
Abstract
The Gram-positive soil bacterium Corynebacterium glutamicum was discovered as a natural overproducer of glutamate about 50 years ago. Linked to the steadily increasing economical importance of this microorganism for production of glutamate and other amino acids, the quest for efficient production strains has been an intense area of research during the past few decades. Efficient production strains were created by applying classical mutagenesis and selection and especially metabolic engineering strategies with the advent of recombinant DNA technology. Hereby experimental and computational approaches have provided fascinating insights into the metabolism of this microorganism and directed strain engineering. Today, C. glutamicum is applied to the industrial production of more than 2 million tons of amino acids per year. The huge achievements in recent years, including the sequencing of the complete genome and efficient post genomic approaches, now provide the basis for a new, fascinating era of research – analysis of metabolic and regulatory properties of C. glutamicum on a global scale towards novel and superior bioprocesses.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- 13C metabolic flux analysis
- Anaplerosis
- Genome-scale model
- Glutamate
- Lysine
- Metabolic engineering
- NADPH
- Systems biology
- Systems metabolic engineering
- Tryptophan
1 Introduction
C. glutamicum was discovered about 50 years ago. Stimulated by the increasing demand for l-glutamate as a flavor enhancer in human nutrition, a screening program in Japan led to the isolation of a soil microorganism, later classified as C. glutamicum, which was able to accumulate l-glutamate in the growth medium [1, 2]. Subsequent analysis revealed that glutamate secretion could be triggered by a limited supply of biotin, opening the possibility for industrial production. The application potential of C. glutamicum in biotechnology soon increased with successful manipulation of key regulatory properties that allowed accumulation of other amino acids such as lysine, threonine, arginine, or ornithine [3]. These findings initiated intensive research over the past decades. One of the key areas of research was the development of efficient production strains of C. glutamicum for which different strategies were pursued. This included classical mutagenesis and selection. With the advent of recombinant DNA technology, targeted genetic engineering of C. glutamicum became available and was successfully used to derive improved production properties. Today, C. glutamicum is applied to the industrial production of glutamate as flavor enhancer in human nutrition (1.5 million tons per year), [4], lysine (850,000 tons per year) [5] as well as tryptophan (10,000 tons per year) as additive in animal feed [6]. The improvement of existing production strains focuses on higher yield and productivity, better stress tolerance, or a broader substrate spectrum. In addition, current research also aims at the creation of efficient processes for biobased production of novel products involving other amino acids, organic acids, or biofuels. In this regard the present chapter highlights the major contributions dealing with investigation and optimization of the metabolic network of C. glutamicum for biotechnology. Hereby, concepts and approaches for systems biology analysis and engineering of metabolic pathways of C. glutamicum are presented and illustrated with actual examples.
2 Metabolism of Corynebacterium glutamicum
The extensive biochemical and physiological analysis of C. glutamicum during the past as reviewed in a recent handbook on C. glutamicum [7] provides a rich source of information on many of the enzymes and pathways present in this organism.
2.1 Nutritional Requirements and Assimilatory Pathways
C. glutamicum can use a multitude of organic compounds as sole carbon source or via coutilization. Sugars such as glucose, fructose, sucrose, or mannose are taken up by a phosphotransferase system [8, 9]. For glucose, an alternative uptake system involving intracellular phosphorylation of glucose by a glucokinase has been identified [10]. In addition, C. glutamicum can grow on different organic acids such as gluconate [11], lactate, [12], acetate [13], propionate [14], or citrate [15]. For growth and also for amino acid overproduction, cells further require nitrogen and sulfur as major elements in addition to carbon. Suitable nitrogen sources are ammonium or organic compounds such as urea or amino acids [16]. For the uptake of ammonium, two alternative systems are present. At high ammonium levels, assimilation is mainly catalyzed by the glutamate dehydrogenase (GDH), whereas a highly affine system comprising glutamine synthetase and GDH is active under low ammonium levels, although at higher energy expenditure [17]. Among suitable sulfur sources, inorganic sulfate is most common. Its assimilation, however, involves a high demand of redox power for reduction to the biologically compatible sulfide [18]. Other sulfur sources assimilated by C. glutamicum are cysteine, sulfonates, or sulfonate esters [18, 19]. With respect to industrial application the most important raw materials are based on molasses (Asia) and starch (America, Europe) applied together with inorganic salts [5]. Thus, the major nutrients utilized are glucose, sucrose and fructose (carbon), ammonia (nitrogen), and sulfate (sulfur).
2.2 Central Carbon Metabolism
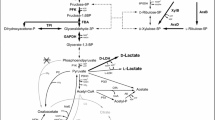
Today the metabolic network of the central metabolism of C. glutamicum involving glycolysis, pentose phosphate pathway (PPP), TCA cycle as well as anaplerotic and gluconeogenetic reactions is well known (Fig. 1). Different enzymes are involved in the interconversion of carbon between TCA cycle (malate/oxaloacetate) and glycolysis (pyruvate/phosphoenolpyruvate). For anaplerotic replenishment of the TCA cycle, C. glutamicum exhibits pyruvate carboxylase [20] and phosphoenolpyruvate (PEP) carboxylase as carboxylating enzymes. Malic enzyme [21] and PEP carboxykinase [22, 23] catalyze decarboxylation reactions from the TCA cycle towards glycolysis. As additional gluconeogenetic enzymes, oxaloacetate decarboxylase [24] and PEP synthetase [25] have been proposed. It has been proposed that cyclic cooperation of these carboxylating and decarboxylating enzymes around the pyruvate node is involved in the regeneration of excess ATP [22, 26, 27]. Different reactions are linked to the supply of NADPH. The major enzymes are glucose 6-phosphate dehydrogenase [28, 29] and 6-phosphogluconate dehydrogenase [28, 30] in the oxidative part of the PPP and the TCA cycle enzyme isocitrate dehydrogenase [31]. In selected cases, NADPH supply might involve malic enzyme [32, 33]. In addition to the central catabolism, anabolic routes have also been elucidated. Through rigorous analysis of the cellular composition, detailed information on the anabolic precursor demand was obtained [34]. These analyses were mainly driven by approaches for metabolic flux analysis in growing C. glutamicum, where anabolic reactions drain carbon, cofactors, and energy from central catabolism and thus have to be considered. In total, about (16.4 mmol NADPH) (g biomass)−1 is required for anabolism. Considering a biomass yield of 0.5 (g dry biomass) (g glucose)−1, which is achieved by C. glutamicum under aerobic conditions, this results in 1.7 mol NADPH (mol glucose)−1 that have to be generated by the NADPH forming reactions in the PPP and the TCA cycle. This substantial anabolic NADPH requirement competes with demand for production pathways, e.g., towards lysine or methionine.

Central metabolic pathways in C. glutamicum
2.3 Biosynthetic Pathways Towards Biotechnology Products
Among the most important amino acids for industrial production are glutamate, the aromatic amino acids, and amino acids belonging to the aspartate family. The biosynthesis of these amino acids is closely linked to the central metabolism (Fig. 2). It is strictly controlled at several steps in C. glutamicum as is typically the case in microbial systems [35]. Obviously, the required precursor metabolites, cofactors and energy have to be supplied in appropriate amounts by the central catabolic routes, thus competing with the cellular requirements for growth. Particular problems with the synthesis of amino acids from the aspartate family are the long biosynthetic pathways and the highly connected network involving intermediates and reactions that are shared at the same time by different biosynthetic routes. Therefore, it is not surprising that multiple regulatory steps are required to ensure the balanced synthesis of all these metabolites for cellular demands (Fig. 2). An important branch point occurs at the level of aspartate semialdehyde, where the biosynthetic pathways separate. Upstream of this node is a key point for flux control of lysine biosynthesis. The responsible enzyme is aspartokinase, which catalyzes the formation of aspartylphosphate from aspartate. It is subjected to feedback inhibition by lysine and threonine [36, 37]. Downstream of this node, C. glutamicum exhibits a dual pathway for lysine biosynthesis, providing an increased flexibility in response to changing environmental conditions [38, 39]. The synthesis of methionine involves complex networks of regulatory interactions and metabolic pathways. Even with over-expressing of almost all genes involved in methionine biosynthesis via deletion of the central repressor McbR, C. glutamicum does not over-produce this amino acid as shown recently, pointing at other (regulatory) mechanisms that are limiting [40]. Due to extended research, complex and efficient feedback inhibition mechanisms have recently been identified in addition to extended transcriptional control [41, 42]. A similar complex picture is yielded for biosynthesis of the aromatic amino acids, where enzyme inhibition by feedback regulation and negative transcriptional control are involved [6, 43].

