
Abstract
The spinel Mn0.5Mg0.5Fe2O4 was obtained via calcining Mn0.5Mg0.5Fe2(C2O4)3·5H2O above 400 °C in air. The precursor and its calcined products were characterized by thermogravimetry and differential scanning calorimetry, Fourier transform FT-IR, X-ray powder diffraction, scanning electron microscopy, energy dispersive X-ray spectrometer, and vibrating sample magnetometer. The results showed that Mn0.5Mg0.5Fe2O4 obtained at 600 °C had a specific saturation magnetization of 46.2 emu g–1. The thermal decomposition of Mn0.5Mg0.5Fe2(C2O4)3·5H2O below 450 °C experienced two steps which involved, at first, the dehydration of five water molecules and then decomposition of Mn0.5Mg0.5Fe2(C2O4)3 into spinel Mn0.5Mg0.5Fe2O4 in air. Based on Starink equation, the values of the activation energies associated with the thermal decomposition of Mn0.5Mg0.5Fe2(C2O4)3·5H2O were determined.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Spinels of the type M2+M 3+2 O4 are always the focus of attention due to their versatility [1–3]. In the case of M3+=Fe3+, the resulting spinel ferrites having a general chemical composition of MFe2O4 (M=Cu, Mn, Mg, Zn, Ni, Co, Ca, etc.) are widely used in the field of high-density information storage, magnetic separation, ferrofluids, catalysts, drug targeting, magnetic resonance imaging, and gas sensor [4–14]. The structure of ferrospinels depends on the outer electron configuration and radius of divalent cation, and distribution of cations at the different sites. For example, ZnFe2O4 is a kind of normal spinel ferrite, NiFe2O4 and CoFe2O4 are inverse spinels, and MnFe2O4 and MnZn ferrites are random spinel. The magnetic moment direction of cations in tetrahedral A-site is opposite with that in octahedral B-site. Therefore, magnetic moment of spinel ferrites can be regulated by distribution of different divalent cations at the different sites.
Nanosized MnFe2O4 is one of the most important magnetic materials. Its properties, such as magnetic behavior and gas-sensing properties, etc., are highly dependent on the synthesis method and doping elements. In recent years, different kinds of MnFe2O4 nanostructured materials have been successfully synthesized, such as nanoparticles [15–17], nanorods [18, 19], and nanofibers [20]. A TEA-assisted route was normally used to obtain the octahedral-like MnFe2O4 crystallites [21].
The doped manganese ferrite can improve its performance. Therefore, doped manganese ferrite caused great concern, and some progress has been made. It was reported that substitutions of Mn2+ with divalent cation led to improved magnetic properties of nanocrystalline ferrites [11, 22–29]. For example, when Mn2+ ions in spinel MnFe2O4 were partially substituted by Co2+ ions, Co0.35Mn0.65Fe2O4 obtained at 700 °C showed a specific saturation magnetization of 71.6 emu g–1 [11]. Rajesh et al. [22] synthesized Mn1–xZnxFe2O4 (x = 0.0, 0.1, 0.3, 0.5, 0.6, 0.7, 0.9) by co-precipitation technique, and studied their magnetic properties. The results showed that the specific saturation magnetization of Mn1–xZnxFe2O4 and Curie temperature (T C) decreased with increasing zinc content in Mn1–xZnxFe2O4. Okasha [27] prepared MgxMn1−xFe2O4 (x = 0.0, 0.15, and 0.25) by γ-irradiation. The results showed that the specific saturation magnetization of MgxMn1−xFe2O4 increased after γ-irradiation, and remanent magnetization decreased after γ-irradiation. However, synthesis research of doped MnFe2O4 still has fewer reports in comparison with that of MnFe2O4. Therefore, new synthesis methods for Mn1−xMxFe2O4 (M = transition metals or alkaline-earth metals) still need to be studied and innovated further. Besides, the kinetics study of thermal decomposition for Mn1−xMxFe2O4 precursor is important to obtain high-quality crystalline doped MnFe2O4 for practical applications.
The aim of this study is to prepare polycrystalline Mn0.5Mg0.5Fe2O4 using MnSO4·H2O, MgSO4·7H2O, FeSO4·7H2O, and Na2C2O4 as raw materials via solid-state reaction at low heating temperatures [6, 11] and to study magnetic properties of Mn0.5Mg0.5Fe2O4, the mechanisms, and kinetics of thermal decomposition of precursor. The kinetics of thermal decomposition of precursor was studied using TG/DTG/DSC techniques. Non-isothermal kinetics of thermal decomposition of precursor was interpreted by Starink equation [30, 31]. The kinetic parameters (E a, A, mechanism) of thermal decomposition of Mn0.5Mg0.5Fe2(C2O4)3·5H2O were discussed for the first time.
