
Abstract
Ventilatory work during heavy endurance exercise has not been thought to influence systemic lactate concentration. We evaluated the effect of maximal isocapnic volitional hyperpnoea upon arterialised venous blood lactate concentration ([lac−]B) during leg cycling exercise at maximum lactate steady state (MLSS). Seven healthy males performed a lactate minimum test to estimate MLSS, which was then resolved using separate 30 min constant power tests (MLSS=207±8 W, mean ± SEM). Thereafter, a 30 min control trial at MLSS was performed. In a further experimental trial, the control trial was mimicked except that from 20 to 28 min maximal isocapnic volitional hyperpnoea was superimposed on exercise. Over 20–28 min minute ventilation, oxygen uptake, and heart rate during the control and experimental trials were 87.3±2.4 and 168.3±7.0 l min−1 (P<0.01), the latter being comparable to that achieved in the maximal phase of the lactate minimum test (171.9±6.8 l min−1), 3.46±0.20 and 3.83 ± 0.20 l min−1 (P<0.01), and 158.5±2.7 and 166.8±2.7 beats min−1 (P<0.05), respectively. From 20 to 30 min of the experimental trial [lac−]B increased from 3.7±0.2 to 4.7±0.3 mmol l−1 (P<0.05). The partial pressure of carbon dioxide in arterialised venous blood increased approximately 3 mmHg during volitional hyperpnoea, which may have attenuated the [lac−]B increase. These results show that, during heavy exercise, respiratory muscle work may affect [lac−]B. We speculate that the changes observed were related to the altered lactate turnover in respiratory muscles, locomotor muscles, or both.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Values of pulmonary ventilation \((\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E})\) up to 250 l min−1 have been recorded during exercise (Secher 1993), which equates to a respiratory muscle power of around 50 W (Milic-Emili 1991). In maximal exercise respiratory muscle work accounts for around 15% of both the total pulmonary oxygen uptake \((\ifmmode\expandafter\dot\else\expandafter\.\fi{V}{\rm O}_{2})\) and cardiac output (Aaron et al. 1992; Harms et al. 1998), causes diaphragmatic fatigue (Wetter and Dempsey 2000), and elicits a respiratory muscle metaboreflex (Harms et al. 1997) in which locomotor muscle perfusion is “stolen” by the respiratory muscles. Furthermore, respiratory muscle training in humans consistently reduces blood lactate concentration ([lac−]B) at identical exercise intensities (reviewed in McConnell and Romer 2004; McConnell and Sharpe 2005).
However, it remains unclear whether the work of breathing affects [lac−]B during endurance exercise. Animal studies suggest that the respiratory muscles engage in lactate consumption during heavy exercise (Fregosi and Dempsey 1986). However, inter-species differences in respiratory muscle fibre type ratios (Edwards and Faulkner 1985) and breathing mechanics during exercise (Bramble and Carrier 1983) make it difficult to extrapolate these observations to exercising humans. Studies on humans show either no change in [lac−]B, or only a modest 0.5 mmol l−1 increase when levels of \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}\) observed during maximal exercise are reproduced voluntarily at rest (Babcock et al. 1995; Martin et al. 1984), or when maximal voluntary isocapnic hyperpnoea was superimposed on exercise at the anaerobic threshold (Engelen et al. 1995). However, these studies were performed under conditions where there was capacity for tissues, e.g. active and resting skeletal muscle, the kidney, and the liver (Brooks 1986), to clear the lactate produced by other metabolically active tissues. Thus, respiratory muscle work may result in additional lactate efflux, but concurrent clearance by other tissues may result in either minimal, or no change in [lac−]B.
We reasoned that increasing the work of breathing whilst simultaneously exercising at maximum lactate steady state (MLSS) would resolve this uncertainty. MLSS represents the highest exercise intensity that can be maintained over time without a continual blood lactate accumulation (McConnell and Sharpe 2005). Thus, due to a limited capacity to match further lactate appearance with an equal rate of lactate clearance during exercise at MLSS, further lactate efflux caused by additional respiratory muscle work should be manifest by an increase in [lac−]B. Accordingly, the present study evaluated the influence of maximal voluntary hyperpnoea on [lac−]B whilst cycling at MLSS.