Metabolic pathways in C. glutamicum for biosynthesis of the aromatic amino acids tryptophan, tyrosine, and phenylalanine (a) and amino acids belonging to the aspartate family including lysine, methionine, threonine, and isoleucine (b). Metabolic regulation by feedback inhibition is indicated by dotted lines
2.4 Genome Sequencing and Metabolic Network Reconstruction
These previous studies contributed significantly to our current understanding of the physiology of C. glutamicum. They each focused on only certain aspects of metabolism and investigated the corresponding reactions as isolated parts, so that an integrated view on metabolism of C. glutamicum as a functional network of highly interconnected reactions could not be provided. Such a holistic view, however, is highly desirable to understand the underlying complex system of metabolic and regulatory networks and derive ideas and concepts on how to engineer them in order to achieve superior production strains. As described above, the synthesis of industrial amino acids competes with cellular metabolism for building blocks and cofactors. As an example, pyruvate as precursor for lysine is potentially involved in more than 150 metabolic reactions of C. glutamicum. Keeping this in mind, we need a global picture of metabolism to understand the link between all these reactions and find the most efficient strategies to modify the flux to achieve optimum network performance. Metabolic flux analysis displays the first systems oriented approach to unravel the physiology of C. glutamicum since it combines experimental data with metabolic network models and allows determining absolute fluxes through larger networks comprising 100–200 reactions of central carbon metabolism [44]. A further milestone towards systems level understanding was the sequencing of the C. glutamicum ATCC 13032 genome by at least three different biotechnological companies (BASF, Degussa, and Kyowa Hakko). The circular genome comprises about 3000 genes with a total size of 3.3 kb [45–47]. Subsequent annotation has helped greatly in elucidating the genetic repertoire. Today, the genomic information of C. glutamicum is publicly available in bioinformatics data bases. Recommendable databases are provided by KEGG (www.genome.jp) or BioCyc (www.biocyc.com) and contain graphical information on genes, proteins, reactions, and pathways which is very useful when linking metabolites, enzymes and reactions to associate metabolism. The availability of genome information enabled new post genome technologies accelerating strain engineering by systems biology approaches. These comprise experimental and computational tools such as transcriptomics [48–51], proteomics [52–54], or in silico pathway modeling [55, 56]. Meanwhile, sequence information is also available for other closely related species providing a detailed overview on the corynebacterial pan-genome and its metabolic pathways. Sequenced strains include the thermotolerant C. efficiens [57], C. glutamicum R [58], and the pathogens C. diphteriae [59], C. jeikeium [60], and C. kroppenstedtii [61].
3 In Silico Metabolic Network Analysis
The engineering approach to analysis and design is using a mathematical or computer model. For target identification, modeling approaches are very useful to extract useful information on metabolic networks, their regulation and capacity. Models can be applied to predict phenotype behavior in response to different environmental or genetic perturbations, integrate complex data sets towards systems oriented understanding of network function, or design organisms with optimal network structure and activity. Overall, a wide variety of computational methods exploiting metabolic models have been developed and applied to C. glutamicum, yielding valuable insights into its metabolism and providing a sound basis for computer-assisted design in metabolic engineering. The type and complexity of the model hereby depends on the aim of the study. Although the ultimate goal of modeling is the development of dynamic models for the complete simulation of cellular systems, the success of such approaches has been severely hampered by the lack of kinetic information [62]. However, it is possible to assess accurately the theoretical capabilities and operative modes of metabolic systems using stoichiometric models.
3.1 Genome-Scale Reconstruction of the Metabolic Network
With the availability of annotated genome sequences, it has become possible to reconstruct genome-scale biochemical reaction networks for microorganisms. Genome-scale models have been reconstructed for almost 20 bacterial species so far [63–65]. Such models bridge the gap between genome-derived biochemical information and metabolic phenotypes and enable straightforward in silico experiments with whole-cell metabolism. Also for C. glutamicum, a genome-scale stoichiometric model was recently created [55]. As for other microorganisms, the construction was carried out in several steps. First a crude model consisting of mass balances for catabolic reactions was assembled from information on the present metabolic pathways and their stoichiometry available in KEGG and BioCyc. Subsequently, the anabolic reactions involved in polymer and biomass synthesis were defined whereby the information was collected from different literature sources. Finally the model was cured and completed by adding missing reactions and integrating information on reaction reversibility. The C. glutamicum model was validated against data found in the literature under different conditions such as different biomass production burdens and growth on different carbon sources. The model comprises 446 reactions and 411 metabolites (Table 1). Overall, it displays a systematic verification and compilation of data from various sources concerning the metabolic network of C. glutamicum and provides a useful basis for in silico studies as well a first step for future integration of complex data sets in systems biology approaches.
3.2 Stoichiometric Modeling
New genome wide stoichiometry based modeling of metabolic pathways is now possible. The stoichiometric data and models available provide the basis for various in silico applications differing in the methodology and algorithms used. Major contributions for C. glutamicum have been obtained from flux balancing [66] and elementary (flux) mode analysis [67]. In short, flux balance analysis is a constraint based method to analyze stoichiometric networks. Through introduction of specific biological constraints the set of various possible solutions for the typically underdetermined metabolic system can be limited to a certain solution space. Within this space, optimal steady-state solutions can be calculated by minimizing or maximizing for objective functions such as growth or overproduction of a metabolite using linear programming techniques [68]. Accordingly, using flux balance analysis, a single solution is found to the optimization problem [69]. Hereby, flux balance analysis can highlight the most efficient pathway through the network in order to achieve the particular objective function. Elementary flux mode analysis systematically enumerates all independent minimal pathways through a network, each a unique elementary mode, that are stoichiometrically and thermodynamically feasible [70]. All possible steady-state flux distributions through the metabolic network are nonnegative linear combinations of the set of elementary modes [71]. This can be exploited to extract key properties from metabolic networks such as maximum network capacity, optimal pathways, network robustness, or phenotype prediction in response to environmental or genetic perturbations [56, 72, 73].
3.2.1 Capacity of the Network of C. glutamicum for Lysine Production
For C. glutamicum stoichiometric network modeling has been utilized to assess the capacity for the production of lysine as one of the major industrial products [5, 11, 55, 74, 75]. The theoretical maximum molar yield of C. glutamicum for lysine production obtained by elementary flux mode analysis is 0.82 mol mol−1 [5]. Under these conditions of zero growth, PPP and malic enzyme supply the required NADPH, whereas the TCA cycle is shut off and lysine is exclusively formed via the dehydrogenase branch (Fig. 3a). This mode would require a metabolic cycle of pyruvate carboxylase, malate dehydrogenase, and malic enzyme acting as a transhydrogenase to convert NADH into NADPH. Biochemical evidence for such a cycle has not been obtained so far, but a similar cyclic pathway involving pyruvate carboxylase, malate dehydrogenase, and PEP carboxykinase has recently been shown to operate in vivo in C. glutamicum [22, 27]. Omitting the transhydrogenase-like cycle from the network results in a slightly lower maximum lysine yield of 0.75 mol mol−1 [77]. Admittedly, all these scenarios imply zero growth which can hardly be realized in a real fermentation. Due to this, the achievable optimum can be expected to be somewhat lower than the value calculated here, but should still be significantly higher than the yield achieved in practice, which is in the range of about 40–50%. Further insight into lysine production has been obtained from flux balance analysis characterizing different in silico mutants and metabolic scenarios [55]. The model described experimental observations from flux analysis of C. glutamicum fairly well, but failed in certain rather important aspects. The reason is that biochemical regulation affecting the fluxes cannot be accounted for and reactions with identical overall stoichiometry cannot be resolved using such stoichiometric approaches.

Metabolic flux distribution of C. glutamicum reflecting maximal theoretical carbon yield of lysine (a) and methionine (b) from glucose. All fluxes are given as relative molar fluxes to the glucose uptake, whereby the relative flux of a reaction is reflected by the thickness of the corresponding arrow. The data are taken from recent simulation studies [5, 76]
3.2.2 Rational Design of C. glutamicum for Methionine Production
Elementary flux mode analysis has been further applied to study possible routes for the production of methionine in C. glutamicum [56]. The maximum theoretical methionine yield on glucose was calculated as 0.59 mol mol−1 (Fig. 3b). Also here, a detailed insight into the metabolic network is obtained. The PPP flux supporting the high yield is even higher than that for optimal lysine production, which reflects the enormous amount of 8 NADPH required per methionine. Additional simulations identified promising genetic targets for improved production such as heterologous expression of a transhydrogenase or of a glycine cleavage system. Moreover, it could be shown that the supply of reduced sulfur is beneficial for high carbon yield. The most effective sulfur source was methanethiol, allowing an almost complete conversion of glucose into methionine with a yield of 0.91 C-mol C-mol−1. Summarizing, stoichiometric modeling studies for C. glutamicum provide a sound basis for strain or process optimization. By calculating the theoretical capacity of an organism for a novel product they also allow useful estimates on the general economical feasibility [56].
