway
When intracellular cystine transport proteins such as erastin are inhibited, intracellular GSH will be exhausted as a result of substrate shortage. This will eventually lead to the inactivation of GPX4, a GSH peroxidase and perhaps the only enzyme that catalyzes liposome peroxide reduction. When cellular redox level exceeds a certain threshold, this will result in lipid peroxidation accumulation that, ultimately, triggers cell ferroptosis [113, 114] (Fig. 2).
GPX4-mediated GSH-dependent ferroptosis can be initiated by directly eliminating GPX4 using inhibitors such as squalene synthase, HMG-CoA reductase, and RSL3 [101]; inputting iron in the form of ferrous iron ions, or consuming GSH. Ferroptosis can also be triggered by inhibiting cystine/glutamate antiporter, namely system xc−, which is a heterodimeric cell surface amino acid antiporter composed of the twelve-pass transmembrane transporter protein SLC7A11 (xCT) and the single-pass transmembrane regulatory protein SLC3A2 [115]. System xc− imports extracellular cysteine proteinase inhibitor 2 (Cys2) in exchange for intracellular GSH [101, 116]. Through inhibiting system xc−, cysteine absorption is inhibited that leads to reduced GSH and GPX4 activity [117].
FSP1-mediated GSH-independent pathway
FSP1 was recently identified as a key component of a non-mitochondrial CoQ antioxidant system that acts in parallel to canonical GPX4-mediated GSH-dependent ferroptosis [108]. In particular, FSP1 myristoylation allocates itself to the plasma membrane and lipid droplets where it reduces coenzyme Q10 (CoQ10) via functioning as an oxidoreductase, and this results in the generation of a lipophilic radical-trapping antioxidant (RTA) that ultimately halts lipid peroxide propagation (Fig. 2).
Ferroptosis in response to metabolic stress associated redox imbalance
Lipid peroxidation is initiated as a result of chain oxidation of unsaturated lipids due to excess ROS accumulation and represents a lipid metabolic disorder [118, 119]. Metabolic stress, in turn, hampers the homoeostasis between fatty acids synthesis and uptake [120, 121].
The tumor suppressor p53 senses metabolic disorder associated redox stress [122] and triggers ferroptosis through p53-mediated suppression on system xc−. By exposing p53-deficient H1299 cells to ROS, cell viability did not alter; yet cells carrying the wildtype p53 underwent a mortality rate as high as 90% under ROS stress; by treating cells with ferrostatin 1 (a ferroptosis inhibitor), the mortality rate of cells dropped about 40% [123], suggesting the role of p53 played in ferroptosis and ROS stress response. The same study also reported a negative correlation between p53 and SLC7A11 [123], implicating that SLC7A11, a core component of system xc−, is a p53 target.
Clinical relevance of ferroptosis in cancer treatment
Erastin, the earliest discovered inhibitor, can effectively inhibit the growth of ovarian tumors cells in mice [124]. Glutathione synthetase inhibitors such as L-Buthionine-sulfoximine (L-BSO) can inhibit breast tumor growth in mice via suppressing GSH formation and thus triggering ferroptosis [125]. Ferroptosis has also been applied in cancer diagnosis. For instance, a paper published in 2020 identified a ferroptosis gene panel composed of 8 genes (ALOX5, CISD1, FTL, CD44, FANCD2, NFE2L2, SLC1A5, GOT1) that is diagnostic of low-grade glioma [126].
Cisplatin has been applied in clinics for treating solid tumors through triggering ferroptosis [127] (Table 1). Sorafenib [128], sulfasalazine [129] and ferumoxytol [130] have been approved by FDA for the treatment of renal cell carcinoma and hepatocellular carcinoma, for the treatment of pancreatic cancer, and for the treatment of leukemia, respectively (Table 1).
Programmed cell death associated with redox imbalance imposed by inflammation
Necroptosis
Basics of necroptosis
Necroptosis is a programmed form of necrosis that is characterized by permeable and rupture of cell membranes, numerous cytoplasmic vacuoles containing cellular remnants, and inflammation [131,132,133]. It is also accompanied with moderate chromatin condensation and clumping, as well as random DNA degradation [131,132,133].
Necroptosis is initiated by the binding of tumor necrosis factor (TNF) or FAS to their receptors that promotes interactions between RIPK1 and RIPK3 [134, 135], followed by activation of these kinases that recruits MLKL to form the necrosome [136] (Fig. 3). RIPK3 phosphorylates MLKL at threonine 357 and serine 358 to enhance its oligomerization; and then MLKL oligomers are translocated from the cytosol to cell membranes to disrupt membrane integrity, triggering necroptosis [137].

Graphic illustration on the crosstalk and differences of 2 example inflammation associated PCD events regarding the mechanism and phenotype
Necroptosis can be detected by examining the loss of membrane integrity through the use of cell-impermeable DNA binding dyes, measuring the release of cellular contents such as LDH, HMGB1 and cyclophilin A via western blot, evaluating mitochondrial potential by fluorescent probes, and observing necroptosis morphology features through electron microscopy [138]. Alternatively, necroptosis can be detected with the aid of necroptosis specific inhibitors such as necrostatin-1 [139].
Necroptosis in response to inflammation associated redox imbalance
Inflammation and oxidative stress are interconnected. It was shown that inflammatory macrophages release proteins in the glutathionylated form such as PRDX2 [140]. Extracellular PRDX2 mediates inflammation in a redox-dependent manner through the release of TNFα, a primary tumor necrosis factor involved in inflammation, from macrophages [140].
In response to inflammation associated oxidative stress, p53 is accumulated in the mitochondrial matrix that enhances the opening of mitochondrial permeability transition pore (PTP) via direct binding of p53 with a PTP regulator cyclophilin D (cypD), and this leads to mitochondrial swelling and necroptosis induction [141]. Besides, p53 transcriptionally upregulates the lncRNA NRF (necrosis-related factor) that enhances PIRK1/PIRK3 translation via repressing miR-873, and elevated RIPK1/RIPK3 is a direct trigger of necroptosis [142].
Clinical relevance of necroptosis in cancer treatment
Triggering necroptosis has been recently proposed as a novel onco-therapeutic strategy, with the feasibility still being controversial. Conventional necroptosis inducers or chemotherapeutic agents can trigger necrosis in many cancer cells especially colorectal cancers and hematopoietic tumors such as leukemia and multiple myeloma [143]. Traditional chemotherapy and molecular targeted drugs such as VEGFR inhibitors and m-TOR inhibitors have recently been identified as cancer necroptosis triggers and approved for clinical trials [144, 145].
Natural products such as shikonin have been shown capable of triggering necroptosis and used in clinics for bladder cancer treatment [146]. Pazopanib [147] and ponatinib [148] have received FDA approval for treating advanced renal cell carcinoma and soft-tissue sarcoma, and for treating chronic myeloid leukemia. respectively (Table 1).
Pyroptosis
Basics of pyroptosis
Pyroptosis is another proinflammatory form of PCD that relies on the enzymatic activity of inflammatory proteases belonging to the cysteine-dependent aspartate-specific protease (caspase) family [149]. Destructed cell membrane structure integrity and release of intracellular substances into the extracellular space are the most notable features of pyroptosis. With the activation of caspases-1, ostioles are formed on cell membrane that leads to the outflow of intracellular contents including pro-inflammatory cytokines, intracellular ions, endogenous ligands, alarmins etc., and finally contributes to cell dissolvement. This feature is similar with caspase-independent PCD such as necroptosis, yet differs significantly from apoptosis that maintains intact cell membrane structure.
Both pyroptosis and necroptosis form pores on the cell membrane surface whereas apoptosis does not. Necroptosis is more like a burst of cells, and cells undergoing pyroptosis become tabular as a result of cytoplasm leak to the extracellular space. Both necroptosis and pyroptosis require the oligomerization and transfer of their execution proteins to the plasma membrane. However, during necroptosis, mixed lineage kinase domain like pseudokinase (MLKL) triggers specific ion influx through inducing the ion selective channel, which leads to cell osmotic swelling and burst; during pyroptosis, gasdermin D (GSDMD) forms holes that lack ion specificity and selectivity, and this does not lead to increased intracellular osmotic pressure nor consequent cell inflate or burst [150].
Pyroptosis can be detected via western blot using antibodies against GSDMD (53 Kd) that is cut into 30 Kd pieces during pyroptosis [151]. It can also be assayed through ELISA by detecting IL-1β and IL-18 that are released into cells when pyroptosis occurs [149]. Pyroptosis can also be detected through detecting the activity of caspase-1 and caspase-4 using the corresponding kits based on spectrophotometry [151].
Pyroptosis can occur via canonical caspase-1 dependent pyroptosis or non-canonical caspase-1 independent pyroptosis.
Caspase-1 dependent pyroptosis
In response to pathogen associated molecular patterns (PAMPs) such as viral pathogens toxins, bacteria, parasites, etc. and danger associated molecular patterns (DAMPs) such as uric acid sodium and silicon dioxide, pattern recognition receptors (PRRs) are triggered to activate caspase-1 (Fig. 3). PRRs can be categorized into two classes based on cellular localization. While C-type lectin receptors (CLRs) and Toll-like receptors (TLRs) are present on cell membranes to sense external stimuli, the nucleotide-binding domain and leucine-rich repeat-containing (NLR) proteins, the AIM2-like receptor (ALR), RIG-I-like receptor (RLR) are localized inside the cell to detect signals issued by PAMPs or DAMPs [152].
The canonical inflammasome or namely pyroptosome that participates in the caspase-1-dependent pathway is composed of apoptosis-associated speck-like protein (ASCs), PRRs such as NLRs and ALRs, and procaspase-1 [153]. ASCs contain a caspase-activation and recruitment domain (CARD) and are recruited by NLRs/ALRs to form a complex called ‘SPECK’ that takes on the most important function of pyroptosome, i.e., activating caspase-1 [153, 154]. The NLR family is further divided into NLR family PYD-containing protein (NLRP) and NLR family CARD-containing protein (NLRC) receptors based on differences in the N-terminal domain composition [153]. Among all NLR family members, NLRP1, NLRP3, NLRC4 are the most intensively studied in pyroptosis, where NLRP3 relies on ASC to recruit procaspase-1, and NLRP1 and NLRC4 do not [155].
Caspase-1 independent pyroptosis
The non-canonical caspase-1 independent pathway is primarily induced by caspase-4/5/11 other than caspase-1. Similar with caspase-1, caspase-4/5/11 also harbor the CARD domain at the N terminal. Caspase-4/5/11 directly interact with lipopolysaccharide (LPS) to form non-canonical pyroptosome that directly lyses the substrate and triggers pyroptosis [156] (Fig. 3). Activated caspase-11 can induce the formation of cell membrane pores through GSDMD cleavage, and the produced N terminus can also activate caspase-1 [157], rendering caspase-11 another trigger of caspase-1 activation and bridging the gap between canonical caspase-1 dependent and non-canonical caspase-1 independent pathways. Caspase-4 and 5 are crucial in caspase-1-independent pyroptosis in humans. While caspase-4 self-activates through direct interactions with LPS, caspase-5 is specifically regulated by IFN–γ and LPS [158, 159].
Caspase-3 has also been reported capable of mediating pyroptosis (Fig. 3B). As caspase-3 is both an executioner of apoptosis and an upstream regulator of pyroptosis [150, 160], pyroptosis and apoptosis are interconnected. In response to TNF-α or chemotherapeutic agents, GSDME is directly cleaved by caspase-3, and the produced GSDME-N fragment takes on a similar function as the activated GSDMD to penetrate cell membrane and trigger pyroptosis [161].
Much more details of non-canonical caspase-1 independent pyroptosis still await to be deciphered.
Pyroptosis in response to inflammation associated redox imbalance
Pyroptosis is typically triggered on pathogen invasion such as viruses, bacteria and parasites [162]. ROS play essential roles in host immune defenses against pathogen invasion [163, 164]. Apart from toxic molecules expressed by pathogens, excessive ROS production by activated immune cells on pathogen invasion creates cytotoxic signals that can be sensed by p53 [163,164,165]. The p53 protein plays essential roles in pyroptosis through up-regulating caspase-1 [166, 167].
Clinical relevance of pyroptosis in cancer treatment
It has been proved that ZnO nanoparticles (ZnO-NPs), ivermectin, etc., and some chemotherapy drugs can all induce pyroptosis in tumor cells. Specifically, high concentrations of ZnO-NPs can activate pyroptosis in lung cancer cells [168].
Drugs capable of triggering pyroptosis and applied in clinics include ivermectin for the treatment of breast cancer [169], α-NETA for the treatment of ovarian [170], and cisplatin for treating solid tumors [171], respectively (Table 1).
Cold atmospheric plasma being redox level controller
Cold atmospheric plasma (CAP), being the fourth state of matter, is composed of ions, electrons, neutral particles and photons [172, 173]. It is featured by its multi-modality nature, with the anti-cancer efficacy being firstly reported in 2007 [174]. Though the underlying mechanism is not fully understood, it has been widely accepted that the selectivity of CAP against cancer cells relies on its roles in modulating cellular redox level [1]. By tuning cell oxidative level, CAP could make malignant cells more easily exceed the cell death threshold without breaking the redox homeostasis of normal cells [175]. Cancer cells have higher baseline ROS levels than healthy cells and are thus more sensitive to elevated ROS production and turnover on CAP treatment. The exceeding of the anti-oxidant capacity in cancer cells forced them undergoing various PCD programs with apoptosis being the most intensively studied [172, 173]. Specifically, interactions between H2O2 and NO2 (components that stably exist in CAP-activated medium) generate peroxynitrite (ONOO−) that can be protonated to peroxynitrous acid (ONOOH) when approaching membrane-associated proton pumps, and decomposed into ·NO2 and ·OH radicals; ·OH reacts with H2O2 to generate peroxynitric acid (O2NOOH) and peroxynitrate (O2NOO−) that lead to the generation of primary singlet oxygen (1O2); primary 1O2 triggers the production of high concentrations of secondary 1O2, with H2O2 and ONOO being the source for sustained secondary 1O2 generation, inactivates the protective membrane-associated catalase in tumor cells, and promotes aquaporin-mediated influx of extracellular H2O2, all of which ultimately initiate redox imbalance triggered apoptosis [1, 176, 177].
