
Abstract
The Gram-positive soil bacterium Corynebacterium glutamicum is a leading workhorse for the biotechnology industry. Worldwide, engineered strains of the microbe synthetize approximately 6 million tons of the amino acids l-glutamate and l-lysine. In addition to these well-known traditional fermentation products, the portfolio of C. glutamicum has impressively expanded over the past years and meanwhile comprises more than 70 natural and non-natural chemicals, materials, fuels, and health-care products. A major challenge to create superior strains for industrial production has always been and still is the design part, i.e. the identification of the most appropriate targets for genomic optimization into a desired phenotype. Metabolic flux analysis is about deciphering the activity of biochemical reactions and pathways of central carbon metabolism. It probably provides the most understandable insight into the core machinery of a living cell. This core part of metabolism displays the controlling centrepiece for synthetizing all the different molecules of recognized industrial value. One can therefore easily understand that metabolic flux analysis had a huge impact on the industrial career of C. glutamicum since its pioneering days in the 1950s, when the microbe was discovered. Initial approaches used simple isotopic tracer studies and mathematical balancing of fermentation data to assess metabolic fluxes in C. glutamicum for the first time. These techniques have been systematically upgraded into comprehensive technologies and enabled a number of seminal studies of fluxes in C. glutamicum, which provided fascinating and unexpected insights into its lifestyle and laid the foundation for metabolically engineered strains, used today in industry. Today, C. glutamicum probably displays the best studied microorganism on the level of metabolic fluxes. New developments promise an even brighter future.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Systems biology
- Metabolic network
- Metabolic engineering
- Industrial strain development
- Metabolite balancing
- Metabolic flux
1 Introduction
Corynebacterium glutamicum, discovered about 70 years ago, is a leading cell factory of the bio-industry. Traditionally, it has been employed as a producer of amino acids. The genetic alteration of metabolic control in the microbe generated multiple candidates for the over-production of a remarkable amino acid spectrum (Ikeda 2003; Becker and Wittmann 2012, 2015; Wendisch 2017). Today, the two major industrial products obtained by C. glutamicum are l-glutamate and l-lysine with an annual global market of approximately 9–10 billion dollar (Becker et al. 2018b; Eggeling and Bott 2015). In addition, strains of C. glutamicum were genetically modified to produce a variety of organic acids (Rohles et al. 2018; Inui et al. 2004b; Becker et al. 2015, 2018a; Tsuge et al. 2015; Okai et al. 2016; Wieschalka et al. 2012), diamines (Kind et al. 2010; Kind and Wittmann 2011; Kind et al. 2011; Becker and Wittmann 2017a; Schneider and Wendisch 2010, 2011), alcohols,(Inui et al. 2004a; Jojima et al. 2015; Smith et al. 2010; Blombach et al. 2011; Yamamoto et al. 2013) and amino-acid related compounds (Becker et al. 2013; Perez-Garcia et al. 2017; Rohles et al. 2016; Sun et al. 2016). The huge industrial relevance has stimulated research for more efficient production strains from early on. Initially, strains were developed classically, using random mutagenesis and selection. With the advent of recombinant DNA technology, strain engineering could be upgraded into a rational approach with iterative optimization of desired properties such as yield, productivity, stress tolerance, and substrate spectrum. However, the obvious complexity of the metabolism of C. glutamicum complicated the design of cell factories for industrial fermentation. It became clear that optimization would strongly benefit from a better knowledge about C. glutamicum, enabling a more straightforward prediction and evaluation of genetic targets (Becker and Wittmann 2015; Ma et al. 2017; Becker et al. 2016; Chae et al. 2017). In this regard, metabolic flux analysis, i.e. fluxomics, appeared perfect to investigate the pathways of C. glutamicum in vivo. Over the years, fluxomics has evolved into the most valuable guide in metabolic engineering among all omics approaches (Wittmann 2010; Becker and Wittmann 2018; Kohlstedt et al. 2010). This chapter presents the major concepts and tools of metabolic flux analysis and highlights prominent application examples for systems biology and metabolic engineering of C. glutamicum. The type strain C. glutamicum ATCC 13032 and thereof derived mutants are in the focus of most flux studies. In addition, there are some reports on fluxes in close relatives including Brevibacterium lactofermentum and Microbacterium ammoniaphilum.
2 Metabolic Network Analysis of C. glutamicum
The complex structure of biological networks typically impedes rational approaches for targeted optimization of cell factories for industrial application. Strategies, which appear promising on a first glance, might quickly be paralyzed by the redundancy of biochemical reactions, an undesired distribution of flux, or the sudden activation of alternative pathways upon genetic perturbation. A profound knowledge of this complex network structure is hence an essential basis for systems metabolic engineering.
2.1 Metabolic Network Reconstruction
Generally, the carbon core metabolism comprises a complex network of approximately 100–200 biochemical reactions, which are closely interconnected. The analysis of the fluxes through these reactions is primarily based on a model of the underlying reaction network. Fortunately, the extensive biochemical and physiological characterization of C. glutamicum during the past decades provides a rich source of information on many of the enzymes and pathways present in this organism (Becker and Wittmann 2017b; Yokota and Lindley 2005; Eikmanns 2005). Approximately 15 years ago, this knowledge was complemented and updated with information from genome sequencing and annotation (Kalinowski et al. 2003; Haberhauer et al. 2001). However, one should be aware that central metabolism is still not totally understood. We continuously discover novel features in practically all studied microbes, which have substantial impact for flux analysis, i.e. regarding enzyme specificity (Nikel et al. 2015) or even the existence of complete metabolic routes (Lange et al. 2017).
The core part of metabolism in C. glutamicum seems well understood although. The microbe possesses the pentose phosphate (PP) pathway, the Emden-Meyerhof-Parnas (EMP) pathway, the tricarboxylic acid (TCA) cycle, the glyoxylic acid shunt, and a rich set of enzymes, which interconnect C3-metabolites of the EMP pathway with C4-metabolites of the TCA cycle (Fig. 1) (Wittmann and Becker 2007; Wittmann and De Graaf 2005; Becker et al. 2016). Metabolic intermediates are withdrawn from these central metabolic pathways to serve as building blocks for biomass formation, which results in continuous anabolic fluxes in growing cells. C. glutamicum requires glucose 6-phosphate, fructose 6-phosphate, ribose 5-phosphate, erythrose 4-phosphate, glyceraldehyde 3-phosphate, pyruvate, phosphoenolpyruvate, acetyl-CoA, oxaloacetate, and 2-oxoglutarate for biomass formation (Wittmann and De Graaf 2005). Due to the structure of the cell wall, diaminopimelate, a direct precursor of l-lysine, is additionally consumed (Wehrmann et al. 1998). Beside carbon precursors, NADPH is required as reducing power for biosynthesis. Overall, 16,400 μmol NADPH is consumed to form 1 g biomass. This amount has to be generated by the NADPH supplying enzymes, glucose 6-phosphate dehydrogenase (Ihnen and Demain 1969; Moritz et al. 2000), 6-phosphogluconate dehydrogenase (Ohnishi et al. 2005; Moritz et al. 2000), isocitrate dehydrogenase (Chen and Yang 2000) and malic enzyme (Dominguez et al. 1998; Becker et al. 2008) (Fig. 1). For most flux studies, knowledge on the cellular composition of C. glutamicum and the anabolic demand for the precursors is therefore important. Corresponding data have been collected in different studies for C. glutamicum MH20-22B in continuous culture using glucose (Marx et al. 1996), for exponentially growing C. glutamicum ATCC17965 using glucose (Cocaign-Bousquet et al. 1996), and for exponentially growing C. glutamicum BS87 (Kind et al. 2013).

Biochemical network of the central metabolism of Corynebacterium glutamicum
In addition, reactions and pathways to secreted products have to be considered. Most important, when studying over-producers, is the biosynthetic pathway for the target product. As example, the biosynthetic pathways of l-lysine and l-glutamate start from oxaloacetate and 2-oxoglutarate, respectively, two intermediates of the carbon core network, and additionally demand for NADPH (4 NADPH per l-lysine and 1 NADPH per l-glutamate). Furthermore, C. glutamicum might secrete minor shares of by-products, such as trehalose, acetate, lactate, succinate, pyruvate, l-alanine, l-glutamate, and l-glycine (Krömer et al. 2004; Becker et al. 2005; Kiefer et al. 2002; Blombach et al. 2007). The observed by-product spectrum determines, which of the corresponding pathways should be taken into account.
For flux analysis, the set of biochemical reactions and pathways has to be arranged into a fully consistent network model. In such a model, the metabolites are stoichiometrically connected by exactly defined enzymatic reactions, which altogether convert substrates into intermediates, and ultimately into products (Vallino and Stephanopoulos 1993; Schilling et al. 1999; Kjeldsen and Nielsen 2009; Shinfuku et al. 2009; Melzer et al. 2009; Zhang et al. 2019; Mei et al. 2016). When designing such networks models, it is important not to base them exclusively on genomic information and stoichiometry, but also take thermodynamic considerations into account, which impact direction and reversibility of flux and constrain feasible flux states (Palsson 2000; Wang et al. 2017). Such constraints are easily implemented into the biochemical model, if the thermodynamic feasibility of the corresponding biochemical reaction is known. This allows, for instance, defining reversible and irreversible reactions in the network. Finally, such a functional biochemical network is the basic prerequisite for the determination of metabolic fluxes.
2.2 Stoichiometric Flux Analysis
Stoichiometric balancing, also called metabolite balancing, is a model-based approach for flux analysis. It is based on a metabolic network of the relevant reactions and applicable to cells under metabolic steady-state, e.g. growing in chemostat, balanced batch cultures, and balanced phases of fed-batch processes (Vallino and Stephanopoulos 1993; Dersch et al. 2016b). In short, a subset of the fluxes of interest is directly measured, e.g. fluxes linked to substrate uptake, biomass formation, and product synthesis (Wittmann and De Graaf 2005; von Kamp et al. 2017). Subsequently, the remaining intracellular fluxes are calculated using the stoichiometric relations of the model. The calculation is straightforward, using linear algebra. Pioneering studies compared metabolic fluxes in C. glutamicum during growth and l-lysine production (Vallino and Stephanopoulos 1993, 1994), as well as under different environmental conditions such as osmotic stress (Varela et al. 2003), and oxygen deprivation (Dominguez et al. 1993). Due to the nature of carbon core metabolism, stoichiometric flux models derived therefrom are usually underdetermined, which is also true for C. glutamicum. Consequently, additional constraints are required to achieve a unique solution of the intracellular flux distribution. These constraints can be provided by balances for ATP, NADH or NADPH, which, however, often are subjected to uncertainties and can even introduce artefacts into the flux estimation (Wittmann and De Graaf 2005; Winter and Krömer 2013; Maarleveld et al. 2013). Alternatively, the underlying network can be simplified, e.g. assuming certain pathways to be inactive (Aiba and Matsuoka 1979). Metabolic fluxes derived from stoichiometric balancing are strongly influenced by the network topology and the chosen assumptions rather than by biological data (Sauer 2006; El Massaoudi et al. 2003), so that care should be taken regarding their interpretation. In addition, important fine structures of the network of C. glutamicum cannot be discriminated at all, using stoichiometric balancing. As example, the two branches of l-lysine biosynthesis both exhibit the same stoichiometry (Wittmann and Becker 2007; Sonntag et al. 1993). The individual fluxes through the parallel routes are therefore not resolvable by this approach (Wittmann and De Graaf 2005). Similarly, the glyoxylate shunt, the bidirectional fluxes around the pyruvate node, and flux reversibility of individual enzymes remain inaccessible (Vallino and Stephanopoulos 1993; Wittmann and De Graaf 2005). Nevertheless, stoichiometric flux analysis was an important approach, particularly in the early days of C. glutamicum research and contributed strongly to our current understanding of the microbe. Moreover, increased knowledge on energy and redox metabolism (Sonderegger et al. 2004) and the consideration of transcriptomics regarding the set of expressed pathways under the studied conditions (Beckers et al. 2016) help to refine network structures and increase model quality and thereby flux accuracy.
3 State-of-Art 13C Metabolic Flux Analysis
The first flux studies of C. glutamicum explored the microbe using isotopic tracers. These tracer substrates are enriched with 13C at specific carbon atom positions (Wittmann 2007; Becker and Wittmann 2014). When fed to the studied cells, they are taken up and metabolized, whereby the 13C label is distributed throughout the network. The carbon transition of each enzymatic reaction thereby specifically defined the fate of the 13C label (Fig. 2). Depending on the tracer chosen, pathway specific labelling signatures are formed, which can be exploited to infer intracellular fluxes. Mass spectrometry and NMR emerged as most prominent techniques to measure 13C data from such isotopic experiments and provide information for flux calculation (Wittmann 2002, 2007; Kiefer et al. 2007; Massou et al. 2007). Pioneering isotope studies generated important knowledge on metabolic properties of C. glutamicum, such as l-histidine biosynthesis (Ishino et al. 1984), and first estimates of the fluxes through the TCA cycle or the glyoxylate shunt (Walker et al. 1982). In addition, they have been successfully used for the identification of unknown metabolites and side-products, resulting from genetic alteration of the metabolism of C. glutamicum (Krömer et al. 2006; Kind et al. 2010). Moreover, isotope experiments provided valuable insights into sulphur (Bolten et al. 2010) and nitrogen metabolism of C. glutamicum (Tesch et al. 1999), allowed to trace the carbon fate from substrates into products (Lessmeier et al. 2015; Bolten et al. 2010; Ishino et al. 1986), provided evidence for the re-uptake of secreted products (Zhao et al. 2012; Rohles et al. 2018), and led to the discovery of novel metabolic pathways (Krömer et al. 2006). Today, selected labelling data are also found useful to obtain model-independent flux estimates (Becker et al. 2010; Klingner et al. 2015; Fürch et al. 2009) and to assess net fluxes in the metabolism of C. glutamicum (Sato et al. 2008; Hoffmann et al. 2018).

In particular, isotopic labelling data are highly valuable to reduce the number of assumptions needed in pure stoichiometric balancing and to obtain fully determined and even overdetermined networks (Sauer 2006; Wittmann and De Graaf 2005). Over the years, this integrated concept has been developed further and further. Today, state-of-art metabolic flux analysis is a comprehensive workflow, which couples sophisticated isotopic experiments with high-resolution 13C labelling analysis and software embedded modelling frameworks for flux computation (Guo et al. 2018; Wittmann 2007; Heux et al. 2017). GC-MS has evolved into the most prominent technique for the 13C measurement due to its high accuracy, sensitivity, speed, and robustness (Wittmann 2002, 2007; Guo et al. 2018). In addition, also NMR has been used for this purpose (de Graaf et al. 2000; Boisseau et al. 2013; Nicolas et al. 2007).
