
Abstract
The precursor of nanocrystalline BiFeO3 was obtained by solid-state reaction at low heat using Bi(NO3)3·5H2O, FeSO4·7H2O, and Na2CO3·10H2O as raw materials. The nanocrystalline BiFeO3 was obtained by calcining the precursor. The precursor and its calcined products were characterized by differential scanning calorimetry (DSC), Fourier transform-infrared spectroscopy (FT-IR), X-ray powder diffraction (XRD), scanning electron microscopy (SEM), and vibrating sample magnetometer (VSM). The data showed that highly crystallization BiFeO3 with rhombohedral structure (space group R3c (161)) was obtained when the precursor was calcined at 873 K for 2 h. The thermal process of the precursor experienced three steps, which involve the dehydration of adsorption water, hydroxide, and decomposition of carbonates at first, and then crystallization of BiFeO3, and at last decomposition of BiFeO3 and formation of orthorhombic Bi2Fe4O9. The mechanism and kinetics of the crystallization process of BiFeO3 were studied using DSC and XRD techniques, the results show that activation energy of the crystallization process of BiFeO3 is 126.49 kJ mol−1, and the mechanism of crystallization process of BiFeO3 is the random nucleation and growth of nuclei reaction.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
BiFeO3 has many unique properties, such as ferroelectricity with high Curie temperature (T C = 820–850 °C) [1, 2] and antiferromagnetic properties below Néel temperature (T N = 350–380 °C) [3, 4]. These excellent properties make BiFeO3 suitable for many applications in the field of radio, television, satellite communication, bubble memory devices, audio–video, and digital recording [5–8], etc. BiFeO3 with pseudocubic or rhombohedral structure shows antiferromagnetic G-type spin configuration along the [111]c or [001]h directions. BiFeO3 has a superimposed incommensurate cycloid spin structure with a periodicity of 620 Å along the [110]h axis at room temperature. This structure cancels the macroscopic magnetization and inhibits observation of the linear ME effect [9–11]. It was reported that cycloid structure of BiFeO3 could be suppressed by decreasing particle size of BiFeO3, and its magnetic moment was enhanced [12, 13]. Doping [14] and preparation of high-quality samples [15] have been generally considered to improve the electrical properties of BiFeO3.
Since BiFeO3 was proposed in 1960s [16], various methods have been developed to synthesize nanocrystalline BiFeO3 compounds, including solid-state reaction at high temperature [17, 18], co-precipitation [19], sonochemical and microemulsion techniques [20], mechanochemical synthesis [21], hydrothermal method [22, 23], combustion synthesis [24, 25], ferrioxalate [26], sol–gel [11, 27, 28], polymeric precursor methods [29, 30], EDTA complexing gel process [31], polyacrylamide gel route [32], molten-salt method [33], and thermal decomposition of the inorganic complex [34], etc. It was found that crystallite diameter and crystalline phases of BiFeO3 associated with magnetic and electrical properties were highly dependent on the synthesis and processing methods. Such as, Yuan et al. [17] obtained crystalline BiFeO3 by solid-state reaction at high temperature, but this technique easily produce impurities Bi x Fe y O1.5x+1.5y−δ(x ≠ y, δ ≥ 0) which results in low electrical resistivity and high porosity in the multi-phase samples. Ke et al. [19] obtained crystalline BiFeO3 by controlling the chemical co-precipitation process. Xu et al. [28] synthesized high purity BiFeO3 with rhombohedral structure by sol–gel process at a temperature as low as 450 °C. However, these two processes easily produce impurities phase Bi2Fe4O9.
The aim of this work is to prepare pure phase nanocrystalline BiFeO3 via solid-state reaction at low heat [35] and to study the kinetics of the crystallization process of BiFeO3 using DSC and XRD technique. Non-isothermal and isothermal kinetics of the crystallization process of BiFeO3 were described by Kissinger [36] and JMA equation [37–39], respectively. Avrami exponent, n, was used to estimate mechanism of crystallization process.
