
Summary
Objectives: To determine the maximum tolerated dose (MTD), toxicities, and suitable dose for weekly 1-h paclitaxel combined with weekly cisplatin and irinotecan to treat advanced gastrointestinal malignancies. Methods: Thirty patients with metastatic or locally advanced (unresectable or recurrent) gastrointestinal solid tumors were enrolled on this single-center, phase I study. Patients were treated with paclitaxel given over 1h at 1 of 4 dose levels (40, 50, 65, or 80 mg/m2). Paclitaxel was followed by fixed doses of cisplatin (30 mg/m2) and irinotecan (50 mg/m2). All treatment was administered sequentially, once a week, in 6-week cycles (4 weeks on, 2 weeks off). Dose-limiting toxicity (DLT) was defined as a 2-week delay in treatment for grade 3 or 4 non-hematologic toxicity, neutropenic fever, a 1-week delay for grade 4 hematologic toxicity, or a 2-week delay for grade 3 hematologic toxicity. Results: Thirty patients were recruited; 28 patients were assessable for safety. Most of the patients (70%) had no prior chemotherapy. The primary first-cycle DLTs were neutropenia, diarrhea, and nausea. Paclitaxel at 65 mg/m2 was defined as the MTD. The most common grade 3–4 toxicities observed during all cycles were neutropenia (57%), febrile neutropenia (11%), diarrhea (29%), fatigue (29%), and nausea (18%). No patients had G-CSF (Neupogen, Amgen Inc., Thousand Oaks, CA) support. Responses were observed in gastric, esophageal, and pancreatic cancers. Conclusion: Paclitaxel at 65 mg/m2, cisplatin (30 mg/m2), and irinotecan (50 mg/m2) given weekly can be safely administered to patients with solid tumor malignancies. To improve cumulative toxicities, a schedule modification was required (3-week cycle; 2-on, 1-off) Neutropenia was the most common DLT. This combination showed substantial activity, particularly in patients with gastric and esophageal adenocarcinoma, and phase II evaluation could be considered.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In 2008, more than 35,000 Americans will be diagnosed with esophageal or gastric cancer and nearly 70% will die of their disease [1]. Despite advances in surgical technique and treatment, 5-year overall survival remains low for both esophageal (14%) and gastric (40%) cancers since metastatic or unresectable disease is found at presentation in more than half of patients [2, 3]. Single-agent or platinum-based combination chemotherapy is a palliative treatment option. Older combination regimens with cisplatin and fluorouracil tend to produce higher response rates, but with excessive toxicities [3–5]. Over the past decade, clinical research has focused on combining cisplatin with newer agents, including paclitaxel (Taxol; Bristol-Meyers Squibb, Princeton, NJ) and irinotecan (CPT-11, Camptosar; Pharmacia Corp., Peapack, NJ). Recent trials in upper gastric cancer support the addition of docetaxel to infusional fluorouracil and cisplatin, although increased response rates and survival come at the cost of significant toxicity [6].
Irinotecan, an inhibitor of topoisomerase-1, has emerged as a significant cytotoxic agent in gastrointestinal and lung cancer. Preclinical studies suggest synergy between irinotecan and cisplatin by interrupting the repair of the platinum-DNA adducts and hence further cancer cell death [7–9]. In a randomized trial in Japanese patients with small cell lung cancer, a survival advantage was observed with this combination [10]. However, these results were not replicated in a similar trial in the United States [11]. In gastric cancer, monthly cisplatin and irinotecan every 2 weeks provided response rates exceeding 40%, but with excessive grade 4 neutropenia [12, 13]. At Memorial Sloan-Kettering Cancer Center, a phase I study evaluated this combination on a weekly, more tolerable schedule, with dose-limiting neutropenia [14]. In the subsequent phase II gastroesophageal cancer trials, response rates were substantial in the first-(57%) and second-line (31%) settings, with minimal toxicities [15–17]. These promising results led to further investigation with concurrent radiation in locally advanced cancer [18–20] and suggested that incorporation of a third agent in metastatic cancer may be feasible.
Paclitaxel, an antimitotic agent, has broad-spectrum antitumor activity, especially in platinum combinations, in several malignancies including ovarian and lung cancer. In upper gastrointestinal cancer, Ajani and colleagues reported the single activity (32%) of paclitaxel given in a 24-h infusion [21]. When 24-h paclitaxel was combined with cisplatin every 3 weeks, response rates were substantial (44%); however, unacceptable neutropenia and hospitalizations limited its use [22]. Paclitaxel has also been studied with the use of a shorter infusion rate (3 h every 3 weeks or 1 h every week), which appears to reduce the myelosuppressive toxicity. In patients with esophageal cancer, weekly 1-h paclitaxel has been shown to be active and well tolerated [23].
