
Abstract
We examined the effects of inspiratory muscle training (IMT) upon volitional hyperpnoea-mediated increases in blood lactate ([lac−]B) during cycling at maximal lactate steady state (MLSS) power, and blood lactate and oxygen uptake kinetics at the onset of exercise. Twenty males formed either an IMT (n = 10) or control group (n = 10). Prior to and following a 6-week intervention, two 30 min trials were performed at MLSS (207 ± 28 W), determined using repeated 30 min constant power trials. The first was a reference trial, whereas during the second trial, from 20 to 28 min, participants mimicked the breathing pattern commensurate with 90% of the maximal incremental exercise test minute ventilation (\( \dot{V}_{\text{E}} \)). Prior to the intervention, the MLSS [lac−]B was 3.7 ± 1.8 and 3.9 ± 1.6 mmol L−1 in the IMT and control groups, respectively. During volitional hyperpnoea, \( \dot{V}_{\text{E}} \) increased from 79.9 ± 9.5 and 76.3 ± 15.4 L min−1 at 20 min to 137.8 ± 15.2 and 135.0 ± 19.7 L min−1 in IMT and control groups, respectively; [lac−]B concurrently increased by 1.0 ± 0.6 (+27%) and 0.9 ± 0.7 mmol L−1 (+25%), respectively (P < 0.05). Following the intervention, maximal inspiratory mouth pressure increased 19% in the IMT group only (P < 0.01). Following IMT only, the increase in [lac−]B during volitional hyperpnoea was abolished (P < 0.05). In addition, the blood lactate (−28%) and phase II oxygen uptake (−31%) kinetics time constants at the onset of exercise and the MLSS [lac−]B (−15%) were reduced (P < 0.05). We attribute these changes to an IMT-mediated increase in the oxidative and/or lactate transport capacity of the inspiratory muscles.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Recently it has emerged that respiratory muscle activity may strongly influence the systemic blood lactate concentration ([lac−]B) (Brown et al. 2008, 2010; Chiappa et al. 2009; Johnson et al. 2006; Renggli et al. 2008; Verges et al. 2007). This notion contradicts the traditional view that the small mass of the respiratory muscles [estimated at around 960 g (Freedman et al. 1983)] and their large oxidative capacity precludes any systemically relevant lactate exchange (Wetter and Dempsey 2000). However, a significant influence of respiratory muscle activity on [lac−]B is supported by three lines of evidence. Firstly, a lower (~1.0 mmol L−1) [lac−]B has been observed during whole-body exercise following pressure-threshold inspiratory muscle training (IMT, a strength-training stimulus) (McConnell and Sharpe 2005; Romer et al. 2002b) and volitional isocapnic hyperpnoea training (endurance-training stimulus) (Leddy et al. 2007; Spengler et al. 1999), with up to 52% of variation in post-training whole-body exercise performance improvement accounted for by the reduced [lac−]B (Romer et al. 2002b).
Secondly, Chiappa et al. (2009) showed that increasing the work of breathing (via pressure-threshold loading) during recovery from maximal exercise significantly accelerated rates of lactate clearance and improved subsequent 30 s maximal cycling performance. The authors suggest that with loading, the inspiratory muscles are capable of net lactate consumption. In support of this notion, we recently demonstrated accelerated lactate recovery kinetics and increased lactate clearance when loading trained (IMT) inspiratory muscles following maximal exercise (Brown et al. 2010).
Thirdly, 10 min of volitional isocapnic hyperpnoea performed at rest typically results in a twofold increase in [lac−]B (Brown et al. 2008; Verges et al. 2007), which is subsequently attenuated following IMT (Brown et al. 2008) and volitional isocapnic hyperpnoea training (Verges et al. 2007). Presumably, once a critical level of hyperpnoea is reached [suggested to be around 70% of maximum voluntary ventilation (MVV) (Brown et al. 2008)] the respiratory muscles engage in significant lactate production and release which exceeds net lactate consumption. However, under resting conditions there is great capacity for other respiratory and non-respiratory muscles and other tissues to remove lactate from the systemic circulation (i.e. the lactate shuttle) (Brooks 1986), which may mask the true influence of respiratory muscle work upon [lac−]B. Accordingly, we developed a protocol whereby maximal volitional isocapnic hyperpnoea was superimposed upon cycling exercise at the maximal lactate steady state (MLSS) (Johnson et al. 2006). Since MLSS represents the highest exercise intensity associated with equivalence in the rates of blood lactate appearance and clearance, the 25% increase in [lac−]B that resulted from the superimposed hyperpnoea represents a truer reflection of net lactate production by the respiratory muscles (Johnson et al. 2006). Thus, if IMT increases the oxidative (Ramirez-Sarmiento et al. 2002) and/or lactate transport capacity of the inspiratory muscles (Brown et al. 2008, 2010), increases in [lac−]B during volitional hyperpnoea superimposed on exercise may be attenuated, although this remains untested.
Training-induced increases in muscle oxidative capacity also elicit alterations in the pulmonary oxygen uptake (\( \dot{V}{\text{O}}_{ 2} \)) response to exercise (Jones and Koppo 2005). The \( \dot{V}{\text{O}}_{ 2} \) response to square wave exercise above the lactate threshold is sequentially characterised by a short (10–20 s) cardiodynamic component (phase I), a primary component comprising an exponential increase in \( \dot{V}{\text{O}}_{ 2} \) over the ensuing 90–180 s (phase II), and a \( \dot{V}{\text{O}}_{ 2} \) slow component describing a continued temporal increase in \( \dot{V}{\text{O}}_{ 2} \) (phase III) (Burnley and Jones 2007). The phase II \( \dot{V}{\text{O}}_{ 2} \) kinetics are primarily limited by metabolic inertia (Burnley and Jones 2007) and may thus be accelerated by an increased intracellular oxygen utilisation due to an IMT-mediated increase in the oxidative capacity of the inspiratory muscles; however, this also remains a conjecture.
Therefore, the aim of this study was to examine the effects of IMT upon increases in [lac−]B when volitional hyperpnoea is superimposed upon exercise at MLSS power, and \( \dot{V}{\text{O}}_{ 2} \) and [lac−]B kinetics at the onset of exercise.
