
Abstract
Cu0.5Mg0.5Fe2O4 precursor was synthesized by solid-state reaction at low heat using CuSO4⋅5H2O, MgSO4⋅6H2O, FeSO4⋅7H2O, and Na2C2O4 as raw materials. The spinel Cu0.5Mg0.5Fe2O4 was obtained via calcining precursor above 300 °C in air. The precursor and its calcined products were characterized by thermogravimetry and differential scanning calorimetry (TG/DSC), Fourier transform FT-IR, X-ray powder diffraction (XRD), scanning electron microscopy (SEM), energy dispersive X-ray spectrometer (EDS), and vibrating sample magnetometer (VSM). The result showed that Cu0.5Mg0.5Fe2O4 obtained at 600 °C had a saturation magnetization of 36.8 emu g−1. The thermal process of Cu0.5Mg0.5Fe2O4 precursor experienced two steps, which involved the dehydration of the five and a half crystal water molecules at first, and then decomposition of Cu0.5Mg0.5Fe2(C2O4)3 into crystalline Cu0.5Mg0.5Fe2O4 in air. Based on the Kissinger equation, the values of the activation energy associated with the thermal process of the precursor were determined to be 85 and 152 kJ mol−1 for the first and second thermal process steps, respectively.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Spinels of the type \(\mbox{M}^{2+}\mbox{M}_{2}^{3+}\mbox{O}_{4}\) (M2+=Cu2+, Mn2+, Mg2+, Zn2+, Ni2+, Co2+, etc.; M3+=Fe3+) have many unique properties, such as high electrical resistivity, high permeability, high Curie temperature, mechanical hardness, large magnetocrystalline anisotropy, high coercivity, chemical stability, and temperate specific saturation magnetization. Therefore, spinel ferrites have been extensively used in many fields, such as high-density information storage, catalysts, ferrofluids, drug targeting, magnetic separation, magnetic resonance imaging, and gas sensor [1–11]. Spinel ferrites have a cubic close-packed structure. The cation occupies two types of interstitial position. One of them is called a tetrahedral A-site with the cation surrounded by four oxygen ions forming tetrahedral coordination. The other interstitial position is known as octahedral B-site with the cation coordinated by six oxygen ions forming octahedral coordination. The ferrospinels can be depicted in the chemical formula \((\mathrm{M}_{1 - \delta} ^{2 +}\mathrm{Fe}_{\delta} ^{3 +} )[\mathrm{M}_{\delta} ^{2 +} \mathrm{Fe}_{2- \delta} ^{3 +} ]\mathrm{O}_{4}^{2 -}\). Here the parentheses and square brackets denote A and B sites, respectively, M represent divalent cation and δ the inversion parameter [1]. The magnetic moment direction of cations in parentheses is opposite with that in square brackets. Therefore, magnetic as well as electrical properties of spinel ferrites depend on the distribution of cations at the different sites as well as preparation conditions. Copper ferrite (CuFe2O4) is very important soft magnetic material. Its properties, such as magnetic behavior and gas-sensing properties, are highly dependent on the synthesis method and doping elements.
To date, various methods have been developed to synthesize CuFe2O4 with cubic structure, including ball-milling [12, 13], sol–gel synthesis [14], coprecipitation [15, 16], combustion synthesis [17], microwave synthesis [18], solid-state reaction at low heat [4, 19], etc. In the synthesis of CuFe2O4, it was found that crystallite diameter, morphology, and crystalline phases of CuFe2O4 associated with its properties were highly dependent on the synthesis method and temperature. For instance, Sun et al. [4] obtained spinel-type CuFe2O4 with a crystallite size of 75 nm by solid-state reaction at low heat when precursor was calcined at 600 °C. Tao et al. [16] studied synthesis of spinel-type CuFe2O4 via sol–gel and coprecipitation methods, respectively. The results showed that spinel-type CuFe2O4 was obtained at 600 °C via calcining precursor from coprecipitation method. However, pure CuFe2O4 was not obtained until 800 °C by a sol–gel process, which might be due to the loose contact between copper and iron ions and the possible low diffusion rate.
