
Abstract
Protocatechuic acid (3,4-dihydroxybenzoic acid; PCA) serves as a building block for polymers and pharmaceuticals. In this study, the biosynthetic pathway for PCA from glucose was engineered in Corynebacterium glutamicum. The pathway to PCA-employed elements of the chorismate pathway by using chorismate-pyruvate lyase (CPL) and 4-hydroxybenzoate hydroxylase (4-HBA hydroxylase). As C. glutamicum has the potential to synthesize the aromatic amino acid intermediate chorismate and possesses 4-HBA hydroxylase, we focused on expressing Escherichia coli CPL in a phenylalanine-producing strain of C. glutamicum ATCC21420. To secrete PCA, the gene (ubiC) encoding CPL from E. coli was expressed in C. glutamicum ATCC 21420 (strain F(UbiC)). The formation of 28.8 mg/L of extracellular 4-HBA (36 h) and 213 ± 29 mg/L of extracellular PCA (80 h) was obtained by the C. glutamicum strain F(UbiC) from glucose. The strain ATCC21420 was also found to produce extracellular PCA. PCA fermentation was performed using C. glutamicum strain F(UbiC) in a bioreactor at the optimized pH of 7.5. C. glutamicum F(UbiC) produced 615 ± 2.1 mg/L of PCA from 50 g/L of glucose after 72 h. Further, fed-batch fermentation of PCA by C. glutamicum F(UbiC) was performed with feedings of glucose every 24 h. The maximum production of PCA (1140.0 ± 11.6 mg/L) was achieved when 117.0 g/L of glucose was added over 96 h of fed-batch fermentation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Petroleum resources are decreasing yearly, and a sustainable supply of chemicals, fuels, and materials is essential to protecting the global climate and avoiding detrimental changes to the environment. Biorefining represents a promising approach for producing building blocks of fuels, chemicals, and bioplastics through microbial fermentation (Hasunuma et al. 2013). Sustainable, so-called green chemistry is desired by the chemical industry as well (US Department of Energy 2004; OECD 2009). Aromatic compounds serve as precursors and scaffold chemicals for synthesizing plastics and fibers, and most are produced using petroleum components. In order to preserve petroleum fuels, and avoid the release of excess CO2 to the environment, β-phenyllactic acid (Fujita et al. 2013), p-hydroxycinnamic acid (Kawai et al. 2013), and cinnamic acid (Noda et al. 2011) are microbially synthesized as building blocks for polymers.
Protocatechuic acid (3,4-dihydroxybenzoic acid; PCA) is a new building block for synthesizing polymer and plastics. The composite polymer of PCA and aniline functions as an electrode with a good electrochemical activity (Sun et al. 1998). The isomer, 2,4-dihydroxybenzoic acid, is also composed of core-shell polymer aerogels (Carrott et al. 2012) and carbon aerogels containing metals (Carrott et al. 2010). A variety of bioplastics can be synthesized from PCA, which is produced from sugar by recombinant microorganisms. PCA is contained in fruits and vegetables and has the potential to be used in pharmaceuticals and functional foods. Recently, PCA has been recognized for its biological activities that are likely responsible for prevention of some chronic-degenerative, cardiovascular, and neurodegenerative diseases (Masella et al. 2012). The pharmacological properties of PCA include its antioxidant, antiaging, anticancer, and antifibrotic activities. PCA also is known to possess antibacterial and antiviral activities (Kakkar and Bais 2014).
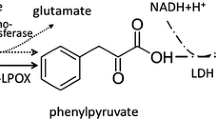
PCA is synthesized by several species of Bacillus, including Bacillus cereus, Bacillus anthracis from siderophore (Wilson et al. 2006; Garner et al. 2004), and Bacillus thuringiensis (Williams et al. 2012). Bacillus thuringiensis ATCC33679 excretes PCA under iron-restricted conditions (Williams et al. 2012; Garner et al. 2004). However, the yields of secreted PCA from Bacillus species are not sufficient to act as polymer building blocks or pharmaceuticals. Therefore, we focused on producing PCA from sugar via the chorismate pathway, which functions to synthesize aromatic amino acids, by increasing the amount of the precursor 4-hydroxybenzoic acid (4-HBA) via expression of the chorismate-pyruvate lyase (CPL; EC 4.1.3.40) in an aromatic amino acid-producing strain of Corynebacterium glutamicum (Fig. 1).

Production of PCA from glucose by recombinant C. glutamicum. Enzymatic reactions catalyzed by chorismate-pyruvate lyase from E. coli and 4-HBA hydroxylase are designed in C. glutamicum
Corynebacterium glutamicum is a non-pathogenic, non-sporulating, non-motile, Gram-positive soil bacterium belonging to the order Actinomycetales, which includes species of Corynebacterium, Nocardia, Rhodococci, and other related microorganisms (George 2001). C. glutamicum is an important industrial microorganism because of its high production of glutamate and amino acids, which are widely used in medicine, animal feed, and food supplements (Leuchtenberger et al. 2005; Hermann 2003). Genetically engineered strains of C. glutamicum are also superior for producing various kinds of organic compounds, including succinic acid as a polymer building block (Okino et al. 2008a), l-lactic acid (Okino et al. 2005) and d-lactic acid (Okino et al. 2008b) for poly lactic acid production, putrescine as a component of bio-based nylon-4,6 (Schneider and Wendisch 2010), cadaverine as a component of bio-based nylon-5,n (Tateno et al. 2009), and gamma-aminobutyric acid as a component of bio-based nylon-4 (Takahashi et al. 2012; Okai et al. 2014).
C. glutamicum is able to grow on PCA as a carbon source (Merkens et al. 2005; Shen and Liu 2005). PCA is catabolized into acetyl-CoA and succinyl-CoA through the β-ketoadipate pathway in C. glutamicum (Fig. S1). The gene sets for PCA catabolism (pobApcaK, pcaHGBC, pcaIJ, pcaRFDG) are conserved in the genome sequence of C. glutamicum-type strain ATCC13032 (Brinkrolf et al. 2006). As the gene for 4-HBA hydroxylase (pobA cg;ncgl1032), which catalyzes the conversion of 4-HBA to PCA, functions in C. glutamicum (Huang et al. 2008), PCA will be produced from sugar when chorismate is sequentially converted to 4-HBA in C. glutamicum.
