
Abstract
Congenital heart defect (CHD) is the most common form of birth deformity and is responsible for substantial morbidity and mortality in humans. Increasing evidence has convincingly demonstrated that genetic defects play a pivotal role in the pathogenesis of CHD. However, CHD is a genetically heterogeneous disorder and the genetic basis underpinning CHD in the vast majority of cases remains elusive. This study was sought to identify the pathogenic mutation in the ISL1 gene contributing to CHD. A cohort of 210 unrelated patients with CHD and a total of 256 unrelated healthy individuals used as controls were registered. The coding exons and splicing boundaries of ISL1 were sequenced in all study subjects. The functional effect of an identified ISL1 mutation was evaluated using a dual-luciferase reporter assay system. A novel heterozygous ISL1 mutation, c.409G > T or p.E137X, was identified in an index patient with congenital patent ductus arteriosus and ventricular septal defect. Analysis of the proband’s pedigree revealed that the mutation co-segregated with CHD, which was transmitted in the family in an autosomal dominant pattern with complete penetrance. The nonsense mutation was absent in 512 control chromosomes. Functional analysis unveiled that the mutant ISL1 protein failed to transactivate the promoter of MEF2C, alone or in synergy with TBX20. This study firstly implicates ISL1 loss-of-function mutation with CHD in humans, which provides novel insight into the molecular mechanism of CHD, implying potential implications for genetic counseling and individually tailored treatment of CHD patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Congenital heart defect (CHD) is a cardiovascular structural deformity that arises from aberrant development of the heart or cardiothoracic great blood vessels during embryogenesis [1]. It is the most common form of birth defect in humans, occurring in approximately 1% of all live newborns, and accounting for almost one-third of all major developmental malformations [1, 2]. Each year about 1.35 million neonates are born with CHD worldwide [1, 2]. Clinically, CHD has been categorized into 25 different types, of which 21 designate specific anatomic or hemodynamic lesions, including patent ductus arteriosus (PDA), ventricular septal defect (VSD), tetralogy of Fallot, atrial septal defect, double outlet right ventricle, transposition of the great arteries, persistent truncus arteriosus, aortic stenosis, coarctation of the aorta, pulmonary stenosis, pulmonary atresia, anomalous pulmonary venous connection and endocardial cushion defect [2]. Although minor cardiovascular defects may resolve spontaneously [2], major abnormalities require timely surgical or catheter-based treatment and otherwise may lead to degraded quality of life [3], decreased exercise performance [4], retarded cerebral development or brain injury [5, 6], pulmonary hypertension [7,8,9,10], thromboembolic or hemorrhagic stroke [11,12,13], metabolic disorder [14], infective endocarditis [15,16,17,18], myocardial fibrosis [19, 20], cardiac dysfunction or heart failure [21,22,23,24,25], arrhythmias [26,27,28,29], and death [30,31,32]. Hence, CHD remains the most prevalent cause of infant defect-related demises, with nearly 24% of infants who died of a birth defect having a cardiovascular defect [2]. Although great progress in surgical treatment of pediatric CHD has allowed more than 90% of livebirths with CHD to survive into adulthood, it brings about an ever-increasing population of adults living with CHD and presently there are more adults with CHD than children with CHD [1]. Moreover, the morbidity and mortality in adult CHD patients are much higher than those in the general population [33,34,35,36,37,38,39,40,41,42]. Despite significant clinical importance, the underlying etiologies of CHD remain largely elusive.
Cardiac morphogenesis is a complex biological process and both genetic and environmental risk factors may interrupt the process, leading to CHD [1, 43,44,45,46]. The well-known environmental risk factors for CHD encompass maternal conditions such as viral infection, diabetes mellitus and autoimmune disorder, and maternal exposures to drugs, tobacco smoke, toxic chemicals or ionizing radiation during the first trimester of pregnancy [46]. However, aggregating evidence underscores the genetic components for CHD, especially for familial CHD, which is transmitted predominantly in an autosomal dominant pattern in the family, though familial transmission of CHD is also observed in other inheritance modes, including autosomal recessive and X-linked modes [1, 43,44,45,46]. Regardless of chromosomal abnormalities such as chromosome 22q11 deletion and trisomy of chromosome 21, mutations in more than 60 genes, including those encoding cardiac transcription factors, signaling molecules, cardiac sarcomeric proteins and chromatin modifiers, have been causally linked to CHD in humans [1, 43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72]. Among these well-established CHD-related genes, most code for cardiac transcription factors, including GATA4, GATA5, GATA6, NKX2-5, CASZ1, HAND1, HAND2, NR2F2, MEF2C, TBX1, TBX5 and TBX20 [1, 43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72]. Nevertheless, CHD is a genetically heterogeneous malady, and the genetic determinants underlying CHD in most patients remain unclear.
As a member of the homeodomain-containing family of transcription factors, ISL1 is highly expressed in the fetal heart and is essential for proper cardiovascular development [73,74,75]. In the mouse, targeted disruption of the Isl1 gene results in embryonic death due to severe developmental defects of the heart, including loss of the outflow tract, right ventricle, and much of the atria [76]. In humans, several single nucleotide polymorphisms of ISL1 have been associated with increased risk of CHD [77, 78]. These observational findings make it rational to screen ISL1 as a prime candidate gene for CHD in patients.
Materials and methods
Study participants
In this study, 210 unrelated patients with CHD were recruited from the Chinese Han population. Among them, there were 118 male and 92 female cases with an average age of 7.1 ± 5.6 years, ranging from 1 to 32 years of age. The available family members of the index patient carrying an identified ISL1 mutation were also enrolled. Two-hundred and fifty-six ethnically-matched individuals, who had no CHD or positive family history of CHD, were enlisted as controls, of whom there were 143 males and 113 females with an average age of 6.8 ± 5.5 years, ranging from 1 to 30 years of age. All study subjects underwent comprehensive clinical appraisal, including detailed medical history, physical examination, electrocardiogram and echocardiogram, as well as cardiac catheterization and/or surgical proceedings for patients. Probands with known chromosomal abnormalities or syndromic CHD, such Holt–Oram syndrome, Turner syndrome and Marfan syndrome, were excluded from the current study. This investigation was conducted in conformity with the ethical principles of the Declaration of Helsinki and was approved by the Research Ethics Committees of the First Affiliated Hospital of Soochow University and Shanghai Chest Hospital, Shanghai Jiao Tong University. Written informed consent was obtained from the guardians of the CHD patients and the control subjects prior to the study.