For flux calculation, data on extracellular fluxes (substrate uptake, product secretion), anabolic fluxes, and 13C labelling patterns are used to globally fit the unknown flux parameters by a computer flux model (Kiefer et al. 2004; Krömer et al. 2004; Wittmann and Heinzle 2002). Hereby, the free flux parameters of interest are varied by an optimization algorithm, until the deviation between experimental and simulated data is minimized (Wittmann 2007). Sophisticated modelling frameworks such as matrix based isotopomer mapping (Wittmann and Heinzle 1999, 2001b; Schmidt et al. 1999), cumomer balancing using the 13C-FLUX software (Wiechert et al. 2001) and the open source package OpenFLUX (Quek et al. 2009) and OpenFLUX2 (Shupletsov et al. 2014) provide convenient software solutions for flux calculation. The platforms are steadily updated and extended to allow a more user-friendly programming and visualization (Shupletsov et al. 2014; Desai and Srivastava 2018; Weitzel et al. 2013; Nöh et al. 2015; He et al. 2016). In addition, they support experimental design of tracer experiments and statistical flux evaluation (Quek et al. 2009; Millard et al. 2014; Nöh et al. 2018).
3.1 Experimental Design
The appropriate combination of tracer substrate and labelling analytes is a critical factor to obtain high quality labelling information for flux estimation (Wittmann and Heinzle 2001b; Yang et al. 2005; Nöh et al. 2018). The selection is case specific and depends on the flux parameters of interest and the studied network. Experimental design has been successfully used to evaluate the suitability of specific tracers (Wittmann and Heinzle 2001b; Yang et al. 2005; Nöh et al. 2018; Crown et al. 2016; Kohlstedt and Wittmann 2019; Millard et al. 2014) and the information content of specific mass isotopomers (Becker et al. 2008; Kohlstedt and Wittmann 2019) for a number of flux problems. Using computer simulations, suitable approaches can be identified also for C. glutamicum, as illustrated for different GC-MS fragment ions and fluxes around the complex network interconnecting glycolysis and TCA cycle (Fig. 3).

Computer-based experimental design for quantification of flux parameters at the pyruvate node of Corynebacterium glutamicum: mass isotopomer distribution of aspartate (m/z 418) with varied flux partitioning between phosphoenolpyruvate carboxylase and pyruvate carboxylase (ΦPEPC), mass isotopomer distribution of valine (m/z 288) with varied exchange flux between phosphoenolpyruvate and oxaloacetate catalyzed by phosphoenolpyruvate carboxylase and phosphoenol-pyruvate carboxykinase (ζPEPC/PEPCK), mass isotopomer distribution of alanine (m/z 260) with varied exchange flux between pyruvate and oxaloacetate catalyzed by pyruvate carboxylase and malic enzyme (ζPC/MalE). The assumed input substrate is an equimolar mixture of [13C6] glucose and [12C6] glucose. The figure is taken from Becker et al. (2008)
In principle, tracer experiments can be carried out on a single carbon source or a substrate mixture, using tracer substrates that are labelled at one or more positions (Becker et al. 2008; Wittmann et al. 2004a; Kiefer et al. 2004; Rados et al. 2014). The optimal labelling degree, position and/or substrate mixture of the tracer substrate can again be identified by a computer-based experimental design. Intensive computational simulations showed that specific tracers are optimal for flux analysis in C. glutamicum and other microbes (Wittmann and Heinzle 2001b; Wiechert and de Graaf 1997; Möllney et al. 1999; Kohlstedt and Wittmann 2019). Flux analysis with [1-13C] glucose, for instance, provides labelling data that sensitively depend on the flux partitioning between the EMP and the PP pathway. The sensitivity allows a precise estimation of the corresponding in vivo fluxes (Wittmann 2007; Wittmann and Heinzle 2001b). On the level of individual carbon atoms, the PP pathway enzyme 6-phosphogluconate dehydrogenase exclusively releases the 13C-labelled C1 atom in the decarboxylation reaction, whereas this atom is retained in the carbon backbone of the pathway intermediates, when glucose is metabolized via the EMP pathway (Fig. 4a). Variation of the flux partitioning ratio thus strongly influences the 13C enrichment in downstream metabolites (Wittmann and De Graaf 2005). At the same time, the degree of reversibility of phosphoglucoisomerase at the entry into the EMP pathway affects the labelling. The measurement of different analytes provides sensitive data to assess both flux parameters in parallel (Wittmann and Heinzle 2001b). Complementary to that, resolution of the exchange fluxes between the TCA cycle and the EMP pathway is optimally based on a mixture of naturally labelled and [13C6] glucose (Wittmann and Heinzle 2001b; Wittmann 2007), as C-C bond assembly and cleavage results in new labelling combinations of TCA cycle metabolites and EMP pathway metabolites that are specifically related to these reactions. Most flux studies of C. glutamicum hence used two parallel tracer experiments on [1-13C] glucose and a mixture of naturally labelled and [13C6] glucose (Becker et al. 2008; Wittmann and Heinzle 2002; Kiefer et al. 2004) or a combination of [1-13C] and [13C6] glucose in one single experiment (Iwatani et al. 2007). Hereby, parallel studies on different tracers result in a higher accuracy and precision of the metabolic flux distribution (Kiefer et al. 2004; Wittmann and Heinzle 2002; Petersen et al. 2000; Krömer et al. 2004; Kohlstedt and Wittmann 2019; Rados et al. 2014).

(a) Strategy for determination of the flux partitioning ratio between glycolysis and pentose phosphate pathway using an isotope study with [1-13C] glucose. (b) Strategy for determination of the flux partitioning between dehydrogenase branch and succinylase branch of the l-lysine biosynthetic pathway of C. glutamicum
In addition, also other tracers have been used to address more specific questions. As stated above, C. glutamicum operates two parallel l-lysine biosynthetic routes. The relative contribution of each branch to the overall l-lysine flux has been determined by tracer experiments on [6-13C] glucose and positional labelling analysis of l-lysine using NMR (Sonntag et al. 1993; Nicolas et al. 2008). Alternatively, the relevant labelling information to determine the l-lysine flux split was obtained in C. glutamicum, using GC-MS analysis of l-alanine, l-lysine and diaminopimelate from tracer studies on [4-13C] glucose or [3-13C] glucose (Fig. 4b) (Becker et al. 2013; Wittmann and Heinzle 2001b). In another prominent example, metabolic flux analysis of sucrose-grown C. glutamicum used three parallel tracer studies on [1-13C glucose] sucrose, [1-13C fructose] sucrose and [13C fructose] sucrose (Wittmann et al. 2004a).
3.2 Labelling Analysis for Flux Estimation
Analysis of 13C labelling patterns is mainly performed by GC-MS, which allows a quantification of the mass isotopomer distribution in a variety of different metabolites (Villas-Boas et al. 2005; Wittmann 2007; Roessner et al. 2000; Chen et al. 1998). Metabolites, accessible by GC-MS for flux analysis are for example amino acids (Wittmann et al. 2002; Wittmann and Heinzle 1999; Christensen et al. 2002; Christensen and Nielsen 1999), organic acids (Adler et al. 2013; Lange et al. 2017), sugars, and sugar derivatives (Kohlstedt and Wittmann 2019; Kiefer et al. 2004). Their analysis requires derivatization, GC separation, electron impact (EI) or chemical ionization (CI), mass separation, and detection (Wittmann 2007; Becker and Wittmann 2014). Due to the high energy input in the ionization process, the analytes are usually fragmented into specific ions. In turn, the formed fragments can be used for identification and labelling analysis (Christensen and Nielsen 1999; Wittmann 2007; Wittmann et al. 2002; Becker and Wittmann 2014).
Proteinogenic amino acids are the most common analytes used to obtain the necessary labelling information for steady-state flux analysis in growing cells (Christensen and Nielsen 1999; Dauner and Sauer 2000). A number of flux studies for C. glutamicum were based on these data (Wittmann and Heinzle 2002; Wittmann et al. 2004b; Krömer et al. 2004; Becker et al. 2008; Buschke et al. 2013; Kind et al. 2013). Hereby, the well-known precursor-amino acid relationship allowed to deduce the 13C-labelling patterns of the respective carbon precursors in intermediary metabolism from that of the amino acids (Fig. 2) (Szyperski 1998; Wittmann 2007). The additional consideration of sugars and sugar derivates in the labelling analysis can enhance flux identifiability, resolution, and accuracy (Kohlstedt and Wittmann 2019; Adler et al. 2013; Lange et al. 2017; Becker et al. 2007). In principle, amino acids can be derivatised by silylation using reagents such as MBDSTFA (N-methyl-N-tert-butyldimethylsilyl-trifluoroacetamide) and MSTFA (N-methyl-N-(trimethylsilyl)-trifluoro acetamid), or alkylation/acylation using MeOH/TMCS (Methanol/Trimethylchlorosilane) and TMSH (Trimethylsulfonium hydroxide) for GC-MS analysis (Wittmann 2007). However, the labelling pattern of amino acids is preferentially quantified from their t-butyl-dimethyl-silyl (TBDMS) derivatives, which yield high quality signals for fragment ions that contain the entire carbon skeleton of the analytes and are therefore particularly useful for metabolic flux analysis. For sugars, the derivatisation typically comprises a combination of oximation and silylation using hydroxylamine and trifluoroacetamides (BSTFA, MSTFA) (Kiefer et al. 2002; Bolten et al. 2009; Wittmann et al. 2007; Wittmann 2007). In addition to GC-MS, other mass spectrometry techniques for 13C labelling analysis comprise LC-MS (Kiefer et al. 2007; Oldiges et al. 2004; van Winden et al. 2005; Noack et al. 2011), CE-MS (Iwatani et al. 2007), GC-C-IRMS (Yuan et al. 2010; Dersch et al. 2016a) and MALDI-TOF MS (Wittmann and Heinzle 2001a). MS techniques provide mass isotopomer data of an investigated molecule. Orthogonal information on the positional labelling of specific carbon atoms within a molecule is obtained, using NMR and 2D-NMR, although at the cost of increased effort and reduced sensitivity (de Graaf et al. 2000; Nicolas et al. 2008; Marx et al. 1996). To some extent, positional labelling information can also be deduced from LC-MS and GC-MS data (Kappelmann et al. 2017; Becker et al. 2013). It depends on the studied metabolic flux problem, which technique and/or combinations thereof should be selected—in terms of costs and effort on one hand and flux resolution and identifiability on the other hand (Millard et al. 2017; Schwechheimer et al. 2018a, b; Sonntag et al. 1993; Rühl et al. 2012).
3.3 Technical Implementation
A spectrum of flux technologies has been developed to enable an application-oriented research of this organism, such as mutant screening at miniaturized scale, routine strain characterization and testing, bioprocess development at laboratory scale, and industrial production at large-scale (Fig. 5).


Current spectrum of 13C metabolic flux technologies and common application field in systems biology and systems metabolic engineering
3.3.1 Routine Flux Analysis of Balanced Cultures at Lab Scale
After years of intense research and development, GC-MS-based 13C metabolic flux analysis has meanwhile evolved into a routine method for determining steady-state fluxes in microbes (Sauer 2006). The approach allows high-resolution of fluxes at high precision and accuracy. The tracer studies are easily performed in shake flasks with culture volumes of 5–25 mL, but can, of course also be conducted in lab scale bioreactors. This technique is usually applied to exponentially growing cells, since these cells are in metabolic and isotopic steady-state (Becker et al. 2008; Winter and Krömer 2013), so that the amino acid labelling from the cell protein provides the necessary information for flux determination. During growth, labelled amino acids are incorporated into biomass, so that their labelling pattern is conserved in the cell protein; they are subsequently easily accessible by simple hydrolysis of the harvested biomass (Kelleher 2001). Moreover, organic acids and sugar derivatives can be analysed to provide complementary labelling data (Kohlstedt and Wittmann 2019; Wittmann and Heinzle 2002; Lange et al. 2017). It should be noticed that the verification of steady-state is an important prerequisite for appropriate analysis. Whereas isotopic steady-state can be validated by constant 13C labelling patterns over time, metabolic steady-state can be inferred from constant kinetics and stoichiometry during the studies process (Rohles et al. 2016; Becker et al. 2008; Bücker et al. 2014).
This type of steady-state stationary flux analysis has proven highly value in guiding systems metabolic engineering of C. glutamicum (Wittmann 2007; Wittmann and Heinzle 2002; Wittmann et al. 2004a; Becker and Wittmann 2018; Becker et al. 2011). Moreover, it provided unique insights into the metabolism of the microbe as response to genetic perturbation (Bartek et al. 2011; Becker et al. 2008; Marx et al. 1997; Kim et al. 2006; Marx et al. 1999), amino acid production (Sonntag et al. 1995; Krömer et al. 2004; Shirai et al. 2006), and the use of different carbon sources, such as sucrose, fructose, xylose, mannitol, acetate and propionate (Wittmann et al. 2004a; Kiefer et al. 2004; Buschke et al. 2013; Hoffmann et al. 2018; Veit et al. 2009; Wendisch et al. 2000; Dominguez et al. 1998).
3.3.2 Miniaturized Flux Analysis for Screening Purposes
Genetic modification of microbes during strain optimization often creates a large number of mutants, which display highly interesting libraries to be screened on the flux level. To this end, approaches were developed for C. glutamicum (Wittmann et al. 2004b) and other microbes (Klingner et al. 2015; Berger et al. 2014; Heux et al. 2014), comprising tracer cultivation at a miniaturized scale for high-throughput (Fischer et al. 2004; Wittmann et al. 2004b; Heux et al. 2017). The established multi-well microtiter plate format allows reproducible, well-controlled cultivation in a volume of approximately 100–1000 μL with online monitoring of dissolved oxygen, offline quantification of substrates, products, and biomass, together with labelling analysis of secreted products, or optionally from hydrolysed biomass, using GC-MS (Wittmann et al. 2004b). In addition to the high number of parallel experiments, the miniaturized scale benefits from a cost advantage since it requires much lower amounts of the costly 13C-labelled substrates. Using such miniaturized approaches, the fluxes through the central metabolic pathways of C. glutamicum can be precisely determined, as demonstrated for different l-lysine producing mutants (Wittmann et al. 2004b). In the meantime a novel platform for automated high-throughput fluxome profiling of bacteria has been established, which combines a robotic system for 13C labelling experiments and sampling with NMR-based isotopic profiling including automated data interpretation (Heux et al. 2014).