Methods
Ethical approval and participants
Following local ethics committee approval from the host institution, 20 healthy non-smoking, and recreationally trained males provided written informed consent to participate in the study (Table 1), which conformed to the standards set by the Declaration of Helsinki. Throughout the study participants adhered to their normal training regimen and did not engage in any strenuous exercise on the day preceding as well as on the day of an exercise test. Each participant completed a 24 h diet record prior to his first exercise trial, which was then repeated prior to all subsequent trials. Participants abstained from alcohol and caffeine for 24 h prior to testing and arrived at the laboratory 2 h post-prandial. All trials for each participant were separated by at least 24 h and performed at a similar time of day and in similar laboratory conditions (control group pre vs. post: temperature: 18.4 ± 3.8 vs. 17.1 ± 2.6°C; relative humidity: 46.8 ± 16.2 vs. 47.7 ± 10.4%; IMT group pre vs. post: temperature: 20.2 ± 2.7 vs. 18.3 ± 3.6°C; relative humidity: 45.4 ± 13.6 vs. 46.2 ± 12.9%).
Experimental design
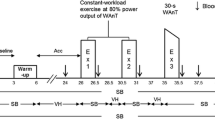
Participants were initially familiarised with all testing procedures and completed pulmonary function and maximal inspiratory mouth pressure (MIP) tests. Subsequently, following MLSS determination participants were matched for 90% maximal exercise minute ventilation (\( \dot{V}_{\text{Emax}} \)), as defined by the highest 30 s average measured during the lactate elevation phase of the lactate minimum test (see MLSS determination below), and divided equally into an IMT or a control group. Prior to and following a 6-week intervention (IMT or without IMT), participants completed in random order: (i) a 30 min reference trial at MLSS power and (ii) a 30 min experimental trial at MLSS power during which from 20 to 28 min, \( \dot{V}_{\text{E}} \) and breathing pattern were volitionally matched to that commensurate with 90% \( \dot{V}_{{{\text{E}}\,{ \max }}} \) (see Experimental trial below). Cycling cadence was displayed upon an electronic display and participants were instructed prior to and regularly during all exercise trials to adopt a constant pedal cadence (Brown et al. 2009).
Pulmonary function and maximal inspiratory mouth pressure
Pulmonary function was assessed using a pneumotachograph (ZAN 600USB, Nspire Health, Oberthulba, Germany) in accordance with published guidelines (ATS/ERS 2005). A hand-held mouth pressure meter (MicroRPM, Micro Medical, Kent, UK) measured MIP as an index of global inspiratory muscle strength. The mouthpiece assembly incorporated a 1 mm orifice to prevent glottic closure during inspiratory efforts. Manoeuvres, performed in an upright standing posture, were initiated from residual volume, and sustained for at least 1 s (Brown et al. 2008, 2010; Johnson et al. 2007). A minimum of five manoeuvres were performed every 30 s; this protocol continued until the highest value recorded from three consecutive measures that differed by no more than 10% or 10 cmH2O (whichever was smallest) was used for subsequent analysis (Brown et al. 2008, 2010). MIP was expressed relative to predicted values, as calculated using the equation of Wilson et al. (1984): MIPPREDICTED = 142 − (1.03 × age). MIP was measured every 2 weeks during the intervention. Following IMT, MIP was measured 48 h following the final training session and on a separate day to exercise testing.
MLSS determination
All exercise trials were performed on different days using an electromagnetically braked cycle ergometer (Excalibur Sport, Lode, Groningen, The Netherlands). MLSS was initially estimated using a previously validated lactate minimum test (Johnson et al. 2009) comprising three consecutive phases: (i) lactate elevation phase comprising maximal incremental exercise; (ii) 8 min recovery phase at 60 W; and (iii) incremental phase comprising five 4 min exercise stages at intensities of 45, 50, 55, 60 and 65% of the maximum power (\( \dot{W}_{ \max } \)) achieved during the lactate elevation phase. During the lactate elevation phase, power output was increased by 10 W every 15 s so that exercise intolerance (cadence <60 revs min−1) occurred in approximately 10 min. The final power defined \( \dot{W}_{ \max } \) and the highest \( \dot{V}{\text{O}}_{ 2} \) recorded over any 30 s period defined \( \dot{V}{\text{O}}_{{ 2 {\text{max}}}} \). During the incremental phase, blood samples were taken in the final seconds of each stage, and the lactate minimum power (estimated MLSS) was calculated as the nadir of a 2nd order polynomial fitting the [lac−]B versus power data. MLSS was subsequently resolved using repeated (typically 2–4) 30 min constant power tests (starting at the lactate minimum power) during which [lac−]B was determined every 3 min from 15 to 30 min. MLSS was defined as the highest cycling power at which [lac−]B did not increase >0.5 mmol L−1 between 15 and 30 min of exercise (Aunola and Rusko 1992). If this criterion was not satisfied, the cycling power for the subsequent 30 min exercise test was, relative to the most recent test, changed by ±2.5% (4–7 W) depending on the direction of change in [lac−]B (Johnson et al. 2009; Johnson et al. 2006; McConnell and Sharpe 2005). The precision to which MLSS was determined was based upon previous work (Johnson et al. 2009; Johnson et al. 2006; McConnell and Sharpe 2005) and was chosen to ensure that a high degree of resolution of MLSS determination was achieved within a relatively short (1–2 weeks) time-frame. Following the 6-week intervention, MLSS was re-assessed in all participants, starting at the pre-intervention MLSS power and using the same criteria and protocol described above.
Reference trial (without volitional hyperpnoea)
Following a 3 min warm-up at 50% MLSS power participants cycled for 30 min at MLSS power. [lac−]B was measured at rest and every 2 min during exercise. Blood gases were measured every 4 min from 0 to 20 min and every 2 min thereafter. Breath-by-breath respiratory variables were averaged over the final 30 s of every 2 min interval. Every 2 min heart rate (HR) was measured using short range telemetry (Polar S610, Polar, Kempele, Finland) and arterial oxygen saturation (SpO2) was estimated using infrared fingertip pulse oximetry (Model 8600, Nonin, Minnesota, USA). Rating of perceived exertion for limb discomfort (RPE) was recorded every 2 min using the Borg 6–20 scale. Rating of perceived dyspnoea (RPD), where participants were asked to rate their breathing “discomfort”, was recorded every 2 min using the modified 0–10 scale (Borg 1982).