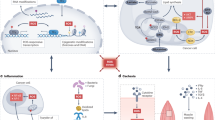
P53 is a pivotal switch controlling cells’ fate towards different PCD programs [178] in response to redox imbalance as a result of various stimuli including DNA damage signal, metabolic stress, and inflammation (Fig. 4). Thus, these diverse types of PCD events are interconnected via this shared controller. Through gene expression profiling and downstream functional studies, CAP was found to be able to activate the expression of p53 pathway-related genes [179], rendering CAP a potential modulator of cells’ life/death fate [1] that is of critical clinical relevance if applied in cancer treatment.

Conceptual illustration on CAP being a redox controller in resembling various stress signals in the trigger of varied PCD events via p53
On the other hand, one stimulus may potentially trigger multiple PCD events. For instance, ivermectin could simultaneously trigger pyroptosis, apoptosis and necroptosis in MDA-MB-231 cells through generating ROS, activating cytoplasmic calcium/calmodulin kinase II (CaMK II), opening mitochondrial permeability transition pore (MPTP) and forming caspase-1 mediated NLRP3 inflammatory corpuscle [180]. Therefore, CAP-induced selectivity against cancer cells may be resulted from multiple PCD programs far beyond just apoptosis that deserves intensive investigations.
Conclusion
PCD events are fundamental to the maintenance of cell redox homeostasis, normal tissue development and human health. Signals triggering redox imbalance and consequently PCD might be originated from DNA damage signaling such as in apoptosis, paraptosis and mitotic catastrophe, might be from metabolic disorder such as in autophagic cell death and ferroptosis, and might be from inflammation such as in necroptosis and pyroptosis. As uncontrolled cell proliferation, metabolic reprogramming and tumor-associated inflammation are essential cancer hallmarks, disordered PCD events as a result of disrupted redox homeostasis are essential for cells to become malignant that, once being under control, might push chaotic cells towards the death state or rewire them towards the healthy state.
CAP could generate and deliver controlled doses of reactive species to cells that selectively triggers redox imbalance in malignant cells. Thus, CAP represents a promising onco-therapeutic approach, alone or being combined with other therapeutic strategies, through selectively inducing PCD events in cancer cells that is not limited to apoptosis. With our incremental understandings on varied types of PCD, underlying mechanism and associated disorders, it is the time to explore other CAP-triggered PCD events beyond apoptosis. Importantly, these PCD events orchestrate the selectivity of CAP against cancer cells through cross-talking due to, e.g., the shared regulator p53. Thus, the efficacy of CAP as a redox controller and an emerging onco-therapeutic strategy is a synergistic result from multiple PCD events that relies on appropriate dosing. Deciphering the precise mechanism underlying the efficacy of CAP against a particular type of cancer type and ultimately translating it into clinics require intensive efforts from both bench and bed sides and involve experts from multiple research domains such as biology, chemistry, physics, bioinformatics, materials and medicine.
References
Dai X et al (2020) Cold atmospheric plasma: a promising controller of cancer cell states. Cancers (Basel) 12(11):3360
Acharya A et al (2010) Redox regulation in cancer: a double-edged sword with therapeutic potential. Oxid Med Cell Longev 3(1):23–34
Raza MH et al (2017) ROS-modulated therapeutic approaches in cancer treatment. J Cancer Res Clin Oncol 143(9):1789–1809
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Hock JM et al (2001) Osteoblast apoptosis and bone turnover. J Bone Miner Res 16(6):975–984
Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26(4):239–257
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35(4):495–516
Smyth PG, Berman SA (2002) Markers of apoptosis: methods for elucidating the mechanism of apoptotic cell death from the nervous system. Biotechniques 32(3):648–50
Pulkkanen KJ et al (2000) False-positive apoptosis signal in mouse kidney and liver detected with TUNEL assay. Apoptosis 5(4):329–333
P.M., Muganda, (2016) Apoptosis methods in toxicology. Humana Press, New York
Zhuang C et al (2020) Oxidative stress induces chondrocyte apoptosis through caspase-dependent and caspase-independent mitochondrial pathways and the antioxidant mechanism of angelica sinensis polysaccharide. Oxid Med Cell Longev 2020:3240820
Siwecka N et al (2019) Dual role of endoplasmic reticulum stress-mediated unfolded protein response signaling pathway in carcinogenesis. Int J Mol Sci 20(18):4354
Wu H et al (2020) Copper sulfate-induced endoplasmic reticulum stress promotes hepatic apoptosis by activating CHOP, JNK and caspase-12 signaling pathways. Ecotoxicol Environ Saf 191:110236
Guo G et al (2015) Induction of apoptosis coupled to endoplasmic reticulum stress through regulation of CHOP and JNK in bone marrow mesenchymal stem cells from patients with systemic lupus erythematosus. J Immunol Res 2015:183738
Yi S et al (2019) Endoplasmic reticulum stress is involved in stress-induced hypothalamic neuronal injury in rats via the PERK-ATF4-CHOP and IRE1-ASK1-JNK pathways. Front Cell Neurosci 13:190
Zou W et al (2008) Coupling of endoplasmic reticulum stress to CDDO-Me-induced up-regulation of death receptor 5 via a CHOP-dependent mechanism involving JNK activation. Cancer Res 68(18):7484–7492
Shen K et al (2015) Cambogin induces caspase-independent apoptosis through the ROS/JNK pathway and epigenetic regulation in breast cancer cells. Mol Cancer Ther 14(7):1738–1749
Kumar S, Boehm J, Lee JC (2003) p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov 2(9):717–726
Christ M et al (1993) Apoptosis induced by oxysterols in murine lymphoma cells and in normal thymocytes. Immunology 78(3):455–460
Schwartzman RA, Cidlowski JA (1993) Apoptosis: the biochemistry and molecular biology of programmed cell death. Endocr Rev 14(2):133–151
Pistritto G et al (2016) Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging (Albany NY) 8(4):603–619
Hassan M et al (2014) Apoptosis and molecular targeting therapy in cancer. Biomed Res Int 2014:150845
Ashkenazi A (2008) Targeting the extrinsic apoptosis pathway in cancer. Cytokine Growth Factor Rev 19(3–4):325–331
Pedersen IM et al (2002) The triterpenoid CDDO induces apoptosis in refractory CLL B cells. Blood 100(8):2965–2972
Kim KB et al (2002) Identification of a novel synthetic triterpenoid, methyl-2-cyano-3,12-dioxooleana-1,9-dien-28-oate, that potently induces caspase-mediated apoptosis in human lung cancer cells. Mol Cancer Ther 1(3):177–184
Ito Y et al (2000) The novel triterpenoid 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid induces apoptosis of human myeloid leukemia cells by a caspase-8-dependent mechanism. Cell Growth Differ 11(5):261–267
Cang S et al (2015) ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J Hematol Oncol 8:129
Smith ML et al (2020) The combinatorial activity of eftozanermin (ABBV-621), a novel and potent TRAIL receptor Agonist fusion protein, in pre-clinical models of hematologic malignancies. Blood 136(Supplement 1):41–41
Seufferlein T et al (2020) Phase I trials in pancreatic cancer. Humana, Cham
Carvajal LA et al (2018) Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci Transl Med 10(436):eaao3003
Konopleva M et al (2020) MDM2 inhibition: an important step forward in cancer therapy. Leukemia 34(Suppl 6):1–17
Zhang X et al (2020) Targeting anti-apoptotic BCL-2 family proteins for cancer treatment. Future Med Chem 12(21):563
Valcourt DM et al (2020) Nanoparticle-Mediated co-delivery of notch-1 antibodies and ABT-737 as a potent treatment strategy for triple-negative breast cancer. ACS Nano 14(3):3378–3388
Jing W et al (2017) APG-1252-12A induces mitochondria-dependent apoptosis through inhibiting the antiapoptotic proteins Bcl-2/Bcl-xl in HL-60 cells. Int J Oncol 51(2):563–572
Decaudin D et al (2020) Preclinical evaluation of drug combinations identifies co-inhibition of Bcl-2/XL/W and MDM2 as a potential therapy in uveal melanoma. Eur J Cancer 126:93–103
Yang C et al (2020) Doxorubicin sensitizes cancer cells to Smac mimetic via synergistic activation of the CYLD/RIPK1/FADD/caspase-8-dependent apoptosis. Apoptosis 25(1):441
Zhao X et al (2017) Focal adhesion kinase inhibitor PF573228 and death receptor 5 agonist lexatumumab synergistically induce apoptosis in pancreatic carcinoma. Tumour Biol 39(5):1010428317699120
Ciuleanu T et al (2016) A randomized, double-blind, placebo-controlled phase II study to assess the efficacy and safety of mapatumumab with sorafenib in patients with advanced hepatocellular carcinoma. Ann Oncol 27:680
Campbell KJ, Swg T (2018) Targeting BCL-2 regulated apoptosis in cancer. Open Biol 8(5):180002
Wong M et al (2012) Navitoclax (ABT-263) reduces Bcl-x(L)-mediated chemoresistance in ovarian cancer models. Mol Cancer Ther 11(4):1026
Luo Q et al (2020) A novel BCL-2 inhibitor APG-2575 exerts synthetic lethality with BTK or MDM2-p53 inhibitor in diffuse large B-cell lymphoma. Oncol Res 28(4):331–344
Yi X et al (2020) AMG-176, an Mcl-1 antagonist, shows preclinical efficacy in chronic lymphocytic leukemia. Clin Cancer Res 26(14):1397
Birsen R et al (2021) APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica. https://doi.org/10.3324/haematol.2020.259531
Goliaei A et al (2020) Multiscale model identifies improved schedule for treatment of Acute Myeloid Leukemia in vitro with the Mcl1 inhibitor AZD5991. CPT 9:561
Kroemer G et al (2009) Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 16(1):3–11
Sperandio S, de Belle I, Bredesen DE (2000) An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci USA 97(26):14376–14381
Sperandio S et al (2004) Paraptosis: mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Differ 11(10):1066–1075
Kessel D (2019) Apoptosis, paraptosis and autophagy: death and survival pathways associated with photodynamic therapy. Photochem Photobiol 95(1):119–125
Gandin V et al (2012) A novel copper complex induces paraptosis in colon cancer cells via the activation of ER stress signalling. J Cell Mol Med 16(1):142–151
Ghosh K et al (2016) Withaferin A induces ROS-mediated paraptosis in human breast cancer cell-lines MCF-7 and MDA-MB-231. PLoS ONE 11(12):e0168488
Nedungadi D et al (2021) Ginger extract activates caspase independent paraptosis in cancer cells via ER stress, mitochondrial dysfunction, AIF translocation and DNA damage. Nutr Cancer 73(1):147–159
Ohlsson C et al (1998) p53 regulates insulin-like growth factor-I (IGF-I) receptor expression and IGF-I-induced tyrosine phosphorylation in an osteosarcoma cell line: interaction between p53 and Sp1. Endocrinology 139(3):1101–1107
Neuberg M et al (1997) The p53/IGF-1 receptor axis in the regulation of programmed cell death. Endocrine 7(1):107–109
Fontana F et al (2020) The emerging role of paraptosis in tumor cell biology: perspectives for cancer prevention and therapy with natural compounds. Biochim Biophys Acta Rev Cancer 1873(2):188338
Sun Q et al (2010) Taxol induces paraptosis independent of both protein synthesis and MAPK pathway. J Cell Physiol 222(2):421–432
Guo WJ et al (2010) Taxol induces concentration-dependent apoptotic and paraptosis-like cell death in human lung adenocarcinoma (ASTC-a-1) cells. J Xray Sci Technol 18(3):293–308
Castedo M et al (2004) Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene 23(25):4362–4370
Nigg EA (2001) Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol 2:21–32
Margottin-Goguet F et al (2003) Prophase destruction of Emi1 by the SCF(betaTrCP/Slimb) ubiquitin ligase activates the anaphase promoting complex to allow progression beyond prometaphase. Dev Cell 4(6):813–826
Bunz F et al (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282(5393):1497–1501
Chen Z et al (2003) Human Chk1 expression is dispensable for somatic cell death and critical for sustaining G2 DNA damage checkpoint. Mol Cancer Ther 2(6):543–548
Yoshikawa R et al (2001) Dual antitumor effects of 5-fluorouracil on the cell cycle in colorectal carcinoma cells: a novel target mechanism concept for pharmacokinetic modulating chemotherapy. Cancer Res 61(3):1029–1037
Barrett RM, Osborne TP, Wheatley SP (2009) Phosphorylation of survivin at threonine 34 inhibits its mitotic function and enhances its cytoprotective activity. Cell Cycle 8(2):278–283
Rello-Varona S et al (2010) An automated fluorescence videomicroscopy assay for the detection of mitotic catastrophe. Cell Death Dis 1:e25
Yun J et al (1999) p53 negatively regulates cdc2 transcription via the CCAAT-binding NF-Y transcription factor. J Biol Chem 274(42):29677–29682
Taylor WR et al (1999) Mechanisms of G2 arrest in response to overexpression of p53. Mol Biol Cell 10(11):3607–3622
Taylor BF et al (2006) p53 suppression of arsenite-induced mitotic catastrophe is mediated by p21CIP1/WAF1. J Pharmacol Exp Ther 318(1):142–151
Fragkos M, Beard P (2011) Mitotic catastrophe occurs in the absence of apoptosis in p53-null cells with a defective G1 checkpoint. PLoS ONE 6(8):e22946
Mirza A et al (2002) Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene 21(17):2613–2622