3.3.3 Metabolic Flux Analysis Under Dynamic Conditions
Notably, the aforementioned techniques for flux estimation are suitable to determine steady-state fluxes. However, industrial fermentation processes are often performed as fed-batch cultures that are characterized by changing conditions. The determination of metabolic fluxes under such dynamic conditions requires specific methods. The key to resolve time-dependent changes in flux is the consideration of intracellular metabolites, which quickly adapt labelling patterns. Suitable metabolites are intracellular amino acids (Krömer et al. 2004; Drysch et al. 2004) and pathway intermediates of the EMP pathway, the PP pathway, and the TCA cycle (Kiefer et al. 2007; Drysch et al. 2004). While the amino acids can be well analysed for 13C labelling by GC-MS (Wittmann et al. 2002; Krömer et al. 2004), free pathway intermediates are accessible using LC-MS (Kiefer et al. 2007; Iwatani et al. 2007; Noack et al. 2011), GC-MS (Kohlstedt et al. 2014), and NMR (Drysch et al. 2004). Previous applications investigated flux variations in response to dynamic culture conditions, such as shifts from growth to L-lysine production (Krömer et al. 2004) or from batch to fed-batch (Iwatani et al. 2007). This technique seems useful to identify metabolic bottlenecks, which specifically result from the changed flux distribution, since these would not be accessible by standard 13C MFA.
3.3.4 Metabolic Flux Analysis of Non-growing Cells
Industrial fermentation processes typically support product synthesis under conditions of limited growth in order to reduce or even abolish anabolism as a competitor for carbon, energy, and reducing power. Typically, industrial production is conducted as fed-batch with an extended nutrient-limited production phase (Kind et al. 2014; Kohlstedt et al. 2018; Rohles et al. 2018). As another example, amino acid production with C. glutamicum traditionally uses auxotrophic mutants with decoupled phases of growth and production (Krömer et al. 2004; Vallino and Stephanopoulos 1993; Drysch et al. 2004). In addition, novel concepts for dynamic metabolic control use genetic switches and metabolic valves to change the metabolic activity from one state that optimally supports growth to another state that is more favourable for production (Lynch 2016). An elaborated respirometric approach for flux estimation has been developed to access metabolic fluxes of such producing, non-growing cells (Yang et al. 2005, 2006a, b). It is based on tracer studies with labelling measurement of carbon dioxide, which is practically always formed during metabolic activity which needs energy and redox power, even in non-growing cells. The technique is thus independent of further product formation or growth. Using a set-up with three parallel experiments on differently labelled tracer substrates, all central metabolic pathways activities of C. glutamicum can be resolved (Yang et al. 2006a). Another study reports on the flux estimation of non-growing succinate-producing C. glutamicum. A tracer combination of [1-13C] glucose, [2-13C] glucose, [1,2-13C] glucose and [U-13C] glucose combined with NMR analysis of secreted products allowed the determination of the central metabolic fluxes of non-growing C. glutamicum under anaerobic condition (Rados et al. 2014).
3.3.5 Metabolic Flux Analysis at Large Scale
Labelling studies for the determination of intracellular pathway activities are usually restricted to small-scale experimental set-ups due to the high cost of the required tracer substrates. However, with regard to industrial production processes it appears attractive to mitigate eventual scale effects by estimating metabolic fluxes also at large scale. This became possible with the development of a sensor-reactor approach allowing flux estimation under varying conditions representative for large-scale fermentation (El Massaoudi et al. 2003). The basic idea is to perform the tracer experiment in a small-scale bioreactor, which is operated in parallel to a large-scale production reactor. The labelling is introduced into the sensor reactor system by [13C6] glucose pulses; key fermentation parameters are assessed in both reactors throughout the course of the experiment to ensure process equivalence. In a case study, labelling data for flux estimation were obtained by NMR analysis of intracellular metabolites (Drysch et al. 2003). The approach was further extended to the determination of time-dependent flux changes that typically occur during fed-batch fermentation (Drysch et al. 2003, 2004).
A smart strategy for metabolic flux analysis at large scale is the use of ultrasensitive technology for labelling analysis. GC combustion isotope ratio mass spectrometry (GC-C-IRMS) can precisely estimate 13C labelling at extremely low enrichment. It is approximately 100–1000-fold more sensitive than common GC-MS or LC-MS approaches (Dersch et al. 2016a). This type of measurement allows to substantially decrease the amount of the applied tracer. GC-C-IRMS allows precise metabolic flux analysis at a reduced labelling degree of the tracer substrate of only 1%, whereby the coupling to a GC enables efficient separation and subsequent analysis of amino acids (Yuan et al. 2010).
4 Metabolic Pathway Engineering in C. glutamicum
With regard to metabolic flux analysis, C. glutamicum is probably the best studies microbe (Wittmann 2010; Wittmann and De Graaf 2005; Bartek et al. 2011; Noack et al. 2011; Hoffmann et al. 2018; Rados et al. 2014). Driven by the strong industrial interest for this bacterium, a number of studies have been conducted over the past years. They provide a very detailed picture and form an excellent knowledge base for systems metabolic engineering of C. glutamicum (Wittmann and Becker 2007; Ikeda 2003).
4.1 Key Fluxes for the Production of l-Lysine and l-Lysine-Derived Chemicals
The feed amino acid l-lysine holds the second largest share in the amino acid business with global sales of approximately 4 billion dollars. Industrial production mainly relies on strains of C. glutamicum, which have been engineered by classical mutagenesis as well as metabolic engineering (Eggeling and Bott 2015; Becker et al. 2018b). In recent years, such strains became more and more attractive as chassis strains for the production of l-lysine derived platform chemicals (Becker et al. 2018b). This chemicals include diaminopentane (Kind et al. 2010; Kind and Wittmann 2011), aminovalerate (Rohles et al. 2016; Shin et al. 2016) and glutarate (Rohles et al. 2018), which serve as building blocks for the production of novel bio-based plastics with advanced material properties, and l-pipecolic acid (Perez-Garcia et al. 2017; Perez-Garcia et al. 2016), a valuable precursor for the pharmaceutical industry. With a stoichiometric demand of 1:1:4 for each molecule of l-lysine, the l-lysine biosynthetic pathway in C. glutamicum requires pyruvate and oxaloacetate as carbon precursors, and NADPH as reducing power (Michal 1999). Similarly, these building blocks are required for all l-lysine-derived compounds. It is obvious that optimized production requires a well-balanced supply of these building blocks.
4.1.1 Engineering of Key Fluxes for Carbon Precursor Supply
Pyruvate and oxaloacetate are both part of a network of carboxylating and decarboxylating reactions, which connect the EMP pathway and the TCA cycle. In C. glutamicum, this complex network comprises pyruvate carboxylase and phosphoenolpyruvate carboxylase as carboxylating enzymes, as well as malic enzyme, PEP carboxykinase and OAA decarboxylase that catalyze decarboxylation reactions from the TCA cycle towards the EMP pathway (Fig. 1) (Eikmanns 2005; Becker and Wittmann 2017b). This enzymatic set-up creates a cyclic reaction network, which controls the anaplerotic net flux into the TCA cycle and, furthermore, allows a redistribution of flux in response to metabolic or genetic disturbance (Becker et al. 2008; Petersen et al. 2000, 2001; Shirai et al. 2007; Sawada et al. 2010; Yanase et al. 2016; Netzer et al. 2004). The general role of the cycle has been attributed to energy maintenance (de Graaf et al. 2001; Sauer et al. 1997), equilibration of metabolite levels around the pyruvate node (Sauer and Eikmanns 2005), and additional NADPH supply under certain conditions (Dominguez et al. 1998; Becker et al. 2008; Netzer et al. 2004). Specifically related to the development of l-lysine overproducing strains, the cycle and its individual enzymes exhibit a key role. Using metabolic flux analysis, a close correlation between the l-lysine flux and the anaplerotic net flux was discovered for different l-lysine producers (Wittmann and De Graaf 2005; Wittmann and Heinzle 2002). This observation was used in different metabolic engineering strategies that aimed at an increased anaplerotic net flux by (1) overexpression of pyruvate carboxylase as major anaplerotic enzyme (Peters-Wendisch et al. 2001; Kind et al. 2011; Becker et al. 2011; Kind et al. 2010; Rohles et al. 2016, 2018; Buschke et al. 2011), (2) overexpression of PEP carboxylase (Sano et al. 1987), (3) deletion of the pck gene encoding the OAA-consuming reaction PEP carboxykinase (Petersen et al. 2001; Kind et al. 2011; Becker et al. 2011; Perez-Garcia et al. 2017, 2018; Jorge et al. 2017b; Rohles et al. 2016, 2018; Kind et al. 2010; Buschke et al. 2011) and (4) implementation of feed-back insensitive variants of pyruvate carboxylase (Ohnishi et al. 2002; Kortmann et al. 2019; Becker et al. 2011; Perez-Garcia et al. 2017, 2018; Jorge et al. 2017b; Rohles et al. 2016, 2018; Kind et al. 2010; Buschke et al. 2011; Kind et al. 2011), and PEP carboxylase (Chen et al. 2014; Yanase et al. 2016; Yokota et al. 2017).
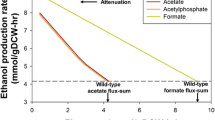
In contrast to the anaplerotic net flux, the flux through pyruvate dehydrogenase and TCA cycle, pathways competing with l-lysine production, was found to decrease with increasing l-lysine flux (Fig. 6). This finding suggested that an attenuation of pyruvate dehydrogenase and TCA cycle could redirect the flux towards anaplerotic carboxylation to support the production of l-lysine and related products. Indeed, l-lysine production was successfully improved by deletion (Blombach et al. 2007) and targeted down-regulation of pdh (Becker et al. 2010; Buchholz et al. 2013). In addition, the down-regulation of the TCA cycle flux at the level of isocitrate dehydrogenase, citrate synthase and succinyl-CoA synthetase substantially improved production performance (Becker et al. 2009; Kind et al. 2013; van Ooyen et al. 2012; Xu et al. 2018; Yokota et al. 2017). Interestingly, perturbation of the metabolism at the level of pyruvate kinase revealed an indifferent effect on l-lysine production, depending on the genetic background of the parent strain (Becker et al. 2008; Shiio et al. 1987; Yanase et al. 2016; Yokota et al. 2017). Only in combination with the expression of a desensitized PEP carboxylase, deletion of the pyruvate kinase gene was beneficial for l-lysine production (Yanase et al. 2016; Yokota et al. 2017).

Metabolic flux correlation between fluxes of L-lysine production, pyruvate dehydrogenase and anaplerotic carboxylation [%] in different strains of Corynebacterium glutamicum. Data represent values from 18 independent 13C flux experiments taken from Becker et al. (2005, 2007, 2008); Krömer et al. (2004); Kiefer et al. (2004); Wittmann and Heinzle (2002); Kim et al. (2006); Marx et al. (1996). The correlation was obtained by paraboloid fitting of the data set
4.1.2 Key Fluxes for NADPH Supply
In addition to carbon, NADPH is required for efficient l-lysine biosynthesis and—in line with this—for all l-lysine-derived chemicals. In C. glutamicum, NADPH is provided by the enzymes glucose 6-phosphate dehydrogenase (G6PDH) and 6-phosphogluconate dehydrogenase, which form the oxidative part of the PP pathway (Ihnen and Demain 1969; Yokota and Lindley 2005), by the TCA cycle enzyme isocitrate dehydrogenase (Chen and Yang 2000), and by malic enzyme (Gourdon et al. 2000; Eikmanns 2005). The relevance of these enzymes for NADPH supply for l-lysine production has been elucidated to great detail by comparative flux studies (Wittmann and Heinzle 2002). The PP pathway is the most important pathway for NADPH supply in glucose-grown C. glutamicum. Improved production performance is accompanied by an increased carbon flux through the PP pathway and a concomitantly reduced flux through the EMP pathway (Fig. 7). The TCA cycle flux, however, decreases with increased l-lysine flux (Wittmann and Heinzle 2002; Becker et al. 2009; Wittmann and De Graaf 2005), unravelling that the cycle rather competes with production for carbon. Accordingly, manipulation of the flux partitioning ratio at the glucose 6-phosphate (G6P) branch point in favour of the PP pathway appeared promising to improve l-lysine production. This was tested by different strategies. The most radical one was a deletion of phosphoglucoisomerase (pgi), forcing the cell to exclusively metabolize carbon via the PP pathway (Marx et al. 2003). This modification improved l-lysine production, but severely impaired growth. Moreover, a pgi deficient mutant would be limited to glucose-based production, because other substrates such as sucrose, glycerol, lactate, fructose, xylose, and arabinose require an active phosphoglucoisomerase for channelling carbon into the PP pathway to form NADPH (Kiefer et al. 2004; Becker et al. 2005; Wittmann et al. 2004a; Buschke et al. 2013; Dominguez et al. 1998).

Metabolic flux correlation between fluxes through L-lysine production, glycolysis and pentose phosphate pathway [%] in different strains of Corynebacterium glutamicum. Data represent values from 18 independent 13C flux experiments taken from Becker et al. (2005, 2007, 2008); Krömer et al. (2004); Kiefer et al. (2004); Wittmann and Heinzle (2002); Kim et al. (2006); Marx et al. (1996). The correlation was obtained by paraboloid fitting of the data set
The strict regulation of the NADPH-providing enzymes G6PDH and 6-phosphogluconate dehydrogenase by NADPH and other metabolites (Moritz et al. 2000) suggested the need to mutate these enzymes in order to relieve feedback inhibition constraints. Implementation of such modified enzyme-variants in a wild type-derived l-lysine producer indeed resulted in increased l-lysine production by increased fluxes through the PP pathway (Ohnishi et al. 2005; Becker et al. 2007). Moreover, over expression of G6PDH by promoter exchange (Becker et al. 2007) and start codon exchange (Becker et al. 2010) influenced the flux split ratio at the G6P node thereby significantly improving l-lysine production. In an advanced l-lysine producer, over expression of G6PDH was realized in its complete tkt-operon-frame thereby additionally upregulating other enzymes of the PP pathway, including transketolase and transaldolase of PP pathway (Becker et al. 2011). Exchange of the natural tkt-promoter by the sod-promoter concomitantly increased PP pathway flux and l-lysine production further (Becker et al. 2011). This strategy was efficiently transferred to diaminopentane-producing strains with substantial improvement of glucose- and xylose-based production (Kind et al. 2014; Buschke et al. 2013).