Experimental trial (with volitional hyperpnoea)
The experimental trial was identical to the reference trial except that from 20 to 28 min, participants volitionally increased \( \dot{V}_{\text{E}} \) by matching the tidal volume (V T), breathing frequency (fR) and duty cycle (T I/T TOT) with that commensurate with 90% \( \dot{V}_{\text{E}} \)max. Pilot work from our laboratory indicated that 90% \( \dot{V}_{\text{E}} \)max was the highest value that could be maintained in a square wave fashion for 8 min whilst exercising at MLSS power. An audio metronome paced fR and real-time visual feedback of \( \dot{V}_{\text{E}} \) and V T were provided (Brown et al. 2008). Following volitional hyperpnoea, participants resumed a spontaneous breathing pattern for the final 2 min of exercise. The volitional hyperpnoea protocol was similar to that used previously by our group (Brown et al. 2008; Johnson et al. 2006) and was designed to mimic the breathing pattern of high-intensity whole body exercise.
Six-week intervention
The IMT group performed 30 consecutive dynamic inspiratory efforts twice daily for 6 weeks using an inspiratory pressure-threshold device (POWERbreathe® Classic series 1st generation, Gaiam, UK). The initial training load was 50% MIP. Participants were encouraged to periodically increase the training load to accommodate improvements in MIP, such that task failure was reached at around the 30th inspiratory effort. Measurement of MIP at 2-week intervals permitted the threshold load of the device to be objectively increased or decreased by the researchers to ensure the training load was equal to 50% MIP. Each inspiratory effort was initiated from residual volume and participants strove to maximise V T. This protocol is known to be effective in eliciting an adaptive response (Brown et al. 2008, 2010; Johnson et al. 2007; McConnell and Sharpe 2005). All participants recorded habitual training in a training diary, and the IMT group also recorded IMT adherence. The control group continued with their habitual training schedule and were not exposed to an intervention. A placebo treatment was not applied to the control group since the study outcome measures, including MLSS power (McConnell and Sharpe 2005) and blood-borne parameters (Brown et al. 2008, 2010) could not be influenced by either motivation or expectation. Participants were informed that they belonged to a control group following the determination of MLSS and, to avoid any possible disadvantage, were afforded the opportunity to undertake IMT upon completion of the study.
Blood sampling and respiratory measurements
Arterialised venous blood was drawn from a dorsal hand vein via an indwelling 21-G cannula. Arterialisation was ensured by immersing the hand in ~ 40°C water for 10 min prior to cannulation and by warming the hand during exercise using an infrared lamp. Blood samples were drawn into a 2 ml pre-heparinised syringe (PICO 50, Radiometer, Copenhagen, Denmark) and analysed immediately (ABL520, Radiometer, Copenhagen, Denmark) for the partial pressure of carbon dioxide (PCO2) and pH, which were corrected for changes in rectal temperature (Squirrel 2020, Grant Instruments, Cambridge, UK). [lac−]B was determined using an automated enzymatic method (Biosen, EKF Diagnostics, Barleben, Denmark). Plasma bicarbonate concentration (\( [ {\text{HCO}}_{ 3}^{ - } ] \)) and base excess of the extracellular fluid (BEECF) were calculated using the Henderson–Hasselbalch and Siggaard–Anderson equations, respectively (for equations see: Brown et al. 2008).
Respiratory variables were measured breath-by-breath (ZAN 600USB, Nspire Health, Oberthulba, Germany). Subjects wore a facemask (model 7400, Hans Rudolph, KS, USA) connected to a flow pneumotachograph and, distally, a two-way non-rebreathing valve (model 2730, Hans Rudolph, Missouri, USA) onto which a 1.5 m length of wide-bore tubing was connected to the inspiratory port. During volitional hyperpnoea, CO2 was added into this tubing to increase FICO2 and retain end-tidal and, consequently, arterialised blood PCO2 at levels commensurate with steady state exercise (Brown et al. 2008; Johnson et al. 2006). The addition of CO2 into the inspiratory line adversely affected the ability of the breath-by-breath analyser to accurately measure \( \dot{V}{\text{CO}}_{ 2} \) and thus RER during volitional hyperpnoea; thus these data are not presented over 22–28 min.
Data and statistical analyses
Pulmonary \( \dot{V}{\text{O}}_{ 2} \) kinetics
The pulmonary \( \dot{V}{\text{O}}_{ 2} \) response to exercise was modelled using a single mono-exponential model (Burnley et al. 2001). Breath-by-breath data were linearly interpolated to provide second-by-second values following the elimination of outlying breaths defined as those ±4 SD of the local mean (Lamarra et al. 1987). Given that each participant performed two square wave transitions from 3 min cycling at 50% MLSS power to 20 min cycling at 100% MLSS power (i.e. 0–20 min during the reference and experimental trials), the two trials were time aligned and averaged to improve the signal-to-noise ratio. This provided for each participant one set of second-by-second data for the pre- and post-intervention trials. The amplitude and time delay of the primary (phase II) \( \dot{V}{\text{O}}_{ 2} \) response were modelled following a 20 s time delay (Ozyener et al. 2001) to exclude the cardiodynamic component (phase I):
where \( \dot{V}{\text{O}}_{ 2} (t ) \) (L min−1) is the \( \dot{V}{\text{O}}_{ 2} \) for a given time (t; min); \( \dot{V}{\text{O}}_{{ 2 , {\text{b}}}} \) is the baseline \( \dot{V}{\text{O}}_{ 2} \) (L min−1) measured during the final minute of the warm-up preceding the onset of exercise; A1 (L min−1) is the amplitude of the exponential curve and defined as the increase in \( \dot{V}{\text{O}}_{ 2} \) from \( \dot{V}{\text{O}}_{{ 2 , {\text{b}}}} \) to the end of phase II; and \( {{\uptau}} \) (s) and TD (s) are the time constant and time delay, respectively. The amplitude of the \( \dot{V}{\text{O}}_{ 2} \) slow component (phase III; A2) was defined as the difference in \( \dot{V}{\text{O}}_{ 2} \) between TD (end of phase II) and that measured during the final 30 s of the 20 min period (Burnley et al. 2001).