Methods
Following the local ethics committee approval and written informed consent, seven non-smoking, physically active male subjects were recruited. The age, height, and body mass of the subjects were (mean ± SEM) 25.6±1.8 years, 179.2±2.4 cm, and 80.3±2.9 kg, respectively. Subjects were instructed not to partake in strenuous exercise the day before test days, during which subjects refrained from consuming caffeine and any substance of nutritional value during the 2 h prior to testing. For each subject tests were performed at a similar time of day.
Experimental design
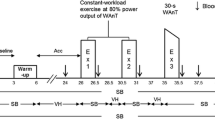
Initially, pulmonary and respiratory muscle function tests were performed. Exercise separated by at least 48 h was performed using an electromagnetically-braked cycle ergometer (Excalibur Sport, Lode, Groningen, The Netherlands). A “lactate minimum” test (Tegtbur et al. 1993) was performed to establish an estimate of the MLSS power, which was then resolved using at least two 30 min constant power tests. Thereafter, subjects performed a 30-min control trial at MLSS. During further experimental trials the control trial was mimicked except that from 20 to 28 min maximal isocapnic volitional hyperpnoea was superimposed. Subjects immobilised their arms during exercise by resting their forearms on time-trial handlebars. A 24 h diet record was completed prior to the control trial and this was then replicated in the 24 h prior to the experimental trial.
Pulmonary and respiratory muscle function
Pulmonary function was assessed using a pneumotachograph spirometer (Compact II, Vitalograph, Buckingham, UK) according to published guidelines (American Thoracic Society 1995). A hand-held mouth pressure meter (P.K. Morgan Ltd, Kent, UK) assessed maximal inspiratory and expiratory mouth pressures (MIP and MEP, respectively) as indices of global inspiratory and expiratory muscle strength, respectively. The mouthpiece assembly incorporated a 1 mm orifice to prevent glottic closure during inspiratory manoeuvres and to reduce the use of buccal muscles during expiratory manoeuvres. Inspiratory and expiratory manoeuvres were performed in an upright standing posture, were initiated from residual volume and total lung capacity, respectively, and were sustained for at least 1 s. Repeat measurements separated by 30–60 s were made until consistent values were produced, and the highest recorded value was used for subsequent analysis.
Blood sampling and analysis, and respiratory measurements
Arterialised venous blood was drawn from a superficial dorsal hand vein via an indwelling 21-G cannula. Arterialisation was ensured by immersing the hand in water at ∼40°C for 10 min prior to cannulation, and by warming the hand during exercise using an infrared lamp. An automated analyser (P-GM7 MicroStat, Analox Instruments, London, UK) was used to measure [lac−]B; prior to analysis blood was stored on ice in microtubes containing a heparin/fluoride/nitrite mixture (Analox Instruments). Blood–gas measurements, including the partial pressure of carbon dioxide (PCO2), pH, and oxygen saturation (SO2), were made immediately using an ABL 520 blood–gas analyser (Radiometer, Copenhagen, Denmark) after drawing a blood sample into a 2 ml pre-heparinised syringe (PICO 50, Radiometer). Blood–gas values were corrected for changes in the rectal temperature during exercise (1000 Series Squirrel, Grant Instruments, Cambridge, UK). Plasma bicarbonate concentration ([HCO −3 ]) was calculated from PCO2 and pH values using the Henderson–Hasselbalch equation:
[HCO −3 ] was subsequently incorporated into the Siggaard-Anderson equation to calculate base excess of the extracellular fluid (BEECF) (Siggaard-Andersen and Fogh-Andersen 1995):
In addition, during the period of volitional hyperpnoea (times 22–28 min, inclusive) metabolic and respiratory components of any acid–base derangements were separated by calculating the standard [HCO −3 ]. The mean PCO2 of minutes 20 and 30 during the experimental trial was taken as the control PCO2. The standard [HCO −3 ] was subsequently incorporated into the Henderson–Hasselbalch equation to partition changes in the pH due to metabolic and respiratory acidosis.
An on-line breath-by-breath respiratory system (Pulmolab EX670, Morgan Medical) measured the respiratory variables during exercise. Subjects wore a facemask (model 7940, Hans Rudolph, Kansas City, Missouri) connected to a turbine flowmeter and, during control and experimental trials, a two-way non-rebreathing valve (model 2730, Hans Rudolph).