Efficient NADPH supply in C. glutamicum is especially challenging, when using substrates, which only support a low flux through the oxidative PP pathway, such as fructose (Kiefer et al. 2004), sucrose (Wittmann et al. 2004a), mannitol (Hoffmann et al. 2018), and xylose (Buschke et al. 2013). To address this issue, different engineering strategies were found valuable: (1) enforced metabolic flux rerouting towards PP pathway, (2) the incorporation of alternative reactions for NADPH supply, and (3) a reduction of the NADPH demand. As example, the amplified expression of the fbp gene, encoding the gluconeogenetic enzyme fructose 1,6-bisphosphatase (FBPase), was highly efficient to push carbon towards the PP pathway (Becker et al. 2005, 2011). This non-obvious target was identified by metabolic flux analysis of C. glutamicum during growth on fructose and sucrose, highlighting this technology as a powerful tool to guide strain optimization (Kiefer et al. 2004; Wittmann et al. 2004a). Overexpression of fbp did not only turn out beneficial for glucose, fructose, and sucrose based production (Becker et al. 2005) but also improved production of l-lysine and l-lysine-derived compounds from other substrates including xylose (Buschke et al. 2013) and silage juice (Neuner et al. 2013). Moreover, this modification is an integral trait of hyper-producing strains, optimized for l-lysine production from mannitol (Hoffmann et al. 2018), and glucose-based production of diaminopentane (Kind et al. 2014), glutarate (Rohles et al. 2018) and aminovalerate (Rohles et al. 2016). Beside the expression level, also kinetic properties of FBPase are relevant for production. Unlike the native C. glutamicum enzyme, the E. coli enzyme variant is insensitive to metabolic regulation by fructose 1-phosphate and fructose 2,6-bisphosphate. Heterologous expression of the E. coli fbp gene supported l-lysine production in glucose (Xu et al. 2014), fructose (Xu et al. 2014), sucrose (Xu et al. 2014) and molasses-based media (Xu et al. 2013).
For mannitol-grown C. glutamicum, an additional metabolic route was established to support a better operation of the PP pathway during growth and production on this sugar alcohol (Hoffmann et al. 2018). Heterologous expression of fructokinase from either E. coli (mak) or Clostridium acetobutylicum (srcK) created a new assimilation pathway allowing higher fluxes through the NADPH-providing reactions of the PP pathway and thus improved l-lysine production (Hoffmann et al. 2018).
Instead of using the PP pathway as the natural NADPH source in C. glutamicum, other studies systematically manipulated the co-factor specificity of homologous enzymes or implanted heterologous enzymes in C. glutamicum to create novel reactions for sufficient NADPH supply. Engineering of the native NAD-dependent glyceraldehyde 3-phosphate dehydrogenase GapA towards the use of NADP as alternative co-factor substantially increased l-lysine production without a significant flux change towards the PP pathway (Bommareddy et al. 2014). Similarly, a replacement of GapA by the heterologous NADPH-generating glyceraldehyde 3-phosphate dehydrogenase GapN of Streptococcus mutans allowed l-lysine production independent of the PP pathway (Takeno et al. 2016). This modification was also the key to success to efficiently use mannitol as alternative carbon source for l-lysine production (Hoffmann et al. 2018). Despite a low operation of the PP pathway, sufficient NADPH supply for l-lysine production was ensured by coupling the supply of this co-factor to the high EMP pathway flux (Hoffmann et al. 2018).
As alternative strategy, enzymes were engineered for co-factor specificity to reduce the NADPH demand for l-lysine biosynthesis. A recent study reports on the mining of NADH-utilizing dehydrogenases, namely aspartate dehydrogenase from Pseudomonas aeruginosa (PaASPDH), aspartate-semialdehyde dehydrogenase from Tistrella mobilis (TmASADH), dihydrodipicolinate reductase from Escherichia coli (EcDHDPR), and diaminopimelate dehydrogenase from Pseudothermotoga thermarum (PtDAPDH), to replace the native C. glutamicum enzyme variants (Wu et al. 2019).
4.1.3 Systems-Wide Flux Engineering for l-Lysine Hyper-production
A seminal study used the power of 13C metabolic flux analysis combined with in silico modelling to systematically redirect the carbon fluxes of C. glutamicum for optimized l-lysine production. The metabolic blueprint of the superior producer based on a differential flux map composed from (1) the in vivo fluxes of the wild type as quantitative measure of the pathway usage as a starting point and (2) a predicted flux scenario for optimal L-lysine production as the ultimate goal (Becker et al. 2011). By iterative implementation of twelve defined genome-based modifications, the carbon flux was stepwise primed for l-lysine production. Under industrial conditions, the final engineered strain LYS-12 produced 120 g L–1 l-lysine within only 30 h at a high yield of 0.55 g g–1. Flux analysis guided strain engineering throughout the genealogy by unravelling the metabolic flux responses towards each of the implemented genetic perturbation. The fluxes provided a rational and quantitative measure to explain the observed changes in the production performance, and to validate the desired rerouting of metabolic fluxes, namely (1) increased l-lysine flux, (2) increased PP pathway flux, (3) increased pyruvate carboxylase flux, (4) decreased PEP carboxykinase flux and (5) decreased TCA cycle flux (Becker et al. 2011).
4.2 Key Fluxes for the Production of l-Glutamate and l-Glutamate Derived Chemicals
Similarly to l-lysine, l-glutamate overproduction is closely linked to the central metabolism. During l-glutamate production, significant amounts of the TCA cycle intermediate 2-oxoglutarate are withdrawn, resulting in the need for anaplerotic replenishment (Shirai et al. 2007; Peters-Wendisch et al. 2001; Sato et al. 2008). In order to support anaplerotic carboxylation, the central glycolytic enzyme pyruvate kinase was deleted in the wild type of C. glutamicum, which improved l-glutamate production under biotin-limited conditions (Sawada et al. 2010). The additional expression of a feedback deregulated PEP carboxylase was especially beneficial for l-glutamate production (Yokota et al. 2017). This is in line with previous findings, which showed that PEP carboxylase is indispensable for l-glutamate production and carries the major anaplerotic flux under these conditions (Sato et al. 2008). Studies on posttranslational modification of PEP carboxylase by acetylation provided reasonable evidence that this enzyme might indeed play a major role for carbon flux redistribution under l-glutamate-producing conditions (Nagano-Shoji et al. 2017). Interestingly, the deletion of pyruvate carboxylase resulted in an improved production of l-glutamate and the thereof derived γ-aminobutyrate (GABA) (Wang et al. 2015), likely related to the activation of PEP carboxykinase. Also targeted overexpression of the encoding ppc gene and deletion of the mdh gene, encoding the OAA consuming malate dehydrogenase, improved GABA production (Shi et al. 2017).
Regarding the l-glutamate precursor 2-oxoglutarate, the activity of isocitrate dehydrogenase (ICD) as 2-oxoglutarate-supplying reaction and the activity of the 2-oxoglutarate dehydrogenase complex (ODHC) as 2-oxoglutarate-consuming reaction have been studied. Metabolic flux analysis revealed that ICD activity does not significantly change during fermentation, while that of the ODHC significantly decreases after the induction of l-glutamate production (Shirai et al. 2005). Artificial amplification of the activities of ICD and l-glutamate dehydrogenase do not influence the fluxes at the 2-oxoglutarate branch point (Shimizu et al. 2003). In the light of this finding, the major role in flux control at the key branch point of 2-oxoglutarate can be attributed to ODHC. In this regard, production of l-glutamate and GABA was successfully improved by deletion of odhA, encoding a subunit of the enzyme complex (Asakura et al. 2007; Wang et al. 2015). A similar effect was attained by odhA antisense RNA expression that leads to a decreased specific activity of ODHC (Kim et al. 2009). A reduced ODHC activity, obtained by start codon engineering (ATG ➔ TTG) of OdhA, also improved production of putrescine (Nguyen et al. 2015) and GABA via the putrescine route (Jorge et al. 2017a). ODHC is subject to a regulatory control mechanism involving the ODHC suppressor protein OdhI and serine/threonine protein kinase G (PknG) (Schultz et al. 2007), whereby the inhibitory effect of OdhI on ODHC can be overcome by PknG-mediated phosphorylation of OdhI at the threonine residue 14 (Schultz et al. 2007; Niebisch et al. 2006). Moreover, posttranslational l-lysine succinylation was found to regulate OdhI activity, which might be of relevance for l-glutamate production (Mizuno et al. 2016; Komine-Abe et al. 2017). Accordingly, perturbation of this regulatory mechanism by overexpression of the odhI gene improve the production of l-glutamate (Kim et al. 2010). Similarly, deletion of the pknG gene or introduction of the point mutation T14A in OdhI—thus stabilizing the inhibitory effect of OdhI on ODHC—successfully increased the production of GABA (Jorge et al. 2017a; Okai et al. 2014) and putrescine (Nguyen et al. 2015).
5 Conclusions and Future Directions
The past years have impressively demonstrated the unique value of metabolic flux analysis to understand molecular and cellular processes in C. glutamicum and upgrade this microbe into efficient cell factories. Future strain optimization will demand even more for efficient flux approaches in order to speed up development times and efficiently extend the application folio of the microbe to non-natural products and raw materials.
One interesting line of research is the holistic interpretation of flux data in combination with other omics data in order to decipher global regulatory mechanisms (Krömer et al. 2004; Kohlstedt et al. 2010, 2014) and to identify genes with hypothetic or unknown function. Initial efforts of multi-omics analysis of C. glutamicum appear promising (Krömer et al. 2004, 2008; Silberbach et al. 2005; Buschke et al. 2013; Buchinger et al. 2009). They promise highly informative data sets to elucidate regulatory mechanisms on a global level and drive ambitious multidimensional optimization (Becker et al. 2018b; Becker and Wittmann 2015, 2018). Clearly, such systematic studies will benefit from the fast progress in miniaturization and automatization of metabolic flux studies, which enable high-throughput (Wittmann et al. 2004b; Heux et al. 2014, 2017). Depending on the desired level of detailedness, metabolic flux analysis could involve systems-wide flux analysis of central metabolic pathways, targeted estimates of flux split ratios, and analysis of characteristic flux fingerprints via isotopic profiling (Heux et al. 2014, 2017; Klingner et al. 2015; Berger et al. 2014; Tang et al. 2012).
With regard to industrial implementation of C. glutamicum, novel approaches for 13C metabolic flux analysis in complex nutrient environments are more and more deepening our understanding of industrial cell factories under production-like conditions (Schwechheimer et al. 2016, 2018b, c; Adler et al. 2013, 2014; Templeton et al. 2017). In this regard, metabolic flux analysis will remain a “bottle opener” for systems-level design of C. glutamicum as efficient cell factory for the production of an array of bio-based goods. Moreover, metabolic flux analysis is more and more applied to gain deeper insights in the metabolism of pathogens and their mechanism of infection and virulence (Bücker et al. 2014; Berger et al. 2014), which is also of high interest for pathogenic Corynebacteria and close relatives such as Mycobacterium tuberculosis.
References
Adler P, Bolten CJ, Dohnt K, Hansen CE, Wittmann C (2013) Core fluxome and metafluxome of lactic acid bacteria under simulated cocoa pulp fermentation conditions. Appl Environ Microbiol 79(18):5670–5681. https://doi.org/10.1128/AEM.01483-13
Adler P, Frey LJ, Berger A, Bolten CJ, Hansen CE, Wittmann C (2014) The key to acetate: metabolic fluxes of acetic acid bacteria under cocoa pulp fermentation-simulating conditions. Appl Environ Microbiol 80(15):4702–4716. https://doi.org/10.1128/AEM.01048-14
Aiba S, Matsuoka M (1979) Identification of metabolic model—citrate production from glucose by Candida lipolytica. Biotechnol Bioeng 21(8):1373–1386. https://doi.org/10.1002/bit.260210806
Asakura Y, Kimura E, Usuda Y, Kawahara Y, Matsui K, Osumi T, Nakamatsu T (2007) Altered metabolic flux due to deletion of odhA causes L-glutamate overproduction in Corynebacterium glutamicum. Appl Environ Microbiol 73(4):1308–1319. https://doi.org/10.1128/AEM.01867-06
Bartek T, Blombach B, Lang S, Eikmanns BJ, Wiechert W, Oldiges M, Nöh K, Noack S (2011) Comparative 13C metabolic flux analysis of pyruvate dehydrogenase complex-deficient, L-valine-producing Corynebacterium glutamicum. Appl Environ Microbiol 77(18):6644–6652. https://doi.org/10.1128/AEM.00575-11
