
Abstract
Congenital heart defect (CHD) represents the most prevalent birth defect, and accounts for substantial morbidity and mortality in humans. Aggregating evidence demonstrates the genetic basis for CHD. However, CHD is a heterogeneous disease, and the genetic determinants underlying CHD in most patients remain unknown. In the present study, a cohort of 186 unrelated cases with CHD and 300 unrelated control individuals were recruited. The coding exons and flanking introns of the MEF2C gene, which encodes a transcription factor crucial for proper cardiovascular development, were sequenced in all study participants. The functional effect of an identified MEF2C mutation was characterized using a dual-luciferase reporter assay system. As a result, a novel heterozygous MEF2C mutation, p.R15C, was detected in an index patient with congenital double outlet right ventricle (DORV) as well as ventricular septal defect. Analysis of the proband’s pedigree showed that the mutation co-segregated with CHD with complete penetrance. The missense mutation, which changed the evolutionarily conserved amino acid, was absent in 300 control individuals. Functional deciphers revealed that the mutant MEF2C protein had a significantly decreased transcriptional activity. Furthermore, the mutation significantly reduced the synergistic activation between MEF2C and GATA4, another transcription factor linked to CHD. This study firstly associates MEF2C loss-of-function mutation with DORV in humans, which provides novel insight into the molecular pathogenesis of CHD, suggesting potential implications for genetic counseling and personalized treatment of CHD patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Congenital heart defect (CHD), a structural malformation caused by abnormal development of the heart or cardiothoracic major blood vessels, represents the most prevalent form of birth defect in humans, accounting for about one-third of all major developmental deformities [1, 2]. Each year there are approximately 1.35 million neonates who are born with CHD worldwide, with an annual incidence of roughly 1% in live births [1, 2]. As a cardiovascular developmental abnormality, CHD is usually classified into 25 distinct clinical types, of which 21 designate specific anatomic or hemodynamic lesions, including ventricular septal defect (VSD), atrial septal defect, patent ductus arteriosus, pulmonary stenosis, tetralogy of Fallot, double outlet right ventricle (DORV), transposition of the great arteries, aortic stenosis, truncus arteriosus, coarctation of the aorta, pulmonary atresia, and endocardial cushion defect [1]. Although minor cardiovascular defects can resolve spontaneously [1], major anomalies may require timely surgical treatment and can lead to poor quality of life [3, 4], reduced exercise capacity [5], delayed cerebral development [6, 7], brain injury [8], arterial thromboembolism [9], infective endocarditis [10, 11], pulmonary hypertension [12,13,14,15], congestive heart failure [16,17,18,19], arrhythmias [20,21,22,23,24], and sudden cardiac death [25,26,27,28,29]. As such, CHD is still the most frequent cause of infant deaths attributable to birth defects, with nearly 24% of infants who died of a birth defect having a heart malformation [1]. Although great advancement made in pediatric care during recent decades has allowed more than 90% of neonates with CHD to survive into adulthood, it results in an increasing number of adults living with CHD, and moreover, the morbidity and mortality in adult CHD patients are much higher compared with the general population [30,31,32]. Despite their significant clinical importance, the etiologies underlying CHD remain largely unclear.
It has been demonstrated that CHD is a complex multifactorial disorder with both environmental and genetic risk factors involved in the pathogenesis of CHD [33,34,35,36]. An expanding list of well-recognized environmental risk factors for CHD include maternal exposures to toxic chemicals, drugs, tobacco smoke, or ionizing radiation during the first trimester of pregnancy and maternal conditions such as viral infection, autoimmune, diabetes, and hypercholesterolemia as well as maternal old age and obesity [35, 36]. However, increasing evidence highlights the genetic determinants for familial CHD, which is predominantly transmitted in an autosomal dominant fashion in the family, though familial transmission of CHD also occurs in other inheritance patterns, encompassing autosomal recessive and X-linked modes [33,34,35,36]. Irrespective of chromosomal abnormalities such as trisomy of chromosome 21 and chromosome 22q11 deletion syndrome [34], an increasing number of mutations in over 60 genes, including those coding for cardiac core transcription factors, cardiac sarcomeric proteins, signaling molecules, and chromatin modifiers, have been associated with syndromic or non-syndromic CHD in humans [33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73]. Among these well-established CHD-causing genes, the overwhelming majority encode cardiac transcription factors, including the homeodomain-containing protein NKX2-5, zinc finger proteins GATA4, GATA5, and GATA6, basic helix–loop–helix transcription factors HAND1 and HAND2, and T-box transcription factors TBX1, TBX5, and TBX20 [48, 74]. Theses transcription factors display partially overlapping expression patterns and share cross-talk functions during cardiovascular morphogenesis, indicating that they comprise a key regulatory network essential for proper heart development [74]. Nevertheless, CHD is of remarkable genetic heterogeneity, and the genetic basis underpinning CHD in most cases remains poorly understood.
As a member of the myocyte enhancer factor-2 (MEF2) family of MADS (MCM1, agamous, deficiens, serum response factor)-box transcription factors that are expressed at high levels in various cells, MEF2C has been implicated in transcriptional regulation of all three muscle lineages, including cardiogenic precursor cells and differentiated cardiomyocytes during embryogenesis [75]. In the mouse, the Mef2c gene is required for normal cardiogenesis, and targeted deletion of the Mef2c gene in mice results in embryonic death due to loss of the right ventricle of the heart, failure of the heart to undergo rightward looping morphogenesis, and diminished expression of several key cardiac-specific genes [75]. Moreover, in mice inactivation of the Mef2c gene in the anterior second heart field, a late differentiating population of cardiac progenitors, leads to a spectrum of outflow tract alignment defects ranging from overriding aorta to DORV and dextro-transposition of the great arteries [76]. These observational results make it justifiable to scan MEF2C as a prime candidate gene for CHD in patients.