CPL catalyzes the pyruvate-eliminating reaction of the C3-position of chorismate to produce 4-HB (Siebert et al. 1994) (Fig. 1). The genes encoding CPL were cloned from Escherichia coli (Nichols and Green 1992), Mycobacterium tuberculosis (Stadthagen et al. 2005), Xanthomonas campestris pv. campestris (Zhou et al. 2013b), and Xanthomonas oryzae pv. oryzae (Zhou et al. 2013a). In E. coli, 4-HBA is an intermediate in ubiquinone biosynthesis and CPL UbiC catalyzes the first reaction from chorismate (Nichols and Green 1992). Extracellular 4-HBA is formed by an ubiC-overexpressed strain of E. coli (Siebert et al. 1994) .
Herein we show that C. glutamicum expressing ubiC from E. coli can produce extracellular PCA from glucose. An aromatic amino acid-producing strain of C. glutamicum (ATCC21420) was found to be a suitable host for PCA production. The fermentation condition using C. glutamicum (ATCC21420) expressing ubiC was optimized in a bioreactor, and fed-batch fermentation of PCA from glucose was achieved by this strain.
Materials and methods
Microorganisms
The bacterial strains and plasmids used in this study are listed in Table 1. Escherichia coli strains were grown in Luria–Bertani (LB) medium (10 g/L tryptone, 5 g/L yeast extract, and 5 g/L sodium chloride) containing 50 μg/mL kanamycin at 37 °C. A phenylalanine producing C. glutamicum strain ATCC 21420 (Brevibacterium lactofermentum strain AJ-3245 (Okumura et al. 1972), derived from C. glutamicum ATCC13869) was acquired from the American Type Culture Collection (ATCC). C. glutamicum ATCC 13032, C. glutamicum ATCC 21420, and all recombinant strains were grown in brain–heart infusion (BHI) medium (Becton, Dickinson and Co., Franklin Lakes, NJ, USA). For the selection of C. glutamicum transformants, BHI medium supplemented with 25 μg/mL kanamycin (Km) and 1.5 % agar was used. The C. glutamicum transformants were precultivated in 5-mL BHI medium containing 25 μg/mL Km in a test tube for 24 h at 30 °C.
Molecular genetic procedures
The genomic sequence of C. glutamicum ATCC 13032 was searched using the genome database of the NCBI (http://www.ncbi.nlm.nih.gov/genome/) and the BLAST server. All genetic manipulations were performed using E. coli SCS110 to avoid DNA methylations, and polymerase chain reactions (PCRs) were conducted using KOD-plus2 DNA polymerase (Toyobo, Osaka, Japan). C. glutamicum–E. coli shuttle vectors with the high constitutive expression (HCE) promoter, pCH, were constructed as reported in our previous study (Tateno et al. 2007). Plasmid DNAs were purified using the Viogene Mini Plus Plasmid DNA Extraction System (Viogene-BioTek Corporation, Taipei Hsin, Taiwan). Genomic DNA from E. coli W3110 grown in LB medium was purified using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA). The CPL gene, ubiC (GeneBank Accession no. X57434), from E. coli was amplified by PCR from the genomic DNA of E. coli W3110 using the following primer pairs: EcoRV-ubiC-Eco-F1 (5′-AATTGATATC ATGTCACACCCCGCGTTAACG-3′, EcoRV restriction site is underlined) and SalI-ubiC-Eco-R1 (5′-AATTGTCGACTTATTAGTACAACGGTGACGCCGG-3′, SalI restriction site is underlined). The amplified DNA fragment was purified from a 1.0 % agarose gel using the Wizard SV Gel and PCR Clean-Up System (Promega) after gel electrophoresis. The purified 498-bp ubiC fragment was digested with EcoRV and SalI (New England Biolabs, MA, USA) and was cloned into pCH to yield pCH-ubiC. DNA sequences were determined using an ABI PRISM 3130xl Genetic Analyzer (Life Technologies, Carlsbad, CA, USA) to confirm the identities and functionalities of the constructs.
The constructed plasmid pCH-ubiC or pCH as a control was individually introduced into C. glutamicum ATCC 21420 and C. glutamicum ATCC 13032. The transformation was conducted by electroporation with a 2.5-kV, 200-μF electric pulse in a 0.2-cm cuvette using a Gene Pulser Xcell (Bio-Rad, Richmond, CA, USA), followed by a heat shock of 46 °C for 6 min. The cells were then incubated in 1-mL BHI medium at 30 °C for 1.5 h. C. glutamicum transformants were selected on BHI agar plates containing 25 μg/mL of Km, and the presence of the ubiC was confirmed by cell-directed PCR using KOD FX. The resultant strains, C. glutamicum ATCC21420 (pCH-ubiC), and C. glutamicum ATCC21420 (pCH), were designed as strains F(UbiC) and F. C. glutamicum ATCC13032 (pCH-ubiC) was designed as C. glutamicum W(UbiC). C. glutamicum W, pCH-harboring C. glutamicum ATCC13032, was constructed in our previous study.
Culture for production of 4-hydroxybenzoic acid and protocatechuic acid by recombinant Corynebacterium strains in a flask
Corynebacterium glutamicum F(UbiC) and F, C. glutamicum W(UbiC) and W were separately cultured at 30 °C in a test tube in 5 mL of BHI medium containing 25 μg/mL of kanamycin and agitated at 180 rpm for 24 h. A starter-culture suspension (5 mL) was added to 500 mL of AY medium containing 25 μg/mL of Km and 40 g/L of glucose in a 3-L shaker (Sakaguchi) flask. The composition of the AY medium was modified by adding 2 g of yeast extract from the AR medium (Kurusu et al. 1990). Fermentation was performed at 30 °C with agitation at 180 rpm in a BioShaker G ∙ BR-200 (TAITEC, Japan). Throughout the 80 h of culture, 1 mL was collected every 24 h, centrifuged at 9000×g for 5 min at 4 °C and filtered through a Whatman Mini-UniPrep Syringeless Filter Device (GE Healthcare Ltd., Buckinghamshire, UK). OD600 was monitored when each aliquot of culture was removed.
RNA extraction and qRT-PCR analysis
Corynebacterium glutamicum strains F(UbiC) and F were separately cultured in 50 mL of AY medium supplemented with 50 g/L glucose in 500-mL shaker flasks. Fermentation was performed at 30 °C for 72 h with an agitation speed of 200 rpm. After every 24 h of fermentation, a 5-mL aliquot of culture was centrifuged at 8000×g for 10 min, washed with RNA Protect Bacteria Reagent (QIAGEN, Germany), and then resuspended in 0.7 mL of RLT buffer (QIAGEN). Glass beads (0.7 g per tube; 0.1 mm diameter; Yasui KIKAI, Osaka) were added to each sample and the cells were disrupted using a Shake Master Neo (Bio Medical Sciences, Tokyo, Japan) by shaking three times at 1500×g for 1 min at 1-min intervals. Following centrifugation at 9000×g for 5 min, the supernatants from the cells of strains F(UbiC) and F were subjected to total RNA extraction using an RNeasy Mini Kit (QIAGEN).