Experimental
Reagent and apparatus
All chemicals were of reagent grade purity. DSC measurements were made using a Netsch 40PC thermogravimetric analyzer. X-ray powder diffraction (XRD) was performed using a Rigaku D/max 2500 V diffractometer equipped with a graphite monochromator and a Cu target. Fourier transform-infrared (FT-IR) spectra of the precursor and its calcined products were recorded on a Nexus 470 FT-IR instrument. The morphology of the calcined samples was examined by S-3400 scanning electron microscopy (SEM). The saturation magnetizations of the calcined sample powders were carried out at room temperature using a magnetic property measurement system (SQUID-MPMS-XL-5).
Preparation of nanocrystalline BiFeO3
The nanocrystalline BiFeO3 with rhombohedral structure was prepared by solid-state reaction at low heat [35] using Bi(NO3)3·5H2O, FeSO4·7H2O, and Na2CO3·10H2O as starting materials. In a typical synthesis, Bi(NO3)3·5H2O (46.52 g), FeSO4·7H2O (26.66 g), Na2CO3·10H2O (68.8 g), and surfactant polyethylene glycol (PEG)-400 (3.0 mL) were put in a mortar, and the mixture was fully ground by hand with a rubbing mallet for 40 min. The grinding velocity was about 90 circles/min, and the strength applied was moderate. The reactant mixture gradually became damp, and then a paste formed quickly. The reaction mixture was kept at 303 K for 1 h. The mixture was washed with deionized water to remove soluble inorganic salts until SO4 2− ion could not be visually detected with a 0.5 mol L−1 BaCl2 solution. The solid was then washed with a small amount of anhydrous ethanol and dried at 363 K for 3 h. Nanocrystalline BiFeO3 was obtained via calcining the precursor above 873 K for 2 h.
Method of determining kinetic parameters
Determination of activation energy and pre-exponential factor by Kissinger method [36]
According to DSC curve and the Kissinger equation (Eq. 1), the activation energy and pre-exponential factor of crystallization of BiFeO3 can be obtained.
where β is the heating rate (K min−1), T P is the peak temperature of DSC curve (K), E a is the activation energy (kJ mol−1) of crystallization process, R is the gas constant (8.314 J mol−1 K−1), and A is the pre-exponential factor. The dependence of \( \ln (\beta /T_{\text{P}}^{2} ) \) on 1/T P must give rise to a straight line. Thus, reaction activation energy E a can be obtained from linear slope (k = –E a/R), and the pre-exponential factor A can be obtained from linear intercept (h = ln (AR/E a)).
Kinetic study of crystallization process by JMA equation [37–39]
Isothermal crystallization process of BiFeO3 could be described by Eq. 2
The double logarithm equation of Eq. 2 can be rewritten in the Eq. 3:
where χ is the crystallized fraction of BiFeO3 at a given temperature time, t, k is the rate constant of crystallization, and n is the Avrami exponent that is related to the crystallization mechanisms. The dependence of ln(−ln(1 − χ)) on ln t must give rise to a straight line. Thus, the Avrami exponent (n) can be obtained from linear slope (that is: linear slope = n), and the rate constant (k) of crystallization can be obtained from linear intercept (h = ln k).
The rate constant (k) can be calculated according to Arrhenius Eq. 4:
where k is the rate constant of crystallization, E a is the activation energy (kJ mol−1), k 0 is the pre-exponential factor, R is the gas constant (8.314 × 10−3 kJ mol−1 K−1), and T is reaction temperature (K).
By a series of transforms, thus Eq. 4 can be rewritten in the Eq. 5:
k 1 and k 2 are the rate constants of crystallization corresponding to reaction temperature T 1 and T 2, respectively. Thus, the activation energy (E a) of the crystallization process of BiFeO3 can be obtained according to Eq. 5.