The doped copper ferrite can improve its performance. Therefore, doped copper ferrite has caused great concern, and some progress has been made. It is reported that substitutions of Cu2+ with other transition metals ions lead to improve magnetic properties and catalytic properties of nanocrystalline ferrites [20–25], such as when Cu2+ ions in spinel CuFe2O4 are partially substituted by Co2+, Cu1−x Co x Fe2O4 obtained by chemical coprecipitation shows that saturation magnetization of sample increases with Co percentage [20]. Banerjee et al. [23] synthesized Zn1−x Cu x Fe2O4 (x=0.0, 0.25, 0.50, 0.75, 1.0) by coprecipitation method, and studied their catalytic properties. The results showed that the conversion of pyridine increases with increasing copper content in Zn1−x Cu x Fe2O4. However, synthesis research of doped CuFe2O4 still has fewer reports in comparison with that of CuFe2O4. Therefore, new synthesis methods for Cu1−x M x Fe2O4 (M=transition metals or alkaline earth metals) still need to be studied and innovated further. Besides, the kinetics study of thermal process for Cu1−x M x Fe2O4 precursor is important to obtain high-quality crystalline doped CuFe2O4 for practical applications.
The aim of this work is to prepare polycrystalline Cu0.5Mg0.5Fe2O4 using CuSO4⋅5H2O, MgSO4⋅7H2O, FeSO4⋅7H2O, and Na2C2O4 as raw materials via solid-state reaction at low heat [6, 11] and to study magnetic properties of Cu0.5Mg0.5Fe2O4, and kinetics of the thermal process of precursor. The kinetics of the thermal process of precursor was studied using TG-DSC techniques. Non-isothermal kinetics of the thermal process of precursor was interpreted by Kissinger equation [11, 26–28]. The kinetic parameters (\(E_{\mathrm{a}}, A\)) of the thermal decomposition of Cu0.5Mg0.5Fe2O4 precursor are discussed for the first time.
2 Experimental
2.1 Reagent and Apparatus
All chemicals were of reagent grade purity (purity>99.9 %). TG/DSC measurements were made using a Netsch 40PC thermogravimetric analyzer. X-ray powder diffraction (XRD) was performed using a Rigaku D/max 2500 V diffractometer equipped with a graphite monochromator and a Cu target. The FT-IR spectra of the precursor and its calcined products were recorded on a Nexus 470 FT-IR instrument. The morphologies of the calcined samples and energy dispersive X-ray spectrometer (EDS) were obtained on S-3400 scanning electron microscopy (SEM). The specific saturation magnetizations (\(M_{\mathrm{s}}\)) of the calcined sample powders were carried out at room temperature using a magnetic property measurement system (SQUID-MPMS-XL-5).
2.2 Preparation of Cu0.5Mg0.5Fe2O4
The Cu0.5Mg0.5Fe2O4 precursor was prepared by solid-state reaction at low heat using CuSO4⋅5H2O, MgSO4⋅7H2O, FeSO4⋅7H2O, and Na2C2O4 as raw materials at first. In a typical synthesis, CuSO4⋅5H2O (12.25 g), MgSO4⋅7H2O (12.09 g), FeSO4⋅7H2O (54.54 g), Na2C2O4(45.34 g), and surfactant polyethylene glycol (PEG)-400 (3.5 mL, 50 vol.%) were put in a mortar, and the mixture was fully ground by hand with a rubbing mallet at room temperature for 35 min. The grinding velocity was about 220 circles/min, and the strength applied was moderate. The reactant mixture gradually became damp, and then a paste formed quickly. The reaction mixture was kept at room temperature for 2 h. The mixture was washed with deionized water to remove soluble inorganic salts until \(\mbox{SO}_{4}^{2-}\) ion could not be visually detected with a 0.5 mol L−1 BaCl2 solution. The solid was then washed with a small amount of anhydrous ethanol and dried at 75 °C for 6 h. The resulting material was subsequently determined to be Cu0.5Mg0.5Fe2(C2O4)3⋅5.5H2O. Nanocrystalline Cu0.5Mg0.5Fe2O4 with cubic structure was obtained via calcining Cu0.5Mg0.5Fe2(C2O4)3⋅5.5H2O above 300 °C in air for 1 h.