Experimental
Reagent and apparatus
All chemicals were of reagent-grade purity (<99.9 %). TG/DSC measurements were taken using a Netzsch Sta 409 PC/PG thermogravimetric analyzer and sample mass was around 11 mg. In order to make ferrous oxalate in the precursor decomposition into Fe2O3 during the heating, and then Fe2O3 reaction with MgO and MnO into Mn0.5Mg0.5Fe2O4, a continuous flow of air (40 mL min–1) was passed into the reaction chamber. The reference material for DSC was Al2O3. X-ray powder diffraction (XRD) was performed using a Rigaku D/max 2,500 V diffractometer equipped with a graphite monochromator and a Cu target. The radiation applied was CuKα (λ = 0.15406 nm), operating at 40 kV and 50 mA. XRD scans were made from 5° to 70° in 2θ with steps of 0.016°. Fourier transform FT-IR spectra of the precursor and its calcined products were recorded on a Nexus 470 Fourier transform FT-IR instrument in KBr pellets in the range of 4,000–400 cm–1. The morphologies of the calcined samples and energy dispersive X-ray spectrometer (EDS) were obtained on S-3400 scanning electron microscopy (SEM). The specific magnetization (Ms) of the calcined sample powder was carried out at room temperature using a magnetic property measurement system (SQUID-MPMS-XL-5).
Preparation of Mn0.5Mg0.5Fe2O4
The Mn0.5Mg0.5Fe2O4 precursor was prepared by solid-state reaction at low heating temperatures using MnSO4·H2O, MgSO4·7H2O, FeSO4·7H2O, and Na2C2O4 as raw materials at first [3]. In a typical synthesis, MnSO4·H2O (8.36 g), MgSO4·7H2O (12.19 g), FeSO4·7H2O (55.00 g), Na2C2O4 (41.57 g), and surfactant polyethylene glycol (PEG)-400 (3.5 mL, 50 vol.%) were put in a mortar, and the mixture was fully ground by hand with a rubbing mallet for 35 min. The grinding velocity was about 220 circles min–1, and the strength applied was moderate. The reactant mixture gradually became damp, and then a paste formed quickly. The reaction mixture was kept at room temperature for 2 h. The mixture was washed with deionized water to remove soluble inorganic salts until SO 2–4 ion could not be visually detected with a 0.5 mol L–1 BaCl2 solution. The solid was then washed with a small amount of anhydrous ethanol and dried at 75 °C for 6 h. The resulting material was subsequently determined to be Mn0.5Mg0.5Fe2(C2O4)3·5H2O. Nanocrystalline Mn0.5Mg0.5Fe2O4 with cubic structure was obtained via calcining Mn0.5Mg0.5Fe2(C2O4)3·5H2O above 400 °C in air.
Method of determining kinetic parameters, and mechanism functions
Determination of activation energy by Starink equation
Activation energy of thermal decomposition of solid compound can be obtained by Starink equation (Eq. 1).
where β i is the heating rate (K min–1), T α is the reaction temperature (K) corresponding to degree of conversion (α) in TG curve, E α is the activation energy (kJ mol–1) of thermal decomposition corresponding to degree of conversion (α), R is the gas constant (8.314 × 10–3 kJ mol–1 K–1). The dependence of \( \ln \left( {\beta_{\text i} /T_{\alpha ,\text i}^{1.92} } \right) \) on 1/T α must give rise to a straight line. Thus, reaction activation energy E α can be obtained from linear slope (−1.0008 E α/R, Eq. 1).
Determination of most probable mechanism functions
The following equation was used to estimate the most correct reaction mechanism of thermal decomposition of Mn0.5Mg0.5Fe2(C2O4)3·5H2O, i.e., g(α) function [32, 33]:
where x = E a/(RT), \( h(x) = \frac{{x^{4} + 18x^{3} + 86x^{2} + 96x}}{{x^{4} + 20x^{3} + 120x^{2} + 240x + 120}} \), and β is the heating rate (K min–1). The conversions α corresponding to multiple rates at the same temperature are put into the left of Eq. (2), combined with 31 types of mechanism functions [33, 34], the slope k and correlation coefficient r 2 are obtained from the plot of ln g(α) versus ln β. The probable mechanism function is the one for which the value of the slope k is near –1.00000 and correlation coefficient r 2 is better.
Calculation of pre-exponential factor A
The pre-exponential factor was estimated from Eq. (3) [31]:
where A is the pre-exponential factor (s–1), β is the heating rate (K min–1), g(α) is the most probably mechanism function determined by Eq. (2), E α is the activation energy (kJ mol–1) of thermal decomposition, R is the gas constant (8.314 × 10–3 kJ mol–1 K–1), and T max is the most rapid decomposition temperature (i.e., peak temperature in DTG curve, K).