Results and discussion
DSC analysis of the precursor
Figure 1 shows the DSC curves of the precursor at four heating rates of 5, 10, 15, and 20 K min−1 from ambient temperature to 1,050 K. The DSC curve shows that the thermal process of the BiFeO3 precursor below 1,050 K experienced three steps. The weak endothermic DSC peak below 450 K is attributed to the dehydration of adsorption water, hydroxide, and decomposition of carbonates. The strong endothermic DSC peak between 600 and 750 K is related to phase transition from a mixture of Bi2O3 and Fe2O3 to rhombohedral phase BiFeO3. The broad and strong exothermic DSC peak, which is located between 800 and 1,000 K, can be assigned to the decomposition of rhombohedral phase BiFeO3. From Fig. 1, the peak temperature of phase transition between 600 and 750 K is the temperature at which it attains its maximum, which is the endothermic peak temperature in the DSC curves. There is an upward shift in T P with increasing heating rate, peak temperatures from heating rate of 5, 10, 15, and 20 K min−1 are 653, 669, 681, and 689 K, respectively.

DSC curves of the BiFeO3 precursor in argon gas at different heating rates: a 5 K min−1, b 10 K min−1, c 15 K min−1, d 20 K min−1
IR spectroscopic analysis of the precursor and its calcined samples
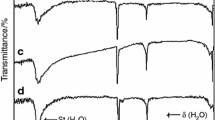
FT-IR spectra of the precursor and calcined samples are shown in Figure 2. From Fig. 2a, the band at 670 cm−1 is the water libration (hindered rotation), while the band at about 3,393 cm−1 is assigned to the stretching O–H vibration of the water molecule [35, 40, 41]. The strong band at 1,384 cm−1 is attributed to v 3 mode of carbonate, and the band at 848 cm−1 is assigned to v 2 mode of carbonate [42].

FT-IR spectra of the precursor and its calcined samples of BiFeO3
From Fig. 2b, FT-IR spectra of two samples obtained at 673 and 773 K are similar. The bands at about 1,384 and 848 cm−1 are attributed to the absorption of CO2 from calcined samples. With the increase of calcined temperature, the band at 1,384 cm−1 becomes weak, and disappers at 973 K. The band of calcined sample at 670 cm−1 is assigned to absorption water from air. The band located at 848 cm−1 appears with more intensity in the spectrum at the highest temperature, the exact cause is not clear.
XRD analysis of the calcined products
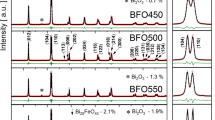
Figure 3a shows the XRD patterns of the calcined products at different temperature for 2 h. The results show that the calcined sample at 673 K is a mixture of tetragonal Bi2O3 and hexagonal Fe2O3. When the precursor was calcined at 773 K for 2 h, a wide and low diffraction pattern, which has a great difference in comparison with that of calcined sample at 673 K, is observed. Except weak part diffraction peaks of tetragonal Bi2O3 are still observed, all other the diffraction peaks in the pattern of sample obtained at 773 K are in agreement with that of rhombohedral BiFeO3, with space group R3c (161), lattice parameters a = 5.588 Å, b = 5.588 Å, c = 13.867 Å, α = β = 90o, γ = 120o, density = 8.311 g cm−3, from PDF card 71-2494. Intensity of diffraction peaks of Bi2O3 decreases with increasing calcination temperature, which indicates that purity of BiFeO3 increases. The sample obtained at 873 K almost becomes pure rhombohedral BiFeO3.

XRD patterns of the calcined products a different temperature for 2 h, b different time at 823 K, c different time at 873 K
However, when the sample is heated at 973 K for 2 h, characteristic diffraction peaks of Bi2Fe4O9 appear, suggesting that the rhombohedral BiFeO3 decomposes into thermodynamically more stable orthorhombic Bi2Fe4O9 at 973 K, which has a few difference in comparison with that reported by Navarro et al. [34] and Carvalho et al. [43]. Such as the thermal decomposition of Bi[Fe(CN)6]·4H2O above 823 K produces Bi2Fe4O9 phase [34], thermal decomposition of precursor from the sol–gel combustion method forms Bi25Fe4O39 phase at 773 K, and BiFeO3 phase decomposes into Bi2Fe4O9 and Bi25FeO39 phases when the precursor is heated at 973 K [43]. The exact cause of difference is not clear.
