
Abstract
The Gram-positive Corynebacterium glutamicum is widely used for fermentative production of amino acids. The world production of l-lysine has surpassed 2 million tons per year. Glucose uptake and phosphorylation by C. glutamicum mainly occur by the phosphotransferase system (PTS) and to lesser extent by inositol permeases and glucokinases. Heterologous expression of the genes for the high-affinity glucose permease from Streptomyces coelicolor and Bacillus subtilis glucokinase fully compensated for the absence of the PTS in Δhpr strains. Growth of PTS-positive strains with glucose was accelerated when the endogenous inositol permease IolT2 and glucokinase from B. subtilis were overproduced with balanced translation initiation rates using plasmid pEKEx3-IolTBest. When the genome-reduced C. glutamicum strain GRLys1 carrying additional in-frame deletions of sugR and ldhA to derepress glycolytic and PTS genes and to circumvent formation of l-lactate as by-product was transformed with this plasmid or with pVWEx1-IolTBest, 18 to 20 % higher volumetric productivities and 70 to 72 % higher specific productivities as compared to the parental strain resulted. The non-proteinogenic amino acid l-pipecolic acid (l-PA), a precursor of immunosuppressants, peptide antibiotics, or piperidine alkaloids, can be derived from l-lysine. To enable production of l-PA by the constructed l-lysine-producing strain, the l-lysine 6-dehydrogenase gene lysDH from Silicibacter pomeroyi and the endogenous pyrroline 5-carboxylate reductase gene proC were overexpressed as synthetic operon. This enabled C. glutamicum to produce l-PA with a yield of 0.09 ± 0.01 g g−1 and a volumetric productivity of 0.04 ± 0.01 g L−1 h−1.To the best of our knowledge, this is the first fermentative process for the production of l-PA from glucose.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
l-lysine is one of the essential amino acids in mammals, which have to intake l-lysine through their diet as they lack l-lysine biosynthesis. l-lysine is produced biotechnologically to meet the annual world market of about 2 million tons (Hermann 2003; Wendisch 2007; Yukawa and Inui 2013). Glucose-based fermentations with C. glutamicum are in place for efficient l-lysine production (Wendisch et al. 2006a, b; Hirasawa et al. 2012; Blombach et al. 2009b, Heider and Wendisch 2015). This microorganism was initially isolated as a natural l-glutamate producer (Abe et al. 1967). C. glutamicum is a non-pathogenic, facultative anaerobic Gram-positive bacterium found in soil, animal feces, fruits, and vegetables with an irregular rod shape (Eggeling and Bott 2005). The physiology, biochemistry, and genetics of l-lysine biosynthesis are well understood and have been employed in rational development of l-lysine producing strains. For example, the l-lysine production strain GRLys1 (Unthan et al. 2015) possesses two gene copies of the key enzyme of l-lysine biosynthesis, aspartokinase (Schrumpf et al. 1992; Cremer et al. 1991; Kalinowski et al. 1991), devoid of feedback inhibition by l-lysine (encoded by lysC T311I), two copies of the l-lysine biosynthesis genes asd, dapA, dapB, ddh, and lysA, and two copies of l-lysine export gene lysE (Vrljic et al. 1996). Side product formation is reduced due to a restricted homoserine dehydrogenase (encoded by hom V59A) (Eikmanns et al. 1991), anaplerosis is improved due to deletion of the PEP carboxykinase gene (Riedel et al. 2001), and a variant of the pyruvate carboxylase gene (Peters-Wendisch et al. 2001) and the strain was cured of prophages CGP1, CGP2, and CGP3 (Baumgart et al. 2013). A wealth of knowledge on l-lysine biosynthesis and production has accumulated (Wendisch et al. 2006a, b; Hirasawa et al. 2012; Blombach et al. 2009a; Heider and Wendisch 2015; Becker and Wittmann 2012; Wendisch 2014; Mitsuhashi 2014; Park et al. 2010).
C. glutamicum takes up and phosphorylates glucose primarily via the phosphoenolpyruvate (PEP)-dependent carbohydrate phosphotransferase system (PTS). In addition, glucose can be imported via the non-phosphorylating inositol permeases IolT1 and IolT2 (Lindner et al. 2011; Ikeda et al. 2011; Bäumchen et al. 2009). Intracellular glucose may be phosphorylated by ATP-dependent or polyphosphate-dependent glucokinases PpgK and Glk (Park et al. 2000; Lindner et al. 2010). While the PTS constitutes a high-affinity uptake system (Km of 14 μM), the permeases IolT1 and IolT2 have about hundred fold lower affinities for glucose (Km values of 2.8 mM and 1.9 mM, respectively). As the ATP- or polyphosphate-dependent glucokinases Glk and PpgK do not consume PEP, the availability of PEP for anabolism e.g., for biosynthesis of aromatic amino acids may increase (Tang et al. 2013; Zhang et al. 2015). Since high initial glucose concentrations are typical for biotechnological processes, use of the glucose permease and glucokinase pathway offers an interesting opportunity to enhance glucose utilization while maintaining high PEP availability. Commensurate with this notion, PpgK was shown to be important only at high glucose concentrations (Lindner et al. 2010). Glucose utilization by C. glutamicum is subject to transcriptional control (Toyoda et al. 2008, 2009a; Teramoto et al. 2011; Park et al. 2010; Gerstmeir et al. 2004). For example, SugR is a general repressor of the PTS, glycolysis and fermentative l-lactate dehydrogenase genes in C. glutamicum (Engels and Wendisch 2007; Engels et al. 2008; Teramoto et al. 2011; Gaigalat et al. 2007). Deletion of sugR accelerated glucose utilization (Blombach et al. 2009b; Bartek et al. 2010; Teramoto et al. 2011), while overexpression of sugR slowed glucose utilization (Engels et al. 2008). Due to co-utilization of protocatechuic acid, single cells of C. glutamicum grow faster with glucose in the highly diluted environments of a Lab Chip than cells in suspension (Unthan et al. 2014). Taken together, glucose uptake and utilization by C. glutamicum wild type as well as by l-lysine producing strains may be relevant targets for strain development.
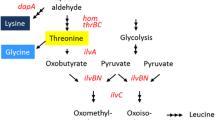
l-lysine can be converted to other value-added compounds such as the diamine cadaverine (Mimitsuka et al. 2007; Tateno et al. 2009; Kind et al. 2014; Oh et al. 2015; Shimizu 2013; Leßmeier et al. 2015), 5-aminovalerate (5AVA) (Park et al. 2013) and l-PA (Fujii et al. 2002; Gatto et al. 2006; Neshich et al. 2013). C. glutamicum has been engineered for cadaverine production but not for production of l-PA. The non-proteinogenic amino acid l-PA is a precursor of immunosuppressants (Maddess et al. 2008), peptide antibiotics (Takayama et al. 1984), or piperidine alkaloids (Clevenstine et al. 1979). In plants, l-PA is a critical regulator of inducible plant immunity, mediating defense amplification and priming (Návarová et al. 2012). Lysine degradation yields l-PA as intermediate. dl-lysine and l-lysine are oxidatively deaminated by stereospecific l-lysine α-oxidases from Pseudomonas putida (Miller and Rodwell 1971) and Trichoderma viride (Kusakabe et al. 1980), respectively, to α-keto-ε-aminocaproate, which spontaneously dehydrates to Δ1-piperideine-2-carboxylate (P2C). P2C reductase yields l-PA from P2C (Payton and Chang 1982). As shown in Fig. 1, l-PA may also be derived from Δ1-piperideine-6-carboxylate (P6C) in an NADPH-dependent reduction reaction catalyzed by pyrroline 5-carboxylate reductase ProC (Fujii et al. 2002), an enzyme of l-proline biosynthesis in many bacteria including C. glutamicum (Ankri et al. 1996). P6C arises from α-aminoadipic semialdehyde (AASA) by spontaneous cyclization (Fig. 1). Four reactions give rise to AASA from l-lysine: (1) l-lysine cyclodeaminase (LCD) from streptomycetes such as S. hygroscopicus that produce polyketide immunosuppressants such as rapamycin or meridiamycin (Molnár et al. 1996); (2) l-lysine 6-aminotransferase (LAT) from Flavobacterium lutencens (Soda et al. 1968); and (3) l-lysine 6-dehydrogenase e.g., from Agrobacterium tumefaciens (Misono et al. 1989) or from Silicibacter pomeroyi (Neshich et al. 2013) (Fig. 1). Furthermore, l-lysine can be converted to AASA via saccharopine by l-lysine α-ketoglutarate reductase and saccharopine dehydrogenase from Rizoctonia leguminicola (Wickwire et al. 1990). l-PA accumulated intracellularly when Escherichia coli overproducing l-lysine 6-dehydrogenase from S. pomeroyi was grown in l-lysine-containing media (Neshich et al. 2013). l-PA was produced from l-lysine in biotransformations with E. coli overproducing either l-lysine α-oxidase from Scomber japonicus (Tani et al. 2015) or LAT (Fujii et al. 2002).