Becker J, Wittmann C (2012) Systems and synthetic metabolic engineering for amino acid production—the heartbeat of industrial strain development. Curr Opin Biotechnol 23(5):718–726. https://doi.org/10.1016/j.copbio.2011.12.025
Becker J, Wittmann C (2014) GC-MS-based 13C metabolic flux analysis. In: Krömer JO, Nielsen LK, Blank LM (eds) Metabolic flux analysis—methods and protocols, Methods in molecular biology. Springer/Humana, New York, pp 165–174. https://doi.org/10.1007/978-1-4939-1170-7_10
Becker J, Wittmann C (2015) Advanced biotechnology: metabolically engineered cells for the bio-based production of chemicals and fuels, materials, and health-care products. Angew Chem Int Ed Engl 54:3328–3350. https://doi.org/10.1002/anie.201409033
Becker J, Wittmann C (2017a) Diamines for bio-based materials. In: Wittmann C, Liao JC (eds) Industrial biotechnology, vol 1. Advanced biotechnology, vol 4. Wiley-VCH, Weinheim, pp 393–404
Becker J, Wittmann C (2017b) Industrial microorganisms: Corynebacterium glutamicum. In: Wittmann C, Liao JC (eds) Industrial biotechnology, vol 1. Advanced biotechnology, vol 3a. Wiley-VCH, Weinheim, pp 183–203
Becker J, Wittmann C (2018) From systems biology to metabolically engineered cells-an omics perspective on the development of industrial microbes. Curr Opin Microbiol 45:180–188. https://doi.org/10.1016/j.mib.2018.06.001
Becker J, Klopprogge C, Zelder O, Heinzle E, Wittmann C (2005) Amplified expression of fructose 1,6-bisphosphatase in Corynebacterium glutamicum increases in vivo flux through the pentose phosphate pathway and lysine production on different carbon sources. Appl Environ Microbiol 71(12):8587–8596. https://doi.org/10.1128/AEM.71.12.8587-8596.2005
Becker J, Klopprogge C, Herold A, Zelder O, Bolten CJ, Wittmann C (2007) Metabolic flux engineering of L-lysine production in Corynebacterium glutamicum—over expression and modification of G6P dehydrogenase. J Biotechnol 132(2):99–109. https://doi.org/10.1016/j.jbiotec.2007.05.026
Becker J, Klopprogge C, Wittmann C (2008) Metabolic responses to pyruvate kinase deletion in lysine producing Corynebacterium glutamicum. Microb Cell Fact 7:8. https://doi.org/10.1186/1475-2859-7-8
Becker J, Klopprogge C, Schröder H, Wittmann C (2009) Metabolic engineering of the tricarboxylic acid cycle for improved lysine production by Corynebacterium glutamicum. Appl Environ Microbiol 75(24):7866–7869. https://doi.org/10.1128/AEM.01942-09
Becker J, Buschke N, Bücker R, Wittmann C (2010) Systems level engineering of Corynebacterium glutamicum—reprogramming translational efficiency for superior production. Eng Lif Sci 10:430–438
Becker J, Zelder O, Haefner S, Schröder H, Wittmann C (2011) From zero to hero—design-based systems metabolic engineering of Corynebacterium glutamicum for L-lysine production. Metab Eng 13(2):159–168. https://doi.org/10.1016/j.ymben.2011.01.003
Becker J, Schäfer R, Kohlstedt M, Harder BJ, Borchert NS, Stöveken N, Bremer E, Wittmann C (2013) Systems metabolic engineering of Corynebacterium glutamicum for production of the chemical chaperone ectoine. Microb Cell Fact 12:110. https://doi.org/10.1186/1475-2859-12-110
Becker J, Lange A, Fabarius J, Wittmann C (2015) Top value platform chemicals: bio-based production of organic acids. Curr Opin Biotechnol 36:168–175. https://doi.org/10.1016/j.copbio.2015.08.022
Becker J, Gießelmann G, Hoffmann SL, Wittmann C (2016) Corynebacterium glutamicum for sustainable bio-production: from metabolic physiology to systems metabolic engineering. In: Zhao H, Zeng AP (eds) Synthetic biology—metabolic engineering, Advances in biochemical engineering/biotechnology. Springer, Heidelberg, pp 217–263
Becker J, Kuhl M, Kohlstedt M, Starck S, Wittmann C (2018a) Metabolic engineering of Corynebacterium glutamicum for the production of cis, cis-muconic acid from lignin. Microb Cell Fact 17(1):115. https://doi.org/10.1186/s12934-018-0963-2
Becker J, Rohles CM, Wittmann C (2018b) Metabolically engineered Corynebacterium glutamicum for bio-based production of chemicals, fuels, materials, and healthcare products. Metab Eng 50:122–141. https://doi.org/10.1016/j.ymben.2018.07.008
Beckers V, Poblete-Castro I, Tomasch J, Wittmann C (2016) Integrated analysis of gene expression and metabolic fluxes in PHA-producing Pseudomonas putida grown on glycerol. Microb Cell Fact 15:73. https://doi.org/10.1186/s12934-016-0470-2
Berger A, Dohnt K, Tielen P, Jahn D, Becker J, Wittmann C (2014) Robustness and plasticity of metabolic pathway flux among uropathogenic isolates of Pseudomonas aeruginosa. PLoS One 9(4):e88368. https://doi.org/10.1371/journal.pone.0088368
Blombach B, Schreiner ME, Moch M, Oldiges M, Eikmanns BJ (2007) Effect of pyruvate dehydrogenase complex deficiency on L-lysine production with Corynebacterium glutamicum. Appl Microbiol Biotechnol 76(3):615–623. https://doi.org/10.1007/s00253-007-0904-1
Blombach B, Riester T, Wieschalka S, Ziert C, Youn JW, Wendisch VF, Eikmanns BJ (2011) Corynebacterium glutamicum tailored for efficient isobutanol production. Appl Environ Microbiol 77(10):3300–3310. https://doi.org/10.1128/AEM.02972-10
Boisseau R, Charrier B, Massou S, Portais JC, Akoka S, Giraudeau P (2013) Fast spatially encoded 3D NMR strategies for 13C-based metabolic flux analysis. Anal Chem 85(20):9751–9757. https://doi.org/10.1021/ac402155w
Bolten CJ, Heinzle E, Müller R, Wittmann C (2009) Investigation of the central carbon metabolism of Sorangium cellulosum: metabolic network reconstruction and quantification of pathway fluxes. J Microbiol Biotechnol 19(1):23–36
Bolten CJ, Schröder H, Dickschat J, Wittmann C (2010) Towards methionine overproduction in Corynebacterium glutamicum—methanethiol and dimethyldisulfide as reduced sulfur sources. J Microbiol Biotechnol 20(8):1196–1203
Bommareddy RR, Chen Z, Rappert S, Zeng AP (2014) A de novo NADPH generation pathway for improving lysine production of Corynebacterium glutamicum by rational design of the coenzyme specificity of glyceraldehyde 3-phosphate dehydrogenase. Metab Eng 25:30–37. https://doi.org/10.1016/j.ymben.2014.06.005
Buchholz J, Schwentner A, Brunnenkan B, Gabris C, Grimm S, Gerstmeir R, Takors R, Eikmanns BJ, Blombach B (2013) Platform engineering of Corynebacterium glutamicum with reduced pyruvate dehydrogenase complex activity for improved production of L-lysine, L-valine, and 2-ketoisovalerate. Appl Environ Microbiol 79(18):5566–5575. https://doi.org/10.1128/AEM.01741-13
Buchinger S, Strösser J, Rehm N, Hänßler E, Hans S, Bathe B, Schomburg D, Krämer R, Burkovski A (2009) A combination of metabolome and transcriptome analyses reveals new targets of the Corynebacterium glutamicum nitrogen regulator AmtR. J Biotechnol 140(1–2):68–74. https://doi.org/10.1016/j.jbiotec.2008.10.009
Bücker R, Heroven AK, Becker J, Dersch P, Wittmann C (2014) The pyruvate—tricarboxylic acid cycle node: a focal point of virulence control in the enteric pathogen Yersinia pseudotuberculosis. J Biol Chem 289(43):30114–30132. https://doi.org/10.1074/jbc.M114.581348
Buschke N, Schröder H, Wittmann C (2011) Metabolic engineering of Corynebacterium glutamicum for production of 1,5-diaminopentane from hemicellulose. Biotechnol J 6(3):306–317. https://doi.org/10.1002/biot.201000304
Buschke N, Becker J, Schäfer R, Kiefer P, Biedendieck R, Wittmann C (2013) Systems metabolic engineering of xylose-utilizing Corynebacterium glutamicum for production of 1,5-diaminopentane. Biotechnol J 8(5):557–570. https://doi.org/10.1002/biot.201200367
Chae TU, Choi SY, Kim JW, Ko YS, Lee SY (2017) Recent advances in systems metabolic engineering tools and strategies. Curr Opin Biotechnol 47:67–82. https://doi.org/10.1016/j.copbio.2017.06.007
Chen R, Yang H (2000) A highly specific monomeric isocitrate dehydrogenase from Corynebacterium glutamicum. Arch Biochem Biophys 383(2):238–245
Chen Z, Landman P, Colmer TD, Adams MA (1998) Simultaneous analysis of amino and organic acids in extracts of plant leaves as tert-butyldimethylsilyl derivatives by capillary gas chromatography. Anal Biochem 259(2):203–211
Chen Z, Bommareddy RR, Frank D, Rappert S, Zeng AP (2014) Deregulation of feedback inhibition of phosphoenolpyruvate carboxylase for improved lysine production in Corynebacterium glutamicum. Appl Environ Microbiol 80(4):1388–1393. https://doi.org/10.1128/AEM.03535-13
Christensen B, Nielsen J (1999) Isotopomer analysis using GC-MS. Metab Eng 1(4):282–290
Christensen B, Gombert AK, Nielsen J (2002) Analysis of flux estimates based on 13C-labelling experiments. Eur J Biochem 269(11):2795–2800
Cocaign-Bousquet M, Guyonvarch A, Lindley ND (1996) Growth rate-dependent modulation of carbon flux through central metabolism and the kinetic consequences for glucose-limited chemostat cultures of Corynebacterium glutamicum. Appl Environ Microbiol 62(2):429–436
Crown SB, Long CP, Antoniewicz MR (2016) Optimal tracers for parallel labeling experiments and 13C metabolic flux analysis: a new precision and synergy scoring system. Metab Eng 38:10–18. https://doi.org/10.1016/j.ymben.2016.06.001
Dauner M, Sauer U (2000) GC-MS analysis of amino acids rapidly provides rich information for isotopomer balancing. Biotechnol Prog 16(4):642–649. https://doi.org/10.1021/bp000058h
Dersch LM, Beckers V, Rasch D, Melzer G, Bolten C, Kiep K, Becker H, Blasing OE, Fuchs R, Ehrhardt T, Wittmann C (2016a) Novel approach for high-throughput metabolic screening of whole plants by stable isotopes. Plant Physiol 171(1):25–41. https://doi.org/10.1104/pp.15.01217
Dersch LM, Beckers V, Wittmann C (2016b) Green pathways: metabolic network analysis of plant systems. Metab Eng 34:1–24. https://doi.org/10.1016/j.ymben.2015.12.001
Desai TS, Srivastava S (2018) FluxPyt: a Python-based free and open-source software for 13C-metabolic flux analyses. PeerJ 6:e4716. https://doi.org/10.7717/peerj.4716
Dominguez H, Nezondet C, Lindley ND, Cocaign M (1993) Modified carbon flux during oxygen limited growth of Corynebacterium glutamicum and the consequences for amino acid overproduction. Biotechnol Lett 15(5):449–454
Dominguez H, Rollin C, Guyonvarch A, Guerquin-Kern JL, Cocaign-Bousquet M, Lindley ND (1998) Carbon-flux distribution in the central metabolic pathways of Corynebacterium glutamicum during growth on fructose. Eur J Biochem 254(1):96–102
Drysch A, El Massaoudi M, Mack C, Takors R, de Graaf AA, Sahm H (2003) Production process monitoring by serial mapping of microbial carbon flux distributions using a novel sensor reactor approach: II-13C-labeling-based metabolic flux analysis and L-lysine production. Metab Eng 5(2):96–107
Drysch A, El Massaoudi M, Wiechert W, de Graaf AA, Takors R (2004) Serial flux mapping of Corynebacterium glutamicum during fed-batch L-lysine production using the sensor reactor approach. Biotechnol Bioeng 85(5):497–505
Eggeling L, Bott M (2015) A giant market and a powerful metabolism: L-lysine provided by Corynebacterium glutamicum. Appl Microbiol Biotechnol 99(8):3387–3394. https://doi.org/10.1007/s00253-015-6508-2
Eikmanns BJ (2005) Central metabolism: tricarboxylic acid cycle and anaplerotic reactions. In: Eggeling L, Bott M (eds) Handbook of Corynebacterium glutamicum. CRC, Boca Raton, pp 241–276
El Massaoudi M, Spelthahn J, Drysch A, de Graaf A, Takors R (2003) Production process monitoring by serial mapping of microbial carbon flux distributions using a novel sensor reactor approach: I-Sensor reactor system. Metab Eng 5(2):86–95
Fischer E, Zamboni N, Sauer U (2004) High-throughput metabolic flux analysis based on gas chromatography-mass spectrometry derived 13C constraints. Anal Biochem 325(2):308–316
Fürch T, Preusse M, Tomasch J, Zech H, Wagner-Döbler I, Rabus R, Wittmann C (2009) Metabolic fluxes in the central carbon metabolism of Dinoroseobacter shibae and Phaeobacter gallaeciensis, two members of the marine Roseobacter clade. BMC Microbiol 9:209. https://doi.org/10.1186/1471-2180-9-209
Gourdon P, Baucher MF, Lindley ND, Guyonvarch A (2000) Cloning of the malic enzyme gene from Corynebacterium glutamicum and role of the enzyme in lactate metabolism. Appl Environ Microbiol 66(7):2981–2987
de Graaf AA, Mahle M, Möllney M, Wiechert W, Stahmann P, Sahm H (2000) Determination of full 13C isotopomer distributions for metabolic flux analysis using heteronuclear spin echo difference NMR spectroscopy. J Biotechnol 77(1):25–35
de Graaf AA, Eggeling L, Sahm H (2001) Metabolic engineering for L-lysine production by Corynebacterium glutamicum. Adv Biochem Eng Biotechnol 73:9–29
Guo W, Sheng J, Feng X (2018) Synergizing 13C Metabolic flux analysis and metabolic engineering for biochemical production. Adv Biochem Eng Biotechnol 162:265–299. https://doi.org/10.1007/10_2017_2
Haberhauer G, Schröder H, Pompejus M, Zelder O, Kröger B (2001) Corynebacterium glutamicum genes encoding proteins involved in membrane synthesis and membrane transport. Patent WO 01/00805 Patent
He L, Wu SG, Zhang M, Chen Y, Tang YJ (2016) WUFlux: an open-source platform for 13C metabolic flux analysis of bacterial metabolism. BMC Bioinformatics 17(1):444. https://doi.org/10.1186/s12859-016-1314-0
Heux S, Poinot J, Massou S, Sokol S, Portais JC (2014) A novel platform for automated high-throughput fluxome profiling of metabolic variants. Metab Eng 25:8–19. https://doi.org/10.1016/j.ymben.2014.06.001