The XRD diffraction patterns for the powders isothermally calcined at 823 and 873 K for various periods of time are shown in Fig. 3b and c, respectively. From Fig. 3b and c, intensity of diffraction peaks increases with increasing calcination time, which indicates that degree of crystallization of BiFeO3 increases with increasing calcination times.
According to the Scherrer formula [41]: D = Kλ/(βcosθ), where D is crystallite diameter, K = 0.89 (the Scherrer constant), λ = 0.15406 nm (wavelength of the X-ray used), β is the width of line at the half-maximum intensity, and θ is the corresponding angle. The resulting crystallite sizes of the products from calcined precursor at the temperatures of 673, 773, 873, and 973 K for 2 h, are 40, 28, 42, and 58 nm, respectively.
SEM analysis of the calcined samples
The morphologies of the calcined samples are shown in Fig. 4. From Fig. 4a and b, it can be seen that the calcined samples at 673 and 773 K are composed of approximately spherical particles, which contain particles having a distribution of small particles (30–50 nm) and large particles (50–200 nm). With the increase of calcining temperature, the calcined samples are aggregated into larger particles further. Figure 4c and d shows the SEM micrographs of samples obtained at 873 and 973 K, respectively. It can be seen that the calcined sample obtained at 873 K can still keep spherical morphology. However, the calcined sample obtained at 973 K become uniform polyhedral grains with particle size of about 400 nm. The average crystallite sizes of calcined samples determined by X-ray diffraction are significantly smaller than the values determined by SEM. This attributed that the values observed by SEM technique give the size of the secondary particles, and the X-ray line broadening analysis discloses only the size of primary particles. In comparison with other methods of synthesis, morphology and size distribution of BiFeO3 have a great difference, such as the powder prepared from combustion method using urea as fuel exhibits uniform and rather isolated agglomerates, and the particle aggregates of the powder prepared using glycine as fuel show a non-homogenous morphology and are strongly interconnected in a kind of three-dimensional, porous skeleton [44]. Sol–gel process can obtain nearly cubic morphology of BiFeO3 with the size of 110–160 nm [31].

SEM micrographs of the calcined samples for 2 h a 673 K, b 773 K, c 873 K, d 973 K
Magnetic properties of the calcined samples
The hysteresis loop of the calcined sample at 873 K is shown in Fig. 5. It can be observed that rhombohedral BiFeO3 exhibits a weak ferromagnetic order at room temperature, and the saturation magnetizations (M s) of the powder is 0.032 emu g−1, which is smaller than values of BiFeO3 samples from other synthesis methods [11, 16, 19]. The larger the particle size, the weaker is the saturation magnetization [11]. The particle size of calcined sample at 873 K is larger than that from other synthesis methods mentioned above, and thus, the calcined sample at 873 K has smaller saturation magnetizations.

Hysteresis loops of the calcined samples at 873 K for 2 h
Kinetics of thermal process of the precursor
In accordance with DSC, FT-IR, and XRD analysis of the precursor and its calcined products mentioned above, thermal process of the precursor below 1,050 K consists of three steps, which can be expressed as follows:
Figure 6 shows Kissinger plot of the crystallization process of BiFeO3. From the slope of the straight lines, the activation energy value of the crystallization process of BiFeO3 is determined to be 132.11 kJ mol−1, and pre-exponential factor A is equal to 7.04 × 109 s−1.

Kissinger plot of the crystallization process of BiFeO3
In accordance with XRD analysis in Fig. 3b and c, the crystallized fraction of BiFeO3 at a given time, t, is calculated via MDI Jade 5.0 software at first, and then the plot of the crystallinity (χ) of BiFeO3 versus ln t is plotted. The dependence of χ on ln t is shown in Fig. 7, the result shows that the dependence of χ on ln t gave a linear relation. Figure 8 shows the dependence of ln[–ln(1 − χ)] on ln t, it is found that the dependence of ln[–ln(1 – χ)] on ln t gives rise to a straight line. In accordance with JMA Eq. 3, the slope and intercept of the straight line can be determined, and then the rate constant (k), the Avrami exponent (n) are obtained, and the activation energy (E a) of the crystallization process of BiFeO3 can be obtained by Eq. 5. Table 1 shows the kinetic parameters of the crystallization process of BiFeO3. From Table 1, it is seen that the activation energy value calculated by the JMA method is close to that obtained by Kissinger method, so the result is credible.