Metabolic pathway for conversion of l-lysine to l-PA in recombinant C. glutamicum. l-lysine 6-dehydrogenase (deaminating) (EC 1.4.1.18) from S. pomeroyi encoded by lysDH and pyrroline 5-carboxylate reductase (EC 1.2.1.41) from C. glutamicum encoded by proC were used. The combined reactions of l-lysine 6-dehydrogenase (EC 1.4.1.18) and the spontaneous cyclization of α-aminoadipic semialdehyde are sometimes referred to as l-lysine:NAD+ oxidoreductase (deaminating, cyclizing) (EC 1.4.1.15)
In this study, l-lysine production from glucose was accelerated by overexpression of endogenous iolT2 encoding inositol permease and by heterologous expression of the glucokinase gene from B. subtilis. In addition, deletion of sugR led to derepression of glycolytic and PTS genes and deletion of ldhA precluded formation of l-lactate as by-product. This strain was used as basis for strain development for fermentative production of l-PA from glucose.
Materials and methods
Bacterial strains, plasmids, and growth conditions
The bacterial strains and plasmids are listed in Table 1. E. coli DH5α (Hanahan 1983) was used for vector construction. The precultivation of C. glutamicum and cultivation of E. coli were carried out in Luria-Bertani (LB) medium (Sambrook et al. 1989) inoculated from a fresh LB agar plate. For growth of C. glutamicum strains, the cells from the preculture were washed in CGXII minimal medium (Eggeling and Bott 2005) without glucose, before 50 ml of main CGXII culture with glucose was inoculated to an optical density of 0.5 (OD600). C. glutamicum precultures and cultures were incubated at 30 °C and 120 rpm in 500 ml baffled flasks. E. coli cultures were incubated at 37 °C and 200 rpm in 500 ml baffled flasks. The final glucose concentration varied between 0.5 % (w/v) and 4 % (w/v). For selection of pVWEx1 and pSH1, 25 μg ml−1 of kanamycin was added to cultures (Peters-Wendisch et al. 2001). For selection of pEKEx3, 100 μg ml−1 of spectinomycin was added to cultures (Stansen et al. 2005).
Construction of expression vectors
Genes were amplified via PCR from genomic DNA of C. glutamicum ATCC 13032 (Kalinowski et al. 2003), Mycobacterium smegmatis mc2 155 (Pimentel-Schmitt et al. 2009), S. coelicolor A3(2) (van Wezel et al. 2005; Imriskova et al. 2005), E. coli K-12 MG 1655 (Meyer et al. 1997), B. subtilis subsp. subtilis str. 168 (Mesak et al. 2004), or S. pomeroyi DSS-3 (DSMZ-15171) (González et al. 2003). Genomic DNA was prepared following the same protocol described previously (Eikmanns et al. 1995). The primers for the vector constructions are listed in Table 2.
Balanced expression of glucose permease and kinase genes
For the balanced expression of glucose permease and kinase genes, a combinatorial approach was performed by combining four ribosomal binding site (AAAGGAGG, AAAGGTGG, AAAGGCGG, and AAAGGGGG) and two transcriptional start site (ATG and GTG) options for the permease genes cloned together with the glucokinase gene, which was not varied. Primers pgk13, pgk14, pgk15, pgk16, pk35, and pk36 (s. Table 2) were used for PCR, and genes were joined via Gibson assembly to yield pEKEx3-glcK Bs -glcP Sco and pEKEx3-glcK Bs -iolT2 Cg vectors (Gibson 2011). These vectors were used to transform C. glutamicum Δhpr, and the best combinations were chosen according to fastest growth in minimal medium with 2 % (w/v) of glucose and different IPTG concentrations. IPTG concentrations of 100 μM were optimal for (pEKEx3-glcK Bs -glcP Sco) and 50 μM for (pEKEx3-glcK Bs-iolT2 Cg).
Deletion of sugR and ldhA
The two-step homologous recombination procedure was used to generate in-frame deletions of ΔsugR and ΔldhA in the strain GRLys1 (Rittmann et al. 2003). For that purpose, the vectors pK19mobsacB-ΔsugR (Engels et al. 2008) and pK19mobsacB-ΔldhA (Blombach et al. 2011) were used as described previously (Engels and Wendisch 2007). The deletions were verified by PCR using the primer pairs ΔsugR-Ver-fw/ΔsugR-Ver-rv and ΔldhA-Ver-fw/ΔldhAR-Ver-rv (Table 2).
Analytical procedures
Extracellular amino acids and carbohydrates were quantified using a high-pressure liquid chromatography system (1200 series, Agilent Technologies Deutschland GmbH, Böblingen, Germany). Cell cultures were centrifuged (13,000×g, 10 min), and the supernatant was used for analysis. For the detection of l-lysine, l-glutamate, l-valine and l-alanine, samples were derivatized with ortho-phthaldialdehyde and for detection of l-PA with fluorenylmethyl chloroformate (FMOC) as described (Schneider and Wendisch 2010). All amino acids were separated on a system consisting of a pre-column (LiChrospher 100 RP18 EC-5μ (40 × 4 mm), CS Chromatographie Service GmbH, Langerwehe, Germany) and a main column (LiChrospher 100 RP18 EC-5 μ (125 × 4 mm), CS Chromatographie Service GmbH), and detected with a fluorescence detector (FLD G1321A, 1200 series, Agilent Technologies). l-Asparagine was used as internal standard. For quantification of carbohydrates, a column for organic acids (300 × 8 mm, 10 μm particle size, 25 Å pore diameter, CS Chromatographie Service GmbH) and a refractive index detector (RID G1362A, 1200 series, Agilent Technologies) was used (Peters-Wendisch et al. 2014).
Transcriptome analysis using DNA microarrays
To identify gene expression changes due to the addition of l-PA to the growth medium, C. glutamicum WT was cultivated in CGXII minimal medium with 4 % (w/v) glucose supplemented with either 200 mM l-PA or 200 mM sodium chloride. Exponentially growing cells were harvested by centrifugation (4000 × g, 10 min, 4 °C) and kept at −80 °C. RNA isolation was performed as described (Wendisch 2003). DNA microarray analysis, synthesis of fluorescently labeled cDNA from total RNA, DNA microarray hybridization, and gene expression analysis were performed as described previously (Ishige et al. 2003; Lange et al. 2003). The data are available as Gene Expression Omnibus GSE79690 data set at http://www.ncbi.nlm.nih.gov/geo/.
Results
Potential of alternative glucose permeases to substitute for the glucose PTS
C. glutamicum not only takes up and phosphorylates glucose mainly via the high-affinity (Km of 14 μM) phosphotransferase system (PTS) but also can use inositol permeases IolT1 and IolT2, which have about hundred fold lower affinities for glucose and require subsequent phosphorylation by glucokinases (Lindner et al. 2011; Ikeda et al. 2015). The latter systems may be important at the high glucose concentrations employed in industrial fermentations. In order to test if uptake and phosphorylation via glucose permeases and glucokinase can fully substitute for the glucose PTS at medium to high glucose concentrations (1 % (w/v) to 4 % (w/v)), glucose permease and glucokinase genes from various organisms were expressed in the PTS-deficient C. glutamicum strain Δhpr (Lindner et al. 2011). The heat-stable protein HPr is an essential component of the PTS system, which is phosphorylated by PTS component EI and, subsequently, phospho-HPr transfers the phosphoryl group to the PTS permease component. The genes for the glucose permease from S. coelicolor A3(2) (GlcPSco) with a Km of 41 μM (van Wezel et al. 2005), the glucose permease from M. smegmatis mc2 155 (GlcPSmeg) with a Km of 19.4 μM (Pimentel-Schmitt et al. 2009), the galactose permease from E. coli MG1655 (GalPEco) with a Km of 10.2 μM (McDonald et al. 1997), and the inositol transporter 2 from C. glutamicum ATCC 13032 (IolT2Cg) with a Km of 1.9 mM (Lindner et al. 2011) were cloned into the IPTG-inducible expression vector pVWEx1 (Peters-Wendisch et al. 2001) and used to transform C. glutamicum Δhpr. In minimal medium with 4 % (w/v) of glucose, C. glutamicum WT (pVWEx1) grew with a growth rate of 0.42 ± 0.01 h−1 (Fig. 2), while C. glutamicum Δhpr(pVWEx1) hardly grew (0.03 ± 0.01 h−1), which is due to low expression of iolT1 and iolT2 in the absence of inositol (Krings et al. 2006). Of the glucose permease genes tested, overexpression of iolT2 Cg supported fastest growth (μ = 0.27 ± 0.01 h−1); however, even after variation of the IPTG concentration used for induction, the wild-type growth rate could not be restored. Thus, overexpression of glucose permease alone was not sufficient to substitute for the PTS in C. glutamicum Δhpr.