Materials and Methods
Ethical Statement
This research was performed in conformity with the ethical principles outlined in the Declaration of Helsinki. The research protocol was reviewed and approved by the local institutional ethical committee of Tongji Hospital, Tongji University, Shanghai, China [Approval No. LL(H)-09-07]. Written informed consent was obtained from the guardians of the CHD patients and the control subjects prior to commencement of the investigation.
Study Population
In all, 186 unrelated patients affected with CHD were enrolled from the Chinese Han population. Among them, there were 101 males and 85 females with a mean age of 5.3 ± 3.8 years, ranging from 0 to 16 years of age. The available close relatives of the index patient carrying an identified MEF2C mutation were also recruited. Phenotypic characteristics of the affected individuals and their family members were derived from detailed clinical records, and medical evaluations based on echocardiography, cardiac catheterization, and/or surgical findings. Probands with known chromosomal abnormalities, other recognized syndromes, or known maternal exposure to significant toxicants during the first trimester of pregnancy were excluded from the present study. A total of 300 healthy ethnically matched volunteers with no history of CHD were enlisted as controls, of whom there were 160 males and 140 females at an average age of 5.1 ± 3.5 years, ranging from 1 to 16 years of age. Cardiac phenotypes of Chinese control volunteers were determined mainly by echocardiography.
Genetic Analysis of MEF2C
Peripheral venous blood samples were drawn from all the study participants and genomic DNA was extracted from blood leukocytes with the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s manual. The referential genomic DNA sequence of the human MEF2C gene (Accession No. NC_000005.10) was derived from the Nucleotide database at the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/nuccore/NC_000005.10?from=88718241&to=88904105&report=genbank&strand=true). With the aid of the online program Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?ORGANISM=9606&INPUT_SEQUENCE=NC_000005.10&LINK_LOC=nuccore&PRIMER5_START=88718241&PRIMER3_END=88904105), the primer pairs to amplify the coding exons and flanking introns of MEF2C by polymerase chain reaction (PCR) were designed as shown in Table 1. The primers for amplification of the 5′-untranslated region (UTR) and 3′-UTR of MEF2C (transcript variant 1) by PCR were designed as shown in Table S1. Amplification of genomic DNA was carried out by PCR using HotStar Taq DNA Polymerase (Qiagen) on a Veriti Thermal Cycler (Applied Biosystems, Waltham, MA, USA) under recommended reagent concentrations and standard reaction conditions. Amplified DNA fragments were fractionated by electrophoresis on a 1.5% agarose gel and isolated with the QIAquick Gel Extraction Kit (Qiagen). Direct PCR sequencing of a purified amplicon was conducted with the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) on an ABI PRISM 3130XL DNA Analyzer (Applied Biosystems). For a detected MEF2C sequence variation, a second independent PCR-sequencing analysis was performed to verify it. The position of an exonic sequence variation was numbered according to the reference sequence of the MEF2C mRNA transcript variant 1 at the Nucleotide database (Accession No. NM_002397.4). In addition, such public databases for human sequence variations as the single nucleotide polymorphism (http://www.ncbi.nlm.nih.gov/SNP), human gene mutation (http://www.hgmd.org), 1000 Genomes (http://www.1000genomes.org/), and Exome Variant Server (http://evs.gs.washington.edu/EVS) databases were consulted to confirm the novelty of an identified MEF2C sequence variance.
Multiple Alignments of MEF2C Protein Sequences Across Species
The MEF2C protein of human was aligned with those of chimpanzee, monkey, dog, cattle, mouse, rat, fowl, zebrafish, fruit fly, mosquito, and frog using the online MUSCLE software (https://www.ncbi.nlm.nih.gov/homologene?cmd=Retrieve&dopt=MultipleAlignment&list_uids=31087).
Prediction of the Pathogenic Potential of a Novel MEF2C Variation
The causative potential of a novel MEF2C variation was predicted by MutationTaster (http://www.mutationtaster.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), PROVEAN (http://provean.jcvi.org/index.php), and SIFT (http://sift.jcvi.org/www/SIFT_enst_submit.html).
Plasmids and Site-Targeted Mutagenesis
Human heart cDNAs were prepared as described previously [42]. The wild-type full-length open read frame of the human MEF2C gene (transcript variant 1; Accession No. NM_002397.4) was amplified by PCR using the pfuUltra high-fidelity DNA polymerase (Stratagene, Santa Clara, CA, USA) and a pair of primers (forward primer: 5′-TGGGCTAGCAGAGAGAGAAGAAAAACGGG-3′; reverse primer: 5′-CCAGCGGCCGCACTAGTAAGTAATAATCTGA-3′). The amplicons were doubly digested by restriction enzymes NheI and NotI (TaKaRa, Dalian, Liaoning, China). The digested product with a length of 1493 bp was separated by 1.5% agarose gel electrophoresis, purified with the QIAquick Gel Extraction Kit (Qiagen), and then inserted into the NheI–NotI sites of the pcDNA3.1 vector (Invitrogen, Carlsbad, CA, USA) to generate a recombinant expression plasmid MEF2C-pcDNA3.1. The non-synonymous variant found in CHD patients in the coding region of MEF2C was introduced into the wild-type MEF2C-pcDNA3.1 plasmid by site-directed mutagenesis using the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene) with a complementary pair of primers according to the manufacturer’s instructions, and was verified by sequencing. The recombinant expression plasmid GATA4-pSSRa and the reporter plasmid ANF-luciferase (ANF-luc), which contains the 2600-bp 5′-untranslated region of the ANF gene and expresses firefly luciferase, were kind gifts provided by Dr. Ichiro Shiojima from Chiba University School of Medicine, Japan.