Plots of the crystallinity (χ) of BiFeO3 versus ln t

Plots of the ln(−ln(1 − χ)) versus ln t
The mechanism of crystallization process can be determined by the value of Avrami exponent (n). Smaller n values indicate that the crystallization is dominated by a surface crystallization mechanism rather than by volume crystallization, and that the crystallization dimension is low. On the other hand, larger n values are expected only in the case of increasing nucleation rates, i.e., n > 2.5 in diffusion-controlled reaction or n > 4 in polymorphic transformation [45]. For the crystallization process of BiFeO3, the value of the Avrami exponent (n) was smaller than 1, which suggests that crystallization process of BiFeO3 is the random nucleation and growth of nuclei reaction [41, 46, 47].
Conclusions
We have successfully synthesized nanocrystalline BiFeO3 using solid-state reaction at low heat. XRD analysis suggests that highly crystallization BiFeO3 with rhombohedral structure is obtained when the precursor is calcined at 873 K for 2 h. Magnetic characterization indicates that rhombohedral BiFeO3 sample exhibits a weak ferromagnetic order at room temperature. The thermal process of the precursor of BiFeO3 in the range of ambient temperature—1,050 K is a complex process, which involves the dehydration of adsorption water, hydroxide, and decomposition of carbonates at first, and then crystallization of rhombohedral BiFeO3, and at last decomposition of BiFeO3 and formation of orthorhombic Bi2Fe4O9. The kinetics of the crystallization process of BiFeO3 was studied using DSC and XRD techniques. The activation energy of crystallization process for the BiFeO3 is 126.49 kJ mol−1. The Avrami exponent, n, is smaller than 1, which suggests that crystallization process of BiFeO3 is the random nucleation and growth of nuclei reaction.
References
Michel C, Moreau JM, Achenbach GD, Gerson R, James WJ. The atomic structure of BiFeO3. Solid State Commun. 1969;7:701–4.
Smolenskii GA, Isupov VA, Agranovskaya AI, Krainik NN. New ferroelectrics of complex composition. Sov Phys Solid State. 1961;2:2651–4.
Smolenskii GA, Yudin VM, Sher ES, Stolypin YE. Antiferromagnetic properties of some perovskites. Sov Phys JETP. 1963;16:622–4.
Moreau JM, Michel C, Gerson R, James WJ. Ferroelectric BiFeO3 X-ray and neutron diffraction study. J Phys Chem Solids. 1971;32:1315–20.
Bucci JD, Robertson BK, James WJ. The precision determination of the lattice parameters and the coefficients of thermal expansion of BiFeO3. J Appl Cryst. 1972;5:187–91.
Kubel F, Schmid H. Structure of a ferroelectric and ferroelastic monodomain crystal of the perovskite BiFeO 3 . Acta Cryst. 1990;46:698–702.
Palkar VR, Pinto R. BiFeO3 thin films: novel effects. J Phys. 2002;58:1003–8.
Wang YP, Zhou L, Zhang MF, Chen XY, Liu JM, Liu ZG. Room-temperature saturated ferroelectric polarization in BiFeO3 ceramics synthesized by rapid liquid phase sintering. Appl Phys Lett. 2004;84:1731–3.
Ederer C, Spaldin NA. Weak ferromagnetism and magnetoelectric coupling in bismuth ferrite. Phys Rev B. 2005;71:060401–4.
Sosnowska I, Neumaier TP, Steichele E. Spiral magnetic ordering in bismuth ferrite. J Phys C Solid State Phys. 1982;15:835–46.