3.3 Dynamic Modeling Approaches
Dynamic metabolic network models theoretically allow a most comprehensive and directed optimization of a pathway or of a whole cell. Therefore the ultimate goal of systems biology is the development of dynamic models for the complete simulation of cellular systems. Such dynamic models, however, require the understanding of all interactions influencing the reaction rates in the network. We are still far from that. For C. glutamicum detailed dynamic models, based on mechanistic equations for the participating enzymes and concentration measurements of the pathway intermediates involved, have been developed at least for the biosynthesis of lysine [78] and valine [79]. Through metabolic control analysis, such models allow the prediction of bottlenecks and of optimal factors, e.g., enzyme concentrations to achieve increased flux. Admittedly, for most pathways of C. glutamicum such kinetic information is still not available. This can be partly overcome by the application of power law or lin-log kinetics in the underlying equations [79, 80]. Dynamic models are also needed for dynamic labeling experiments for flux analysis as has been shown for E. coli [81]. The current progress in method development for metabolomics of C. glutamicum will surely stimulate the future generation of dynamic models for such organisms by providing extended data sets on intracellular metabolite concentrations required to derive in vivo kinetics [82–85]. For many years, dynamic macroscopic models have been used to describe dynamic phenomena of growth and production and derive control or feeding strategies for optimized bioprocesses. A recent example deals with fed-batch production of valine [79]. Such overall phenomenological models are surely useful but do not provide any detailed description of network activities [86].
4 Analysis of Metabolic Fluxes
In recent years, powerful approaches were developed which allow the quantification of small molecule fluxes through metabolic networks, i.e., in vivo reaction rates. Among the achievements obtained from flux analysis are the identification of novel pathways, the elucidation of metabolic control, identification of targets for rational strain engineering in biotechnology, and first insights into design principles of metabolic networks in systems biology studies. Concerning C. glutamicum, metabolic flux analysis provided a fascinating view of its metabolic pathways and offered new possibilities for rational strain engineering as recently reviewed [34, 87].
4.1 Tools and Concepts
4.1.1 Conventional Metabolic Flux Analysis by Stoichiometric Balancing
First flux estimates through larger parts of its metabolism were based on constraining assumed reaction networks with measurement of uptake and production rates [74]. A number of studies utilized stoichiometric balancing to assess the flexibility of the metabolic network [11, 75] and to investigate the influence of environmental conditions such as dissolved oxygen level [88], salt content [89], or nutrient status [12, 90, 91]. However, this conventional approach cannot yield reliable information about parallel or bidirectional reactions and has to rely on balances for NADH or NADPH, which may not be accurate [34]. Moreover, it is limited to derive new conclusions since the results are strongly based on the taken assumptions and not on data [92].
4.1.2 State-of-the-Art 13C Metabolic Flux Analysis and Current Developments
Recent developments combining stoichiometric networks with additional intracellular information from 13C-isotopomer analysis have overcome these limitations and display the state-of-the-art in metabolic flux analysis. Hereby, the feeding of 13C labeled substrates to the growing cells leads to a distribution of the 13C label throughout the metabolic network. The resulting labeling pattern in the intermediary metabolites and other cellular constituents depends on the well defined and well known carbon transfer of the involved reactions and their particular fluxes. As such, the labeling patterns contain the key information about the fluxes and can be used for their estimation. For label quantification, MS or NMR techniques are available, whereby MS has evolved to the preferred method due to its high accuracy, sensitivity, speed, and robustness [93, 94]. The most common method hereby exploits the 13C pattern in proteinogenic amino acids as a rich source of labeling information for the determination of steady-state fluxes in growing cells [95, 96]. Respirometric flux analysis, conceptually based on sole labeling measurement of CO2 extended the application of flux analysis to nongrowing cells [97–99]. Additionally, labeling measurement of intracellular metabolites has also received increasing attraction since it offers the possibility to resolve fluxes under dynamic conditions [81, 100]. After a decade of intense research and development, such 13C-based flux methods can routinely track steady-state fluxes in microbes [92]. Convenient software tools can be used to calculate the intracellular fluxes from the labeling data utilizing either global parameter fitting from the entire labeling data set [101] or the estimation of local flux ratios from selected labeling patterns [102].
4.2 Metabolic Fluxes in C. glutamicum
With respect to metabolic fluxes, C. glutamicum is probably the most extensively studied organism so far [34]. Metabolic flux analysis in this organism has become a fundamental tool of strain engineering, since it provides otherwise not accessible key data on cellular function and regulation. The first pioneering studies of C. glutamicum using stable isotopes to elucidate metabolic properties were initiated soon after its isolation [103]. Focusing each on selected reaction, these labeling studies identified the dual lysine pathway in C. glutamicum [104] or provided first estimates on central pathways, i.e., citric acid cycle, anaplerosis, and glyoxylate shunt [105]. The major findings on metabolic fluxes and their regulation in C. glutamicum result from the fully integrated approaches comprising stoichiometric and isotopomer balancing with 13C labeling and thus are quite recent (Table 2).
4.2.1 Metabolic Fluxes of Precursor Metabolism
The synthesis of relevant amino acids such as lysine, threonine, methionine, or glutamate demands for precursor compounds stemming from the TCA cycle. This has stimulated intensive research on fluxes involved in replenishment of the TCA cycle, involving pyruvate carboxylase, PEP carboxylase as carboxylating enzymes, as well as malic enzyme and PEP carboxykinase that catalyze decarboxylation reactions from the TCA cycle towards glycolysis. Metabolic flux analysis unraveled the metabolism around the pyruvate node in great detail (Fig. 4a). It could be shown that carboxylating and decarboxylating enzymes are active at the same time, forming a cyclic pathway for interconversion of C4 metabolites of the TCA cycle and C3 metabolites of glycolysis [27]. This cyclic pathway is highly flexible, as cells can redistribute the flux depending on the metabolic burden (Fig. 4b–d). The role of this cycle has been attributed to regeneration of excess ATP under certain conditions [121, 122] or equilibration of metabolite levels around the pyruvate node [123]. Additionally, a contribution to NADPH metabolism has been suggested [12, 124]. Moreover, flux studies identified the enzymes around the pyruvate node as key targets for metabolic engineering of the precursor supply in C. glutamicum. An increase of flux through the lysine pathway is linked to an increase of flux through the carboxylating enzymes, providing the lysine precursor oxaloacetate (Fig. 5a). In contrast, the flux through the decarboxylating enzymes, withdrawing oxaloacetate, is negatively correlated with lysine production (Fig. 5b). In light of these findings, amplification of the carboxylating and deletion of the decarboxylating enzymes became a promising strategy to enhance the anaplerotic net flux for improved production.

Metabolic fluxes around the pyruvate node of C. glutamicum under different physiological conditions: Growth of the wild type C. glutamicum ATCC 13032 in continuous culture on glucose [27] (a), batch cultivation of the lysine producing strain C. glutamicum ATCC 13032 lysCfbr (b), its pyruvate kinase deficient variant C. glutamicum ATCC 13032 lysCfbr Δpyk [120] (c), and glutamate production in batch culture by C. glutamicum AJ-1511 (ATCC13869) on glucose [118] (d). All fluxes are given as relative molar fluxes to entry flux into the PEP pool

Metabolic flux analysis of precursor supply and production of lysine (a, b) and glutamate (c) in different C. glutamicum strains investigated under various cultivation conditions by 13C flux analysis: [112] (white circles), [125] (gray circles), [126] (black circles), [127] (white squares), [111] (gray squares), [111] (black squares), [128] (white triangles), [116] (gray triangles), [26] (black triangles), [129] (white diamonds), [115] (gray diamonds), [120] (black diamonds), [76] (white hexagons), and [118] (gray hexagons). The flux reversibility at the pyruvate node is defined as the ratio of the back flux to the anaplerotic net flux [130]
Beyond lysine, the close link of anaplerotic replenishment of the TCA cycle and production could be also observed for the formation of glutamate, demanding for the TCA cycle precursor α-ketoglutarate (Fig. 5c). Regarding the supply of α-ketoglutarate as product precursor, flux analysis revealed that the specific activity of isocitrate dehydrogenase (ICDH) and GDH did not show large changes throughout the fermentation, while that of the α-ketoglutarate dehydrogenase complex (ODHC) significantly decreased upon induction of glutamate production, clearly leading to flux redistribution (Shirai et al. 2005). The results suggest that ODHC plays the largest role in controlling flux at the key branch point of α-ketoglutarate from the view of metabolic flux.
