
Abstract
Purpose
We conducted a phase 1 study to determine the maximum tolerated dose and the recommended dose of gemcitabine/nab-paclitaxel/S-1 combination chemotherapy in patients with unresectable pancreatic cancer.
Methods
We enrolled patients aged 20 years or older with unresectable pancreatic cancer and who had not been treated with chemotherapy or radiation therapy. Gemcitabine and nab-paclitaxel were administered on days 1 and 8, and S-1 was administered orally twice daily for 2 weeks, repeated every 3 weeks. The starting dose was level 0 [gemcitabine 700 mg/m2, nab-paclitaxel 90 mg/m2, S-1 60/80/100 mg/day (< 1.25 m2/1.25–1.50 m2/ > 1.5 m2)]. Dose-limiting toxicities were determined during the first course, and a classical 3 + 3 dose finding design was planned.
Results
From March 2018 to October 2019, 20 patients were enrolled. At dose level 0, three of six patients experienced dose-limiting toxicities; one grade 3 skin rash on day 8, and two grade 3 or 4 neutropenia on day 8. At dose level-1 (gemcitabine 600 mg/m2, nab-paclitaxel 90 mg/m2, and S-1 50/70/80 mg/day), two of twelve patients experienced dose-limiting toxicities, all of which were grade 3 neutropenia on day 8. The most frequently observed toxicity during eight courses was neutropenia. Other treatment-related adverse events were mild. Eleven out of 19 (58%) patients achieved partial response.
Conclusion
We defined the maximum tolerated dose and the recommended dose for combination therapy with gemcitabine/nab-paclitaxel/S-1 as dose level-1. Considering the observed response rate, further studies are warranted in order to determine the efficacy of this regimen (UMIN-CTR 000030007).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The prognosis of patients with pancreatic cancer is one of the worst among all malignancies, with an estimated 5-year survival rate of less than 5% [1]. Many cases are diagnosed with locally advanced or distant metastatic disease [2], and the number of deaths due to pancreatic cancer has increased for both men and women over the past decade [3]. One of the reasons for the high mortality rate of pancreatic cancer is that there are no effective chemotherapies to treat the disease. To improve the prognosis of pancreatic cancer, the development of a highly active chemotherapy regimen is necessary.
The standard chemotherapies for unresectable pancreatic cancer are gemcitabine plus nab-paclitaxel combination therapy or FOLFIRINOX therapy. Gemcitabine plus nab-paclitaxel is superior to gemcitabine alone (median overall survival 8.5 vs 6.7 months, hazard ratio (HR) 0.72) [4]; FOLFIRINOX is also superior to gemcitabine (median overall survival 11.1 vs 6.8 months, HR 0.57) [5]. Additionally, S-1 (an oral fluoropyrimidine prodrug) is non-inferior to gemcitabine (median overall survival 9.7 vs 8.8 months, HR 0.96) [6]. Based on these studies, gemcitabine, nab-paclitaxel, 5-fluorouracil, irinotecan, oxaliplatin, and S-1 can be considered key-drugs, and combinations of these drugs are usually used for unresectable pancreatic cancer. Although FOLFIRINOX has a promising HR, its efficacy is limited by the high rate of febrile neutropenia it induces, and the regimen is not considered tolerable in the Japanese population [7, 8]. Therefore, it is usually used as a modified regimen with dose reduction, but superiority of modified FOLFIRINOX to gemcitabine has not been demonstrated in randomized studies. On the other hand, the combination of gemcitabine plus nab-paclitaxel also causes cumulative peripheral neuropathy, leading to treatment interruption or dose reduction [4, 9].
