
Abstract
Intracellular precursor supply is a critical factor for amino acid productivity of Corynebacterium glutamicum. To test for the effect of improved pyruvate availability on l-lysine production, we deleted the aceE gene encoding the E1p enzyme of the pyruvate dehydrogenase complex (PDHC) in the l-lysine-producer C. glutamicum DM1729 and characterised the resulting strain DM1729-BB1 for growth and l-lysine production. Compared to the host strain, C. glutamicum DM1729-BB1 showed no PDHC activity, was acetate auxotrophic and, after complete consumption of the available carbon sources glucose and acetate, showed a more than 50% lower substrate-specific biomass yield (0.14 vs 0.33 mol C/mol C), an about fourfold higher biomass-specific l-lysine yield (5.27 vs 1.23 mmol/g cell dry weight) and a more than 40% higher substrate-specific l-lysine yield (0.13 vs 0.09 mol C/mol C). Overexpression of the pyruvate carboxylase or diaminopimelate dehydrogenase genes in C. glutamicum DM1729-BB1 resulted in a further increase in the biomass-specific l-lysine yield by 6 and 56%, respectively. In addition to l-lysine, significant amounts of pyruvate, l-alanine and l-valine were produced by C. glutamicum DM1729-BB1 and its derivatives, suggesting a surplus of precursor availability and a further potential to improve l-lysine production by engineering the l-lysine biosynthetic pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Corynebacterium glutamicum is an aerobic, Gram-positive organism that grows on a variety of sugars and organic acids. The organism is used for the fermentative production of amino acids, such as l-glutamate and l-lysine (Kelle et al. 2005; Kimura 2005; Leuchtenberger et al. 2005), recently amounting to more than 1,500,000 and 750,000 t/a, respectively (Shimizu and Hirasawa 2007; Wittmann and Becker 2007). These impressing production numbers became possible because of tremendous advances in fermentation technology on the one side and in sophisticated strain improvement on the other (Sahm et al. 2000; de Graaf et al. 2001; Pfefferle et al. 2003; Ikeda 2003; Kelle et al. 2005). However, due to the growing world market for amino acids (Leuchtenberger et al. 2005), there is an increasing interest in further strain development and optimisation of efficient fermentative l-lysine production processes.
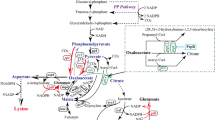
As shown in Fig. 1, C. glutamicum synthesises l-lysine in a split pathway from oxaloacetate via l-aspartate, l-aspartate semialdehyde and d, l-diaminopimelate as the main intermediates (Schrumpf et al. 1991). At high ammonium concentrations (and thus, under l-lysine production conditions), the organism uses the diaminopimelate dehydrogenase (DDH in Fig. 1) variant of the pathway, whereas the succinyl transferase (in Fig. 1) variant is only operative at low ammonium concentrations (Sonntag et al. 1993; reviewed in Sahm et al. 2000). Four moles of reduced form nicotinamide adenine dinucleotide phosphate (NADPH) are required for the synthesis of 1 mol l-lysine from oxaloacetate. Key enzymes of the l-lysine biosynthetic pathway in C. glutamicum are aspartate kinase (ASK), which in the wild-type background of C. glutamicum, shows concerted feedback inhibition by l-lysine and l-threonine (Shiio and Miyajima 1969; Nakayama et al. 1966; Kalinowski et al. 1991), and dihydrodipicolinate synthase (DPS), which condenses pyruvate with l-aspartate semialdehyde and competes with homoserine dehydrogenase for this latter substrate (Cremer et al. 1988, 1991). Overexpression of the respective genes, lysC and dapA, and especially, (over)expression of lysC alleles coding for feedback-resistant aspartate kinases (ASKsFBR) improved l-lysine production significantly (Eggeling et al. 1998; Cremer et al. 1991; Schrumpf et al. 1992). Furthermore, the limitation of the intracellular C flux towards l-threonine (Ohnishi et al. 2002) and the augmentation of the NADPH supply led to an improvement of l-lysine production. The latter has been accomplished by expression of the membrane-bound transhydrogenase genes from Escherichia coli in C. glutamicum (Kabus et al. 2007) and by redirection of the C flux from glycolysis to the pentose phosphate pathway by disruption of the phosphoglucose isomerase gene pgi (Marx et al. 2003) or by introduction of a mutant allele encoding a feedback-resistant 6-phosphogluconate dehydrogenase (Ohnishi et al. 2005).