Luciferase Assays
HeLa cells were incubated in Dulbecco’s modified Eagle’s media supplemented with 10% fetal bovine serum as well as 100 µg/ml streptomycin and 100 U/ml penicillin at 37 °C and in an incubator with an atmosphere of 5% CO2. Cells were seeded in 12-well plates 24 h before being transfected with various plasmids using the Lipofectamine 2000® reagent (Invitrogen) according to the manufacturer’s protocol. Additionally, the pGL4.75 (Promega, Madison, WI, USA) vector expressing a renilla luciferase was co-transfected into the cells as an internal control for transfection efficiency. For transient transfection experiments, HeLa cells were transfected with 1.0 µg of wild-type MEF2C-pcDNA3.1, 1.0 µg of R15C-mutant MEF2C-pcDNA3.1, 0.5 µg of wild-type MEF2C-pcDNA3.1, or 0.5 µg of wild-type MEF2C-pcDNA3.1 together with 0.5 µg of R15C-mutant MEF2C-pcDNA3.1 was used, in combination with 1.0 µg of ANF-luc and 0.04 µg of pGL4.75 (Promega). In order to evaluate the ability of the mutant MEF2C to transcriptionally activate the ANF promoter in synergy with GATA4, the same amount (0.4 µg) of expression plasmid DNA (empty pcDNA3.1, wild-type MEF2C-pcDNA3.1, R15C-mutant MEF2C-pcDNA3.1 or GATA4-pSSRa) was used alone or together, in the presence of 1.0 µg of ANF-luc and 0.04 µg of pGL4.75. The empty plasmid pcDNA3.1 was used as a negative control. Cells were cultured at 37 °C and harvested 36 h after transfection. Luciferase activity of the lysates was measured using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. Firefly luciferase data were normalized to the transfection control (renilla luciferase readings). For each construct, a minimum of three independent experiments were each performed in triplicate, and the results were expressed as mean ± standard deviation (SD).
Statistical Analysis
Statistical analyses were performed using the SPSS software package for Windows, version 17.0 (SPSS Inc., Chicago, IL, USA). Continuous variables were expressed as mean ± SD. Categorical variables were given as a number and percentage. Comparison of continuous variables between two groups was made using Student’s unpaired t test; whereas categorical variables were compared with Pearson’s χ2 test or Fisher’s exact test, when appropriate. A two-sided p < 0.05 indicated significant difference.
Results
Clinical Features of the Study Participants
In the current study, 186 unrelated CHD patients were clinically evaluated in contrast to 300 unrelated healthy control individuals. The patients and controls were matched in ethnicity, gender, and age. All the cases had echocardiogram-documented CHD, of whom approximately 19% had a positive family history of CHD. The control subjects were healthy with a negative family history of CHD, and their echocardiograms showed normal cardiovascular images with no evidence of structural heart defects. The baseline clinical characteristics of the patients with CHD are summarized in Table 2.
Identification of a Novel MEF2C Mutation
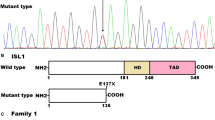
By sequence analysis of the MEF2C gene in 186 unrelated patients affected with CHD, a missense mutation was detected in a female patient who was 1 year old, with a mutational prevalence nearly 0.54%. Specifically, a substitution of thymine for cytosine at the first nucleotide of codon 15 (c.43C>T), predicting the transition of arginine at amino acid position 15 to cysteine (p.R15C), was discovered in a girl with DORV and VSD, who had a positive family history of CHD. The DNA sequencing electropherograms displaying the heterozygous MEF2C mutation of c.43C>T as well as its wild-type control sequence are shown in Fig. 1a. A schematic diagram of the MEF2C protein indicating the key structural domains and location of the mutation identified in this study is shown in Fig. 1b. The missense mutation was neither detected in the 300 control subjects nor reported in the single nucleotide polymorphism, human gene mutation, 1000 Genomes, or Exome Variant Server database (queried again in October 22 2017). Genetic screen of the mutation carrier’s family members available showed that the mutation was present in all the affected family members, but absent in unaffected family members. Analysis of the index patient’s pedigree revealed that the mutation co-segregated with CHD, which was transmitted in an autosomal dominant pattern in the family with complete penetrance. The pedigree structure of the proband’s family is shown in Fig. 1c. The phenotypic characteristics of the proband’s living affected family members are shown in Table 3.

Novel MEF2C mutation associated with congenital heart defects. a Sequence electropherograms showing the heterozygous MEF2C mutation as well as its wild-type control. The arrow points to the heterozygous nucleotides of C/T in the proband (mutant type) or the homozygous nucleotides of C/C in a control individual (wild type). The rectangle marks the nucleotides comprising a codon of MEF2C. b Schematic diagram depicting the structural domains of the MEF2C protein and the location of the mutation linked to congenital heart defects. The mutation identified in patients with congenital heart defects is noted above the structural domains. NH2 amino terminus; MADS, MCM1, agamous, deficiens, serum response factor; MEF2 myocyte enhancer factor 2; TAD transcriptional activation domain; NLS nuclear location signal; COOH carboxyl terminus. c Pedigree structure of the family with congenital heart defects. The family was designated as family 1. Family members are identified by generations and numbers. Square indicates male family member; circle, female member; closed symbol, affected member; open symbol, unaffected member; arrow, proband; “+,” carrier of the heterozygous missense mutation; “–,” non-carrier
MEF2C Sequence Variation Predicted to Be Conserved and Disease-Causing
Alignment of multiple MEF2C protein sequences from various species exhibited that the altered arginine residue at amino acid position 15 was completely conserved evolutionarily (Fig. 2). Additionally, the identified MEF2C sequence variation was predicted to be pathogenic with a p value of 1.000 by MutationTaster, probably damaging with a score of 0.999 (sensitivity: 0.09; specificity: 0.99) by PolyPhen-2, deleterious with a PROVEAN score of − 7.203 by PROVEAN, and damaging with a SIFT score of 0 and a median information content of 3.4 by SIFT.