Given the paucity of chemotherapeutic options for unresectable pancreatic cancer, there is a clear need to develop a safer and more efficacious new regimen. Previous phase 2 trials showed the effectiveness of nab-paclitaxel plus S-1 combination therapy for advanced pancreatic cancer [10, 11]. On the other hand, the FUGA-BT randomized phase 3 trial showed that S-1 combined with gemcitabine was non-inferior to the combination of gemcitabine plus cisplatin in patients with biliary tract cancer [12]. We hypothesized that combining S-1 with gemcitabine plus nab-paclitaxel therapy would have an additive effect on efficacy. We, therefore, conducted the current phase 1 study to determine the maximum tolerated dose (MTD) and the recommended dose (RD) of the gemcitabine/nab-paclitaxel/S-1 triplet combination chemotherapy in patients with unresectable pancreatic cancer (GeNeS1S).
Materials and methods
Study design
This study was a prospective, multi-center, phase 1 study with a 3 + 3 dose escalation design. The study protocol was approved by the institutional review board of each institution, and written informed consent was obtained from all participants. This study was conducted in accordance with the declaration of Helsinki, the principles of Good Clinical Practice and all applicable regulations. This clinical study was registered in the University hospital Medical Information Network-Clinical Trial Registry (UMIN-CTR), identification number 000030007.
Eligibility criteria
We recruited patients with unresectable (locally advanced or metastatic) pancreatic cancer. Eligibility criteria were as follows: a histologically or cytologically confirmed diagnosis of pancreatic cancer, aged 20 years or older at registration, Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, one or more measurable lesions according to the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, no previous treatment (radiotherapy, chemotherapy and immunotherapy) for advanced disease, adequate bone marrow function (neutrophil count ≥ 1500/mm3, hemoglobin ≥ 9.0 g/dL, platelet count ≥ 100,000/mm3), liver function (total bilirubin ≤ 1.5 times the upper limit of normal (ULN), aspartate aminotransferase (AST) and alanine aminotransferase (ALT) ≤ 3 times the ULN), renal function (creatinine clearance ≥ 60 ml/min), no serious complications, adequate oral intake, and grade 1 or less peripheral neuropathy. Previous neoadjuvant or adjuvant chemotherapy was allowed if the treatment had ended more than 6 months before recurrence, and biliary drainage (percutaneous, endoscopic or laparotomy) was also allowed. Exclusion criteria were as follows: interstitial pneumonitis or pulmonary fibrosis, uncontrollable diabetes, angina or myocardial infarction within 3 months, severe infection or suspected severe infection with fever, pregnant women, breastfeeding and cases in which women were attempting to become pregnant, severe drug allergies, psychosis or psychiatric symptoms that were considered to pose difficulties during study participation, moderate or higher ascites or pleural effusion, and watery diarrhea that was difficult to control.
Treatment
The starting dose (dose level 0) was gemcitabine 700 mg/m2 and nab-paclitaxel 90 mg/m2 on days 1 and 8 combined with S-1 60/80/100 mg/day (< 1.25 m2/1.25–1.50 m2/ > 1.5 m2) orally twice daily for 2 weeks, repeated every 3 weeks. The treatment period for this study was 24 weeks (8 courses). For the administration on day 1, the criteria to start each course were: neutrophil count ≥ 1500/mm3, hemoglobin ≥ 8.0 g/dl, platelet count ≥ 100,000/mm3, total bilirubin ≤ 1.5 times the ULN, AST and ALT ≤ 3 times the ULN, creatinine clearance ≥ 50 ml/min, non-hematological toxicity related to treatment drug ≤ grade 2, and no fever suspected of infection. Similarly, the administration criteria of day 8 were as follows: neutrophil count ≥ 1000/mm3, hemoglobin ≥ 8.0 g/dl, platelet count ≥ 70,000/mm3, total bilirubin ≤ 1.5 times the ULN (≤ 3.0 times the ULN for Gilbert Syndrome), AST and ALT ≤ 3 times the ULN, serum creatinine concentration ≤ 1.5 mg/dl, non-hematological toxicity related to treatment drugs ≤ grade 2, and the absence of fever that would indicate an ongoing infection. If the patient did not meet these criteria, the administration of gemcitabine and nab-paclitaxel scheduled on day 8 was skipped and S-1 on day 8 and thereafter was withheld. One week later, the next course of chemotherapy was initiated.