Heux S, Berges C, Millard P, Portais JC, Letisse F (2017) Recent advances in high-throughput 13C-fluxomics. Curr Opin Biotechnol 43:104–109. https://doi.org/10.1016/j.copbio.2016.10.010
Hoffmann SL, Jungmann L, Schiefelbein S, Peyriga L, Cahoreau E, Portais JC, Becker J, Wittmann C (2018) Lysine production from the sugar alcohol mannitol: design of the cell factory Corynebacterium glutamicum SEA-3 through integrated analysis and engineering of metabolic pathway fluxes. Metab Eng. https://doi.org/10.1016/j.ymben.2018.04.019
Ihnen ED, Demain AL (1969) Glucose-6-phosphate dehydrogenase and its deficiency in mutants of Corynebacterium glutamicum. J Bacteriol 98(3):1151–1158
Ikeda M (2003) Amino acid production processes. Adv Biochem Eng Biotechnol 79:1–35
Inui M, Kawaguchi H, Murakami S, Vertes AA, Yukawa H (2004a) Metabolic engineering of Corynebacterium glutamicum for fuel ethanol production under oxygen-deprivation conditions. J Mol Microbiol Biotechnol 8(4):243–254. https://doi.org/10.1159/000086705
Inui M, Murakami S, Okino S, Kawaguchi H, Vertes AA, Yukawa H (2004b) Metabolic analysis of Corynebacterium glutamicum during lactate and succinate productions under oxygen deprivation conditions. J Mol Microbiol Biotechnol 7(4):182–196. https://doi.org/10.1159/000079827
Ishino S, Yamaguchi K, Shirahata K, Araki K (1984) Involvement of meso-α,ε-diaminopimelate D-dehydrogenase in lysine biosynthesis in Corynebacterium glutamicum. Agric Biol Chem 48(10):2557–2560
Ishino S, Kuga T, Yamaguchi K, Shirahata K, Araki K (1986) 13C NMR-studies of histidine fermentation with a Corynebacterium-glutamicum mutant. Agric Biol Chem 50(2):307–310. https://doi.org/10.1080/00021369.1986.10867392
Iwatani S, Van Dien S, Shimbo K, Kubota K, Kageyama N, Iwahata D, Miyano H, Hirayama K, Usuda Y, Shimizu K, Matsui K (2007) Determination of metabolic flux changes during fed-batch cultivation from measurements of intracellular amino acids by LC-MS/MS. J Biotechnol 128(1):93–111
Jojima T, Noburyu R, Sasaki M, Tajima T, Suda M, Yukawa H, Inui M (2015) Metabolic engineering for improved production of ethanol by Corynebacterium glutamicum. Appl Microbiol Biotechnol 99(3):1165–1172. https://doi.org/10.1007/s00253-014-6223-4
Jorge JM, Nguyen AQ, Perez-Garcia F, Kind S, Wendisch VF (2017a) Improved fermentative production of gamma-aminobutyric acid via the putrescine route: systems metabolic engineering for production from glucose, amino sugars, and xylose. Biotechnol Bioeng 114(4):862–873. https://doi.org/10.1002/bit.26211
Jorge JMP, Perez-Garcia F, Wendisch VF (2017b) A new metabolic route for the fermentative production of 5-aminovalerate from glucose and alternative carbon sources. Bioresour Technol 245 (Pt B):1701–1709. https://doi.org/10.1016/j.biortech.2017.04.108
Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A, Dusch N, Eggeling L, Eikmanns BJ, Gaigalat L, Goesmann A, Hartmann M, Huthmacher K, Krämer R, Linke B, McHardy AC, Meyer F, Möckel B, Pfefferle W, Pühler A, Rey DA, Rückert C, Rupp O, Sahm H, Wendisch VF, Wiegrabe I, Tauch A (2003) The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J Biotechnol 104(1-3):5–25
Kappelmann J, Klein B, Geilenkirchen P, Noack S (2017) Comprehensive and accurate tracking of carbon origin of LC-tandem mass spectrometry collisional fragments for 13C-MFA. Anal Bioanal Chem 409(9):2309–2326. https://doi.org/10.1007/s00216-016-0174-9
Kelleher JK (2001) Flux estimation using isotopic tracers: common ground for metabolic physiology and metabolic engineering. Metab Eng 3(2):100–110
Kiefer P, Heinzle E, Wittmann C (2002) Influence of glucose, fructose and sucrose as carbon sources on kinetics and stoichiometry of lysine production by Corynebacterium glutamicum. J Ind Microbiol Biotechnol 28(6):338–343. https://doi.org/10.1038/sj/jim/7000252
Kiefer P, Heinzle E, Zelder O, Wittmann C (2004) Comparative metabolic flux analysis of lysine-producing Corynebacterium glutamicum cultured on glucose or fructose. Appl Environ Microbiol 70(1):229–239
Kiefer P, Nicolas C, Letisse F, Portais JC (2007) Determination of carbon labeling distribution of intracellular metabolites from single fragment ions by ion chromatography tandem mass spectrometry. Anal Biochem 360(2):182–188
Kim H-M, Heinzle E, Wittmann C (2006) Deregulation of aspartokinase by single nucleotide exchange leads to global flux rearrangement in the central metabolism of Corynebacterium glutamicum. J Microbiol Biotechnol 16(8):1174–1179
Kim J, Hirasawa T, Sato Y, Nagahisa K, Furusawa C, Shimizu H (2009) Effect of odhA overexpression and odhA antisense RNA expression on Tween-40-triggered glutamate production by Corynebacterium glutamicum. Appl Microbiol Biotechnol 81(6):1097–1106. https://doi.org/10.1007/s00253-008-1743-4
Kim J, Fukuda H, Hirasawa T, Nagahisa K, Nagai K, Wachi M, Shimizu H (2010) Requirement of de novo synthesis of the OdhI protein in penicillin-induced glutamate production by Corynebacterium glutamicum. Appl Microbiol Biotechnol 86(3):911–920. https://doi.org/10.1007/s00253-009-2360-6
Kind S, Wittmann C (2011) Bio-based production of the platform chemical 1,5-diaminopentane. Appl Microbiol Biotechnol 91(5):1287–1296. https://doi.org/10.1007/s00253-011-3457-2
Kind S, Jeong WK, Schröder H, Wittmann C (2010) Systems-wide metabolic pathway engineering in Corynebacterium glutamicum for bio-based production of diaminopentane. Metab Eng 12(4):341–351. https://doi.org/10.1016/j.ymben.2010.03.005
Kind S, Kreye S, Wittmann C (2011) Metabolic engineering of cellular transport for overproduction of the platform chemical 1,5-diaminopentane in Corynebacterium glutamicum. Metab Eng 13(5):617–627. https://doi.org/10.1016/j.ymben.2011.07.006
Kind S, Becker J, Wittmann C (2013) Increased lysine production by flux coupling of the tricarboxylic acid cycle and the lysine biosynthetic pathway—metabolic engineering of the availability of succinyl-CoA in Corynebacterium glutamicum. Metab Eng 15:184–195. https://doi.org/10.1016/j.ymben.2012.07.005
Kind S, Neubauer S, Becker J, Yamamoto M, Völkert M, Abendroth GV, Zelder O, Wittmann C (2014) From zero to hero—production of bio-based nylon from renewable resources using engineered Corynebacterium glutamicum. Metab Eng 25:113–123. https://doi.org/10.1016/j.ymben.2014.05.007
Kjeldsen KR, Nielsen J (2009) In silico genome-scale reconstruction and validation of the Corynebacterium glutamicum metabolic network. Biotechnol Bioeng 102(2):583–597. https://doi.org/10.1002/bit.22067
Klingner A, Bartsch A, Dogs M, Wagner-Döbler I, Jahn D, Simon M, Brinkhoff T, Becker J, Wittmann C (2015) Large-scale 13C flux profiling reveals conservation of the Entner-Doudoroff pathway as a glycolytic strategy among marine bacteria that use glucose. Appl Environ Microbiol 81(7):2408–2422. https://doi.org/10.1128/AEM.03157-14
Kohlstedt M, Wittmann C (2019) GC-MS based 13C metabolic flux analysis resolves the cyclic glucose metabolism of Pseudomonas putida KT2440 and Pseudomonas aeruginosa PAO1. Metab Eng 54:35–53
Kohlstedt M, Becker J, Wittmann C (2010) Metabolic fluxes and beyond-systems biology understanding and engineering of microbial metabolism. Appl Microbiol Biotechnol 88(5):1065–1075. https://doi.org/10.1007/s00253-010-2854-2
Kohlstedt M, Sappa PK, Meyer H, Maass S, Zaprasis A, Hoffmann T, Becker J, Steil L, Hecker M, van Dijl JM, Lalk M, Mader U, Stülke J, Bremer E, Völker U, Wittmann C (2014) Adaptation of Bacillus subtilis carbon core metabolism to simultaneous nutrient limitation and osmotic challenge: a multi-omics perspective. Environ Microbiol 16(6):1898–1917. https://doi.org/10.1111/1462-2920.12438
Kohlstedt M, Starck S, Barton N, Stolzenberger J, Selzer M, Mehlmann K, Schneider R, Pleissner D, Rinkel J, Dickschat JS, Venus J, van Duuren JBJH, Wittmann C (2018) From lignin to nylon: cascaded chemical and biochemical conversion using metabolically engineered Pseudomonas putida. Metab Eng 47:279–293. https://doi.org/10.1016/j.ymben.2018.03.003
Komine-Abe A, Nagano-Shoji M, Kubo S, Kawasaki H, Yoshida M, Nishiyama M, Kosono S (2017) Effect of lysine succinylation on the regulation of 2-oxoglutarate dehydrogenase inhibitor, OdhI, involved in glutamate production in Corynebacterium glutamicum. Biosci Biotechnol Biochem 81(11):2130–2138. https://doi.org/10.1080/09168451.2017.1372182
Kortmann M, Mack C, Baumgart M, Bott M (2019) Pyruvate carboxylase variants enabling improved lysine production from glucose identified by biosensor-based high-throughput fluorescence-activated cell sorting screening. ACS Synth Biol 8(2):274–281. https://doi.org/10.1021/acssynbio.8b00510
Krömer JO, Sorgenfrei O, Klopprogge K, Heinzle E, Wittmann C (2004) In-depth profiling of lysine-producing Corynebacterium glutamicum by combined analysis of the transcriptome, metabolome, and fluxome. J Bacteriol 186(6):1769–1784
Krömer JO, Heinzle E, Schröder H, Wittmann C (2006) Accumulation of homolanthionine and activation of a novel pathway for isoleucine biosynthesis in Corynebacterium glutamicum McbR deletion strains. J Bacteriol 188(2):609–618. https://doi.org/10.1128/JB.188.2.609-618.2006
Krömer JO, Bolten CJ, Heinzle E, Schröder H, Wittmann C (2008) Physiological response of Corynebacterium glutamicum to oxidative stress induced by deletion of the transcriptional repressor McbR. Microbiology 154(Pt 12):3917–3930. https://doi.org/10.1099/mic.0.2008/021204-0
Lange A, Becker J, Schulze D, Cahoreau E, Portais JC, Haefner S, Schröder H, Krawczyk J, Zelder O, Wittmann C (2017) Bio-based succinate from sucrose: high-resolution 13C metabolic flux analysis and metabolic engineering of the rumen bacterium Basfia succiniciproducens. Metab Eng 44:198–212. https://doi.org/10.1016/j.ymben.2017.10.003
Lessmeier L, Pfeifenschneider J, Carnicer M, Heux S, Portais JC, Wendisch VF (2015) Production of carbon-13-labeled cadaverine by engineered Corynebacterium glutamicum using carbon-13-labeled methanol as co-substrate. Appl Microbiol Biotechnol 99(23):10163–10176. https://doi.org/10.1007/s00253-015-6906-5
Lynch MD (2016) Into new territory: improved microbial synthesis through engineering of the essential metabolic network. Curr Opin Biotechnol 38:106–111. https://doi.org/10.1016/j.copbio.2016.01.009
Ma Q, Zhang Q, Xu Q, Zhang C, Li Y, Fan X, Xie X, Chen N (2017) Systems metabolic engineering strategies for the production of amino acids. Synth Syst Biotechnol 2(2):87–96. https://doi.org/10.1016/j.synbio.2017.07.003
Maarleveld TR, Khandelwal RA, Olivier BG, Teusink B, Bruggeman FJ (2013) Basic concepts and principles of stoichiometric modeling of metabolic networks. Biotechnol J 8(9):997–1008. https://doi.org/10.1002/biot.201200291
Marx A, de Graaf AA, Wiechert W, Eggeling L, Sahm H (1996) Determination of the fluxes in the central metabolism of Corynebacterium glutamicum by nuclear magnetic resonance spectroscopy combined with metabolite balancing. Biotechnol Bioeng 49(2):111–129. https://doi.org/10.1002/(SICI)1097-0290(19960120)49:2<111::AID-BIT1>3.0.CO;2-T
Marx A, Striegel K, de Graaf AA, Sahm H, Eggeling L (1997) Response of the central metabolism of Corynebacterium glutamicum to different flux burdens. Biotechnol Bioeng 56(2):168–180. https://doi.org/10.1002/(SICI)1097-0290(19971020)56:2<168::AID-BIT6>3.0.CO;2-N
Marx A, Eikmanns BJ, Sahm H, de Graaf AA, Eggeling L (1999) Response of the central metabolism in Corynebacterium glutamicum to the use of an NADH-dependent glutamate dehydrogenase. Metab Eng 1(1):35–48. https://doi.org/10.1006/mben.1998.0106
Marx A, Hans S, Möckel B, Bathe B, de Graaf AA, McCormack AC, Stapleton C, Burke K, O’Donohue M, Dunican LK (2003) Metabolic phenotype of phosphoglucose isomerase mutants of Corynebacterium glutamicum. J Biotechnol 104(1-3):185–197
Massou S, Nicolas C, Letisse F, Portais JC (2007) Application of 2D-TOCSY NMR to the measurement of specific 13C-enrichments in complex mixtures of 13C-labeled metabolites. Metab Eng 9(3):252–257. https://doi.org/10.1016/j.ymben.2007.03.001
Mei J, Xu N, Ye C, Liu L, Wu J (2016) Reconstruction and analysis of a genome-scale metabolic network of Corynebacterium glutamicum S9114. Gene 575(2 Pt 3):615–622. https://doi.org/10.1016/j.gene.2015.09.038
Melzer G, Esfandabadi ME, Franco-Lara E, Wittmann C (2009) Flux design: in silico design of cell factories based on correlation of pathway fluxes to desired properties. BMC Syst Biol 3:120. https://doi.org/10.1186/1752-0509-3-120
Michal G (1999) Biochemical pathways. Wiley, Chichester
Millard P, Sokol S, Letisse F, Portais JC (2014) IsoDesign: a software for optimizing the design of 13C-metabolic flux analysis experiments. Biotechnol Bioeng 111(1):202–208. https://doi.org/10.1002/bit.24997
Millard P, Cahoreau E, Heuillet M, Portais JC, Lippens G (2017) 15N-NMR-based approach for amino acids-based 13C-metabolic flux analysis of metabolism. Anal Chem 89(3):2101–2106. https://doi.org/10.1021/acs.analchem.6b04767
Mizuno Y, Nagano-Shoji M, Kubo S, Kawamura Y, Yoshida A, Kawasaki H, Nishiyama M, Yoshida M, Kosono S (2016) Altered acetylation and succinylation profiles in Corynebacterium glutamicum in response to conditions inducing glutamate overproduction. Microbiologyopen 5(1):152–173. https://doi.org/10.1002/mbo3.320
Möllney M, Wiechert W, Kownatzki D, de Graaf AA (1999) Bidirectional reaction steps in metabolic networks: IV. Optimal design of isotopomer labeling experiments. Biotechnol Bioeng 66(2):86–103