Multiple alignments of MEF2C proteins from various species. Alignment of multiple MEF2C proteins across species displayed that the altered arginine at amino acid 15 was completely conserved evolutionarily
Diminished Transcriptional Activity of MEF2C Caused by the Mutation
Previous experiments have demonstrated that MEF2C transcriptionally activates the ANF promoter alone or in synergy with GATA4 in HeLa cells [77]. As shown in Fig. 3, the same amount (1.0 µg) of wild-type and R15C-mutant MEF2C-pcDNA3.1 plasmids transcriptionally activated the ANF promoter by ~ 16- and ~ 3 -folds, respectively. When 0.5 µg of wild-type MEF2C-pcDNA3.1 was used alone or together with 0.5 µg of R15C-mutant MEF2C-pcDNA3.1, the induced transcriptional activation of the ANF promoter was ~ 7 or ~ 8 folds. These data indicate that the R15C-mutant MEF2C has a significantly decreased transcriptional activity.

Diminished transcriptional activity of MEF2C resulted from the mutation. Activation of the ANF promoter-driven luciferase in HeLa cells by wild-type MEF2C or R15C-mutant MEF2C (R15C), alone or together, showed significantly decreased transcriptional activation by the mutant protein. Experiments were performed in triplicate, and means with standard deviations are shown. **t = 9.59826, p = 0.00066; *t = 4.76792, p = 0.00885, when compared with wild-type MEF2C (1.0 µg)
Reduced Synergistic Transcriptional Activity Between MEF2C Mutant and GATA4
As shown in Fig. 4, in the presence of 0.4 µg of wild-type GATA4, the same amount (0.4 µg) of wild-type and mutant MEF2C activated the ANF promoter by ~ 52- and ~ 17-folds, respectively, indicating that the MEF2C mutant has a significantly decreased synergistic transcriptional activity with GATA4.

Decreased synergistic transcriptional activity between R15C-mutant MEF2C and GATA4. In the presence of GATA4, activation of the ANF promoter-driven luciferase reporter in HeLa cells by wild-type MEF2C or R15C-mutant MEF2C (R15C) showed significantly reduced synergistic transcriptional activity caused by the mutation. Experiments were carried out in triplicate, and mean and standard deviations are shown. *t = 11.614, p = 0.00031, when compared with the wild-type counterpart
Discussion
In this study, a novel heterozygous mutation (c.43C>T or p.R15C) in the MEF2C gene was identified in a family with DORV and VSD. The missense mutation, which was absent in the 600 control chromosomes, co-segregated with CHD in the family with complete penetrance. Functional data showed that the R15C-mutant MEF2C protein had a significantly reduced transcriptional activity. Therefore, it is very likely that mutated MEF2C predisposes to DORV and VSD in this family.
In vertebrates, there are four members of the MEF2 family, including MEF2A, MEF2B, MEF2C, and MEF2D, of which MEF2C and MEF2B are firstly expressed in the cardiac mesoderm at approximately embryonic day 7.5; while MEF2D and MEF2A are expressed after birth for 24 h [78]. MEF2C proteins are expressed ubiquitously during embryonic period of eukaryote organisms to regulate tissue-specific gene expression in many types of cells, such as cardiac muscle, skeletal muscle, neural, chondroid, immune, and endothelial cells [78]. In humans, MEF2C maps on chromosome 5q14.3, encoding multiple isoforms of proteins, including isoform 1 with 473 amino acids. The human MEF2C protein possesses five functionally important structural domains, including MADS, MEF2, TAD1, TAD2, and NLS [78]. The highly conserved MADS domain at the N-terminus of MEF2C comprises 56 amino acids, and its main role is to mediate DNA binding, dimerization, and co-factor interactions. Adjacent to the MADS domain is the MEF2 domain which consists of 30 highly conserved amino acids, starting from amino acid 57 to amino acid 86, and is important in mediating dimerization and DNA binding. The transcriptional activation domains TAD1 and TAD2 are responsible for activating transcription, while the nuclear localization signal NLS domain at the C-terminus of MEF2C is required for nuclear translocation of the protein [78]. In the present study, the mutation identified in CHD patients was located at the MADS domain of MEF2C, and thus was anticipated to impair the transactivational function of MEF2C mainly by interfering with its binding to the promoters of target genes, including the ANF gene abundantly expressed in the embryonic hearts [77]. Functional assays showed that the R15C-mutant MEF2C protein had significantly diminished transcriptional activation of the ANF promoter alone or in synergy with GATA4. These findings imply that haploinsufficiency resulted from a MEF2C mutation is potentially an alternative pathological mechanism of CHD.
Notably, previous studies have related mutations in more than 60 genes, including those encoding cardiac transcription factors, to CHD in humans [33,34,35,36, 48]. In this study, as previously described, we have screened several other cardiac transcription factors in the index patient who harbored an identified MEF2C mutation, including NKX2-5 [66], NKX2-6 [63], GATA4 [46], GATA5 [56], GATA6 [71], HAND1 [62], HAND2 [51], TBX1 [79], TBX5 [80], TBX20 [41], PITX2 [59], CASZ1 [42], NR2F2 [54] and MESP1 [69], and found no pathogenic mutations. Nevertheless, we can’t exclude the possibility that other genes may also contribute to the pathogenesis of CHD.
Additionally, functional data provided are rather robust, but unfortunately with two caveats. Firstly, data were obtained in HeLa cells rather than in primary cultures of cardiomyocyte, and therefore its relevance to cardiac function was minimal. In cultured cardiomyocytes, the inducibility levels of target genes might be smaller but more relevant from the biological point of view. Secondly, it was hard to reconcile that impaired transactivation of ANF might be linked to CHD. These functional experiments were proof of concept only. In fact it is highly desirable to repeat similar transcriptional activation experiments using more relevant targets. Similarly, it would be interesting if competition assays with other MEF2 family members are provided given the fact that the identified mutation might alter dimerization.
It is interesting that MEF2C mutations have been found in patients with neurodevelopmental disorder, which is characteristic of intellectual disability with inability to speak, hypotonia, stereotypic movement, and epilepsy [81]. Besides, MEF2C mutations have also been involved in dilated cardiomyopathy [82] and other congenital heart defects, including patent ductus arteriosus, VSD, pulmonary atresia, and pulmonary stenosis [53, 83]. In the present research, a novel MEF2C mutation was identified in patients with DORV. Therefore, this research expands the phenotypic spectrum linked to MEF2C mutation.