The following indication of toxicities led to dose reduction by one step (gemcitabine to 600 mg/m2, nab-paclitaxel to 80 mg/m2): neutropenia (grade 4), thrombocytopenia (grade 3), and febrile neutropenia. For grade 3 or higher non-hematological toxicity related to each drug, the dose of the relevant drug was reduced by one step. Stepwise reduction of gemcitabine (700 → 600 → 500 mg/m2) and nab-paclitaxel (90 → 80 → 70 → 60 mg/m2) was allowed. For S-1, the dose was reduced to 50/70/80 mg/day. If further dose reduction was necessary, study treatment was discontinued. The protocol treatment was discontinued when any of the following occurred: disease progression, grade 4 non-hematological adverse events, or delay in schedule of 4 weeks or more due to an adverse event. The dose level was set in eight stages from −3 to 3 (Table 1). Up to six patients at each level were enrolled, and the dose level was increased or decreased according to the frequency of dose-limiting toxicity (DLT).
Definition of DLTs, MTD and RD
Dose-limiting toxicities (DLTs) were determined during the first course. With regard to S-1, administration of 75% or more of the S-1 doses was required for DLT evaluation. If > 25% the planned dose of S-1 was not taken due to a toxicity, it was deemed a DLT. The DLTs were defined as drug-related adverse events according to the Common Terminology Criteria for Adverse Events (CTCAE) version 4.0 as one or more of the following events: ≥ grade 3 neutropenia with fever above 38.3 °C, grade 4 neutropenia lasting 7 days or more, grade 4 thrombocytopenia, ≥ grade 3 non-hematologic toxicities, delay in course of more than 14 days due to an adverse event, or need to withhold study treatment on day 8 due to an adverse event.
The standard 3 + 3 method for dose finding was used. If DLT was not observed in three patients, the dose was escalated to the next level. If DLT was observed in one or two of three patients, three additional patients were enrolled at that level. Thus, if DLT was observed in only one or two of six patients, the dose level was escalated. If all three initial patients (or if three or more of six patients) experienced DLT, the dose level was de-escalated.
The MTD was defined as the highest dose level that produced the frequency of DLT ≤ 33%. The RD was determined by taking into consideration the toxicity and tolerability observed across the entire study.
Pretreatment and follow-up evaluation
Pretreatment evaluation included each patient’s medical history and physical examination, imaging tests using contrast-enhanced computed tomography or magnetic resonance imaging, blood tests, electrocardiogram, and chest X-rays. Creatinine clearance was calculated using the Cockcroft-Gault formula.
During the DLT evaluation period, 21 days of the first course, physical examination and blood tests were performed on days 1, 8, and 15. After the DLT evaluation period (from the second course), the tests were scheduled on day 1 and day 8. Tumor markers (CEA and CA19-9) were measured at the time patients were enrolled in the study and every month thereafter. Toxicity was evaluated using the CTCAE v4.0 throughout the study. Imaging tests were planned for every 2 months after the start of treatment and objective response rate (ORR) was assessed according to the RECIST version 1.1. Additional imaging tests were performed if clinically indicated or at the discretion of the treating physician.
Results
Patient characteristics
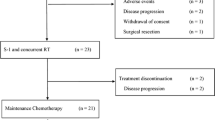
From March 2018 to October 2019, 20 patients were enrolled from two facilities (Table 2). According to National Comprehensive Cancer Network (NCCN) Guidelines 2017 Version 3, 19 patients had primary unresectable pancreatic cancer and one patient had locally recurrent disease after curative operation. No one received neoadjuvant or adjuvant chemotherapy.