The PEP–pyruvate–oxaloacetate node in C. glutamicum and the biosynthetic pathway of l-lysine via the diaminopimelate dehydrogenase (DDH)
Aside from engineering the biosynthetic l-lysine pathway starting from oxaloacetate and aside from improving NADPH regeneration, optimisation of precursor availability has been found to be crucial for efficient production of l-lysine. Both oxaloacetate and pyruvate are central metabolic precursors for l-lysine formation (see Fig. 1), and their supply should be balanced for optimal l-lysine production. First indications for a positive effect of improved oxaloacetate and pyruvate supply on l-lysine production can be deduced from the generation and analysis of undefined l-lysine-producing mutants of Brevibacterium flavum with low citrate synthase activity or low pyruvate dehydrogenase complex (PDHC) activity (Shiio et al. 1982, 1984). Menkel et al. (1989) showed that addition of fumarate to the growth medium and thus increasing the oxaloacetate availability led to a higher l-lysine yield with a producer strain. Later, it has been shown that increasing the carbon (C) flux from pyruvate to oxaloacetate by either overexpression of the pyruvate carboxylase (PCx) gene (pyc) or inactivation of the phosphoenolpyruvate (PEP) carboxykinase gene (pck) is highly beneficial for the production of l-lysine by C. glutamicum (Peters-Wendisch et al. 2001; Riedel et al. 2001; Petersen et al. 2001). In accordance with these results, Ohnishi et al. (2002) obtained a significant increase in l-lysine accumulation when they introduced a pyc allele (pyc P458S), probably encoding a deregulated enzyme, into a l-lysine-producing C. glutamicum strain. These findings made it conceivable that improvement of the availability of the precursor pyruvate might also prove beneficial for efficient production of l-lysine and, therefore, decreasing or abolishing the PDHC activity by genetic engineering represented an attractive approach for further optimisation of l-lysine production by C. glutamicum.
Recently, we identified and functionally characterised the E1p subunit of the PDHC in C. glutamicum and showed that activity of this complex is essential for growth of this organism on glucose, unless supplemented with acetate (Schreiner et al. 2005). Moreover, we engineered C. glutamicum for the production of l-valine from glucose by inactivation of the PDHC and additional overexpression of genes encoding the l-valine biosynthetic enzymes (Blombach et al. 2007). In the present study, we investigate the effect of PDHC inactivation in the genetically defined lysine-producer C. glutamicum DM1729 on growth and l-lysine formation.
Materials and methods
Bacterial strains, plasmids, oligonucleotides and culture conditions
All bacterial strains and plasmids and their relevant characteristics and sources, and the oligonucleotides used in this study are listed in Table 1.
E. coli was grown aerobically in a 2xTY complex medium (Sambrook et al. 2001) at 37°C as 50-ml cultures in 500-ml baffled Erlenmeyer flasks on a rotary shaker at 120 rpm. Pre-cultures of the different C. glutamicum strains were grown in 3.7% (w/v) brain heart infusion (BHI) medium (Merck) containing 0.5% (w/v) acetate. For growth and amino acid fermentations in shake flasks, the cells of an overnight pre-culture were washed with 0.9% (w/v) NaCl and inoculated into CGXII minimal medium (Eikmanns et al. 1991) to give an initial optical density at 600 nm (OD600) of about 1. The medium contained 111 and 222 mM glucose [i.e. 2 or 4% (w/v), respectively] and 170 mM potassium acetate [corresponds to 1% (w/v) acetate]. When indicated in “Results”, 0.5% (w/v) BHI powder (Merck) was added to the medium. The plasmid-carrying strains were grown in the presence of kanamycin (50 μg ml−1). C. glutamicum was grown aerobically at 30°C as 50-ml cultures in 500-ml baffled Erlenmeyer flasks on a rotary shaker at 120 rpm. The batch fermentations were performed at 30°C as 250-ml cultures in a Fedbatch-pro fermentation system from DASGIP (Jülich, Germany). The fermentation conditions for aeration and pH control were described in Blombach et al. (2007).
DNA preparation and transformation
The isolation of plasmids from E. coli and C. glutamicum was performed as described before (Eikmanns et al. 1994). Plasmid transfer into C. glutamicum was carried out by electroporation, recombinant strains were selected on Luria-Bertani/BHI supplemented broth agar plates containing kanamycin (50 μg ml−1; van der Rest et al. 1999).
Construction of C. glutamicum DM1729-BB1
Inactivation of the chromosomal aceE gene in the defined l-lysine producer C. glutamicum DM1729 was performed as described previously for aceE inactivation of C. glutamicum WT (Schreiner et al. 2005). The deletion at the chromosomal locus was verified by polymerase chain reaction (PCR) using primers delaceE1/delaceE5.3 and by Southern blot analysis. For the latter, an aceE-specific 580-bp DNA probe was generated from chromosomal DNA of C. glutamicum WT by PCR with the primers delaceE1 and delaceE3 and hybridised to SalI-restricted and size-fractionated chromosomal DNA from C. glutamicum DM1729 and the ΔaceE-derivative DM1729-BB1. Labelling, hybridisation, washing and detection were conducted using the nonradioactive DNA labelling and detection kit and the instructions from Roche Diagnostics (Penzberg, Germany). As expected, the hybridisation resulted in a signal of 8.3 kb with DNA from strain DM1729 and in a signal of 6.2 kb in the C. glutamicum DM1729-BB1.