Influence of sugar permease gene overexpression on growth of C. glutamicum Δhpr in glucose minimal medium. Growth rates in minimal medium with 4 % (w/v) glucose at the indicated IPTG concentrations are given for strains WT(pVWEx1), Δhpr(pVWEx1), Δhpr(pVWEx1-glcP Sco), Δhpr(pVWEx1-glcP Smeg), Δhpr(pVWEx1-galP Eco), and Δhpr(pVWEx1-iolT Cg) as means and standard deviations of three replicates
Combined overexpression of glucokinase and glucose permease genes
C. glutamicum possesses ATP-dependent glucokinase Glk (Park et al. 2000) and polyphosphate/ATP-dependent glucokinase PpgK with a Km for glucose of 1 mM (Lindner et al. 2010). Combined overexpression of ppgK with either iolT1 or iolT2 in a PTS-negative strain led to slightly slower glucose utilization rates than in the parent strain (Lindner et al. 2011). Thus, combinations of alternative glucokinases with glucose permeases were tested for their potential to fully compensate for the lack of the PTS. Genes encoding glucokinase from E. coli K-12 MG1655 (GlkEco) with Km of 0.78 mM (Meyer et al. 1997), the glucokinase from S. coelicolor A3(2) (GlkSco) with Km of 1.4 mM (Imriskova et al. 2005), the glucokinase from B. subtilis subsp. Subtilis str. 168 (GlcKBs) with Km of 0.24 mM (Mesak et al. 2004), the glucokinase (GlkCg) and the polyphosphate (PolyP)/ATP-dependent glucokinase from C. glutamicum (PpgKCg) with Km of 1 mM (Lindner et al. 2010) were cloned into the IPTG-inducible expression vector pEKEx3 (Stansen et al. 2005). The resulting vectors were used to transform C. glutamicum Δhpr(pVWEx1- iolT2 Cg), Δhpr(pVWEx1-galP Eco), and Δhpr(pVWEx1-glcP Sco), respectively. All strains were grown in CGXII minimal medium with 4 % (w/v) glucose (Fig. 3). The fastest growing strains were also cultivated at lower glucose concentrations (Fig. 3). At 1 % (w/v) and 0.5 % (w/v) glucose, none of the combinations allowed growth as fast as the wild-type control. With 4 % (w/v) glucose, the combination of IolT2Cg and GlcKBs allowed faster growth (μ = 0.35 h−1) than the wild-type control (μ = 0.32 h−1). With 2 % (w/v) glucose, the combination of GlcPSco/GlcKBs grew faster (μ = 0.37 h−1) than the wild-type control (μ = 0.32 h−1).

Influence of sugar permease and glucokinase gene overexpression on growth of C. glutamicum Δhpr in glucose minimal medium. Growth rates in minimal medium with the indicated glucose concentration (4 %, 2 %, 1 % or 0.5 % (w/v)) are given as means of three replicates. The strain C. glutamicum WT(pVWEx1)(pEKEx3) was used as positive control (labeled WT in the figure). The strain Δhpr(pVWEx1)(pEKEx3) was used as negative control (labeled Δhpr in the figure). Combinations of the indicated glucokinases and permeases were tested in Δhpr with permease genes expressed from plasmid pVWEx1 and glucokinase genes expressed from plasmid pEKEx3. A growth rate of zero is depicted if growth was not tested
Varying translation initiation rates for balanced glucokinase and glucose permease gene expression
Since overexpression of genes for permeases and other membrane proteins often perturbs growth, various translation initiation sequences for expression of the permease genes glcP Sco and iolT2 Cg were tested for combined expression with glcK Bs in one vector. Expression of glcK Bs was kept constant, and translation initiation rates of the permease gene (glcP Sco or iolT2 Cg) were varied by modifying ribosomal binding sites (RBS) and the translational start codons (TLS). The pool of vectors carrying different RBS and TLS sequences was used to transform Δhpr, and 24 clones for each combination were assayed for growth in minimal medium containing 2 % (w/v) glucose. The fastest-growing strains were selected and the inserts of the vectors were sequenced. Vector pEKEx3-ScoBest carried the RBS sequence of AAAGGTGG and the TLS sequence of GTG (Fig. 4a) for expression of glcK Bs and glcP Sco, and vector pEKEx3-IolTBest carried the RBS sequence of AAAGGGGG and the TLS sequence of GTG (Fig. 4b) for expression of glcK Bs and iolT2 Cg.

Influence of varied ribosome binding site and translational start codon sequences of the permease gene glcP Sco (a) or iolT Cg (b) for expression in combination with glucokinase gene glcK Bs on growth in minimal medium with 2 % (w/v) glucose. Growth rates of 24 clones with varied glcP Sco (a) or iolT Cg (b) ribosome binding site and translational start codon sequences expressed in strain Δhpr in combination with glcK Bs are indicated. The translational start codon (underlined) and ribosome binding site (in italics) sequences for the fastest growing clones (highlighted in dark gray) are depicted, and differences to the consensus ribosome binding site are given in bold. The respective plasmids were named pEKEx3-ScoBest and pEKEx3-IolTBest, respectively
To assay the effect of vectors pEKEx3-ScoBest and pEKEx3-IolTBest on glucose utilization by PTS-positive C. glutamicum, both vectors were used to transform C. glutamicum WT and growth, glucose consumption, and by-products formation in minimal medium with 2 % (w/v) glucose were compared to the empty vector carrying control strain (Table 3). Under these conditions, both WT(pEKEx3-ScoBest) and WT(pEKEx3-IolTBest) grew faster than the control strain with growth rates of 0.35 and 0.37 h−1, respectively, which were 9 and 15 % higher than that of WT(pEKEx3) (μ = 0.32 h−1). l-Lactate formation increased by about 30 % for WT(pEKEx3-ScoBest) and by about 50 % for WT(pEKEx3-IolTBest) as compared to WT(pEKEx3) (about 6 mM; Table 3 (A)). Since vectors pEKEx3-ScoBest and pEKEx3-IolTBest accelerated growth in glucose minimal medium by PTS-positive (WT) and PTS-negative (Δhpr) strains, both were used in subsequent experiments with the l-lysine-producing C. glutamicum strain GRLys1.
However, C. glutamicum WT transformed with the empty vector pEKEx3 (replication origin of pBL1, SpecR) grew slower than WT carrying the empty vector pVWEx1 (replication origin of pHM1519, KanR) reflecting different burdens by these plasmids. To exclude vector specific effects, vectors pVWEx1-IolTBest and pVWEx1-ScoBest were constructed and used to transform C. glutamicum WT. In line with the results obtained with pEKEx3-ScoBest and pEKEx3-IolTBest (Table 3 (A)), strains WT(pVWEx1-IolTBest) and WT(pVWEx1-ScoBest) grew faster in glucose minimal medium and showed improved glucose consumption rates as compared to WT(pVWEx1) (Table 3 (B) and Fig. S1).
Engineering C. glutamicum strain GRLys1 for increased l-lysine productivity
The l-lysine-producing strain GRLys1 was transformed with vectors pEKEx3, pEKEx3-ScoBest, and pEKEx3-IolTBest, and growth, Yx/s, lactate, and l-lysine production were monitored in 2 % (w/v) glucose minimal medium. GRLys1(pEKEx3) showed a growth rate of 0.30 ± 0.01 h−1, while GRLys1 (pEKEx3-ScoBest) and GRLys1 (pEKEx3-IolTBest) showed growth rates of 0.34 ± 0.01 and 0.35 ± 0.01 h−1, respectively (Fig. 5). However, the l-lysine titer decreased in comparison to GRLys1(pEKEx3) (21 ± 1 mM) as 20 ± 1 mM l-lysine was produced by GRLys1(pEKEx3-ScoBest) and 18 ± 1 mM by GRLys1(pEKEx3-IolTBest) (Fig. 5). Thus, faster growth reduced l-lysine formation. A similar trend is often observed in l-lysine strain development, i.e., an increase of the l-lysine titer is often accompanied by a reduction of the growth rate.

Growth and l-lysine production of various strains derived from C. glutamicum GRLys1. Growth rates, biomass yields (Yx/s), l-lactate titers, l-lysine titers, l-lysine volumetric productivities, and l-lysine-specific productivities are given as means and standard deviations of three replicates. The parameters of the control strain GRLys1(pEKEx3) carrying empty vector pEKEx3 are highlighted in black. The parameters of the control strain GRLys1(pVWEx1) carrying empty vector pVWEx1 are highlighted in dark gray. Parameters obtained for the strains that carry pEKEx3-ScoBest or pEKEx3-IolTBest are shown in white, while data for strains that carry pVWEx1-ScoBest or pEKEx3-IolTBest are shown in light gray. nd not detected
To test if relieving glycolytic genes from transcriptional repression improved growth rates and l-lysine titers, the repressor gene sugR was deleted because deletion of sugR in C. glutamicum has been shown previously to increase expression of glycolysis genes and glucose utilization (Engels et al. 2008; Toyoda et al. 2008). The resulting strain GRLys1ΔsugR was transformed with the vectors pEKEx3-ScoBest, pEKEx3-IolTBest, as well as the empty vector pEKEx3. The absence of sugR led to higher titer and volumetric productivities of 30 ± 1 mM and 0.19 ± 0.01 g L−1 h−1, respectively, as compared to GRLys1(pEKEx3) (Fig. 5). However, deletion of sugR reduced the growth rate to 0.20 ± 0.01 h−1 (Fig. 5).
As consequence of overexpression of glcP Sco and glcK Bs in GRLys1ΔsugR, the resulting strain GRLys1ΔsugR(pEKEx3-ScoBest) grew faster (μ = 0.27 ± 0.01 h−1), but produced less l-lysine (25 ± 1 mM), which resulted in a volumetric productivity (0.19 ± 0.01 g L−1 h−1) comparable to the empty vector control GRLys1ΔsugR(pEKEx3) (Fig. 5). On the other hand, overexpression of iolT2 Cg and glcK Bs in GRLys1ΔsugR improved l-lysine titer by 57 % to 33 ± 1 mM and volumetric productivity by 22 % to 0.22 ± 0.01 g L−1 h−1 as compared to GRLys1(pEKEx3) (Fig. 5). Taken together, C. glutamicum GRLys1ΔsugR(pEKEx3-IolTBest) showed the fastest l-lysine production of the strains constructed here.