4.2.2 Metabolic Fluxes of NADPH Metabolism
The synthesis of most amino acids demands NADPH so that efficient supply of this cofactor is highly relevant in production processes. In C. glutamicum, glucose 6-phosphate dehydrogenase, 6-phosphogluconate dehydrogenase, isocitrate dehydrogenase, and malic enzyme catalyze NADPH generating reactions, whereas NADPH consuming reactions comprise growth with a stoichiometric demand of 16.4 mmol NADPH (g biomass)−1 [34] and overproduction. From 13C metabolic flux studies of lysine producing strains under different physiological conditions, remarkable insights into function and regulation of the NADPH metabolism of C. glutamicum could be obtained. The NADPH metabolism of C. glutamicum is highly flexible, adjusting to the physiological growth state [126], the nutrient status [13, 32, 111], or the genetic background [108, 131]. In most cases this results in an apparent NADPH excess, pointing at so far unassigned reactions which consume NADPH. Possible, but not validated, candidates are NADPH oxidase [132] or cyclic fluxes around the pyruvate node involving malic enzyme [27, 124]. Towards higher production, this apparent excess diminishes and turns into an apparent NADPH limitation [5]. Extrapolating these findings to industrial producers with much higher yields makes a limitation of production by NADPH very likely and strongly suggests the pathways involved in NADPH supply as promising targets for metabolic engineering of C. glutamicum. Hereby, stoichiometric investigation of the lysine network almost 20 years ago predicted that an increased lysine yield is linked to an increased flux through the PPP [133]. The importance of the PPP for efficient lysine production was later shown by metabolic flux analysis. Studies, investigating different strains under different conditions, revealed a close link of lysine production flux with the flux through the PPP (Fig. 6a). In contrast to the PPP, the contribution of isocitrate dehydrogenase decreases with increasing lysine production (Fig. 6b). The role of malic enzyme is still not completely clear. Its contribution to NADPH supply has been demonstrated for growth on fructose [32] as well as in a pyruvate kinase deficient lysine production strain [120]. However, overexpression of the malic enzyme gene did not result in improved lysine production independent from the carbon source tested [134].

Metabolic flux analysis linking lysine production with NADPH metabolism involving the pentose phosphate pathway (a) and isocitrate dehydrogenase (b) in different C. glutamicum strains investigated under various cultivation conditions by 13C flux analysis: [125] (white circles), [129] (gray circles), [126] (black circles), [127] (white squares), [112] (gray squares), [126] (black squares), [128] (white triangles), [116] (gray triangles), [26, 76] (black triangles), [115] (white diamonds), and [120] (gray diamonds)
5 Metabolic Pathway Engineering
In C. glutamicum the biosynthesis of amino acids is strictly regulated through feedback inhibition. Therefore, overproduction requires, first of all, the removal of all metabolic control mechanisms. Moreover, the amplification of biosynthetic pathway genes is a logical step of strain improvement, especially in the presence of transcriptional repression. Moreover, balanced supply of precursor compounds and cofactors has to be addressed to achieve efficient production of the amino acid. For amino acids, these supporting pathways are part of the highly interconnected network of central carbon metabolism, so that modifications will likely interfere with growth related metabolic reactions. Due to these various hurdles, classical mutagenesis and selection did not yield high levels of production, although efforts have been continuing now for almost four decades [6]. The development of recombinant DNA technologies, allowing targeted genetic modification, has initiated intensive research towards rational optimization of C. glutamicum, resulting in remarkable progress in production efficiency [30, 135, 136]. In the following, the advances in metabolic pathway engineering of C. glutamicum are highlighted. Due to the strong impact of raw material costs, optimization of the conversion yield was at the focus of many studies in order to improve the economy of the production process [137].
5.1 Metabolic Engineering of Lysine Production
5.1.1 Metabolic Engineering of Lysine Biosynthesis
A number of studies have addressed the optimization of lysine production by direct modification of enzymes of the biosynthetic pathway (Fig. 7). The high relevance of engineering these enzymes is underlined by the fact that today every single gene of the lysine biosynthetic pathway is covered with patents by the major industrial players in the field [137]. The release of aspartate kinase, controlling the pathway flux, from feedback inhibition is one of the most important targets. Different point mutations in the lysC gene, i.e., its regulatory β-subunit, result in feedback resistant enzyme variants and increase lysine production [36, 138, 140]. Also, overexpression of aspartate kinase is beneficial for production [141]. Another relevant target is the dapA gene, encoding dihydrodipicolinate synthase. Amplified expression, increasing lysine was realized using plasmids [138, 142–144] as well as mutation of the promoter sequence [145]. Also, overproduction of diaminopimelate epimerase (DapF) and succinyl-aminoketopimelate transaminase (DapC), two enzymes of the succinylase branch, was beneficial for lysine formation [137]. Following the discovery of the lysine exporter (LysE), the subsequent overexpression of the lysE gene resulted in an increased lysine secretion rate [146–148]. The recently performed expression of lysE from C. glutamicum in a Methylophilus methylotrophus lysine producing strain was also shown to improve lysine production from methanol by this organism [149].

Metabolic engineering strategies for optimization of lysine production in C. glutamicum. Highlighted are modifications for improved supply of precursors and NADPH, for increased flux through lysine biosynthesis and secretion and for extension of the substrate spectrum. The optimization comprises decrease of flux (thick dotted arrow), via deletion or attenuation of genes (white box with dotted line), increase of flux (thick solid arrow) via amplification of genes (white box with solid line), modification of metabolic control for feedback regulated enzymes (dark grey box with solid line) and the introduction of new reactions (grey arrows) via heterologous expression of foreign genes (white box with grey solid line)
5.1.2 Metabolic Engineering of Precursor Supply
The importance of anaplerotic enzymes for supply of the lysine precursor oxaloacetate stimulated intensive metabolic engineering activities (Fig. 7). Among the most striking findings was the identification of pyruvate carboxylase as the major anaplerotic enzyme in C. glutamicum [27]. Subsequently, overexpression of its gene has been shown to improve lysine production [150]. Knowing the importance of pyruvate carboxylase for lysine production, the point mutation P458S, identified in a classically derived producer, was introduced into the pyc gene and also resulted in strong increase of lysine production [136]. Moreover, also overexpression of PEP carboxylase is beneficial for the formation of amino acids of the aspartate family [151]. In order to reduce the back flux from the TCA cycle and the withdrawal of oxaloacetate to glycolysis, deletion of PEP carboxykinase resulted in a significant improvement of lysine production [22]. For malic enzyme no clear effects could be observed so far. Neither deletion nor overexpression of the corresponding gene influenced the metabolism of C. glutamicum on sugars markedly [21, 152].
5.1.3 Metabolic Engineering of NADPH Supply
As revealed by metabolic flux analysis, the PPP is the major pathway for supply of NADPH required in high amounts for lysine biosynthesis. Due to this, different approaches have aimed at increasing flux through the PPP (Fig. 7). Metabolic flux studies on glucose, fructose, and sucrose identified fructose 1,6-bisphosphatase as a non-obvious target [110, 111]. Subsequent amplified expression of the fbp gene indeed increased lysine yield on glucose, fructose, and sucrose up to about 40% [128]. Hereby, the mutant with overexpression of fructose 1,6-bisphosphatase exhibited 10% enhanced PPP flux. A second major target approached was glucose 6-phosphate dehydrogenase. Overexpression of the encoding zwf gene in the feedback-deregulated lysine producing strain C. glutamicum ATCC13032 lysCfbr resulted in increased lysine production on different carbon sources including the two major industrial sugars, glucose and sucrose [116]. In further successful examples, an increased PPP flux was achieved by modifying the regulatory properties of enzymes in the PPP, i.e., partially releasing glucose 6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase from allosteric inhibition. The substitution A243T in the zwf gene encoding for glucose 6-phosphate dehydrogenase [116, 153] and the substitution S361F in the gnd gene encoding for 6-phosphogluconate dehydrogenase [30] both led to significantly increased lysine titer. The modifications cause an increase in the PPP flux which probably resulted from positively changed kinetics of the enzymes. For glucose based processes, disruption of glycolysis via deletion of phosphoglucose isomerase forces the cell to metabolize the substrate completely via the PPP, and indeed results in increased lysine production [108]. This strategy, however, is only applicable for glucose based processes, since other substrates such as sucrose, glycerol, fructose, xylose, or arabinose require an active phosphoglucose isomerase for channeling carbon into the PPP [111, 128].