Based on our experience with phase I/II trials with cisplatin–irinotecan combinations in gastrointestinal cancers and the non-overlapping mechanisms of paclitaxel, we undertook this phase I trial to see if this three-drug combination is feasible. The primary objectives were: (1) to determine the maximum tolerated dose (MTD) and dose-limiting toxicities (DLTs) for the combination of weekly paclitaxel given over 1h, with weekly cisplatin (30 mg/m2) and irinotecan (50 mg/m2) each given over 30 min; (2) to determine the qualitative and quantitative toxicities of this combination; and (3) to seek preliminary evidence of therapeutic activity with this combination regimen in several different gastrointestinal malignancies.
Patients and methods
Patient selection
To be eligible for this trial, all patients had to meet the following criteria: (1) histologic proof of a locally advanced (unresectable or recurrent) or metastatic solid tumor malignancy; (2) bi-dimensionally measurable or evaluable disease was preferable but not required; (3) one prior chemotherapy regimen was allowed in the first cohort (the protocol was amended after cohort 2 to exclude any patients with prior chemotherapy); (4) prior radiation was allowed, provided at least 4 weeks had elapsed and the radiation portal did not include the pelvis; (5) ≥18 years of age; (6) Karnofsky performance status [KPS] of ≥70%; (7) signed consent; and (8) adequate organ function as documented by laboratory studies (white blood count ≥3,000/μL, neutrophils ≥1,500/μL, platelets ≥100,000/μL, serum bilirubin ≤1.5 mg/dL, serum creatinine ≤1.5 mg/dL, and AST (SGOT) ≤3× upper reference value, except when abnormal value was due to liver metastases, in which case ≤5× upper reference value was acceptable).
Patients were excluded from this trial for the following criteria: (1) active or uncontrolled infection; (2) brain metastases or carcinomatous meningitis; (3) interstitial pulmonary fibrosis; (4) unstable angina or grade III/IV NYHA cardiac disease; (5) uncontrolled diabetes mellitus (blood glucose ≥250 mg/dL); (6) hypercalcemia (serum Ca ≥12.0 mg/dL); (7) pregnant or lactating women; (8) history of Gilbert’s disease; (9) history of a seizure disorder and on antiepileptic medications; or (9) unable to comply with the protocol or to undergo the specified follow-up for safety or effectiveness. All patients were required to read, agree to, and sign a statement of informed consent prior to enrollment, as approved by the Institutional Review Board at our institution.
Treatment plan
All chemotherapy was administered in the outpatient setting, and all agents were obtained commercially. Paclitaxel (Bristol-Myers Squibb, Inc., Princeton, NJ) was administrated as an intravenous (IV) infusion over 60 min, followed by IV cisplatin over 30 min, and then IV irinotecan (Pharmacia Corp., Peapack, NJ) over 30 min. Paclitaxel was dose escalated (40–50–65–80 mg/m2) with fixed doses of cisplatin (30 mg/m2) and irinotecan (50 mg/m2). All chemotherapy was given once weekly for 4 weeks, followed by a 2-week break. Each treatment cycle lasted 6 weeks.
To prevent hypersensitivity reaction, patients were pre-treated with dexamethasone (20 mg IV once), diphenhydramine (50 mg IV once), and cimetidine (300 mg IV once) just prior to paclitaxel. For antiemetic control, granisetron (2 mg oral) was given prior to paclitaxel. Recognition and management of treatment-related diarrhea was outlined for each patient. All patients were instructed to begin loperamide at the earliest sign of diarrhea that occurred more than 12 h after receiving irinotecan. Loperamide dosing was as follows: 4 mg at the onset of diarrhea, then 2 mg every 2 h as needed until resolution of diarrhea ≥12 h. Loperamide was not taken prophylactically. Atropine (0.5–1 mg IV bolus) was given for diarrhea or abdominal cramping that occurred within 1h of receiving irinotecan. If a patient required atropine, all subsequent irinotecan doses were preceded by atropine. To prevent renal dysfunction, patients were advised to maintain an oral intake ≥1L/m2 on the day of treatment and were hydrated with 5% dextrose in normal saline at a minimum of 250 mL/h for 2 h prior to cisplatin.