Genetic analysis of ISL1
Peripheral venous blood specimens were collected from all the study subjects. Genomic DNA was purified from blood leukocytes with the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. The genomic DNA sequence of the human ISL1 gene (accession no. NG_023040.1) was derived from the Nucleotide database at the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/nuccore/NG_023040.1?from=5001&to=16607&report=genbank). With the help of the online program Primer-BLAT (https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?ORGANISM=9606&INPUT_SEQUENCE=NG_023040.1&LINK_LOC=nuccore&PRIMER5_START=5001&PRIMER3_END=16607), the primers to amplify the coding regions and splicing boundaries of ISL1 by polymerase chain reaction (PCR) were designed as given in Table 1. Genomic DNA of interest was performed by PCR using HotStar Taq DNA Polymerase (Qiagen) on a Veriti Thermal Cycler (Life Technologies, Carlsbad, CA, USA) under recommended reagent concentrations and reaction conditions. PCR sequencing of the purified amplicons was carried out with the BigDye® Terminator v3.1 Cycle Sequencing Kit (Life Technologies) on an ABI PRISM 3130 XL DNA Analyzer (Life Technologies). The position of an exonic sequence variation was numbered in accordance with the reference sequence of the ISL1 mRNA transcript at the Nucleotide database (https://www.ncbi.nlm.nih.gov/nuccore/NM_002202.2), with an accession number of NM_002202.2. Additionally, the Single Nucleotide Polymorphism database (https://www.ncbi.nlm.nih.gov/snp/?term=ISL1), the Exome Variant Server database (http://evs.gs.washington.edu/EVS), the 1000 Genomes Project database (http://www.internationalgenome.org/), and the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/all.php) were queried to verify the novelty of an identified ISL1 sequence variance.
Plasmid constructs and site-directed mutagenesis
RNA isolation and cDNA preparation from human heart tissue were prepared as previously described [79]. The wild-type full-length open read frame of the human ISL1 gene (accession no. NM_002202.2) was amplified by PCR using the pfuUltra high-fidelity DNA polymerase (Stratagene, Santa Clara, CA, USA) and a specific pair of primers (forward primer: 5′-TGGGCTAGCAACCACCATTTCACTGTGG-3′; reverse primer: 5′-TGGGCGGCCGCAAAATACAGAATGAATGTTC-3′). The amplified product was doubly digested by restriction enzymes NheI and NotI (TaKaRa, Dalian, Liaoning, China). The digested product with a length of 1118 base pairs was separated by 1.0% agarose gel electrophoresis, isolated using the GeneJET™ gel extraction kit (Life Technologies), and then inserted into the NheI-NotI sites of the pcDNA3.1 plasmid (Invitrogen, Carlsbad, CA, USA) to generate a recombinant expression plasmid ISL1-pcDNA3.1. The nonsense mutation detected in CHD patients was introduced into the wild-type ISL1-pcDNA3.1 plasmid by site-targeted mutagenesis using the QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene) with a complementary pair of primers following the manufacturer’s descriptions and was checked by sequencing. For generation of the reporter plasmid MEF2C-luciferase (MEF2C-luc), which expresses firefly luciferase, a 1387-bp promoter region of the MEF2C gene (nucleotides 2584–3970; accession No. AY324098) was subcloned into the promoterless pGL3-Basic vector (Promega, Madison, WI, USA) as described previously [80]. The expression plasmid TBX20-pcDNA3.1 was constructed as described previously [58].
Cell transfection and luciferase assay
CHO and 10T1/2 cells were cultured in Dulbecco’s modified Eagle’s media supplemented with 10% fetal bovine serum as well as 100 U/ml penicillin and 100 μg/ml streptomycin in an incubator with an atmosphere of 5% CO2 at 37 °C. Cells were seeded at a density of 1 × 105 cells per well in 12-well plates. After 48 h, transfection was performed using the Lipofectamine 2000® reagent (Invitrogen) following the manufacturer’s manual. For transient transfection experiments, CHO cells were transfected with 1.0 μg of wild-type ISL1-pcDNA3.1, 1.0 μg of E137X-mutant ISL1-pcDNA3.1, 1.0 μg of wild-type ISL1-pcDNA3.1 plus 1.0 μg of E137X-mutant ISL1-pcDNA3.1, 0.5 μg of wild-type ISL1-pcDNA3.1, or 0.5 μg of wild-type ISL1-pcDNA3.1 in combination with 0.5 μg of E137X-mutant ISL1-pcDNA3.1, in the presence of 1.0 μg of MEF2C-luc and 0.04 μg of pGL4.75 (Promega). The pGL4.75 vector expressing a renilla luciferase was co-transfected into the cells as an internal control for transfection efficiency. For the negative control, the empty plasmid pcDNA3.1 was used. In order to explore the synergistic effect between ISL1 and TBX20 on the MEF2C promoter, 10T1/2 cells were co-transfected with 0.25 μg of wild-type ISL1-pcDNA3.1, or 0.25 μg of E137X-mutant ISL1-pcDNA3.1, or 0.25 μg of wild-type TBX20-pcDNA3.1, or 0.25 μg of wild-type ISL1-pcDNA3.1 plus 0.25 μg of wild-type TBX20-pcDNA3.1, or 0.25 μg of E137X-mutant ISL1-pcDNA3.1 plus 0.25 μg of wild-type TBX20-pcDNA3.1, in the presence of 1.0 μg of MEF2C-luc and 0.04 μg of pGL4.75 (Promega). Cells were incubated at 37 °C and harvested 48 h after transfection. Luciferase activity of the cell lysates was determined in a luminometer with the Dual-Luciferase Reporter Assay System (Promega) following the manufacturer’s instructions. The results were expressed as the ratios of the activities of firefly luciferase (pGL3-Basic) to renilla luciferase (pGL4.75). For each expression plasmid, three independent experiments were each performed in triplicate and the MEF2C promoter activity was expressed as mean ± standard deviation (SD).
In order to test whether the mutant ISL1 protein is properly expressed by the reconstructed E137X-mutant ISL1-pcDNA3.1 plasmid, 1.0 μg of E243X-mutant ISL1-pcDNA3.1 or 1.0 μg of wild-type ISL1-pcDNA3.1 was transfected into the cultured CHO cells with the Lipofectamine 2000® reagent (Invitrogen). Cells were collected 36 h after transient transfection and total mRNAs were extracted using TRIzol Reagent (Invitrogen). Reverse transcription (RT) of mRNA coding for ISL1 protein was carried out with SuperScript™ IV First-Strand Synthesis System (Invitrogen) and the ISL1-specific primer (5′-CACCATGGGAGTTCCTGTCA-3′). Amplification of cDNA by PCR was performed with HotStar Taq DNA Polymerase (Qiagen) and a pair of primers specific to ISL1 (forward primer: 5′-CATCGAGTGTTTCCGCTGTG-3′; backward primer: 5′-CACCATGGGAGTTCCTGTCA-3′; product size: 496 bp). The amplicons were quantified by electrophoresis analysis on 1.2% agarose gel stained with ethidium bromide.