3 Determination of Kinetic Parameters of Thermal Process
According to DSC curves and the Kissinger equation (Eq. (1)) [26], the activation energy of thermal process of the Cu0.5Mg0.5Fe2O4 precursor can be obtained,
where β is the heating rate (K min−1), \(T_{\mathrm{P}}\) is the peak temperature (K) in DSC curve, \(E_{\mathrm{a}}\) is the activation energy (kJ mol−1) of thermal process, R is the gas constant (8.314×10−3 kJ mol−1 K−1), and A is the pre-exponential factor. According to Eq. (1), the plot of \(\ln(\beta /T_{\mathrm{P}}^{2})\) versus \(1/T_{\mathrm{P}}\) can be obtained by a linear regression of least-square method. The dependence of \(\ln(\beta /T_{\mathrm{P}}^{2})\) on \(1/T_{\mathrm{P}}\) must give a good linear relation. Thus, reaction activation energy \(E_{\mathrm{a}}\) can be obtained from linear slope (\(-E_{\mathrm{a}}/R)\), and the pre-exponential factor A can be obtained from linear intercept (\(\ln(\mathit{AR}/E_{\mathrm{a}})\)).
4 Results and Discussion
4.1 TG/DSC Analysis of Precursor
Figure 1 shows the TG/DSC curves of the synthetic product at four different heating rates in air, respectively.

TG/DSC curves of Cu0.5Mg0.5Fe2(C2O4)3⋅5.5H2O at different heating rates in air
The TG/DSC curves show that thermal process of the Cu0.5Mg0.5Fe2(C2O4)3⋅5.5H2O below 700 °C occurs in two well-defined steps. For heating rate of 10 °C min−1, the first step starts at 50 °C, ends at 202 °C, and characterized by an endothermic DSC peak at about 202 °C, which can be attributed to dehydration of the five and a half crystal water molecules from Cu0.5Mg0.5Fe2(C2O4)3⋅5.5H2O and formation of Cu0.5Mg0.5Fe2(C2O4)3. The observed mass loss in the TG curve is 19.53 %, which is in good agreement with 19.1 % theoretic mass loss of dehydration of the five and a half crystal water molecules from Cu0.5Mg0.5Fe2(C2O4)3⋅5.5H2O. The second decomposition step begins at 202 °C, and ends at 400 °C, which involves an exothermic process with a strong DSC peak at 255 °C, attributed to the decomposition of Cu0.5Mg0.5Fe2(C2O4)3 in air and formation of Cu0.5Mg0.5Fe2O4. The corresponding observed mass loss in the TG curve is 37.29 %, which close to 38.55 % theoretic mass loss of reaction of Cu0.5Mg0.5Fe2(C2O4)3 with two O2 molecules. No other exothermic DSC peak that is ascribed to crystallization of Cu0.5Mg0.5Fe2O4, which indicates that exothermic DSC peak of crystallization of Cu0.5Mg0.5Fe2O4 is overlapped with that from decomposition of Cu0.5Mg0.5Fe2(C2O4)3. The peak temperature of thermal process is the temperature at which it attains its maximum. From Fig. 1b, there is an upward shift in \(T_{\mathrm{P}}\) with increasing heating rate, peak temperatures from heating rate of 10, 15, 20, and 25 °C min−1 are 202, 209, 216, and 221 °C for first step, and 255, 261, 266, and 268 °C for second step, respectively.