DLTs
One patient developed DLT among three patients assigned to level 0, and three additional patients were treated at this level. Among these six patients, one experienced grade 3 skin rash on day 8 and two experienced grade 3 or 4 neutropenia on day 8, which lead to withholding the study treatment, and the dose was decreased to level-1 [gemcitabine 600 mg/m2, nab-paclitaxel 90 mg/m2, S-1 60/80/100 mg/day [< 1.25 m2/1.25–1.50 m2/ > 1.5 m)]. At the reduced dose level, DLTs were observed in two of the first three patients, all of which were grade 3 neutropenia on day 8; three additional patients were treated at this level without DLTs. To confirm the safety of this treatment, eight additional patients were treated as an expansion cohort at dose level-1. However, two patients with biliary stent were not assessable for DLTs because of early treatment withdrawal due to cholangitis without neutropenia. DLT was not observed in the other six patients (Table 3). Eventually, at dose level-1, DLTs were observed in two of twelve (17%) assessable patients, and this dose was determined as the MTD.
Toxicity and RD
The median treatment period of the 20 patients was 21.9 weeks (2.7–26.0). The most frequently observed toxicity during 8 courses of treatment was neutropenia (Table 4). Grade 3 febrile neutropenia was observed in 33% at dose level 0 during the second course of treatment. No febrile neutropenia was observed at the reduced dose.
Non-hematological adverse events were mild. At dose level 0, grade 3 anorexia, skin rash, and peripheral neuropathy were found in 17% of patients. At dose level-1, one grade 3 cholangitis that was not related to the study treatment developed after the first course. Neither peripheral neuropathy nor other grade 3 or 4 non-hematological adverse events were observed. Four of 14 patients developed grade 1 peripheral neuropathy when treated at dose level-1. Gastrointestinal toxicities including nausea/vomiting, anorexia, and diarrhea were also mild at dose level-1. There were no treatment-related deaths.
Considering the frequency of DLTs and adverse events throughout eight courses, we recommend dose level-1 for further studies.
Efficacy
Nineteen of 20 patients were evaluable for response. Eleven (58%) showed confirmed partial response (PR), one achieved PR without confirmation, seven had stable disease (SD), and none of them had progressive disease at the first evaluation. Median time to response was 2.0 months (Table 5). At a median follow-up period of 16.4 months (6.0–29.5), the median progression-free survival was 7.6 months (95% CI 4.3–10.5) (Fig. 1), and the median overall survival was not reached yet.

Kaplan–Meier curve for progression-free survival. The median progression-free survival was 7.6 months (95% confidence interval 4.3–10.5)
Discussion
This study investigated the MTD and the RD of the gemcitabine/nab-paclitaxel/S-1 triplet combination chemotherapy in patients with unresectable pancreatic cancer. We determined that the MTD was dose level-1 [gemcitabine 600 mg/m2, nab-paclitaxel 90 mg/m2 on days 1 and 8, and S-1 50/70/80 mg/day (< 1.25 m2/1.25–1.50 m2/ > 1.5 m2) on days 1–14] with a DLT frequency of 17%. At this dose, grade 3 or higher adverse events were hardly observed during the entire treatment course, and preliminary evaluation of antitumor activity revealed a relatively high (58%) confirmed response rate. Based on these results, we recommend this dose level for further investigation.
Our regimen using dose level-1 is well tolerated. Nab-paclitaxel (one of the key drugs used to treat advanced pancreatic cancer) causes cumulative peripheral neuropathy, which can reduce quality of life or lead to the cessation of treatment. The MPACT phase 3 clinical trial showed that grade 3 or higher peripheral neuropathy was found at a frequency of 17% [4]. In the current trial, grade 3 or higher peripheral neuropathy was found in only one patient treated at dose level 0, and no patients developed grade 2 or higher peripheral neuropathy at the recommended dose level-1. Other Grade 3 or higher non-hematological adverse events such as fatigue, vomiting, and diarrhea were documented in ≤ 5% of cases following this triplet chemotherapy. In contrast, they were observed in ≥ 10% of cases after gemcitabine plus nab-paclitaxel combination therapy or FOLFIRINOX therapy, which are standard treatments for unresectable pancreatic cancer [4, 5]. In gemcitabine plus nab-paclitaxel combination therapy, 1000 mg/m2 of gemcitabine and 125 mg/m2 of nab-paclitaxel are administered on days 1, 8, and 15 every 4 weeks, and the dose intensity is 750 mg/m2/week for gemcitabine and 94 mg/m2/week for nab-paclitaxel. On the other hand, at dose level-1 in our GeNeS1S trial, gemcitabine 600 mg/m2 and nab-paclitaxel 90 mg/m2 were administered on days 1 and 8, in combination with S-1 50/70/80 mg/day (< 1.25 m2/1.25–1.50 m2/ > 1.5 m2) orally twice daily for 2 weeks, repeated every 3 weeks. The dose intensities of gemcitabine, nab-paclitaxel, and S-1 were 400 mg/m2/week, 60 mg/m2/week, and 233/327/373 mg/week, respectively. Lower doses of gemcitabine and nab-paclitaxel resulted in a lower incidence of grade 3 or higher non-hematological adverse events such as peripheral neuropathy.