Determination of MLSS power
The lactate minimum test protocol entailed three consecutive phases: (1) a maximal, incremental peak pulmonary oxygen uptake \((\ifmmode\expandafter\dot\else\expandafter\.\fi{V}{\rm O}_{2} \hbox{peak})\) test; (2) 8 min of cycling at 60 W to maximise [lac−]B; and (3) an incremental test consisting of five consecutive 4 min cycling stages at intensities of 45, 50, 55, 60, and 65% of the maximum power \((\ifmmode\expandafter\dot\else\expandafter\.\fi{W} \hbox{max})\) achieved during the maximal phase. During the maximal phase cycling power was increased every 15 s by a constant increment that was chosen for each subject in order to elicit exercise intolerance in approximately 10 min. The maximal phase was terminated when, despite verbal encouragement, cycling cadence could not be maintained above 60 rpm. The power at which exercise intolerance ensued defined \(\ifmmode\expandafter\dot\else\expandafter\.\fi{W}{\rm max},\) and the highest \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}{\rm O}_{2}\) and \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}\) recorded in any 30 s period defined the \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}{\rm O}_{2}\) peak and maximal exercise \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}\) \((\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}\hbox{max}).\) During the incremental test [lac−]B was determined during the final seconds of each stage, and the values were plotted against the corresponding power. The nadir (i.e. estimated MLSS power) of a second order polynomial curve fitted to the plot of [lac−]B against power was calculated through differentiation of the quadratic equation.
The MLSS power was subsequently resolved through each subject performing at least two constant power trials of 30 min duration (preceded by a 3 min warm-up at 50% of the prescribed power). [lac−]B was determined every 2 min from 16 to 30 min, inclusive, and MLSS was defined as the highest power at which a significant, positive gradient of a linear regression line fitted through the plot of [lac−]B against time was not observed. The gradient of the linear regression was evaluated relative to zero as:
where b is the gradient of the line, S x is the standard deviation of x (time), and S yx is the standard error of the estimate. The critical significance level for this analysis was set at α=0.10. Subsequent trials were performed at a power 2.5% above or below the initial power until MLSS was verified.
Control trial
Once the MLSS power was established a 30 min control trial at this intensity was performed. Exercise was preceded by a 3 min warm-up period at 50% MLSS power. [lac−]B was determined every 2 min during exercise. Blood–gas variables were determined every 4 min from 2 to 18 min, inclusive, and then every 2 min thereafter. Respiratory variables were averaged over the final minute of every 2 min interval. Heart rate (HR) was recorded continuously during exercise by using short-range telemetry (Polar Accurex Plus, Polar, Kempele, Finland).
Experimental trial
The experimental trial mimicked the control trial (including power and cadence). However, following 19.5 min of exercise a 1.5 m length of tubing was connected to the inspiratory line of the two-way valve. Inserted halfway along this tubing was a small-bore tube connected, via a CO2 rotameter, to a 100% CO2 certified gas cylinder (BOC Gases, Guilford, UK). Following 20 min of exercise subjects were instructed and verbally encouraged to volitionally elevate \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}\) to the highest sustainable level for a duration of 8 min. To maintain isocapnia during volitional hyperpnoea, end-tidal CO2 (ETCO2) was held constant by adjusting the flow of CO2 into the inspiratory circuit. Subjects were given real-time visual feedback of \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E},\) and were encouraged to maintain this at a maximal level. Subjects returned to spontaneous breathing for the remaining 2 min of exercise.
Statistical analyses
Statistical analyses were performed independently over two time periods: 12–20 min (steady state period) and 20–30 min (intervention period). A repeated measures factorial ANOVA was applied to establish differences between the control and experimental trials in the magnitude of change over time in all dependent variables. When a significant F ratio was detected, differences between trials at equal times were determined using paired t-tests or a one-way ANOVA for repeated measures, and differences over time within each trial were determined using a one-way ANOVA for repeated measures. Differences in the breathing pattern between that associated with \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}{\rm max}\) and that demonstrated at each measurement time during volitional hyperpnoea were established using a one-way ANOVA for repeated measures. One-way ANOVA analyses were followed by Tukey’s HSD post hoc analysis. Pearson’s product moment correlation coefficient (r) was calculated to determine the degree of association between selected variables. The critical significance level was set at α=0.05. Results are presented as means ± standard error of the mean (SEM) unless otherwise indicated. Statistical analyses were performed using the 11.0 release version of SPPS for Windows (SPSS Inc., Chicago, IL, USA).