Statistical analysis
For statistical analyses, the SPSS software package for Windows, version 17.0 (SPSS Inc., Chicago, IL, USA) was used. Continuous variables between two groups were compared with Student’s unpaired t test, whereas categorical variables between two groups were compared with Pearson’s χ2 test or Fisher’s exact test, when appropriate. A two-sided P < 0.05 was considered statistically different.
Results
Clinical characteristics of the study population
In the present study, 210 unrelated CHD patients were clinically investigated in contrast to 256 unrelated control individuals without CHD. Patients were matched with controls for age, gender and ethnicity. All the patients had echocardiogram-documented CHD, of whom about 20% had a positive family history of CHD. The control individuals had no family history of CHD and their echocardiograms showed normal cardiac images without evidence of structural cardiac defects. The demographic and baseline clinical features of the CHD patients are summarized in Table 2.
Identification of a novel ISL1 mutation
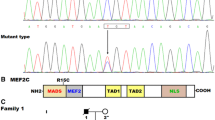
The entire coding exons and exon–intron boundaries as well as partial 3′- and 5′-untranslated regions of the ISL1 gene were analyzed by direct sequencing in a cohort of 210 patients with CHD and a nonsense mutation was found in an index patient who was five years old. Specifically, a substitution of thymine for guanine at the first nucleotide of codon 137 (c.409G > T), predicting the transition of the codon encoding glutamic acid at amino acid position 137 to a premature termination codon (p.E137X), was detected in a girl with PDA and VSD, who had a positive family history of CHD. The sequence electropherograms of the heterozygous ISL1 mutation of c.409G > T and its wild-type control are displayed in Fig. 1A. The schematic diagrams of the E137X-mutant and wild-type ISL1 proteins indicating the key structural domains and location of the mutation identified in this study are exhibited in Fig. 1B. The nonsense mutation was neither detected in the 256 control individuals nor reported in the Single Nucleotide Polymorphism, Exome Variant Server, 1000 Genomes Project, and Human Gene Mutation databases (queried again in June 29, 2018). Genetic scan of the mutation carrier′s family members available revealed that the mutation was present in all the affected family members, but absent in unaffected family members. Analysis of the proband′s pedigree unveiled that the mutation co-segregated with CHD, which was transmitted as an autosomal dominant trait in the family with complete penetrance. The pedigree structure of the proband’s family is shown in Fig. 1C. The phenotypic features of the proband’s affected family members are shown in Table 3.

Novel ISL1 mutation associated with congenital heart defects. (A) Sequence electropherograms displaying the heterozygous ISL1 mutation and its homozygous wild-type control. The arrow points to the heterozygous nucleotides of G/T in the proband (mutant type) or the homozygous nucleotides of G/G in a control subject (wild type). The rectangle denotes the nucleotides comprising a codon of ISL1. (B) Schematic diagrams showing the structural domains of the wild-type and E137X-mutant ISL1 protein and the location of the mutation causally linked to congenital heart defects. The mutation identified in patients with congenital heart defects is marked above the structural domains. NH2 means amino terminus; HD, homeodomain; TAD, transcriptional activation domain; COOH, carboxyl terminus. (C) Pedigree structure of the family with congenital heart defects. The family was designated as family 1. Family members are identified by generations and numbers. The proband is indicated by an arrow. Squares indicate male family members and circles indicate female members. Phenotypes are indicated as: right half black symbols, patent ductus arteriosus (PDA); left half shaded symbols, ventricular septal defect (VSD); the symbol with a diagonal, the dead member. “+” means a carrier of the heterozygous nonsense mutation; “–”, a non-carrier
Failure to transactivate the promoter of MEF2C by the mutant ISL1 protein
Previous studies have validated that ISL1 transcriptionally activates the MEF2C promoter in vivo and in vitro, alone or in synergy with TBX20 [80,81,82]. As shown in Fig. 2, the same amount (1.0 μg) of wild-type and E137X-mutant ISL1-pcDNA3.1 plasmids transcriptionally activated the MEF2C promoter in CHO cells by ~ 10 folds and ~ onefolds, respectively. When 1.0 μg of wild-type ISL1-pcDNA3.1 and 1.0 μg of E137X-mutant ISL1-pcDNA3.1 were co-transfected, the induced transcriptional activation of the MEF2C promoter was ~ ninefold. Additionally, when 0.5 μg of wild-type ISL1-pcDNA3.1 together with 0.5 μg of empty ISL1-pcDNA3.1 or 0.5 μg of E137X-mutant ISL1-pcDNA3.1 was used, the induced transcriptional activation of the MEF2C promoter was ~ sixfold or ~ fivefold. These data suggest that the E137X-mutant ISL1 has neither transcriptional activation of target genes nor dominant-negative effect on its wild-type counterpart.

Inability to transcriptionally activate the MEF2C promoter by the mutant ISL1. Activation of the MEF2C promoter driven luciferase in CHO cells by wild-type ISL1 or E127X-mutant ISL1 (E137X), alone or together, showed no transcriptional activation by the mutant protein. Three independent transfection experiments were performed in triplicate. The results are represented by means with standard deviations. ** indicates t = 14.7009, p = 0.00012; * indicates t = 6.76641, p = 0.00249, when compared with wild-type ISL1 (1.0 μg)
As shown in Fig. 3, the same amount (0.25 μg) of wild-type and E137X-mutant ISL1-pcDNA3.1 plasmids transcriptionally activated the MEF2C promoter in 10T1/2 cells by ~ eightfolds and ~ onefold, respectively. In the presence of 0.25 μg of TBX20-pcDNA3.1, the induced synergistic transcriptional activity by the same amount (0.25 μg) of wild-type or E137X-mutant ISL1-pcDNA3.1 plasmid was ~ 28-fold or ~ tenfold. These data indicate that the E137X mutation disrupts the synergistic transcriptional activation between ISL1 and TBX20.

Synergic transcriptional activation between ISL1 and TBX20 disrupted by the mutation. In the presence of TBX20, activation of the MEF2C promoter driven luciferase in 10T1/2 cells by wild-type ISL1 or E127X-mutant ISL1 (E137X) showed significantly reduced transcriptional activity by the mutant protein. Three independent transfection experiments were performed in triplicate. The results are represented by means with standard deviations. * indicates t = 9.52952, p = 0.000677, when compared with wild-type counterpart
Besides, quantitative RT-PCR was performed and the results demonstrated that both wild-type ISL1-pcDNA3.1 and E137X-mutant ISL1-pcDNA3.1 were equally efficient in transcribing ISL1 mRNAs, indicating that wild-type ISL1-pcDNA3.1 and E137X-mutant ISL1-pcDNA3.1 produce the same amounts of ISL1 proteins (data not shown).