5.2 Metabolic Engineering of Aromatic Amino Acid Production
Remarkable progress was also achieved for the synthesis of aromatic amino acids in C. glutamicum [6]. Effective approaches comprised the modification of terminal pathways leading to removal of undesired control mechanisms, engineering of the PPP for increased supply of precursors, and transport engineering leading to reduced intracellular pools. Among the most striking findings was the overexpression of the tkt gene in the PPP encoding transketolase which strongly enhanced tryptophan production by improved supply of the precursor E4P [154, 155]. To avoid accumulation of indole, the last intermediate in the tryptophan pathway, due to a limiting supply of l-serine, the availability of l-serine was improved by amplification of the serA gene for 3-phosphoglycerate dehydrogenase, the key enzyme in the l-serine biosynthesis [156]. A key to increased production was also the deletion of transport systems for aromatic amino acids impairing their uptake from the medium [6].
5.3 Metabolic Engineering of Glutamate Production
Concerning synthesis of glutamate in C. glutamicum which demands efficient replenishment of the TCA cycle, pyruvate carboxylase was identified as a major anaplerotic enzyme [118, 150]. The flux through pyruvate carboxylase strongly increased under glutamate production induced by Tween 40 addition, while PEP carboxylase and PEP carboxykinase fluxes remained constant. An impressive set of studies investigated the metabolism within the TCA cycle linked to glutamate production in great detail [4, 117, 157, 158]. Successful strategies to increase glutamate production involved deletion of odhA, encoding a key protein of the complex [159] or attenuation of odhA expression via the use of antisense RNA [160].
5.4 Utilization of Alternative Substrates
The major industrial carbon sources for fermentation processes with C. glutamicum are cane molasses, beet molasses, sucrose, and dextrose [3]. Since the carbon source is the major cost factor in industrial lysine production [137] and prices for molasses and raw sugar have increased significantly in recent years, many attempts have been aimed at extending the substrate spectrum of C. glutamicum to exploit alternative raw materials. Today, C. glutamicum has been engineered to utilize a large number of different substrates (Fig. 7). The future use of the corresponding raw materials in industrial fermentations of C. glutamicum, however, requires further intensive efforts in metabolic and process engineering. So far, key contributions enable the utilization of the pentose sugars which C. glutamicum naturally cannot metabolize. These sugars, namely xylose and arabinose, display significant fractions in agricultural residues and other lignocellulosic biomass recently receiving increasing interest as a cheap and most abundant raw material for biobased production [161]. Through heterologous expression of the E. coli genes xylA and xylB C. glutamicum was able to consume xylose as sole source of carbon [162]. In substrate mixtures, glucose-mediated regulation still exerts a measurable influence on xylose consumption kinetics. Similarly, arabinose utilization was also achieved, whereby the genes araA, araB, and araD were also derived from E. coli [163]. An interesting study recently investigated the tolerance of C. glutamicum to toxic compounds such as furfurals or phenols, typically present in lignocellulosic raw materials [164]. These compounds cause significant inhibition of growth. For growth-arrested production processes this is not a critical point, but open questions remain to be elucidated for the majority of bioprocesses with C. glutamicum which typically are fed-batch processes with important phases of growth of biomass to achieve efficient production. Introducing the S. griseus amy gene on an expression vector into a lysine producing strain of C. glutamicum allowed synthesizing and secreting of α-amylase into the culture broth [165]. Although some high-molecular-weight degradation products remained in the culture broth, the recombinant strain effectively used soluble starch as carbon and energy substrate for growth and also for lysine production. This could allow the direct conversion of starch into desired products and the avoidance of cost intensive hydrolysation pretreatment currently required when feeding starch hydrolysates from corn, wheat, or cassava for production [166]. C. glutamicum cannot utilize glycerol, a stoichiometric by-product of biodiesel production. By heterologous expression of Escherichia coli glycerol utilization genes, C. glutamicum was engineered to grow on glycerol [167]. The engineered strains were able to produce glutamate efficiently as well as lysine. Recent attempts to enable utilization of lactose by heterologous coexpression of genes from Lactococci have shown the basic feasibility, but are still linked to suboptimal performance of the obtained strains [168]. At present, metabolic engineering has significantly broadened the substrate spectrum of C. glutamicum.
5.5 Global Strain Engineering Through Applied Systems Biology
The experience of the past clearly shows that detailed quantitative knowledge of metabolic physiology is required for rational design of superior production strains [169]. Especially for the optimization of amino acid production by C. glutamicum, characterized by a close connection between central metabolism and product biosynthetic pathways, understanding of global metabolic regulation has turned out to be crucial [5]. In this light, systems biology approaches elucidating cell physiology on a global level, displaying powerful strategies. Metabolic flux analysis may be regarded as a first systems approach to C. glutamicum since it combines measurement data with metabolic network models of biosynthetic and central metabolic pathways and predicts metabolic engineering strategies based on such systems oriented insights. Hence, these systems biology approaches per se are not new concepts to design and improve C. glutamicum. However, the availability of the genome sequence and the rapid progress in postgenomics methods such as transcriptomics, proteomics, fluxomics, and metabolomics today allow one to study its metabolic and regulatory properties on a truly global level, opening a new era of industrial strain improvement [44]. Rational strain improvement was initially done on a gene-by-gene basis but was recently put on the genome level by comparing the genome sequence of producer and wild-type to identify relevant mutations. The success of this strategy, named “genome breeding” was successfully demonstrated for a C. glutamicum l-lysine producer [136]. From an engineering perspective this strategy worked, but it is still based on trial and error, so that a large number of targets identified from the genome sequence have to be tried. Moreover, it requires an already efficient production strain for target identification, which in the case of lysine took decades of development and is not available for other processes. Here, new omics tools from functional genomics can help to identify key regulators and guide rational strain engineering. Impressive progress has been made toward this goal, especially in the frame of systems biological study of C. glutamicum [44]. Transcriptome analysis through DNA microarrays [44, 170] allows global expression profiling of C. glutamicum. The proof of value of this technique for rational strain engineering has recently been shown, when novel targets for improved lysine production could be identified from selected transcriptome studies [171, 172]. Similarly, the analysis of the proteome, based on 2-D gel electrophoresis [52–54, 173] has also provided some insights into metabolic processes such as nitrogen starvation [174] or the utilization of citrate [15]. Among all systems oriented approaches, 13C metabolic flux analysis has clearly contributed the most to our current detailed picture of the C. glutamicum metabolism. Among the achievements obtained from flux analysis are the identification of novel pathways, the elucidation of metabolic control, identification of targets for rational strain engineering in biotechnology, and first insights into design principles of the metabolic network. Hereby, metabolic flux analysis provided quantitative data which directly reflect the phenotype of the investigated C. glutamicum strain, whereas other omics approaches often do not allow a direct conclusion on the active pathways determining the phenotypic behavior [76, 175]. Recent developments provide the next level of systems biology studies, the parallel investigation of C. glutamicum on levels of gene expression, proteins, metabolites, and fluxes providing important links between the different functional components of cellular physiology. First examples of such systems-oriented studies already reveal a great potential [76, 119, 176]. Such approaches are especially promising for the targeted multidimensional alteration of complex regulatory networks towards better tolerance of production strains to high temperature or salt levels, or extreme pH values [137], but also reveal a great potential for efficient design of novel bioprocesses. Similarly, systems based metabolic engineering approaches have also created impressive progress in developing superior strains of Escherichia coli, the second major industrial amino acid producer [177]. As an example, production of valine [178] or threonine [179] using E. coli could be substantially optimized.
5.6 Towards Novel Products
In light of the established fermentation on cheap substrates [5], its known genome sequence [47] and the availability of genetic engineering tools C. glutamicum is regarded as key candidate to develop production strain for various other products. In addition to the above-mentioned traditional products, C. glutamicum has been recently engineered to accumulate a great variety of different compounds (Table 3) Due to impressive work in recent years, C. glutamicum is applicable to produce efficiently amino acids, organic acids, diamines, or biofuels, and the coming years will surely see a further broadening of its potential.