Moritz B, Striegel K, De Graaf AA, Sahm H (2000) Kinetic properties of the glucose-6-phosphate and 6-phosphogluconate dehydrogenases from Corynebacterium glutamicum and their application for predicting pentose phosphate pathway flux in vivo. Eur J Biochem 267(12):3442–3452
Nagano-Shoji M, Hamamoto Y, Mizuno Y, Yamada A, Kikuchi M, Shirouzu M, Umehara T, Yoshida M, Nishiyama M, Kosono S (2017) Characterization of lysine acetylation of a phosphoenolpyruvate carboxylase involved in glutamate overproduction in Corynebacterium glutamicum. Mol Microbiol 104(4):677–689. https://doi.org/10.1111/mmi.13658
Netzer R, Krause M, Rittmann D, Peters-Wendisch PG, Eggeling L, Wendisch VF, Sahm H (2004) Roles of pyruvate kinase and malic enzyme in Corynebacterium glutamicum for growth on carbon sources requiring gluconeogenesis. Arch Microbiol 182(5):354–363. https://doi.org/10.1007/s00203-004-0710-4
Neuner A, Wagner I, Sieker T, Ulber R, Schneider K, Peifer S, Heinzle E (2013) Production of L-lysine on different silage juices using genetically engineered Corynebacterium glutamicum. J Biotechnol 163(2):217–224. https://doi.org/10.1016/j.jbiotec.2012.07.190
Nguyen AQ, Schneider J, Reddy GK, Wendisch VF (2015) Fermentative production of the diamine putrescine: system metabolic engineering of Corynebacterium glutamicum. Metabolites 5(2):211–231. https://doi.org/10.3390/metabo5020211
Nicolas C, Kiefer P, Letisse F, Krömer J, Massou S, Soucaille P, Wittmann C, Lindley ND, Portais JC (2007) Response of the central metabolism of Escherichia coli to modified expression of the gene encoding the glucose-6-phosphate dehydrogenase. FEBS Lett 581(20):3771–3776. https://doi.org/10.1016/j.febslet.2007.06.066
Nicolas C, Becker J, Sanchou L, Letisse F, Wittmann C, Portais JC, Massou S (2008) Measurement of isotopic enrichments in 13C-labelled molecules by 1D selective zero-quantum filtered TOCSY NMR experiments. Cr Chim 11(4–5):480–485. https://doi.org/10.1016/j.crci.2007.06.018
Niebisch A, Kabus A, Schultz C, Weil B, Bott M (2006) Corynebacterial protein kinase G controls 2-oxoglutarate dehydrogenase activity via the phosphorylation status of the OdhI protein. J Biol Chem 281(18):12300–12307. https://doi.org/10.1074/jbc.M512515200
Nikel PI, Chavarria M, Fuhrer T, Sauer U, de Lorenzo V (2015) Pseudomonas putida KT2440 strain metabolizes glucose through a cycle formed by enzymes of the Entner-Doudoroff, Embden-Meyerhof-Parnas, and pentose phosphate pathways. J Biol Chem 290(43):25920–25932. https://doi.org/10.1074/jbc.M115.687749
Noack S, Nöh K, Moch M, Oldiges M, Wiechert W (2011) Stationary versus non-stationary 13C-MFA: a comparison using a consistent dataset. J Biotechnol 154(2-3):179–190. https://doi.org/10.1016/j.jbiotec.2010.07.008
Nöh K, Droste P, Wiechert W (2015) Visual workflows for 13C-metabolic flux analysis. Bioinformatics 31(3):346–354. https://doi.org/10.1093/bioinformatics/btu585
Nöh K, Niedenfuhr S, Beyss M, Wiechert W (2018) A Pareto approach to resolve the conflict between information gain and experimental costs: multiple-criteria design of carbon labeling experiments. PLoS Comput Biol 14(10):e1006533. https://doi.org/10.1371/journal.pcbi.1006533
Ohnishi J, Mitsuhashi S, Hayashi M, Ando S, Yokoi H, Ochiai K, Ikeda M (2002) A novel methodology employing Corynebacterium glutamicum genome information to generate a new L-lysine-producing mutant. Appl Microbiol Biotechnol 58(2):217–223
Ohnishi J, Katahira R, Mitsuhashi S, Kakita S, Ikeda M (2005) A novel gnd mutation leading to increased L-lysine production in Corynebacterium glutamicum. FEMS Microbiol Lett 242(2):265–274. https://doi.org/10.1016/j.femsle.2004.11.014
Okai N, Takahashi C, Hatada K, Ogino C, Kondo A (2014) Disruption of pknG enhances production of gamma-aminobutyric acid by Corynebacterium glutamicum expressing glutamate decarboxylase. AMB Express 4:20. https://doi.org/10.1186/s13568-014-0020-4
Okai N, Miyoshi T, Takeshima Y, Kuwahara H, Ogino C, Kondo A (2016) Production of protocatechuic acid by Corynebacterium glutamicum expressing chorismate-pyruvate lyase from Escherichia coli. Appl Microbiol Biotechnol 100(1):135–145. https://doi.org/10.1007/s00253-015-6976-4
Oldiges M, Kunze M, Degenring D, Sprenger GA, Takors R (2004) Stimulation, monitoring, and analysis of pathway dynamics by metabolic profiling in the aromatic amino acid pathway. Biotechnol Prog 20(6):1623–1633
Palsson B (2000) The challenges of in silico biology. Nat Biotechnol 18(11):1147–1150
Perez-Garcia F, Peters-Wendisch P, Wendisch VF (2016) Engineering Corynebacterium glutamicum for fast production of L-lysine and L-pipecolic acid. Appl Microbiol Biotechnol 100(18):8075–8090. https://doi.org/10.1007/s00253-016-7682-6
Perez-Garcia F, Max Risse J, Friehs K, Wendisch VF (2017) Fermentative production of L-pipecolic acid from glucose and alternative carbon sources. Biotechnol J 12(7). https://doi.org/10.1002/biot.201600646
Perez-Garcia F, Jorge JMP, Dreyszas A, Risse JM, Wendisch VF (2018) Efficient production of the dicarboxylic acid glutarate by Corynebacterium glutamicum via a novel synthetic pathway. Front Microbiol 9:2589. https://doi.org/10.3389/fmicb.2018.02589
Petersen S, de Graaf AA, Eggeling L, Mollney M, Wiechert W, Sahm H (2000) In vivo quantification of parallel and bidirectional fluxes in the anaplerosis of Corynebacterium glutamicum. J Biol Chem 275(46):35932–35941. https://doi.org/10.1074/jbc.M908728199
Petersen S, Mack C, de Graaf AA, Riedel C, Eikmanns BJ, Sahm H (2001) Metabolic consequences of altered phosphoenolpyruvate carboxykinase activity in Corynebacterium glutamicum reveal anaplerotic regulation mechanisms in vivo. Metab Eng 3(4):344–361. https://doi.org/10.1006/mben.2001.0198
Peters-Wendisch PG, Schiel B, Wendisch VF, Katsoulidis E, Möckel B, Sahm H, Eikmanns BJ (2001) Pyruvate carboxylase is a major bottleneck for glutamate and lysine production by Corynebacterium glutamicum. J Mol Microbiol Biotechnol 3(2):295–300
Quek LE, Wittmann C, Nielsen LK, Krömer JO (2009) OpenFLUX: efficient modelling software for 13C-based metabolic flux analysis. Microb Cell Fact 8:25. https://doi.org/10.1186/1475-2859-8-25
Rados D, Turner DL, Fonseca LL, Carvalho AL, Blombach B, Eikmanns BJ, Neves AR, Santos H (2014) Carbon flux analysis by 13C nuclear magnetic resonance to determine the effect of CO2 on anaerobic succinate production by Corynebacterium glutamicum. Appl Environ Microbiol 80(10):3015–3024. https://doi.org/10.1128/AEM.04189-13
Roessner U, Wagner C, Kopka J, Trethewey RN, Willmitzer L (2000) Technical advance: simultaneous analysis of metabolites in potato tuber by gas chromatography-mass spectrometry. Plant J 23(1):131–142
Rohles CM, Giesselmann G, Kohlstedt M, Wittmann C, Becker J (2016) Systems metabolic engineering of Corynebacterium glutamicum for the production of the carbon-5 platform chemicals 5-aminovalerate and glutarate. Microb Cell Fact 15(1):154. https://doi.org/10.1186/s12934-016-0553-0
Rohles CM, Gläser L, Kohlstedt M, Giesselmann G, Pearson S, del Campo A, Becker J, Wittmann C (2018) A bio-based route to the carbon-5 chemical glutaric acid and to bionylon-6,5 using metabolically engineered Corynebacterium glutamicum. Green Chem 20(20):4662–4674. https://doi.org/10.1039/c8gc01901k
Rühl M, Rupp B, Nöh K, Wiechert W, Sauer U, Zamboni N (2012) Collisional fragmentation of central carbon metabolites in LC-MS/MS increases precision of 13C metabolic flux analysis. Biotechnol Bioeng 109(3):763–771. https://doi.org/10.1002/bit.24344
Sano K, Ito K, Miwa K, Nakamori S (1987) Amplification of the phosphoenol pyruvate carboxylase gene of Brevibacterium lactofermentum to improve amino acid production. Agric Biol Chem 51(2):597–599
Sato H, Orishimo K, Shirai T, Hirasawa T, Nagahisa K, Shimizu H, Wachi M (2008) Distinct roles of two anaplerotic pathways in glutamate production induced by biotin limitation in Corynebacterium glutamicum. J Biosci Bioeng 106(1):51–58. https://doi.org/10.1263/jbb.106.51
Sauer U (2006) Metabolic networks in motion: 13C-based flux analysis. Mol Syst Biol 2:62. https://doi.org/10.1038/msb4100109
Sauer U, Eikmanns BJ (2005) The PEP-pyruvate-oxaloacetate node as the switch point for carbon flux distribution in bacteria. FEMS Microbiol Rev 29(4):765–794. https://doi.org/10.1016/j.femsre.2004.11.002
Sauer U, Hatzimanikatis V, Bailey JE, Hochuli M, Szyperski T, Wuthrich K (1997) Metabolic fluxes in riboflavin-producing Bacillus subtilis. Nat Biotechnol 15(5):448–452
Sawada K, Zen-in S, Wada M, Yokota A (2010) Metabolic changes in a pyruvate kinase gene deletion mutant of Corynebacterium glutamicum ATCC 13032. Metab Eng 12(4):401–407. https://doi.org/10.1016/j.ymben.2010.01.004
Schilling CH, Schuster S, Palsson BO, Heinrich R (1999) Metabolic pathway analysis: basic concepts and scientific applications in the post-genomic era. Biotechnol Prog 15(3):296–303
Schmidt K, Norregaard LC, Pedersen B, Meissner A, Duus JO, Nielsen JO, Villadsen J (1999) Quantification of intracellular metabolic fluxes from fractional enrichment and 13C-13C coupling constraints on the isotopomer distribution in labeled biomass components. Metab Eng 1(2):166–179. https://doi.org/10.1006/mben.1999.0114
Schneider J, Wendisch VF (2010) Putrescine production by engineered Corynebacterium glutamicum. Appl Microbiol Biotechnol 88(4):859–868. https://doi.org/10.1007/s00253-010-2778-x
Schneider J, Wendisch VF (2011) Biotechnological production of polyamines by bacteria: recent achievements and future perspectives. Appl Microbiol Biotechnol 91(1):17–30. https://doi.org/10.1007/s00253-011-3252-0
Schultz C, Niebisch A, Gebel L, Bott M (2007) Glutamate production by Corynebacterium glutamicum: dependence on the oxoglutarate dehydrogenase inhibitor protein OdhI and protein kinase PknG. Appl Microbiol Biotechnol 76(3):691–700. https://doi.org/10.1007/s00253-007-0933-9
Schwechheimer SK, Park EY, Revuelta JL, Becker J, Wittmann C (2016) Biotechnology of riboflavin. Appl Microbiol Biotechnol. https://doi.org/10.1007/s00253-015-7256-z
Schwechheimer SK, Becker J, Peyriga L, Portais JC, Sauer D, Müller R, Hoff B, Haefner S, Schröder H, Zelder O, Wittmann C (2018a) Improved riboflavin production with Ashbya gossypii from vegetable oil based on 13C metabolic network analysis with combined labeling analysis by GC/MS, LC/MS, 1D, and 2D NMR. Metab Eng 47:357–373. https://doi.org/10.1016/j.ymben.2018.04.005
Schwechheimer SK, Becker J, Peyriga L, Portais JC, Wittmann C (2018b) Metabolic flux analysis in Ashbya gossypii using 13C-labeled yeast extract: industrial riboflavin production under complex nutrient conditions. Microb Cell Fact 17(1):162. https://doi.org/10.1186/s12934-018-1003-y
Schwechheimer SK, Becker J, Wittmann C (2018c) Towards better understanding of industrial cell factories: novel approaches for 13C metabolic flux analysis in complex nutrient environments. Curr Opin Biotechnol 54:128–137. https://doi.org/10.1016/j.copbio.2018.07.001
Shi F, Zhang M, Li Y (2017) Overexpression of ppc or deletion of mdh for improving production of gamma-aminobutyric acid in recombinant Corynebacterium glutamicum. World J Microbiol Biotechnol 33(6):122. https://doi.org/10.1007/s11274-017-2289-3
Shiio I, Yokota A, Sugimoto SI (1987) Effect of pyruvate kinase deficiency on l-lysine productivities of mutants with feedback-resistant aspartokinases. Agric Biol Chem 51(9):2485–2493
Shimizu H, Tanaka H, Nakato A, Nagahisa K, Kimura E, Shioya S (2003) Effects of the changes in enzyme activities on metabolic flux redistribution around the 2-oxoglutarate branch in glutamate production by Corynebacterium glutamicum. Bioprocess Biosyst Eng 25(5):291–298. https://doi.org/10.1007/s00449-002-0307-8
Shin JH, Park SH, Oh YH, Choi JW, Lee MH, Cho JS, Jeong KJ, Joo JC, Yu J, Park SJ, Lee SY (2016) Metabolic engineering of Corynebacterium glutamicum for enhanced production of 5-aminovaleric acid. Microb Cell Fact 15(1):174. https://doi.org/10.1186/s12934-016-0566-8
Shinfuku Y, Sorpitiporn N, Sono M, Furusawa C, Hirasawa T, Shimizu H (2009) Development and experimental verification of a genome-scale metabolic model for Corynebacterium glutamicum. Microb Cell Fact 8:43
Shirai T, Nakato A, Izutani N, Nagahisa K, Shioya S, Kimura E, Kawarabayasi Y, Yamagishi A, Gojobori T, Shimizu H (2005) Comparative study of flux redistribution of metabolic pathway in glutamate production by two coryneform bacteria. Metab Eng 7(2):59–69
Shirai T, Matsuzaki K, Kuzumoto M, Nagahisa K, Furusawa C, Shioya S, Shimizu H (2006) Precise metabolic flux analysis of coryneform bacteria by gas chromatography-mass spectrometry and verification by nuclear magnetic resonance. J Biosci Bioeng 102(5):413–424. https://doi.org/10.1263/jbb.102.413
Shirai T, Fujimura K, Furusawa C, Nagahisa K, Shioya S, Shimizu H (2007) Study on roles of anaplerotic pathways in glutamate overproduction of Corynebacterium glutamicum by metabolic flux analysis. Microb Cell Fact 6:19. https://doi.org/10.1186/1475-2859-6-19
Shupletsov MS, Golubeva LI, Rubina SS, Podvyaznikov DA, Iwatani S, Mashko SV (2014) OpenFLUX2: 13 C-MFA modeling software package adjusted for the comprehensive analysis of single and parallel labeling experiments. Microb Cell Fact 13(1):152. https://doi.org/10.1186/s12934-014-0152-x