For quantitative reverse transcription-PCR (qRT-PCR) analysis, RNA-direct SYBR Green real-time PCR Master Mix (Toyobo) was used. A 100-ng aliquot of total RNA was used in 10-μL reaction mixtures for the amplification of ubiC, while 1 ng of total RNA was used for the amplification of the 16S ribosomal RNA (rRNA) gene. Oligonucleotide primers used for qRT-PCR analysis are shown in Table S1. Primers ubiC-100-F and ubiC-378-R were used for the amplification of ubiC, and primers Cg16S-F1 and Cg16S-R1 were used to amplify the 16S rRNA gene. qRT-PCR was performed using Stratagene Mx3000P (Agilent Technologies, CA, USA). The cycle parameters were as follows: one cycle of 96 °C for 30 s, 61 °C for 20 min, and 95 °C for 30 s, followed by 45 cycles of 95 °C for 15 s, 55 °C for 15 s, and 74 °C for 30 s. The relative abundance of the target messenger RNA (mRNA) was quantified based on cycle threshold values. To standardize the results, the relative abundance of 16S rRNA was used as the internal standard. The mRNA levels of ubiC at 24, 48, and 72 h post-inoculation were quantified by qRT-PCR analysis, and are presented relative to the value obtained at 24 h of cultivation.
Culture, preparation of subcellular fractions, and CPL activity assay
Corynebacterium glutamicum strains FUbiC and F were separately cultured in 50 mL of AY medium containing 50 g/L glucose and 25 μg/mL Km in 500-mL shaker flasks. Fermentation was performed at 30 °C for 72 h with an agitation speed of 200 rpm. A 10-mL aliquot of culture was removed every 24 h and centrifuged at 8000×g for 10 min at 4 °C. The resulting cell pellet was washed twice with 50 mM Tris–HCl (pH 7.5), resuspended in 0.7 mL of the same buffer, and then 0.7 g of glass beads were added to the tube. The cells were disrupted as described earlier, and then centrifuged at 9000×g for 5 min. The supernatants (cytoplasmic fractions) were used for the CPL activity assay. Protein concentrations were determined by Bradford assay (Bradford 1976) using bovine serum albumin as the standard.
CPL activity was assayed using a coupled assay protocol as described by Siebert et al. (Siebert et al. 1994). Pyruvate, a product of the CPL reaction, was coupled with the NADH oxidation in a second reaction by lactate dehydrogenase. The reactants used in the assay were as follows: 500 μM chorismate from Enterobacter aerogenes (Sigma-Aldrich, St. Louis, MI, USA), 5 U/mL l-lactate dehydrogenase from chicken heart (Oriental Yeast Co., Tokyo, Japan), 200 μM NADH, and 200 mM NaCl in 50 mM Tris–HCl (pH 7.5). A 10-μg aliquot of each of the cytoplasmic fractions from C. glutamicum strains F(UbiC) and F were added to 0.1 mL of the reaction mixture. Control reaction mixtures were prepared without chorismate. The reactions were performed at 37 °C for 10 min. The decrease of absorbance at 340 nm was measured using an Envision 2104 Multiplate Reader (PerkinElmer, Inc., Waltham, MA, USA).
Production of PCA from glucose by recombinant C. glutamicum strains in a bioreactor
Corynebacterium glutamicum F(UbiC) and F were separately cultured at 30 °C in a test tube in 5 mL of BHI medium containing 25 μg/mL of Km and agitated at 180 rpm for 24 h. A starter culture suspension (4 mL) was added to 400 mL of BY medium containing Km in a 1-L baffled flask for 24 h. The pH of the BY medium was not adjusted. Flask cultivation was performed at 30 °C with an agitation speed of 180 rpm in a BioShaker (G-BR-200). After 24 h, 2 g of each of the cells was collected by centrifugation at 8000×g for 10 min using a refrigerated centrifuge (model 7780) (Kubota, Tokyo, Japan), dissolved in 5 mL of Y medium, and transferred to 100 mL of Y medium (2 % wet cell weight) containing Km. PCA fermentation was performed in A 100 Ml bioreactor in a 250-mL vessel (BioJr 8 Microbio; ABLE Corp., Tokyo, Japan). The 50 g/L of glucose was added to the 100 mL of Y medium, and the fermentation was performed at an agitation speed of 400 rpm for 72 h. The composition of the Y medium was 10 g peptone, 5 g yeast extract, 5 g sodium chloride, 0.2 mg biotin, and 0.2 mg thiamine, per liter. The pH was maintained at 6.0, 7.0, and 7.5 in the medium by feeding an ammonium solution, and the fermentation was performed for 72 h. For the PCA production from glucose, the aeration rate was set at 0.2 vvm, and the pH was maintained at 7.5 during the fermentation. Throughout the culture, 1 mL was collected every 24 h, centrifuged at 9000×g for 5 min at 4 °C, and filtered.
Analysis of PCA production and sugar consumption
The concentrations of PCA and 4-HBA in the culture supernatants were analyzed using a Prominence high-performance liquid chromatography system (HPLC) (Shimadzu, Kyoto, Japan) equipped with a Cosmosil Cholester column (5 μm, 150 mm × 4.6 mm I.D.; Nacalai Tesque, Kyoto, Japan). The mobile phase (20 % methanol [v/v] containing 0.1 % formate [v/v]) was delivered at 1.0 mL/min, and the column was maintained at 30 °C. PCA and 4-HBA (Wako Chemicals, Japan) (0.1–1.0 mM) served as standards. The absorbance at 254 nm of PCA and 4-HBA was monitored using a UV Detector SPD20A (Shimadzu). Glucose was analyzed using a Prominence HPLC System equipped with an SPR-Pb column (0.5 μm, 250 mm × 4.0 mm I.D., Shimadzu). Water was used as the mobile phase and was delivered at 0.6 mL/min to the column maintained at 80 °C. Elution of glucose was monitored using an RID-10A refractometer (Shimadzu).
Mass spectrometry
Corynebacterium glutamicum F(UbiC) was cultured in AY medium containing 50 g/L of glucose and 25 μg/ml of kanamycin for 3 days (80 h) at 30 °C in a test tube agitated at 180 rpm. Aromatic compounds in the supernatant were separated using the HPLC system described above. The peak fraction (0.5 mL) eluting at 6.4 min from the Cosmosil Cholester column was collected in a tube, dried using a CentriVap Concentrator (Labconco Corporation, Kansas City, MO, USA), and dissolved in 100 μL of Milli-Q water.