C. glutamicum GRLys1ΔsugR(pEKEx3-IolTBest) grew to lower biomass concentrations (3.2 ± 0.1 gCDW L−1) than GRLys1(pEKEx3) (5.3 ± 0.1 gCDW L−1) and produced more lactate (27 ± 2 mM) as by-product than GRLys1(pEKEx3) (4 ± 1 mM). Increased lactate formation is due to deletion of sugR and was also observed with C. glutamicum GRLys1ΔsugR(pEKEx3) that accumulated 22 ± 1 mM lactate. Since l-lactate is produced from pyruvate by NAD-dependent l-lactate dehydrogenase LdhA (Toyoda et al. 2009b), ldhA was deleted in strain GRLys1ΔsugR(pEKEx3-IolTBest). The resulting strain GRLys1ΔsugRΔldhA(pEKEx3-IolTBest) did not accumulate l-lactate but produced l-lysine with a lower titer (31 ± 1 mM) and a lower volumetric productivity (0.21 ± 0.01 mM g L−1 h−1) than the parental LdhA-positive strain (Fig. 5). Since GRLys1ΔsugRΔldhA(pEKEx3-IolTBest) and GRLys1ΔsugR(pEKEx3-IolTBest) grew to biomass concentrations of 3.2 ± 0.1 and 3.7 ± 0.1 gCDW L−1, respectively, deletion of ldhA improved biomass formation rather than l-lysine production. To exclude vector-specific effects, pVWEx1-based vectors were also tested (Fig. S1). C. glutamicum GRLys1(pVWEx1) showed comparable growth rate, biomass yield, l-lysine titer, volumetric and specific productivity as GRLys1(pEKEx3) (Fig. 5). Moreover, the production parameters obtained with strains GRLys1ΔsugR(pVWEx1-IolTBest) and GRLys1ΔsugRΔldhA(pVWEx1-IolTBest), respectively, were comparable to those of GRLys1ΔsugR(pEKEx3-IolTBest) and GRLys1ΔsugRΔldhA(pEKEx3-IolTBest) (Fig. 5).
Engineering C. glutamicum for production of l-PA
In order to test how accelerated l-lysine production can be translated into production of a l-lysine-derived value-added chemical, strains overproducing the non-proteinogenic amino acid l-PA, a precursor of immunosuppressants, peptide antibiotics, or piperidine alkaloids, were constructed. l-PA is not synthesized by C. glutamicum which is unable to catabolize l-lysine (Vrljic et al. 1996). However, l-PA was rapidly accumulated from the growth medium by corynebacteria but did not accumulate under hyperosmolar conditions (Frings et al. 1993). To determine the response of C. glutamicum to l-PA, the effect of extracellularly added l-PA on growth and global gene expression was analyzed. Growth of C. glutamicum WT in 4 % (w/v) glucose minimal medium was hardly affected by addition of l-PA, and an inhibition constant (K i) of about 3.5 M was extrapolated (Fig. 6a). The gene expression response to 200 mM l-PA added to the medium (Table 4) revealed increased expression of genes of carbon metabolism such as rpe (ribulose-5-phosphate-3-epimerase), pck (phosphoenolpyruvate carboxykinase), vanR (repressor of the vanillate utilization operon), and cdaS (cyclomaltodextrinase). On the other hand, malP coding for maltodextrin phosphorylase and lldD encoding quinone-dependent l-lactate dehydrogenases for l-lactate utilization (Stansen et al. 2005) showed decreased expression. The gene cg0569 encoding a cation transporting P-type ATPase, one of the 50 most abundant membrane proteins in C. glutamicum (Burkovski 2008), was highly expressed in the presence of l-PA. Neither the gene expression response nor the growth response to l-PA indicated that product inhibition may limit l-PA production by C. glutamicum.

Influence of l-PA on growth of C. glutamicum WT (a) and l-lysine and l-PA titers produced by various strains derived from C. glutamicum GRLys1 (b). a l-PA was added to the medium before inoculation. b Means and standard deviations of three replicates are given
A synthetic pathway for conversion of l-lysine to l-PA was employed (Fig. 1). l-Lysine can be oxidatively deaminated by l-lysine 6-dehydrogenase to yield α-aminoadipic semialdehyde, which after spontaneous cyclization to 1-piperidine 6-carboxylic acid can be reduced by pyrroline 5-carboxylate reductase to l-PA. While C. glutamicum possesses pyrroline 5-carboxylate reductase ProC (Ankri et al. 1996), corynebacterial genomes lack genes encoding l-lysine 6-dehydrogenase (Neshich et al. 2013). Accordingly, a vector for overexpression of endogenous proC and for expression of heterologous l-lysine 6-dehydrogenase gene lysDH from S. pomeroyi under the control of the constitutive C. glutamicum promoter pTuf was constructed and used to transform GRLys1(pEKEx3), GRLys1ΔsugR(pEKEx3-IolTBest), and GRLys1ΔsugRΔldhA(pEKEx3-IolTBest). While the empty vector carrying control strain GRLys1(pEKEx3)(pSH1) produced l-lysine but not l-PA in 2 % (w/v) glucose minimal medium, the strains carrying pSH1-lysDH-proC accumulated l-PA besides l-lysine after 48 h (Fig. 6b).
Notably, the four strains accumulated l-lysine to comparable concentrations (about 20 mM), and l-PA was produced in addition to l-lysine by strains GRLys1(pEKEx3)(pSH1-lysDH-proC), GRLys1ΔsugR(pEKEx3-IolTBest)(pSH1-lysDH-proC) and GRLys1ΔsugRΔldhA(pEKEx3-IolTBest)(pSH1-lysDH-proC) (Fig. 6b). This result may indicate that conversion of l-lysine to l-PA lowers the intracellular l-lysine concentration below a threshold relevant for genetic or allosteric regulation by l-lysine, as a similar metabolic pull effect was observed for the production of cadaverine employing l-lysine decarboxylase in Bacillus methanolicus (Naerdal et al. 2015). Indeed, the presence of pSH1-lysDH-proC in C. glutamicum WT(pEKEx3) reduced the growth rate from 0.32 ± 0.01 h−1 observed for WT(pEKEx3)(pSH1) to 0.17 ± 0.01 h−1 observed for WT(pSH1-lysDH-proC)(pEKEx3) (data not shown). While under these conditions l-PA was not produced, the availability of l-lysine for growth may have been perturbed.
The l-PA product titers achieved with GRLys1(pEKEx3)(pSH1-lysDH-proC), GRLys1ΔsugR(pEKEx3-IolTBest)(pSH1-lysDH-proC), and GRLys1ΔsugRΔldhA(pEKEx3-IolTBest)(pSH1-lysDH-proC), respectively, were 8 ± 1, 11 ± 1, and 14 ± 1 mM, respectively. Thus, while l-lysine could not be converted to l-PA completely, it is possible to produce l-PA in the grams per liter range using recombinant C. glutamicum.
Discussion
l-Lysine productivity of C. glutamicum on glucose media was improved here (a) by overexpressing genes for uptake and phosphorylation of glucose in addition to the endogenous PTS, (b) by relieving glycolysis genes from SugR-mediated repression, and (c) avoiding formation of l-lactate as by-product through disruption of ldhA. Subsequently, l-PA production was established.
In nature, C. glutamicum rarely encounters the high glucose concentrations prevalent in the early phases of biotechnological batch and fed-batch fermentations. While a high-affinity system for glucose utilization such as the PTS is favorable under most natural conditions, its capacity may limit glucose utilization under conditions of industrial l-lysine production. In line with this notion, replacing the endogenous PTS under low glucose conditions was achieved with the high-affinity glucose permease of S. coelicolor A3(2) (GlcPSco), which constitutes the major glucose uptake system of this organism (van Wezel et al. 2005). Intracellular glucose was phosphorylated by GlcK from B. subtilis, a high-affinity glucokinase with a Km for glucose of 0.24 mM (Mesak et al. 2004). Coupling of iolT2 overexpression with overexpression of endogenous glk or ppgK was less efficient (Fig. 3), which may be explained by lower affinities of the endogenous glucokinases and commensurates with earlier findings that e.g., deletion of ppgK only entails a growth disadvantage at high glucose concentrations (Lindner et al. 2010).