Silvestri R (2013) New prospects for vinblastine analogues as anticancer agents. J Med Chem 56(3):625–627
Gidding CE et al (1999) Vincristine revisited. Crit Rev Oncol Hematol 29(3):267–287
Mekhail TM, Markman M (2002) Paclitaxel in cancer therapy. Expert Opin Pharmacother 3(6):755–766
Cortes JE, Pazdur R (1995) Docetaxel. J Clin Oncol 13(10):2643–2655
Frost A et al (2012) Phase i study of the Plk1 inhibitor BI 2536 administered intravenously on three consecutive days in advanced solid tumours. Curr Oncol 19(1):28–35
Gumireddy K et al (2005) ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell 7(3):275–286
Mak JPY et al (2015) Pharmacological inactivation of CHK1 and WEE1 induces mitotic catastrophe in nasopharyngeal carcinoma cells. Oncotarget 6(25):21074–21084
Glick D, Barth S, Macleod KF (2010) Autophagy: cellular and molecular mechanisms. J Pathol 221(1):3–12
Cao Y, Klionsky DJ (2007) Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res 17(10):839–849
Ganley IG et al (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 284(18):12297–12305
Shimizu S et al (2004) Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 6(12):1221–1228
Liu Y, Levine B (2015) Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ 22(3):367–376
Orhon I, Reggiori F (2017) Assays to monitor autophagy progression in cell cultures. Cells 6(3):20
Jin S, White E (2007) Role of autophagy in cancer: management of metabolic stress. Autophagy 3(1):28–31
Hay N (2016) Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer 16(10):635–649
Cairns RA, Mak TW (2016) The current state of cancer metabolism. Nat Rev Cancer 16:613–614
Panieri E, Santoro MM (2016) ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis 7(6):e2253
Maiuri MC et al (2010) Autophagy regulation by p53. Curr Opin Cell Biol 22(2):181–185
Feng Z et al (2005) The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA 102(23):8204–8209
Feng Z et al (2007) The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res 67(7):3043–3053
Budanov AV, Karin M (2008) p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 134(3):451–460
Criollo A, Dessen P, Kroemer G (2009) DRAM: a phylogenetically ancient regulator of autophagy. Cell Cycle 8(15):2319–2320
Wang EY et al (2013) p53 mediates autophagy and cell death by a mechanism contingent on Bnip3. Hypertension 62(1):70–77
Yee KS et al (2009) PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ 16(8):1135–1145
Maiuri MC et al (2007) BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L). Autophagy 3(4):374–376
Tripathi R, Ash D, Shaha C (2014) Beclin-1-p53 interaction is crucial for cell fate determination in embryonal carcinoma cells. J Cell Mol Med 18(11):2275–2286
White E (2012) Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 12(6):401–410
Zhang Y et al (2015) The utility of chloroquine in cancer therapy. Curr Med Res Opin 31(5):1009–1013
Spiliotaki M et al (2021) Dynamic changes of CTCs in patients with metastatic HR(+)/HER2() breast cancer receiving salvage treatment with everolimus/exemestane. Cancer Chemother Pharmacol 87:277
Atkins M (2004) Randomized phase II study of multiple dose levels of CCI-779, a Novel mammalian target of rapamycin kinase inhibitor, in patients with advanced refractory renal cell carcinoma. J Clin Oncol 22:909
Truong DH, Vu KHL, Pham TPD (2021) Delivery systems for vorinostat in cancer treatment: an updated review. J Drug Deliv Sci Technol. https://doi.org/10.1016/j.jddst.2021.102334
Dixon SJ et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149(5):1060–1072
Stockwell BR et al (2017) Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171(2):273–285
Liang C et al (2019) Recent progress in ferroptosis inducers for cancer therapy. Adv Mater 31(51):e1904197
Song X et al (2020) Role of GPX4-mediated ferroptosis in the sensitivity of triple negative breast cancer cells to gefitinib. Front Oncol 10:597434
Maiorino M, Conrad M, Ursini F (2018) GPx4, Lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid Redox Signal 29(1):61–74
Doll S et al (2019) FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575(7784):693–698
Hadian K (2020) Ferroptosis suppressor protein 1 (FSP1) and coenzyme Q10 cooperatively suppress ferroptosis. Biochemistry 59(5):637–638
Bersuker K et al (2019) The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575(7784):688–692
Yuan H et al (2016) Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun 478(3):1338–1343
Liu Z et al (2020) Systematic analysis of the aberrances and functional implications of ferroptosis in cancer. iScience 23(7):101302
Feng H et al (2020) Transferrin receptor is a specific ferroptosis marker. Cell Rep 30(10):3411–3423
Martinez AM, Kim A, Yang WS (2020) Detection of ferroptosis by BODIPY 581/591 C11. Methods Mol Biol 2108:125–130
Stockwell BR, Jiang X, Gu W (2020) Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol 30(6):478–490
Ursini F, Maiorino M (2020) Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med 152:175–185
Sato H et al (1999) Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem 274(17):11455–11458
Dixon SJ et al (2014) Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3:e02523
Yang WS, Stockwell BR (2008) Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 15(3):234–245
Wilhelm J (1990) Metabolic aspects of membrane lipid peroxidation. Acta Univ Carol Med Monogr 137:1–53
Lyamzaev KG et al (2020) Novel fluorescent mitochondria-targeted probe mitoclox reports lipid peroxidation in response to oxidative stress in vivo. Oxid Med Cell Longev 2020:3631272
Rohrig F, Schulze A (2016) The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer 16(11):732–749
Munir R et al (2019) Lipid metabolism in cancer cells under metabolic stress. Br J Cancer 120(12):1090–1098
Jiang L et al (2015) Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle 14(18):2881–2885
Jiang L et al (2015) Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520(7545):57–62
Basuli D et al (2017) Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene 36(29):4089–4099
Harris IS et al (2015) Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27(2):211–222
Liu Y et al (2020) Ferroptosis in low-grade glioma: a new marker for diagnosis and prognosis. Med Sci Monit 26:e921947
Ghosh S (2019) Cisplatin: The first metal based anticancer drug. Bioorg Chem 88:102925
Escudier B, Worden F, Kudo M (2019) Sorafenib: key lessons from over 10 years of experience. Expert Rev Anticancer Ther 19(2):177–189
Lo M et al (2010) Potential use of the anti-inflammatory drug, sulfasalazine, for targeted therapy of pancreatic cancer. Curr Oncol 17(3):9–16
Su Y, Zhao B, Zhou L, Zhang Z, Shen Y, Lv H, AlQudsy LH, Shang P (2020) Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett 483:127–136
Liu C et al (2018) Necroptosis: a novel manner of cell death, associated with stroke (Review). Int J Mol Med 41(2):624–630
Galluzzi L, Kroemer G (2008) Necroptosis: a specialized pathway of programmed necrosis. Cell 135(7):1161–1163
Linkermann A, Green DR (2014) Necroptosis. N Engl J Med 370(5):455–465
Laster SM, Wood JG, Gooding LR (1988) Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol 141:2629–2634
Matsumura H et al (2000) Necrotic death pathway in Fas receptor signaling. J Cell Biol 151(6):1247–1256
Moriwaki K, Chan FK (2014) Necrosis-dependent and independent signaling of the RIP kinases in inflammation. Cytokine Growth Factor Rev 25(2):167–174
Sun L et al (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148(1–2):213–227
Yan G, Elbadawi M, Efferth T (2020) Multiple cell death modalities and their key features. World Acad Sci J 2:39–48
Vanden Berghe T et al (2013) Determination of apoptotic and necrotic cell death in vitro and in vivo. Methods 61(2):117–129
Salzano S et al (2014) Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc Natl Acad Sci USA 111(33):12157–12162
Vaseva AV et al (2012) p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 149(7):1536–1548
Wang K et al (2016) The long noncoding RNA NRF regulates programmed necrosis and myocardial injury during ischemia and reperfusion by targeting miR-873. Cell Death Differ 23(8):1394–1405
Su Z et al (2016) Cancer therapy in the necroptosis era. Cell Death Differ 23(5):748–756
Martin-Sanchez D et al (2018) Cell death-based approaches in treatment of the urinary tract-associated diseases: a fight for survival in the killing fields. Cell Death Dis 9(2):118
Razaghi A et al (2018) Negative regulators of cell death pathways in cancer: perspective on biomarkers and targeted therapies. Apoptosis 23(2):93–112
Wang Y et al (2018) PKM2 inhibitor shikonin overcomes the cisplatin resistance in bladder cancer by inducing necroptosis. Int J Biol Sci 14(13):1883–1891
Miyamoto S et al (2018) Drug review: pazopanib. Jpn J Clin Oncol 48(6):503–513
Tan FH et al (2019) Ponatinib: a novel multi-tyrosine kinase inhibitor against human malignancies. Onco Targets Ther 12:635–645
vande Walle L, Lamkanfi M (2016) Pyroptosis. Curr Biol 26(13):R568–R572
Frank D, Vince JE (2019) Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ 26(1):99–114
Yu J et al (2020) Induction of programmed necrosis: a novel anti-cancer strategy for natural compounds. Pharmacol Ther 214:107593
Lamkanfi M, Dixit VM (2014) Mechanisms and functions of inflammasomes. Cell 157(5):1013–1022
Malik A, Kanneganti TD (2017) Inflammasome activation and assembly at a glance. J Cell Sci 130(23):3955–3963
Kesavardhana S, Kanneganti TD (2017) Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int Immunol 29(5):201–210
He Y, Hara H, Nunez G (2016) Mechanism and regulation of NLRP3 Inflammasome activation. Trends Biochem Sci 41(12):1012–1021
Yi YS (2017) Caspase-11 non-canonical inflammasome: a critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology 152(2):207–217
Shalini S et al (2015) Old, new and emerging functions of caspases. Cell Death Differ 22(4):526–539
Lin XY, Choi MS, Porter AG (2000) Expression analysis of the human caspase-1 subfamily reveals specific regulation of the CASP5 gene by lipopolysaccharide and interferon-gamma. J Biol Chem 275(51):39920–39926
Matikainen S, Nyman TA, Cypryk W (2020) Function and regulation of noncanonical caspase-4/5/11 inflammasome. J Immunol 204(12):3063–3069
Rogers C et al (2017) Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun 8:14128
Wang Y et al (2017) Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547(7661):99–103
Jenni AH et al (2018) Cytosolic Recognition of microbes and pathogens: inflammasomes in action. Microbiol Mol Biol Rev 82:e00015
Sorci G, Faivre B (2009) Inflammation and oxidative stress in vertebrate host-parasite systems. Philos Trans R Soc Lond B Biol Sci 364(1513):71–83
Pohanka M (2013) Role of oxidative stress in infectious diseases. A review. Folia Microbiol (Praha) 58(6):503–513
Ivanov AV, Bartosch B, Isaguliants MG (2017) Oxidative stress in infection and consequent disease. Oxid Med Cell Longev 2017:3496043
Gupta S et al (2001) Direct transcriptional activation of human caspase-1 by tumor suppressor p53. J Biol Chem 276(14):10585–10588
Zhang T et al (2019) Transcription factor p53 suppresses tumor growth by prompting pyroptosis in non-small-cell lung cancer. Oxid Med Cell Longev 2019:8746895
Song J et al (2013) Pyroptosis induced by zinc oxide nanoparticles in A549 cells. Wei Sheng Yan Jiu 42(2):273–276
Melotti A et al (2014) The river blindness drug Ivermectin and related macrocyclic lactones inhibit WNT-TCF pathway responses in human cancer. EMBO Mol Med 6(10):1263–1278
Mengjiao W et al (2019) A PLK1 kinase inhibitor enhances the chemosensitivity of cisplatin by inducing pyroptosis in oesophageal squamous cell carcinoma. EBioMedicine 41:244
Zhang CC et al (2019) Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis 24:312
Bauer G (2019) The synergistic effect between hydrogen peroxide and nitrite, two long-lived molecular species from cold atmospheric plasma, triggers tumor cells to induce their own cell death. Redox Biol 26:101291
Bauer G et al (2019) Dynamics of singlet oxygen-triggered, rons-based apoptosis induction after treatment of tumor cells with cold atmospheric plasma or plasma-activated medium. Sci Rep 9(1):13931
Laroussi M, Akan T (2010) Arc-Free atmospheric pressure cold plasma jets: a review. Plasma Process Polym 4(9):777–788
Graves DB (2012) The emerging role of reactive oxygen and nitrogen species in redox biology and some implications for plasma applications to medicine and biology. J Phys D Appl Phys 45(26):263001
Bauer G (2018) Signal amplification by tumor cells: clue to the understanding of the antitumor effects of cold atmospheric plasma and plasma-activated medium. In: IEEE transactions on radiation & plasma medical sciences, pp 1–1
Bauer G, Graves DB (2016) Mechanisms of selective antitumor action of cold atmospheric plasma-derived reactive oxygen and nitrogen species. Plasma Process Polym. https://doi.org/10.1002/ppap.201600089
Ranjan A, Iwakuma T (2016) Non-canonical cell death induced by p53. Int J Mol Sci 17(12):2068
Shi L et al (2017) Gene expression profiling and functional analysis reveals that p53 pathway-related gene expression is highly activated in cancer cells treated by cold atmospheric plasma-activated medium. PeerJ 5:e3751
Draganov D et al (2015) Modulation of P2X4/P2X7/Pannexin-1 sensitivity to extracellular ATP via Ivermectin induces a non-apoptotic and inflammatory form of cancer cell death. Sci Rep 5:16222