In conclusion, this study firstly associates MEF2C loss-of-function mutation with DORV in humans, which provides novel insight into the molecular mechanism underpinning CHD, suggesting potential implications for genetic counseling and personalized management of the patients with MEF2C-related CHD.
References
Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American Heart Association Statistics Committee and Stroke Statistics Subcommittee (2017) Heart disease and stroke statistics—2017 update: a report from the American Heart Association. Circulation 135:e146–e603
Postma AV, Bezzina CR, Christoffels VM (2016) Genetics of congenital heart disease: the contribution of the noncoding regulatory genome. J Hum Genet 61:13–19
Ernst MM, Marino BS, Cassedy A, Piazza-Waggoner C, Franklin RC, Brown K, Wray J (2017) Biopsychosocial predictors of quality of life outcomes in pediatric congenital heart disease. Pediatr Cardiol. https://doi.org/10.1007/s00246-017-1730-6
Kahr PC, Radke RM, Orwat S, Baumgartner H, Diller GP (2015) Analysis of associations between congenital heart defect complexity and health-related quality of life using a meta-analytic strategy. Int J Cardiol 199:197–203
Gomes-Neto M, Saquetto MB, da Silva e Silva CM, Conceição CS, Carvalho VO (2016) Impact of exercise training in aerobic capacity and pulmonary function in children and adolescents after congenital heart disease surgery: a systematic review with meta-analysis. Pediatr Cardiol 37:217–224
Morton PD, Ishibashi N, Jonas RA (2017) Neurodevelopmental abnormalities and congenital heart disease: insights into altered brain maturation. Circ Res 120:960–977
Peyvandi S, De Santiago V, Chakkarapani E, Chau V, Campbell A, Poskitt KJ, Xu D, Barkovich AJ, Miller S, McQuillen P (2016) Association of prenatal diagnosis of critical congenital heart disease with postnatal brain development and the risk of brain injury. JAMA Pediatr 170:e154450
Marelli A, Miller SP, Marino BS, Jefferson AL, Newburger JW (2016) Brain in congenital heart disease across the lifespan: the cumulative burden of injury. Circulation 133:1951–1962
Jensen AS, Idorn L, Thomsen C, von der Recke P, Mortensen J, Sørensen KE, Thilén U, Nagy E, Kofoed KF, Ostrowski SR, Søndergaard L (2015) Prevalence of cerebral and pulmonary thrombosis in patients with cyanotic congenital heart disease. Heart 101:1540–1546
Diller GP, Baumgartner H (2017) Endocarditis in adults with congenital heart disease: new answers-new questions. Eur Heart J 38:2057–2059
Kuijpers JM, Koolbergen DR, Groenink M, Peels KC, Reichert CL, Post MC, Bosker HA, Wajon EM, Zwinderman AH, Mulder BJ, Bouma BJ (2017) Incidence, risk factors, and predictors of infective endocarditis in adult congenital heart disease: focus on the use of prosthetic material. Eur Heart J 38:2048–2056
Li G, Li Y, Tan XQ, Jia P, Zhao J, Liu D, Wang T, Liu B (2017) Plasma growth differentiation factor-15 is a potential biomarker for pediatric pulmonary arterial hypertension associated with congenital heart disease. Pediatr Cardiol 38:1620–1626
Li G, Tang L, Jia P, Zhao J, Liu D, Liu B (2016) Elevated plasma connective tissue growth factor levels in children with pulmonary arterial hypertension associated with congenital heart disease. Pediatr Cardiol 37:714–721
Müller J, Heck PB, Ewert P, Hager A (2017) Noninvasive screening for pulmonary hypertension by exercise testing in congenital heart disease. Ann Thorac Surg 103:1544–1549
van der Feen DE, Bartelds B, de Boer RA, Berger RM (2017) Pulmonary arterial hypertension in congenital heart disease: translational opportunities to study the reversibility of pulmonary vascular disease. Eur Heart J 38:2034–2041
Budts W, Roos-Hesselink J, Rädle-Hurst T, Eicken A, McDonagh TA, Lambrinou E, Crespo-Leiro MG, Walker F, Frogoudaki AA (2016) Treatment of heart failure in adult congenital heart disease: a position paper of the Working Group of Grown-Up Congenital Heart Disease and the Heart Failure Association of the European Society of Cardiology. Eur Heart J 37:1419–1427
Hinton RB, Ware SM (2017) Heart failure in pediatric patients with congenital heart disease. Circ Res 120:978–994
Nandi D, Rossano JW, Wang Y, Jerrell JM (2017) Risk factors for heart failure and Its costs among children with complex congenital heart disease in a medicaid cohort. Pediatr Cardiol 38:1672–1679
Stout KK, Broberg CS, Book WM, Cecchin F, Chen JM, Dimopoulos K, Everitt MD, Gatzoulis M, Harris L, Hsu DT, Kuvin JT, Law Y, Martin CM, Murphy AM, Ross HJ, Singh G, Spray TL, American Heart Association Council on Clinical Cardiology, Council on Functional Genomics and Translational Biology, and Council on Cardiovascular Radiology and Imaging (2016) Chronic heart failure in congenital heart disease: a scientific statement from the American Heart Association. Circulation 133:770–801