At 4 % (w/v) glucose, however, overexpression of the low affinity permease IolT2 in combination with overexpression of glcK from B. subtilis proved beneficial as a 10 % higher growth rate than the control strain C. glutamicum WT(pEKEx3) was reached (Fig. 3). This was accompanied by a higher specific glucose utilization rate (Qs), lower final biomass concentrations, and higher titers of l-lactate as by-product (Table 3). The problem of l-lactate formation was enhanced when sugR was deleted. This effect may be called overflow metabolism and is often observed when glycolysis capacity exceeds other parts of metabolism, in E. coli e.g., the TCA cycle (Veit et al. 2007) or the respiratory chain (Vemuri et al. 2006). This limitation may be due to transcriptional control by global regulators such as ArcA (Perrenoud and Sauer 2005). Overexpression of genes encoding glycolytic enzymes as applied in E. coli (Xie et al. 2014; Seol et al. 2015), B. subtilis (Liu et al. 2014), or S. coelicolor (Borodina et al. 2008) was also used for C. glutamicum, e.g., to improve the production of high value compounds like amino acids (Yamamoto et al. 2012; Reddy and Wendisch 2014), alcohols (Jojima et al. 2015; Yamamoto et al. 2013), or the organic acid d-lactate (Tsuge et al. 2015). Under these conditions or when a repressor gene of glycolysis genes was deleted, i.e., that of the DeoR-type transcriptional regulator SugR, high titers of l-lactate were formed as side effect of faster glucose utilization (Engels and Wendisch 2007; Engels et al. 2008; Teramoto et al. 2011). Likewise, sugR deletion accelerated l-valine production, but at the expense of increased l-lactate formation (Blombach et al. 2009a). Similarly, deletion of sugR in the l-lysine-producing strain GRLys1 increased l-lactate formation (22 ± 1 mM as compared to 4 ± 1 mM; Fig. 5). Consequently, the fermentative l-lactate dehydrogenase gene ldhA was deleted in GRLys1ΔsugR(pEKEx3-IolTBest) and l-lactate formation was abolished. This entailed more biomass formation rather than more l-lysine production since the final cell dry weight increased by 15 % while the l-lysine titer was slightly reduced. However, the volumetric l-lysine productivity remained almost unchanged (Fig. 5). Taken together, GRLys1ΔsugRΔldhA(pEKEx3-IolTBest) and GRLys1ΔsugRΔldhA(pVWEx1-IolTBest) are suitable strains for fast production of l-lysine and l-lysine derivatives from glucose without formation of l-lactate as by-product.
l-PA production from glucose using recombinant C. glutamicum strains was established here (Fig. 6). Chemical methods to obtain optically pure l-PA (and derivatives thereof) have been described and include diazotation of l-lysine or electrochemical oxidation of protected l-lysine, but also conversion of protected l-aspartic acid or l-glutamic acid derivatives to l-PA molecules substituted at the 4- or 5-position (Couty 1999). However, these reaction sequences are often long, e.g., 11 steps from l-glutamic acid to 4-oxopiperidine, and show low yields even for the one-step conversion of l-lysine to l-PA using disodium nitrosyl-pentacyanoferrate(II) as oxidant (Couty 1999). l-PA can be generated from racemic pipecolic acid via resolution by fractional crystallization of diastereomeric salts or via dynamic kinetic resolution by stereoselective amidases (Eichhorn et al. 1997), but it is difficult to achieve 100 % enantiomeric excess. In nature, l-PA occurs in l-lysine-catabolizing bacteria, e.g., in S. pomeroyi, and its biosynthesis is known to be induced in a l-lysine-dependent manner under high salt conditions (Neshich et al. 2013). l-PA accumulated intracellularly (to about 0.6 μg mg−1 protein) when E. coli BL21 expressing the l-lysine 6-dehydrogenase gene from S. pomeroyi was grown in l-lysine-containing medium (Neshich et al. 2013). Biotransformation of l-lysine to l-PA has been developed using E. coli overproducing LAT (about 3.9 g L−1 of optically pure l-PA (Fujii et al. 2002). Additional overexpression of the E. coli gene for lysine uptake, lysP, accelerated l-PA formation from l-lysine about five fold (Fujii et al. 2002). Biotransformation of racemic dl-lysine with E. coli overproducing l-lysine α-oxidase from S. japonicus yielded high titers (about 45 g L−1) of optically pure l-PA (Tani et al. 2015). The functionality of l-lysine cyclodeaminase from Actinoplanes friuliensis has been shown in C. glutamicum (Wagner et al. 2010), and the enzyme from Streptomyces. pristinaespiralis has been used in biotransformations of l-lysine to l-PA with recombinant E. coli (Tsotsou and Barbirato 2007). However, since l-lysine cyclodeaminases show low turnover numbers (about 0.6 s−1), only 4.5 mM l-PA was produced per hour using 1 g L−1 pure enzyme (Tsotsou and Barbirato 2007). All biotransformations with isolated enzymes or whole cells use l- or dl-lysine as substrate for conversion to l-PA, while to the best of our knowledge, a fermentative route for production of l-PA from glucose is presented here for the first time.
l-PA production from glucose proceeded with a yield of 0.09 ± 0.01 g g−1, and a volumetric productivity of 0.04 ± 0.01 g L−1 h−1 by the recombinant strains developed here (Fig. 6). Metabolic engineering often targets transport of products and/or by-products, which pertains to export of l-PA, re-uptake of l-PA, and export of l-lysine here. l-PA has been described to rapidly accumulate from the growth medium by corynebacteria; however, l-PA did not accumulate under hyperosmolar conditions (Frings et al. 1993). Avoiding l-PA re-uptake may not be of priority for l-PA production by C. glutamicum since the l-PA concentration did neither diminish once it was produced nor when added extracellularly (data not shown). On the other hand, improving l-PA export may be relevant to increase volumetric productivity since, for instance, lysine producing strains such as DM1933 and GRLys1 contain multiple copies of the lysine export gene lysE (Unthan et al. 2015). It is tempting to speculate that l-PA is exported by l-proline export systems since both heterocyclic secondary amino acids only differ in ring size. Genes and proteins for l-proline import are known (the l-proline-specific uptake system PutP, the proline/ectoine uptake system ProP, and the ectoine/proline/glycine betaine carrier EctP (Peter et al. 1998), but l-proline export has only been characterized biochemically. Osmoregulated channel(s) are responsible for efflux of various solutes upon sudden decrease in osmotic pressure and were shown to preferentially mediate efflux of the compatible solutes glycine betaine and l-proline (Ruffert et al. 1997). l-Proline and glycine betaine export occurred even in the absence of the genes mscL and yggB, which encode mechanosensitive channels (Nottebrock et al. 2003). YggB functions as major l-glutamate export system, its C-terminal domain is important for gating hysteresis in a voltage-dependent manner (Nakayama et al. 2016), and certain amino acid exchanges in YggB proved beneficial for l-glutamate export (Nakamura et al. 2007). By contrast, a genetic strategy to improve l-proline export is not evident as long as proteins/genes important for l-proline export remain unknown.
Full conversion of l-lysine to l-PA was not observed. Since conversion of l-lysine to l-PA occurs intracellularly, l-PA production suffers from l-lysine export by GRLys1 that overexpresses lysE from two chromosomal copies. Export of l-lysine is well studied and is due to a single protein, LysE (Vrljic et al. 1996). Thus, deletion of lysE in the l-PA-producing strain may be of advantage for l-PA production. The sum of l-lysine production and l-PA production was higher than production of l-lysine alone, which may be explained by a metabolic pull effect. Export of l-lysine and conversion of l-lysine to l-PA may lower the intracellular l-lysine concentration below a threshold relevant for negative genetic or allosteric regulation by l-lysine. A similar metabolic pull effect was observed for the production of cadaverine from l-lysine employing l-lysine decarboxylase in methylotrophic B. methanolicus (Naerdal et al. 2015). On the other hand, strains with increasing l-lysine productivity, i.e., GRLys1(pEKEx3), GRLys1ΔsugR(pEKEx3-IolTBest), and GRLys1ΔsugRΔldhA(pEKEx3-IolTBest), showed increasing l-PA production after transformation with pSH1-lysDH-proC (8 ± 1 mM, 11 ± 1 mM, and 14 ± 1 mM, Fig. 6), thus, supply of l-lysine as precursor positively influenced l-PA production. It can be expected that transformation of industrial l-lysine-producing strains with pSH1-lysDH-proC is a viable strategy to reach higher l-PA titers.