Determination of PDHC enzyme activity
To determine the specific PDHC activities in cell extracts, C. glutamicum DM1729 and DM1729-BB1 were grown in BHI medium containing 1% (w/v) glucose and 1% (w/v) acetate and harvested in the exponential growth phase (OD600 of about 7), washed twice in 100 mM Tris–HCl, pH 7.2, 3 mM l-cysteine, 10 mM MgCl2 and resuspended in 0.5 ml of the same buffer. Cell disruption was performed as described by Schreiner et al. (2005) with a RiboLyser™ at 4°C. The protein concentrations of the cell extracts were determined by the Bradford method using bovine serum albumin as standard.
PDHC enzyme activities were determined photometrically according to the method described by Guest and Creaghan (1974). One unit of activity is defined as 1 μmol reduced form nicotinamide adenine dinucleotide formed per minute at 30°C.
Analytics
For quantification of substrates and products, 1-ml samples were taken from the cultures, centrifuged at 13,600×g (10 min), and the supernatant was used for determination of amino acid, glucose and/or organic acid concentrations in the culture fluid. The amino acid concentrations were determined by reversed-phase high-pressure liquid chromatography as described before (Blombach et al. 2007). Glucose, acetate and l-lactate concentrations were determined by enzymatic tests from Roche Diagnostics. The pyruvate concentrations were determined enzymatically according to Bergmeyer (1983).
Online analysis of the oxygen and carbon dioxide content of the exhaust gas was performed using the GA4 gas analyser from DASGIP (Jülich, Germany).
The carbon evolution rate [CER; given in mol/(l × h)] was determined by the following equation:
V N, the mol volume of the ideal gas (l/mol) at standard conditions; V R, the working volume of the bioreactor (l); v g,in, the volumetric inlet air flow (l/h) at standard conditions; and \( Y^{{{\text{in}}}}_{{{\text{O}}_{{\text{2}}} }} \), \( Y^{{{\text{in}}}}_{{{\text{CO}}_{{\text{2}}} }} \) and \( Y^{{{\text{out}}}}_{{{\text{O}}_{{\text{2}}} }} \), \( Y^{{{\text{out}}}}_{{{\text{CO}}_{{\text{2}}} }} \), the molecular fractions of oxygen and carbon dioxide in the inlet and outlet air, respectively.
Determination of the ratio CDW per OD600
For determination of the cell dry weight (CDW) per OD600, cells of a BHI pre-culture of C. glutamicum DM1729 and DM1729-BB1 were washed with 0.9% (w/v) NaCl and inoculated into CGXII medium containing 4% (w/v) glucose, 1% (w/v) acetate and 0.5% (w/v) BHI powder to give an initial OD600 of about 1. Cells of both strains were harvested by centrifugation (5,000 rpm, 4°C, 20 min) in the exponential phase (OD600 of about 9) and in the stationary phase (OD600 for DM1729 of about 60 and for DM1729-BB1 of about 14). After two washing steps with water, the cells were dried for 24 h at 105°C, and the CDW was determined. For both DM1729 and DM1729-BB1, an OD600 of 1 corresponded to 0.3 g CDW l−1. According to Liebl (2005), the C content of the C. glutamicum CDW amounts to 41% (w/w).
Substrate-specific biomass and product yields in the parallel fermentation system
For analysis of the substrate-specific biomass and product yields, C. glutamicum DM1729 and DM1729-BB1 were grown in CGXII medium containing 4% (w/v) glucose, 1% (w/v) acetate and 0.5% (w/v) BHI. The yields were calculated by relating the total initial C content of the culture (i.e. C in initial CDW, glucose and acetate) to the total C recovered in CDW, amino acids and organic acids after complete consumption of all C sources. Before calculating the final biomass and product yields, we subtracted (from the recovered C) the amount of biomass and product C derived from BHI, amounting to 1.7 and 0.5% of the total recovered C for C. glutamicum DM1729 and DM1729-BB1, respectively. The final biomass and product yields are given as mol C/mol C.