Ai Y et al (2015) Synthesis of CDDO-amino acid-nitric oxide donor trihybrids as potential antitumor agents against both drug-sensitive and drug-resistant colon cancer. J Med Chem 58(5):2452–2464
Ball MS et al (2020) CDDO-Me alters the tumor microenvironment in estrogen receptor negative breast cancer. Sci Rep 10(1):6560
Chaudhari N et al (2017) CDDO and ATRA instigate differentiation of IMR32 human neuroblastoma cells. Front Mol Neurosci 10:310
Hermann C et al (2020) Bardoxolone-Methyl (CDDO-Me) impairs tumor growth and induces radiosensitization of oral squamous cell carcinoma cells. Front Pharmacol 11:607580
Ito Y et al (2001) The novel triterpenoid CDDO induces apoptosis and differentiation of human osteosarcoma cells by a caspase-8 dependent mechanism. Mol Pharmacol 59(5):1094–1099
Khurana N et al (2020) Bardoxolone-Methyl (CDDO-Me) suppresses androgen receptor and its splice-variant AR-V7 and enhances efficacy of enzalutamide in prostate cancer cells. Antioxidants (Basel) 9(1):68
Konopleva M et al (2002) Novel triterpenoid CDDO-Me is a potent inducer of apoptosis and differentiation in acute myelogenous leukemia. Blood 99(1):326–335
Leal AS et al (2016) The triterpenoid CDDO-imidazolide reduces immune cell infiltration and cytokine secretion in the KrasG12D;Pdx1-Cre (KC) mouse model of pancreatic cancer. Carcinogenesis 37(12):1170–1179
Qin D et al (2016) CDDO-Me reveals USP7 as a novel target in ovarian cancer cells. Oncotarget 7(47):77096–77109
To C et al (2015) Dimethyl fumarate and the oleanane triterpenoids, CDDO-imidazolide and CDDO-methyl ester, both activate the Nrf2 pathway but have opposite effects in the A/J model of lung carcinogenesis. Carcinogenesis 36(7):769–781
Townson JL et al (2011) The synthetic triterpenoid CDDO-Imidazolide suppresses experimental liver metastasis. Clin Exp Metastasis 28(3):309–317
Wang YY et al (2016) The therapeutic response of CDDO-Me in the esophageal squamous cell carcinoma (ESCC) cells is mediated by CaMKIIα. Am J Transl Res 8(4):1695–1707
Zhao Y et al (2015) Nanoparticle delivery of CDDO-Me remodels the tumor microenvironment and enhances vaccine therapy for melanoma. Biomaterials 68:54–66
Bate-Eya LT et al (2016) High efficacy of the BCL-2 inhibitor ABT199 (venetoclax) in BCL-2 high-expressing neuroblastoma cell lines and xenografts and rational for combination with MCL-1 inhibition. Oncotarget 7(19):27946–27958
Bodo J et al (2016) Acquired resistance to venetoclax (ABT-199) in t(14;18) positive lymphoma cells. Oncotarget 7(43):70000–70010
Boidol B et al (2017) First-in-human response of BCL-2 inhibitor venetoclax in T-cell prolymphocytic leukemia. Blood 130(23):2499–2503
Deeks ED (2016) Venetoclax: first global approval. Drugs 76(9):979–987
Jiang H et al (2019) Venetoclax as a single agent and in combination with PI3K-MTOR1/2 kinase inhibitors against ibrutinib sensitive and resistant mantle cell lymphoma. Br J Haematol 184(2):298–302
Kanate AS, Vos J, Chargualaf MJ (2019) Venetoclax for refractory myeloid sarcoma. J Oncol Pract 15(7):413–415
Konopleva M et al (2016) Efficacy and biological correlates of response in a phase II Study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov 6(10):1106–1117
Lok SW et al (2019) A phase ib dose-escalation and expansion study of the BCL2 Inhibitor venetoclax combined with tamoxifen in ER and BCL2-positive metastatic breast cancer. Cancer Discov 9(3):354–369
Matulis SM et al (2019) Functional profiling of venetoclax sensitivity can predict clinical response in multiple myeloma. Leukemia 33(5):1291–1296
Nguyen TH et al (2019) Fenretinide via NOXA induction, enhanced activity of the BCL-2 inhibitor venetoclax in high BCL-2-expressing neuroblastoma preclinical models. Mol Cancer Ther 18(12):2270–2282
O’Steen S et al (2017) Venetoclax synergizes with radiotherapy for treatment of B-cell lymphomas. Cancer Res 77(14):3885–3893
Patil N, Went RG (2019) Venetoclax is an option in B-cell prolymphocytic leukaemia following progression on B-cell receptor pathway inhibitors. Br J Haematol 186(4):e80–e82
Sasi BK et al (2019) Inhibition of SYK or BTK augments venetoclax sensitivity in SHP1-negative/BCL-2-positive diffuse large B-cell lymphoma. Leukemia 33(10):2416–2428
Schieber M, Ma S (2019) The expanding role of venetoclax in chronic lymphocytic leukemia and small lymphocytic lymphoma. Blood Lymphat Cancer 9:9–17
Quezada MJ et al (2020) BCL2L10 Is overexpressed in melanoma downstream of STAT3 and promotes cisplatin and ABT-737 resistance. Cancers (Basel) 13(1):78
Lomovsky A et al (2020) Melatonin can modulate the effect of navitoclax (ABT-737) in HL-60 cells. Antioxidants (Basel) 9(11):1143
Liu M et al (2020) Synergistic co-delivery of diacid metabolite of norcantharidin and ABT-737 based on folate-modified lipid bilayer-coated mesoporous silica nanoparticle against hepatic carcinoma. J Nanobiotechnol 18(1):114
Masilamani AP et al (2020) An Anti-PSMA immunotoxin reduces Mcl-1 and Bcl2A1 and specifically induces in combination with the BAD-Like BH3 mimetic ABT-737 Apoptosis in prostate cancer cells. Cancers (Basel) 12(6):1648
Florent R et al (2020) Bim, puma and noxa upregulation by naftopidil sensitizes ovarian cancer to the BH3-mimetic ABT-737 and the MEK inhibitor trametinib. Cell Death Dis 11(5):380
Shahverdi M et al (2020) Gene therapy with MiRNA-mediated targeting of Mcl-1 promotes the sensitivity of non-small cell lung cancer cells to treatment with ABT-737. Asian Pac J Cancer Prev 21(3):675–681
Hsin IL et al (2019) The application of arsenic trioxide in ameliorating ABT-737 target therapy on uterine cervical cancer cells through unique pathways in cell death. Cancers (Basel) 12(1):108
Wang Q, Hao S (2019) A-1210477, a selective MCL-1 inhibitor, overcomes ABT-737 resistance in AML. Oncol Lett 18(5):5481–5489
Yu X et al (2019) Synergistic antitumor effects of 9.2.27-PE38KDEL and ABT-737 in primary and metastatic brain tumors. PLoS ONE 14(1):e0210608
Remy J et al (2019) Inhibition of PIM1 blocks the autophagic flux to sensitize glioblastoma cells to ABT-737-induced apoptosis. Biochim Biophys Acta Mol Cell Res 1866(2):175–189
Ou YC et al (2018) Aspirin restores ABT-737-mediated apoptosis in human renal carcinoma cells. Biochem Biophys Res Commun 502(2):187–193
Crassini K et al (2018) MEK1/2 inhibition by binimetinib is effective as a single agent and potentiates the actions of Venetoclax and ABT-737 under conditions that mimic the chronic lymphocytic leukaemia (CLL) tumour microenvironment. Br J Haematol 182(3):360–372
Štefaniková A et al (2017) Cyclin-dependent kinase 2 inhibitor SU9516 increases sensitivity of colorectal carcinoma cells Caco-2 but not HT29 to BH3 mimetic ABT-737. Gen Physiol Biophys 36(5):539–547
Wu X et al (2017) Cooperation of IRAK1/4 inhibitor and ABT-737 in nanoparticles for synergistic therapy of T cell acute lymphoblastic leukemia. Int J Nanomed 12:8025–8034
Zhang F et al (2017) ABT-737 potentiates cisplatin-induced apoptosis in human osteosarcoma cells via the mitochondrial apoptotic pathway. Oncol Rep 38(4):2301–2308
Kasai S et al (2017) Bcl-2/Bcl-x(L) inhibitor ABT-737 sensitizes pancreatic ductal adenocarcinoma to paclitaxel-induced cell death. Oncol Lett 14(1):903–908
Zhang H et al (2016) Enhanced anticancer effect of ABT-737 in combination with naringenin on gastric cancer cells. Exp Ther Med 11(2):669–673
Broecker-Preuss M, Becher-Boveleth N, Mann K (2016) Cell death induction by the indirubin derivative 7BIO and the BH3 mimetic drugs ABT-737 and GX15-070 in medullary thyroid carcinoma cells. Exp Clin Endocrinol Diabetes 124(5):324–330
Jane EP et al (2016) Dinaciclib, a Cyclin-dependent kinase inhibitor promotes proteasomal degradation of Mcl-1 and enhances ABT-737-mediated cell death in malignant human glioma cell lines. J Pharmacol Exp Ther 356(2):354–365
Shin JA et al (2015) Targeting ERK1/2-bim signaling cascades by BH3-mimetic ABT-737 as an alternative therapeutic strategy for oral cancer. Oncotarget 6(34):35667–35683
Mani J et al (2016) Knockdown of BAG3 sensitizes bladder cancer cells to treatment with the BH3 mimetic ABT-737. World J Urol 34(2):197–205
Lee SJ et al (2012) Inhibition of Bcl-xL by ABT-737 enhances chemotherapy sensitivity in neurofibromatosis type 1-associated malignant peripheral nerve sheath tumor cells. Int J Mol Med 30(2):443–450
Allaman-Pillet N et al (2013) The Bcl-2/Bcl-XL inhibitor ABT-737 promotes death of retinoblastoma cancer cells. Ophthalmic Genet 34(1–2):1–13
Fang H et al (2011) Synergistic activity of fenretinide and the Bcl-2 family protein inhibitor ABT-737 against human neuroblastoma. Clin Cancer Res 17(22):7093–7104
Levesley J et al (2011) RASSF1A and the BH3-only mimetic ABT-737 promote apoptosis in pediatric medulloblastoma cell lines. Neuro Oncol 13(12):1265–1276
Bodet L et al (2011) ABT-737 is highly effective against molecular subgroups of multiple myeloma. Blood 118(14):3901–3910
Yi H et al (2020) Bcl-2/Bcl-xl inhibitor APG-1252-M1 is a promising therapeutic strategy for gastric carcinoma. Cancer Med 9(12):4197–4206
Wang J et al (2017) APG-1252-12A induces mitochondria-dependent apoptosis through inhibiting the antiapoptotic proteins Bcl-2/Bcl-xl in HL-60 cells. Int J Oncol 51(2):563–572
Seipel K et al (2018) MDM2- and FLT3-inhibitors in the treatment of FLT3-ITD acute myeloid leukemia, specificity and efficacy of NVP-HDM201 and midostaurin. Haematologica 103(11):1862–1872
Runckel K et al (2018) The SMAC mimetic LCL-161 displays antitumor activity in preclinical models of rituximab-resistant B-cell lymphoma. Blood Adv 2(23):3516–3525
Zhao X et al (2011) Reactive oxygen species is essential for cycloheximide to sensitize lexatumumab-induced apoptosis in hepatocellular carcinoma cells. PLoS ONE 6(2):e16966
Belyanskaya LL et al (2007) Human agonistic TRAIL receptor antibodies Mapatumumab and Lexatumumab induce apoptosis in malignant mesothelioma and act synergistically with cisplatin. Mol Cancer 6:66
Luster TA et al (2009) Mapatumumab and lexatumumab induce apoptosis in TRAIL-R1 and TRAIL-R2 antibody-resistant NSCLC cell lines when treated in combination with bortezomib. Mol Cancer Ther 8(2):292–302
Marini P et al (2006) Combined treatment of colorectal tumours with agonistic TRAIL receptor antibodies HGS-ETR1 and HGS-ETR2 and radiotherapy: enhanced effects in vitro and dose-dependent growth delay in vivo. Oncogene 25(37):5145–5154
Menoret E et al (2006) Mcl-1L cleavage is involved in TRAIL-R1- and TRAIL-R2-mediated apoptosis induced by HGS-ETR1 and HGS-ETR2 human mAbs in myeloma cells. Blood 108(4):1346–1352
Mukherjee N et al (2020) MCL1 inhibitors S63845/MIK665 plus Navitoclax synergistically kill difficult-to-treat melanoma cells. Cell Death Dis 11(6):443
Pullarkat VA et al (2021) Venetoclax and navitoclax in combination with chemotherapy in patients with relapsed or refractory acute lymphoblastic leukemia and lymphoblastic lymphoma. Cancer Discov. https://doi.org/10.1158/2159-8290.CD-20-1465
Gadsden NJ et al (2021) Palbociclib renders human papilloma virus-negative head and neck squamous cell carcinoma vulnerable to the senolytic agent navitoclax. Mol Cancer Res 19(5):862–873
Zhang X et al (2020) KRT-232 and navitoclax enhance trametinib’s anti-Cancer activity in non-small cell lung cancer patient-derived xenografts with KRAS mutations. Am J Cancer Res 10(12):4464–4475
Chteinberg E et al (2020) Navitoclax combined with Alpelisib effectively inhibits Merkel cell carcinoma cell growth in vitro. Ther Adv Med Oncol 12:1758835920975621
Zoeller JJ et al (2020) Navitoclax enhances the effectiveness of EGFR-targeted antibody-drug conjugates in PDX models of EGFR-expressing triple-negative breast cancer. Breast Cancer Res 22(1):132
de Vos S et al (2021) Safety and efficacy of navitoclax, a BCL-2 and BCL-X(L) inhibitor, in patients with relapsed or refractory lymphoid malignancies: results from a phase 2a study. Leuk Lymphoma 62(4):810–818
Morimoto Y et al (2020) Bcl-2/Bcl-xL inhibitor navitoclax increases the antitumor effect of Chk1 inhibitor prexasertib by inducing apoptosis in pancreatic cancer cells via inhibition of Bcl-xL but not Bcl-2. Mol Cell Biochem 472(1–2):187–198
Ding J et al (2020) Ultra pH-sensitive polymeric nanovesicles co-deliver doxorubicin and navitoclax for synergetic therapy of endometrial carcinoma. Biomater Sci 8(8):2264–2273
Ommer J et al (2020) Aurora a kinase inhibition destabilizes PAX3-FOXO1 and MYCN and synergizes with navitoclax to induce rhabdomyosarcoma cell death. Cancer Res 80(4):832–842
Jeong JH et al (2019) Combination treatment with the BRAF(V600E) inhibitor vemurafenib and the BH3 mimetic navitoclax for BRAF-mutant thyroid carcinoma. Thyroid 29(4):540–548
Stamelos VA et al (2013) Navitoclax augments the activity of carboplatin and paclitaxel combinations in ovarian cancer cells. Gynecol Oncol 128(2):377–382
Ackler S et al (2012) Navitoclax (ABT-263) and bendamustine ± rituximab induce enhanced killing of non-Hodgkin’s lymphoma tumours in vivo. Br J Pharmacol 167(4):881–891
Yi X et al (2020) AMG-176, an Mcl-1 antagonist, shows preclinical efficacy in chronic lymphocytic leukemia. Clin Cancer Res 26(14):3856–3867
De La Rosa J et al (2021) APR-246 combined with 3-deazaneplanocin A, panobinostat or temozolomide reduces clonogenicity and induces apoptosis in glioblastoma cells. Int J Oncol 58(3):312–330