Discussion
In this research, a novel heterozygous mutation (c.409G > T or p.E137X) in the ISL1 gene was identified in a family with PDA and VSD. The nonsense mutation, which was absent in the 512 control chromosomes, co-segregated with CHD in the family with complete penetrance. Functional analysis demonstrated that the E137X-mutant ISL1 protein had no transcriptional activity. Therefore, it is very likely that genetically compromised ISL1 contributes to PDA and VSD in this family.
In humans, ISL1 maps on chromosome 5q11.1, encoding a protein with 349 amino acids. The encoded ISL1 protein, as a member of the homeodomain family of transcription factors, binds to the enhancer region of the target genes, playing a key role in regulating expression of target genes, which are central to the heart development [73,74,75,76]. The human ISL1 protein has two functionally important structural domains, including homeodomain (HD) and transcriptional activation domain (TAD) [82, 83]. The highly conserved HD domain comprises 60 amino acids (amino acids 181–240) and its main functional role is to bind to consensus DNA sequence in the promoter of a target gene. Adjacent to the HD domain is the TAD domain which consists of 109 highly conserved amino acids, starting from amino acid 241 to amino acid 349, and is responsible for activating transcription [82, 83]. In the present study, the mutation identified in CHD patients was predicted to generate a truncated protein with only 136 amino-terminal amino acids left, lacking the DNA-binding and transcriptional activation domains and, thus, was anticipated to nullify its transcriptional activation of target genes, including the MEF2C gene that has been associated with CHD [59, 65, 84]. Functional deciphers demonstrated that the E137X-mutant ISL1 protein had no transcriptional activation of the MEF2C promoter. Furthermore, the mutation disrupted the synergic activation between ISL1 and TBX20, another cardiac core transcriptional factor that has been causally linked to CHD [58]. Importantly, co-immunoprecipitation experiments performed in cultivated HeLa cells demonstrated the physical interactions between ISL1 and TBX20, and reporter gene analyses revealed that TBX20 could potently activate the MEF2C and NKX2-5 promoters alone or synergistically with ISL1 [81]. Besides, as an important transcriptional co-regulator of ISL1, TBX20 was also involved in directly up-regulating the expression of other key cardiac genes, including PITX2, FGF10 and MYH7, singly or in synergy with ISL1, NKX2-5 and GATA4 [81]. These findings indicate that haploinsufficiency resulted from an ISL1 mutation is likely to be an alternative pathological mechanism of CHD.
Previous investigations have revealed that a premature translation termination codon may cause degradation of mRNA in many types of organisms and cell lines by a mechanism named as nonsense-mediated mRNA decay (NMD), a translation-dependent, multi-step process that monitors and degrades irregular or faulty mRNAs [79, 85]. In the present research, the nonsense mutation in ISL1 yielded a premature translation termination codon; therefore, the mutant ISL1 mRNAs were likely to undergo NMD, although not all nonsense mutations triggered NMD [86]. At present, we could not validate NMD in the mutation carriers due to the unavailability of their cardiac tissue specimens, where the mutant ISL1 protein might be expressed. Even if the mutant mRNAs underwent NMD, the overall abundance of ISL1 mRNAs would be decreased by a half, leading to haploinsufficiency, which was in line with our functional results. Notably, downstream intron or pre-mRNA splicing, which causes the deposition of a multi-protein complex, termed as the exon-junction-complex, 20–24 nucleotides upstream of each exon–exon junction, is required for the degradation of mRNA containing a premature translation termination codon by the NMD mechanism. Hence, NMD could not occur in the context of cDNA constructs [79, 85].
In addition, recent researches have associated several common polymorphisms of the ISL1 gene with an increased risk of CHD [77, 78]. Stevens and colleagues [77] in stage 1 made a case–control analysis of 30 polymorphisms mapping to the ISL1 locus in 300 pediatric patients with complex CHD and 2201 healthy children, and discovered that eight polymorphisms (rs6867206, rs4865656, rs6869844, rs2115322, rs6449600, IVS1 + 17C > T, rs1017, rs6449612) in and flanking ISL1 were significantly associated with complex CHD. To independently validate their findings, in stage 2 they performed a replication study of these candidate polymorphisms in 1044 new cases and 3934 independent controls and confirmed the association of these polymorphisms within and around ISL1 with increased risk of non-syndromic CHD [77]. To determine the association of genetic polymorphisms in and near the ISL1 gene with CHD in the Chinese Han population, Luo and partners [78] analyzed nine polymorphisms (rs6867206, rs6869844, rs3762977, rs1017, rs6449612, rs4865656, rs2115322, rs6449600, rs150104955) of ISL1 in 233 patients with CHD and 288 healthy subjects, and found that one polymorphism (rs1017) in ISL1 was significantly associated with simple CHD. These data provide strong evidence that ISL1 plays an important role in the development of heart and pathogenesis of CHD.
Notably, previous investigations have causally linked over 60 genes, including those coding for cardiac transcription factors, to CHD in humans [1, 43,44,45,46]. In the current study, as previously described, we have analyzed several other cardiac transcription factors in the index patient who carried an identified ISL1 mutation, including GATA4 [64], GATA5 [87], GATA6 [72], TBX1 [69], TBX5 [88], TBX20 [58], HAND1 [52, 53], HAND2 [89], NKX2-5 [54], MEF2C [59, 65], PITX2 [90], CASZ1 [91], NR2F2 [63] and MESP1 [60], and detected no pathogenic mutations. Nevertheless, we cannot rule out the possibility that other genes may also contribute to the pathogenesis of CHD.
In conclusion, this study firstly associates ISL1 loss-of-function mutation with CHD in humans, which adds novel insight to the molecular pathogenesis of CHD, suggesting potential implications for genetic counseling and individualized treatment of the patients with CHD.