Results
Construction and analysis of a PDHC-deficient derivative of C. glutamicum DM1729
To study the influence of inactivation of the PDHC on l-lysine production, we deleted the aceE gene encoding the E1p subunit of the PDHC in the lysine-producer C. glutamicum DM1729, resulting in C. glutamicum DM1729-BB1. The parental strain DM1729 originates from C. glutamicum WT and has undergone three allelic exchanges (pyc P458S; hom V59A; lysC T311I), which previously have been described to transform C. glutamicum WT to a l-lysine producer (Ohnishi et al. 2002; Georgi et al. 2005). Successful deletion of aceE in C. glutamicum DM1729-BB1 was verified (see “Materials and methods”), and the parental strain C. glutamicum DM1729 and the deletion mutant DM1729-BB1 were tested for PDHC activity and growth.
PDHC activities were determined in extracts of cells grown in BHI medium containing glucose and acetate. C. glutamicum DM1729 showed specific PDHC activities of 64 ± 6 mU mg protein−1, whereas C. glutamicum DM1729-BB1 mutant was devoid of any detectable PDHC activity (<1 mU mg protein−1). These results show that deletion of the aceE gene in C. glutamicum DM1729 led to complete inactivation of the PDHC.
For comparative analysis of the growth of C. glutamicum DM1729 and DM1729-BB1, shake flask cultivations were performed with minimal medium containing different substrates. C. glutamicum DM1729-BB1 was unable to grow in minimal medium containing glucose, unless supplemented with acetate (data not shown). In minimal medium containing 2% (w/v) glucose plus 1% (w/v) acetate, C. glutamicum DM1729 had a short lag-phase and then grew exponentially with a maximal growth rate of 0.34 h−1 to an OD600 of 34 (10.2 g CDW/l) within 16 h. C. glutamicum DM1729-BB1 showed a maximal growth rate of 0.19 h−1 and an OD600 of about 8 (2.4 g CDW/l) after 24 h (Fig. 2). When the medium containing glucose and acetate additionally was supplemented with 0.5% (w/v) of BHI powder, both C. glutamicum DM1729 and DM1729-BB1 showed no lag-phase and grew exponentially with growth rates of 0.44 h−1 to maximal ODs600 of about 36 (10.8 g CDW/l) and 16 (4.8 g CDW/l), respectively (Fig. 2).

Growth of C. glutamicum DM1729 and DM1729-BB1 in minimal medium containing 2% glucose and 1% acetate (filled square DM1729; filled circle DM1729-BB1) or 2% glucose, 1% acetate and 0.5% BHI (open square DM1729; open circle DM1729-BB1)
Fermentation pattern and yields of C. glutamicum DM1729 and DM1729-BB1
For characterisation of the product pattern of C. glutamicum DM1729 and DM1729-BB1, shake flask fermentations in minimal medium containing 4% (w/v) glucose, 1% (w/v) acetate and 0.5% (w/v) BHI were performed, and growth, glucose and acetate utilisation and pyruvate and amino acid accumulation were monitored in the course of the experiment. C. glutamicum DM1729 grew exponentially within 20 h to an OD600 of about 62 (18.6 g CDW/l), metabolised glucose and acetate simultaneously and produced 23 mM l-lysine with a resulting biomass-specific yield (Y P/X) of 1.23 mmol/(g CDW) (Fig. 3a and Table 2). Apart from l-lysine, no other amino acids and no acetate, pyruvate or l-lactate were observed in the culture fluid of C. glutamicum DM1729. The PDHC-deficient mutant DM1729-BB1 also showed exponential growth in the first 6 to 8 h of fermentation to an OD600 of about 14 (4.2 g CDW/l) and metabolised glucose and acetate simultaneously (Fig. 3b). With depletion of acetate, C. glutamicum DM1729-BB1 stopped growth but continued to metabolise glucose and to form amino acids and pyruvate. After complete consumption of glucose, C. glutamicum DM1729-BB1 reached a final OD600 of about 19 (5.7 g CDW/l) and produced 30 mM l-lysine with a Y P/X of 5.27 mmol/(g CDW) (Fig. 3b and Table 2). In addition to l-lysine, C. glutamicum DM1729-BB1 excreted about 14 mM l-valine, 16 mM l-alanine and up to 65 mM pyruvate into the culture broth (Fig. 3b).