4.2 IR Spectroscopic Analysis of Cu0.5Mg0.5Fe2(C2O4)3⋅5.5H2O and Its Calcined Samples
The FT-IR spectra of Cu0.5Mg0.5Fe2(C2O4)3⋅5.5H2O and of its calcined sample are shown in Fig. 2.

FT-IR spectra of Cu0.5Mg0.5Fe2(C2O4)3⋅5.5H2O and its calcined samples
The Cu0.5Mg0.5Fe2(C2O4)3⋅5.5H2O exhibits a strong and broad band at about 3393 cm−1 that can be assigned to the stretching OH vibration of the water molecule. The strong band which appears at 1647 cm−1 in the spectrum of the precursor can be ascribed to the bending mode of the HOH. The bands at 1356 cm−1 can be assigned to either the appearance of new M–OC2O3 (M=Cu, Mg, Fe) bonds and/or to the combinations of OH librations and lattice modes [6, 11, 19, 29]. The band at 1356 cm−1 shifts to 1444 cm−1 when sample is calcined at 400 °C, which indicates that Cu0.5Mg0.5Fe2(C2O4)3 finishes the decomposition of \(\mbox{C}_{2}\mbox{O}_{4}^{2-}\). The absorption band of the samples obtained above 300 °C, which appears at about 3400 cm−1, is attributed to the adsorption of water from air.
4.3 XRD Analysis of Cu0.5Mg0.5Fe2(C2O4)3⋅5.5H2O and Its Calcined Samples
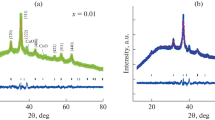
Figure 3 shows the XRD patterns of Cu0.5Mg0.5Fe2(C2O4)3⋅5.5H2O dried at 75 °C and the products resulting from calcination at different temperatures in air for 1 h.

XRD patterns of Cu0.5Mg0.5Fe2(C2O4)3⋅5.5H2O and its calcined samples at different temperatures in air for 1 h
From Fig. 3a, characteristic diffraction peaks of crystalline compound from the precursor are observed, which indicates that the Cu0.5Mg0.5Fe2(C2O4)3⋅5.5H2O obtained at 75 °C is a crystalline with higher crystallinity. The diffraction peaks in the pattern can be indexed to be in agreement with the orthorhombic FeC2O4⋅2H2O from PDF card 23-0293, with space group 12/a(15). No diffraction peaks of copper oxalate and magnesium oxalate, such as CuC2O4, CuC2O4⋅xH2O, MgC2O4, and MgC2O4⋅xH2O are observed, which implies that Cu2+ and Mg2+ ions enter lattice of FeC2O4⋅2H2O, and CuC2O4, CuC2O4⋅xH2O, MgC2O4, and MgC2O4⋅xH2O form a solid solution with FeC2O4⋅2H2O. It is explained by the fact that Cu2+ ion (72 pm), Mg2+ ion (65 pm), and Fe2+ ion (76 pm) have same electric charge and similar ionic radius. When the precursor is calcined at 300 °C for 1 h, characteristic diffraction peaks of crystalline compound are observed, all the diffraction peaks in the pattern are in agreement with that of cubic MgFe2O4 [space group Fd-3m(227), PDF card 36-0398] and cubic CuFe2O4 [space group Fd-3m(227), PDF card 77-0010], which indicates that MgFe2O4 and CuFe2O4 in the calcined product Cu0.5Mg0.5Fe2O4 form a solid solution. Intensity of diffraction peaks of cubic Cu0.5Mg0.5Fe2O4 increases with increasing calcination temperature, which indicates that crystallite sizes of cubic Cu0.5Mg0.5Fe2O4 increases with increasing calcination temperature. However, diffraction peaks of impurities, such as CuO, and MgO, increase with increasing calcination temperature, which implies that crystallinity of Cu0.5Mg0.5Fe2O4 decreases with increasing calcination temperature.