With regard to hematological adverse events, grade 3 or higher hematological toxicities had occurred about 80% at dose level-1. However, no grade 4 toxicities were observed at this dose level, and grade 3 neutropenia was documented due to mandatory blood tests on day 15 in the first course. Grade 3 neutropenia on day 8 led to dose holding and was deemed a DLT according to the protocol. While this prevented dose escalation, withholding the treatment on day 8 resulted in no febrile neutropenia and chemotherapy could be continued without interruption at dose level-1.
In a previous phase 1 study of gemcitabine/nab-paclitaxel/S-1 combination as neo-adjuvant chemotherapy for patients with locally advanced pancreatic cancer (the GAS trial), gemcitabine, and nab-paclitaxel were administered on day 1, and S-1 was given on days 1–7; this was repeated every 2 weeks [13]. Although the frequency of DLT was relatively low, a response rate of 31% did not justify this neoadjuvant triplet chemotherapy regimen [13]. Hypothesizing that further dose escalation or modification of the administration schedule might be necessary to show antitumor activity, we performed the current GeNeS1S trial. The dose intensities in our study were slightly lower than those of the GAS trial for gemcitabine (400 vs 500 mg/m2/week) and nab-paclitaxel (60 vs 63 mg/m2/week). Despite this, we observed a higher response rate (58% compared to 31% in GAS). We infer that the improved efficacy in our study might be due to the slightly higher dose intensity of S-1 in this study (233/327/373 mg/week) than that in the GAS study (210/280/350 mg/week) as well as the longer exposure period to S-1 (2 weeks) compared to GAS study (one week). The S-1 dose and administration schedule might play a more important role among the three drugs in terms of antitumor effect, as suggested by a previous study [14]. Despite it being a triplet chemotherapeutic approach, the advantage of our current regimen is that it is well tolerated at dose level-1 and appears to exert a high antitumor activity. However, we enrolled a small number of Japanese patients, and further clinical trials are required in order to confirm the efficacy and tolerability.
In conclusion, chemotherapy regimen of gemcitabine 600 mg/m2 and nab-paclitaxel 90 mg/m2 on days 1 and 8 combined with oral S-1 50/70/80 mg/day (< 1.25 m2/1.25–1.50 m2/ > 1.5 m2) on days 1–14, repeated every 3 weeks is well tolerated and recommended for patients with unresectable pancreatic cancer. This triplet regimen appears efficacious and has fewer side effects, although further clinical studies to validate our observations are warranted. In this regard, we are now conducting a phase 2 study of this combination treatment for unresectable pancreatic cancer.