Results and discussion
TG/DTG/DSC analysis of the precursor
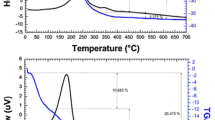
Figure 1 shows the TG/DTG/DSC curves of Mn0.5Mg0.5Fe2(C2O4)3·5H2O at four heating rates of 5, 10, 15, and 20 K min–1 in air.

TG/DTG/DSC curves of Mn0.5Mg0.5Fe2(C2O4)3·5H2O at different heating rates in air
The TG/DTG/DSC curves show that thermal decomposition of the Mn0.5Mg0.5Fe(C2O4)3·5H2O below 450 °C occured in two well-defined steps. For heating rate of 10 K min–1, the first step starts at 40 °C, ends at 205 °C, and is characterized by a weak DTG peak at about 201 °C and a strong endothermic DSC peak at 207 °C, which can be attributed to dehydration of the five crystal water molecules from Mn0.5Mg0.5Fe2(C2O4)3·5H2O (mass loss: observed, 17.82 %; theo., 17.82 %). The second decomposition step begins at 205 °C, ends at 449 °C, and is characterized by a strong DTG peak at 230 °C and a strong DSC exothermic peak at 266 °C, attributed to decomposition of Mn0.5Mg0.5Fe2(C2O4)3 into cubic Mn0.5Mg0.5Fe2O4 in air by one step, and of six CO2 (mass loss: observed, 39.39 %; theo., 39.57 %). No other exothermic DSC peak that was ascribed to crystallization of Mn0.5Mg0.5Fe2O4 below 450 °C except exothermic DSC peak at 266 °C, which indicated that exothermic DSC peak of crystallization of Mn0.5Mg0.5Fe2O4 was overlapped with exothermic DSC peak from decomposition of Mn0.5Mg0.5Fe2(C2O4)3 in air. In other words, The decomposition of Mn0.5Mg0.5Fe2(C2O4)3 and crystallization of cubic Mn0.5Mg0.5Fe2O4 were almost finished at the same temperature. Crystalline Mn0.5Mg0.5Fe2O4 can be obtained at lower calcination temperature, which was attributed to Mn2+, Mg2+, and Fe2+ oxalates that have similar lower decomposition temperature, and ultra-fine the oxide particles with high activity formed by thermal decomposition of Mn0.5Mg0.5Fe2(C2O4)3·5H2O can be easily in situ self-assembled into crystalline Mn0.5Mg0.5Fe2O4.
IR spectroscopic analysis of Mn0.5Mg0.5Fe2(C2O4)3·5H2O and its calcined samples
The FT-IR spectra of Mn0.5Mg0.5Fe2(C2O4)3·5H2O and its calcined sample are shown in Figure 2. The Mn0.5Mg0.5Fe2(C2O4)3·5H2O exhibited a strong and broad band at about 3,373 cm–1, which can be assigned to the stretching OH vibration of the water molecule. The strong band, which appeared at 1,653 cm–1 in the spectrum of the precursor, can be ascribed to the bending mode of the HOH [3, 6, 35–38]. The weak band at about 828 cm–1 was the water libration (hindered rotation). The band at 1,322 cm–1 can be assigned to either the appearance of new M-OC2O3 (M=Mn, Mg, Fe) bonds and/or to the combinations of OH librations and lattice modes [39, 40]. The band at about 609 cm–1 was assigned to the Fe–O stretching vibration of the Fe–O tetrahedron when sample was calcined at 400 °C [41]. The absorption band of the samples obtained above 200 °C, which appeared at about 3,400 cm–1, was attributed to the adsorption of water from air.

FT-IR spectra of Mn0.5Mg0.5Fe2(C2O4)3·5H2O and its calcined samples
XRD and SEM analysis of Mn0.5Mg0.5Fe2(C2O4)3·5H2O and its calcined samples
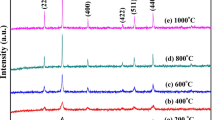
Figure 3 shows the XRD patterns of Mn0.5Mg0.5Fe2(C2O4)3·5H2O dried at 75 °C and the products resulting from calcination at different temperatures in air for 1 h.