Holst KA, Said SM, Nelson TJ, Cannon BC, Dearani JA (2017) Current interventional and surgical management of congenital heart disease: specific focus on valvular disease and cardiac arrhythmias. Circ Res 120:1027–1044
Khairy P (2016) Ventricular arrhythmias and sudden cardiac death in adults with congenital heart disease. Heart 102:1703–1709
Loomba RS, Aggarwal S, Gupta N, Buelow M, Alla V, Arora RR, Anderson RH (2016) Arrhythmias in adult congenital patients with bodily isomerism. Pediatr Cardiol 37:330–337
Lüscher TF (2016) Frontiers in congenital heart disease: pulmonary hypertension, heart failure, and arrhythmias. Eur Heart J 37:1407–1409
McLeod CJ, Warnes C (2016) Recognition and management of arrhythmias in adult congenital heart disease. Curr Opin Cardiol 31:117–123
Diller GP, Baumgartner H (2016) Sudden cardiac death during exercise in patients with congenital heart disease: the exercise paradox and the challenge of appropriate counselling. Eur Heart J 37:627–629
Diller GP, Kempny A, Alonso-Gonzalez R, Swan L, Uebing A, Li W, Babu-Narayan S, Wort SJ, Dimopoulos K, Gatzoulis MA (2015) Survival prospects and circumstances of death in contemporary adult congenital heart disease patients under follow-up at a large tertiary center. Circulation 13:2118–2125
Engelings CC, Helm PC, Abdul-Khaliq H, Asfour B, Bauer UM, Baumgartner H, Kececioglu D, Körten MA, Diller GP, Tutarel O (2016) Cause of death in adults with congenital heart disease—an analysis of the German National Register for Congenital Heart Defects. Int J Cardiol 211:31–36
Jortveit J, Eskedal L, Hirth A, Fomina T, Døhlen G, Hagemo P, Tell GS, Birkeland S, Øyen N, Holmstrøm H (2016) Sudden unexpected death in children with congenital heart defects. Eur Heart J 37:621–626
Koyak Z, de Groot JR, Bouma BJ, Zwinderman AH, Silversides CK, Oechslin EN, Budts W, Van Gelder IC, Mulder BJ, Harris L (2017) Sudden cardiac death in adult congenital heart disease: can the unpredictable be foreseen? Europace 19:401–406
Bouma BJ, Mulder BJ (2017) Changing landscape of congenital heart disease. Circ Res 120:908–922
Mandalenakis Z, Rosengren A, Skoglund K, Lappas G, Eriksson P, Dellborg M (2017) Survivorship in children and young adults with congenital heart disease in Sweden. JAMA Int 177:224–230
Williams RG (2016) Late causes of death after congenital heart defects: a population-based study from Finland. J Am Coll Cardiol 68:499–501
Andersen TA, Troelsen Kde L, Larsen LA (2014) Of mice and men: molecular genetics of congenital heart disease. Cell Mol Life Sci 71:1327–1352
Blue GM, Kirk EP, Giannoulatou E, Sholler GF, Dunwoodie SL, Harvey RP, Winlaw DS (2017) Advances in the genetics of congenital heart disease: a clinician’s guide. J Am Coll Cardiol 69:859–870
Edwards JJ, Gelb BD (2016) Genetics of congenital heart disease. Curr Opin Cardiol 31:235–241
Fahed AC, Gelb BD, Seidman JG, Seidman CE (2013) Genetics of congenital heart disease: the glass half empty. Circ Res 112:707–720
Boyle L, Wamelink MM, Salomons GS, Roos B, Pop A, Dauber A, Hwa V, Andrew M, Douglas J, Feingold M, Kramer N, Saitta S, Retterer K, Cho MT, Begtrup A, Monaghan KG, Wynn J, Chung WK (2016) Mutations in TKT are the cause of a syndrome including short stature, developmental delay, and congenital heart defects. Am J Hum Genet 98:1235–1242
Cao Y, Wang J, Wei C, Hou Z, Li Y, Zou H, Meng M, Wang W, Jiang L (2016) Genetic variations of NKX2-5 in sporadic atrial septal defect and ventricular septal defect in Chinese Yunnan population. Gene 575:29–33
Chen J, Qi B, Zhao J, Liu W, Duan R, Zhang M (2016) A novel mutation of GATA4 (K300T) associated with familial atrial septal defect. Gene 575:473–477
Ellesøe SG, Johansen MM, Bjerre JV, Hjortdal VE, Brunak S, Larsen LA (2016) Familial atrial septal defect and sudden cardiac death: identification of a novel NKX2-5 mutation and a review of the literature. Congenit Heart Dis 11:283–290
Huang RT, Wang J, Xue S, Qiu XB, Shi HY, Li RG, Qu XK, Yang XX, Liu H, Li N, Li YJ, Xu YJ, Yang YQ (2017) TBX20 loss-of-function mutation responsible for familial tetralogy of Fallot or sporadic persistent truncus arteriosus. Int J Med Sci 14:323–332
Huang RT, Xue S, Wang J, Gu JY, Xu JH, Li YJ, Li N, Yang XX, Liu H, Zhang XD, Qu XK, Xu YJ, Qiu XB, Li RG, Yang YQ (2016) CASZ1 loss-of-function mutation associated with congenital heart disease. Gene 595:62–68
Li FF, Deng X, Zhou J, Yan P, Zhao EY, Liu SL (2016) Characterization of human bone morphogenetic protein gene variants for possible roles in congenital heart disease. Mol Med Rep 14:1459–1464
Li L, Wang J, Liu XY, Liu H, Shi HY, Yang XX, Li N, Li YJ, Huang RT, Xue S, Qiu XB, Yang YQ (2017) HAND1 loss-of-function mutation contributes to congenital double outlet right ventricle. Int J Mol Med 39:711–718