According to the Scherrer formula [11]: D=Kλ/(βcosθ), where D is crystallite diameter, K=0.89 (the Scherrer constant), λ=0.15406 nm (wavelength of the X-ray used), β is the width of line at the half-maximum intensity, and θ is the corresponding angle. The resulting crystallite sizes of the products from calcining precursor at the temperatures of 400, 600, 700, and 800 °C for 1 h, are 30, 40, 48, and 52 nm, respectively.
4.4 SEM and EDS Analysis of Calcined Samples
The morphologies and EDS spectrum of the calcined samples are shown in Fig. 4. From Fig. 4a, it can be seen that the calcined sample obtained at 600 °C is composed of approximately spherical particles, and there is a soft agglomeration phenomenon in the particles of Cu0.5Mg0.5Fe2O4 sample, which contains particles having a distribution of small particles (70–100 nm) and large particles (100–200 nm). With the increase of calcination temperature, the calcined sample is aggregated into larger polyhedral grains further. Figure 4c shows the SEM micrograph of sample obtained at 800 °C. It can be seen that the particle sizes of calcined sample obtained at 800 °C are between 250 and 500 nm. The average crystallite sizes of the calcined samples determined by X-ray diffraction are significantly smaller than the values determined by SEM. This is attributed that values observed by SEM technique give the size of the secondary particles, and the X-ray line broadening analysis discloses only the size of primary particles. EDS spectrum of the calcined product at 700 °C shows that the atomic percentages of Cu, Mg, and Fe are 15.36 %, 16.01 %, and 68.63 %, respectively. In other words, that mole ratio of Cu:Mg:Fe in calcined product is equal to 0.47:0.49:2.10, which is close to the value of the pre-design and synthesis.

SEM and EDS analysis of the calcined sample: (a) 600 °C, (b) 700 °C, and (c) 800 °C
4.5 Magnetic Properties of Calcined Samples
Figure 5 shows the hysteresis loops of the Cu0.5Mg0.5Fe2O4 particles obtained at different calcination temperatures. From Fig. 5, it can be observed that specific saturation magnetizations of powders calcined at 400, 600, 700, and 800 °C for 1 h are 25.4, 36.8, 32.5, and 31.7 emu g−1, respectively. That is, specific saturation magnetization increases between 400 and 600 °C with increasing calcination temperature. In other words, the larger the crystallite size of the particles, the larger is the specific saturation magnetizations. The larger \(M_{\mathrm{s}}\) values associated with larger crystallite sizes can be explained as follow: First, surface distortions due to the interaction of the transition metal ions with the oxygen atoms in the spinel lattice of Cu0.5Mg0.5Fe2O4 can reduce the net magnetic moment in the particle. When the calcination temperature increases, the crystallite size of particles increases, and surface distortions is reduced, so \(M_{\mathrm{s}}\) value increases with increasing calcination temperature. Second, the magnetocrystalline anisotropy of the particles is dependent on the crystallinity of Cu0.5Mg0.5Fe2O4. The higher calcination temperature, the larger is crystallinity of particles, which reduces magnetocrystalline anisotropy distortion, and increases magnetic moment within the particles of Cu0.5Mg0.5Fe2O4 [3, 11]. Compared with magnetic properties of CuFe2O4 (600 °C, 33.5 emu g−1) [19] and MgFe2O4 (600 °C, 30.4 emu g−1) [6], it can be seen that Cu0.5Mg0.5Fe2O4 obtained at 600 °C exhibits higher specific saturation magnetizations than CuFe2O4 and MgFe2O4 obtained at same calcination temperature, which implies that Cu2+, and Mg2+ ions in Cu0.5Mg0.5Fe2O4 have a synergistic effect in improving the saturation magnetization of Cu0.5Mg0.5Fe2O4. The reason can be explained as follow: First, when Cu2+ ions with magnetic moment are partially substituted by Mg2+ ions with zero magnetic moment, Mg2+ and Cu2+ ions occupy tetrahedral A-site together, which reduces the tetrahedral A-site magnetic moment, and results that magnetic moment of Cu0.5Mg0.5Fe2O4 is greater than that of CuFe2O4. Second, the magnetic moment of MgFe2O4 is less than that of Cu0.5Mg0.5Fe2O4, which is attributed that part of Mg2+ ions occupy octahedral B-site in MgFe2O4, and part of Fe3+ ions occupy tetrahedral A-site. This causes that magnetic moment of tetrahedral A-site increases, and magnetic moment of octahedral B-site decreases, so that the net magnetic moment of MgFe2O4 is decreased.