Results
Baseline pulmonary and respiratory muscle function values are shown in Table 1. \(\ifmmode\expandafter\dot\else\expandafter\.\fi{W}{\rm max}\) and \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}{\rm O}_{2}\) peak achieved during the maximal phase of the lactate minimum test were 367±16 W and 3.87±0.16 l min−1, respectively. The MLSS power was 207±8 W (range=233–172).
Breathing pattern
During the steady state period (12–20 min) no significant differences in the breathing pattern were observed between trials (Fig. 1). However, volitional hyperpnoea resulted in \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}\) being higher in the experimental trial at all times from 22 to 28 min, inclusive. The increase in \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}\) during volitional hyperpnoea was primarily achieved via an increase in respiratory frequency (f R), which, compared to the control trial, was higher at all times from 22 to 28 min, inclusive (P<0.01). The pattern of change in tidal volume (V T) over 22–28 min was not different between trials. There was a trend for the duty cycle to be lower throughout volitional hyperpnoea, although an interaction effect was only observed at 26 min (P<0.05).

Ventilatory response to exercise at MLSS power in control (filled circle) and experimental (open circle) trials (mean ± SEM). \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{E}\) minute ventilation; V T tidal volume; f R respiratory frequency; T I /T tot inspiratory time/total breath time (duty cycle). *Interaction (P<0.05). **Interaction (P<0.01)
Breathing pattern: experimental trial vs. \( \ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{{\text{E}}} \max \)
The mean \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}\) during volitional hyperpnoea (169.6±7.1 l min−1) was not different (P=0.48) from \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}{\rm max}\) (171.9±6.8 l min−1). The V T at each measurement during volitional hyperpnoea was not different from the 3.29±0.27 l corresponding to \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}{\rm max }.\) However, following 4 min of volitional hyperpnoea f R was greater than that corresponding to \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}{\rm max }\) (69.0±7.3 vs. 53.6±5.2 breaths min−1) and differences existed up to 28 min (P<0.05). The duty cycle, inspiratory flow rate, and expiratory flow rate during volitional hyperpnoea were not different from the 0.49±0.01, 6.01±0.30, and 5.68±0.38 l s−1, respectively, corresponding to \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}{\rm max}.\)
Blood parameters
During control and experimental trials the rectal temperature increased similarly from 37.4±0.1 and 37.6±0.1°C at 2 min to 38.3±0.1 and 38.5±0.1°C at 30 min, respectively. Changes in SO2 during exercise were also not different between trials. From 10 to 30 min of exercise SO2 remained relatively constant and averaged 93.6±0.6 and 92.8±1.1% during control and experimental trials, respectively. Changes in all the remaining blood parameters during the steady state period were not different between or within trials (Fig. 2). All blood parameters also showed no change over time in the control trial from 20 to 30 min. During the control trial [lac−]B increased transiently during the first 10 min, after which it remained at a steady state of 3.7±0.1 mmol l−1. During the first 20 min of the experimental trial changes in [lac−]B were not different from that observed during the control trial. However, during volitional hyperpnoea and the last 2 min of exercise, [lac−]B increased by 0.20±0.05 mmol l−1 min−1. Following 6 min of volitional hyperpnoea [lac−]B had increased above the steady state (P<0.01), and at the end of the experimental trial [lac−]B was 4.7±0.3 mmol l−1, which represented a 24.9±5.5% increase from the steady state (P<0.01). Differences in [lac−]B were observed between trials at 26, 28, and 30 min (P<0.05).

Changes in blood lactate concentration ([lac−]B) and blood acid–base balance during exercise at MLSS power in control (filled circle) and experimental (open circle) trials (mean ± SEM). PCO 2 partial pressure of carbon dioxide in arterialised venous blood; [HCO − 3 ] plasma bicarbonate concentration; BE ECF base excess of the extracellular fluid; [H + ] hydrogen ion concentration. Open triangles denote standard [HCO −3 ] and changes in pH (and thus [H+]) according to the standard [HCO −3 ] and control PCO2. *Interaction (filled circle vs. open circle) (P<0.05). **Interaction (filled circle vs. open circle) (P<0.01). †Interaction (filled circle vs. open circle) (P<0.05)
A degree of hypercapnia was evident during volitional hyperpnoea, in that PCO2 increased from 43.4±1.5 mmHg at 20 min to 46.3±2.12 mmHg at 24 min (P<0.05). Thereafter, PCO2 remained relatively constant up to 28 min. PCO2 at 30 min (43.2±1.6 mmHg) during the experimental trial was not different from that observed at 20 min. Differences in PCO2 were observed between trials from 22 to 28 min, inclusive (P<0.05 and 0.01 for 22–24 and 26–28 min, respectively).