XRD patterns of Mn0.5Mg0.5Fe2(C2O4)3·5H2O and its calcined samples at different temperatures in air for 1 h
From Fig. 3 characteristic diffraction peaks of crystalline compound from the precursor were observed, which indicated that Mn0.5Mg0.5Fe2(C2O4)3·5H2O obtained at 75 °C was a crystal with higher crystallinity. The diffraction peaks in the pattern can be indexed to be in agreement with the orthorhombic FeC2O4·2H2O from PDF card 23-0293, with space group I2/a(15). No diffraction peaks of manganese and magnesium oxalates, such as MnC2O4, MnC2O4·xH2O, MgC2O4, and MgC2O4·xH2O, were observed, which implied that Mn2+ and Mg2+ ions entered lattice of FeC2O4·2H2O. MnC2O4, MnC2O4·xH2O, MgC2O4, and MgC2O4·xH2O formed a solid solution with FeC2O4·2H2O. It is explained by the fact that Mn2+ ion (80 pm), Mg2+ ion (65 pm), and Fe2+ ion (76 pm) have same electric charge and similar ionic radius. When Mn0.5Mg0.5Fe2(C2O4)3·5H2O was calcined at 300 °C for 1 h, a part of characteristic diffraction peaks of cubic MgFe2O4 were observed. When Mn0.5Mg0.5Fe2(C2O4)3·5H2O was calcined at 400 °C, all the diffraction peaks in the pattern of sample were in agreement with that of cubic MgFe2O4, with space group Fd-3 m(227), lattice parameters: a = b = c = 0.8387 nm, α = β = γ = 90°, density = 4.502 g cm–3, from PDF card 36-0398. No diffraction peaks of crystalline MnFe2O4 were observed, which implied that MnFe2O4 and MgFe2O4 formed a solid solution. When sample was calcined at 800 °C, a part of characteristic diffraction peaks of rhombohedral Fe2O3 appeared, which implied that Mn0.5Mg0.5Fe2O4 was partly decomposed into Fe2O3.
According to the Scherrer formula [6]: D = Kλ/(βcosθ), where D is crystallite diameter, K = 0.89 (the Scherrer constant), λ = 0.15406 nm (wavelength of the X-ray used), β is the width of line at the half-maximum intensity, and θ is the corresponding angle. The resulting crystallite sizes of the products from calcining precursor at the temperatures of 400, 600, 700, and 800 °C in air for 1 h were 26, 41, 41, and 35 nm, respectively. Crystallite diameter of the product obtained at 800 °C was smaller than that of the samples obtained at 600 and 700 °C, which was attributed to Mn0.5Mg0.5Fe2O4 that was decomposed into Fe2O3 particles with smaller crystallite diameter at 800 °C. The crystallinity of cubic Mn0.5Mg0.5Fe2O4 can be evaluated via MDI Jade 5.0 software, the results showed that crystallinity of cubic Mn0.5Mg0.5Fe2O4 obtained at 400, 600, 700, and 800 °C were 98.73, 99.44, 99.51, and 92.59 %, respectively.
The morphologies and EDS spectrum of the calcined samples are shown in Fig. 4. From Fig. 4a it can be seen that the calcined sample obtained at 600 °C was composed of polyhedral particles, which contained particles having a distribution of small particles (100–200 nm) and large particles (200–300 nm). With the increase of calcination temperature, the crystallite in calcined sample was aggregated into larger grains further. Figure 4b and c show the SEM micrographs of samples obtained at 700 and 800 °C, respectively. The calcined sample at 700 °C still kept polyhedral morphology, particles sizes are mainly between 300 and 400 nm. However, the calcined sample obtained at 800 °C became platelets grains with particle size of between 300 and 1,000 nm. The average crystallite sizes of the calcined samples determined by XRD were significantly smaller than the values determined by SEM. This was attributed to values observed by SEM technique that gave the size of the secondary particles, which were composed of several or many crystallites by soft reunion, and the X-ray line broadening analysis disclosed only the size of single crystallite. Figure 4d shows the EDS spectrum of product obtained at 700 °C in air, mole ratio of Mn:Mg:Fe was equal to 0.51:0.48:2.0, which was close to the value of the pre-design and synthesis.