Jia DC, Xu JH, Ke H, Wang W, Zhou Y. Structure and multiferroic properties of BiFeO3 powders. J Eur Ceram Soc. 2009;29:3099–103.
Park TJ, Papaefthymiou GC, Viescas AJ, Moodenbaugh AR, Wong SS. Size-dependent magnetic properties of single-crystalline multiferroic BiFeO3 nanoparticles. Nano Lett. 2007;7:766–72.
Mazumder R, Sujatha Devi P, Bhattacharya D, Choudhury P, Sen A, Raja M. Ferromagnetism in nanoscale BiFeO3. Appl Phys Lett. 2007;91:062510–2.
Lee YH, Wu JM, Lai CH. Influence of La doping in multiferroic properties of BiFeO3 thin films. Appl Phys Lett. 2006;88:042903–5.
Lebeugle D, Colson D, Forget A, Viret M. Very large spontaneous electric polarization in BiFeO3 single crystals at room temperature and its evolution under cycling fields. Appl Phys Lett. 2007;91:022907–9.
Jiang QH, Nan CW, Wang Y, Liu YH, Shen ZJ. Synthesis and properties of multiferroic BiFeO3 ceramics. J Electroceram. 2008;21:690–3.
Yuan GL, Or SW, Wang YP, Liu ZG, Liu JM. Preparation and multi-properties of insulated single-phase BiFeO3 ceramics. Solid State Commun. 2006;138:76–81.
Choudhary RNP, Pradhan DK, Bonilla GE, Katiyar RS. Effect of La-substitution on structural and dielectric properties of Bi(Sc1/2Fe1/2)O3 ceramics. J Alloys Compd. 2007;437:220–4.
Ke H, Wang W, Wang YB, Xu JH, Jia DC, Lu Z, Zhou Y. Factors controlling pure-phase multiferroic BiFeO3 powders synthesized by chemical co-precipitation. J Alloys Compd. 2011;509:2192–7.
Das N, Majumdar R, Sen A, Maiti HS. Nanosized bismuth ferrite powder prepared through sonochemical and microemulsion techniques. Mater Lett. 2007;61:2100–4.
Szafraniak I, Polomska M, Hilczer B, Pietraszko A, Kepiński L. Characterization of BiFeO3 nanopowder obtained by mechanochemical synthesis. J Eur Ceram Soc. 2007;27:4399–402.
Basu S, Pal M, Chakravorty D. Magnetic properties of hydrothermally synthesized BiFeO3 nanoparticles. J Mag Mag Mater. 2008;320:3361–5.
Cho CM, Noh JH, Cho IS, An JS, Hong KS, Kim JY. Low-temperature hydrothermal synthesis of pure BiFeO3 nanopowders using triethanolamine and their applications as visible-light photocatalysts. J Am Ceram Soc. 2008;91:3753–5.
Fruth V, Mitoseriu L, Berger D, Ianculescu A, Matei C, Preda S, Zaharescu M. Preparation and characterization of BiFeO3 ceramic. Prog Solid State Chem. 2007;35:193–202.
Farhadi S, Zaidi M. Bismuth ferrite (BiFeO3) nanopowder prepared by sucrose-assisted combustion method: a novel and reusable heterogeneous catalyst for acetylation of amines, alcohols and phenols under solvent-free conditions. J Mol Catal A Chem. 2009;299:18–25.
Ghosh S, Dasgupta S, Sen A, Himadri Sekhar Maiti HS. Low temperature synthesis of bismuth ferrite nanoparticles by a ferrioxalate precursor method. Mater Res Bull. 2005;40:2073–9.
Kim JK, Kim SS, Kim WJ. Sol–gel synthesis and properties of multiferroic BiFeO3. Mater. Lett. 2005;59:4006–9.
Xu JH, Ke H, Jia DC, Wang W, Zhou Y. Low-temperature synthesis of BiFeO3 nanopowders via a sol–gel method. J Alloys Compd. 2009;472:473–7.
Popa M, Crespo D, Calderon-Moreno JM, Preda S. Synthesis and structural characterization of single-phase BiFeO3 powders from a polymeric precursor. J Am Ceram Soc. 2007;90:2723–7.