References
Hidalgo M (2010) Pancreatic cancer. N Engl J Med 362:1605–1617
Matsuno S, Egawa S, Fukuyama S et al (2004) Pancreatic Cancer Registry in Japan: 20 years of experience. Pancreas 28:219–230
llic M, llic I, (2016) Epidemiology of pancreatic cancer. World J Gastroenterol 22:9694–9705
Von Hoff DD, Ervin T, Arena FP et al (2013) Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 369:1691–1703
Thierry C, Francoise D, Marc Y et al (2011) FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364:1817–1825
Ueno H, Ioka T, Ikeda M et al (2013) Randomized phase III study of gemcitabine plus s-1, s-1 alone, or gemcitabine alone in patients with locally advanced and metastatic pancreatic cancer in Japan and Taiwan: GEST study. J Clin Oncol 31:1640–1648
Okusaka T, Ikeda M, Fukutomi A et al (2014) Phase II study of FOLFIRINOX for chemotherapy-naive Japanese patients with metastatic pancreatic cancer. Cancer Sci 105:1321–1326
Sasaki Y, Hamada K, Kaneta T et al (2015) Is repeating FOLFIRINOX in the original dosage and treatment schedule tolerable in Japanese patients with pancreatic cancer? Cancer Sci 106:1100
Ueno H, Ikeda M, Ueno M et al (2016) Phase I/II study of nab- paclitaxel plus gemcitabine for chemotherapy-naive Japanese patients with metastatic pancreatic cancer. Cancer Chemother Pharmacol 77:595–603
Yan S, Sui Z, Quanli H et al (2017) Nab-paclitaxel plus S-1 in advanced pancreatic adenocarcinoma (NPSPAC): a single arm, single center, phase II trial. Oncotarget 8:92401–92410
Wen Z, Chunxia D, Yongkun S et al (2018) Nab-paclitaxel plus S-1 as first-line followed by S-1 maintenance for advanced pancreatic adenocarcinoma: a single-arm phase II trial. Cancer Chemother Pharmacol 82:655–660
Morizane C, Okusaka T, Mizusawa J et al (2019) Combination gemcitabine plus S-1 versus gemcitabine plus cisplatin for advanced/recurrent biliary tract cancer: the FUGA-BT (JCOG1113) randomized phase III clinical trial. Ann Oncol 30:1950–1958
Kondo N, Murakami Y, Uemura K et al (2017) A phase 1 study of gemcitabine/nab-paclitaxel/S-1 (GAS) combination neoadjuvant chemotherapy for patients with locally advanced pancreatic adenocarcinoma. Cancer Chemother Pharmacol 79:775–781
Tanaka H, Kanda M, Morita S et al (2017) Randomized phase II study of daily and alternate-day administration of S-1 for advanced gastric cancer (JFMC43-1003). Int J Clin Oncol 22:1052–1059
Funding
No funding was received for conducting this study.
Author information
Authors and Affiliations
Contributions
All authors have contributed significantly, and all authors are in agreement with the content of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Masanori Toyoda has received a honoraria from Taiho Pharmaceutical Co., Ltd. Hironaga Satake has received a research grant from Taiho Pharmaceutical Co., Ltd, a honoraria from Eli Lilly Japan Co., Ltd. and Taiho Pharmaceutical Co., Ltd. Hisateru Yasui has received a honoraria from Eli Lilly Japan Co., Ltd. and Taiho Pharmaceutical Co., Ltd. Hironobu Minami has received a research grant and personal fees from Eli Lilly Japan Co., Ltd. and Taiho Pharmaceutical Co., Ltd. The remaining authors have no conflict of interest to declare.
Ethical approval
This study was conducted in compliance with the ethical principles of the Declaration of Helsinki, the principles of Good Clinical Practice and all applicable regulations. The study protocol was approved by the institutional review board of each participating institution, and written informed consent was obtained from all participants.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Consent for publication
Patients signed informed consent regarding publishing their data.
Availability of data and material
The content of this manuscript has not been published or submitted for publication elsewhere and is not under consideration elsewhere.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sai, S., Toyoda, M., Tobimatsu, K. et al. Phase 1 study of Gemcitabine/Nab-paclitaxel/S-1 in patients with unresectable pancreatic cancer (GeNeS1S trial). Cancer Chemother Pharmacol 87, 65–71 (2021). https://doi.org/10.1007/s00280-020-04174-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-020-04174-1