Erythropoietin (Procrit, Amgen Inc., Thousand Oaks, CA) was permitted per institutional guidelines. Filgrastim (G-CSF, [Neupogen, Amgen Inc., Thousand Oaks, CA]) use was not permitted during cycle 1, as hematologic toxicity or treatment delays due to hematologic toxicity defined DLT. After cycle 1, filgrastim could be used at the discretion of the treating physician.
Dose escalation and definition of MTD and DLT
Three to six patients were accrued at each dose level until DLT was reached. DLT was defined based on toxicities in the first cycle of treatment. If a DLT was experienced in one out of three patients, then another cohort of three patients were evaluated prior to dose escalation. A maximum of six patients were enrolled in each cohort. If fewer than one out of the three or two out of the six patients experienced a DLT, then the next cohort of patients were treated at the next higher dose level of paclitaxel (25% dose escalation). However, prior to any dose escalation, all patients in the cohort had to have completed one full cycle (6 weeks) of therapy. If more than one out of the three, or two out of the six, patients experienced a DLT, then no further dose escalation was made and that level was considered to have exceeded the MTD. The level immediately preceding that level was designated as the MTD. Up to a total of ten patients were treated at the MTD to further define any toxicities that may have subsequently occurred with this regimen. Of these ten patients, at least three with stable disease or response were to receive at least three cycles (18 weeks) of therapy to detect any long-term toxicity associated with this drug combination.
Toxicities were graded according to the National Cancer Institute (NCI) Common Toxicity Criteria, version 1.0. Fatigue and asthenia were graded by the Cancer and Leukemia Group B (CALGB) criteria. Hematologic DLT was any grade 4 toxicity (absolute neutrophil count [ANC] <500/μL, platelets <25,000/μL) lasting longer than 7 days, neutropenic fever (≥38.1°C with ANC <1,000/μL), or dose delay lasting greater than 2 weeks. Renal DLT was defined as serum creatinine >1.8 mg/dl lasting longer than 2 weeks. Gastrointestinal DLT was defined as grade 3 diarrhea lasting longer than 2 days (despite intensive loperamide therapy), grade 4 diarrhea, grade 3 stomatitis lasting longer than 7 days (despite intensive viscous lidocaine therapy), or grade 4 stomatitis. Grade 3 or 4 peripheral neuropathy lasting longer than 2 weeks or grade 4 fatigue lasting longer than 1 week was defined as a DLT. Other non-hematologic DLTs (except nausea and vomiting) must have been grade 3/4 and determined treatment related by the principal investigator.
Dose modifications
Prior to each weekly treatment, complete blood count and serum creatinine were performed. For cycle 1, treatment could proceed on Day 1 if: ANC ≥1,500/μL, platelets ≥100,000/μL, and creatinine ≤1.5 mg/dL. After week 1 in cycle 1, treatment could proceed if: ANC ≥1,000/μL, platelets ≥75,000/μL, and creatinine ≤1.8 mg/dL. If these parameters were not met, all treatment was delayed for 1 week, up to 2 weeks in total. If renal function or blood counts did not improve after a 2-week delay, DLT was defined and the patient was removed from the study.
Dose reductions were not permitted during cycle 1, as toxicity defined DLT. Dose reductions during cycle 2 or thereafter were permitted for hematologic and non-hematologic toxicity that did not improve after a 2-week treatment delay. Paclitaxel was adjusted for hematologic/neurosensory toxicity and fatigue; irinotecan for hematologic and gastrointestinal toxicity; and cisplatin for renal toxicity and nausea. If a treatment delay was required, all drugs were held until recovery occurred. Dose adjustments were as follows:
Cisplatin
If on Day 1 the serum creatinine was >1.5 and ≤2.0 mg/dL, then cisplatin was administered at dose level 1 (15 mg/m2). At any time, if creatinine was >2.0 mg/dL, all three chemotherapy agents were held. If creatine remained >2.0 mg/dL after 2 weeks, then cisplatin and irinotecan were discontinued indefinitely and patients proceeded to single-agent weekly paclitaxel at 80 mg/m2/week. For grade 3 or 4 nausea lasting more than 3 days, cisplatin alone was reduced at 5 mg/m2 intervals.