During the control trial [HCO −3 ] remained at a steady state of 21.7±0.6 mmol l−1 over 20–30 min. Conversely, a continual decline was observed during and following volitional hyperpnoea, such that [HCO −3 ] recorded at 30 min was lower than that observed at 20 min (20.9±0.6 vs. 22.3±0.5 mmol l−1, P<0.01). However, although there was an interaction effect (trial × time) for [HCO −3 ] over 20–30 min of exercise (P<0.05), paired t-tests did not reveal significant differences between trials because the data were disordinal (i.e. crossing over). There was, however, a greater decline in the standard [HCO −3 ] compared to [HCO −3 ] during volitional hyperpnoea. During the experimental trial standard [HCO −3 ] from 22 to 30 min, inclusive, was lower than that recorded at 20 min (P<0.01). Standard [HCO −3 ] was also lower than [HCO −3 ] recorded in both control and experimental trials at 24 and 28 min (P<0.05).
The ANOVA analysis for BEECF revealed an interaction effect (trial × time) over 20–30 min of exercise (P<0.05); however, because the data were disordinal, differences between trials at each sample time were not significant. From 20 to 30 min BEECF during the control trial was maintained at a steady state of −3.23±0.69 mEq l−1. In contrast, a continual decline in BEECF was observed during volitional hyperpnoea. Specifically, BEECF decreased from −2.89±0.43 mEq l−1 at 20 min to −3.89±0.51 mEq l−1 at 26 min (P<0.01), and further reductions were evident for the remainder of the exercise.
[H+] increased during volitional hyperpnoea from 46.4±0.8 nmol l−1 (pH=7.33±0.01) at 20 min to 51.5±1.4 nmol l−1 (pH=7.29±0.01) at 28 min (P<0.01). [H+] declined during the final 2 min of exercise as subjects returned to spontaneous breathing, although values were still elevated above that recorded at 20 min (P<0.01). Also shown on the [H+] and pH figures are changes in these variables when calculated using the standard [HCO −3 ] and control PCO2. The [H+] calculated from the standard [HCO −3 ] and control PCO2 (i.e. the metabolically derived [H+]) increased during the experimental trial from 46.3±0.1 nmol l−1 (pH=7.33±0.01) at 20 min to 49.2±1.3 nmol l−1 (pH=7.31±0.01) at 30 min (P<0.01). [H+] was greater than the metabolically derived [H+] at 24 (P<0.01) and 26–28 min (P<0.05), inclusive, and it was also greater than that recorded during the control trial at 22 (P<0.05), and 24–30 min (P<0.01), inclusive. The metabolically derived [H+] at 28 min was greater than that recorded during the control trial (P<0.05).
Pulmonary gas exchange and heart rate responses
Changes in ETCO2 throughout exercise were not different between trials (Fig. 3). Changes in \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}{\rm O}_{2}\) over the steady state period were also not different between trials. During the control trial, \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}{\rm O}_{2}\) increased up to 12 min, after which it remained at a steady state of 3.46±0.20 l min−1 for the remainder of the exercise. However, during volitional hyperpnoea there was a notable increase in \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}{\rm O}_{2}\) above the steady state. Differences in \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}{\rm O}_{2}\) were observed between trials at all times from 22 to 28 min, inclusive. Once elevated at 22 min, \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}{\rm O}_{2}\) remained unchanged for the remainder of volitional hyperpnoea. The mean \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}{\rm O}_{2}\) during volitional hyperpnoea was 3.83±0.20 l min−1; thus the oxygen cost of the additional hyperpnoea was 4.3±0.8 ml O2 per l min−1 of \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}.\) Compared to the control trial, HR was higher in the experimental trial at 20 min (162.1±2.1 vs. 157±2.8 beats min−1, P<0.05). The mean HR during volitional hyperpnoea was greater than that recorded over the same time period during the control trial (166.8±2.7 vs. 158.5±2.7 beats min−1, P<0.05). Additionally, compared to the control trial, HR was higher during volitional hyperpnoea at all times from 24 to 28 min (P<0.01), inclusive, and at 30 min (P<0.05).