Li N, Subrahmanyan L, Smith E, Yu X, Zaidi S, Choi M, Mane S, Nelson-Williams C, Bahjati M, Kazemi M, Hashemi M, Fathzadeh M, Narayanan A, Tian L, Montazeri F, Mani M, Begleiter ML, Coon BG, Lynch HT, Olson EN, Zhao H, Ruland J, Lifton RP, Mani A (2016) Mutations in the histone modifier PRDM6 are associated with isolated nonsyndromic patent ductus arteriosus. Am J Hum Genet 98:1082–1091
Li RG, Xu YJ, Wang J, Liu XY, Yuan F, Huang RT, Xue S, Li L, Liu H, Li YJ, Qu XK, Shi HY, Zhang M, Qiu XB, Yang YQ (2017) GATA4 loss-of-function mutation and the congenitally bicuspid aortic valve. Am J Cardiol. https://doi.org/10.1016/j.amjcard.2017.11.012
Li X, Shi L, Xu M, Zheng X, Yu Y, Jin J (2017) RCAN1 mutation and functional characterization in children with sporadic congenital heart disease. Pediatr Cardiol. https://doi.org/10.1007/s00246-017-1746-y
Li YJ, Yang YQ (2017) An update on the molecular diagnosis of congenital heart disease: focus on loss-of-function mutations. Expert Rev Mol Diagn 17:393–401
Liu D, Liu QQ, Guan LH, Jiang X, Zhou DX, Beghetti M, Qu JM, Jing ZC (2016) BMPR2 mutation is a potential predisposing genetic risk factor for congenital heart disease associated pulmonary vascular disease. Int J Cardiol 211:132–136
Liu S, Su Z, Tan S, Ni B, Pan H, Liu B, Wang J, Xiao J, Chen Q (2017) Functional analyses of a novel CITED2 nonsynonymous mutation in Chinese Tibetan patients with congenital heart disease. Pediatr Cardiol 38:1226–1231
Lu CX, Gong HR, Liu XY, Wang J, Zhao CM, Huang RT, Xue S, Yang YQ (2016) A novel HAND2 loss-of-function mutation responsible for tetralogy of Fallot. Int J Mol Med 37:445–451
Priest JR, Osoegawa K, Mohammed N, Nanda V, Kundu R, Schultz K, Lammer EJ, Girirajan S, Scheetz T, Waggott D, Haddad F, Reddy S, Bernstein D, Burns T, Steimle JD, Yang XH, Moskowitz IP, Hurles M, Lifton RP, Nickerson D, Bamshad M, Eichler EE, Mital S, Sheffield V, Quertermous T, Gelb BD, Portman M, Ashley EA (2016) De novo and rare variants at multiple loci support the oligogenic origins of atrioventricular septal heart defects. PLoS Genet 12:e100596
Qiao XH, Wang F, Zhang XL, Huang RT, Xue S, Wang J, Qiu XB, Liu XY, Yang YQ (2017) MEF2C loss-of-function mutation contributes to congenital heart defects. Int J Med Sci 14:1143–1153
Qiao XH, Wang Q, Wang J, Liu XY, Xu YJ, Huang RT, Xue S, Li YJ, Zhang M, Qu XK, Li RG, Qiu XB, Yang YQ (2017) A novel NR2F2 loss-of-function mutation predisposes to congenital heart defect. Eur J Med Genet. https://doi.org/10.1016/j.ejmg.2017.12.003
Shanshen E, Rosenberg J, Van Bergen AH (2017) Identification of novel congenital heart disease candidate genes using chromosome microarray. Pediatr Cardiol. https://doi.org/10.1007/s00246-017-1741-3
Shi LM, Tao JW, Qiu XB, Wang J, Yuan F, Xu L, Liu H, Li RG, Xu YJ, Wang Q, Zheng HZ, Li X, Wang XZ, Zhang M, Qu XK, Yang YQ (2014) GATA5 loss-of-function mutations associated with congenital bicuspid aortic valve. Int J Mol Med 33:1219–1226
Sifrim A, Hitz MP, Wilsdon A, Breckpot J, Turki SH, Thienpont B, McRae J, Fitzgerald TW, Singh T, Swaminathan GJ, Prigmore E, Rajan D, Abdul-Khaliq H, Banka S, Bauer UM, Bentham J, Berger F, Bhattacharya S, Bu’Lock F, Canham N, Colgiu IG, Cosgrove C, Cox H, Daehnert I, Daly A, Danesh J, Fryer A, Gewillig M, Hobson E, Hoff K, Homfray T, INTERVAL Study, Kahlert AK, Ketley A, Kramer HH, Lachlan K, Lampe AK, Louw JJ, Manickara AK, Manase D, McCarthy KP, Metcalfe K, Moore C, Newbury-Ecob R, Omer SO, Ouwehand WH, Park SM, Parker MJ, Pickardt T, Pollard MO, Robert L, Roberts DJ, Sambrook J, Setchfield K, Stiller B, Thornborough C, Toka O, Watkins H, Williams D, Wright M, Mital S, Daubeney PE, Keavney B, Goodship J, UK10K Consortium, Abu-Sulaiman RM, Klaassen S, Wright CF, Firth HV, Barrett JC, Devriendt K, FitzPatrick DR, Brook JD, Deciphering Developmental Disorders Study, Hurles ME (2016) Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet 48:1060–1065
Sun YM, Wang J, Qiu XB, Yuan F, Li RG, Xu YJ, Qu XK, Shi HY, Hou XM, Huang RT, Xue S, Yang YQ (2016) A HAND2 loss-of-function mutation causes familial ventricular septal defect and pulmonary stenosis. G3 6:987–992
Sun YM, Wang J, Qiu XB, Yuan F, Xu YJ, Li RG, Qu XK, Huang RT, Xue S, Yang YQ (2016) PITX2 loss-of-function mutation contributes to tetralogy of Fallot. Gene 577:258–264
Tong YF (2016) Mutations of NKX2.5 and GATA4 genes in the development of congenital heart disease. Gene 588:86–94
Wang F, Wang H, Wang L, Zhou S, Chang M, Zhou J, Dou Y, Wang Y, Shi X (2016) Association between single nucleotide polymorphisms in NFATC1 signaling pathway genes and susceptibility to congenital heart disease in the Chinese population. Pediatr Cardiol 37:1548–1561
Wang J, Hu XQ, Guo YH, Gu JY, Xu JH, Li YJ, Li N, Yang XX, Yang YQ (2017) HAND1 loss-of-function mutation causes tetralogy of Fallot. Pediatr Cardiol 38:547–557