SEM and EDS analysis of the calcined sample: SEM: a 600 °C, b 700 °C, and c 800 °C; EDS, d 700 °C
Magnetic properties of the calcined samples
Figure 5 shows the hysteresis loops of Mn0.5Mg0.5Fe2O4 particles obtained at different calcination temperatures. From Fig. 5 it can be observed that specific saturation magnetizations of powders calcined at 400, 600, 700, and 800 °C for 1 h were 34.7, 46.2, 45.9, and 31.5 emu g–1, respectively. That is, specific saturation magnetization of Mn0.5Mg0.5Fe2O4 powders increases between 400 and 600 °C with increasing calcination temperature at first, and then decreases. In other words, the larger the crystallite size of the particles, the larger is the specific saturation magnetizations. The larger Ms values associated with larger crystallite sizes can be explained as follow: First, surface distortions due to the interaction of the transition metal ions with the oxygen atoms in the spinel lattice of Mn0.5Mg0.5Fe2O4 can reduce the net magnetic moment in the particle. When the calcination temperature increases, the crystallite size of particles increases, and surface distortions is reduced, so Ms value increases with increasing calcination temperature. Second, the magnetocrystalline anisotropy of the particles is dependent on the crystallinity of Mn0.5Mg0.5Fe2O4. The higher calcination temperature, the larger is crystallinity of particles, which reduces magnetocrystalline anisotropy distortion, and increases magnetic moment within the particles of Mn0.5Mg0.5Fe2O4 [3, 11, 42]. Compared with magnetic properties of MnFe2O4 (500 °C, 9.5 emu g–1; 700 °C, 41.5 emu g–1) [5] and MgFe2O4 (600 °C, 30.4 emu g–1; 700 °C, 35.7 emu g–1) [6], it can be seen that Mn0.5Mg0.5Fe2O4 powders exhibits higher specific saturation magnetizations than MnFe2O4 and MgFe2O4 obtained at same calcination temperature, which implies that Mn2+ and Mg2+ ions in Mn0.5Mg0.5Fe2O4 have a synergistic effect in improving the specific saturation magnetization of Mn0.5Mg0.5Fe2O4. However, specific saturation magnetization of Mn0.5Mg0.5Fe2O4 powders decreases above 600 °C with increasing calcination temperature, which is attributed to Mn0.5Mg0.5Fe2O4 that was decomposed into Fe2O3 particles with weak magnetization above 700 °C.

Hysteresis loops for Mn0.5Mg0.5Fe2O4 samples obtained at different temperatures in air for 1 h
Figure 6 shows effect of calcination temperature on coercivity and remanence of Mn0.5Mg0.5Fe2O4 powders. The results showed that coercivities (Hc) and remanences (Mr) of samples decreased with increasing calcination temperature between 400 and 700 °C.

Effect of calcination temperature on coercivity and remanence of samples
Kinetics of thermal decomposition of Mn0.5Mg0.5Fe2(C2O4)3·5H2O
In accordance with TG/DTG/DSC, IR, and XRD analysis of Mn0.5Mg0.5Fe2(C2O4)3·5H2O and its calcined products mentioned above, thermal decomposition of Mn0.5Mg0.5Fe2(C2O4)3·5H2O below 450 °C in air consists of two steps, which can be expressed, respectively, as follows:
According to non-isothermal method, the basic data of α and T were collected from the TG curves of thermal decomposition of Mn0.5Mg0.5Fe2(C2O4)3·5H2O at various heating rates (5, 10, 15, and 20 K min–1). According to Eq. (1), the isoconversional calculation procedure of Starink equation was used. The corresponding Starink lines for different decomposition steps were obtained at different conversion degrees α and different heating rates β at first, and then reaction activation energy E α can be obtained from linear slope (−1.0008 E α/R). The results are shown in Table 1.
From Table 1, the activation energy changes for the step 1 with α are higher than 10 %, and that for the step 2 with α are lower than 10 %, so that we draw a conclusion that thermal decomposition of Mn0.5Mg0.5Fe2(C2O4)3·5H2O for step 1 is multi-step reaction mechanisms. In other words, dehydration of Mn0.5Mg0.5Fe2(C2O4)3·5H2O experiencing simple dehydration reactions of two or more steps; and reaction of Mn0.5Mg0.5Fe2(C2O4)3 with O2 into Mn0.5Mg0.5Fe2O4 is simple reaction mechanisms [43–45]. The activation energy of the step 1 is higher than that of the step 2, which implies that the step 1 of the thermal decomposition of Mn0.5Mg0.5Fe2(C2O4)3·5H2O may be interpreted as a ‘‘slow’’ stage, while step 2 may be interpreted as ‘‘fast’’ stage.
Figure 7 shows the curves of α versus t and dα versus t for step 2. From Fig. 7 it can be seen that step 2 consists of two models, that is, accelerating model between 0.0 and 2.71 min, and sigmoidal model between 2.71 and 24.5 min [31]. In accelerating model, rate increases continuously with increasing the extent of conversion and reaches its maximum at the end of the process, the result shows that rate of the accelerating stage reaches its maximum at 2.61 min. In sigmoidal model (sometimes also called autocatalytic), initial and final stages demonstrate the accelerating and decelerating behavior, respectively, so that the process rate reaches its maximum at some values of the extent of conversion, the result shows that rate for sigmoidal model reaches its maximum at about 4.64 min.