Changes in pulmonary oxygen uptake \((\ifmmode\expandafter\dot\else\expandafter\.\fi{V}{\rm O}_{2}),\) end-tidal CO2 (ETCO2) and HR during exercise at MLSS power in control (filled circle) and experimental (open circle) trials (mean ± SEM). *Interaction (P<0.05). **Interaction (P<0.01)
Correlations between variables
The relative change in [lac−]B during volitional hyperpnoea was not correlated with either the relative or absolute increase in \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E},\) nor was it correlated with any index of pulmonary or respiratory muscle function, or endurance training status \((\ifmmode\expandafter\dot\else\expandafter\.\fi{V}{\rm O}_{2} \text{peak and } \ifmmode\expandafter\dot\else\expandafter\.\fi{W}\hbox{max}). \) There was also no correlation between the changes in [lac−]B and those in the parameters of acid–base balance. However, the relative increase in [lac−]B was correlated with both the relative change in duty cycle (r=0.95) and the fraction of MVV12 utilised (r=0.94) (P<0.01).
Discussion
We examined the effects of maximal voluntary hyperpnoea on the [lac−]B response to cycle ergometry exercise at MLSS. In agreement with our hypothesis, [lac−]B increased (+24.9%) during exercise at MLSS when \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}\) was elevated to a level comparable with that observed in maximal exercise. These findings suggest that an increase in respiratory muscle work is capable of influencing [lac−]B during heavy exercise.
Our findings are in contrast to reports that have shown minimal or no change in [lac−]B when maximal exercise \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}\) is reproduced voluntarily under resting conditions (Babcock et al. 1995; Martin et al. 1984). However, unlike at rest, at MLSS there is limited capacity to counter further lactate appearance because the system is operating at a point where the rates of lactate appearance and removal are in equilibrium. However, [lac−]B does not change when the work of breathing is increased (via an inspiratory resistance), or decreased (via proportional assist ventilation), during maximal exercise (Harms et al. 1997). There are two possible explanations for this: firstly, that the recruitment patterns of the respiratory muscles under conditions of external loading and voluntary hyperpnoea differ (McCool et al. 1992) and result in different lactate responses; and secondly, during maximal exercise any influence the respiratory muscles have upon [lac−]B may be overshadowed by the action of the much larger locomotor muscle mass.
It was beyond the scope of this study to identify the source(s) of the increased [lac−]B during volitional hyperpnoea. However, a potential mechanism for a change in lactate kinetics in the locomotor muscles may reside in the respiratory muscle metaboreflex that occurs during heavy endurance exercise under conditions of fatiguing inspiratory muscle work (Harms et al. 1997). The subsequent decrease in leg blood flow could either attenuate lactate uptake by locomotor muscles by reducing the extra-to-intramuscular lactate concentration gradient, and/or increase the lactate efflux if local oxygen delivery/extraction were compromised. However, inspiratory muscle loading during maximal exercise caused no change in the arterial and femoral venous [lac−]B, even in the presence of a reflex reduction in the locomotor muscle blood flow of 7.1% (Harms et al. 1997); this suggests that the locomotor muscle is probably not the source of the increased systemic lactate that we observed. Alternatively, vasoconstriction in other lactate consuming tissues, e.g. the liver (Nielsen et al. 2002), may have reduced lactate clearance during volitional hyperpnoea.
Thus, at least part of the increase in [lac−]B during volitional hyperpnoea originated from the respiratory muscles. Though the diaphragm, because of its small mass (about 0.5% of body mass), is unlikely to influence [lac−]B (Wetter and Dempsey 2000), maximal exercise hyperpnoea requires substantial activity by many other accessory muscles, all of which may contribute to the lactate turnover. The relative change in [lac−]B during volitional hyperpnoea was correlated with the ratio of \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}\) to MVV12. Using the greatest fraction of MVV12, and hence the respiratory muscle power, may result in greater metabolic perturbations (i.e. lactate production). The relative change in duty cycle during volitional hyperpnoea was also correlated with changes in [lac−]B. Raising duty cycle increases the fraction of each breath for which the perfusion vessels of the inspiratory muscles are occluded (Bellemare et al. 1983), which may influence lactate exchange between the inspiratory muscles and blood.