Selbach SM, Einarsrud MA, Tybell T, Grande T. Synthesis of BiFeO3 by wet chemical methods. J Am Ceram Soc. 2007;90:3430–4.
Wei J, Xue DS. Low-temperature synthesis of BiFeO3 nanoparticles by ethylenediaminetetraacetic acid complexing sol–gel process. Mater Res Bull. 2008;43:3368–73.
Xian T, Yang H, Shen X, Jiang JL, Wei ZQ, Feng WJ. Preparation of high-quality BiFeO3 nanopowders via a polyacrylamide gel route. J Alloys Compd. 2009;480:889–92.
He XB, Lian Gao L. Synthesis of pure phase BiFeO3 powders in molten alkali metal nitrates. Ceram. Int. 2009;35:975–8.
Navarro MC, Lagarrigue MC, De Paoli JM, Carbonio RE, Gómez MI. A new method of synthesis of BiFeO3 prepared by thermal decomposition of Bi[Fe(CN)6]·4H2O. J Therm Anal Calorim. 2010;102:655–60.
Wu XH, Wu WW, Li SS, Cui XM, Liao S. Kinetics and thermodynamics of thermal decomposition of NH4NiPO4·6H2O. J Therm Anal Calorim. 2011;103:805–12.
Kissinger HE. Reaction kinetics in differential thermal analysis. Anal Chem. 1957;29:1702–6.
Avrami M. Kinetics of phase change. I general theory. J Chem Phys. 1939;7:1103–12.
Avrami M. Kinetics of phase change. II transformation-time relations for random distribution of nuclei. J Chem Phys. 1940;8:212–24.
Avrami M. Granulation, phase change, and microstructure kinetics of phase change. III. J Chem Phys. 1941;9:177–84.
Wu XH, Wu WW, Liu C, Li SS, Liao S, Cai JC. Synthesis of layered sodium manganese phosphate via low-heating solid-state reaction and its properties. Chin J Chem. 2010;28:2394–8.
Liu C, Wu XH, Wu WW, Cai JC, Liao S. Preparation of nanocrystalline LiMnPO4 via a simple and novel method and its isothermal kinetics of crystallization. J Mater Sci. 2011;46:2474–8.
Li ZJ, Shen XQ, Feng X, Wang PY, Wu ZS. Non-isothermal kinetics studies on the thermal decomposition of zinc hydroxide carbonate. Thermochim Acta. 2005;438:102–6.
Carvalho TT, Tavares PB. Synthesis and thermodynamic stability of multiferroic BiFeO3. Mater Lett. 2008;62:3984–6.
Carmen Paraschiv B. Jurca, Adelina Ianculescu, and Oana Carp. Synthesis of nanosized bismuth ferrite (BiFeO3) by a combustion method starting from Fe(NO3)3·9H2O–Bi(NO3)3·9H2O–glycine or urea systems. J Therm Anal Calorim. 2008;94:411–6.
Takei T, Kameshima Y, Yasumori A, Okada K. Crystallization kinetics of mullite from Al2O3–SiO2 glasses under non-isothermal conditions. J Eur Ceram Soc. 2001;21:2487–93.
Johnson BR, Kriven WM, Schneider J. Crystal structure development during devitrification of quenched mullite. J Eur Ceram Soc. 2001;21:2541–62.
Boonchom B, Danvirutai C. Kinetics and thermodynamics of thermal decomposition of synthetic AlPO4·2H2O. J Therm Anal Calorim. 2009;98:771–7.
Acknowledgements
This study was financially supported by the Guangxi Natural Scientific Foundation of China (Grant No. 2011GXNSFA018036), and the Guangxi Science and Technology Agency Research Item of China (Grant No. 0992001-5).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wu, X., Wu, W., Cui, X. et al. Preparation of nanocrystalline BiFeO3 via a simple and novel method and its kinetics of crystallization. J Therm Anal Calorim 107, 625–632 (2012). https://doi.org/10.1007/s10973-011-1483-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-011-1483-z