Growth, substrate consumption and product (pyruvate and l-amino acid) accumulation during a representative shake flask batch cultivation of aC. glutamicum DM1729 and b DM1729-BB1 in minimal medium initially containing glucose (4%), acetate (1%) and BHI (0.5%). Diamond growth; filled square glucose; open square acetate; triangle pyruvate; open circlel-alanine; filled circlel-valine; plus signl-lysine. Three independent fermentations were performed, all three showing comparable results
We also studied the fermentation patterns, the biomass-specific l-lysine yields (Y P/X), the substrate-specific biomass yields (Y X/S) and product yields (Y P/S) of C. glutamicum DM1729 and DM1729-BB1 in minimal medium containing glucose, acetate and BHI in a controlled parallel fermentation system providing a constant pH of 7.0 and a constant oxygen concentration of 30% saturation. Both strains started growth with nearly identical growth rates of 0.4 h−1 and reached ODs600 of about 51 (15.3 g CDW/l; DM1729) and 16 (4.8 g CDW/l; DM1729-BB1) after 17 and 8 h, respectively. After depletion of acetate in the medium (at t = 8 h), C. glutamicum DM1729-BB1 but not C. glutamicum DM1729 showed a severe drop in the CER from about 41 to 7.5 mmol/(l × h). l-Lysine production continued until both acetate and glucose were exhausted and the final l-lysine concentrations in the C. glutamicum DM1729 and DM1729-BB1 cultures were 26 and 36 mM, respectively, with a Y P/X of 1.7 and 6.3 mmol/g CDW. Additionally and as observed before in the shake flasks, C. glutamicum DM1729-BB1 excreted significant amounts of l-valine, l-alanine and pyruvate. As shown in Table 3, the l-lysine Y X/S of C. glutamicum DM1729-BB1 was more than twofold lower, whereas all Y P/Ss (given as mol C/mol C) and the l-lysine productivity [mmol l-lysine/(g CDW × h)] were significantly higher than those of C. glutamicum DM1729.
l-Lysine production with C. glutamicum DM1729 and DM1729-BB1 derivatives overexpressing l-lysine biosynthetic genes
As shown above, C. glutamicum DM1729-BB1 excreted aside from l-lysine also pyruvate, l-alanine and l-valine into the medium. This observation prompted us to try to direct the C flux more efficiently towards l-lysine by additional overexpression of the PCx gene and of l-lysine biosynthetic genes. For this purpose, we transformed C. glutamicum DM1729-BB1 with plasmids pVWEx1-pyc, pJC33, pJC40 and pJC50. These plasmids carry the genes encoding PCx, ASKFBR, DDH and ASKFBR plus DPS, respectively (see Fig. 1). l-Lysine shake flask fermentations with the recombinant C. glutamicum DM1729-BB1 strains were carried out, and Table 2 shows the final ODs600, the extracellular l-lysine and the pyruvate concentrations after complete consumption of glucose and acetate and the overall l-lysine yields (Y P/X). Independent of the plasmid, all C. glutamicum DM1729-BB1 derivatives showed about the same or slightly lower final ODs600 when compared to the parental strain. The C. glutamicum DM1729-BB1 strains with plasmids pJC33 and pJC50, both carrying the lysCFBR gene encoding the ASKFBR, showed severely reduced l-lysine and severely enhanced pyruvate accumulation when compared to the host strain. In contrast to the former two DM1729-BB1 derivatives, the C. glutamicum DM1729-BB1 strains with plasmids pVWEx1-pyc and pJC40 produced significantly more l-lysine (17 and 57%, respectively) and less pyruvate (15% both) than the plasmid-free C. glutamicum DM1729-BB1. However, the two strains still excreted about 55 mM pyruvate and 15 to 20 mM l-valine and l-alanine into the medium.
Discussion
As previously described for the aceE deletion derivative of C. glutamicum WT (Schreiner et al. 2005) and in contrast to the parental strain DM1729, C. glutamicum DM1729-BB1 showed no PDHC activity and was unable to grow in minimal medium containing glucose as sole C and energy source. In minimal medium containing glucose plus acetate, C. glutamicum DM1729-BB1 showed slower growth and a lower final OD600 than the parental strain DM1729. Slower growth on glucose and acetate was not observed with C. glutamicum WT ΔaceE when compared to the WT strain (Schreiner et al. 2005), and therefore, the aceE deletion obviously is unfavourable for the growth when combined with the allelic exchanges introduced into C. glutamicum DM1729. It might well be that the acetylcoenzyme A (acetyl-CoA) provision from acetate (via acetate kinase and phosphotransacetylase, see Fig. 1) together with an oxaloacetate drain off towards l-lysine limits the citrate synthase reaction and, in consequence, limits the synthesis of amino acids derived from glutamate. In favour of this hypothesis, we could partly relieve the slower growth of C. glutamicum DM1729-BB1 by adding 3 mM of glutamate to the medium (data not shown). However, although the slower growth rate could be completely relieved by addition of small amounts of BHI, the differences in the final ODs600 of C. glutamicum DM1729 and DM1729-BB1 remained. The stop of growth and, thus, the lower final biomass of strain DM1729-BB1 at a given substrate (glucose + acetate) concentration can be attributed to the exhaustion of acetate. Upon depletion of acetate in the growth medium, acetyl-CoA formation within the PDHC-negative cells drops down, and therefore, biosynthetic pathways starting from acetyl-CoA (e.g. fatty acid biosynthesis) and also the TCA cycle become almost completely inactive. The latter conclusion is indicated by the observations that CO2 formation and O2 uptake rates of the C. glutamicum DM1729-BB1 culture (but not those of the C. glutamicum DM1729 culture) severely dropped down after consumption of acetate.