Liang Y et al (2019) APR-246 alone and in combination with a phosphatidylserine-targeting antibody inhibits lung metastasis of human triple-negative breast cancer cells in nude mice. Breast Cancer (Dove Med Press) 11:249–259
Fransson Å et al (2016) Strong synergy with APR-246 and DNA-damaging drugs in primary cancer cells from patients with TP53 mutant High-Grade Serous ovarian cancer. J Ovarian Res 9(1):27
Deben C et al (2016) APR-246 (PRIMA-1(MET)) strongly synergizes with AZD2281 (olaparib) induced PARP inhibition to induce apoptosis in non-small cell lung cancer cell lines. Cancer Lett 375(2):313–322
Krayem M et al (2016) p53 Reactivation by PRIMA-1(Met) (APR-246) sensitises (V600E/K)BRAF melanoma to vemurafenib. Eur J Cancer 55:98–110
Liu DS et al (2015) APR-246 potently inhibits tumour growth and overcomes chemoresistance in preclinical models of oesophageal adenocarcinoma. Gut 64(10):1506–1516
Aryee DN et al (2013) Variability in functional p53 reactivation by PRIMA-1(Met)/APR-246 in Ewing sarcoma. Br J Cancer 109(10):2696–2704
Saha MN et al (2013) PRIMA-1Met/APR-246 displays high antitumor activity in multiple myeloma by induction of p73 and Noxa. Mol Cancer Ther 12(11):2331–2341
Li XL et al (2015) PRIMA-1met (APR-246) inhibits growth of colorectal cancer cells with different p53 status through distinct mechanisms. Oncotarget 6(34):36689–36699
Tron AE et al (2018) Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun 9(1):5341
Wen X et al (2021) Application of taxol nanomicelles with Lyp-1 target in targeted 805–813 therapy of colon cancer. J Nanosci Nanotechnol 21(2):805–813
Tian Y et al (2021) Triptolide inhibits epithelial-mesenchymal transition phenotype through the p70S6k/GSK3/β-catenin signaling pathway in taxol-resistant human lung adenocarcinoma. Transl Lung Cancer Res 10(2):1007–1019
Qu F et al (2021) Circular RNA circ_0006168 enhances Taxol resistance in esophageal squamous cell carcinoma by regulating miR-194-5p/JMJD1C axis. Cancer Cell Int 21(1):273
Ma X et al (2021) Down-regulation of autophagy-associated protein increased acquired radio-resistance bladder cancer cells sensitivity to taxol. Int J Radiat Biol 97(4):507–516
Li T et al (2021) Formononetin ameliorates the drug resistance of taxol resistant triple negative breast cancer by inhibiting autophagy. Am J Transl Res 13(2):497–514
Huang R, Zhu L, Zhang Y (2020) XIST lost induces ovarian cancer stem cells to acquire taxol resistance via a KMT2C-dependent way. Cancer Cell Int 20:436
Duan Y et al (2020) LncRNA HOTAIR contributes Taxol-resistance of hepatocellular carcinoma cells via activating AKT phosphorylation by down-regulating miR-34a. Biosci Rep 40(7):BSR20201627
Gach-Janczak K et al (2020) A new hybrid δ-lactone induces apoptosis and potentiates anticancer activity of taxol in HL-60 human leukemia cells. Molecules 25(7):1479
Park SY et al (2018) Role of DDX53 in taxol-resistance of cervix cancer cells in vitro. Biochem Biophys Res Commun 506(3):641–647
Duan S et al (2018) Effects of taxol resistance gene 1 on the cisplatin response in gastric cancer. Oncol Lett 15(6):8287–8294
Dong JR et al (2013) Inhibition of nemo-like kinase increases taxol sensitivity in laryngeal cancer. Asian Pac J Cancer Prev 14(12):7137–7141
Ledwitch K et al (2013) Taxol: efficacy against oral squamous cell carcinoma. Mini Rev Med Chem 13(4):509–521
Kelling J et al (2003) Suppression of centromere dynamics by taxol in living osteosarcoma cells. Cancer Res 63(11):2794–2801
Allman R, Errington RJ, Smith PJ (2003) Delayed expression of apoptosis in human lymphoma cells undergoing low-dose taxol-induced mitotic stress. Br J Cancer 88(10):1649–1658
Wang F et al (2003) Anti-invasion and anti-angiogenesis effect of taxol and camptothecin on melanoma cells. J Asian Nat Prod Res 5(2):121–129
Kim JH et al (2003) Differential sensitivity of taxol-induced apoptosis in U2OS and SaOS2 osteogenic sarcoma cells. Cancer Res Treat 35(2):148–153
Reinecke P et al (2000) Multidrug resistance phenotype and paclitaxel (Taxol) sensitivity in human renal carcinoma cell lines of different histologic types. Cancer Invest 18(7):614–625
Lieu CH et al (1998) Role of mitogen-activated protein kinase in taxol-induced apoptosis in human leukemic U937 cells. Cell Growth Differ 9(9):767–776
Plasswilm L, Kirschner M, Sauer R (1996) Concurrent taxol and split-course accelerated radiotherapy for advanced head and neck cancer. Strahlenther Onkol 172(10):573–579
Saghatelyan T et al (2020) Efficacy and safety of curcumin in combination with paclitaxel in patients with advanced, metastatic breast cancer: a comparative, randomized, double-blind, placebo-controlled clinical trial. Phytomedicine 70:153218
Howells LM et al (2019) Curcumin combined with FOLFOX Chemotherapy is safe and tolerable in patients with metastatic colorectal cancer in a randomized phase IIa trial. J Nutr 149(7):1133–1139
Dhillon N et al (2008) Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res 14(14):4491–4499
Caruso Bavisotto C et al (2020) Curcumin affects HSP60 folding activity and levels in neuroblastoma cells. Int J Mol Sci 21(2):661
Riaz IB, Hussain SA (2021) Perioperative treatment in muscle-invasive bladder cancer: analysis of secondary endpoints in a randomized trial comparing gemcitabine and cisplatin versus dose-dense methotrexate, vinblastine, adriamycin, and cisplatin. Eur Urol 79(2):222–224
Knörr F et al (2020) Stem cell transplantation and vinblastine monotherapy for relapsed pediatric anaplastic large cell lymphoma: results of the international, prospective ALCL-relapse trial. J Clin Oncol 38(34):3999–4009
Böll B et al (2019) Doxorubicin, vinblastine, dacarbazine and lenalidomide for older Hodgkin lymphoma patients: final results of a German Hodgkin Study Group (GHSG) phase-I trial. Br J Haematol 185(1):42–52
Brady SF et al (2002) Design and synthesis of a pro-drug of vinblastine targeted at treatment of prostate cancer with enhanced efficacy and reduced systemic toxicity. J Med Chem 45(21):4706–4715
McConkey DJ et al (2016) A prognostic gene expression signature in the molecular classification of chemotherapy-naïve urothelial cancer is predictive of clinical outcomes from neoadjuvant chemotherapy: a phase 2 trial of dose-dense methotrexate, vinblastine, doxorubicin, and cisplatin with bevacizumab in urothelial cancer. Eur Urol 69(5):855–862
Arnold EJ et al (2011) Clinical trial of vinblastine in dogs with transitional cell carcinoma of the urinary bladder. J Vet Intern Med 25(6):1385–1390
Keller AM et al (2004) Randomized phase III trial of pegylated liposomal doxorubicin versus vinorelbine or mitomycin C plus vinblastine in women with taxane-refractory advanced breast cancer. J Clin Oncol 22(19):3893–3901
Albrecht W et al (2004) Randomized Phase II trial assessing estramustine and vinblastine combination chemotherapy vs estramustine alone in patients with progressive hormone-escaped metastatic prostate cancer. Br J Cancer 90(1):100–105
Paccagnella A et al (2004) Cisplatin versus carboplatin in combination with mitomycin and vinblastine in advanced non small cell lung cancer. A multicenter, randomized phase III trial. Lung Cancer 43(1):83–91
Haas NB et al (2003) Vinblastine and estramustine phosphate in metastatic renal cell carcinoma: a phase II trial of the Fox Chase Network. Cancer 98(9):1837–1841
Atkins MB et al (2002) A phase II pilot trial of concurrent biochemotherapy with cisplatin, vinblastine, temozolomide, interleukin 2, and IFN-alpha 2B in patients with metastatic melanoma. Clin Cancer Res 8(10):3075–3081
Ramírez-Amador V et al (2002) Intralesional vinblastine vs. 3% sodium tetradecyl sulfate for the treatment of oral Kaposi’s sarcoma. A double blind, randomized clinical trial. Oral Oncol 38(5):460–7
Siefker-Radtke AO et al (2002) Phase III trial of fluorouracil, interferon alpha-2b, and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in metastatic or unresectable urothelial cancer. J Clin Oncol 20(5):1361–1367
Kikkawa F et al (2000) Randomized trial of cisplatin and carboplatin versus cisplatin, vinblastine and bleomycin in ovarian cancer. Gynecol Obstet Invest 50(4):269–274
Long HJ 3rd et al (1995) Phase II trial of methotrexate, vinblastine, doxorubicin, and cisplatin in advanced/recurrent endometrial carcinoma. Gynecol Oncol 58(2):240–243
Lovett D et al (1991) A phase II trial of carboplatin and vinblastine in the treatment of advanced squamous cell carcinoma of the esophagus. Cancer 67(2):354–356
Wozniak AJ et al (1991) A randomized trial of cisplatin, vinblastine, and bleomycin versus vinblastine, cisplatin, and etoposide in the treatment of advanced germ cell tumors of the testis: a Southwest Oncology Group study. J Clin Oncol 9(1):70–76
El-Sayed S et al (1990) Phase II trial of carboplatin and vinblastine in advanced squamous-cell carcinoma of the head and neck. Cancer Chemother Pharmacol 26(6):464–466
Cowan JD et al (1988) Phase II trial of five day intravenous infusion vinblastine sulfate in patients with diffuse malignant mesothelioma: a Southwest Oncology Group study. Invest New Drugs 6(3):247–248
Costa G et al (1963) Clinical trial of vinblastine in multiple myeloma. Cancer Chemother Rep 27:87–89
Xu J et al (2021) Anlotinib, vincristine, and irinotecan for advanced ewing sarcoma after failure of standard multimodal therapy: a two-cohort, phase Ib/II trial. Oncologist. https://doi.org/10.1002/onco.13726
Bell EH et al (2020) Comprehensive genomic analysis in NRG oncology/RTOG 9802: A Phase III trial of radiation versus radiation plus procarbazine, lomustine (CCNU), and vincristine in high-risk low-grade glioma. J Clin Oncol 38(29):3407–3417
Shimada K et al (2020) Rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone combined with high-dose methotrexate plus intrathecal chemotherapy for newly diagnosed intravascular large B-cell lymphoma (PRIMEUR-IVL): a multicentre, single-arm, phase 2 trial. Lancet Oncol 21(4):593–602
Cafferty FH et al (2020) Long-term outcomes with intensive induction chemotherapy (carboplatin, bleomycin, vincristine and cisplatin/bleomycin, etoposide and cisplatin) and standard bleomycin, etoposide and cisplatin in poor prognosis germ cell tumours: a randomised phase II trial (ISRCTN53643604). Eur J Cancer 127:139–149
Schiller GJ et al (2018) High-Dose vincristine sulfate liposome injection, for advanced, relapsed, or refractory philadelphia chromosome-negative acute lymphoblastic leukemia in an adolescent and young adult subgroup of a phase 2 clinical trial. J Adolesc Young Adult Oncol 7(5):546–552
Advani RH et al (2016) A phase II study of cyclophosphamide, etoposide, vincristine and prednisone (CEOP) Alternating with Pralatrexate (P) as front line therapy for patients with peripheral T-cell lymphoma (PTCL): final results from the T- cell consortium trial. Br J Haematol 172(4):535–544
Lazaryan A et al (2014) Mature results of MM-011: a phase I/II trial of liposomal doxorubicin, vincristine, dexamethasone, and lenalidomide combination therapy followed by lenalidomide maintenance for relapsed/refractory multiple myeloma. Am J Hematol 89(4):349–354
DuBois SG et al (2016) Phase I study of the aurora a kinase inhibitor alisertib in combination with irinotecan and temozolomide for patients with relapsed or refractory neuroblastoma: a NANT (new approaches to neuroblastoma therapy) trial. J Clin Oncol 34(12):1368–1375
Mascarenhas L et al (2010) Randomized phase II window trial of two schedules of irinotecan with vincristine in patients with first relapse or progression of rhabdomyosarcoma: a report from the Children’s Oncology Group. J Clin Oncol 28(30):4658–4663
Nagatoshi Y et al (2010) Randomized trial to compare LSA2L2-type maintenance therapy to daily 6-mercaptopurine and weekly methotrexate with vincristine and dexamethasone pulse for children with acute lymphoblastic leukemia. Pediatr Blood Cancer 55(2):239–247
Daliani DD et al (2003) Phase II trial of cyclophosphamide, vincristine, and dexamethasone in the treatment of androgen-independent prostate carcinoma. Cancer 97(3):561–567
Hussein MA et al (2002) A phase II trial of pegylated liposomal doxorubicin, vincristine, and reduced-dose dexamethasone combination therapy in newly diagnosed multiple myeloma patients. Cancer 95(10):2160–2168
Cocconi G et al (2002) Randomized trial comparing cyclophosphamide, methotrexate, and 5-fluorouracil (CMF) with rotational CMF, epirubicin and vincristine as primary chemotherapy in operable breast carcinoma. Cancer 95(2):228–235
Chang TC et al (2000) Randomized trial of neoadjuvant cisplatin, vincristine, bleomycin, and radical hysterectomy versus radiation therapy for bulky stage IB and IIA cervical cancer. J Clin Oncol 18(8):1740–1747
Medical Research Council Brain Tumour Working Party (2001) Randomized trial of procarbazine, lomustine, and vincristine in the adjuvant treatment of high-grade astrocytoma: a Medical Research Council trial. J Clin Oncol 19(2):509–18
Sarris AH et al (2000) Liposomal vincristine in relapsed non-Hodgkin’s lymphomas: early results of an ongoing phase II trial. Ann Oncol 11(1):69–72
Northfelt DW et al (1998) Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi’s sarcoma: results of a randomized phase III clinical trial. J Clin Oncol 16(7):2445–2451