Paclitaxel
For grade 3 neutropenia or thrombocytopenia, paclitaxel was held for 1week and continued at the same dose if hematologic parameters were met. Dose was reduced by one dose level if: (1) parameters were met only after a 2-week break, (2) grade 4 hematologic toxicity, (3) neutropenic fever, or (4) bleeding due to thrombocytopenia. For grade 3–4 neuropathy, paclitaxel was held for 1–2 weeks. If improved to grade 2, paclitaxel was restarted and reduced one dose level. If persistent, the patient was removed from the study. For grade 4 fatigue, paclitaxel was reduced by one dose level.
Irinotecan
Dose attentuations for irinotecan were made for grade 3–4 hematologic toxicity and grade 4 fatigue only after maximal dose reductions for paclitaxel (40 mg/m2) occurred. Dose reductions for grade 3–4 diarrhea or mucositis were made from 50 to 40 mg/m2, and if toxicity persisted, patients were removed from study.
Patient evaluation
Before treatment, patients underwent a full medical history and physical examination. Baseline testing included laboratory data (complete blood cell count, serum chemistry, coagulation profile, and pregnancy test for women of childbearing age), urinalysis, an ECG, and radiographic studies for baseline tumor measurements (chest x-ray, CT or MRI scan, and/or esophagram). For the first two cycles (12 weeks), patients were examined and interviewed by a physician weekly prior to each treatment to assess for interval toxicity. At cycle 3 and thereafter, patients were evaluated by a physician every other week (weeks 1 and 3) and by a registered nurse the remaining weeks (weeks 2 and 4). Clinic visits during the 2-week rest period were made only if the patient required medical attention. A complete blood cell count and serum creatinine was drawn prior to every treatment.
Patients had repeated tumor evaluations, after cycle 1 (week 6), after cycle 2 (week 12), and then after every subsequent two cycles (12 weeks) of treatment. Tumor response was assessed by the same imaging method at baseline, usually a CT scan, according to the modified World Health Organization criteria (i.e., measurable disease, non-measurable disease but assessable disease, non-assessable disease, complete response, partial response, minor response, stable disease, and progressive disease [24]. Time to response, duration of response, time to tumor progression, and survival data were recorded.
Patients were removed from the study if any toxicity endpoints were reached, if there was evidence of disease progression, for a major protocol violation, or if a number of dose attentuations were made (i.e., paclitaxel <40 mg/m2, irinotecan <40 mg/m2).
Results
Patient characteristics
A total of 30 patients were enrolled between April 1999 and April 2001 (demographics outlined in Table 1). The median age was 54 years, with good baseline performance status; 25 patients (83%) had 80–90% KPS, and five patients (17%) had 70% KPS. The majority of the patients were men (87%), in part due to the gender differences in prevalence of gastroesophageal cancer.
All 30 patients had a stage IV cancer diagnosis, defined as distant nodal or soft tissue metastasis. All patients had either a gastrointestinal or hepatobiliary cancer—16 patients (54%) with adenocarcinoma of the esophagus, GE junction or stomach; two patients (7%) with squamous cell carcinoma of the esophagus; seven patients (23%) with pancreatic cancer; and one patient (3%) with hepatocellular carcinoma. Adenocarcinoma of unknown origin was diagnosed in 4 patients (13%), although all four were suspected to be pancreaticobiliary primaries. The predominant site of measurable disease was the liver (67%) and lymph nodes (60%).
Most of the patients (70%) had never undergone any prior chemotherapy. Six patients (20%) had undergone one chemotherapy regimen. Three patients (10%) had undergone two prior regimens (one patients with gemcitabine alone then gemcitabine plus cisplatin, one patient with gemcitabine alone then gemcitabine plus 5-FU, and one patient with oral Xeloda then oral thalidomide); these three patients were entered into the protocol given excellent performance status and organ function. Two patients (7%) had undergone an attempt at primary surgical resection and one patient (3%) had radiation therapy with gemcitabine for locally advanced unresectable pancreatic cancer.
Dose escalation and determination of first-cycle MTD and DLT
Table 2 lists the number of patients treated at each dose level, the number of patients who progressed during the first cycle, and a description and number of patients with a first-cycle DLT.
At the first dose level (40 mg/m2), three patients were initially enrolled. One patient with pancreatic cancer developed rapid disease progression with a gastric outlet obstruction and was removed from the protocol. One patient developed significant dehydration with a DLT (prolonged grade 3 nausea and grade 3 hyponatremia). The cohort was expanded by five patients. One additional patient with pancreatic cancer had early progression with obstructive jaundice requiring a biliary stent, as well as grade 3 neutropenia. Since only one of the six patients (17%) who completed one full cycle developed a DLT (nausea, hyponatremia), it was decided that this dose level was tolerable.