M–H (magnetization–hysteresis) loops of Cu0.5Mg0.5Fe2O4 samples obtained at different temperatures in air for 1 h
When precursor is calcined above 600 °C, specific saturation magnetization decreases with increasing calcination temperature, which is attributed that Cu0.5Mg0.5Fe2O4 is decomposed into CuO, MgO, and Fe2O3 particles with weak magnetic intensity above 600 °C. In other words, crystalline Cu0.5Mg0.5Fe2O4 is thermodynamically more stable at 600 °C.
4.6 Kinetics of Thermal Process of Precursor
In accordance with TG/DSC, FT-IR, XRD, and EDS analysis of Cu0.5Mg0.5Fe2(C2O4)3⋅5.5H2O and its calcined products mentioned above, thermal process of the precursor below 700 °C consists of two steps, which can be expressed as follows:


Figure 6 shows Kissinger plots of the thermal process of the precursor for the first and second thermal steps. From the slopes of the straight lines, the activation energy values of two thermal process of the precursor are determined to be 85, and 152 kJ mol−1 for the first, and second thermal process steps, respectively (Table 1). The step 2 exhibits higher activation energy value in comparison with steps 1, which suggests that thermal decomposition of Cu0.5Mg0.5Fe2(C2O4)3 into Cu0.5Mg0.5Fe2O4 is the rate-limiting step of the thermal process of the precursor.

Kissinger plots of thermal processes of the precursor for the first and second steps
5 Conclusions
We have successfully synthesized nanocrystalline Cu0.5Mg0.5Fe2O4 with cubic structure via a novel and simple method. XRD analysis shows that precursor is a solid solution containing MFe2(C2O4)3⋅5.5H2O (M=Cu, and Mg). Crystalline Cu0.5Mg0.5Fe2O4 with cubic structure is obtained via calcining precursor above 300 °C in air for 1 h. Magnetic characterization indicates that the specific saturation magnetization of Cu0.5Mg0.5Fe2O4 obtained at 600 °C is 36.8 emu g−1. Cu2+ and Mg2+ ions in Cu0.5Mg0.5Fe2O4 have a synergistic effect in improving the specific saturation magnetization of Cu0.5Mg0.5Fe2O4. The thermal process of the precursor in the range of ambient temperature to 700 °C experiences two steps, which involves the dehydration of the five and a half waters of crystallization at first, and then the decomposition of Cu0.5Mg0.5Fe2(C2O4)3 into cubic Cu0.5Mg0.5Fe2O4. The kinetics of the thermal process of the precursor is studied using DSC technique. The values of the activation energy associated with thermal process of the precursor are 85 and 152 kJ mol−1 for the first and second steps, respectively.