Shiio et al. (1984) already showed a positive effect of low PDHC activity on the performance of a l-lysine-producing B. flavum mutant. However, this mutant was obtained by several rounds of random mutagenesis, and the conclusion concerning a direct relation between PDHC activity and l-lysine production was questionable. Our results of the comparative shake flask and fermenter experiments with C. glutamicum DM1729 and its PDHC-deficient derivative DM1729-BB1 demonstrate that inactivation of PDHC in this strain led to improved l-lysine production. After complete consumption of the C sources, the final l-lysine titers, the Y P/X and the Y P/S values of C. glutamicum DM1729-BB1 were increased by 30 to 40%, by 200 to 300% and by about 40%, respectively, when compared to C. glutamicum DM1729. It should be noted, however, that Y P/X and Y P/S in both strains were very similar (±10%) in the first 8 h of the fermentations, i.e. as long as the media contained acetate. The higher overall l-lysine yields were due to the fact that after depletion of acetate the PDHC-deficient mutant stopped to form biomass and to oxidise the substrate to CO2, but continued to produce l-lysine as long as glucose was available. In contrast and as indicated by the constant growth and the CERs, the parental strain DM1729 used a significant proportion of the available substrate for biomass formation and respiration. As shown recently for l-valine production with C. glutamicum WT ΔaceE (Blombach et al. 2007), it can be expected that increasing the initial glucose concentration and/or fed-batch cultivation of C. glutamicum DM1729-BB1 further improves l-lysine production.
In addition to l-lysine, C. glutamicum DM1729-BB1 excreted pyruvate, l-alanine and l-valine (the latter two derived from pyruvate) into the culture broth, indicating that l-lysine production in this strain is limited by the reactions from pyruvate to l-lysine and not by pyruvate availability. This hypothesis is corroborated by our results obtained with C. glutamicum DM1729-BB1 carrying the PCx and the DDH genes on plasmid. Both strains produced more l-lysine and less pyruvate; however, they still excreted pyruvate into the medium. This result shows that, even in the strains with enhanced C flux towards l-lysine, there is still a surplus of pyruvate, which in principle should be available for l-lysine production. However, the results also show that, in C. glutamicum DM1729-BB1, the oxaloacetate supply on the one and the DDH reaction on the other hand are bottlenecks for l-lysine-overproduction. Whereas overexpression of the PCx gene and thus improvement of oxaloacetate supply has previously been shown to be advantageous for l-lysine production by an undefined l-lysine-producing C. glutamicum strain (DG52-5; Peters-Wendisch et al. 2001; Petersen et al. 2001), a positive effect of the overexpression of the DDH gene on l-lysine production has not been observed in other l-lysine producers so far (Cremer et al. 1991). The improvement of l-lysine production by ddh overexpression might be restricted to the ΔaceE background, as a C. glutamicum DM1729 derivative carrying pJC40 showed the same accumulation of l-lysine (and pyruvate) as the host strain DM1729 (data not shown). However, the results of the fermentations with plasmid-carrying derivatives of C. glutamicum DM1729-BB1 indicate that a balanced supply of pyruvate as well as of oxaloacetate is important for l-lysine production and they indicate a further potential to optimise l-lysine production by engineering the l-lysine biosynthetic pathway from oxaloacetate and pyruvate as precursors.
Surprisingly, the C. glutamicum DM1729-BB1 strains with plasmids carrying the gene encoding the ASKFBR showed lower l-lysine and higher pyruvate accumulation than the plasmid-free host strain. As elevation of ASKFBR in C. glutamicum DM1729 (data not shown) and also in other l-lysine-producing strains led to improvement of l-lysine production (see “Introduction”), the shift in the product spectrum was unexpected and can only be explained by, so far, unknown regulatory devices.
Further approaches to improve l-lysine production with C. glutamicum DM1729 will aim at the construction of acetate-prototrophic derivatives with low specific PDHC activity and sophisticated sets or combinations of plasmid-bound l-lysine biosynthetic genes. Such defined derivatives of C. glutamicum DM1729 should not depend on acetate or BHI for optimal growth, should rapidly grow to high cell concentrations and may prove useful as cost-effective biocatalysts for the production of l-lysine or of pyruvate itself and/or of other products derived from this intermediate.