References
Kinoshita S, Shigezo U, Shimono M (1957) J Gen Appl Microbiol 3:193
Udaka S (1960) J Bacteriol 79:754
Ikeda M (2003) Adv Biochem Eng Biotechnol 79:1
Schultz C, Niebisch A, Gebel L, Bott M (2007) Appl Microbiol Biotechnol 76:691
Wittmann C, Becker J (2007) The L-lysine story: from metabolic pathways to industrial production. In: Wendisch VF (ed) Amino acid biosynthesis – pathways, regulation and metabolic engineering. Springer, Berlin
Ikeda M (2006) Appl Microbiol Biotechnol 69:615
Eggeling L, Bott M (2005) Handbook of Corynebacterium glutamicum. CRC Press, Boca Raton
Dominguez H, Lindley ND (1996) Appl Environ Microbiol 62(10):3878
Moon MW, Kim HJ, Oh TK, Shin CS, Lee JS, Kim SJ, Lee JK (2005) FEMS Microbiol Lett 244:259
Park SY, Kim HK, Yoo SK, Oh TK, Lee JK (2000) FEMS Microbiol Lett 188:209
Vallino JJ, Stephanopoulos G (1994) Biotechnol Prog 10:327
Cocaign-Bousquet M, Lindley ND (1995) Enzyme Microb Technol 17:260
Wendisch VF, de Graaf AA, Sahm H, Eikmanns BJ (2000) J Bacteriol 182:3088
Claes WA, Pühler A, Kalinowski J (2002) J Bacteriol 184:2728
Polen T, Schluesener D, Poetsch A, Bott M, Wendisch VF (2007) FEMS Microbiol Lett 273:109
Burkovski A (2003) FEMS Microbiol Rev 27:617
Meier-Wagner J, Nolden L, Jakoby M, Siewe R, Kramer R, Burkovski A (2001) Microbiology 147:135
Lee H-S (2005) Sulfur metabolism and its regulation. In: Eggeling L, Bott M (eds) Handbook of Corynebacterium glutamicum. CRC Press, Taylor and Francis, Boca Raton
Rückert C, Koch DJ, Rey DA, Albersmeier A, Mormann S, Pühler A, Kalinowski J (2005) BMC Genomics 6:121
Peters-Wendisch PG, Kreutzer C, Kalinowski J, Patek M, Sahm H, Eikmanns BJ (1998) Microbiology 144(Pt 4):915
Gourdon P, Baucher MF, Lindley ND, Guyonvarch A (2000) Appl Environ Microbiol 66:2981
Riedel C, Rittmann D, Dangel P, Mockel B, Petersen S, Sahm H, Eikmanns BJ (2001) J Mol Microbiol Biotechnol 3:573
Jetten M, Sinskey AJ (1993) FEMS Microbiol Lett 111:183
Jetten MS, Sinskey AJ (1995) Antonie Van Leeuwenhoek 67:221
Jetten MS, Pitoc GA, Follettie MT, Sinskey AJ (1994) Appl Microbiol Biotechnol 41:47
Marx A, de Graaf A, Wiechert W, Eggeling L, Sahm H (1996) Biotechnol Bioeng 49(2):111
Petersen S, de Graaf AA, Eggeling L, Möllney M, Wiechert W, Sahm H (2000) J Biol Chem 275:35932
Moritz B, Striegel K, De Graaf AA, Sahm H (2000) Eur J Biochem 267:3442
Ihnen ED, Demain AL (1969) J Bacteriol 98:1151
Ohnishi J, Katahira R, Mitsuhashi S, Kakita S, Ikeda M (2005) FEMS Microbiol Lett 242:265
Audette GF, Quail JW, Hayakawa K, Bai C, Chen R, Delbaere LT (1999) Acta Crystallogr D Biol Crystallogr 55:1584
Dominguez H, Rollin C, Guyonvarch A, Guerquin-Kern JL, Cocaign-Bousquet M, Lindley ND (1998) Eur J Biochem 254:96
Kim HM, Heinzle E, Wittmann C (2006) J Microbiol Biotechnol 16:1174
Wittmann C, de Graaf A (2005) Metabolic flux analysis in Corynebacterium glutamicum. In: Eggeling L, Bott M (eds) Handbook of Corynebacterium glutamicum. CRC Press, Boca Raton
Michal G (1999) Biochemical pathways. Wiley, Chichester
Kalinowski J, Cremer J, Bachmann B, Eggeling L, Sahm H, Puhler A (1991) Mol Microbiol 5:1197
Malumbres M, Martin JF (1996) FEMS Microbiol Lett 143:103
Schrumpf B, Schwarzer A, Kalinowski J, Pühler A, Eggeling L, Sahm H (1991) J Bacteriol 173:4510
Sonntag K, Eggeling L, De Graaf AA, Sahm H (1993) Eur J Biochem 213:1325
Krömer JO, Heinzle E, Schröder H, Wittmann C (2006) J Bacteriol 188:609
Rey DA, Pühler A, Kalinowski J (2003) J Biotechnol 103:51
Rey DA, Nentwich SS, Koch DJ, Rückert C, Puhler A, Tauch A, Kalinowski J (2005) Mol Microbiol 56:871
Ikeda M, Katsumata R (1992) Appl Environ Microbiol 58:781
Wendisch VF, Bott M, Kalinowski J, Oldiges M, Wiechert W (2006) J Biotechnol 124:74
Haberhauer G, Schröder H, Pompejus M, Zelder O, Kröger B (2001) Patent WO 01/00805
Ikeda M, Nakagawa S (2003) Appl Microbiol Biotechnol 62:99
Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A, Dusch N, Eggeling L, Eikmanns BJ, Gaigalat L, Goesmann A, Hartmann M, Huthmacher K, Krämer R, Linke B, McHardy AC, Meyer F, Mockel B, Pfefferle W, Pühler A, Rey DA, Rückert C, Rupp O, Sahm H, Wendisch VF, Wiegrabe I, Tauch A (2003) J Biotechnol 104:5
Glanemann C, Loos A, Gorret N, Willis LB, O’Brien XM, Lessard PA, Sinskey AJ (2003) Appl Microbiol Biotechnol 61:61
Hayashi M, Ohnishi J, Mitsuhashi S, Yonetani Y, Hashimoto S, Ikeda M (2006) Biosci Biotechnol Biochem 70:546
Silberbach M, Burkovski A (2006) J Biotechnol 126:101
Huser AT, Becker A, Brune I, Dondrup M, Kalinowski J, Plassmeier J, Puhler A, Wiegrabe I, Tauch A (2003) J Biotechnol 106:269
Schluesener D, Fischer F, Kruip J, Rogner M, Poetsch A (2005) Mapping the membrane proteome of Corynebacterium glutamicum. Proteomics 5:1317–1330
Bendt AK, Burkovski A, Schaffer S, Bott M, Farwick M, Hermann T (2003) Proteomics 3:1637
Hermann T, Pfefferle W, Baumann C, Busker E, Schaffer S, Bott M, Sahm H, Dusch N, Kalinowski J, Puhler A, Bendt AK, Kramer R, Burkovski A (2001) Electrophoresis 22:1712
Kjeldsen KR, Nielsen J (2009) Biotechnol Bioeng 102:583
Krömer JO, Wittmann C, Schröder H, Heinzle E (2006) Metabolic pathway analysis for rational design of L-methionine production by Escherichia coli and Corynebacterium glutamicum. Metab Eng 8(4):353–369
Nishio Y, Nakamura Y, Kawarabayasi Y, Usuda Y, Kimura E, Sugimoto S, Matsui K, Yamagishi A, Kikuchi H, Ikeo K, Gojobori T (2003) Genome Res 13:1572
Yukawa H, Omumasaba CA, Nonaka H, Kos P, Okai N, Suzuki N, Suda M, Tsuge Y, Watanabe J, Ikeda Y, Vertes AA, Inui M (2007) Microbiology 153:1042
Cerdeno-Tarraga AM, Efstratiou A, Dover LG, Holden MT, Pallen M, Bentley SD, Besra GS, Churcher C, James KD, De Zoysa A, Chillingworth T, Cronin A, Dowd L, Feltwell T, Hamlin N, Holroyd S, Jagels K, Moule S, Quail MA, Rabbinowitsch E, Rutherford KM, Thomson NR, Unwin L, Whitehead S, Barrell BG, Parkhill J (2003) Nucleic Acids Res 31:6516