References
Maqsood, A., Faraz, A.: J. Supercond. Nov. Magn. doi:10.1007/s10948-011-1343-x
Msomi, J.Z., Moyo, T., Abdallah, H.M.I.: J. Supercond. Nov. Magn. doi:10.1007/s10948-011-1235-0
Abdallah, H.M.I., Moyo, T., Msomi, J.Z.: J. Supercond. Nov. Magn. 24, 669–673 (2011)
Sun, Z.P., Liu, L., Jia, D.Z., Pan, W.Y.: Sens. Actuators B, Chem. 125, 144–148 (2007)
Li, J.J., Yuan, H.M., Li, G.D., Liu, Y.J., Leng, J.S.: J. Magn. Magn. Mater. 322, 3396–3400 (2010)
Wu, X.H., Wu, W.W., Zhou, K.W., Cui, X.M., Liao, S.: J. Therm. Anal. Calorim. doi:10.1007/s10973-011-1968-9
Li, F.S., Wang, H.B., Wang, L., Wang, J.B.: J. Magn. Magn. Mater. 309, 295–299 (2007)
Wu, W.W., Cai, J.C., Wu, X.H., Li, Y.N., Liao, S.: Rare Metals. doi:10.1007/s12598-011-0439-6
Satyanarayana, L., Madhusudan Reddy, K., Manorama, S.V.: Mater. Chem. Phys. 82, 21–26 (2003)
Zhang, K., Holloway, T., Pradhan, A.K.: J. Magn. Magn. Mater. 323, 1616–1622 (2011)
Wu, W.W., Cai, J.C., Wu, X.H., Liao, S., Huang, A.G.: Powder Technol. 215– 216, 200–205 (2012)
Goya, G.F., Rechenberg, H.R.: Nanostruct. Mater. 10, 1001–1011 (1998)
Jiang, J.Z., Goya, G.F., Rechenberg, H.R.: J. Phys., Condens. Matter 11, 4063–4078 (1999)
Bomio, M., Lavela, P., Tirado, J.L.: J. Solid State Electrochem. 12, 729–737 (2008)
Pandya, P.B., Joshi, H.H., Kulkarni, R.G.: J. Mater. Sci. Lett. 10, 474–476 (1991)
Tao, S.W., Gao, F., Liu, X.Q., Sørensen, O.T.: Mater. Sci. Eng. B, Solid-State Mater. Adv. Technol. 77, 172–176 (2000)
Zhang, Y., Stangle, G.C.: J. Mater. Res. 9, 1997–2004 (1994)
Vanetsev, A.S., Ivanov, V.K., Tret’yakov, Yu.D.: Dokl., Phys. Chem. 387, 332–334 (2002)
Wu, X.H., Zhou, K.W., Wu, W.W., Cui, X.M., Li, Y.N.: J. Therm. Anal. Calorim. doi:10.1007/s10973-011-2104-6
Mathew, T., Shylesh, S., Reddy, S.N., Sebastian, C.P., Date, S.K., Rao, B.S., Kulkarni, S.D.: Catal. Lett. 93, 155–163 (2004)
Birajdar, D.S., Devatwal, U.N., Jadhav, K.M.: J. Mater. Sci. 37, 1443–1448 (2002)
Gabal, M.A., Ahmed, M.A.: J. Mater. Sci. 40, 387–398 (2005)
Banerjee, M., Verma, N., Prasad, R.: J. Mater. Sci. 42, 1833–1837 (2007)
Kenfack, F., Langbein, H.: J. Mater. Sci. 41, 3683–3693 (2006)
Varalaxmi, N., Siva Kumar, K.V.: J. Mater. Sci., Mater. Electron. 22, 555–560 (2011)
Kissinger, H.E.: Anal. Chem. 29, 1702–1706 (1957)
Danvirutai, C., Noisong, P., Youngme, S.: J. Therm. Anal. Calorim. 100, 117–124 (2010)
Boonchom, B., Danvirutai, C.: J. Therm. Anal. Calorim. 98, 771–777 (2009)
Vlaev, L., Nedelchev, N., Gyurova, K., Zagorcheva, M.: J. Anal. Appl. Pyrolysis 81, 253–262 (2008)
Acknowledgements
This study was financially supported by the National Nature Science Foundation of China (Grant No. 21161002) and the Guangxi Nature Science Foundation of China (Grant No. 2011GXNSFA018036).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Huang, J., Su, P., Wu, W. et al. Preparation of Magnetic Cu0.5Mg0.5Fe2O4 Nanoparticles and Kinetics of Thermal Process of Precursor. J Supercond Nov Magn 25, 1971–1977 (2012). https://doi.org/10.1007/s10948-012-1545-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-012-1545-x