References
Bergmeyer HU (1983) Methods of enzymatic analysis, vol VI, 3rd edn. Verlag Chemie, Weinheim, pp 59–66
Blombach B, Schreiner ME, Holatko J, Bartek T, Oldiges M, Eikmanns BJ (2007) l-Valine production with pyruvate dehydrogenase complex-deficient Corynebacterium glutamicum. Appl Environ Microbiol, in press
Cremer J, Treptow C, Eggeling L, Sahm H (1988) Regulation of the enzymes of lysine biosynthesis in Corynebacterium glutamicum. J Gen Microbiol 134:3221–3229
Cremer J, Eggeling L, Sahm H (1991) Control of the lysine biosynthesis sequence in Corynebacterium glutamicum as analyzed by overexpression of the individual corresponding genes. Appl Environ Microbiol 57:1746–1752
de Graaf AA, Eggeling L, Sahm H (2001) Metabolic engineering for l-lysine production by Corynebacterium glutamicum. Adv Biochem Eng Biotechnol 73:9–29
Eggeling L, Oberle S, Sahm H (1998) Improved l-lysine yield with Corynebacterium glutamicum: use of dapA resulting in increased flux combined with growth limitation. Appl Microbiol Biotechnol 49:24–30
Eikmanns BJ, Metzger M, Reinscheid D, Kircher M, Sahm H (1991) Amplification of three threonine biosynthesis genes in Corynebacterium glutamicum and its influence on carbon flux in different strains. Appl Microbiol Biotechnol 34:617–622
Eikmanns BJ, Thum-Schmitz N, Eggeling L, Lüdtke KU, Sahm H (1994) Nucleotide sequence, expression and transcriptional analysis of the Corynebacterium glutamicum gltA gene encoding citrate synthase. Microbiology 140:1817–1828
Georgi T, Rittmann D, Wendisch VF (2005) Lysine and glutamate production by Corynebacterium glutamicum on glucose, fructose and sucrose: roles of malic enzyme and fructose-1,6-bisphosphatase. Metab Eng 7:291–301
Guest JR, Creaghan IT (1974) Further studies with lipoamide dehydrogenase mutants of Escherichia coli K12. J Gen Microbiol 81:237–245
Hanahan D (1985) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580
Ikeda M (2003) Amino acid production process. Adv Biochem Eng Biotechnol 79:1–35
Kabus A, Georgi T, Wendisch VF, Bott M (2007) Expression of the Escherichia coli pntAB genes encoding a membrane-bound transhydrogenase in Corynebacterium glutamicum improves l-lysine. Appl Microbiol Biotechnol, in press
Kalinowski J, Cremer J, Bachmann B, Eggeling L, Sahm H, Puhler A (1991) Genetic and biochemical analysis of the aspartokinase from Corynebacterium glutamicum. Mol Microbiol 5:1197–1204
Kelle R, Hermann T, Bathe B (2005) l-lysine production: In: Eggeling L, Bott M (eds) Handbook of Corynebacterium glutamicum. CRC Press, Boca Raton, pp 465–488
Kimura E (2005) l-Glutamate production. In: Eggeling L, Bott M (eds) Handbook of Corynebacterium glutamicum. CRC Press, Boca Raton, pp 439–463
Leuchtenberger W, Huthmacher K, Drauz K (2005) Biotechnological production of amino acids and derivates: current status and prospects. Appl Microbiol Biotechnol 69:1–8
Liebl W (2005) Corynebacterium taxonomy. In: Eggeling L, Bott M (eds) Handbook of Corynebacterium glutamicum. CRC Press, Boca Raton, pp 9–34
Marx A, Hans S, Mockel B, Bathe B, de Graaf AA (2003) Metabolic phenotype of phosphoglucose isomerase mutants of Corynebacterium glutamicum. J Biotechnol 104:185–197
Menkel E, Thierbach G, Eggeling L, Sahm H (1989) Influence of increased aspartate availability on lysine formation by a recombinant strain of Corynebacterium glutamicum and utilization of fumarate. Appl Environ Microbiol 55:684–688
Nakayama K, Tanaka H, Hagino H, Kinoshita S (1966) Studies on lysine fermentation. Part V. Concerted feedback inhibition of aspartokinase and the absence of lysine inhibition on aspartic semialdehyde–pyruvate condensation in Micrococcus glutamicus. Agric Biol Chem 30:611–616
Ohnishi J, Mitsuhashi S, Hayashi M, Ando S, Yokoi H, Ochiai K, Ikeda M (2002) A novel methodology employing Corynebacterium glutamicum genome information to generate a new l-lysine-producing mutant. Appl Microbiol Biotechnol 58:217–223