Samuel LM et al (1994) Phase II trial of procarbazine, vincristine and lomustine (POC) chemotherapy in metastatic cutaneous malignant melanoma. Eur J Cancer 30(14):2054
Tamura K et al (1987) Phase II trial of cis-diamminedichloroplatinum (cis-platinum), vincristine, and peplomycin for advanced squamous cell carcinoma. J Surg Oncol 35(4):241–244
Taylor SGT, Desai SA, DeWys WD (1978) Phase II trial of a combination of cyclosphamide, vincristine, and methotrexate in advanced colorectal carcinoma. Cancer Treat Rep 62(8):1203–5
Maia ALC et al (2018) Vincristine-loaded hydroxyapatite nanoparticles as a potential delivery system for bone cancer therapy. J Drug Target 26(7):592–603
Zheng Y et al (2021) Efficacy and safety of cetuximab plus cisplatin alone or in combination with paclitaxel in patients with head and neck squamous cell carcinoma: a randomized trial. Cancer Control 28:1073274821997444
Yuan Y et al (2021) Phase II trial of neoadjuvant carboplatin and nab-paclitaxel in patients with triple-negative breast cancer. Oncologist 26(3):e382–e393
Yoneshima Y et al (2021) Phase 3 trial comparing nanoparticle albumin-bound paclitaxel with docetaxel for previously treated advanced NSCLC. J Thorac Oncol. https://doi.org/10.1016/j.jtho.2021.03.027
Sundar R et al (2021) Machine-learning model derived gene signature predictive of paclitaxel survival benefit in gastric cancer: results from the randomised phase III SAMIT trial. Gut. https://doi.org/10.1136/gutjnl-2021-324060
Sai S et al (2021) Phase 1 study of Gemcitabine/Nab-paclitaxel/S-1 in patients with unresectable pancreatic cancer (GeNeS1S trial). Cancer Chemother Pharmacol 87(1):65–71
Falandry C et al (2021) Efficacy and safety of first-line single-agent carboplatin vs carboplatin plus paclitaxel for vulnerable older adult women with ovarian cancer: a randomized clinical trial. JAMA Oncol 7:853
Sridhar SS et al (2020) Efficacy and safety of nab-paclitaxel vs paclitaxel on survival in patients with platinum-refractory metastatic urothelial cancer: the Canadian Cancer Trials Group BL.12 randomized clinical trial. JAMA Oncol 6(11):1751–1758
Urbonas V et al (2019) Paclitaxel with or without trametinib or pazopanib in advanced wild-type BRAF melanoma (PACMEL): a multicentre, open-label, randomised, controlled phase II trial. Ann Oncol 30(2):317–324
Shroff RT et al (2019) Gemcitabine, cisplatin, and nab-paclitaxel for the treatment of advanced biliary tract cancers: a phase 2 clinical trial. JAMA Oncol 5(6):824–830
Pignata S et al (2019) The MITO CERV-2 trial: A randomized phase II study of cetuximab plus carboplatin and paclitaxel, in advanced or recurrent cervical cancer. Gynecol Oncol 153(3):535–540
Xia Y et al (2018) A phase II trial of concurrent chemoradiotherapy with weekly paclitaxel and carboplatin in advanced oesophageal carcinoma. Int J Clin Oncol 23(3):458–465
Wong YN et al (2018) Phase 2 study of weekly paclitaxel plus estramustine in metastatic hormone-refractory prostate carcinoma: ECOG-ACRIN Cancer Research Group (E1898) Trial. Clin Genitourin Cancer 16(2):e315–e322
Simpkins F et al (2015) A phase II trial of paclitaxel, carboplatin, and bevacizumab in advanced and recurrent endometrial carcinoma (EMCA). Gynecol Oncol 136(2):240–245
Grivas PD et al (2013) A phase II trial of neoadjuvant nab-paclitaxel, carboplatin, and gemcitabine (ACaG) in patients with locally advanced carcinoma of the bladder. Urology 82(1):111–117
Ito Y et al (2012) Clinical trial of weekly paclitaxel chemotherapy for papillary thyroid carcinoma with squamous cell carcinoma component. Endocr J 59(9):839–844
Cianfrocca M et al (2011) Pilot study evaluating the interaction between paclitaxel and protease inhibitors in patients with human immunodeficiency virus-associated Kaposi’s sarcoma: an Eastern Cooperative Oncology Group (ECOG) and AIDS Malignancy Consortium (AMC) trial. Cancer Chemother Pharmacol 68(4):827–833
Tew WP et al (2009) Phase I trial of weekly cisplatin, irinotecan and paclitaxel in patients with advanced gastrointestinal cancer. Invest New Drugs 27(4):366–373
Bylow KA et al (2009) Phase II trial of carboplatin and paclitaxel in papillary renal cell carcinoma. Clin Genitourin Cancer 7(1):39–42
Abbasi MR et al (2003) Phase I trial of fludarabine and paclitaxel in non-Hodgkin’s lymphoma. Med Oncol 20(1):53–58
Pivot X et al (2002) Phase II trial of paclitaxel-epirubicin in patients with recurrent soft-tissue sarcoma. Am J Clin Oncol 25(6):561–564
Hinton S et al (2002) Phase II study of paclitaxel plus gemcitabine in refractory germ cell tumors (E9897): a trial of the Eastern Cooperative Oncology Group. J Clin Oncol 20(7):1859–1863
Tan EH et al (1999) Phase II trial of a paclitaxel and carboplatin combination in Asian patients with metastatic nasopharyngeal carcinoma. Ann Oncol 10(2):235–237
Zong B et al (2021) HORMAD1 promotes docetaxel resistance in triple negative breast cancer by enhancing DNA damage tolerance. Oncol Rep 46(1):1–15
Hu Y et al (2020) Co-delivery of docetaxel and curcumin via nanomicelles for enhancing anti-ovarian cancer treatment. Int J Nanomed 15:9703–9715
Korphaisarn K et al (2021) Efficacy of combination docetaxel and nintedanib in advanced non-small cell lung cancer in Thailand: a multicenter study. Front Oncol 11:572740
Maj-Hes A et al (2021) Multiple Docetaxel retreatments without prednisone for metastatic castration-resistant prostate cancer in the docetaxel-only era: effects on PSA kinetics and survival. Adv Ther. https://doi.org/10.1007/s12325-021-01778-8
Colevas AD, Posner MR (1998) Docetaxel in head and neck cancer: a review. Am J Clin Oncol 21(5):482–486
Ajani JA (2002) Docetaxel for gastric and esophageal carcinomas. Oncology (Williston Park) 16(6 Suppl 6):89–96
Doxetaxel: new indication (2006) Prostate cancer: a few more weeks. Prescrire Int. 15(81):6–7
Fouladian P et al (2021) Drug-loaded, polyurethane coated nitinol stents for the controlled release of docetaxel for the treatment of oesophageal cancer. Pharmaceuticals (Basel) 14(4):311
Chiu HI, Lim V (2021) Wheat germ agglutinin-conjugated disulfide cross-linked alginate nanoparticles as a docetaxel carrier for colon cancer therapy. Int J Nanomed 16:2995–3020
Liang C et al (2021) Π electron-stabilized polymeric micelles potentiate docetaxel therapy in advanced-stage gastrointestinal cancer. Biomaterials 266:120432
Steinberg RL et al (2020) Multi-Institution evaluation of sequential gemcitabine and docetaxel as rescue therapy for nonmuscle invasive bladder cancer. J Urol 203(5):902–909
Koussis H et al (2015) Complete response to weekly carboplatin-docetaxel chemotherapy in a 91-year-old woman with anaplastic thyroid cancer. Am J Otolaryngol 36(2):268–272
Guo J et al (2015) RASSF10 suppresses colorectal cancer growth by activating P53 signaling and sensitizes colorectal cancer cell to docetaxel. Oncotarget 6(6):4202–4213
Oberic L et al (2011) Docetaxel- and 5-FU-concurrent radiotherapy in patients presenting unresectable locally advanced pancreatic cancer: a FNCLCC-ACCORD/0201 randomized phase II trial’s pre-planned analysis and case report of a 5.5-year disease-free survival. Radiat Oncol 6:124
Grover A et al (2014) Brain-targeted delivery of docetaxel by glutathione-coated nanoparticles for brain cancer. AAPS PharmSciTech 15(6):1562–1568
Cheng CY et al (2018) BI2536 induces mitotic catastrophe and radiosensitization in human oral cancer cells. Oncotarget 9(30):21231–21243
Lian G et al (2018) BI2536, a potent and selective inhibitor of polo-like kinase 1, in combination with cisplatin exerts synergistic effects on gastric cancer cells. Int J Oncol 52(3):804–814
Breitenbuecher F et al (2017) Comprehensive biomarker analyses in patients with advanced or metastatic non-small cell lung cancer prospectively treated with the polo-like kinase 1 inhibitor BI2536. Oncol Res Treat 40(7–8):435–439
Prashanth Kumar BN et al (2015) BI2536–A PLK inhibitor augments paclitaxel efficacy in suppressing tamoxifen induced senescence and resistance in breast cancer cells. Biomed Pharmacother 74:124–132
Wagenblast J et al (2013) Antitumoral effect of PLK-1-inhibitor BI2536 in combination with cisplatin and docetaxel in squamous cell carcinoma cell lines of the head and neck. Mol Clin Oncol 1(2):286–290
Nuthalapati S et al (2012) Preclinical pharmacokinetic and pharmacodynamic evaluation of novel anticancer agents, ON01910.Na (Rigosertib, EstybonTM and ON013105, for brain tumor chemotherapy. Pharm Res 29(9):2499–2511
Hoffmann MJ et al (2021) Downregulation of cell cycle and checkpoint genes by class I HDAC inhibitors limits synergism with G2/M checkpoint inhibitor MK-1775 in bladder cancer cells. Genes (Basel) 12(2):260
Qi W et al (2014) CHK1 plays a critical role in the anti-leukemic activity of the wee1 inhibitor MK-1775 in acute myeloid leukemia cells. J Hematol Oncol 7:53
Pokorny JL et al (2015) The efficacy of the wee1 inhibitor MK-1775 combined with temozolomide is limited by heterogeneous distribution across the blood-brain barrier in glioblastoma. Clin Cancer Res 21(8):1916–1924
Rajeshkumar NV et al (2011) MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res 17(9):2799–2806
Barbosa RSS et al (2019) Sequential combination of bortezomib and WEE1 inhibitor, MK-1775, induced apoptosis in multiple myeloma cell lines. Biochem Biophys Res Commun 519(3):597–604
Chen G et al (2017) Suppression of Sirt1 sensitizes lung cancer cells to WEE1 inhibitor MK-1775-induced DNA damage and apoptosis. Oncogene 36(50):6863–6872
Kreahling JM et al (2013) Wee1 inhibition by MK-1775 leads to tumor inhibition and enhances efficacy of gemcitabine in human sarcomas. PLoS ONE 8(3):e57523
Fukudo M, Ishibashi K, Kitada M (2021) Real-world pharmacokinetics and pharmacodynamics of everolimus in metastatic breast cancer. Invest New Drugs. https://doi.org/10.1007/s10637-021-01131-4
Cui J et al (2021) Everolimus regulates the activity of gemcitabine-resistant pancreatic cancer cells by targeting the Warburg effect via PI3K/AKT/mTOR signaling. Mol Med 27(1):38
Fazio N et al (2021) Relationship between metabolic toxicity and efficacy of everolimus in patients with neuroendocrine tumors: a pooled analysis from the randomized, phase 3 RADIANT-3 and RADIANT-4 trials. Cancer. https://doi.org/10.1002/cncr.33540
Wiele AJ et al (2021) Lenvatinib with or without everolimus in patients with metastatic renal cell carcinoma after immune checkpoint inhibitors and vascular endothelial growth factor receptor-tyrosine kinase inhibitor therapies. Oncologist 26:476
Pan S et al (2021) Significant benefit of everolimus in a patient with urothelial bladder cancer harboring a rare M1043I mutation of PIK3CA. Invest New Drugs. https://doi.org/10.21203/rs.3.rs-216259/v1
Janku F et al (2020) Safety and efficacy of vorinostat plus sirolimus or everolimus in patients with relapsed refractory hodgkin lymphoma. Clin Cancer Res 26(21):5579–5587
Hellyer JA et al (2020) Everolimus in the treatment of metastatic thymic epithelial tumors. Lung Cancer 149:97–102
Kramer B et al (2020) Tyrosine kinase inhibitors and everolimus reduce IGF1R expression in HPV16-positive and -negative squamous cell carcinoma. Anticancer Res 40(7):3847–3855
Miklja Z et al (2020) Everolimus improves the efficacy of dasatinib in PDGFRα-driven glioma. J Clin Invest 130(10):5313–5325
Liu H et al (2020) MEK inhibition overcomes everolimus resistance in gastric cancer. Cancer Chemother Pharmacol 85(6):1079–1087
Nogova L et al (2020) Sorafenib and everolimus in patients with advanced solid tumors and KRAS-mutated NSCLC: a phase I trial with early pharmacodynamic FDG-PET assessment. Cancer Med 9(14):4991–5007
Hoeg RT et al (2020) A phase I study of everolimus and bendamustine in patients with relapsed/refractory lymphoid hematologic malignancies. Clin Lymphoma Myeloma Leuk 20(7):453–458
George DJ et al (2020) Phase 2 clinical trial of TORC1 inhibition with everolimus in men with metastatic castration-resistant prostate cancer. Urol Oncol 38(3):79.e15-79.e22
Davis KA et al (2019) Use of cardiac MRI to assess antitumor efficacy of everolimus in sporadic cardiac rhabdomyoma. Pediatrics 143(6):e20182495
Ciołczyk-Wierzbicka D et al (2019) mTOR inhibitor Everolimus-induced apoptosis in melanoma cells. J Cell Commun Signal 13(3):357–368
Kuki I et al (2018) Efficacy and safety of everolimus in patients younger than 12 months with congenital subependymal giant cell astrocytoma. Brain Dev 40(5):415–420
Hanna GJ et al (2018) Genomic Correlates of response to everolimus in aggressive radioiodine-refractory thyroid cancer: a phase II study. Clin Cancer Res 24(7):1546–1553
Witzig TE et al (2018) Adjuvant everolimus in high-risk diffuse large B-cell lymphoma: final results from the PILLAR-2 randomized phase III trial. Ann Oncol 29(3):707–714
Tiong IS et al (2018) Phase Ib study of the mTOR inhibitor everolimus with low dose cytarabine in elderly acute myeloid leukemia. Leuk Lymphoma 59(2):493–496
Ozeki M et al (2017) Everolimus for treatment of pseudomyogenic hemangioendothelioma. J Pediatr Hematol Oncol 39(6):e328–e331
He K et al (2016) BRAFV600E-dependent Mcl-1 stabilization leads to everolimus resistance in colon cancer cells. Oncotarget 7(30):47699–47710
Calleja-Algarra A et al (2017) Everolimus: “Killing 2 birds with one stone” in a liver recipient with Kaposi sarcoma. Med Clin (Barc) 148(3):e17