During exercise, there is a close agreement between measurements of [lac−]B, pH, and PCO2 taken from arterial and arterialised venous blood (Forster et al. 1972; McLoughlin et al. 1992). However, due to the delay in acquiring PCO2 measurements from the blood–gas analyser we attempted to maintain isocapnia by controlling ETCO2. Despite achieving this an approximate 6% increase, from the steady state, in PCO2 was observed and this may have affected [lac−]B. The greater decline in standard [HCO −3 ] compared to [HCO −3 ] during volitional hyperpnoea, along with the changes in pH calculated using the actual and standard [HCO −3 ] and control PCO2, is indicative of a mixed respiratory and metabolic acidosis. However, during exercise, an increase in PCO2 results in a reduction in [lac−]B (Ehrsam et al. 1982; Graham et al. 1986; Graham and Wilson 1983). Graham et al. (1982) showed that a 15% increase in PCO2 (achieved through hypercapnic breathing) during exercise at 65% \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}{\rm O}_{2}{\rm max}\) resulted in a 1 mmol l−1 reduction in [lac−]B. Thus, respiratory acidosis was likely to have led to a slight underestimate of the increase in [lac−]B.
Respiratory acidosis increases the cardiac output (Kiely et al. 1996) and skeletal muscle blood flow (Nielsen et al. 2001), which create potentially confounding influences upon [lac−]B. This stimulus to increase leg blood flow during volitional hyperpnoea may counter the potential effects of the respiratory muscle metaboreflex, or it may cause increased lactate efflux from locomotor muscles due to an increased muscle lactate gradient (Pilegaard et al. 1995). Respiratory acidosis also increases cerebral blood flow (Nielsen et al. 2001), which together with greater cerebral activity caused by volitional hyperpnoea may have promoted greater lactate uptake by the brain (Dalsgaard et al. 2004).
Our findings indicate that respiratory muscle work influences [lac−]B during heavy endurance exercise. This observation is consistent with responses to respiratory muscle training, in which [lac−]B during whole-body endurance exercise is attenuated (McConnell and Romer 2004). McConnell and Sharpe (2005) reported an approximate 1 mmol l−1 reduction in the MLSS [lac−]B following a 6 week pressure-threshold inspiratory muscle training regimen, although the corresponding cycling power remained unchanged. The implications of these findings are that the respiratory muscles may affect the [lac−]B of even steady state submaximal exercise in which \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E} \) may approximate only 50% of the maximal exercise \(\ifmmode\expandafter\dot\else\expandafter\.\fi{V}_{\rm E}.\)
References
Aaron EA, Seow KC, Johnson BD, Dempsey JA (1992) Oxygen cost of exercise hyperpnoea: implications for performance. J Appl Physiol 72:1818–1825
American Thoracic Society (1995) Standardization of spirometry, 1994 update. Am J Respir Crit Care Med 152:1107–1136
Babcock MA, Pegelow DF, McClaran SR, Suman OE, Dempsey JA (1995) Contribution of diaphragmatic power output to exercise-induced diaphragm fatigue. J Appl Physiol 78:1710–1719
Bellemare F, Wight D, Lavigne CM, Grassino A (1983) Effect of tension and timing of contraction on the blood flow of the diaphragm. J Appl Physiol 54:1597–1606
Bramble DM, Carrier DR (1983) Running and breathing in mammals. Science 219:251–256
Brooks GA (1986) The lactate shuttle during exercise and recovery. Med Sci Sports Exerc 18:360–368
Cotes JE (1993) Lung function: assessment and application in medicine, 5th edn. Blackwell, London
Dalsgaard MK, Volianitis S, Yoshiga CC, Dawson EA, Secher NH (2004) Cerebral metabolism during upper and lower body exercise. J Appl Physiol 97:1733–1739
Edwards RHT, Faulkner JA (1985) Structure and function of the respiratory muscles. In: Roussos C, Macklem PT (ed) The thorax. Marcel Dekker Inc., New York, pp 297–326
Ehrsam RE, Heigenhauser GJ, Jones NL (1982) Effect of respiratory acidosis on metabolism in exercise. J Appl Physiol 53:63–69