Ohnishi J, Katahira R, Mitsuhashi S, Kakita S, Ikeda M (2005) A novel gnd mutation leading to increased l-lysine production in Corynebacterium glutamicum. FEMS Microbiol Lett 242:265–274
Petersen S, Mack C, de Graaf AA, Riedel C, Eikmanns BJ, Sahm H (2001) Metabolic consequences of altered phosphoenolpyruvate carboxykinase activity in Corynebacterium glutamicum reveal anaplerotic mechanisms in vivo. Metab Eng 3:344–361
Peters-Wendisch PG, Schiel B, Wendisch VF, Katsoulidis E, Mockel B, Sahm H, Eikmanns BJ (2001) Pyruvate carboxylase is a major bottleneck for glutamate and lysine production by Corynebacterium glutamicum. J Mol Microbiol Biotechnol 3:295–300
Pfefferle W, Möckel B, Bathe B, Marx A (2003) Biotechnological manufacture of lysine. Adv Biochem Eng Biotechnol 79:59–112
Riedel C, Rittmann D, Dangel P, Möckel B, Sahm H, Eikmanns BJ (2001) Characterization, expression, and inactivation of the phosphoenolpyruvate carboxykinase gene from Corynebacterium glutamicum and significance of the enzyme for growth and amino acid production. J Mol Microbiol Biotechnol 3:573–583
Sahm H, Eggeling L, de Graaf AA (2000) Pathway analysis and metabolic engineering in Corynebacterium glutamicum. Biol Chem 381:899–910
Sambrook J, Russel DW, Irwin N, Janssen UA (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
Schreiner ME, Fiur D, Holátko J, Pátek M, Eikmanns BJ (2005) E1 enzyme of the pyruvate dehydrogenase complex in Corynebacterium glutamicum: molecular analysis of the gene and phylogenetic aspects. J Bacteriol 187:6005–6018
Schrumpf B, Schwarzer A, Kalinowski J, Pühler A, Eggeling L, Sahm H (1991) A functional split pathway for lysine biosynthesis in Corynebacterium glutamicum. J Bacteriol 173:4510–4516
Schrumpf B, Eggeling L, Sahm H (1992) Isolation and prominent characteristics of an l-lysine hyperproducing strain of Corynebacterium glutamicum. Appl Microbiol Biotechnol 37:566–571
Shiio I, Miyajima R (1969) Concerted inhibition and its reversal by end products of aspartate kinase in Brevibacterium flavum. J Biochem (Tokyo) 5:849–859
Shiio I, Ozaki H, Ujigawa-Takeda K (1982) Production of aspartic acid and lysine by citrate synthase mutants of Brevibacterium flavum. Agric Biol Chem 46:101–107
Shiio I, Toride Y, Sugimoto S (1984) Production of lysine by pyruvate dehydrogenase mutants of Brevibacterium flavum. Agric Biol Chem 48:3091–3098
Shimizu H, Hirasawa T (2007) Production of glutamate and glutamate-related amino acids: molecular mechanism analysis and metabolic engineering. In: Wendisch VF (ed) Amino acid biosynthesis—pathways, regulation and metabolic engineering. Springer, Heidelberg, Germany, in press
Sonntag K, Eggeling L, de Graaf AA, Sahm H (1993) Flux partitioning in the split pathway of lysine synthesis in Corynebacterium glutamicum: quantification by 13C- and 1H-NMR spectroscopy. Eur J Biochem 213:1325–1331
van der Rest ME, Lange C, Molenaar D (1999) A heat shock following electroporation induces highly efficient transformation of Corynebacterium glutamicum with xenogenic plasmid DNA. Appl Microbiol Biotechnol 52:541–545
Wittmann C, Becker J (2007) The l-Lysine story: from metabolic pathways to industrial production. In: Wendisch VF (ed) Amino acid biosynthesis—pathways, regulation and metabolic engineering. Springer, Heidelberg, Germany, in press
Acknowledgement
We thank Brigitte Bathe for providing C. glutamicum DM1729 and Lothar Eggeling for providing plasmids pJC33, pJC40 and pJC50. The support of the Fachagentur Nachwachsende Rohstoffe of the BMVEL (grant 04NR004/22000404) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
This study is dedicated to Prof. Dr. Hermann Sahm on the occasion of his 65th birthday.
Rights and permissions
About this article
Cite this article
Blombach, B., Schreiner, M.E., Moch, M. et al. Effect of pyruvate dehydrogenase complex deficiency on l-lysine production with Corynebacterium glutamicum . Appl Microbiol Biotechnol 76, 615–623 (2007). https://doi.org/10.1007/s00253-007-0904-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-007-0904-1