Silberbach M, Schäfer M, Hüser AT, Kalinowski J, Pühler A, Krämer R, Burkovski A (2005) Adaptation of Corynebacterium glutamicum to ammonium limitation: a global analysis using transcriptome and proteome techniques. Appl Environ Microbiol 71(5):2391–2402. https://doi.org/10.1128/AEM.71.5.2391-2402.2005
Smith KM, Cho KM, Liao JC (2010) Engineering Corynebacterium glutamicum for isobutanol production. Appl Microbiol Biotechnol 87(3):1045–1055. https://doi.org/10.1007/s00253-010-2522-6
Sonderegger M, Jeppsson M, Hahn-Hagerdal B, Sauer U (2004) Molecular basis for anaerobic growth of Saccharomyces cerevisiae on xylose, investigated by global gene expression and metabolic flux analysis. Appl Environ Microbiol 70(4):2307–2317
Sonntag K, Eggeling L, De Graaf AA, Sahm H (1993) Flux partitioning in the split pathway of lysine synthesis in Corynebacterium glutamicum. Quantification by 13C- and 1H-NMR spectroscopy. Eur J Biochem 213(3):1325–1331
Sonntag K, Schwinde J, deGraaf AA, Marx A, Eikmanns BJ, Wiechert W, Sahm H (1995) 13C NMR studies of the fluxes in the central metabolism of Corynebacterium glutamicum during growth and overproduction of amino acids in batch cultures. Appl Microbiol Biotechnol 44(3–4):489–495
Sun H, Zhao D, Xiong B, Zhang C, Bi C (2016) Engineering Corynebacterium glutamicum for violacein hyper production. Microb Cell Fact 15(1):148. https://doi.org/10.1186/s12934-016-0545-0
Szyperski T (1998) C-13-NMR, MS and metabolic flux balancing in biotechnology research. Q Rev Biophys 31(1):41–106. https://doi.org/10.1017/S0033583598003412
Takeno S, Hori K, Ohtani S, Mimura A, Mitsuhashi S, Ikeda M (2016) l-Lysine production independent of the oxidative pentose phosphate pathway by Corynebacterium glutamicum with the Streptococcus mutans gapN gene. Metab Eng 37:1–10. https://doi.org/10.1016/j.ymben.2016.03.007
Tang JK, You L, Blankenship RE, Tang YJ (2012) Recent advances in mapping environmental microbial metabolisms through 13C isotopic fingerprints. J R Soc Interface 9(76):2767–2780. https://doi.org/10.1098/rsif.2012.0396
Templeton N, Smith KD, McAtee-Pereira AG, Dorai H, Betenbaugh MJ, Lang SE, Young JD (2017) Application of 13C flux analysis to identify high-productivity CHO metabolic phenotypes. Metab Eng 43(Pt B):218–225. https://doi.org/10.1016/j.ymben.2017.01.008
Tesch M, de Graaf AA, Sahm H (1999) In vivo fluxes in the ammonium-assimilatory pathways in Corynebacterium glutamicum studied by 15N nuclear magnetic resonance. Appl Environ Microbiol 65(3):1099–1109
Tsuge Y, Yamamoto S, Kato N, Suda M, Vertes AA, Yukawa H, Inui M (2015) Overexpression of the phosphofructokinase encoding gene is crucial for achieving high production of D-lactate in Corynebacterium glutamicum under oxygen deprivation. Appl Microbiol Biotechnol 99(11):4679–4689. https://doi.org/10.1007/s00253-015-6546-9
Vallino JJ, Stephanopoulos G (1993) Metabolic flux distributions in Corynebacterium glutamicum during growth and lysine overproduction. Biotechnol Bioeng 41(6):633–646. https://doi.org/10.1002/bit.260410606
Vallino JJ, Stephanopoulos G (1994) Carbon flux distributions at the glucose-6-phosphate branch point in Corynebacterium glutamicum during lysine overproduction. Biotechnol Prog 10(3):327–334. https://doi.org/10.1021/Bp00027a014
van Ooyen J, Noack S, Bott M, Reth A, Eggeling L (2012) Improved L-lysine production with Corynebacterium glutamicum and systemic insight into citrate synthase flux and activity. Biotechnol Bioeng 109(8):2070–2081. https://doi.org/10.1002/bit.24486
van Winden WA, van Dam JC, Ras C, Kleijn RJ, Vinke JL, van Gulik WM, Heijnen JJ (2005) Metabolic-flux analysis of Saccharomyces cerevisiae CEN.PK113-7D based on mass isotopomer measurements of 13C-labeled primary metabolites. FEMS Yeast Res 5(6–7):559–568
Varela C, Agosin E, Baez M, Klapa M, Stephanopoulos G (2003) Metabolic flux redistribution in Corynebacterium glutamicum in response to osmotic stress. Appl Microbiol Biotechnol 60(5):547–555
Veit A, Rittmann D, Georgi T, Youn JW, Eikmanns BJ, Wendisch VF (2009) Pathway identification combining metabolic flux and functional genomics analyses: acetate and propionate activation by Corynebacterium glutamicum. J Biotechnol 140(1–2):75–83. https://doi.org/10.1016/j.jbiotec.2008.12.014
Villas-Boas SG, Mas S, Akesson M, Smedsgaard J, Nielsen J (2005) Mass spectrometry in metabolome analysis. Mass Spectrom Rev 24(5):613–646
von Kamp A, Thiele S, Hädicke O, Klamt S (2017) Use of CellNetAnalyzer in biotechnology and metabolic engineering. J Biotechnol 261:221–228. https://doi.org/10.1016/j.jbiotec.2017.05.001
Walker TE, Han CH, Kollman VH, London RE, Matwiyoff NA (1982) 13C nuclear magnetic resonance studies of the biosynthesis by Microbacterium ammoniaphilum of L-glutamate selectively enriched with carbon-13. J Biol Chem 257(3):1189–1195
Wang N, Ni Y, Shi F (2015) Deletion of odhA or pyc improves production of gamma-aminobutyric acid and its precursor L-glutamate in recombinant Corynebacterium glutamicum. Biotechnol Lett 37(7):1473–1481. https://doi.org/10.1007/s10529-015-1822-4
Wang L, Dash S, Ng CY, Maranas CD (2017) A review of computational tools for design and reconstruction of metabolic pathways. Synth Syst Biotechnol 2(4):243–252. https://doi.org/10.1016/j.synbio.2017.11.002
Wehrmann A, Phillipp B, Sahm H, Eggeling L (1998) Different modes of diaminopimelate synthesis and their role in cell wall integrity: a study with Corynebacterium glutamicum. J Bacteriol 180(12):3159–3165
Weitzel M, Nöh K, Dalman T, Niedenfuhr S, Stute B, Wiechert W (2013) 13CFLUX2—high-performance software suite for 13C-metabolic flux analysis. Bioinformatics 29(1):143–145. https://doi.org/10.1093/bioinformatics/bts646
Wendisch VF (2017) Microbial production of amino acid-related compounds. Adv Biochem Eng Biotechnol 159:255–269. https://doi.org/10.1007/10_2016_34
Wendisch VF, de Graaf AA, Sahm H, Eikmanns BJ (2000) Quantitative determination of metabolic fluxes during coutilization of two carbon sources: comparative analyses with Corynebacterium glutamicum during growth on acetate and/or glucose. J Bacteriol 182(11):3088–3096
Wiechert W, de Graaf A (1997) Bidirectional reaction steps in metabolic networks: I. Modeling and simulation of carbon isotope labeling experiments. Biotechnol Bioeng 55(1):102–117
Wiechert W, Möllney M, Petersen S, de Graaf AA (2001) A universal framework for 13C metabolic flux analysis. Metab Eng 3(3):265–283
Wieschalka S, Blombach B, Eikmanns BJ (2012) Engineering Corynebacterium glutamicum for the production of pyruvate. Appl Microbiol Biotechnol 94(2):449–459. https://doi.org/10.1007/s00253-011-3843-9
Winter G, Krömer JO (2013) Fluxomics—connecting ‘omics analysis and phenotypes. Environ Microbiol 15(7):1901–1916. https://doi.org/10.1111/1462-2920.12064
Wittmann C (2002) Metabolic flux analysis using mass spectrometry. Adv Biochem Eng Biotechnol 74:39–64
Wittmann C (2007) Fluxome analysis using GC-MS. Microb Cell Fact 6:6. https://doi.org/10.1186/1475-2859-6-6
Wittmann C (2010) Analysis and engineering of metabolic pathway fluxes in Corynebacterium glutamicum. Adv Biochem Eng Biotechnol 120:21–49. https://doi.org/10.1007/10_2009_58
Wittmann C, Becker J (2007) The L-lysine story. From metabolic pathways to industrial production. In: Wendisch VF (ed) Amino acid biosynthesis—pathways, regulation and metabolic engineering, Microbiology monographs, vol 5. Springer, Berlin, pp 40–68
Wittmann C, De Graaf AA (2005) Metabolic flux analysis in Corynebacterium glutamicum. In: Eggeling L, Bott M (eds) Handbook of Corynebacterium glutamicum. CRC, Boca Raton, pp 277–304
Wittmann C, Heinzle E (1999) Mass spectrometry for metabolic flux analysis. Biotechnol Bioeng 62(6):739–750
Wittmann C, Heinzle E (2001a) Application of MALDI-TOF MS to lysine-producing Corynebacterium glutamicum: a novel approach for metabolic flux analysis. Eur J Biochem 268(8):2441–2455
Wittmann C, Heinzle E (2001b) Modeling and experimental design for metabolic flux analysis of lysine-producing Corynebacteria by mass spectrometry. Metab Eng 3(2):173–191. https://doi.org/10.1006/mben.2000.0178
Wittmann C, Heinzle E (2002) Genealogy profiling through strain improvement by using metabolic network analysis: metabolic flux genealogy of several generations of lysine-producing Corynebacteria. Appl Environ Microbiol 68(12):5843–5859
Wittmann C, Hans M, Heinzle E (2002) In vivo analysis of intracellular amino acid labelings by GC/MS. Anal Biochem 307(2):379–382
Wittmann C, Kiefer P, Zelder O (2004a) Metabolic fluxes in Corynebacterium glutamicum during lysine production with sucrose as carbon source. Appl Environ Microbiol 70(12):7277–7287. https://doi.org/10.1128/AEM.70.12.7277-7287.2004
Wittmann C, Kim HM, Heinzle E (2004b) Metabolic network analysis of lysine producing Corynebacterium glutamicum at a miniaturized scale. Biotechnol Bioeng 87(1):1–6. https://doi.org/10.1002/bit.20103
Wittmann C, Weber J, Betiku E, Krömer J, Bohm D, Rinas U (2007) Response of fluxome and metabolome to temperature-induced recombinant protein synthesis in Escherichia coli. J Biotechnol 132(4):375–384. https://doi.org/10.1016/j.jbiotec.2007.07.495
Wu W, Zhang Y, Liu D, Chen Z (2019) Efficient mining of natural NADH-utilizing dehydrogenases enables systematic cofactor engineering of lysine synthesis pathway of Corynebacterium glutamicum. Metab Eng 52:77–86. https://doi.org/10.1016/j.ymben.2018.11.006
Xu J, Zhang J, Guo Y, Zai Y, Zhang W (2013) Improvement of cell growth and L-lysine production by genetically modified Corynebacterium glutamicum during growth on molasses. J Ind Microbiol Biotechnol 40(12):1423–1432. https://doi.org/10.1007/s10295-013-1329-8
Xu JZ, Zhang JL, Guo YF, Jia QD, Zhang WG (2014) Heterologous expression of Escherichia coli fructose-1,6-bisphosphatase in Corynebacterium glutamicum and evaluating the effect on cell growth and L-lysine production. Prep Biochem Biotechnol 44(5):493–509. https://doi.org/10.1080/10826068.2013.833115
Xu JZ, Wu ZH, Gao SJ, Zhang W (2018) Rational modification of tricarboxylic acid cycle for improving L-lysine production in Corynebacterium glutamicum. Microb Cell Fact 17(1):105. https://doi.org/10.1186/s12934-018-0958-z
Yamamoto S, Suda M, Niimi S, Inui M, Yukawa H (2013) Strain optimization for efficient isobutanol production using Corynebacterium glutamicum under oxygen deprivation. Biotechnol Bioeng 110(11):2938–2948. https://doi.org/10.1002/bit.24961
Yanase M, Aikoh T, Sawada K, Ogura K, Hagiwara T, Imai K, Wada M, Yokota A (2016) Pyruvate kinase deletion as an effective phenotype to enhance lysine production in Corynebacterium glutamicum ATCC13032: redirecting the carbon flow to a precursor metabolite. J Biosci Bioeng 122(2):160–167. https://doi.org/10.1016/j.jbiosc.2015.12.023
Yang TH, Heinzle E, Wittmann C (2005) Theoretical aspects of 13C metabolic flux analysis with sole quantification of carbon dioxide labeling. Comput Biol Chem 29(2):121–133. https://doi.org/10.1016/j.compbiolchem.2005.02.005
Yang TH, Wittmann C, Heinzle E (2006a) Respirometric 13C flux analysis—Part II: in vivo flux estimation of lysine-producing Corynebacterium glutamicum. Metab Eng 8(5):432–446. https://doi.org/10.1016/j.ymben.2006.03.002
Yang TH, Wittmann C, Heinzle E (2006b) Respirometric 13C flux analysis, Part I: design, construction and validation of a novel multiple reactor system using on-line membrane inlet mass spectrometry. Metab Eng 8(5):417–431. https://doi.org/10.1016/j.ymben.2006.03.001
Yokota A, Lindley ND (2005) Central metabolism: sugar uptake and conversion. In: Eggeling L, Bott M (eds) Handbook of Corynebacterium glutamicum. CRC, Boca Raton, pp 215–240
Yokota A, Sawada K, Wada M (2017) Boosting anaplerotic reactions by pyruvate kinase gene deletion and phosphoenolpyruvate carboxylase desensitization for glutamic acid and lysine production in Corynebacterium glutamicum. Adv Biochem Eng Biotechnol 159:181–198. https://doi.org/10.1007/10_2016_31
Yuan Y, Yang TH, Heinzle E (2010) 13C metabolic flux analysis for larger scale cultivation using gas chromatography-combustion-isotope ratio mass spectrometry. Metab Eng 12(4):392–400. https://doi.org/10.1016/j.ymben.2010.02.001
Zhang Y, Shang X, Wang B, Hu Q, Liu S, Wen T (2019) Reconstruction of tricarboxylic acid cycle in Corynebacterium glutamicum with a genome-scale metabolic network model for trans-4-hydroxyproline production. Biotechnol Bioeng 116(1):99–109. https://doi.org/10.1002/bit.26818
Zhao Z, Ding JY, Ma WH, Zhou NY, Liu SJ (2012) Identification and characterization of gamma-aminobutyric acid uptake system GabPCg (NCgl0464) in Corynebacterium glutamicum. Appl Environ Microbiol 78(8):2596–2601. https://doi.org/10.1128/AEM.07406-11
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Becker, J., Wittmann, C. (2020). Pathways at Work: Metabolic Flux Analysis of the Industrial Cell Factory Corynebacterium glutamicum . In: Inui, M., Toyoda, K. (eds) Corynebacterium glutamicum. Microbiology Monographs, vol 23. Springer, Cham. https://doi.org/10.1007/978-3-030-39267-3_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-39267-3_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-39266-6
Online ISBN: 978-3-030-39267-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)