References
Zaidi S, Brueckner M (2017) Genetics and genomics of congenital heart disease. Circ Res 120:923–940
Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O’Flaherty M, Palaniappan LP, Pandey A, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee (2018) Heart disease and stroke statistics—2018 update: a report from the American Heart Association. Circulation 137:e67–e492
Ernst MM, Marino BS, Cassedy A, Piazza-Waggoner C, Franklin RC, Brown K, Wray J (2018) Biopsychosocial predictors of quality of life outcomes in pediatric congenital heart disease. Pediatr Cardiol 39:79–88
Banks L, Rosenthal S, Manlhiot C, Fan CS, McKillop A, Longmuir PE, McCrindle BW (2017) Exercise capacity and self-efficacy are associated with moderate-to-vigorous intensity physical activity in children with congenital heart disease. Pediatr Cardiol 38:1206–1214
Morton PD, Ishibashi N, Jonas RA (2017) Neurodevelopmental abnormalities and congenital heart disease: insights into altered brain maturation. Circ Res 120:960–977
Peyvandi S, Chau V, Guo T, Xu D, Glass HC, Synnes A, Poskitt K, Barkovich AJ, Miller SP, McQuillen PS (2018) Neonatal brain injury and timing of neurodevelopmental assessment in patients with congenital heart disease. J Am Coll Cardiol 71:1986–1996
Zorzanelli L, Maeda N, Clavé M, Thomaz A, Galas F, Rabinovitch M, Lopes A (2017) Relation of cytokine profile to clinical and hemodynamic features in young patients with congenital heart disease and pulmonary hypertension. Am J Cardiol 119:119–125
van der Feen DE, Bartelds B, de Boer RA, Berger RM (2017) Pulmonary arterial hypertension in congenital heart disease: translational opportunities to study the reversibility of pulmonary vascular disease. Eur Heart J 38:2034–2041
Schwartz SS, Madsen N, Laursen HB, Hirsch R, Olsen MS (2018) Incidence and mortality of adults with pulmonary hypertension and congenital heart disease. Am J Cardiol 121:1610–1616
Muneuchi J, Ochiai Y, Masaki N, Okada S, Iida C, Sugitani Y, Ando Y, Watanabe M (2018) Pulmonary arterial compliance is a useful predictor of pulmonary vascular disease in congenital heart disease. Heart Vessels. https://doi.org/10.1007/s00380-018-1263-9
Lanz J, Brophy JM, Therrien J, Kaouache M, Guo L, Marelli AJ (2015) Stroke in adults with congenital heart disease: incidence, cumulative risk, and predictors. Circulation 132:2385–2394
Masuda K, Ishizu T, Niwa K, Takechi F, Tateno S, Horigome H, Aonuma K (2017) Increased risk of thromboembolic events in adult congenital heart disease patients with atrial tachyarrhythmias. Int J Cardiol 234:69–75
Giang KW, Mandalenakis Z, Dellborg M, Lappas G, Eriksson P, Hansson PO, Rosengren A (2018) Long-term risk of hemorrhagic stroke in young patients with congenital heart disease. Stroke 49:1155–1162
Forte A, Grossi M, Bancone C, Cipollaro M, De Feo M, Hellstrand P, Persson L, Nilsson BO, Della Corte A (2018) Polyamine concentration is increased in thoracic ascending aorta of patients with bicuspid aortic valve. Heart Vessels 33:327–339
Diller GP, Baumgartner H (2017) Endocarditis in adults with congenital heart disease: new answers-new questions. Eur Heart J 38:2057–2059
Mylotte D, Rushani D, Therrien J, Guo L, Liu A, Guo K, Martucci G, Mackie AS, Kaufman JS, Marelli A (2017) Incidence, predictors, and mortality of infective endocarditis in adults with congenital heart disease without prosthetic valves. Am J Cardiol 120:2278–2283
Kuijpers JM, Koolbergen DR, Groenink M, Peels KC, Reichert CL, Post MC, Bosker HA, Wajon EM, Zwinderman AH, Mulder BJ, Bouma BJ (2017) Incidence, risk factors, and predictors of infective endocarditis in adult congenital heart disease: focus on the use of prosthetic material. Eur Heart J 38:2048–2056
Tutarel O, Alonso-Gonzalez R, Montanaro C, Schiff R, Uribarri A, Kempny A, Grübler MR, Uebing A, Swan L, Diller GP, Dimopoulos K, Gatzoulis MA (2018) Infective endocarditis in adults with congenital heart disease remains a lethal disease. Heart 104:161–165
Ciepłucha A, Trojnarska O, Kociemba A, Łanocha M, Barczynski M, Rozmiarek S, Kramer L, Pyda M (2018) Clinical aspects of myocardial fibrosis in adults with Ebstein’s anomaly. Heart Vessels 33:1076–1085
Lluri G, Renella P, Finn JP, Vorobiof G, Aboulhosn J, Deb A (2017) Prognostic significance of left ventricular fibrosis in patients with congenital bicuspid aortic valve. Am J Cardiol 120:1176–1179
Miyazaki A, Sakaguchi H, Noritake K, Hayama Y, Negishi J, Kagisaki K, Yasuda K, Ichikawa H, Ohuchi H (2017) Interventricular dyssynchrony in a patient with a biventricular physiology and a systemic right ventricle. Heart Vessels 32:234–239
Shiina Y, Inai K, Takahashi T, Taniguchi K, Watanabe E, Fukushima K, Niwa K, Nagao M (2018) Inter- and intra-ventricular dyssynchrony in the systemic right ventricle is a surrogate marker of major cardiac events in mildly symptomatic patients. Heart Vessels 33:1086–1093
Hinton RB, Ware SM (2017) Heart failure in pediatric patients with congenital heart disease. Circ Res 120:978–994
Isogai T, Matsui H, Tanaka H, Kohyama A, Fushimi K, Yasunaga H (2018) Clinical features and peripartum outcomes in pregnant women with cardiac disease: a nationwide retrospective cohort study in Japan. Heart Vessels 33:918–930
Shaddy RE, George AT, Jaecklin T, Lochlainn EN, Thakur L, Agrawal R, Solar-Yohay S, Chen F, Rossano JW, Severin T, Burch M (2018) Systematic literature review on the incidence and prevalence of heart failure in children and adolescents. Pediatr Cardiol 39:415–436