References
Abe S, Takayama K-I, Kinoshita S (1967) Taxonomical studies on glutamic acid-producing bacteria. J Gen Appl Microbiol 13:279–301. doi:10.2323/jgam.13.279
Ankri S, Serebrijski I, Reyes O, Leblon G (1996) Mutations in the Corynebacterium glutamicum proline biosynthetic pathway: a natural bypass of the proA step. J Bacteriol 178:4412–4419
Bartek T, Rudolf C, Kerssen U, Klein B, Blombach B, Lang S, Eikmanns BJ, Oldiges M (2010) Studies on substrate utilisation in L-valine-producing Corynebacterium glutamicum strains deficient in pyruvate dehydrogenase complex. Bioprocess Biosyst Eng 33:873–883. doi:10.1007/s00449-010-0410-1
Bäumchen C, Krings E, Bringer S, Eggeling L, Sahm H (2009) Myo-inositol facilitators IolT1 and IolT2 enhance D-mannitol formation from D-fructose in Corynebacterium glutamicum. FEMS Microbiol Lett 290:227–235. doi:10.1111/j.1574-6968.2008.01425.x
Baumgart M, Unthan S, Rückert C, Sivalingam J, Grünberger A, Kalinowski J, Bott M, Noack S, Frunzke J (2013) Construction of a prophage-free variant of Corynebacterium glutamicum ATCC 13032 for use as a platform strain for basic research and industrial biotechnology. Appl Environ Microbiol 79:6006–6015. doi:10.1128/AEM.01634-13
Becker J, Wittmann C (2012) Bio-based production of chemicals, materials and fuels-Corynebacterium glutamicum as versatile cell factory. Curr Opin Biotechnol 23:631–640. doi:10.1016/j.copbio.2011.11.012
Blombach B, Arndt A, Auchter M, Eikmanns BJ (2009a) L-valine production during growth of pyruvate dehydrogenase complex-deficient Corynebacterium glutamicum in the presence of ethanol or by inactivation of the transcriptional regulator SugR. Appl Environ Microbiol 75:1197–1200. doi:10.1128/AEM.02351-08
Blombach B, Hans S, Bathe B, Eikmanns BJ (2009b) Acetohydroxyacid synthase, a novel target for improvement of L-lysine production by Corynebacterium glutamicum. Appl Environ Microbiol 75:419–427. doi:10.1128/AEM.01844-08
Blombach B, Riester T, Wieschalka S, Ziert C, Youn J-W, Wendisch VF, Eikmanns BJ (2011) Corynebacterium glutamicum tailored for efficient isobutanol production. Appl Environ Microbiol 77:3300–3310. doi:10.1128/AEM.02972-10
Borodina I, Siebring J, Zhang J, Smith CP, van Keulen G, Dijkhuizen L, Nielsen J (2008) Antibiotic overproduction in Streptomyces coelicolor A3(2) mediated by phosphofructokinase deletion. J Biol Chem 283:25186–25199. doi:10.1074/jbc.M803105200
Burkovski A (ed.). 2008. Corynebacteria: genomics and molecular biology. Horizon Scientific, Haverhill
Clevenstine EC, Walter P, Harris TM, Broquist HP (1979) Biosynthesis of slaframine, (1S,6S,8aS)-1-acetoxy-6-aminooctahydroindolizine, a parasympathomimetic alkaloid of fungal origin. 4. Metabolic fate of ethyl pipecolylacetate, 1,3-dioxooctahydroindolizine, and 1-hydroxyoctahydroindolizine in Rhizoctonia leguminicola. Biochemistry (Mosc) 18:3663–3667
Couty F (1999) Asymmetric syntheses of pipecolic acid and derivatives. Amino Acids 16:297–320
Cremer J, Eggeling L, Sahm H (1991) Control of the lysine biosynthesis sequence in Corynebacterium glutamicum as analyzed by overexpression of the individual corresponding genes. Appl Environ Microbiol 57:1746–1752
Eggeling L, Bott M (eds) (2005) Handbook of Corynebacterium glutamicum. CRC Press, Boca Raton
Eichhorn E, Roduit J-P, Shaw N, Heinzmann K, Kiener A (1997) Preparation of (S)-piperazine-2-carboxylic acid, (R)-piperazine-2-carboxylic acid, and (S)-piperidine-2-carboxylic acid by kinetic resolution of the corresponding racemic carboxamides with stereoselective amidases in whole bacterial cells. Tetrahedron Asymmetry 8:2533–2536. doi:10.1016/S0957-4166(97)00256-5
Eikmanns BJ, Metzger M, Reinscheid D, Kircher M, Sahm H (1991) Amplification of three threonine biosynthesis genes in Corynebacterium glutamicum and its influence on carbon flux in different strains. Appl Microbiol Biotechnol 34:617–622
Eikmanns BJ, Rittmann D, Sahm H (1995) Cloning, sequence analysis, expression, and inactivation of the Corynebacterium glutamicum icd gene encoding isocitrate dehydrogenase and biochemical characterization of the enzyme. J Bacteriol 177:774–782
Engels V, Wendisch VF (2007) The DeoR-type regulator SugR represses expression of ptsG in Corynebacterium glutamicum. J Bacteriol 189:2955–2966. doi:10.1128/JB.01596-06
Engels V, Lindner SN, Wendisch VF (2008) The global repressor SugR controls expression of genes of glycolysis and of the L-lactate dehydrogenase LdhA in Corynebacterium glutamicum. J Bacteriol 190:8033–8044. doi:10.1128/JB.00705-08
Frings E, Kunte HJ, Galinski EA (1993) Compatible solutes in representatives of the genera Brevibacterium and Corynebacterium: occurrence of tetrahydropyrimidines and glutamine. FEMS Microbiol Lett 109:25–32. doi:10.1111/j.1574-6968.1993.tb06138.x
Fujii T, Mukaihara M, Agematu H, Tsunekawa H (2002) Biotransformation of L-lysine to L-pipecolic acid catalyzed by L-lysine 6-aminotransferase and pyrroline-5-carboxylate reductase. Biosci Biotechnol Biochem 66:622–627
Gaigalat L, Schlüter J-P, Hartmann M, Mormann S, Tauch A, Pühler A, Kalinowski J (2007) The DeoR-type transcriptional regulator SugR acts as a repressor for genes encoding the phosphoenolpyruvate:sugar phosphotransferase system (PTS) in Corynebacterium glutamicum. BMC Mol Biol 8:104. doi:10.1186/1471-2199-8-104
Gatto GJ, Boyne MT, Kelleher NL, Walsh CT (2006) Biosynthesis of pipecolic acid by RapL, a lysine cyclodeaminase encoded in the rapamycin gene cluster. J Am Chem Soc 128:3838–3847. doi:10.1021/ja0587603
Gerstmeir R, Cramer A, Dangel P, Schaffer S, Eikmanns BJ (2004) RamB, a novel transcriptional regulator of genes involved in acetate metabolism of Corynebacterium glutamicum. J Bacteriol 186:2798–2809
Gibson DG (2011) Enzymatic assembly of overlapping DNA fragments. Methods Enzymol 498:349–361. doi:10.1016/B978-0-12-385120-8.00015-2
González JM, Covert JS, Whitman WB, Henriksen JR, Mayer F, Scharf B, Schmitt R, Buchan A, Fuhrman JA, Kiene RP, Moran MA (2003) Silicibacter pomeroyi sp. nov. and Roseovarius nubinhibens sp. nov., dimethylsulfoniopropionate-demethylating bacteria from marine environments. Int J Syst Evol Microbiol 53:1261–1269. doi:10.1099/ijs.0.02491-0
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580
Heider SAE, Wendisch VF (2015) Engineering microbial cell factories: metabolic engineering of Corynebacterium glutamicum with a focus on non-natural products. Biotechnol J 10:1170–1184. doi:10.1002/biot.201400590
Hermann T (2003) Industrial production of amino acids by coryneform bacteria. J Biotechnol 104:155–172
Hirasawa T, Kim J, Shirai T, Furusawa C, Shimizu H (2012) Molecular mechanisms and metabolic engineering of glutamate overproduction in Corynebacterium glutamicum. Subcell Biochem 64:261–281. doi:10.1007/978-94-007-5055-5_13
Ikeda M, Mizuno Y, Awane S, Hayashi M, Mitsuhashi S, Takeno S (2011) Identification and application of a different glucose uptake system that functions as an alternative to the phosphotransferase system in Corynebacterium glutamicum. Appl Microbiol Biotechnol 90:1443–1451. doi:10.1007/s00253-011-3210-x
Ikeda M, Noguchi N, Ohshita M, Senoo A, Mitsuhashi S, Takeno S (2015) A third glucose uptake bypass in Corynebacterium glutamicum ATCC 31833. Appl Microbiol Biotechnol 99:2741–2750. doi:10.1007/s00253-014-6323-1
Imriskova I, Arreguín-Espinosa R, Guzmán S, Rodriguez-Sanoja R, Langley E, Sanchez S (2005) Biochemical characterization of the glucose kinase from Streptomyces coelicolor compared to Streptomyces peucetius var. caesius. Res Microbiol 156:361–366. doi:10.1016/j.resmic.2004.11.001
Ishige T, Krause M, Bott M, Wendisch VF, Sahm H (2003) The phosphate starvation stimulon of Corynebacterium glutamicum determined by DNA microarray analyses. J Bacteriol 185:4519–4529
Jojima T, Noburyu R, Sasaki M, Tajima T, Suda M, Yukawa H, Inui M (2015) Metabolic engineering for improved production of ethanol by Corynebacterium glutamicum. Appl Microbiol Biotechnol 99:1165–1172. doi:10.1007/s00253-014-6223-4
Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A, Dusch N, Eggeling L, Eikmanns BJ, Gaigalat L, Goesmann A, Hartmann M, Huthmacher K, Krämer R, Linke B, McHardy AC, Meyer F, Möckel B, Pfefferle W, Pühler A, Rey DA, Rückert C, Rupp O, Sahm H, Wendisch VF, Wiegräbe I, Tauch A (2003) The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J Biotechnol 104:5–25
Kalinowski J, Cremer J, Bachmann B, Eggeling L, Sahm H, Pühler A (1991) Genetic and biochemical analysis of the aspartokinase from Corynebacterium glutamicum. Mol Microbiol 5:1197–1204