Blood lactate kinetics
The [lac−]B response to exercise was modelled using a single mono-exponential function (Beneke 2003). For each participant, the [lac−]B measured during the reference and experimental trials (from 0 to 20 min only; i.e. excluding volitional hyperpnoea) was time aligned and averaged, thus producing one set of [lac−]B against time data per-participant for pre- and post-intervention comparisons:
where La(t) (mmol L−1) is the [lac−]B for a given time (t; min); La(0) (mmol L−1) is the [lac−]B at the onset of exercise; and A (mmol L−1) and τ (s) are the amplitude, defined as the increase in [lac−]B from La(0) to steady state (where Δ[lac−]B/Δt = 0), and the time constant of the response, respectively.
Statistical analysis
In order to compare the discrete physiological responses between steady state exercise and volitional hyperpnoea, the reference and the experimental trials were averaged and analysed over two time periods: a steady state period from 12 to 20 min (as it takes approximately 12 min to reach a steady state in [lac−]B) and a volitional hyperpnoea period from 22 to 28 min (Johnson et al. 2006). Pre- and post-intervention results and group interactions were assessed using one-way or two-way ANOVA with repeated measures and Tukey’s HSD post-hoc analysis. Interactions were defined for “group” (IMT vs. control), “trial” (reference [without hyperpnoea] vs. experimental [with hyperpnoea]) and “time” (12–20 min [steady state period] or 22–28 min [volitional hyperpnoea period]). Pearson product-moment correlation coefficients were calculated to assess the relationship between selected variables. A priori α was set at P ≤ 0.05. Results are presented as mean ± SD. All mathematical modelling was performed using commercially available software (GraphPad Prism Version 5.01, GraphPad software, Inc. CITY, USA). Statistical analysis was performed using SPSS for Windows (SPSS, Chicago, IL, USA).
Results
With the exception of age and predicted MIP and MVV10, there were no between-group differences in any variable prior to the intervention, and all responses remained unchanged in the control group following the intervention.
Inspiratory muscle strength
Baseline pulmonary function and MIP were within normal limits (Table 1). Training compliance was excellent in the IMT group (96 ± 4% of training sessions completed). Inspection of training diaries revealed habitual training remained constant in both groups. MIP was unchanged in the control group following the intervention period (pre vs. post: 149 ± 25 vs. 147 ± 27 cmH2O). Conversely, MIP increased in the IMT group from 164 ± 23 cmH2O at baseline to 174 ± 24 (P < 0.05), 187 ± 23 (P < 0.01) and 194 ± 21 cmH2O (+19%) (P < 0.01) following 2, 4 and 6 weeks, respectively. Baseline absolute MIP was negatively correlated with the relative IMT-mediated increase in MIP (r = −0.79, P < 0.05), but not the absolute IMT-mediated increase in MIP (r = −0.464, P = 0.177).
MLSS power
With the exception of one control participant in whom MLSS power decreased by 2.5% (−7 W), MLSS power was unchanged following the intervention in both groups. MLSS power was 210 ± 27 and 210 ± 27 W pre- and post-IMT, respectively, and 205 ± 29 and 204 ± 27 W in the control group pre- and post-intervention.
Respiratory responses to MLSS and volitional hyperpnoea
Volitional hyperpnoea was 90.3 ± 9.6% \( \dot{V}_{\text{Emax}} \) (76 ± 19% MVV10) in the IMT group and 91.2 ± 4.9% \( \dot{V}_{\text{Emax}} \) (81 ± 15% MVV10) in the control group (for absolute \( \dot{V}_{\text{E}} ,\,\dot{V}{\text{O}}_{ 2} \) and \( \dot{V}{\text{CO}}_{ 2} \) see Fig. 1; Tables 2 and 3). \( \dot{V}_{\text{E}} \) and breathing pattern during both the reference and the experimental trials were not different between groups before or after the intervention period. \( \dot{V}{\text{O}}_{ 2} ,\,\dot{V}{\text{CO}}_{ 2} \) and RER throughout the reference trial and \( \dot{V}{\text{O}}_{ 2} \) and \( \dot{V}{\text{CO}}_{ 2} \) throughout the experimental trial were not different between groups and remained unchanged following the intervention (Tables 2, 3). Therefore, the breathing challenge was repeated with considerable accuracy following the intervention. Parameters of the pulmonary \( \dot{V}{\text{O}}_{ 2} \) response to exercise are shown in Table 4. A ~14 s reduction in τ 1 was observed after IMT (group × time interaction, P < 0.05); all other parameters of the \( \dot{V}{\text{O}}_{ 2} \) response to exercise remained unchanged following the intervention in both groups. The reduction in τ 1 following IMT was not correlated with the absolute (r = −0.014, P = 0.974) or relative (r = 0.206, P = 0.625) change in baseline MIP following IMT.

Pre-IMT breathing pattern during the reference and experimental trials. Data post-IMT and pre- and post-intervention in the control group not shown since they were not different from the data illustrated above. Time 0 min reflects the end of a 3 min warm-up at 50% MLSS power. *Different from steady state period (12–20 min) (P < 0.01)
Blood lactate concentration
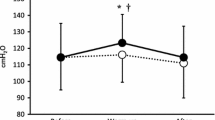
Prior to IMT the MLSS [lac−]B was 3.7 ± 1.8 mmol L−1 in the reference trial (12–30 min) and 4.0 ± 2.2 mmol L−1 (12–20 min) in the experimental trial. Following IMT [lac−]B over 12–30 min during the reference trial was reduced (main effect trial, P < 0.05) by 0.7 ± 1.7 mmol L−1 (−8%) and over 12–20 min of the experimental trial by 0.9 ± 1.8 mmol L−1 (−15%) (main effect trial, P < 0.05) (Fig. 2). Furthermore, the amplitude (A) of the increase in [lac−]B at the onset of exercise, plateau in MLSS [lac−]B (where Δ[lac−]B/Δt = 0) and the time constant (τ) of the increase in [lac−]B from exercise onset to the plateau in MLSS [lac−]B (i.e. the steady state) were reduced after IMT (P < 0.05) (Table 5).