Labombarda F, Hamilton R, Shohoudi A, Aboulhosn J, Broberg CS, Chaix MA, Cohen S, Cook S, Dore A, Fernandes SM, Fournier A, Kay J, Macle L, Mondésert B, Mongeon FP, Opotowsky AR, Proietti A, Rivard L, Ting J, Thibault B, Zaidi A, Khairy P, AARCC (2017) Increasing prevalence of atrial fibrillation and permanent atrial arrhythmias in congenital heart disease. J Am Coll Cardiol 70:857–865
McLeod CJ (2017) Acute arrhythmias in adults with congenital heart disease. Heart 103:1380–1388
Holst KA, Said SM, Nelson TJ, Cannon BC, Dearani JA (2017) Current interventional and surgical management of congenital heart disease: specific focus on valvular disease and cardiac arrhythmias. Circ Res 120:1027–1044
Hernández-Madrid A, Paul T, Abrams D, Aziz PF, Blom NA, Chen J, Chessa M, Combes N, Dagres N, Diller G, Ernst S, Giamberti A, Hebe J, Janousek J, Kriebel T, Moltedo J, Moreno J, Peinado R, Pison L, Rosenthal E, Skinner JR, Zeppenfeld K; ESC Scientific Document Group (2018) Arrhythmias in congenital heart disease: a position paper of the European Heart Rhythm Association (EHRA), Association for European Paediatric and Congenital Cardiology (AEPC), and the European Society of Cardiology (ESC) Working Group on Grown-up Congenital heart disease, endorsed by HRS, PACES, APHRS, and SOLAECE. Europace https://doi.org/10.1093/europace/eux380
Koyak Z, de Groot JR, Bouma BJ, Zwinderman AH, Silversides CK, Oechslin EN, Budts W, Van Gelder IC, Mulder BJ, Harris L (2017) Sudden cardiac death in adult congenital heart disease: can the unpredictable be foreseen? Europace 19:401–406
Moore B, Yu C, Kotchetkova I, Cordina R, Celermajer DS (2018) Incidence and clinical characteristics of sudden cardiac death in adult congenital heart disease. Int J Cardiol 254:101–106
Lynge TH, Jeppesen AG, Winkel BG, Glinge C, Schmidt MR, Søndergaard L, Risgaard B, Tfelt-Hansen J (2018) Nationwide study of sudden cardiac death in people with congenital heart defects aged 0 to 35 years. Circ Arrhythm Electrophysiol 11:e005757
Izumi G, Senzaki H, Takeda A, Yamazawa H, Takei K, Furukawa T, Inai K, Shinohara T, Nakanishi T (2017) Significance of right atrial tension for the development of complications in patients after atriopulmonary connection Fontan procedure: potential indicator for Fontan conversion. Heart Vessels 32:850–855
Saito C, Fukushima N, Fukushima K, Matsumura G, Ashihara K, Hagiwara N (2017) Factors associated with aortic root dilatation after surgically repaired ventricular septal defect. Echocardiography 34:1203–1209
Shinkawa T, Chipman C, Holloway J, Tang X, Gossett JM, Imamura M (2017) Single center experience of aortic bypass graft for aortic arch obstruction in children. Heart Vessels 32:76–82
Henmi R, Ejima K, Yagishita D, Iwanami Y, Nishimura T, Takeuchi D, Toyohara K, Shoda M, Hagiwara N (2017) Long-term efficacy of implantable cardioverter defibrillator in repaired tetralogy of Fallot—role of anti-tachycardia pacing. Circ J 81:165–171
Kim J, Kuwata S, Kurishima C, Iwamoto Y, Ishido H, Masutani S, Senzaki H (2018) Importance of dynamic central venous pressure in Fontan circulation. Heart Vessels 33:664–670
Takahashi T, Shiina Y, Nagao M, Inai K (2018) Stroke volume ratio derived from magnetic resonance imaging as an indicator of interventricular dyssynchrony predicts future cardiac event in patients with biventricular Fontan circulation. Heart Vessels. https://doi.org/10.1007/s00380-018-1217-2
Hasegawa T, Masuda M, Okumura M, Arai H, Kobayashi J, Saiki Y, Tanemoto K, Nishida H, Motomura N (2017) Trends and outcomes in neonatal cardiac surgery for congenital heart disease in Japan from 1996 to 2010. Eur J Cardiothorac Surg 51:301–307
Bouma BJ, Mulder BJ (2017) Changing landscape of congenital heart disease. Circ Res 120:908–922
Mandalenakis Z, Rosengren A, Skoglund K, Lappas G, Eriksson P, Dellborg M (2017) Survivorship in children and young adults with congenital heart disease in Sweden. JAMA Intern Med 177:224–230
Hayabuchi Y, Ono A, Homma Y, Kagami S (2018) Pulmonary annular motion velocity reflects right ventricular outflow tract function in children with surgically repaired congenital heart disease. Heart Vessels 33:316–326
Blue GM, Kirk EP, Giannoulatou E, Sholler GF, Dunwoodie SL, Harvey RP, Winlaw DS (2017) Advances in the genetics of congenital heart disease: a clinician’s guide. J Am Coll Cardiol 69:859–870
Akhirome E, Walton NA, Nogee JM, Jay PY (2017) The complex genetic basis of congenital heart defects. Circ J 81:629–634
Li YJ, Yang YQ (2017) An update on the molecular diagnosis of congenital heart disease: focus on loss-of-function mutations. Expert Rev Mol Diagn 17:393–401
Fahed AC, Gelb BD, Seidman JG, Seidman CE (2013) Genetics of congenital heart disease: the glass half empty. Circ Res 112:707–720
Wang X, Chang WL, Chen CA, Rosenfeld JA, Al Shamsi A, Al-Gazali L, McGuire M, Mew NA, Arnold GL, Qu C, Ding Y, Muzny DM, Gibbs RA, Eng CM, Walkiewicz M, Xia F, Plon SE, Lupski JR, Schaaf CP, Yang Y (2017) Germline mutations in ABL1 cause an autosomal dominant syndrome characterized by congenital heart defects and skeletal malformations. Nat Genet 49:613–617
Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC, Hung WC, Haider S, Zhang J, Knight J, Bjornson RD, Castaldi C, Tikhonoa IR, Bilguvar K, Mane SM, Sanders SJ, Mital S, Russell MW, Gaynor JW, Deanfield J, Giardini A, Porter GA Jr, Srivastava D, Lo CW, Shen Y, Watkins WS, Yandell M, Yost HJ, Tristani-Firouzi M, Newburger JW, Roberts AE, Kim R, Zhao H, Kaltman JR, Goldmuntz E, Chung WK, Seidman JG, Gelb BD, Seidman CE, Lifton RP, Brueckner M (2017) Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet 49:1593–1601