Kind S, Neubauer S, Becker J, Yamamoto M, Völkert M, von Abendroth G, Zelder O, Wittmann C (2014) From zero to hero - production of bio-based nylon from renewable resources using engineered Corynebacterium glutamicum. Metab Eng 25:113–123. doi:10.1016/j.ymben.2014.05.007
Krings E, Krumbach K, Bathe B, Kelle R, Wendisch VF, Sahm H, Eggeling L (2006) Characterization of myo-inositol utilization by Corynebacterium glutamicum: the stimulon, identification of transporters, and influence on L-lysine formation. J Bacteriol 188:8054–8061. doi:10.1128/JB.00935-06
Kusakabe H, Kodama K, Kuninaka A, Yoshino H, Misono H, Soda K (1980) A new antitumor enzyme, L-lysine alpha-oxidase from Trichoderma Viride purification and enzymological properties. J Biol Chem 255:976–981
Lange C, Rittmann D, Wendisch VF, Bott M, Sahm H (2003) Global expression profiling and physiological characterization of Corynebacterium glutamicum grown in the presence of L-valine. Appl Environ Microbiol 69:2521–2532
Leßmeier L, Pfeifenschneider J, Carnicer M, Heux S, Portais J-C, Wendisch VF (2015) Production of carbon-13-labeled cadaverine by engineered Corynebacterium glutamicum using carbon-13-labeled methanol as co-substrate. Appl Microbiol Biotechnol. doi:10.1007/s00253-015-6906-5
Lindner SN, Knebel S, Pallerla SR, Schoberth SM, Wendisch VF (2010) Cg2091 encodes a polyphosphate/ATP-dependent glucokinase of Corynebacterium glutamicum. Appl Microbiol Biotechnol 87:703–713. doi:10.1007/s00253-010-2568-5
Lindner SN, Seibold GM, Henrich A, Krämer R, Wendisch VF (2011) Phosphotransferase system-independent glucose utilization in Corynebacterium glutamicum by inositol permeases and glucokinases. Appl Environ Microbiol 77:3571–3581. doi:10.1128/AEM.02713-10
Liu D-F, Ai G-M, Zheng Q-X, Liu C, Jiang C-Y, Liu L-X, Zhang B, Liu Y-M, Yang C, Liu S-J (2014) Metabolic flux responses to genetic modification for shikimic acid production by Bacillus subtilis strains. Microb Cell Factories 13:40. doi:10.1186/1475-2859-13-40
Maddess ML, Tackett MN, Ley SV (2008) Total synthesis studies on macrocyclic pipecolic acid natural products: FK506, the antascomicins and rapamycin. Prog Drug Res Fortschritte Arzneimittelforschung Prog Rech Pharm 66(13):15–186
McDonald TP, Walmsley AR, Henderson PJ (1997) Asparagine 394 in putative helix 11 of the galactose-H+ symport protein (GalP) from Escherichia coli is associated with the internal binding site for cytochalasin B and sugar. J Biol Chem 272:15189–15199
Mesak LR, Mesak FM, Dahl MK (2004) Bacillus subtilis GlcK activity requires cysteines within a motif that discriminates microbial glucokinases into two lineages. BMC Microbiol 4:6. doi:10.1186/1471-2180-4-6
Meyer D, Schneider-Fresenius C, Horlacher R, Peist R, Boos W (1997) Molecular characterization of glucokinase from Escherichia coli K-12. J Bacteriol 179:1298–1306
Miller DL, Rodwell VW (1971) Metabolism of basic amino acids in Pseudomonas putida. Catabolism of lysine by cyclic and acyclic intermediates. J Biol Chem 246:2758–2764
Mimitsuka T, Sawai H, Hatsu M, Yamada K (2007) Metabolic engineering of Corynebacterium glutamicum for cadaverine fermentation. Biosci Biotechnol Biochem 71:2130–2135. doi:10.1271/bbb.60699
Misono H, Hashimoto H, Uehigashi H, Nagata S, Nagasaki S (1989) Properties of L-lysine epsilon-dehydrogenase from Agrobacterium tumefaciens. J Biochem (Tokyo) 105:1002–1008
Mitsuhashi S (2014) Current topics in the biotechnological production of essential amino acids, functional amino acids, and dipeptides. Curr Opin Biotechnol 26:38–44. doi:10.1016/j.copbio.2013.08.020
Molnár I, Aparicio JF, Haydock SF, Khaw LE, Schwecke T, König A, Staunton J, Leadlay PF (1996) Organisation of the biosynthetic gene cluster for rapamycin in Streptomyces hygroscopicus: analysis of genes flanking the polyketide synthase. Gene 169:1–7
Naerdal I, Pfeifenschneider J, Brautaset T, Wendisch VF (2015) Methanol-based cadaverine production by genetically engineered Bacillus methanolicus strains. Microb Biotechnol 8:342–350. doi:10.1111/1751-7915.12257
Nakamura J, Hirano S, Ito H, Wachi M (2007) Mutations of the Corynebacterium glutamicum NCgl1221 gene, encoding a mechanosensitive channel homolog, induce L-glutamic acid production. Appl Environ Microbiol 73:4491–4498. doi:10.1128/AEM.02446-06
Nakayama Y, Becker M, Ebrahimian H, Konishi T, Kawasaki H, Krämer R, Martinac B (2016) The impact of the C-terminal domain on the gating properties of MscCG from Corynebacterium glutamicum. Biochim Biophys Acta 1858:130–138. doi:10.1016/j.bbamem.2015.10.010
Návarová H, Bernsdorff F, Döring A-C, Zeier J (2012) Pipecolic acid, an endogenous mediator of defense amplification and priming, is a critical regulator of inducible plant immunity. Plant Cell 24:5123–5141. doi:10.1105/tpc.112.103564
Neshich IAP, Kiyota E, Arruda P (2013) Genome-wide analysis of lysine catabolism in bacteria reveals new connections with osmotic stress resistance. ISME J 7:2400–2410. doi:10.1038/ismej.2013.123
Nottebrock D, Meyer U, Krämer R, Morbach S (2003) Molecular and biochemical characterization of mechanosensitive channels in Corynebacterium glutamicum. FEMS Microbiol Lett 218:305–309
Oh YH, Choi JW, Kim EY, Song BK, Jeong KJ, Park K, Kim I-K, Woo HM, Lee SH, Park SJ (2015) Construction of synthetic promoter-based expression cassettes for the production of cadaverine in recombinant Corynebacterium glutamicum. Appl Biochem Biotechnol 176:2065–2075. doi:10.1007/s12010-015-1701-4
Park SJ, Kim EY, Noh W, Park HM, Oh YH, Lee SH, Song BK, Jegal J, Lee SY (2013) Metabolic engineering of Escherichia coli for the production of 5-aminovalerate and glutarate as C5 platform chemicals. Metab Eng 16:42–47. doi:10.1016/j.ymben.2012.11.011
Park SY, Kim HK, Yoo SK, Oh TK, Lee JK (2000) Characterization of glk, a gene coding for glucose kinase of Corynebacterium glutamicum. FEMS Microbiol Lett 188:209–215
Park S-Y, Moon M-W, Subhadra B, Lee J-K (2010) Functional characterization of the glxR deletion mutant of Corynebacterium glutamicum ATCC 13032: involvement of GlxR in acetate metabolism and carbon catabolite repression. FEMS Microbiol Lett 304:107–115. doi:10.1111/j.1574-6968.2009.01884.x
Payton CW, Chang YF (1982) delta1-piperideine-2-carboxylate reductase of Pseudomonas putida. J Bacteriol 149:864–871
Perrenoud A, Sauer U (2005) Impact of global transcriptional regulation by ArcA, ArcB, Cra, Crp, Cya, Fnr, and Mlc on glucose catabolism in Escherichia coli. J Bacteriol 187:3171–3179. doi:10.1128/JB.187.9.3171-3179.2005
Peter H, Weil B, Burkovski A, Krämer R, Morbach S (1998) Corynebacterium glutamicum is equipped with four secondary carriers for compatible solutes: identification, sequencing, and characterization of the proline/ectoine uptake system, ProP, and the ectoine/proline/glycine betaine carrier, EctP. J Bacteriol 180:6005–6012
Peters-Wendisch P, Götker S, Heider SA, Komati Reddy G, Nguyen AQ, Stansen KC, Wendisch VF (2014) Engineering biotin prototrophic Corynebacterium glutamicum strains for amino acid, diamine and carotenoid production. J Biotechnol 192 Pt B:346–354. doi:10.1016/j.jbiotec.2014.01.023
Peters-Wendisch PG, Schiel B, Wendisch VF, Katsoulidis E, Möckel B, Sahm H, Eikmanns BJ (2001) Pyruvate carboxylase is a major bottleneck for glutamate and lysine production by Corynebacterium glutamicum. J Mol Microbiol Biotechnol 3:295–300
Pimentel-Schmitt EF, Jahreis K, Eddy MP, Amon J, Burkovski A, Titgemeyer F (2009) Identification of a glucose permease from Mycobacterium smegmatis mc2 155. J Mol Microbiol Biotechnol 16:169–175. doi:10.1159/000119546
Reddy GK, Wendisch VF (2014) Characterization of 3-phosphoglycerate kinase from Corynebacterium glutamicum and its impact on amino acid production. BMC Microbiol 14:54. doi:10.1186/1471-2180-14-54
Riedel C, Rittmann D, Dangel P, Möckel B, Petersen S, Sahm H, Eikmanns BJ (2001) Characterization of the phosphoenolpyruvate carboxykinase gene from Corynebacterium glutamicum and significance of the enzyme for growth and amino acid production. J Mol Microbiol Biotechnol 3:573–583
Rittmann D, Schaffer S, Wendisch VF, Sahm H (2003) Fructose-1,6-bisphosphatase from Corynebacterium glutamicum: expression and deletion of the fbp gene and biochemical characterization of the enzyme. Arch Microbiol 180:285–292. doi:10.1007/s00203-003-0588-6
Ruffert S, Lambert C, Peter H, Wendisch VF, Krämer R (1997) Efflux of compatible solutes in Corynebacterium glutamicum mediated by osmoregulated channel activity. Eur J Biochem FEBS 247:572–580