Dihydroxybenzoic acids were analyzed using a liquid chromatography–triple quadrupole mass spectrometry (LC-QqQ-MS) system (LC, 1260 Infinity; MS, Agilent Technologies 6460 Triple Quad LC/MS; Agilent Technologies) controlled with MassHunter Data Collection software version 04.01 (Agilent Technologies). The aromatic compounds from the supernatant were analyzed using an LC 1260 Infinity System equipped with a Cosmosil Cholester column (5 μm, 100 mm × 2 mm I.D., Nacalai Tesque). The column was equilibrated with 20 % methanol (v/v) containing 0.1 % formate (v/v) delivered at 0.3 mL/min at 30 °C. PCA (3,4-DHBA); 2,3-dihydroxybenzoic acid (2,3-DHBA); 2,4-dihydroxybenzoic acid (2,4-DHBA); and 2,5-dihydroxybenzoic acid (2,5-DHBA) (1 μL, 10 μM each) served as standards. Mass spectrometry was performed using a 6460 Triple Quad LC/MS set to negative-ion scan mode at a capillary voltage of 4000 V. The temperature of the electrospray ionization gas was 350 °C, and the nebulizer gas flowed at 10 L/min at 50 psi. Other conditions were as follows: fragmentor voltage 120 V; sheath gas 400 °C; sheath-gas flow rate 12 L/min; and nozzle voltage 1000 V. The total ion chromatogram was monitored throughout the analysis. The peak areas were determined using MassHunter Qualitative Analysis version B.05.00 (Agilent Technologies).
Fed-batch production of PCA from glucose by C. glutamicum strains
To increase secreted PCA from glucose, fed-batch cultivation of C. glutamicum F(UbiC) was performed for 120 h). C. glutamicum F(UbiC) and C. glutamicum F were cultivated in BY medium in a flask for 24 h, collected by centrifugation, and harvested in the bioreactor (MiniJar8 microbio). The cells (wet wt 2 g) were collected and transferred to 80 mL of Y medium in 250-mL vessels. The fermentation was started with an agitation speed of 400 rpm at 30 °C by adding 50 g/L of glucose. The aeration speed in the bioreactors was set at 0.2 vvm, and the pH of the medium was maintained at pH 7.5 throughout the fermentation. The initial concentration of glucose was 50 g/L, and an extra 25 g/L of glucose was fed to each reactor at 24, 48, and 72 h of cultivation. Extracellular PCA and glucose were assayed as described above.
Results
Expression of the ubiC gene in C. glutamicum
The pathway for producing PCA from sugar in C. glutamicum was designed as shown in Fig. 1. The BLAST search showed that the CPL gene, the first enzyme involved in synthesizing PCA from chorismate, was not conserved in C. glutamicum. On the other hand, the gene set required for PCA catabolism (pobApcaK;_ncgl1032-1031, pcaHGBC; ncgl2315-2305, pcaIJ; ncgl2307-23066, pcaRFDG;_ncgl2308-2311) is conserved in C. glutamicum ATCC13032. PCA is converted into acetyl-CoA and succinyl-CoA through the β-ketoadipate pathway in C. glutamicum (Fig. S1) (Brinkrolf et al. 2006). Therefore, the 4-HBA hydroxylase (pobA cg; ncgl 1032) was chosen as the second enzyme to produce PCA from chorismate (Fig. 1). Because CPL catalyzes the reaction from chorismate to PCA (Fig. 1), we focused on high-expressing E. coli ubiC in C. glutamicum to produce secreted PCA from glucose. A phenylalanine-producing C. glutamicum, strain ATCC 21420, was used as a host for expression of CPL. The ubiC gene amplified from the genomic DNA of E. coli W3110 was inserted into pCH under the control of the HCE promoter to yield pCH-ubiC. pCH-ubiC was individually introduced into C. glutamicum ATCC21420 and C. glutamicum ATCC 13032, and the resultant recombinant strain was designated C. glutamicum F(UbiC) and C. glutamicum W(UbiC), respectively. As a control, pCH was introduced into C. glutamicum ATCC 21420 to generate C. glutamicum F. C. glutamicum W; C. glutamicum ATCC13032 harboring pCH was also used as a control.
To analyze the formation of extracellular PCA, C. glutamicum strains F(UbiC), F, W(UbiC), and W was cultured aerobically in AY medium containing 40 g/L of glucose. HPLC analysis showed the formation of PCA in C. glutamicum F(UbiC) and F after 24 h. The PCA standard and the culture supernatant peak eluted at 6.4 min.
To verify the expression of ubiC in C. glutamicum strain F(UbiC), qRT-PCR analysis was performed. C. glutamicum strains F(UbiC) and F were separately cultured in 50 mL of AY medium containing 50 g/L glucose for 72 h at 30 °C. The levels of ubiC mRNA expression were quantified at 24, 48, and 72 h post-inoculation. ubiC expression was observed in C. glutamicum F(UbiC) cells throughout the 3-day cultivation period (Fig. S2). The levels of ubiC expression by F(UbiC) were 3.7- and 11.6-fold higher at 48 and 72 h, respectively, than that at 24 h post-inoculation. No ubiC expression was observed in C. glutamicum F at any point during the cultivation (Fig. S2).
To examine the expression of CPL in C. glutamicum F(UbiC), the CPL activity of C. glutamicum strains F(UbiC) and F was monitored during the fermentation using AY medium containing 50 g/L glucose (Fig. S3). CPL activity was detected from the intracellular fractions of C. glutamicum F(UbiC) from 24 to 72 h post-inoculation. No CPL activity was detected for strain F (Fig. S3). The maximum CPL activity (10.81 ± 0.18 nmol/mg/min) of strain F(UbiC) was observed after 48 h of cultivation. CPL activity was observed upon overexpression of ubiC in C. glutamicum F(UbiC).
4-Hydroxybenzoic acid production by recombinant C. glutamicum strains
To verify the expression of ubiC, C. glutamicum F(UbiC) and C. glutamicum W(UbiC) were separately cultured for 80 h using 0.5 L of AY medium containing 40 g/L of glucose in a 3-L shaker flask. At the same time, C. glutamicum F and C. glutamicum W served as controls (Fig. 2a, b). The formation of 4-HBA was observed in the culture supernatant of C. glutamicum F(UbiC), and 28.8 mg/L of 4-HBA was produced as indicated by the peak at 36 h of cultivation (Fig. 2a). At the same time, a small amount of 4-HBA (16.3 mg/L) was observed in the culture supernatant of C. glutamicum W(UbiC) at 12 h of cultivation (Fig. 2b). The growth of the ubiC-expressing C. glutamicum F and W strains were similar compared with their control strains (Fig. 2a, b). Thus, ubiC was successfully expressed in C. glutamicum strains. C. glutamicum ATCC21420 was the preferred host for expression of CPL (ubiC).