Liu X, Yagi H, Saeed S, Bais AS, Gabriel GC, Chen Z, Peterson KA, Li Y, Schwartz MC, Reynolds WT, Saydmohammed M, Gibbs B, Wu Y, Devine W, Chatterjee B, Klena NT, Kostka D, de Mesy Bentley KL, Ganapathiraju MK, Dexheimer P, Leatherbury L, Khalifa O, Bhagat A, Zahid M, Pu W, Watkins S, Grossfeld P, Murray SA, Porter GA Jr, Tsang M, Martin LJ, Benson DW, Aronow BJ, Lo CW (2017) The complex genetics of hypoplastic left heart syndrome. Nat Genet 49:1152–1159
Tan TY, Gonzaga-Jauregui C, Bhoj EJ, Strauss KA, Brigatti K, Puffenberger E, Li D, Xie L, Das N, Skubas I, Deckelbaum RA, Hughes V, Brydges S, Hatsell S, Siao CJ, Dominguez MG, Economides A, Overton JD, Mayne V, Simm PJ, Jones BO, Eggers S, Le Guyader G, Pelluard F, Haack TB, Sturm M, Riess A, Waldmueller S, Hofbeck M, Steindl K, Joset P, Rauch A, Hakonarson H, Baker NL, Farlie PG (2017) Monoallelic BMP2 variants predicted to result in haploinsufficiency cause craniofacial, skeletal, and cardiac features overlapping those of 20p12 deletions. Am J Hum Genet 101:985–994
Hempel M, Casar Tena T, Diehl T, Burczyk MS, Strom TM, Kubisch C, Philipp M, Lessel D (2017) Compound heterozygous GATA5 mutations in a girl with hydrops fetalis, congenital heart defects and genital anomalies. Hum Genet 136:339–346
Wang J, Hu XQ, Guo YH, Gu JY, Xu JH, Li YJ, Li N, Yang XX, Yang YQ (2017) HAND1 loss-of-function mutation causes tetralogy of Fallot. Pediatr Cardiol 38:547–557
Li L, Wang J, Liu XY, Liu H, Shi HY, Yang XX, Li N, Li YJ, Huang RT, Xue S, Qiu XB, Yang YQ (2017) HAND1 loss-of-function mutation contributes to congenital double outlet right ventricle. Int J Mol Med 39:711–718
Xu YJ, Qiu XB, Yuan F, Shi HY, Xu L, Hou XM, Qu XK, Liu X, Huang RT, Xue S, Yang YQ, Li RG (2017) Prevalence and spectrum of NKX2.5 mutations in patients with congenital atrial septal defect and atrioventricular block. Mol Med Rep 15:2247–2254
Chen HX, Zhang X, Hou HT, Wang J, Yang Q, Wang XL, He GW (2017) Identification of a novel and functional mutation in the TBX5 gene in a patient by screening from 354 patients with isolated ventricular septal defect. Eur J Med Gene 60:385–390
Ramond F, Duband S, Croisille P, Cavé H, Teyssier G, Adouard V, Touraine R (2017) Expanding the cardiac spectrum of Noonan syndrome with RIT1 variant: left main coronary artery atresia causing sudden death. Eur J Med Genet 60:299–302
Guo C, Wang Q, Wang Y, Yang L, Luo H, Cao XF, An L, Qiu Y, Du M, Ma X, Li H, Lu C (2017) Exome sequencing reveals novel IRXI mutation in congenital heart disease. Mol Med Rep 15:3193–3197
Huang RT, Wang J, Xue S, Qiu XB, Shi HY, Li RG, Qu XK, Yang XX, Liu H, Li N, Li YJ, Xu YJ, Yang YQ (2017) TBX20 loss-of-function mutation responsible for familial tetralogy of Fallot or sporadic persistent truncus arteriosus. Int J Med Sci 14:323–332
Qiao XH, Wang F, Zhang XL, Huang RT, Xue S, Wang J, Qiu XB, Liu XY, Yang YQ (2017) MEF2C loss-of-function mutation contributes to congenital heart defects. Int J Med Sci 14:1143–1153
Zhang M, Li FX, Liu XY, Huang RT, Xue S, Yang XX, Li YJ, Liu H, Shi HY, Pan X, Qiu XB, Yang YQ (2017) MESP1 loss–of–function mutation contributes to double outlet right ventricle. Mol Med Rep 16:2747–2754
Vaidya A, Flores SK, Cheng ZM, Nicolas M, Deng Y, Opotowsky AR, Lourenço DM Jr, Barletta JA, Rana HQ, Pereira MA, Toledo RA, Dahia PLM (2018) EPAS1 Mutations and Paragangliomas in Cyanotic Congenital Heart Disease. N Engl J Med 378:1259–1261
Bashamboo A, Eozenou C, Jorgensen A, Bignon-Topalovic J, Siffroi JP, Hyon C, Tar A, Nagy P, Sólyom J, Halász Z, Paye-Jaouen A, Lambert S, Rodriguez-Buritica D, Bertalan R, Martinerie L, Rajpert-De Meyts E, Achermann JC, McElreavey K (2018) Loss of function of the nuclear receptor NR2F2, encoding COUP-TF2, causes testis development and cardiac defects in 46, XX children. Am J Hum Genet 102:487–493
Qiao XH, Wang Q, Wang J, Liu XY, Xu YJ, Huang RT, Xue S, Li YJ, Zhang M, Qu XK, Li RG, Qiu XB, Yang YQ (2018) A novel NR2F2 loss-of-function mutation predisposes to congenital heart defect. Eur J Med Genet 61:197–203
Li RG, Xu YJ, Wang J, Liu XY, Yuan F, Huang RT, Xue S, Li L, Liu H, Li YJ, Qu XK, Shi HY, Zhang M, Qiu XB, Yang YQ (2018) GATA4 loss-of-function mutation and the congenitally bicuspid aortic valve. Am J Cardiol 121:469–474
Lu CX, Wang W, Wang Q, Liu XY, Yang YQ (2018) A novel MEF2C loss-of-function mutation associated with congenital double outlet right ventricle. Pediatr Cardiol 39:794–804
Yokoyama R, Kinoshita K, Hata Y, Abe M, Matsuoka K, Hirono K, Kano M, Nakazawa M, Ichida F, Nishida N, Tabata T (2018) A mutant HCN4 channel in a family with bradycardia, left bundle branch block, and left ventricular noncompaction. Heart Vessels 33:802–819
Li X, Shi L, Xu M, Zheng X, Yu Y, Jin J (2018) RCAN1 mutation and functional characterization in children with sporadic congenital heart disease. Pediatr Cardiol 39:226–235
Nijak A, Alaerts M, Kuiperi C, Corveleyn A, Suys B, Paelinck B, Saenen J, Van Craenenbroeck E, Van Laer L, Loeys B, Verstraeten A (2018) Left ventricular non-compaction with Ebstein anomaly attributed to a TPM1 mutation. Eur J Med Genet 61:8–10
Zhang M, Li FX, Liu XY, Hou JY, Ni SH, Wang J, Zhao CM, Zhang W, Kong Y, Huang RT, Xue S, Yang YQ (2018) TBX1 loss-of-function mutation contributes to congenital conotruncal defects. Exp Ther Med 15:447–453
Jaouadi A, Tabebi M, Abdelhedi F, Abid D, Kamoun F, Chabchoub I, Maatoug S, Doukali H, Belghuith N, Ksentini MA, Keskes LA, Triki C, Hachicha M, Kamoun S, Kamoun H (2018) A novel TBX1 missense mutation in patients with syndromic congenital heart defects. Biochem Biophys Res Commun 499:563–569