Wang J, Mao JH, Ding KK, Xu WJ, Liu XY, Qiu XB, Li RG, Qu XK, Xu YJ, Huang RT, Xue S, Yang YQ (2015) A novel NKX2.6 mutation associated with congenital ventricular septal defect. Pediatr Cardiol 36:646–656
Wang X, Chang WL, Chen CA, Rosenfeld JA, Al Shamsi A, Al-Gazali L, McGuire M, Mew NA, Arnold GL, Qu C, Ding Y, Muzny DM, Gibbs RA, Eng CM, Walkiewicz M, Xia F, Plon SE, Lupski JR, Schaaf CP, Yang Y (2017) Germline mutations in ABL1 cause an autosomal dominant syndrome characterized by congenital heart defects and skeletal malformations. Nat Genet 49:613–617
Werner P, Latney B, Deardorff MA, Goldmuntz E (2016) MESP1 mutations in patients with congenital heart defects. Hum Mutat 37:308–314
Xie X, Shi X, Xun X, Rao L (2016) Association of NKX2-5 genetic polymorphisms with the risk of congenital heart disease: a meta-analysis. Pediatr Cardiol 37:953–961
Xu YJ, Qiu XB, Yuan F, Shi HY, Xu L, Hou XM, Qu XK, Liu X, Huang RT, Xue S, Yang YQ, Li RG (2017) Prevalence and spectrum of NKX2.5 mutations in patients with congenital atrial septal defect and atrioventricular block. Mol Med Rep 15:2247–2254
Yoshida A, Morisaki H, Nakaji M, Kitano M, Kim KS, Sagawa K, Ishikawa S, Satokata I, Mitani Y, Kato H, Hamaoka K, Echigo S, Shiraishi I, Morisaki T (2016) Genetic mutation analysis in Japanese patients with non-syndromic congenital heart disease. J Hum Genet 61:157–162
Zhang M, Li FX, Liu XY, Huang RT, Xue S, Yang XX, Li YJ, Liu H, Shi HY, Pan X, Qiu XB, Yang YQ (2017) MESP1 loss of function mutation contributes to double outlet right ventricle. Mol Med Rep 16:2747–2754
Zhao CM, Sun B, Song HM, Wang J, Xu WJ, Jiang JF, Qiu XB, Yuan F, Xu JH, Yang YQ (2016) TBX20 loss-of-function mutation associated with familial dilated cardiomyopathy. Clin Chem Lab Med 54:325–332
Zheng GF, Wei D, Zhao H, Zhou N, Yang YQ, Liu XY (2012) A novel GATA6 mutation associated with congenital ventricular septal defect. Int J Mol Med 29:1065–1071
Zhou YM, Dai XY, Huang RT, Xue S, Xu YJ, Qiu XB, Yang YQ (2016) A novel TBX20 loss of function mutation contributes to adult onset dilated cardiomyopathy or congenital atrial septal defect. Mol Med Rep 14:3307–3314
Zhou YM, Dai XY, Qiu XB, Yuan F, Li RG, Xu YJ, Qu XK, Huang RT, Xue S, Yang YQ (2016) HAND1 loss-of-function mutation associated with familial dilated cardiomyopathy. Clin Chem Lab Med 54:1161–1167
McCulley DJ, Black BL (2012) Transcription factor pathways and congenital heart disease. Curr Top Dev Biol 100:253–277
Lin Q, Schwarz J, Bucana C, Olson EN (1997) Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science 276:1404–1407
Barnes RM, Harris IS, Jaehnig EJ, Sauls K, Sinha T, Rojas A, Schachterle W, McCulley DJ, Norris RA, Black BL (2016) MEF2C regulates outflow tract alignment and transcriptional control of Tdgf1. Development 143:774–779
Morin S, Charron F, Robitaille L, Nemer M (2000) GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J 19:2046–2055
Dong C, Yang XZ, Zhang CY, Liu YY, Zhou RB, Cheng QD, Yan EK, Yin DC (2017) Myocyte enhancer factor 2C and its directly-interacting proteins: a review. Prog Biophys Mol Biol 126:22–30
Pan Y, Wang ZG, Liu XY, Zhao H, Zhou N, Zheng GF, Qiu XB, Li RG, Yuan F, Shi HY, Hou XM, Yang YQ (2015) A novel TBX1 loss-of-function mutation associated with congenital heart disease. Pediatr Cardiol 36:1400–1410
Guo DF, Li RG, Yuan F, Shi HY, Hou XM, Qu XK, Xu YJ, Zhang M, Liu X, Jiang JQ, Yang YQ, Qiu XB (2016) TBX5 loss-of-function mutation contributes to atrial fibrillation and atypical Holt-Oram syndrome. Mol Med Rep 13:4349–4356
Rocha H, Sampaio M, Rocha R, Fernandes S, Leão M (2016) MEF2C haploinsufficiency syndrome: report of a new MEF2C mutation and review. Eur J Med Genet 59:478–482
Yuan F, Qiu ZH, Wang XH, Sun YM, Wang J, Li RG, Liu H, Zhang M, Shi HY, Zhao L, Jiang WF, Liu X, Qiu XB, Qu XK, Yang YQ (2017) MEF2C loss-of-function mutation associated with familial dilated cardiomyopathy. Clin Chem Lab Med. https://doi.org/10.1515/cclm-2017-0461
Kodo K, Nishizawa T, Furutani M, Arai S, Ishihara K, Oda M, Makino S, Fukuda K, Takahashi T, Matsuoka R, Nakanishi T, Yamagishi H (2012) Genetic analysis of essential cardiac transcription factors in 256 patients with non-syndromic congenital heart defects. Circ J 76:1703–1711
Acknowledgements
We sincerely thank the study subjects for their dedication to the research. This work was financially supported by grants from the National Natural Science Foundation of China (81470372) and the Natural Science Foundation of Shanghai, China (16ZR1432500).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interests.
Electronic Supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lu, CX., Wang, W., Wang, Q. et al. A Novel MEF2C Loss-of-Function Mutation Associated with Congenital Double Outlet Right Ventricle. Pediatr Cardiol 39, 794–804 (2018). https://doi.org/10.1007/s00246-018-1822-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-018-1822-y