Production of extracellular 4-HBA by C. glutamicum strains expressing ubiC. C. glutamicum strains F(UbiC), C. glutamicum F, C. glutamicum W(UbiC) and C. glutamicum W were individually cultured in 0.5 L of AY medium containing 40 g/L of glucose by using a 3-L shake flask for 80 h, and the extracellular 4-HBA was monitored every 12 h. a Extracellular 4-HBA of C. glutamicum F(UbiC) (closed circles) and F (open circles). OD600 of C. glutamicum F(UbiC) (closed diamonds) and F (open diamonds) were monitored at the same time. Data points represent the mean and the error from two independent experiments. b Extracellular 4-HBA of C. glutamicum W(UbiC) (closed triangles) and W (open triangles). OD600 of C. glutamicum W(UbiC) (closed diamonds) and W (open diamonds) were monitored at the same time
Protocatechuic acid production from glucose by recombinant C. glutamicum strains
PCA concentrations were monitored using CPL-expressing C. glutamicum strains and their control strains cultured in 0.5 L of AY medium containing 40 g/L of glucose (Fig. 3), over the same time course as for 4-HBA (Fig. 2). C. glutamicum F(UbiC), C. glutamicum F, C. glutamicum W(UbiC) and C. glutamicum W were separately cultured in 0.5 L of AY medium containing 40 g/L of glucose, and the concentrations of extracellular PCA were monitored (Fig. 3). The formation of PCA was observed in the supernatant of recombinant C. glutamicum strains F(UbiC) and F. C. glutamicum strains F(UbiC) and F produced 213 ± 29 and 114 mg/L of extracellular PCA after 80 h of cultivation, respectively (Fig. 3). The formation of a small amount of PCA was observed in the culture supernatant of C. glutamicum W(UbiC) (Fig. 3). C. glutamicum strain F(UbiC) successfully produced extracellular PCA, and strain F also exhibited formation of PCA.

Protocatechuic acid production by recombinant C. glutamicum strains. Extracellular PCA of C. glutamicum F(UbiC) (closed circles), C. glutamicum F (open circles), C. glutamicum W(UbiC) (closed triangles), and C. glutamicum W (open triangles) were monitored. The four strains were cultured in 0.5 L of AY medium containing 40 g/L of glucose by using a 3-L shake flask for 80 h, and the extracellular PCA was monitored every 12 h. Data represent the means and the standard error from two independent experiments
Mass spectrometric verification of PCA synthesis by C. glutamicum F(UbiC)
To verify the synthesis of PCA by C. glutamicum F(UbiC), the dihydroxybenzoic acid fractions from the 3 days (80 h) culture supernatant were isolated and analyzed using LC-QqQ-MS in the negative ionization mode. Total ion chromatograms and the MS/MS product ion spectra are shown in Fig. 4a–e, f–g, respectively. The peak supernatant fraction (Fig. 4a) and the PCA (3,4-DHBA) standard (Fig. 4b) eluted at 2.79 min without overlapping the 2,3-DHBA (Fig. 4c); 2,4-DHBA (Fig. 4d); or 2,5-DHBA (Fig. 4e) peaks. For MS/MS analysis, the fragmentor voltage was set to 120 V to monitor the parent and fragment ions of the dihydroxybenzoic acids. The product obtained from the culture supernatant eluting at 2.79 min was the deprotonated parent ion [M-H]− (m/z 153.0) of PCA (molecular weight, 154.02) (Fig. 4f). The product ion of [M-H]− (m/z 109.0) for PCA was also detected (Fig. 4f). The mass spectra of the culture supernatant peak (Fig. 4f) and the PCA standard (Fig. 4g) were identical. Thus, PCA was selectively produced by C. glutamicum F(UbiC).

Chromatographic analysis of PCA produced by C. glutamicum F(UbiC) and the dihydroxybenzoic acid standards (a–e). Total ion chromatograms of dihydroxybenzoic acids produced by C. glutamicum F(UbiC) (a); 3,4-DHBA(PCA) (b); 2,3-DHBA (c); 2,4-DHBA (d); and 2,5-DHBA (e). Standard chromatograms were generated by injecting 2 μL of each dihydroxy acid (10 μL). Mass spectrometric analysis of PCA produced by C. glutamicum F(UbiC) (f, 2.79 min peak of A) and the 3,4-dihydroxybenzoic acid standard (g, 2.79 min peak of B) are shown
Effect of pH on the production of PCA by C. glutamicum F(UbiC)
To examine the effect of pH on the production of PCA, fermentation was performed from glucose by recombinant C. glutamicum using bioreactors. C. glutamicum F(UbiC) was cultured in a flask using BHI medium for 24 h, and the cells were collected and transferred to 100 mL of Y medium in the bioreactor. The fermentation was started with an agitation speed of 400 rpm at 30 °C by adding 50 g/L of glucose. The aeration speed was set at 0.2 vvm, and the pH of the medium was adjusted to 6.0, 7.0, and 7.5 by adding aqueous ammonium during the fermentation. C. glutamicum F(UbiC) produced 224.3 ± 34.6, 422.4 ± 23.1, and 470.9 ± 3.7 mg/L of PCA at pH 6.0, 7.0, and 7.5 after 48 h of fermentation. C. glutamicum F(UbiC) showed maximum productivity of PCA and maximum glucose consumption at pH 7.5 (Fig. 5).

Effect of pH on the production of PCA by C. glutamicum F(UbiC). Fermentation of PCA from glucose by C. glutamicum F(UbiC) was carried out in bioreactors using Y medium containing 50 g/L glucose and kanamycin. The pH of the medium was maintained at pH 6.5 (squares), 7.0 (triangles), and 7.5 (circles) by adding ammonium solution. The fermentations were performed for 72 h. The PCA concentration (closed symbols) and glucose consumption (open symbols) were determined. Data represent the mean and standard error from three independent experiments
PCA production from glucose by recombinant C. glutamicum strains
For the production of PCA from glucose, fermentation was performed using C. glutamicum F(UbiC) and C. glutamicum F at the optimum pH of 7.5 in the bioreactors (Fig. 6a). C. glutamicum F(UbiC) consumed 55.7 ± 0.08 g/L of glucose, and produced 615.6 ± 2.1 mg/L of extracellular PCA after 72 h of cultivation. The maximum volumetric productivity of PCA from glucose by C. glutamicum F(UbiC) reached 360.53 ± 18.7 mg/L/day (24 h). The yield of PCA from glucose by C. glutamicum F(UbiC) reached 2.35 % mol/mol-glucose after 24 h. C. glutamicum F consumed 53.8 ± 0.13 g/L of glucose, and produced 241.4 ± 7.1 mg/L of PCA after 72 h (Fig. 6a). The growth of the two strains was not significantly different. The formation of extracellular 4-HBA was observed in C. glutamicum F(UbiC) with a peak at 28 h (Fig. 6b).