At the second dose level (50 mg/m2), seven patients were enrolled. In the first three patients, two developed a DLT—1 with grade 4 febrile neutropenia and the other with prolonged grade 3 diarrhea requiring hospitalization. Because these patients had prior chemotherapy treatment, the protocol was amended to exclude prior chemotherapy at this point. To clarify the toxicity profile, an additional four patients were treated at this dose level and none of them experienced a DLT. Since only two (29%) out of seven developed a first-cycle DLT, the dose level was escalated as none of five patients without prior chemotherapy had a DLT.
Three patients were treated at the next dose level (65 mg/m2) and none experienced a DLT. The paclitaxel dose was escalated to the next level for three patients (80 mg/m2). One patient required hospitalization for grade 4 febrile neutropenia (DLT). He also developed grade 3 syncope with sinus bradycardia requiring a pacemaker. The second patient had prolonged grade 3 neutropenia (DLT) with grade 3 diarrhea, and the third patient developed grade 3 neutropenia (DLT). Since two (67%) of the three developed a DLT, it was felt that the MTD had been exceeded.
An additional 9 patients were accrued at the prior paclitaxel dose level (65 mg/m2). Four (33%) out of the 12 patients developed a first-cycle DLT—three patients with prolonged grade 3–4 neutropenia without hospitalization and 1 with prolonged grade 3 diarrhea. This dose level (65 mg/m2) was felt to represent the MTD and the recommended phase II dose level for future trials.
Within this last cohort (paclitaxel 65 mg/m2), 7 (58%) out of 12 patients had a 1-week dose delay at the third treatment week for most cycles due to neutropenia. Therefore, the recommended schedule was changed to 2 weeks on, 1 week off, times two, for each 6-week cycle.
Treatment administration and toxicities during all cycles of treatment (cumulative toxicity)
The 28 patients assessable for safety received a total of 74 cycles of therapy (4 treatments per cycle per patient). The median number of cycles was 1.5 (range, 1–9). In total, 14 patients received only one cycle (four progression of disease [PDs], ten DLTs), five patients had two cycles (three PDs, one toxicity, one went to surgical resection, given near complete response), one patient had three cycles (referred for surgery and taken off study), two patients had four cycles (one PD, one toxicity), two patients had five cycles (one PD, one toxicity), three patients had six cycles (two PDs, one toxicity), and one patient had nine cycles (went to operating room given minimal disease). Eighteen patients (64%) had dose delays longer than 7 days owing to treatment toxicity, primarily myelosuppression.
Table 3 lists the overall incidence of grade 3/4 toxicities for all evaluable patients treated on this trial, and Table 4 lists the median and ranges of hematologic nadirs for each paclitaxel dose level. Grade 3/4 myelosuppression was common with neutropenia (57%) and anemia (11%). Grade 3/4 thrombocytopenia was not observed. Three patients (11%) had neutropenic fever. None of the patients received colony-stimulating factors or erythropoietin during the study. Grade 3 fatigue (29%), diarrhea (29%), and nausea (18%) were consistent with those expected in patients receiving the drugs individually, in particular irinotecan. Eight patients (29%) required hospitalization due to severe toxicity, typically neutropenic fever or diarrhea. One patient developed sinus bradycardia and required a cardiac pacemaker. Increased creatinine and ototoxicity was not observed in any patients. No patient died during or within 30 days of receiving treatment on this study.
Efficacy
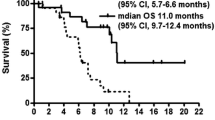
Although toxicity is the primary endpoint of this phase I study, radiographic response was determined. Objective tumor response, as a partial radiographic response, was observed in 43% (13/30) of patients; 92% (12/13) had no prior chemotherapy. Complete response was not observed. Eight (27%) patients had stable disease, and nine (30%) had progressive disease. In relation to underlying diagnosis, partial response occurred in patients with gastric adenocarcinoma (63%, 5/8), esophageal or GE junction adenocarcinoma (50%, 4/8), esophageal squamous carcinoma (50%, 1/2), and unknown primary (50%, 2/4). One response was seen in a patient with pancreatic cancer (14%, 1/7).