Guo H et al (2016) Everolimus exhibits anti-tumorigenic activity in obesity-induced ovarian cancer. Oncotarget 7(15):20338–20356
Mego M et al (2016) Phase II study of everolimus in refractory testicular germ cell tumors. Urol Oncol 34(3):122.e17–22
Mendes J et al (2016) L744,832 and everolimus induce cytotoxic and cytostatic effects in non-hodgkin lymphoma cells. Pathol Oncol Res 22(2):301–309
Kim DW et al (2014) A multicenter phase II study of everolimus in patients with progressive unresectable adenoid cystic carcinoma. BMC Cancer 14:795
Liu C et al (2020) Apatinib combined with CCI-779 inhibits the proliferation and migration of small cell lung cancer NCI-H446 cells in vitro. Zhongguo Fei Ai Za Zhi 23(4):216–222
Li S et al (2013) The novel mTOR inhibitor CCI-779 (temsirolimus) induces antiproliferative effects through inhibition of mTOR in Bel-7402 liver cancer cells. Cancer Cell Int 13:30
Ito D et al (2006) In vivo antitumor effect of the mTOR inhibitor CCI-779 and gemcitabine in xenograft models of human pancreatic cancer. Int J Cancer 118(9):2337–2343
Teachey DT et al (2006) The mTOR inhibitor CCI-779 induces apoptosis and inhibits growth in preclinical models of primary adult human ALL. Blood 107(3):1149–1155
Witzig TE et al (2005) Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol 23(23):5347–5356
Wu L, Birle DC, Tannock IF (2005) Effects of the mammalian target of rapamycin inhibitor CCI-779 used alone or with chemotherapy on human prostate cancer cells and xenografts. Cancer Res 65(7):2825–2831
Chan S et al (2005) Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol 23(23):5314–5322
Gallicchio MA, van Sinderen M, Bach LA (2003) Insulin-like growth factor binding protein-6 and CCI-779, an ester analogue of rapamycin, additively inhibit rhabdomyosarcoma growth. Horm Metab Res 35(11–12):822–827
Thallinger C et al (2007) Comparison of a treatment strategy combining CCI-779 plus DTIC versus DTIC monotreatment in human melanoma in SCID mice. J Invest Dermatol 127(10):2411–2417
Lin CY et al (2021) Vorinostat combined with brigatinib overcomes acquired resistance in EGFR-C797S-mutated lung cancer. Cancer Lett 508:76–91
van den Bijgaart RJE et al (2020) Anti-GD2 antibody and Vorinostat immunocombination therapy is highly effective in an aggressive orthotopic neuroblastoma model. Oncoimmunology 9(1):1817653
Brown S et al (2021) Bortezomib, Vorinostat, and dexamethasone combination therapy in relapsed myeloma: results of the phase 2 MUK four trial. Clin Lymphoma Myeloma Leuk 21(3):154-161.e3
Ahmed AA, Neidle S (2020) A G-Quadruplex-binding small molecule and the HDAC inhibitor SAHA (Vorinostat) act synergistically in gemcitabine-sensitive and resistant pancreatic cancer cells. Molecules 25(22):5407
Zhao G et al (2020) Local delivery of minocycline and vorinostat targets the tumor microenvironment to inhibit the recurrence of glioma. Onco Targets Ther 13:11397–11409
Porcu P et al (2021) Quality of life effect of the anti-CCR4 monoclonal antibody mogamulizumab versus vorinostat in patients with cutaneous T-cell lymphoma. Clin Lymphoma Myeloma Leuk 21(2):97–105
Abdel-Ghany S et al (2020) Vorinostat-loaded titanium oxide nanoparticles (anatase) induce G2/M cell cycle arrest in breast cancer cells via PALB2 upregulation. 3 Biotech 10(9):407
Kang DW et al (2021) Phospholipase D1 is upregulated by vorinostat and confers resistance to vorinostat in glioblastoma. J Cell Physiol 236(1):549–560
Rodriguez CP et al (2020) A phase II trial of pembrolizumab and vorinostat in recurrent metastatic head and neck squamous cell carcinomas and salivary gland cancer. Clin Cancer Res 26(4):837–845
Spurgeon SE et al (2019) Phase 1–2 study of vorinostat (SAHA), cladribine and rituximab (SCR) in relapsed B-cell non-Hodgkin lymphoma and previously untreated mantle cell lymphoma. Br J Haematol 186(6):845–854
Ding L et al (2018) Targeting the autophagy in bone marrow stromal cells overcomes resistance to vorinostat in chronic lymphocytic leukemia. Onco Targets Ther 11:5151–5170
Yazbeck V et al (2018) A Phase II trial of bortezomib and vorinostat in mantle cell lymphoma and diffuse large B-cell lymphoma. Clin Lymphoma Myeloma Leuk 18(9):569-575.e1
Liao B et al (2018) Vorinostat enhances the anticancer effect of oxaliplatin on hepatocellular carcinoma cells. Cancer Med 7(1):196–207
Ghiaseddin A et al (2018) Phase II study of bevacizumab and vorinostat for patients with recurrent World Health Organization grade 4 malignant glioma. Oncologist 23(2):157-e21
Bernhart E et al (2017) Histone deacetylase inhibitors vorinostat and panobinostat induce G1 cell cycle arrest and apoptosis in multidrug resistant sarcoma cell lines. Oncotarget 8(44):77254–77267
Luu T et al (2018) Phase IB trial of ixabepilone and vorinostat in metastatic breast cancer. Breast Cancer Res Treat 167(2):469–478
Ma X et al (2017) Targeting CD146 in combination with vorinostat for the treatment of ovarian cancer cells. Oncol Lett 13(3):1681–1687
Kwak TW et al (2015) Antitumor activity of vorinostat-incorporated nanoparticles against human cholangiocarcinoma cells. J Nanobiotechnol 13:60
Lee SJ et al (2014) Transactivation of bad by vorinostat-induced acetylated p53 enhances doxorubicin-induced cytotoxicity in cervical cancer cells. Exp Mol Med 46(2):e76
Bergadà L et al (2013) Combination of Vorinostat and caspase-8 inhibition exhibits high anti-tumoral activity on endometrial cancer cells. Mol Oncol 7(4):763–75
Phillip CJ et al (2012) Genistein cooperates with the histone deacetylase inhibitor vorinostat to induce cell death in prostate cancer cells. BMC Cancer 12:145
Claerhout S et al (2011) Gene expression signature analysis identifies vorinostat as a candidate therapy for gastric cancer. PLoS ONE 6(9):e24662
Uematsu S et al (2021) Acceptability and feasibility of S-1 plus cisplatin adjuvant chemotherapy for completely resected non-small cell lung cancer: an open-label, single arm, multicenter, phase 2 trial. J Thorac Dis 13(4):2224–2232
Rosenberg JE et al (2021) Randomized phase III trial of gemcitabine and cisplatin with bevacizumab or placebo in patients with advanced urothelial carcinoma: results of CALGB 90601 (Alliance). J Clin Oncol. https://doi.org/10.1200/JCO.21.00286
Garcia-Soto AE et al (2021) Phase 1 trial of nelfinavir added to standard cisplatin chemotherapy with concurrent pelvic radiation for locally advanced cervical cancer. Cancer 127:2279
Lv X et al (2021) Induction chemotherapy with lobaplatin and fluorouracil versus cisplatin and fluorouracil followed by chemoradiotherapy in patients with stage III-IVB nasopharyngeal carcinoma: an open-label, non-inferiority, randomised, controlled, phase 3 trial. Lancet Oncol 22(5):716–726
Chan CY et al (2021) A dose-finding trial for hyperthermic intraperitoneal cisplatin in gynecological cancer patients receiving hyperthermic intraperitoneal chemotherapy. Front Oncol 11:616264
Challapalli A et al (2021) A single-arm phase II trial of neoadjuvant cabazitaxel and cisplatin chemotherapy for muscle-invasive transitional cell carcinoma of the urinary bladder. Clin Genitourin Cancer. https://doi.org/10.1016/j.clgc.2021.02.001
Bellon JR et al (2021) A phase 1 dose-escalation trial of radiation therapy and concurrent cisplatin for stage II and III triple-negative breast cancer. Int J Radiat Oncol Biol Phys. https://doi.org/10.1016/j.ijrobp.2021.03.002
Ju WT et al (2021) Phase III trial of docetaxel cisplatin 5-fluorouracil induction chemotherapy for resectable oral cancer suggests favorable pathological response as a surrogate endpoint for good therapeutic outcome. Cancer Commun (Lond) 41(3):279–283
Fachini AMD et al (2021) Long-term outcomes of concomitant cisplatin plus radiotherapy versus radiotherapy alone in patients with stage IIIB squamous cervical cancer: a randomized controlled trial. Gynecol Oncol 160(2):379–383
Kurokawa Y et al (2021) Docetaxel plus S-1 versus cisplatin plus S-1 in unresectable gastric cancer without measurable lesions: a randomized phase II trial (HERBIS-3). Gastric Cancer 24(2):428–434
Yan P et al (2020) Raltitrexed versus 5-fluorouracil with cisplatin and concurrent radiotherapy for locally advanced nasopharyngeal carcinoma: an open labeled, randomized, controlled, and multicenter clinical trial. Cancer Med 9(17):6166–6172
Rao S et al (2020) International rare cancers initiative multicenter randomized phase II trial of cisplatin and fluorouracil versus carboplatin and paclitaxel in advanced anal cancer: interAAct. J Clin Oncol 38(22):2510–2518
Jones R et al (2020) A randomised Phase II trial of carboplatin and gemcitabine ± vandetanib in first-line treatment of patients with advanced urothelial cell cancer not suitable to receive cisplatin. BJU Int 126(2):292–299
Boudewijns S et al (2020) Autologous monocyte-derived DC vaccination combined with cisplatin in stage III and IV melanoma patients: a prospective, randomized phase 2 trial. Cancer Immunol Immunother 69(3):477–488
Makimoto A et al (2019) Magnesium supplementation therapy to prevent cisplatin-induced acute nephrotoxicity in pediatric cancer: a protocol for a randomized phase 2 trial. Contemp Clin Trials Commun 16:100440
Tsao AS et al (2019) Phase II trial of cediranib in combination with cisplatin and pemetrexed in chemotherapy-naïve patients with unresectable malignant pleural mesothelioma (SWOG S0905). J Clin Oncol 37(28):2537–2547
Scagliotti GV et al (2019) Nintedanib in combination with pemetrexed and cisplatin for chemotherapy-naive patients with advanced malignant pleural mesothelioma (LUME-Meso): a double-blind, randomised, placebo-controlled phase 3 trial. Lancet Respir Med 7(7):569–580
Chen Y et al (2019) Comparing paclitaxel plus fluorouracil versus cisplatin plus fluorouracil in chemoradiotherapy for locally advanced esophageal squamous cell cancer: a randomized, multicenter, phase III clinical trial. J Clin Oncol 37(20):1695–1703
Gillison ML et al (2019) Radiotherapy plus cetuximab or cisplatin in human papillomavirus-positive oropharyngeal cancer (NRG Oncology RTOG 1016): a randomised, multicentre, non-inferiority trial. Lancet 393(10166):40–50
Reni M et al (2018) Nab-paclitaxel plus gemcitabine with or without capecitabine and cisplatin in metastatic pancreatic adenocarcinoma (PACT-19): a randomised phase 2 trial. Lancet Gastroenterol Hepatol 3(10):691–697
Park S et al (2013) A prospective phase II trial of induction chemotherapy with docetaxel/cisplatin for Masaoka stage III/IV thymic epithelial tumors. J Thorac Oncol 8(7):959–66
Meyer T et al (2013) A randomised phase II/III trial of 3-weekly cisplatin-based sequential transarterial chemoembolisation vs embolisation alone for hepatocellular carcinoma. Br J Cancer 108(6):1252–9
Bible KC et al (2012) A phase 2 trial of flavopiridol (Alvocidib) and cisplatin in platin-resistant ovarian and primary peritoneal carcinoma: MC0261. Gynecol Oncol 127(1):55–62
Arrieta Ó et al (2012) First-line chemotherapy with liposomal doxorubicin plus cisplatin for patients with advanced malignant pleural mesothelioma: phase II trial. Br J Cancer 106(6):1027–32
Chua DT et al (2012) Phase II trial of capecitabine plus cisplatin as first-line therapy in patients with metastatic nasopharyngeal cancer. Head Neck 34(9):1225–30
Landrum LM et al (2011) Phase II trial of intraperitoneal cisplatin combined with intravenous paclitaxel in patients with ovarian, primary peritoneal and fallopian tube cancer. Gynecol Oncol 122(3):527–31
Feldman DR et al (2007) Phase II trial of ixabepilone in patients with cisplatin-refractory germ cell tumors. Invest New Drugs 25(5):487–90
Dantas AA et al (2006) Topotecan, Ara-C, cisplatin and prednisolone (Toposhap) for patients with refractory and relapsing lymphomas: results of a phase II trial. Acta Haematol 116(4):275–8
Pierga JY et al (1997) Phase II trial of doxorubicin, 5-fluorouracil, etoposide, and cisplatin in advanced or recurrent endometrial carcinoma. Gynecol Oncol 66(2):246–9
Feun LG et al (1986) Phase II trial of intracarotid BCNU and cisplatin in primary malignant brain tumors. Cancer Drug Deliv 3(2):147–56
Feun LG et al (1984) Phase-I trial of intracarotid BCNU and cisplatin in patients with malignant intracerebral tumors. Cancer Drug Deliv 1(3):239–45
Brenner J et al (1982) Phase II trial of cisplatin (CPDD) in previously treated patients with advanced soft tissue sarcoma. Cancer 50(10):2031–3
Lin JC et al (2021) Trilostane, a 3β-hydroxysteroid dehydrogenase inhibitor, suppresses growth of hepatocellular carcinoma and enhances anti-cancer effects of sorafenib. Invest New Drugs. https://doi.org/10.1007/s10637-021-01132-3
Lu Z et al (2021) Combined Anti-cancer effects of platycodin D and sorafenib on androgen-independent and PTEN-deficient prostate cancer. Front Oncol 11:648985
Szarek M et al (2021) Q-TWiST Analysis of tivozanib versus sorafenib in patients with advanced renal cell carcinoma in the TIVO-3 study. Clin Genitourin Cancer. https://doi.org/10.1016/j.clgc.2021.03.018
Wu CH et al (2021) Sorafenib induces apoptosis and inhibits NF-κB-mediated anti-apoptotic and metastatic potential in osteosarcoma cells. Anticancer Res 41(3):1251–1259
Röllig C et al (2021) Sorafenib or placebo in patients with newly diagnosed acute myeloid leukaemia: long-term follow-up of the randomized controlled SORAML trial. Leukemia. https://doi.org/10.1038/s41375-021-01148-x