Tauch A, Kaiser O, Hain T, Goesmann A, Weisshaar B, Albersmeier A, Bekel T, Bischoff N, Brune I, Chakraborty T, Kalinowski J, Meyer F, Rupp O, Schneiker S, Viehoever P, Pühler A (2005) J Bacteriol 187:4671
Tauch A, Schneider J, Szczepanowski R, Tilker A, Viehoever P, Gartemann KH, Arnold W, Blom J, Brinkrolf K, Brune I, Gotker S, Weisshaar B, Goesmann A, Droge M, Puhler A (2008) J Biotechnol 136:22
Varma A, Palsson BO (2001) Nat Biotechnol 12:994
Feist AM, Herrgard MJ, Thiele I, Reed JL, Palsson BO (2009) Nat Rev Microbiol 7:129
Feist AM, Palsson BO (2008) Nat Biotechnol 26:659
Price ND, Reed JL, Palsson BO (2004) Nat Rev Microbiol 2:886
Schilling CH, Edwards JS, Letscher D, Palsson BO (2000) Biotechnol Bioeng 71:286
Schuster S, Dandekar T, Fell DA (1999) Trends Biotechnol 17:53
Edwards JS, Covert M, Palsson B (2002) Environ Microbiol 4:133
Papin JA, Stelling J, Price ND, Klamt S, Schuster S, Palsson BO (2004) Trends Biotechnol 22:400
Schuster S, Fell DA, Dandekar T (2000) Nat Biotechnol 18:326
Gagneur J, Klamt S (2004) BMC Bioinformatics 5:175
Carlson R, Fell D, Srienc F (2002) Biotechnol Bioeng 79:121
Carlson R, Srienc F (2004) Biotechnol Bioeng 85:1
Vallino JJ, Stephanopoulos G (1993) Biotechnol Bioeng 41:633
Vallino JJ, Stephanopoulos G (1994) Biotechnol Prog 10:320
Krömer JO, Sorgenfrei O, Klopprogge K, Heinzle E, Wittmann C (2004) J Bacteriol 186:1769
Kiss RD, Stephanopoulos G (1991) Biotechnol Prog 7:501
Yang C, Hua Q, Shimizu K (1999) J Biosci Bioeng 88:393
Magnus JB, Hollwedel D, Oldiges M, Takors R (2006) Biotechnol Prog 22:1071
Ruijter GJ, Bax M, Patel H, Flitter SJ, van de Vondervoort PJ, de Vries RP, vanKuyk PA, Visser J (2003) Eukaryot Cell 2:690
Nöh K, Grönke K, Luo B, Takors R, Oldiges M, Wiechert W (2007) J Biotechnol 129:249
Bolten C, Kiefer P, Letisse F, Portais JC, Wittmann C (2007) Anal Chem 79:3843–3849
Borner J, Buchinger S, Schomburg D (2007) Anal Biochem 367:143
Strelkov S, von Elstermann M, Schomburg D (2004) Biol Chem 385:853
Wittmann C, Krömer JO, Kiefer P, Binz T, Heinzle E (2004) Anal Biochem 327:135
Gayen K, Venkatesh KV (2007) J Ind Microbiol Biotechnol 34:363
Iwatani S, Yamada Y, Usuda Y (2008) Biotechnol Lett 30:791
Dominguez H, Nezondet C, Lindley ND, Cocaign M (1993) Biotechnol Lett 15(5):449
Varela C, Agosin E, Baez M, Klapa M, Stephanopoulos G (2003) Appl Microbiol Biotechnol 60:547
Cocaign-Bousquet M, Guyonvarch A, Lindley ND (1996) Appl Environ Microbiol 62(2):429
Pons A, Dussap CG, Pequignot C, Gros JB (1996) Biotechnol Bioeng 51(2):177
Sauer U (2006) Mol Syst Biol 2:62
Wittmann C, Hans M, Heinzle E (2002) Anal Biochem 307:379
Wittmann C (2007) Microb Cell Fact 6:6
Christensen B, Nielsen J (1999) Metab Eng 1:282
Dauner M, Sauer U (2000) Biotechnol Prog 16:642
Yang TH, Heinzle E, Wittmann C (2005) Comput Biol Chem 29:121
Yang TH, Wittmann C, Heinzle E (2006) Metab Eng 8:417
Yang TH, Wittmann C, Heinzle E (2006) Metab Eng 8:432
Iwatani S, Van Dien S, Shimbo K, Kubota K, Kageyama N, Iwahata D, Miyano H, Hirayama K, Usuda Y, Shimizu K, Matsui K (2007) J Biotechnol 128:93
Wiechert W (2002) J Biotechnol 94:37
Fischer E, Zamboni N, Sauer U (2004) Anal Biochem 325:308
Shiio I, Otsuka SI, Tsunoda T (1960) J Biochem 47:414
Ishino S, Yamaguchi K, Shirahata K, Araki K (1984) Agric Biol Chem 48(10):2557
Walker TE, Han CH, Kollman VH, London RE, Matwiyoff NA (1982) J Biol Chem 257:1189
Drysch A, El Massaoudi M, Mack C, Takors R, de Graaf AA, Sahm H (2003) Metab Eng 5:96
Klapa MI, Aon JC, Stephanopoulos G (2003) Eur J Biochem 270:3525
Marx A, Hans S, Mockel B, Bathe B, de Graaf AA (2003) J Biotechnol 104:185
Drysch A, El Massaoudi M, Wiechert W, de Graaf AA, Takors R (2004) Biotechnol Bioeng 85:497
Kiefer P, Heinzle E, Zelder O, Wittmann C (2004) Appl Environ Microbiol 70:229
Wittmann C, Kiefer P, Zelder O (2004) Appl Environ Microbiol 70:7277
Wittmann C, Kim HM, Heinzle E (2004) Biotechnol Bioeng 87:1
Becker J, Heinzle E, Klopprogge C, Zelder O, Wittmann C (2005) Appl Environ Microbiol 71:8587–8596
Yang TH, Wittmann C, Heinzle E (2006) Metab Eng (submitted for publication)
Kim HM, Heinzle E, Wittmann C (2006) J Microbiol Biotechnol 16:1174
Becker J, Klopprogge C, Herold A, Zelder O, Bolten CJ, Wittmann C (2007) J Biotechnol
Shirai T, Nakato A, Izutani N, Nagahisa K, Shioya S, Kimura E, Kawarabayasi Y, Yamagishi A, Gojobori T, Shimizu H (2005) Metab Eng 7:59
Shirai T, Fujimura K, Furusawa C, Nagahisa K, Shioya S, Shimizu H (2007) Microb Cell Fact 6:19
Krömer JO, Bolten CJ, Heinzle E, Schröder H, Wittmann C (2008) Microbiology 154:3917
Becker J, Klopprogge C, Wittmann C (2008) Microb Cell Fact 7:8
Sauer U, Hatzimanikatis V, Bailey JE, Hochuli M, Szyperski T, Wüthrich K (1997) Nat Biotechnol 15:448
Wittmann C, Heinzle E (2001) Biotechnol Bioeng 72:642
Sauer U, Eikmanns BJ (2005) FEMS Microbiol Rev 29:765
de Graaf AA (2000) Metabolic flux analysis of Corynebacterium glutamicum. In: Schügerl K, Bellgard KH (eds) Bioreaction engineering. Springer, Berlin
Wittmann C, Heinzle E (2002) Appl Environ Microbiol 68:5843
Marx A, Striegel K, de Graaf A, Sahm H, Eggeling L (1997) Biotechnol Bioeng 56(2):168
Wittmann C, Heinzle E (2001) Eur J Biochem 268:2441
Becker J, Klopprogge C, Zelder O, Heinzle E, Wittmann C (2005) Appl Environ Microbiol 71:8587
Sonntag K, Schwinde J, de Graaf A, Marx A, Eikmanns B, Wiechert W, Sahm H (1995) Appl Microbiol Biotechnol 44:489
Wittmann C, Heinzle E (2001) Metab Eng 3:173
Marx A, Eikmanns BJ, Sahm H, de Graaf AA, Eggeling L (1999) Metab Eng 1:35
Matsushita K, Otofuji A, Iwahashi M, Toyama H, Adachi O (2001) FEMS Microbiol Lett 204:271
Kiss RD, Stephanopoulos G (1992) Biotechnol Bioeng 39:565
Georgi T, Rittmann D, Wendisch VF (2005) Metab Eng 7:291
Sahm H, Eggeling L, de Graaf AA (2000) Biol Chem 381:899
Ohnishi J, Mitsuhashi S, Hayashi M, Ando S, Yokoi H, Ochiai K, Ikeda M (2002) Appl Microbiol Biotechnol 58:217