Discussion
Combination chemotherapy with a platinum agent has become a standard of care in the palliative treatment of upper gastrointestinal malignancies. Metastatic gastric cancer trials have shown a survival benefit with chemotherapy compared to best supportive care alone [3]. In addition, randomized trials have indicated that adding a third agent to the combination of fluorouracil and cisplatin, either epirubicin or docetaxel, may modestly improve response rates, time to progression, and survival with greater therapy-related toxicity [25, 26]. Toxicity for the new proposed standard regimen combining docetaxel with fluorouracil and cisplatin (DCF) can be prohibitive, including high rates of hematologic and gastrointestinal toxicity, and such a regimen may not be feasible to administer in many patients.
Nonetheless, median survival remains less than 1 year, and hence, more active, tolerable regimens are needed. Paclitaxel and irinotecan are highly active single agents in the treatment of metastatic gastroesophageal cancers [21, 26]. Cisplatin-containing regimens with either irinotecan or paclitaxel are showing promising activity in gastroesophageal cancer [12, 15, 17, 22, 27]; however, all three drugs have not been combined in this population. We undertook this phase I trial to see if this three-drug combination is feasible.
We combined weekly, dose-escalation, paclitaxel over 1h with the conventionally used weekly cisplatin and irinotecan [15, 17], and limited our cohort to patients with upper gastrointestinal and hepatobiliary malignancies. In our 28 evaluable patients, first cycle DLTs were expected and consisted primarily of neutropenia with prolonged grade 3–4 neutropenia (five patients, 18%) and febrile neutropenia (two patients, 7%). In addition, grade 4 diarrhea (two patients, 7%) and grade 3 nausea (one patient, 3%) were seen, likely from the irinotecan and to a lesser degree cisplatin, which has been seen at these doses. Two patients were removed from the trial after only two treatments due to rapid disease progression—obstructive jaundice and gastric outlet obstruction. These were not felt to be due to the therapy and were therefore not included as true DLTs.
The recommended phase II dose level for future trials was paclitaxel 65 mg/m2; however, neutropenia requiring dose delays was still significant. In this dose cohort, four (33%) out of the 12 patients developed a first-cycle DLT—three patients with prolonged grade 3–4 neutropenia without hospitalization and one with prolonged grade 3 diarrhea. Most patients (58%) had a 1-week dose delay at the third treatment week for most cycles, which did allow better tolerability. Given this observation, we suggest a modified schedule of this regimen in this cohort (paclitaxel 65 mg/m2) to 2 weeks on, 1 week off, times two, for each 6-week cycle. Only one patient required hospitalization despite a high incidence of grade 3–4 neutropenia (50%). In addition, no grade 3–4 neuropathy was observed as has been described in other trials with cisplatin–paclitaxel (24- and 3-h infusions) [28, 29]. This however was likely due to the relatively short duration of therapy in most patients.
We also observed substantial activity for this three-drug regimen. The overall response rate of 43% is remarkable, especially since up to a third of patients had prior chemotherapy. As expected, we observed most activity in the patients with upper gastrointestinal cancers—gastric adenocarcinoma (63%), esophageal/GE junction adenocarcinoma (50%), and esophageal squamous carcinoma (50%). Remarkably, activity was seen in a patient with pancreatic cancer (14%).
These results are consistent with pervious trials with platinum, taxane, and a topoisomerase inhibitor (irinotecan or topotecan). Another three-drug regimen with weekly docetaxel, cisplatin, and irinotecan in gastroesophageal cancer has also reported preliminary findings of frequent grade 3–4 toxicity (diarrhea [44%] and neutropenia [22%]), which markedly improved with a reduction in the irinotecan to 50 mg/m2 (grade 3–4 toxicity: diarrhea [6%], neutropenia [6%]). Antitumor activity was substantial (greater than 50%) and did not appear comprised with irinotecan dose reduction [30]. In non-small cell lung cancer, a regimen of carboplatin (area under the curve [AUC], 5), paclitaxel (175 mg/m2), and irinotecan (100 mg/m2) every 3 weeks has been described in two trials, which had similar neutropenic rates to our trial (50–78%) [31, 32]. Platinum-containing regimens with three agents have been described in various cohorts with similar toxicity profiles and high response rates [33–37].
In summary, this single-center phase I trial has identified a regimen of paclitaxel 65 mg/m2, cisplatin 30 mg/m2, and irinotecan 50 mg/m2 given weekly on a 2-week on, 1-week off schedule. DLT with this regimen was primarily neutropenia, although diarrhea and nausea were observed. The tolerability and activity of this regimen warrants further exploration, especially in upper gastrointestinal malignancies.