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, New York
Schneider J, Wendisch VF (2010) Putrescine production by engineered Corynebacterium glutamicum. Appl Microbiol Biotechnol 88:859–868. doi:10.1007/s00253-010-2778-x
Schrumpf B, Eggeling L, Sahm H (1992) Isolation and prominent characteristics of an L-lysine hyperproducing strain of Corynebacterium glutamicum. Appl Microbiol Biotechnol 37:566–571. doi:10.1007/BF00240726
Seol E, Sekar BS, Raj SM, Park S (2015) Co-production of hydrogen and ethanol from glucose by modification of glycolytic pathways in Escherichia coli–from Embden-Meyerhof-Parnas pathway to pentose phosphate pathway. Biotechnol J. doi:10.1002/biot.201400829
Shimizu H (2013) Systems metabolic engineering for the production of bio-nylon precursor. Biotechnol J 8:513–514. doi:10.1002/biot.201300097
Soda K, Misono H, Yamamoto T (1968) L-lysine:alpha-ketoglutarate aminotransferase. I. Identification of a product, delta-1-piperideine-6-carboxylic acid. Biochemistry (Mosc) 7:4102–4109
Stansen C, Uy D, Delaunay S, Eggeling L, Goergen J-L, Wendisch VF (2005) Characterization of a Corynebacterium glutamicum lactate utilization operon induced during temperature-triggered glutamate production. Appl Environ Microbiol 71:5920–5928. doi:10.1128/AEM.71.10.5920-5928.2005
Takayama S, Isogai A, Nakata M, Suzuki H, Suzuki A (1984) Structure of Cyl-1, a novel cyclotetrapeptide from Cylindrocladium scoparium. Agric Biol Chem 48:839–842. doi:10.1080/00021369.1984.10866232
Tang J, Zhu X, Lu J, Liu P, Xu H, Tan Z, Zhang X (2013) Recruiting alternative glucose utilization pathways for improving succinate production. Appl Microbiol Biotechnol 97:2513–2520. doi:10.1007/s00253-012-4344-1
Tani Y, Miyake R, Yukami R, Dekishima Y, China H, Saito S, Kawabata H, Mihara H (2015) Functional expression of L-lysine α-oxidase from Scomber japonicus in Escherichia coli for one-pot synthesis of L-pipecolic acid from DL-lysine. Appl Microbiol Biotechnol 99:5045–5054. doi:10.1007/s00253-014-6308-0
Tateno T, Okada Y, Tsuchidate T, Tanaka T, Fukuda H, Kondo A (2009) Direct production of cadaverine from soluble starch using Corynebacterium glutamicum coexpressing alpha-amylase and lysine decarboxylase. Appl Microbiol Biotechnol 82:115–121. doi:10.1007/s00253-008-1751-4
Teramoto H, Inui M, Yukawa H (2011) Transcriptional regulators of multiple genes involved in carbon metabolism in Corynebacterium glutamicum. J Biotechnol 154:114–125. doi:10.1016/j.jbiotec.2011.01.016
Toyoda K, Teramoto H, Inui M, Yukawa H (2008) Expression of the gapA gene encoding glyceraldehyde-3-phosphate dehydrogenase of Corynebacterium glutamicum is regulated by the global regulator SugR. Appl Microbiol Biotechnol 81:291–301. doi:10.1007/s00253-008-1682-0
Toyoda K, Teramoto H, Inui M, Yukawa H (2009a) Involvement of the LuxR-type transcriptional regulator RamA in regulation of expression of the gapA gene, encoding glyceraldehyde-3-phosphate dehydrogenase of Corynebacterium glutamicum. J Bacteriol 191:968–977. doi:10.1128/JB.01425-08
Toyoda K, Teramoto H, Inui M, Yukawa H (2009b) The ldhA gene, encoding fermentative L-lactate dehydrogenase of Corynebacterium glutamicum, is under the control of positive feedback regulation mediated by LldR. J Bacteriol 191:4251–4258. doi:10.1128/JB.00303-09
Tsotsou GE, Barbirato F (2007) Biochemical characterisation of recombinant Streptomyces pristinaespiralis L-lysine cyclodeaminase. Biochimie 89:591–604. doi:10.1016/j.biochi.2006.12.008
Tsuge Y, Yamamoto S, Kato N, Suda M, Vertès AA, Yukawa H, Inui M (2015) Overexpression of the phosphofructokinase encoding gene is crucial for achieving high production of D-lactate in Corynebacterium glutamicum under oxygen deprivation. Appl Microbiol Biotechnol 99:4679–4689. doi:10.1007/s00253-015-6546-9
Unthan S, Baumgart M, Radek A, Herbst M, Siebert D, Brühl N, Bartsch A, Bott M, Wiechert W, Marin K, Hans S, Krämer R, Seibold G, Frunzke J, Kalinowski J, Rückert C, Wendisch VF, Noack S (2015) Chassis organism from Corynebacterium glutamicum–a top-down approach to identify and delete irrelevant gene clusters. Biotechnol J 10:290–301. doi:10.1002/biot.201400041
Unthan S, Grünberger A, van Ooyen J, Gätgens J, Heinrich J, Paczia N, Wiechert W, Kohlheyer D, Noack S (2014) Beyond growth rate 0.6: what drives Corynebacterium glutamicum to higher growth rates in defined medium. Biotechnol Bioeng 111:359–371. doi:10.1002/bit.25103
Van Wezel GP, Mahr K, König M, Traag BA, Pimentel-Schmitt EF, Willimek A, Titgemeyer F (2005) GlcP constitutes the major glucose uptake system of Streptomyces coelicolor A3(2). Mol Microbiol 55:624–636. doi:10.1111/j.1365-2958.2004.04413.x
Veit A, Polen T, Wendisch VF (2007) Global gene expression analysis of glucose overflow metabolism in Escherichia coli and reduction of aerobic acetate formation. Appl Microbiol Biotechnol 74:406–421. doi:10.1007/s00253-006-0680-3
Vemuri GN, Altman E, Sangurdekar DP, Khodursky AB, Eiteman MA (2006) Overflow metabolism in Escherichia coli during steady-state growth: transcriptional regulation and effect of the redox ratio. Appl Environ Microbiol 72:3653–3661. doi:10.1128/AEM.72.5.3653-3661.2006
Vrljic M, Sahm H, Eggeling L (1996) A new type of transporter with a new type of cellular function: L-lysine export from Corynebacterium glutamicum. Mol Microbiol 22:815–826
Wagner N, Steinkämper A, Biener R, Schwartz D (2010) Heterologous production of L-pipecolic acid in Corynebacterium glutamicum. J Biotechnol 150:400–400. doi:10.1016/j.jbiotec.2010.09.523
Wendisch VF (2003) Genome-wide expression analysis in Corynebacterium glutamicum using DNA microarrays. J Biotechnol 104:273–285
Wendisch VF (ed) (2007) Amino acid biosynthesis ∼ pathways, regulation and metabolic engineering. Springer Berlin Heidelberg, Berlin, Heidelberg
Wendisch VF (2014) Microbial production of amino acids and derived chemicals: synthetic biology approaches to strain development. Curr Opin Biotechnol 30:51–58. doi:10.1016/j.copbio.2014.05.004
Wendisch VF, Bott M, Eikmanns BJ (2006a) Metabolic engineering of Escherichia coli and Corynebacterium glutamicum for biotechnological production of organic acids and amino acids. Curr Opin Microbiol 9:268–274. doi:10.1016/j.mib.2006.03.001
Wendisch VF, Bott M, Kalinowski J, Oldiges M, Wiechert W (2006b) Emerging Corynebacterium glutamicum systems biology. J Biotechnol 124:74–92. doi:10.1016/j.jbiotec.2005.12.002
Wickwire BM, Harris CM, Harris TM, Broquist HP (1990) Pipecolic acid biosynthesis in Rhizoctonia leguminicola. I. The lysine saccharopine, delta 1-piperideine-6-carboxylic acid pathway. J Biol Chem 265:14742–14747
Xie X, Liang Y, Liu H, Liu Y, Xu Q, Zhang C, Chen N (2014) Modification of glycolysis and its effect on the production of L-threonine in Escherichia coli. J Ind Microbiol Biotechnol 41:1007–1015. doi:10.1007/s10295-014-1436-1
Yamamoto S, Gunji W, Suzuki H, Toda H, Suda M, Jojima T, Inui M, Yukawa H (2012) Overexpression of genes encoding glycolytic enzymes in Corynebacterium glutamicum enhances glucose metabolism and alanine production under oxygen deprivation conditions. Appl Environ Microbiol 78:4447–4457. doi:10.1128/AEM.07998-11
Yamamoto S, Suda M, Niimi S, Inui M, Yukawa H (2013) Strain optimization for efficient isobutanol production using Corynebacterium glutamicum under oxygen deprivation. Biotechnol Bioeng 110:2938–2948. doi:10.1002/bit.24961
Yukawa H, Inui M (eds) (2013) Corynebacterium glutamicum, microbiology monographs. Springer, Berlin, Heidelberg
Zhang B, Zhou N, Liu Y-M, Liu C, Lou C-B, Jiang C-Y, Liu S-J (2015) Ribosome binding site libraries and pathway modules for shikimic acid synthesis with Corynebacterium glutamicum. Microb Cell Factories 14:71. doi:10.1186/s12934-015-0254-0
Acknowledgments
The authors thank Sabine Heider and Annalena Wille for help with plasmid construction. This work was supported in part by a fellowship from the CLIB Graduate Cluster to Fernando Pérez-García and by EVONIK (Halle, Germany).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ehical approval
The research performed did not involve human participants and/or animals.
Electronic supplementary material
ESM 1
(PDF 173 kb)
Rights and permissions
About this article

Cite this article
Pérez-García, F., Peters-Wendisch, P. & Wendisch, V.F. Engineering Corynebacterium glutamicum for fast production of l-lysine and l-pipecolic acid. Appl Microbiol Biotechnol 100, 8075–8090 (2016). https://doi.org/10.1007/s00253-016-7682-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7682-6