Kelle R, Hermann T, Bathe B (2005) L-Lysine production. In: Eggeling L, Bott M (eds) Handbook of Cornyebacterium glutamicum. CRC Press, Taylor and Francis, Boca Raton
Cremer J, Eggeling L, Sahm H (1991) Appl Environ Microbiol 57(6):1746
Follettie MT, Peoples OP, Agoropoulou C, Sinskey AJ (1993) J Bacteriol 175:4096
Sugimoto M, Tanaka A, Suzuki T, Matsui H, Nakamori S, Takagi H (1997) Biosci Biotechnol Biochem 61:1760
Jetten MS, Follettie MT, Sinskey AJ (1995) Appl Microbiol Biotechnol 43:76
Bonnassie S, Oreglia J, Sicard AM (1990) Nucleic Acids Res 18:6421
Eggeling L, Oberle S, Sahm H (1998) Appl Microbiol Biotechnol 49:24
Pisabarro A, Malumbres M, Mateos LM, Oguiza JA, Martin JF (1993) J Bacteriol 175:2743
Vasicova P, Patek M, Nesvera J, Sahm H, Eikmanns B (1999) J Bacteriol 181:6188
Bellmann A, Vrljic M, Patek M, Sahm H, Kramer R, Eggeling L (2001) Microbiology 147:1765
Vrljic M, Garg J, Bellmann A, Wachi S, Freudl R, Malecki MJ, Sahm H, Kozina VJ, Eggeling L, Saier MH Jr, Eggeling L, Saier MH Jr (1999) J Mol Microbiol Biotechnol 1:327
Vrljic M, Sahm H, Eggeling L (1996) Mol Microbiol 22:815
Gunji Y, Yasueda H (2006) J Biotechnol 127:1
Peters-Wendisch PG, Schiel B, Wendisch VF, Katsoulidis E, Mockel B, Sahm H, Eikmanns BJ (2001) J Mol Microbiol Biotechnol 3:295
Sano K, Ito K, Miwa K, Nakamori S (1987) Agric Biol Chem 51(2):597
Netzer R, Peters-Wendisch P, Eggeling L, Sahm H (2004) Appl Environ Microbiol 70:7148
Zelder O, Pompejus M, Schröder H, Kröger B, Klopprogge C, Haberhauer G (2005) UP Office, 2005/0014235, A1. US
Ikeda M, Katsumata R (1999) Appl Environ Microbiol 65:2497
Ikeda M, Okamoto K, Katsumata R (1999) Appl Microbiol Biotechnol 51:201
Ikeda M, Nakanishi K, Kino K, Katsumata R (1994) Biosci Biotechnol Biochem 58:674
Niebisch A, Kabus A, Schultz C, Weil B, Bott M (2006) J Biol Chem 281:12300
Shimizu H, Tanaka H, Nakato A, Nagahisa K, Kimura E, Shioya S (2003) Bioprocess Biosyst Eng 25:291
Asakura Y, Kimura E, Usuda Y, Kawahara Y, Matsui K, Osumi T, Nakamatsu T (2007) Appl Environ Microbiol 73:1308
Kim J, Hirasawa T, Sato Y, Nagahisa K, Furusawa C, Shimizu H (2009) Appl Microbiol Biotechnol 81:1097–1106
Aristidou A, Penttila M (2000) Curr Opin Biotechnol 11:187
Kawaguchi H, Vertes AA, Okino S, Inui M, Yukawa H (2006) Appl Environ Microbiol 72:3418
Kawaguchi H, Sasaki M, Vertes AA, Inui M, Yukawa H (2008) Appl Microbiol Biotechnol 77:1053
Sakai S, Tsuchida Y, Okino S, Ichihashi O, Kawaguchi H, Watanabe T, Inui M, Yukawa H (2007) Appl Environ Microbiol 73:2349
Seibold G, Auchter M, Berens S, Kalinowski J, Eikmanns BJ (2006) J Biotechnol 124:381
Tateno T, Fukuda H, Kondo A (2007) Appl Microbiol Biotechnol 74:1213
Rittmann D, Lindner SN, Wendisch VF (2008) Appl Environ Microbiol 74:6216
Barrett E, Stanton C, Zelder O, Fitzgerald G, Ross RP (2004) Appl Environ Microbiol 70:2861
Koffas M, Stephanopoulos G (2005) Curr Opin Biotechnol 16:361
Wendisch VF (2003) J Biotechnol 104:273
Sindelar G, Wendisch VF (2007) Appl Microbiol Biotechnol 76:677
Hayashi M, Mizoguchi H, Ohnishi J, Mitsuhashi S, Yonetani Y, Hashimoto S, Ikeda M (2006) Appl Microbiol Biotechnol 72:783
Schaffer S, Burkovski A (2005) Proteomics. In: Eggeling L, Bott M (eds) Handbook of Corynebacterium glutamicum. CRC Press, Boca Raton
Schmid R, Uhlemann EM, Nolden L, Wersch G, Hecker R, Hermann T, Marx A, Burkovski A (2000) FEMS Microbiol Lett 187:83
Schilling O, Frick O, Herzberg C, Ehrenreich A, Heinzle E, Wittmann C, Stulke J (2007) Appl Environ Microbiol 73:499
Silberbach M, Schafer M, Huser AT, Kalinowski J, Puhler A, Kramer R, Burkovski A (2005) Appl Environ Microbiol 71:2391
Park JH, Lee SY (2008) Curr Opin Biotechnol 19:454
Park JH, Lee KH, Kim TY, Lee SY (2007) Proc Natl Acad Sci USA 104:7797
Lee KH, Park JH, Kim TY, Kim HU, Lee SY (2007) Mol Syst Biol 3:149
Kimura E (2003) Adv Biochem Eng Biotechnol 79:37
Colon GE, Nguyen TT, Jetten MS, Sinskey AJ, Stephanopoulos G (1995) Appl Microbiol Biotechnol 43:482
Guillouet S, Rodal AA, An GH, Gorret N, Lessard PA, Sinskey AJ (2001) Appl Microbiol Biotechnol 57:667
Park SD, Lee JY, Sim SY, Kim Y, Lee HS (2007) Metab Eng 9:327
Stolz M, Peters-Wendisch P, Etterich H, Gerharz T, Faurie R, Sahm H, Fersterra H, Eggeling L (2007) Appl Environ Microbiol 73:750
Peters-Wendisch P, Stolz M, Etterich H, Kennerknecht N, Sahm H, Eggeling L (2005) Appl Environ Microbiol 71:7139
Sahm H, Eggeling L, Eikmanns B, Kramer R (1996) Ann N Y Acad Sci 782:25
Blombach B, Schreiner ME, Bartek T, Oldiges M, Eikmanns BJ (2008) Appl Microbiol Biotechnol 79:471
Hüser AT, Chassagnole C, Lindley ND, Merkamm M, Guyonvarch A, Elisakova V, Patek M, Kalinowski J, Brune I, Pühler A, Tauch A (2005) Appl Environ Microbiol 71:3255
Mimitsuka T, Sawai H, Hatsu M, Yamada K (2007) Biosci Biotechnol Biochem 71:2130
Okino S, Noburyu R, Suda M, Jojima T, Inui M, Yukawa H (2008) Appl Microbiol Biotechnol 81:459
Okino S, Suda M, Fujikura K, Inui M, Yukawa H (2008) Appl Microbiol Biotechnol 78:449
Inui M, Kawaguchi H, Murakami S, Vertes AA, Yukawa H (2004) J Mol Microbiol Biotechnol 8:243
Acknowledgments
The author gratefully acknowledges support by the BMBF-Grant “Biobased Polyamides through Fermentation” (No 0315239A) within the initiative Bioindustry21.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Dedicated to Prof. Dr. Dietmar Hempel on the occasion of his 65th birthday
Rights and permissions
Copyright information
© 2010 Springer
About this chapter
Cite this chapter
Wittmann, C. (2010). Analysis and Engineering of Metabolic Pathway Fluxes in Corynebacterium glutamicum . In: Wittmann, C., Krull, R. (eds) Biosystems Engineering I. Advances in Biochemical Engineering / Biotechnology, vol 120. Springer, Berlin, Heidelberg. https://doi.org/10.1007/10_2009_58
Download citation
DOI: https://doi.org/10.1007/10_2009_58
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-14230-7
Online ISBN: 978-3-642-14231-4
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)