References
Jemal A, Siegel R, Ward E et al (2008) Cancer statistics, 2008. CA Cancer J Clin 58:71–96. doi:10.3322/CA.2007.0010
Enzinger PC, Mayer RJ (2003) Esophageal cancer. N Engl J Med 349:2241–2252. doi:10.1056/NEJMra035010
Shah MA, Schwartz GK (2004) Treatment of metastatic esophagus and gastric cancer. Semin Oncol 31:574–587. doi:10.1053/j.seminoncol.2004.04.013
Bleiberg H, Conroy T, Paillot B et al (1997) Randomised phase II study of cisplatin and 5-fluorouracil (5-FU) versus cisplatin alone in advanced squamous cell oesophageal cancer. Eur J Cancer 33:1216–1220. doi:10.1016/S0959-8049(97)00088-9
Barone C, Cassano A, Landriscina M et al (2000) Bolus and infusional 5-fluorouracil combined with cisplatin in advanced gastric cancer. Oncol Rep 7:1305–1309
Van Custem E, Moiseyenko V, Tjulandin S et al (2006) Phase 3 study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol 24:4991–4997. doi:10.1200/JCO.2006.06.8429
Fukuda M, Nishio K, Kanzawa F et al (1996) Synergism between cisplatin and topoisomerase I inhibitors, NB-506 and SN-38, in human small cell lung cancer cells. Cancer Res 56:789–793
Masumoto N, Nakano S, Esaki T et al (1995) Sequence-dependent modulation of anticancer drug activities by 7-ethyl-10-hydroxycamptothecin in an HST-1 human squamous carcinoma cell line. Anticancer Res 15:405–409
Kano Y, Suzuki K, Akutsu M (1992) Effects of CPT-11 in combination with other anti-cancer agents in culture. Int J Cancer 50:604–610. doi:10.1002/ijc.2910500420
Noda K, Nishiwaki Y, Kawahara M, Japan Clinical Oncology Group et al (2002) Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small-cell lung cancer. N Engl J Med 346:85–91. doi:10.1056/NEJMoa003034
Hanna N, Bunn PA, Langer C et al (2006) Randomized phase III trial comparing irinotecan/cisplatin with etoposide/cisplatin in patients with previously untreated extensive-stage disease small-cell lung cancer. J Clin Oncol 24:2038–2043. doi:10.1200/JCO.2005.04.8595
Boku N, Ohtsu A, Shimada Y et al (1999) Phase II study of a combination of irinotecan and cisplatin against metastatic gastric cancer. J Clin Oncol 17:319–323
Shirao K, Shimada Y, Kondo H et al (1997) Phase I-II study of irinotecan hydrochloride combined with cisplatin in patients with advanced gastric cancer. J Clin Oncol 15:921–927
Saltz LB, Spriggs D, Schaaf LJ et al (1998) Phase I clinical and pharmacologic study of weekly cisplatin combined with weekly irinotecan in patients with advanced solid tumors. J Clin Oncol 16:3858–3865
Ilson DH, Saltz L, Enzinger P et al (1999) Phase II trial of weekly irinotecan plus cisplatin in advanced esophageal cancer. J Clin Oncol 17:3270–3275
Ajani JA, Baker J, Pisters PW et al (2002) Irinotecan/cisplatin in advanced, treated gastric or gastroesophageal junction carcinoma. Oncology (Williston Park) 16:16–18
Ajani JA, Baker J, Pisters PW et al (2002) CPT-11 plus cisplatin in patients with advanced, untreated gastric or gastroesophageal junction carcinoma: results of a phase II study. Cancer 94:641–646. doi:10.1002/cncr.10279
Ilson DH, Bains M, Kelsen DP et al (2003) Phase I trial of escalating-dose irinotecan given weekly with cisplatin and concurrent radiotherapy in locally advanced esophageal cancer. J Clin Oncol 21:2926–2932. doi:10.1200/JCO.2003.02.147
Ajani JA, Walsh G, Komaki R et al (2004) Preoperative induction of CPT-11 and cisplatin chemotherapy followed by chemoradiotherapy in patients with locoregional carcinoma of the esophagus or gastroesophageal junction. Cancer 100:2347–2354. doi:10.1002/cncr.20284
Tew W, Minsky B, Bains M et al (2005) Phase II trial of preoperative combined modality therapy for esophageal carcinoma: induction cisplatin–irinotecan followed by concurrent cisplatin–irinotecan and radiotherapy. Proc Am Soc Clin Oncol 23:4017 abst