Production of PCA from glucose by C. glutamicum strains F(UbiC) and F. Time course for the production of PCA from glucose by C. glutamicum F(UbiC) (closed symbols) and F (open symbols). C. glutamicum F(UbiC) and C. glutamicum F was cultivated in BY medium in a flask for 24 h, and the cells (wet wt 2 g) were transferred to 100 mL of Y medium. The 50 g/L of glucose was added to the medium, and the fermentations were performed in bioreactors with an agitation speed of 400 rpm for 72 h at 30 °C. The aeration speed was set at 0.2 vvm, and the pH of the medium was maintained at 7.5 throughout the fermentation. a Extracellular PCA (circles) and glucose (diamonds). b Extracellular 4-HBA (triangles) and OD600 (squares) were determined. Data represent the mean and standard error from three independent experiments
Fed-batch production of PCA from glucose by C. glutamicum F(UbiC) and F
To increase the secreted PCA from glucose, the fed-batch cultivation of C. glutamicum F(UbiC) and C. glutamicum F was performed for 120 h (Fig. 7). The initial concentration of glucose was set at 50 g/L, and an extra 25 g/L of glucose was added to each reactor at 24, 48, and 72 h of cultivation (Fig. 7). C. glutamicum F(UbiC) produced 1140.0 ± 11.6 mg/L of extracellular PCA after 96 h using fed-batch cultivation (Fig. 7). The total glucose consumed by C. glutamicum F(UbiC) was 117.0 g/L for 96 h, and the molar yield of PCA from glucose was 1.50 % mol/mol. The maximum volumetric productivity of PCA by C. glutamicum F(UbiC) was 551.9 mg/L/day after 24 h of cultivation. Finally, the productivity of PCA by C. glutamicum F(UbiC) reached 1168.1 ± 27.4 mg/L after 120 h (Fig. 7). C. glutamicum F produced 466.1 ± 18.6 mg/L of PCA after 120 h (Fig. 7). The total glucose consumed by C. glutamicum F was 115.4 g/L over 120 h, and the molar yield of PCA from glucose was 0.46 % mol/mol (120 h) under this fed-batch condition.

Fed-batch production of PCA from glucose by C. glutamicum strains F(UbiC) and F. C. glutamicum F(UbiC) (closed symbols) and C. glutamicum F (open symbols) were cultivated in BY medium in a flask for 24 h, collected, and harvested in the bioreactor. The cells (wet wt 2 g) were collected and transferred to 80 mL of Y medium, and the fermentation was started with an agitation speed of 400 rpm at 30 °C by adding 50 g/L of glucose. The aeration speeds in the bioreactors were set at 0.2 vvm, and the pH was maintained at pH 7.5. An extra 25 g/L of glucose was fed to each reactor at 24, 48, and 72 h of cultivation. Extracellular PCA (circles) and glucose (squares) were determined. Data represent the mean and the standard error from three independent experiments
Discussion
Extracellular PCA was successfully produced from glucose by a recombinant C. glutamicum strain F(UbiC) expressing ubiC derived from E. coli W3110. C. glutamicum F(UbiC) successfully produced 615.6 mg/L of extracellular PCA from glucose after 3 days of cultivation (Fig. 6), and 1140.0 mg/L of PCA after 4 days of fed-batch cultivation (Fig. 7). This is the first report, to our knowledge, of the production of extracellular PCA from glucose by C. glutamicum expressing CPL. PCA is excreted by the siderophore bacterium B. thuringiensis (Williams et al. 2012) or B. anthracis strain Sterne (Garner et al. 2004) under iron-restricted conditions. A total of 160 μg of PCA/ culture OD unit was excreted by B. anthracis strain Sterne (Garner et al. 2004), In Bacillus, PCA is formed from 3-dehydroshikimate (3-DHS) in the aromatic biosynthesis pathway, and the 3-dehydroshikimate dehydratase (3-DHSD) gene (asbF) was cloned from B. thuringiensis ATCC33679 (Williams et al. 2012). Recently, PCA was produced as a precursor of cis, cis-muconic acid by a recombinant strain of Saccharomyces cerevisiae. The pathway converting 3-DHS to shikimate was blocked, and codon-optimized 3-DSHD from B. thuringiensis was introduced into S. cerevisiae, yielding 0.14 g/L of PCA after 120 h of reaction (Weber et al. 2012). However, the yields of secreted PCA from these organisms were not enough as a starter to synthesize polymer building block or pharmaceuticals.
Corynebacterium glutamicum can catabolize a wide range of aromatic compounds, including 4HBA, and PCA (Shen and Liu 2005). PCA is also used to supplement minimal medium to improve iron uptake (Liebl et al. 1989) and accelerate growth of C. glutamicum when used as a co-substrate with glucose (Unthan et al. 2014). In C. glutamicum, PCA is assimilated in the β-ketoadipate pathway, leading to the tricarboxylic acid cycle intermediates acetyl-CoA and succinyl-CoA. As 4-HB hydroxylase (PobAcg) catalyzes the conversion reaction of 4HBA to PCA, and is functionally involved in 4HBA assimilation (Huang et al. 2008), the route from chorismate to PCA was designed by heterologous expression of CPL in C. glutamicum. Notably, C. glutamicum ATCC 21420 was found to produce extracellular PCA (Fig. 3). C. glutamicum F produced 241.2 mg/L of extracellular PCA in the medium after 72 h of flask cultivation (Fig. 5). It was suggested that strain F has another PCA producing enzyme because extracellular 4HBA was not produced during the fermentation (Fig. 6b). By overexpressing ubiC of E. coli for the production of PCA in C. glutamicum strain F, extracellular PCA was increased (Fig. 6). Currently, we are attempting the whole genome sequencing of strain ATCC 21420 to identify the synthetic pathway of PCA.