Engelen MP, Casaburi R, Rucker R, Carithers E (1995) Contribution of the respiratory muscles to the lactic acidosis of heavy exercise. Chest 108:1246–1251
Forster HV, Dempsey JA, Thomson J, Vidruk E, DoPico GA (1972) Estimation of arterial PO 2, PCO 2, pH, and lactate from arterialised venous blood. J Appl Physiol 32:134–137
Fregosi RF, Dempsey JA (1986) Effects of exercise in normoxia and acute hypoxia on respiratory muscle metabolites. J Appl Physiol 60:1274–1283
Graham TE, Wilson B (1983). Effects of hypercapnia and hyperoxia on metabolism during exercise. Med Sci Sports Exerc 15:514–519
Graham TE, Wilson BA, Sample M, Van Dijk J, Goslin B (1982) The effects of hypercapnia on the metabolic response to steady-state exercise. Med Sci Sports Exerc 14:286–291
Graham TE, Barclay JK, Wilson BA (1986) Skeletal muscle lactate release and glycolytic intermediates during hypercapnia. J Appl Physiol 60:568–575
Harms CA, Babcock MA, McClaran SR, Pegelow DF, Nickele GA, Nelson WB, Dempsey JA (1997) Respiratory muscle work compromises leg blood flow during maximal exercise. J Appl Physiol 82:1573–1583
Harms CA, Wetter TJ, McClaran SR, Pegelow DF, Nickele GA, Nelson WB, Hanson P, Dempsey JA (1998) Effects of respiratory muscle work on cardiac output and its distribution during maximal exercise. J Appl Physiol 85:609–618
Kiely DG, Cargill RI, Lipworth BJ (1996) Effects of hypercapnia on haemodynamic, inotropic, lusitropic, and electrophysiologic indices in humans. Chest 109:1215–1221
Martin BJ, Chen HI, Kolka MA (1984) Anaerobic metabolism of the respiratory muscles during exercise. Med Sci Sports Exerc 16:82–86
McConnell AK, Romer LM (2004) Respiratory muscle training in healthy humans: resolving the controversy. Int J Sports Med 25:284–293
McConnell AK, Sharpe GR (2005) The effect of inspiratory muscle training upon maximum lactate steady-state and blood lactate concentration. Eur J Appl Physiol 94:277–284
McCool FD, Hershenson MB, Tzelepis GE, Kikuchi Y, Leith DE (1992) Effect of fatigue on maximal inspiratory pressure-flow capacity. J Appl Physiol 73:36–43
McLoughlin P, Popham P, Linton RA, Bruce RC, Band DM (1992) Use of arterialised venous blood sampling during incremental exercise tests. J Appl Pysiol 73:937–940
Milic-Emili J (1991) Work of breathing. In: Crystal RG, West JB (ed) The lung: scientific foundations. Raven Press, New York, pp 1065–1075
Nielsen HB, Boesen M, Secher NH (2001) Near-infrared spectroscopy determined brain and muscle oxygenation during exercise with normal and resistive breathing. Acta Physiol Scand 171:63–70
Nielsen HB, Clemmesen JO, Skak C, Ott P, Secher NH (2002) Attenuated hepatosplanchnic uptake of lactate during intense exercise in humans. J Appl Physiol 92:1677–1683
Pilegaard H, Bangsbo J, Henningsen P, Juel C, Richter EA (1995) Effect of blood flow on muscle lactate release studied in perfused rat hindlimb. Am J Physiol 269:1044–1051
Secher NH (1993) Physiological and biomechanical aspects of rowing. Sports Med 15:24–42
Siggaard-Andersen O, Fogh-Andersen N (1995) Base excess or buffer base (strong ion difference) as measure of a non-respiratory acid-base disturbance. Acta Anaesthesiol Scand 107:267–271
Tegtbur U, Busse MW, Braumann KM (1993) Estimation of an individual equilibrium between lactate production and catabolism during exercise. Med Sci Sports Exerc 25:620–627
Wetter TJ, Dempsey JA (2000) Pulmonary system and endurance exercise. In: Shephard RJ, Åstrand P-O (eds) Endurance in sport. Blackwell science, London, pp 52–67
Wilson SH, Cooke NT, Edwards RHT, Spiro SG (1984) Predicted normal values for maximal respiratory pressures in Caucasian adults and children. Thorax 39:535–538
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Johnson, M.A., Sharpe, G.R. & McConnell, A.K. Maximal voluntary hyperpnoea increases blood lactate concentration during exercise. Eur J Appl Physiol 96, 600–608 (2006). https://doi.org/10.1007/s00421-005-0098-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00421-005-0098-0