Blood lactate concentration ([lac−]B) during the reference and experimental trials for the IMT group only. Pre- and post-intervention data for the control group were not different from pre-IMT. Time 0 min represents the end of a 3 min warm-up at 50% MLSS power. *Different from steady state period (12–20 min) (P < 0.05); †post-IMT experimental trial different from pre-IMT experimental trial (P < 0.05)
Prior to IMT, volitional hyperpnoea caused a 1.0 ± 0.6 mmol L−1 (27%) increase in [lac−]B (relative to 12–20 min) (P < 0.05). Following IMT, the increase in [lac−]B was abolished (Fig. 2), thus at 30 min, [lac−]B was 1.8 ± 1.3 mmol L−1 (−26%) lower than that measured during the pre-IMT experimental trial (group × trial × time interaction; P < 0.01). Both the absolute (r = 0.75, P < 0.05) and relative (r = 0.66, P < 0.05) increases in baseline MIP following IMT were correlated with the absolute reduction in [lac−]B observed at min 30 of the experimental trial (Fig. 3).

Relationship between the absolute (left) and relative (right) IMT-induced change in MIP and the reduction in [lac−]B at 30 min during the experimental trials
Acid–base balance
Temporal changes in acid–base balance for the IMT group are shown in Fig. 4. Volitional hyperpnoea increased [H+] (P < 0.05) and this response was unchanged following IMT. From 22 to 28 min of the reference trial, there was a trend (P = 0.07) for [H+] to be lower after IMT.

Pre- and post-IMT acid–base parameters. Data pre- and post-intervention in the control group not shown since they were not different from pre-IMT. Time 0 min reflects the end of a 3 min warm-up at 50% MLSS power. § Trial × time interaction for post-IMT reference trial (P < 0.05); † post-IMT experimental trial different from pre-IMT experimental trial (P < 0.05); §§ trial × time interaction for post-IMT experimental trial (P < 0.05)
Approximately 10 min following the onset of exercise, \( [ {\text{HCO}}_{ 3}^{ - } ] \) and BEECF remained constant with time during the pre-intervention reference trial in both groups. Following IMT and during the reference trial, BEECF was greater from 22 to 28 min (trial × time interaction; P < 0.05). Prior to the intervention, volitional hyperpnoea caused a reduction in \( [ {\text{HCO}}_{ 3}^{ - } ] \) and BEECF (P < 0.05) in both groups by 30 min, and these responses were unchanged in the control group following the intervention. Conversely, following IMT, the reduction in [HCO3 −] and BEECF during volitional hyperpnoea was attenuated (trial × time interaction; P < 0.05; Fig. 4).
PCO2 was similar between groups and between trials prior to and following the intervention (Fig. 4). PCO2 during volitional hyperpnoea and over the equivalent time period of the reference trial was not different, thus isocapnia was maintained successfully throughout volitional hyperpnoea.
Heart rate and arterial oxygen saturation
Over 12–20 min of the pre-IMT reference trial, HR (145 ± 11 beats min−1) and SpO2 (95.3 ± 1.2%) were not different from that measured over 12–20 and 22–28 min of all other trials, except for the volitional hyperpnoea period during the experimental trials. Specifically, prior to the intervention in the IMT group, HR increased during volitional hyperpnoea to 160 ± 15 beats min−1 (P < 0.05); this increase was attenuated following IMT (157 ± 14 beats min−1) (trial × time interaction, P < 0.05). Prior to IMT, the SpO2 over 12–20 min of the experimental trial was 94.6 ± 1.3% and increased to 95.8 ± 1.2% during volitional hyperpnoea (P < 0.05), which was unchanged following IMT.
Perceptual responses
There were no differences between groups for RPD measured during the reference trial prior to and following the intervention. Similarly, prior to the intervention, there were no differences between groups for RPD measured over 12–20 min of the experimental trial (see Tables 2, 3 for data from the IMT group). During volitional hyperpnoea (22–28 min), RPD increased significantly in both groups (P < 0.05) and this response was unchanged following the intervention in the control group. Conversely, following IMT, RPD during volitional hyperpnoea was lower by 1.6 ± 1.8 AU (trial × time interaction, P < 0.05, Table 3). RPE responses during the reference trial were unchanged in both groups following the intervention. RPE responses during the experimental trial were also unchanged in the control group following the intervention. Conversely, following IMT, RPE during the experimental trial was lower over 12–20 min (12.7 ± 0.9 vs. 11.7 ± 1.1, P < 0.05) and 22–28 min (13.1 ± 1.4 vs. 12.0 ± 1.0, P = 0.07).
Discussion
The novel finding of this study was that 6 weeks of IMT abolished the increase in [lac−]B observed when volitional hyperpnoea was superimposed on cycling exercise at the MLSS power. IMT also attenuated the accompanying volitional hyperpnoea-induced increases in RPD, HR and acid–base disturbance. We have also demonstrated for the first time accelerated [lac−]B kinetics and, in agreement with a previous study (Bailey et al. 2010), accelerated \( \dot{V}{\text{O}}_{ 2} \) kinetics at the onset of exercise following IMT.
The increase in [lac−]B observed when volitional hyperpnoea was superimposed on exercise is consistent with previous studies in which the breathing challenge was performed during exercise (Johnson et al. 2006) or at rest (Brown et al. 2008; Verges et al. 2007). The ~25% increase in [lac−]B caused by the relatively modest (~60 L min−1) increase in \( \dot{V}_{\text{E}} \) during volitional hyperpnoea suggests that the respiratory muscles were contributing to systemic lactate exchange. In contrast, despite a larger absolute increase in \( \dot{V}_{\text{E}} \) (~100 L min−1) Babcock et al. (1995) failed to observe an increase in [lac−]B- during ~4.5 min of volitional hyperpnoea performed at rest, although a greater lactate clearance capacity under resting conditions (Brooks 1986) and the brevity of the breathing challenge may have precluded measurable changes in [lac−]B (Brown et al. 2008). Spengler et al. (2000) also failed to observe an increase in [lac−]B during ~43 min of volitional isocapnic hyperpnoea performed at rest. However, further to the limitations associated with volitional hyperpnoea performed at rest (see above), the \( \dot{V}_{\text{E}} \) (~122 L min−1, 62% of MVV) was probably below the critical level (around 70% MVV) (Brown et al. 2008) at which the respiratory muscles engage in significant lactate production and release. Nevertheless, a limitation of the present study was that the work of breathing specifically associated with exercise hyperpnoea, i.e. the pressure–volume characteristics of each breath (Babcock et al. 1995), was not mimicked. Therefore, since the work of breathing (particularly that associated with expiration) during volitional hyperpnoea exceeds that associated with an identical, spontaneous \( \dot{V}_{\text{E}} \) during exercise (Klas and Dempsey 1989), the net increase in respiratory muscle lactate production in our experimental condition may overestimate that of exercise. Conversely, it is also possible that the increased [lac−]B during volitional hyperpnoea superimposed on exercise may have been partially countered by an increase in lactate consumption by highly oxidative respiratory muscles. Nonetheless, our primary and novel finding that the volitional hyperpnoea-mediated increase in [lac−]B was abolished following IMT is a stark illustration of the inspiratory muscles’ plasticity and capacity to affect systemic blood chemistry.