The aromatic amino acid intermediate chorismate is synthesized via the shikimate pathway in microorganisms and plants (Bentley 1990). The aromatic amino acid-producing strain of C. glutamicum was supposed to possess an improved flux of chorismate in the shikimate pathway. C. glutamicum ATCC 21420, which is resistant to the phenylalanine analog beta-amino-beta-phenylpropionic acid, produces 2.3 g/L of phenylalanine (Okumura et al. 1972). As shown in Fig. 2b, C. glutamicum F(UbiC) produced extracellular 4-HBA (28 mg/L), while C. glutamicum W(UbiC) produced only a small amount of 4-HBA (Fig. 2a). This suggested that the internal level of chorismate was higher in C. glutamicum strain F than in strain W. C. glutamicum F(UbiC). A total of 213 mg/L of extracellular PCA was obtained from strain F after 80 h of cultivation, while W(UbiC) only formed a small amount of PCA (Fig. 3). Thus, C. glutamicum strain ATCC 21420 is a suitable host for the expression of CPL to produce PCA. In the bioreactor, C. glutamicum F(UbiC) produced a maximum yield of PCA at pH 7.5 (Fig. 5), which is the optimum condition for E. coli CPL. Under the optimized conditions, C. glutamicum F(UbiC) produced 1168.1 ± 11.6 mg/L of PCA after 5 days of fed-batch cultivation using glucose (Fig. 7). To our knowledge, this is the highest yield for the production of PCA from glucose. The advantage of producing PCA using C. glutamicum ATCC21420 expressing CPL is that it involves the direct fermentation of PCA from cheap sugar. As PCA serves as a building block for synthesizing functional polymers, the method for producing PCA from sugar will make the downstream synthesis of polymers very cost effective.
PCA also has the potential to be used in pharmaceuticals and functional foods. PCA may be involved in the control of oxidative stress and inflammation. The antioxidant and anti-inflammatory activities of PCA were analyzed in vitro and in vivo (Semaming et al. 2015). PCA has both antiapoptotic and proapoptotic effects on cells, characteristics which are likely responsible for its roles in human disease prevention (Masella et al. 2012). PCA also has antibacterial and antiviral activities (Kakkar and Bais 2014). As C. glutamicum is recognized as a generally regarded as safe (GRAS) microorganism, a system for producing PCA using engineered C. glutamicum can also be applied to the production of PCA as a component of functional foods and pharmaceuticals.
Corynebacterium glutamicum has the ability to utilize the lignin degradation product ferulic acid and vanillin as carbon sources (Merkens et al. 2005). However, as the parent strain of C. glutamicum cannot utilize cellulosic substrates, we developed an expression system to produce an endoglucanase in C. glutamicum (Tsuchidate et al. 2011), which we plan to test for its ability to produce PCA from the abundant material lignocellulose. In the future, a system for producing PCA from various types of biomass including lignocellulose should be applicable as a way to synthesize building blocks for polymers and pharmaceuticals.
In conclusion, we expressed CPL from E. coli in an aromatic amino acid-producing strain (ATCC21420) of C. glutamicum. The recombinant C. glutamicum strain expressing UbiC successfully produced extracellular PCA from glucose. The CPL expression system using C. glutamicum should prove useful for producing PCA as a building block of plastics and also as a component of pharmaceuticals.
References
Bentley R (1990) The shikimate pathway—a metabolic tree with many branches. Crit Rev Biochem Mol Biol 25:307–384
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Brinkrolf K, Brune I, Tauch A (2006) Transcriptional regulation of catabolic pathways for aromatic compounds in Corynebacterium glutamicum. Genet Mol Res 5:773–789
Carrott PJM, Marques LM, Carrott MMLR (2010) Characterisation of the porosity of polymer and carbon aerogels containing Fe, Ni or Cu prepared from 2,4-dihydroxybenzoic acid by n-nonane pre-adsorption and density functional theory. Microporous Mesoporous Mater 131:75–81
Carrott PJM, Marques LM, Carrott MMLR (2012) Core-shell polymer aerogels prepared by co-polymerisation of 2,4-dihydroxybenzoic acid, resorcinol and formaldehyde. Microporous Mesoporous Mater 158:170–174
Fujita T, Nguyen HD, Ito T, Zhou S, Osada L, Tateyama S, Kaneko T, Takaya N (2013) Microbial monomers custom-synthesized to build true bio-derived aromatic polymers. Appl Microbiol Biotechnol 97:8887–8894
Garner BL, Arceneaux JE, Byers BR (2004) Temperature control of a 3,4-dihydroxybenzoate (protocatechuate)-based siderophore in Bacillus anthracis. Curr Microbiol 49:89–94
George M (2001) Burgey’s manual of systematic bacteriology, 2nd edn. Springer, New York
Hasunuma T, Okazaki F, Okai N, Hara KY, Ishii J, Kondo A (2013) A review of enzymes and microbes for lignocellulosic biorefinery and the possibility of their application to consolidated bioprocessing technology. Bioresour Technol 135:513–522
Hermann T (2003) Industrial production of amino acids by Coryneform bacteria. J Biotechnol 104:155–172
Huang Y, Zhao KX, Shen XH, Jiang CY, Liu SJ (2008) Genetic and biochemical characterization of a 4-hydroxybenzoate hydroxylase from Corynebacterium glutamicum. Appl Microbiol Biotechnol 78:75–83
Kakkar S, Bais S (2014) A review on protocatechuic acid and its pharmacological potential. ISRN Pharmacol 2014: Article ID 952943, 1–9. doi:10.1155/2014/952943
Kawai Y, Noda S, Ogino C, Takeshima Y, Okai N, Tanaka T, Kondo A (2013) p-hydroxycinnamic acid production directly from cellulose using endoglucanase- and tyrosine ammonia lyase-expressing Streptomyces lividans. Microb Cell Factories 12:45
Kurusu Y, Kainuma M, Inui M, Satoh Y, Yukawa H (1990) Electroporation-transformation system for Coryneform bacteria by auxotrophic complementation. Agric Biol Chem 54:443–447
Leuchtenberger W, Huthmacher K, Drauz K (2005) Biotechnological production of amino acids and derivatives: current status and prospects. Appl Microbiol Biotechnol 69:1–8
Liebl W, Klamer R, Schleifer KH (1989) Requirement of chelating compounds for the growth of Corynebacterium glutamicum in synthetic media. Appl Microbiol Biotechnol 32:205–210
Masella R, Santangelo C, D’Archivio M, Li Volti G, Giovannini C, Galvano F (2012) Protocatechuic acid and human disease prevention: biological activities and molecular mechanisms. Curr Med Chem 19:2901–2917
Merkens H, Beckers G, Wirtz A, Burkovski A (2005) Vanillate metabolism in Corynebacterium glutamicum. Curr Microbiol 51:59–65
Nichols BP, Green JM (1992) Cloning and sequencing of Escherichia coli ubiC and purification of chorismate lyase. J Bacteriol 174:5309–5316
Noda S, Miyazaki T, Miyoshi T, Miyake M, Okai N, Tanaka T, Ogino C, Kondo A (2011) Cinnamic acid production using Streptomyces lividans expressing phenylalanine ammonia lyase. J Ind Microbiol Biotechnol 38:643–648
OECD (2009) The bioeconomy to 2030. Organization of economic co-operation and development.