Guo T et al (2020) Photodynamic therapy in combination with sorafenib for enhanced immunotherapy of lung cancer. J Biomed Nanotechnol 16(8):1219–1228
Kim JY et al (2020) Sorafenib increases tumor treating fields-induced cell death in glioblastoma by inhibiting STAT3. Am J Cancer Res 10(10):3475–3486
Spiegelberg D et al (2020) The novel anti-cmet antibody seeMet 12 potentiates sorafenib therapy and radiotherapy in a colorectal cancer model. Front Oncol 10:1717
Celano M et al (2020) Quercetin improves the effects of sorafenib on growth and migration of thyroid cancer cells. Endocrine 67(2):496–498
Srivastava A, Moorthy A (2019) Sorafenib induces synergistic effect on inhibition of vemurafenib resistant melanoma growth. Front Biosci (Schol Ed) 11:193–202
Hubbard JM et al (2016) Phase I trial of FOLFIRI in combination with sorafenib and bevacizumab in patients with advanced gastrointestinal malignancies. Invest New Drugs 34(1):96–103
Agarwal S, Elmquist WF (2012) Insight into the cooperation of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) at the blood-brain barrier: a case study examining sorafenib efflux clearance. Mol Pharm 9(3):678–84
Jane EP, Premkumar DR, Pollack IF (2006) Coadministration of sorafenib with rottlerin potently inhibits cell proliferation and migration in human malignant glioma cells. J Pharmacol Exp Ther 319(3):1070–80
Siu LL et al (2006) Phase I trial of sorafenib and gemcitabine in advanced solid tumors with an expanded cohort in advanced pancreatic cancer. Clin Cancer Res 12(1):144–51
Wrighton KH (2019) Desmoid tumours stalled by sorafenib. Nat Rev Clin Oncol 16(4):209
Shamaa MM (2021) Sulfasalazine synergistically enhances the inhibitory effects of imatinib against hepatocellular carcinoma (HCC) cells by targeting NFκB, BCR/ABL, and PI3K/AKT signaling pathway-related proteins. FEBS Open Biol 11(3):588–597
Yu H et al (2020) Identification of target genes related to sulfasalazine in triple-negative breast cancer through network pharmacology. Med Sci Monit 26:e926550
Zheng Z et al (2020) The X(c)(-) inhibitor sulfasalazine improves the anti-cancer effect of pharmacological vitamin C in prostate cancer cells via a glutathione-dependent mechanism. Cell Oncol (Dordr) 43(1):95–106
Ogihara K et al (2019) Sulfasalazine could modulate the CD44v9-xCT system and enhance cisplatin-induced cytotoxic effects in metastatic bladder cancer. Cancer Sci 110(4):1431–1441
Mooney MR et al (2019) Anti-tumor effect of sulfasalazine in neuroblastoma. Biochem Pharmacol 162:237–249
Kim EH et al (2018) CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett 432:180–190
Nagane M et al (2018) Sulfasalazine, an inhibitor of the cystine-glutamate antiporter, reduces DNA damage repair and enhances radiosensitivity in murine B16F10 melanoma. PLoS ONE 13(4):e0195151
Shitara K et al (2017) Phase 1 study of sulfasalazine and cisplatin for patients with CD44v-positive gastric cancer refractory to cisplatin (EPOC1407). Gastric Cancer 20(6):1004–1009
Ignarro RS et al (2016) Sulfasalazine intensifies temozolomide cytotoxicity in human glioblastoma cells. Mol Cell Biochem 418(1–2):167–78
Takayama T et al (2016) Potential of sulfasalazine as a therapeutic sensitizer for CD44 splice variant 9-positive urogenital cancer. Med Oncol 33(5):45
Ng TSC et al (2021) Detecting immune response to therapies targeting PDL1 and BRAF by using ferumoxytol MRI and macrin in anaplastic thyroid cancer. Radiology 298(1):123–132
Barajas RF Jr et al (2020) Distinguishing extravascular from intravascular ferumoxytol pools within the brain: proof of concept in patients with treated glioblastoma. AJNR Am J Neuroradiol 41(7):1193–1200
Zhao J et al (2018) Anti-tumor macrophages activated by ferumoxytol combined or surface-functionalized with the TLR3 agonist poly (I : C) promote melanoma regression. Theranostics 8(22):6307–6321
Turkbey B et al (2015) A phase I dosing study of ferumoxytol for MR lymphography at 3 T in patients with prostate cancer. AJR Am J Roentgenol 205(1):64–9
Hedgire SS et al (2014) Enhanced primary tumor delineation in pancreatic adenocarcinoma using ultrasmall super paramagnetic iron oxide nanoparticle-ferumoxytol: an initial experience with histopathologic correlation. Int J Nanomed 9:1891–6
Yang W et al (2021) Shikonin differentially regulates glucose metabolism via PKM2 and HIF1α to overcome apoptosis in a refractory HCC cell line. Life Sci 265:118796
Wang W et al (2021) Shikonin is a novel and selective IMPDH2 inhibitor that target triple-negative breast cancer. Phytother Res 35(1):463–476
Markowitsch SD et al (2021) Shikonin reduces growth of docetaxel-resistant prostate cancer cells mainly through necroptosis. Cancers (Basel) 13(4):882
Liu C et al (2021) Shikonin inhibits cholangiocarcinoma cell line QBC939 by regulating apoptosis, proliferation, and invasion. Cell Transplant 30:963689720979162
Zhang N et al (2020) Shikonin induces colorectal carcinoma cells apoptosis and autophagy by targeting galectin-1/JNK signaling axis. Int J Biol Sci 16(1):147–161
Zhang J, Zhou J, Xiao S (2020) Shikonin inhibits growth, invasion and glycolysis of nasopharyngeal carcinoma cells through inactivating the phosphatidylinositol 3 kinase/AKT signal pathway. Anticancer Drugs 31(9):932–941
Zhang DF et al (2020) Effect of shikonin on the proliferation and apoptosis of human ovarian cancer cell SKOV3: a protocol of systematic review and meta-analysis. Medicine (Baltimore) 99(22):e20450
Zang F et al (2020) Shikonin suppresses NEAT1 and Akt signaling in treating paclitaxel-resistant non-small cell of lung cancer. Mol Med 26(1):28
Tang Q et al (2020) Regulations of miR-183-5p and snail-mediated shikonin-reduced epithelial-mesenchymal transition in cervical cancer cells. Drug Des Devel Ther 14:577–589
Sweeney SR et al (2020) Identification of a synergistic combination of dimethylaminoparthenolide and shikonin alters metabolism and inhibits proliferation of pediatric precursor-B cell acute lymphoblastic leukemia. Mol Carcinog 59(4):399–411
Guo N et al (2019) Shikonin inhibits proliferation and induces apoptosis in glioma cells via downregulation of CD147. Mol Med Rep 19(5):4335–4343
Du W et al (2019) Shikonin potentiates paclitaxel antitumor efficacy in esophageal cancer cells via the apoptotic pathway. Oncol Lett 18(3):3195–3201
Zhang Y et al (2018) Shikonin inhibites migration and invasion of thyroid cancer cells by downregulating DNMT1. Med Sci Monit 24:661–670
Su Y et al (2018) Shikonin-mediated up-regulation of miR-34a and miR-202 inhibits retinoblastoma proliferation. Toxicol Res (Camb) 7(5):907–912
Yang Q et al (2017) Shikonin promotes adriamycin-induced apoptosis by upregulating caspase-3 and caspase-8 in osteosarcoma. Mol Med Rep 16(2):1347–1352
Min R et al (2008) Growth inhibition and induction of apoptosis in human oral squamous cell carcinoma Tca-8113 cell lines by Shikonin was partly through the inactivation of NF-kappaB pathway. Phytother Res 22(3):407–15
Wu Z et al (2004) p53-mediated cell cycle arrest and apoptosis induced by shikonin via a caspase-9-dependent mechanism in human malignant melanoma A375–S2 cells. J Pharmacol Sci 94(2):166–76
Chowdhury S et al (2021) A Phase I/II study to assess the safety and efficacy of pazopanib and pembrolizumab combination therapy in patients with advanced renal cell carcinoma. Clin Genitourin Cancer. https://doi.org/10.1016/j.clgc.2021.04.007
Yamada H et al (2021) lncRNA HAR1B has potential to be a predictive marker for pazopanib therapy in patients with sarcoma. Oncol Lett 21(6):455
Yakobson A et al (2021) Epithelioid hemangioendothelioma and epithelioid hemangioma: pazopanib as a potential salvage therapy. Case Rep Oncol 14(1):309–317
Cui Y et al (2020) Overexpression of NDRG2 promotes the therapeutic effect of pazopanib on ovarian cancer. J Recept Signal Transduct Res. https://doi.org/10.1080/10799893.2020.1831536
Küronya Z et al (2020) Predictive Markers of first line pazopanib treatment in kidney cancer. Pathol Oncol Res 26(4):2475–2481
Chiabotto G et al (2020) Pazopanib and trametinib as a synergistic strategy against osteosarcoma: preclinical activity and molecular insights. Cancers (Basel) 12(6):1519
Puliafito I et al (2020) Occurrence of abscesses during treatment with pazopanib in metastatic renal cancer: a case report. J Med Case Rep 14(1):7
Di Desidero T et al (2019) Effects of pazopanib monotherapy vs. pazopanib and topotecan combination on anaplastic thyroid cancer cells. Front Oncol 9:1202
George DJ et al (2019) Phase 1b trial of docetaxel, prednisone, and pazopanib in men with metastatic castration-resistant prostate cancer. Prostate 79(15):1752–1761
Kessler T et al (2019) Phase II clinical trial of pazopanib in patients with acute myeloid leukemia (AML), relapsed or refractory or at initial diagnosis without an intensive treatment option (PazoAML). Ann Hematol 98(6):1393–1401
Ganjoo KN et al (2014) A multicenter phase II study of pazopanib in patients with advanced gastrointestinal stromal tumors (GIST) following failure of at least imatinib and sunitinib. Ann Oncol 25(1):236–40
Ahn HK et al (2013) Phase II study of pazopanib monotherapy in metastatic gastroenteropancreatic neuroendocrine tumours. Br J Cancer 109(6):1414–9
Juliachs M et al (2013) Effectivity of pazopanib treatment in orthotopic models of human testicular germ cell tumors. BMC Cancer 13:382
Kumar S et al (2013) Tumor dynamics in response to antiangiogenic therapy with oral metronomic topotecan and pazopanib in neuroblastoma xenografts. Transl Oncol 6(4):493–503
Cohen Y et al (2013) A case of metastatic adamantinoma responding to treatment with pazopanib. Acta Oncol 52(6):1229–30
Brady J et al (2013) An open-label study of the safety and tolerability of pazopanib in combination with FOLFOX6 or CapeOx in patients with colorectal cancer. Invest New Drugs 31(5):1228–35
Kidoguchi K et al (2021) Efficacy and safety of ponatinib for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: a case series from a single institute. Int J Hematol. https://doi.org/10.1007/s12185-021-03156-0
Castagnetti F et al (2021) Dosing strategies for improving the risk-benefit profile of ponatinib in patients with chronic myeloid leukemia in chronic phase. Front Oncol 11:642005
Liu C et al (2019) Ponatinib inhibits proliferation and induces apoptosis of liver cancer cells, but its efficacy is compromised by its activation on PDK1/Akt/mTOR signaling. Molecules 24(7):1363
Tan FH et al (2018) Ponatinib Inhibits multiple signaling pathways involved in STAT3 signaling and attenuates colorectal tumor growth. Cancers (Basel) 10(12):526
Zhang J et al (2014) The effects of ponatinib, a multi-targeted tyrosine kinase inhibitor, against human U87 malignant glioblastoma cells. Onco Targets Ther 7:2013–9
De Falco V et al (2013) Ponatinib (AP24534) is a novel potent inhibitor of oncogenic RET mutants associated with thyroid cancer. J Clin Endocrinol Metab 98(5):E811-9
Ren M et al (2013) Novel FGFR inhibitor ponatinib suppresses the growth of non-small cell lung cancer cells overexpressing FGFR1. Oncol Rep 29(6):2181–90
Rabben HL et al (2021) Computational drug repositioning and experimental validation of ivermectin in treatment of gastric cancer. Front Pharmacol 12:625991
Zhang X et al (2020) Ivermectin augments the in vitro and in vivo efficacy of cisplatin in epithelial ovarian cancer by suppressing Akt/mTOR signaling. Am J Med Sci 359(2):123–129
Liu J et al (2019) Ivermectin induces autophagy-mediated cell death through the AKT/mTOR signaling pathway in glioma cells. Biosci Rep 39(12):BSR20192489
Deng F et al (2018) Suppressing ROS-TFE3-dependent autophagy enhances ivermectin-induced apoptosis in human melanoma cells. J Cell Biochem 120:1702
Wang K et al (2016) Ivermectin induces PAK1-mediated cytostatic autophagy in breast cancer. Autophagy 12(12):2498–2499
Damian D, Rogers M (2003) Demodex infestation in a child with leukaemia: treatment with ivermectin and permethrin. Int J Dermatol 42(9):724–6
Qiao L et al (2019) α-NETA induces pyroptosis of epithelial ovarian cancer cells through the GSDMD/caspase-4 pathway. FASEB J 33(11):12760–12767
Funding
This study was funded by the National Natural Science Foundation of China (Grant No. 81972789), Fundamental Research Funds for the Central Universities (Grant No. JUSRP22011), Technology Development Funding of Wuxi (Grant No. WX18IVJN017), Zhengzhou Major Project for Collaborative Innovation(18XTZX12007). These funding sources have no role in the writing of the manuscript or the decision to submit it for publication.
Author information
Authors and Affiliations
Contributions
X.F. Dai initiated this project and drafted the manuscript. D.J. Wang helped in literature searching, figure drafting and table preparation. J.Y. Zhang and X.F. Dai financed this project. All authors have read and approved the submission of this paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no completing interest.
Informed Consent
All authors agree with the content and are consent for its publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dai, X., Wang, D. & Zhang, J. Programmed cell death, redox imbalance, and cancer therapeutics. Apoptosis 26, 385–414 (2021). https://doi.org/10.1007/s10495-021-01682-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-021-01682-0