Our findings suggest that following IMT, the relative work performed by the inspiratory muscles during volitional hyperpnoea was reduced (due to the 19% increase in inspiratory muscle strength) resulting in a reduction in net lactate production (Brown et al. 2008; Johnson et al. 2006). Hypertrophy of the diaphragm (Downey et al. 2007) and external intercostals (Ramirez-Sarmiento et al. 2002) may have decreased the relative work performed by the inspiratory muscles, thus reducing/delaying the recruitment of less efficient, lactate producing inspiratory muscle fibres (Marcinik et al. 1991). Moderate intensity, high repetition strength training may also increase type I muscle fibre prevalence, mitochondrial enzyme activity (Sale et al. 1990), and expression of sarcolemmal and mitochondrial membrane-bound monocarboxylate transporters (Juel et al. 2004), thus promoting reduced production/increased clearance of lactate. Intriguingly, an increased prevalence of type I fibres and oxidative enzymes has been observed in the external intercostals of COPD patients following 5 weeks of IMT (Ramirez-Sarmiento et al. 2002).
An alternative/additional explanation for our findings is that prior to IMT the increased [lac−]B during volitional hyperpnoea was, in part, due to reduced locomotor muscle blood flow secondary to a respiratory muscle metaboreflex (Dempsey et al. 2006). The latter is activated by the accumulation of fatigue-related metabolites, which stimulate respiratory muscle metaboreceptors, thus promoting sympathetically mediated vasoconstrictor outflow. A reduction in locomotor muscle perfusion may have resulted in increased type II motor unit recruitment within the locomotor muscles, thus promoting greater lactate production and less lactate clearance by leg muscle. Following IMT, an attenuation of metabolite accumulation in the inspiratory muscles (Brown et al. 2008, 2010) may have blunted the inspiratory muscle metaboreflex (Witt et al. 2007) thereby preserving locomotor muscle blood flow and attenuating the impact of locomotor muscle on [lac−]B. However, this notion is challenged by the observation of an unchanged arterial-femoral venous [lac−]B despite (relative to a control trial) an 11% change in leg blood flow due to ±50% change in the work of breathing during maximal exercise (Harms et al. 1997).
The IMT-mediated decrease in the MLSS [lac−]B, without a corresponding change in MLSS power, concurs with previous findings (McConnell and Sharpe 2005), and may be related, in part, to the reduced phase II time constant of the pulmonary \( \dot{V}{\text{O}}_{ 2} \) kinetics. There is growing evidence that the respiratory muscles per se may influence \( \dot{V}{\text{O}}_{ 2} \) kinetics. Specifically, low intensity inspiratory muscle loading (15 cmH2O) increases the phase II \( \dot{V}{\text{O}}_{ 2} \) time constant (Keslacy et al. 2005), and the \( \dot{V}{\text{O}}_{ 2} \) slow component during heavy exercise is partly explained (up to 24%) by the increased \( \dot{V}_{\text{E}} \) (Candau et al. 1998). Furthermore, the \( \dot{V}{\text{O}}_{ 2} \) slow component is increased with inspiratory muscle loading (15 cmH2O) (Carra et al. 2003) and decreased with unloading (heliox breathing) (Cross et al. 2010). Recently, 4 weeks of IMT was shown to reduce the \( \dot{V}{\text{O}}_{ 2} \) slow component during severe and maximal, but not moderate, intensity exercise (Bailey et al. 2010), although despite a shorter overall time constant for \( \dot{V}{\text{O}}_{ 2} \), the phase II time constant was unchanged. This discrepancy with the present study could be due, in part, to between-study differences in IMT duration (4 weeks in Bailey et al. vs. 6 weeks in the present study). Specifically, although the 17% increase in MIP observed by Bailey et al. (2010) was similar to the 19% increase observed in the present study, increased MIP per se is not correlated with changes in \( \dot{V}{\text{O}}_{ 2} \) kinetics (present study; Bailey et al. 2010) or performance enhancement (Griffiths and McConnell 2007; Johnson et al. 2007). Conversely, we suggest that changes in physiological responses to exercise following IMT are largely the result of IMT-mediated increases in the oxidative capacity of the inspiratory muscles (see below), and that the additional 28 IMT sessions performed in the present study (compared to Bailey et al. 2010) resulted in greater inspiratory muscle adaptation (Henriksson 2000; Staron et al. 1994), as reflected, in part, by the reduced phase II time constant.
A reduced phase II \( \dot{V}{\text{O}}_{ 2} \) time constant is commonly observed, albeit to a greater extent (up to 50%) following whole-body endurance training (Burnley and Jones 2007). This is attributed to increased intracellular oxygen utilisation (i.e. reduced metabolic inertia) most likely due to an increased prevalence of type I muscle fibres and increased intracellular-oxidative enzyme activity (Jones and Koppo 2005). These adaptations may well occur within the inspiratory muscles following IMT, thus reducing metabolic inertia within the inspiratory muscles. Consequently, increased lactate utilisation by inspiratory muscles at exercise onset (which is also likely to be magnified by the greater expression of membrane-bound lactate transport proteins) may “reset” the MLSS [lac−]B without modifying the MLSS power. Such adaptations may also explain the attenuated metabolic acidosis associated with both exercise and volitional hyperpnoea (see Fig. 4). Improved \( \dot{V}{\text{O}}_{ 2} \) dynamics have also been linked with increased exercise tolerance (Burnley and Jones 2007), thus faster phase II \( \dot{V}{\text{O}}_{ 2} \) kinetics, and hence reduced oxygen deficit, may partly explain our previous observation of an IMT-mediated increase in anaerobic work capacity and hence cycling time trial performance (Johnson et al. 2007).