Okai N, Takahashi C, Hatada K, Ogino C, Kondo A (2014) Disruption of pknG enhances production of gamma-aminobutyric acid by Corynebacterium glutamicum expressing glutamate decarboxylase. AMB Express 4:20
Okino S, Inui M, Yukawa H (2005) Production of organic acids by Corynebacterium glutamicum under oxygen deprivation. Appl Microbiol Biotechnol 68:475–480
Okino S, Noburyu R, Suda M, Jojima T, Inui M, Yukawa H (2008a) An efficient succinic acid production process in a metabolically engineered Corynebacterium glutamicum strain. Appl Microbiol Biotechnol 81:459–464
Okino S, Suda M, Fujikura K, Inui M, Yukawa H (2008b) Production of D-lactic acid by Corynebacterium glutamicum under oxygen deprivation. Appl Microbiol Biotechnol 78:449–454
Okumura S, Otsuka S, Yamanoi A, Yoshinaga F, Honda T, Kubota K, Tsuchida T (1972) Method for producing phenylalanine by fermentation. US Patent 3,660,235.
Schneider J, Wendisch VF (2010) Putrescine production by engineered Corynebacterium glutamicum. Appl Microbiol Biotechnol 88:859–868
Semaming Y, Pannengpetch P, Chattipakorn SC, Chattipakorn N (2015) Pharmacological properties of protocatechuic acid and its potential roles as complementary medicine. Evid Based Complement Alternat Med 2015: Article ID 593902, 1-11. doi:10.1155/2015/593902
Shen X, Liu S (2005) Key enzymes of the protocatechuate branch of the beta-ketoadipate pathway for aromatic degradation in Corynebacterium glutamicum. Sci China C Life Sci 48:241–249
Siebert M, Severin K, Heide L (1994) Formation of 4-hydroxybenzoate in Escherichia coli: characterization of the ubiC gene and its encoded enzyme chorismate pyruvate-lyase. Microbiology 140(Pt 4):897–904
Stadthagen G, Kordulakova J, Griffin R, Constant P, Bottova I, Barilone N, Gicquel B, Daffe M, Jackson M (2005) p-hydroxybenzoic acid synthesis in Mycobacterium tuberculosis. J Biol Chem 280:40699–40706
Sun JJ, Zhou DM, Fang HQ, Chen HY (1998) The electrochemical copolymerization of 3,4-dihydroxybenzoic acid and aniline at microdisk gold electrode and its amperometric determination for ascorbic acid. Talanta 45:851–856
Takahashi C, Shirakawa J, Tsuchidate T, Okai N, Hatada K, Nakayama H, Tateno T, Ogino C, Kondo A (2012) Robust production of gamma-amino butyric acid using recombinant Corynebacterium glutamicum expressing glutamate decarboxylase from Escherichia coli. Enzym Microb Technol 51:171–176
Tateno T, Fukuda H, Kondo A (2007) Direct production of l-lysine from raw corn starch by Corynebacterium glutamicum secreting Streptococcus bovis alpha-amylase using cspB promoter and signal sequence. Appl Microbiol Biotechnol 77:533–541
Tateno T, Okada Y, Tsuchidate T, Tanaka T, Fukuda H, Kondo A (2009) Direct production of cadaverine from soluble starch using Corynebacterium glutamicum coexpressing alpha-amylase and lysine decarboxylase. Appl Microbiol Biotechnol 82:115–121
Tsuchidate T, Tateno T, Okai N, Tanaka T, Ogino C, Kondo A (2011) Glutamate production from beta-glucan using endoglucanase-secreting Corynebacterium glutamicum. Appl Microbiol Biotechnol 90:895–901
Unthan S, Grunberger A, van Ooyen J, Gatgens J, Heinrich J, Paczia N, Wiechert W, Kohlheyer D, Noack S (2014) Beyond growth rate 0.6: what drives Corynebacterium glutamicum to higher growth rates in defined medium. Biotechnol Bioeng 111:359–371
US Department of Energy (2004) Top value added chemicals from biomass, volume I - results of screening for potential candidates from sugars and synthesis gas. T.Werpy and G. Petersen. the Pacific Northwest National Laboratory (PNNL).
Weber C, Bruckner C, Weinreb S, Lehr C, Essl C, Boles E (2012) Biosynthesis of cis, cis-muconic acid and its aromatic precursors, catechol and protocatechuic acid, from renewable feedstocks by Saccharomyces cerevisiae. Appl Environ Microbiol 78:8421–8430
Williams KM, Martin WE, Smith J, Williams BS, Garner BL (2012) Production of protocatechuic acid in Bacillus thuringiensis ATCC33679. Int J Mol Sci 13:3765–3772
Wilson MK, Abergel RJ, Raymond KN, Arceneaux JE, Byers BR (2006) Siderophores of Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis. Biochem Biophys Res Commun 348:320–325
Zhou L, Huang TW, Wang JY, Sun S, Chen G, Poplawsky A, He YW (2013a) The rice bacterial pathogen Xanthomonas oryzae pv. oryzae produces 3-hydroxybenzoic acid and 4-hydroxybenzoic acid via xanB2 for use in xanthomonadin, ubiquinone, and exopolysaccharide biosynthesis. Mol Plant Microbe Interact 26:1239–1248
Zhou L, Wang JY, Wu J, Wang J, Poplawsky A, Lin S, Zhu B, Chang C, Zhou T, Zhang LH, He YW (2013b) The diffusible factor synthase XanB2 is a bifunctional chorismatase that links the shikimate pathway to ubiquinone and xanthomonadins biosynthetic pathways. Mol Microbiol 87:80–93
Acknowledgments
This work was partially supported by Special Coordination Funds for Promoting Science and Technology, Creation of Innovation Centers for Advanced Interdisciplinary Research Areas (Innovative Bioproduction Kobe) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We appreciate help from Drs. Fumio Matsuda, Fumiyoshi Okazaki, Satoshi Wakai and Shimpei Aikawa for discussions regarding this work. We thank Ms. Michiru Miyake for technical assistance.
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Compliance with ethical standards
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 272 kb)
Rights and permissions
About this article

Cite this article
Okai, N., Miyoshi, T., Takeshima, Y. et al. Production of protocatechuic acid by Corynebacterium glutamicum expressing chorismate-pyruvate lyase from Escherichia coli . Appl Microbiol Biotechnol 100, 135–145 (2016). https://doi.org/10.1007/s00253-015-6976-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6976-4