Curves of α versus t and dα versus t at heating rate of 10 K min–1
The appropriate temperatures corresponding to conversions for β = 5, 10, 15, and 20 K min–1 were determined at first and then conversions corresponding to an appropriate temperature for β = 5, 10, 15, and 20 K min–1 were put into 31 types of mechanism functions [34]. The slope k, correlation coefficient r 2, and intercept B of linear regression of ln g(α) versus ln β were obtained. The two mechanism functions of better correlation coefficient r 2 were determined to be probable mechanism functions at first, and then several temperatures were randomly chosen to calculate the slope k, correlation coefficient r 2, and intercept B of the two probable mechanism functions by the same method. Mechanism function, in which the value of k was closest to −1.00000 and the correlation coefficient r 2 was higher, was chosen as mechanism function of thermal decomposition of Mn0.5Mg0.5Fe2(C2O4)3·5H2O. The results showed that probable mechanism function integral form of thermal decomposition of Mn0.5Mg0.5Fe2(C2O4)3·5H2O for step 2 was determined to be g(α) = [–ln(1–α)]1/3. Rate-determining mechanism for step 2 was assumed to be random nucleation.
The pre-exponential factor was obtained from Eq. (3), inserting the most probable mechanism function g(α), β, E α, R, and T max values. The results showed that the pre-exponential factor of thermal decomposition of Mn0.5Mg0.5Fe2(C2O4)3·5H2O for steps 2 was determined to be A = 3.03 × 106 s–1.
Conclusions
We have successfully synthesized spinel Mn0.5Mg0.5Fe2O4 via calcining Mn0.5Mg0.5Fe2(C2O4)3·5H2O in air. XRD analysis showed that precursor was a solid solution containing MFe2(C2O4)3·5H2O (M=Mn, and Mg). Mn0.5Mg0.5Fe2O4 with cubic structure was obtained when the precursor was calcined above 400 °C in air for 1 h. Magnetic characterization indicated that the specific saturation magnetization of Mn0.5Mg0.5Fe2O4 obtained at 600 °C was 46.2 emu g–1. Mn2+ and Mg2+ ions in Mn0.5Mg0.5Fe2O4 have a synergistic effect in improving the specific saturation magnetization of Mn0.5Mg0.5Fe2O4. The thermal decomposition of Mn0.5Mg0.5Fe2(C2O4)3·5H2O in air in the range of ambient temperature to 450 °C experienced two steps which involved, at first, the dehydration of five water molecules, and then decomposition of Mn0.5Mg0.5Fe2(C2O4)3 into spinel Mn0.5Mg0.5Fe2O4 in air. Crystalline Mn0.5Mg0.5Fe2O4 can be obtained at lower calcination temperature, which was attributed to Mn2+, Mg2+, and Fe2+ oxalates that have similar lower decomposition temperature, and ultra-fine the oxide particles with high activity can be easily in situ self-assembled into crystalline Mn0.5Mg0.5Fe2O4. The kinetics of the thermal decomposition of Mn0.5Mg0.5Fe2(C2O4)3·5H2O was studied using TG/DTG/DSC technique. The values of the activation energies associated with the thermal decomposition of Mn0.5Mg0.5Fe2(C2O4)3·5H2O were determined to be 92.4 ± 20.6 and 83.0 ± 4.0 kJ mol–1 for the first and second thermal decomposition steps, respectively.
References
Marinca TF, Chicinas I, Isnard O. Influence of the heat treatment conditions on the formation of CuFe2O4 from mechanical milled precursors oxides. J Therm Anal Calorim. 2012;110:301–7.
Gabal MA, Ahmed MA. structural, electrical and magnetic properties of copper-cadmium ferrites prepared from metal oxalates. J Mater Sci. 2005;40:387–98.
Wu WW, Li YN, Zhou KW, Wu XH, Liao S, Wang Q. Nanocrystalline Zn0.5Ni0.5Fe2O4: preparation and kinetics of thermal process of precursor. J Therm Anal Calorim. 2012;110:1143–51.
Sun ZP, Liu L, Jia DZ, Pan WY. Simple synthesis of CuFe2O4 nanoparticles as gas-sensing materials. Sens Actuators B. 2007;125:144–8.
Li JJ, Yuan HM, Li GD, Liu YJ, Leng JS. Cation distribution dependence of magnetic properties of sol–gel prepared MnFe2O4 spinel ferrite nanoparticles. J Magn Magn Mater. 2010;322:3396–400.
Wu XH, Wu WW, Zhou KW, Cui XM, Liao S. Products and non-isothermal kinetics of thermal decomposition of MgFe2(C2O4)3·6H2O. J Therm Anal Calorim. 2012;110:781–7.
Li FS, Wang HB, Wang L, Wang JB. Magnetic properties of ZnFe2O4 nanoparticles produced by a low-temperature solid-state reaction method. J Magn Magn Mater. 2007;309:295–9.