The ~30% reduction in RPD during volitional hyperpnoea following IMT concurs with observations made during whole body exercise (Romer et al. 2002a; Volianitis et al. 2001) and isolated breathing challenges (Verges et al. 2007; Verges et al. 2009). Whilst the breathing challenge was identical following IMT the lower relative load on the inspiratory muscles, due to a greater force generating capacity (i.e. increased MIP), may have reduced central corollary discharge for a given \( \dot{V}_{\text{E}} \) (Redline et al. 1991). Repeated IMT bouts may also alter mechanosensitive type III and chemosensitive type IV nerve afferents within the inspiratory muscles (Sinoway et al. 1996), thus also reducing ascending afferent feedback from the inspiratory muscles. These changes may elicit performance benefits as during heavy exercise with high \( \dot{V}_{\text{E}} \) reduced dyspnoea due to IMT is correlated with increased exercise tolerance (Romer et al. 2002b).
Finally, IMT also attenuated the increase in HR during volitional hyperpnoea which is consistent with observations made during fatiguing resistive inspiratory muscle loading performed whilst at rest (Witt et al. 2007). Although repeated mechanical deformations of the inspiratory muscles during IMT may desensitise mechanically sensitive type III afferents (Sinoway et al. 1996), Witt et al. (2007) argue that an IMT-mediated reduction in HR is primarily due to an increased oxidative capacity of the inspiratory muscles, which attenuates metabolite accumulation within the inspiratory muscles, thereby reducing phrenic afferent discharge of both type III and IV nerve endings. The lower [lac−]B and attenuated disturbance in acid–base balance during volitional hyperpnoea following IMT observed in the present study supports this notion.
In conclusion, increases in [lac−]B when volitional hyperpnoea was superimposed on exercise at MLSS power were abolished following 6 weeks of IMT. Furthermore, IMT resulted in faster phase II \( \dot{V}{\text{O}}_{ 2} \) and [lac−]B kinetics at the onset of exercise and a lower MLSS [lac−]B. Although the precise mechanisms underpinning these changes remain unknown, there is mounting evidence supporting an IMT-mediated increase in the oxidative and/or lactate transport capacity of the inspiratory muscles. The relevance of these findings are twofold: firstly, we have further illustrated that the trained inspiratory muscles have a significant effect upon systemic lactate and pulmonary \( \dot{V}{\text{O}}_{ 2} \) kinetics; and secondly, that these mechanisms may explain, in part, the improvements in whole body exercise tolerance noted in previous studies following specific IMT.
References
ATS/ERS (2005) Standardization of spirometry: ATS/ERS Task Force. Eur Respir J 26:319–338
Aunola S, Rusko H (1992) Does anaerobic threshold correlate with maximal lactate steady-state? J Sports Sci 10(4):309–323
Babcock MA, Pegelow DF, McClaran SR et al (1995) Contribution of diaphragmatic power output to exercise-induced diaphragm fatigue. J Appl Physiol 78(5):1710–1719
Bailey SJ, Romer LM, Kelly J et al (2010) Inspiratory muscle training enhances pulmonary O2 uptake kinetics and high-intensity exercise tolerance in humans. J Appl Physiol 109(2):457–468
Beneke R (2003) Methodological aspects of maximal lactate steady state-implications for performance testing. Eur J Appl Physiol 89(1):95–99
Borg GA (1982) Psychophysical bases of perceived exertion. Med Sci Sports Exerc 14(5):377–381
Brooks GA (1986) The lactate shuttle during exercise and recovery. Med Sci Sports Exerc 18(3):360–368
Brown PI, Sharpe GR, Johnson MA (2008) Inspiratory muscle training reduces blood lactate concentration during volitional hyperpnoea. Eur J Appl Physiol 104(1):111–117
Brown PI, Johnson MA, Sharpe GR (2009) Cycling cadence influences physiological parameters at maximal lactate steady state. J Sport Sci 26 (S2):S32
Brown PI, Sharpe GR, Johnson MA (2010) Loading of trained inspiratory muscles speeds lactate recovery kinetics. Med Sci Sports Exerc 42(6):1103–1112
Burnley M, Jones AM (2007) Oxygen uptake kinetics as a determinant of sports performance. Eur J Sport Sci 7:63–79
Burnley M, Doust JH, Carter H et al (2001) Effects of prior exercise and recovery duration on oxygen uptake kinetics during heavy exercise in humans. Exp Physiol 86(3):417–425
Candau R, Belli A, Millet GY et al (1998) Energy cost and running mechanics during a treadmill run to voluntary exhaustion in humans. Eur J Appl Physiol Occup Physiol 77(6):479–485
Carra J, Candau R, Keslacy S et al (2003) Addition of inspiratory resistance increases the amplitude of the slow component of O2 uptake kinetics. J Appl Physiol 94(6):2448–2455
Chiappa GR, Ribeiro JP, Alves CN et al (2009) Inspiratory resistive loading after all-out exercise improves subsequent performance. Eur J Appl Physiol 106(2):297–303
Cross TJ, Sabapathy S, Schneider DA et al (2010) Breathing He-O2 attenuates the slow component of O2 uptake kinetics during exercise performed above the respiratory compensation threshold. Exp Physiol 95(1):172–183
Dempsey JA, Romer L, Rodman J et al (2006) Consequences of exercise-induced respiratory muscle work. Respir Physiol Neurobiol 151(2–3):242–250
Downey AE, Chenoweth LM, Townsend DK et al (2007) Effects of inspiratory muscle training on exercise responses in normoxia and hypoxia. Respir Physiol Neurobiol 156(2):137–146
Freedman S, Cooke NT, Moxham J (1983) Production of lactic acid by respiratory muscles. Thorax 38(1):50–54
Griffiths LA, McConnell AK (2007) The influence of inspiratory and expiratory muscle training upon rowing performance. Eur J Appl Physiol 99(5):457–466