Ajani JA, Ilson DH, Daugherty K et al (1994) Activity of taxol in patients with squamous cell carcinoma and adenocarcinoma of the esophagus. J Natl Cancer Inst 86:1086–1091. doi:10.1093/jnci/86.14.1086
Ilson DH, Forastiere A, Arquette M et al (2000) A phase II trial of paclitaxel and cisplatin in patients with advanced carcinoma of the esophagus. Cancer J 6:316–323
Ilson D, Wadleigh S, Leichman L et al (2007) Paclitaxel given by weekly one hour infusion in advanced esophageal cancer. Ann Oncol 18:898–902. doi:10.1093/annonc/mdm004
Therasse P, Arbuck SG, Eisenhauer EA et al (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216. doi:10.1093/jnci/92.3.205
Ross P, Nicolson M, Cunningham D et al (2002) Prospective randomized trial comparing mitomycin, cisplatin, and protracted venous-infusion fluorouracil (PVI 5-FU) With epirubicin, cisplatin, and PVI 5-FU in advanced esophagogastric cancer. J Clin Oncol 20:1996–2004. doi:10.1200/JCO.2002.08.105
Lin L, Hecht J (2000) A Phase II trial of irinotecan in patients with advanced adenocarcinoma of the GE junction. Proc Am Soc Clin Oncol 19:1130 abstr
Petrasch S, Welt A, Reinacher A et al (1998) Chemotherapy with cisplatin and paclitaxel in patients with locally advanced, recurrent or metastatic oesophageal cancer. Br J Cancer 78:511–514
Gordon AN, Stringer CA, Matthews CM et al (1997) Phase I dose escalation of paclitaxel in patients with advanced ovarian cancer receiving cisplatin: rapid development of neurotoxicity is dose-limiting. J Clin Oncol 15:1965–1973
Wasserheit C, Frazein A, Oratz R et al (1996) Phase II trial of paclitaxel and cisplatin in women with advanced breast cancer: an active regimen with limiting neurotoxicity. J Clin Oncol 14:1993–1999 Erratum in: J Clin Oncol 14:3175
Enzinger PC, Clark J, Ryan D et al (2004) Phase II study of docetaxel, cisplatin, and irinotecan in advanced esophageal and gastric cancer. Proc Am Soc Clin Oncol 22:4040 abstr
Socinski MA, Sandler AB, Miller LL et al (2001) Phase I trial of the combination of irinotecan, paclitaxel, and carboplatin in patients with advanced non-small-cell lung cancer. J Clin Oncol 19:1078–1087
Socinski MA, Sandler AB, Israel VK et al (2002) Phase II trial of irinotecan, paclitaxel and carboplatin in patients with previously untreated Stage IIIB/IV nonsmall cell lung carcinoma. Cancer 95:1520–1527. doi:10.1002/cncr.10852
Frasci G, Nicolella G, Comella P et al (2001) A weekly regimen of cisplatin, paclitaxel and topotecan with granulocyte-colony stimulating factor support for patients with extensive disease small cell lung cancer: a phase II study. Br J Cancer 84:1166–1171. doi:10.1054/bjoc.2001.1741
Frasci G, Panza N, Comella P et al (1999) Cisplatin–topotecan–paclitaxel weekly administration with G-CSF support for ovarian and small-cell lung cancer patients: a dose-finding study. Ann Oncol 10:355–358. doi:10.1023/A:1008301222560
Gonzalez MS, Calvo E, Gassent JM et al (2003) Treatment of advanced non-small cell lung cancer (NSCLC) with a bi-weekly schedule of irinotecan (I), paclitaxel (T) and cisplatinum (P). A feasibility study. Proc Am Soc Clin Oncol 22:2816 abstr
Herben VM, Panday VR, Richel DJ et al (1999) Phase I and pharmacologic study of the combination of paclitaxel, cisplatin, and topotecan administered intravenously every 21days as first-line therapy in patients with advanced ovarian cancer. J Clin Oncol 17:747–755
D'Adamo DR, Bains M, Minsky B et al (2003) A phase I trial of paclitaxel, cisplatin, irinotecan and concurrent radiation therapy in locally advanced esophageal cancer. Proc Am Soc Clin Oncol 22:1401 abstr
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tew, W.P., Radovich, D., O’Reilly, E. et al. Phase I trial of weekly cisplatin, irinotecan and paclitaxel in patients with advanced gastrointestinal cancer. Invest New Drugs 27, 366–373 (2009). https://doi.org/10.1007/s10637-008-9194-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-008-9194-4