Wu WW. C, Wu XH, Li YN, Liao S. Magnetic properties and crystallization kinetics of Zn0.5Ni0.5Fe2O4. Rare Met. 2011;30:621–6.
Satyanarayana L, Madhusudan Reddy K, Manorama SV. Nano-sized spinel NiFe2O4: a novel material for the detection of liquefied petroleum gas in air. Mater Chem Phys. 2003;82:21–6.
Zhang K, Holloway T, Pradhan AK. Magnetic behavior of nanocrystalline CoFe2O4. J Magn Magn Mater. 2011;323:1616–22.
Wu WW, Cai JC, Wu XH, Liao S, Huang AG. Co0.35Mn0.65Fe2O4 magnetic particles: preparation and kinetics research of thermal process of the precursor. Powder Technol. 2012;215–216:200–5.
John Berchmans L, Karthikeyan R, Helan M, Berchmans Sheela, Ŝepelak V, Becker KD. Mechanochemical synthesis and electrochemical characterization of nano crystalline calcium ferrite. Catal Lett. 2011;141:1451–7.
Maqsood A, Faraz A. Synthesis, structural, electrical and magnetic characterization of Mn0.5Mg0.5−x Ni x Fe2O4 spinel Nanoferrites. J Supercond Nov Magn. 2011. doi:10.1007/s10948-011-1343-x.
Huang JW, Su P, Wu WW, Li YN, Wu XH, Liao S. Preparation of magnetic Cu0.5Mg0.5Fe2O4 nanoparticles and kinetics of thermal process of precursor. J Supercond Nov Magn. 2012;25:1971–7.
Wang WW. Microwave-induced polyol-process synthesis of MIIFe2O4 (M = Mn, Co) nanoparticles and magnetic property. Mater Chem Phys. 2008;108:227–31.
Tromsdorf UI, Bigall NC, Kaul MG, Bruns OT, Nikolic MS, Mollwitz B, Sperling RA, Reimer R, Hohenberg H, Parak WJ, Förster S, Beisiegel U, Adam G, Weller H. Size and surface effects on the MRI, relaxivity of manganese ferrite nanoparticle contrast agents. Nano Lett. 2007;7:2422–7.
Liu XM, Yang G, Fu SY. mass synthesis of manocrystalline spinel ferrites by a polymer-pyrolysis route. Mater Sci Eng C. 2007;27:750–5.
Zhen L, He K, Xu CY, Shao WZ. Synthesis and characterization of single-crystalline MnFe2O4 nanorods via a surfactant-free hydrothermal route. J Magn Magn Mater. 2008;320:2672–5.
Wang J, Chen QW, Hou BY, Peng ZM. Synthesis and magnetic properties of single-crystals of MnFe2O4 nanorods. Eur J Inorg Chem. 2004;35:1165–8.
Ju YW, Park JH, Jung HR, Cho SJ, Lee WJ. Electrospun MnFe2O4 nanofibers: preparation and morphology. Compos Sci Technol. 2008;68:1704–9.
Zhang DE, Zhang XJ, Ni XM, Song JM, Zheng HG. Low-temperature fabrication of MnFe2O4 octahedrons: magnetic and electrochemical properties. Chem Phys Lett. 2006;426:120–3.
Iyer R, Desai R, Upadhyay RV. Low temperature synthesis of nanosized Mn1–xZnxFe2O4 ferrites and their characterizations. Bull Mater Sci. 2009;32:141–7.
Faraz A, Saqib M, Ahmad NM, Fazal-ur-Rehman, Maqsood A, Usman M, Mumtaz A, Hassan MA. Synthesis, structural, and magnetic characterization of Mn1−xNixFe2O4 spinel nanoferrites. J Supercond Nov Magn. 2011. doi: 10.1007/s10948-011-1212-7.
Sharma SK, Ravi Kumar, Siva Kumar VV, Knobel M, Reddy VR, Gupta A, Singh M. Role of electronic energy loss on the magnetic properties of Mg0.95Mn0.05Fe2O4 nanoparticles. Nucl Instrum Methods Phys Res B. 2006;248:37–41.
Mansour SF, Elkestawy MA. A comparative study of electric properties of nano-structured and bulk Mn–Mg spinel ferrite. Ceram Int. 2011;37:1175–80.
Mansour SF. Structural and magnetic investigations of sub-nano Mn–Mg ferrite prepared by wet method. J Magn Magn Mater. 2011;323:1735–40.
Okasha N. Enhancement of magnetization of Mg–Mn nanoferrite by γ-irradiation. J Alloys Compd. 2010;490:307–10.