Harms CA, Babcock MA, McClaran SR et al (1997) Respiratory muscle work compromises leg blood flow during maximal exercise. J Appl Physiol 82(5):1573–1583
Henriksson J (2000) Cellular metabolism and endurance. In: Shephard RJ, Astrand PO (eds) Endurance in sport. Blackwell Science, London, pp 118–135
Johnson MA, Sharpe GR, McConnell AK (2006) Maximal voluntary hyperpnoea increases blood lactate concentration during exercise. Eur J Appl Physiol 96(5):600–608
Johnson MA, Sharpe GR, Brown PI (2007) Inspiratory muscle training improves cycling time-trial performance and anaerobic work capacity but not critical power. Eur J Appl Physiol 101(6):761–770
Johnson MA, Sharpe GR, Brown PI (2009) Investigations of the lactate minimum test. Int J Sports Med 30(6):448–454
Jones AM, Koppo K (2005) Effect of training on VO2 kinetics and performance. In: Jones AM, Poole DC (eds) Oxygen uptake kinetics in sport exercise and medicine. Routledge, London, pp 373–397
Juel C, Holten MK, Dela F (2004) Effects of strength training on muscle lactate release and MCT1 and MCT4 content in healthy and type 2 diabetic humans. J Physiol 556(Pt 1):297–304
Keslacy S, Matecki S, Carra J et al (2005) Effect of inspiratory threshold loading on ventilatory kinetics during constant-load exercise. Am J Physiol Regul Integr Comp Physiol 289(6):R1618–R1624
Klas JV, Dempsey JA (1989) Voluntary versus reflex regulation of maximal exercise flow: volume loops. Am Rev Respir Dis 139(1):150–156
Lamarra N, Whipp BJ, Ward SA et al (1987) Effect of interbreath fluctuations on characterizing exercise gas exchange kinetics. J Appl Physiol 62(5):2003–2012
Leddy JJ, Limprasertkul A, Patel S et al (2007) Isocapnic hyperpnea training improves performance in competitive male runners. Eur J Appl Physiol 99(6):665–676
Marcinik EJ, Potts J, Schlabach G et al (1991) Effects of strength training on lactate threshold and endurance performance. Med Sci Sports Exerc 23(6):739–743
McConnell AK, Sharpe GR (2005) The effect of inspiratory muscle training upon maximum lactate steady-state and blood lactate concentration. Eur J Appl Physiol 94(3):277–284
Ozyener F, Rossiter HB, Ward SA et al (2001) Influence of exercise intensity on the on- and off-transient kinetics of pulmonary oxygen uptake in humans. J Physiol 533(Pt 3):891–902
Ramirez-Sarmiento A, Orozco-Levi M, Guell R et al (2002) Inspiratory muscle training in patients with chronic obstructive pulmonary disease: structural adaptation and physiologic outcomes. Am J Respir Crit Care Med 166(11):1491–1497
Redline S, Gottfried SB, Altose MD (1991) Effects of changes in inspiratory muscle strength on the sensation of respiratory force. J Appl Physiol 70(1):240–245
Renggli AS, Verges S, Notter DA et al (2008) Development of respiratory muscle contractile fatigue in the course of hyperpnoea. Respir Physiol Neurobiol 164(3):366–372
Romer LM, McConnell AK, Jones DA (2002a) Effects of inspiratory muscle training on time-trial performance in trained cyclists. J Sports Sci 20(7):547–562
Romer LM, McConnell AK, Jones DA (2002b) Effects of inspiratory muscle training upon recovery time during high intensity, repetitive sprint activity. Int J Sports Med 23(5):353–360
Sale DG, MacDougall JD, Jacobs I et al (1990) Interaction between concurrent strength and endurance training. J Appl Physiol 68(1):260–270
Sinoway L, Shenberger J, Leaman G et al (1996) Forearm training attenuates sympathetic responses to prolonged rhythmic forearm exercise. J Appl Physiol 81(4):1778–1784
Spengler CM, Roos M, Laube SM et al (1999) Decreased exercise blood lactate concentrations after respiratory endurance training in humans. Eur J Appl Physiol Occup Physiol 79(4):299–305
Spengler CM, Knopfli-Lenzin C, Birchler K et al (2000) Breathing pattern and exercise endurance time after exhausting cycling or breathing. Eur J Appl Physiol 81(5):368–374
Staron RS, Karapondo DL, Kraemer WJ et al (1994) Skeletal muscle adaptations during early phase of heavy-resistance training in men and women. J Appl Physiol 76(3):1247–1255
Verges S, Lenherr O, Haner AC et al (2007) Increased fatigue resistance of respiratory muscles during exercise after respiratory muscle endurance training. Am J Physiol Regul Integr Comp Physiol 292(3):R1246–R1253
Verges S, Renggli AS, Notter DA et al (2009) Effects of different respiratory muscle training regimes on fatigue-related variables during volitional hyperpnoea. Respir Physiol Neurobiol 169(3):282–290
Volianitis S, McConnell AK, Koutedakis Y et al (2001) Inspiratory muscle training improves rowing performance. Med Sci Sports Exerc 33(5):803–809
Wetter TJ, Dempsey JA (2000) Pulmonary system and endurance exercise. In: Shephard RJ, Astrand PO (eds) Endurance in sport. Blackwell Science, London, pp 52–67
Wilson SH, Cooke NT, Edwards RH et al (1984) Predicted normal values for maximal respiratory pressures in caucasian adults and children. Thorax 39(7):535–538
Witt JD, Guenette JA, Rupert JL et al (2007) Inspiratory muscle training attenuates the human respiratory muscle metaboreflex. J Physiol 584(Pt 3):1019–1028
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Susan A. Ward.
Rights and permissions
About this article
Cite this article
Brown, P.I., Sharpe, G.R. & Johnson, M.A. Inspiratory muscle training abolishes the blood lactate increase associated with volitional hyperpnoea superimposed on exercise and accelerates lactate and oxygen uptake kinetics at the onset of exercise. Eur J Appl Physiol 112, 2117–2129 (2012). https://doi.org/10.1007/s00421-011-2185-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00421-011-2185-8