Pu T, Liu Y, Xu R, Li F, Chen S, Sun K (2018) Identification of ZFPM2 mutations in sporadic conotruncal heart defect patients. Mol Genet Genomics 293:217–223
Xu YJ, Di RM, Qiao Q, Li XM, Huang RT, Xue S, Liu XY, Wang J, Yang YQ (2018) GATA6 loss-of-function mutation contributes to congenital bicuspid aortic valve. Gene 663:115–120
Laugwitz KL, Moretti A, Lam J, Gruber P, Chen Y, Woodard S, Lin LZ, Cai CL, Lu MM, Reth M, Platoshyn O, Yuan JX, Evans S, Chien KR (2005) Postnatal isl1 + cardioblasts enter fully differentiated cardiomyocyte lineages. Nature 433:647–653
Moretti A, Caron L, Nakano A, Lam JT, Bernshausen A, Chen Y, Qyang Y, Bu L, Sasaki M, Martin-Puig S, Sun Y, Evans SM, Laugwitz KL, Chien KR (2006) Multipotent embryonic isl1 + progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell 127:1151–1165
Bu L, Jiang X, Martin-Puig S, Caron L, Zhu S, Shao Y, Roberts DJ, Huang PL, Domian IJ, Chien KR (2009) Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature 460:113–117
Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, Evans S (2003) Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell 5:877–889
Stevens KN, Hakonarson H, Kim CE, Doevendans PA, Koeleman BP, Mital S, Raue J, Glessner JT, Coles JG, Moreno V, Granger A, Gruber SB, Gruber PJ (2010) Common variation in ISL1 confers genetic susceptibility for human congenital heart disease. PLoS ONE 5:e10855
Luo ZL, Sun H, Yang ZQ, Ma YH, Gu Y, He YQ, Wei D, Xia LB, Yang BH, Guo T (2014) Genetic variations of ISL1 associated with human congenital heart disease in Chinese Han people. Genet Mol Res 13:1329–1338
Sun YM, Wang J, Xu YJ, Wang XH, Yuan F, Liu H, Li RG, Zhang M, Li YJ, Shi HY, Zhao L, Qiu XB, Qu XK, Yang YQ (2018) ZBTB17 loss-of-function mutation contributes to familial dilated cardiomyopathy. Heart Vessels 33:722–732
Dodou E, Verzi MP, Anderson JP, Xu SM, Black BL (2004) Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development 131:3931–3942
Takeuchi JK, Mileikovskaia M, Koshiba-Takeuchi K, Heidt AB, Mori AD, Arruda EP, Gertsenstein M, Georges R, Davidson L, Mo R, Hui CC, Henkelman RM, Nemer M, Black BL, Nagy A, Bruneau BG (2005) Tbx20 dose-dependently regulates transcription factor networks required for mouse heart and motoneuron development. Development 132:2463–2474
Friedrich FW, Dilanian G, Khattar P, Juhr D, Gueneau L, Charron P, Fressart V, Vilquin JT, Isnard R, Gouya L, Richard P, Hammoudi N, Komajda M, Bonne G, Eschenhagen T, Dubourg O, Villard E, Carrier L (2013) A novel genetic variant in the transcription factor Islet-1 exerts gain of function on myocyte enhancer factor 2C promoter activity. Eur J Heart Fail 15:267–276
Zhang H, Wang WP, Guo T, Yang JC, Chen P, Ma KT, Guan YF, Zhou CY (2009) The LIM-homeodomain protein ISL1 activates insulin gene promoter directly through synergy with BETA2. J Mol Biol 392:566–577
Yuan F, Qiu ZH, Wang XH, Sun YM, Wang J, Li RG, Liu H, Zhang M, Shi HY, Zhao L, Jiang WF, Liu X, Qiu XB, Qu XK, Yang YQ (2018) MEF2C loss-of-function mutation associated with familial dilated cardiomyopathy. Clin Chem Lab Med 56:502–511
Gong Q, Zhou Z (2018) Nonsense-mediated mRNA decay of hERG mutations in long QT syndrome. Methods Mol Biol 1684:37–49
Inácio A, Silva AL, Pinto J, Ji X, Morgado A, Almeida F, Faustino P, Lavinha J, Liebhaber SA, Romão L (2004) Nonsense mutations in close proximity to the initiation codon fail to trigger full nonsense-mediated mRNA decay. J Biol Chem 279:32170–32180
Huang RT, Xue S, Xu YJ, Zhou M, Yang YQ (2014) Somatic GATA5 mutations in sporadic tetralogy of Fallot. Int J Mol Med 33:1227–1235
Guo DF, Li RG, Yuan F, Shi HY, Hou XM, Qu XK, Xu YJ, Zhang M, Liu X, Jiang JQ, Yang YQ, Qiu XB (2016) TBX5 loss-of-function mutation contributes to atrial fibrillation and atypical Holt-Oram syndrome. Mol Med Rep 13:4349–4356
Lu CX, Gong HR, Liu XY, Wang J, Zhao CM, Huang RT, Xue S, Yang YQ (2016) A novel HAND2 loss-of-function mutation responsible for tetralogy of Fallot. Int J Mol Med 37:445–451
Sun YM, Wang J, Qiu XB, Yuan F, Xu YJ, Li RG, Qu XK, Huang RT, Xue S, Yang YQ (2016) PITX2 loss-of-function mutation contributes to tetralogy of Fallot. Gene 577:258–264
Qiu XB, Qu XK, Li RG, Liu H, Xu YJ, Zhang M, Shi HY, Hou XM, Liu X, Yuan F, Sun YM, Wang J, Huang RT, Xue S, Yang YQ (2017) CASZ1 loss-of-function mutation contributes to familial dilated cardiomyopathy. Clin Chem Lab Med 55:1417–1425
Acknowledgments
We are sincerely thankful to the study participants for their dedication to the investigation.
Funding
This study was funded by the grants from the National Natural Science Foundation of China (81470372, 81400244, and 81370400), the Medicine Guided Program of Shanghai, China (17411971000), the Experimental Animal Program of Shanghai, China (17140902400), the Clinical Research Plan of SHDC, Shanghai, China (16CR3005A), the Project Foundation of Health and Family Planning Commission of Shanghai, China (M20170348), and the Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that no conflict of interest exists.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study or their legal guardians.
Rights and permissions
About this article

Cite this article
Ma, L., Wang, J., Li, L. et al. ISL1 loss-of-function mutation contributes to congenital heart defects. Heart Vessels 34, 658–668 (2019). https://doi.org/10.1007/s00380-018-1289-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00380-018-1289-z