Maqsood A, Faraz A. Synthesis, structural, electrical and magnetic characterization of Mn0.5Mg0.5−xNixFe2O4 spinel nanoferrites. J Supercond Nov Magn. 2012;25:1025–33.
Msomi JZ, Moyo T, Abdallah HMI. Magnetic properties of MgxMn1−xFe2O4 nanoferrites. J Supercond Nov Magn. 2012. doi:10.1007/s10948-011-1235-0.
Starink MJ. The determination of activation energy from linear heating rate experiments: a comparison of the accuracy of isoconversion methods. Thermochim Acta. 2003;404:163–76.
Vyazovkin S, Burnham AK, Criado JM, Pérez-Maqueda LA, Popescu C, Sbirrazzuoli N. ICTAC Kinetics Committee recommendations for performing kinetic computations on thermal analysis data. Thermochim Acta. 2011;520:1–19.
Vlaev L, Nedelchev N, Gyurova K, Zagorcheva M. A comparative study of non-isothermal kinetics of decomposition of calcium oxalate monohydrate. J Anal Appl Pyrol. 2008;81:253–62.
Liqing L, Donghua C. Application of iso-temperature method of multiple rate to kinetic analysis: Dehydration for calcium oxalate monohydrate. J Therm Anal Calorim. 2004;78:283–93.
Jiang HY, Wang JG, Wu SQ, Wang BS, Wang ZZ. Pyrolysis kinetics of phenol–formaldehyde resin by non-isothermal thermogravimetry. Carbon. 2010;48:352–8.
Wu XH, Wu WW, Cui XM, Liao S. Preparation of nanocrystalline BiFeO3 via a simple and novel method and its kinetics of crystallization. J Therm Anal Calorim. 2012;107:625–32.
Boonchom B, Danvirutai C, Youngme S, Maensiri S. Simple synthesis, magnetic properties, and nonisothermal decomposition kinetics of Fe(H2PO4)2·2H2O. Ind Eng Chem Res. 2008;47:7642–7.
Deb N. Solid-state thermal decomposition of heterobimetallic oxalate coordination compounds, zinc(II) tetraaquatris (oxalato) lanthanate(III)hexahydrate and cadmium(II) heptaaquatris(oxalato)lanthanate(III)tetrahydrate. J Therm Anal Calorim. 2012. http://dx.doi.org/10.1007/s10973-012-2437-9.
Deb N. Some heterobimetallic oxalate coordination precursors of lanthanum(III) of the type, M3[La(C2O4)3(H2O) m ]2·nH2O (M = Mn(II), Co(II), Ni(II) and Cu(II)). J Thermal Anal Calorim. 2012;107:561–71.
Donia AM. Synthesis, identification and thermal analysis of coprecipitates of silver-(cobalt, nickel, copper and zinc) oxalate. Polyhedron. 1997;16:3013–31.
Goel SP, Mehrotra PN. IR and thermal studies on lithium oxomolybdenum (VI) oxalate. J Therm Anal. 1985;30:145–51.
Berbenni V, Milanese C, Bruni G, Girella A, Marini A. Synthesis of YFeO3 by thermal decomposition of mechanically activated mixtures Y(CH3COO)3·4H2O–FeC2O4·2H2O. Thermochim Acta. 2011;521:218–23.
Jiang CT, Liu RJ, Shen XQ, Zhu L, Song FZ. Ni0.5Zn0.5Fe2O4 nanoparticles and their magnetic properties and adsorption of bovine serum albumin. Powder Technol. 2011;211:90–4.
Budrugeac P, Muşat V, Segal E. Non-isothermal kinetic study on the decomposition of Zn acetate-based sol-gel precursor. J Therm Anal Calorim. 2007;88:699–702.
Chaiyo N, Muanghlua R, Niemcharoen S, Boonchom B, Seeharaj P, Vittayakorn N. Non-isothermal kinetics of the thermal decomposition of sodium oxalate Na2C2O4. J Therm Anal Calorim. 2012;107:1023–9.
Huang JW, Su P, Wu WW, Li YN, Wu XH, Tao L. Preparation of nanocrystalline BiFeO3 and kinetics of thermal process of precursor. J Therm Anal Calorim. 2012;. doi:10.1007/s10973-012-2524-y.
Acknowledgements
This study was financially supported by the National Nature Science Foundation of China (Grant no. 21161002) and the Guangxi Nature Science Foundation of China (Grant no. 2011GXNSFA018036).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Zhou, K., Wu, W., Li, Y. et al. Preparation of magnetic nanocrystalline Mn0.5Mg0.5Fe2O4 and kinetics of thermal decomposition of precursor. J Therm Anal Calorim 114, 205–212 (2013). https://doi.org/10.1007/s10973-012-2927-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-012-2927-9