
Abstract
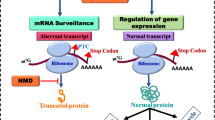
Long QT syndrome type 2 (LQT2) is caused by mutations in the human ether-à-go-go related gene (hERG), which encodes the Kv11.1 potassium channel in the heart. Over 30% of identified LQT2 mutations are nonsense or frameshift mutations that introduce premature termination codons (PTCs). Contrary to intuition, the predominant consequence of LQT2 nonsense and frameshift mutations is not the production of truncated proteins, but rather the degradation of mutant mRNA by nonsense-mediated mRNA decay (NMD), an RNA surveillance mechanism that selectively eliminates the mRNA transcripts that contain PTCs. In this chapter, we describe methods to study NMD of hERG nonsense and frameshift mutations in long QT syndrome.
Similar content being viewed by others

References
Warmke JW, Ganetzky B (1994) A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci U S A 91:3438–3442
Sanguinetti MC, Jiang C, Curran ME, Keating MT (1995) A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81:299–307
Trudeau MC, Warmke JW, Ganetzky B, Robertson GA (1995) HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 269:92–95
Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA, January CT (1998) Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J 74:230–241
Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT (1995) A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80:795–803
Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT (2000) Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102:1178–1185
Gong Q, Stump MR, Zhou Z (2014) Position of premature temination codons determines susceptibility of hERG mutations to nonsense mediated mRNA decay in long QT syndrome. Gene 529:190–197
Gong Q, Zhang L, Vincent GM, Horne BD, Zhou Z (2007) Nonsense mutations in hERG cause a decrease in mutant mRNA transcripts by nonsense-mediated mRNA decay in human long-QT syndrome. Circulation 116:17–24
Bhuiyan ZA, Momenah TS, Gong Q, Amin AS, Ghamdi SA, Carvalho JS, Homfray T, Mannens MM, Zhou Z, Wilde AA (2008) Recurrent intrauterine fetal loss due to near absence of HERG: clinical and functional characterization of a homozygous nonsense HERG Q1070X mutation. Heart Rhythm 5:553–561
Gong Q, Stump MR, Zhou Z (2011) Inhibition of nonsense-mediated mRNA decay by antisense morpholino oligonucleotides restores functional expression of hERG nonsense and frameshift mutations in long-QT syndrome. J Mol Cell Cardiol 50:223–229
Zarraga IG, Zhang L, Stump MR, Gong Q, Vincent GM, Zhou Z (2011) Nonsense-mediated mRNA decay caused by a frameshift mutation in a large kindred of type 2 long QT syndrome. Heart Rhythm 8:1200–1206
Stump MR, Gong Q, Packer JD, Zhou Z (2012) Early LQT2 nonsense mutation generates N-terminally truncated hERG channels with altered gating properties by the reinitiation of translation. J Mol Cell Cardiol 53:725–733
Stump MR, Gong Q, Zhou Z (2013) LQT2 nonsense mutations generate trafficking defective NH2-terminally truncated channels by the reinitiation of translation. Am J Physiol Heart Circ Physiol 305:H1397–H1404
Kuzmiak HA, Maquat LE (2006) Applying nonsense-mediated mRNA decay research to the clinic: progress and challenges. Trends Mol Med 12:306–316
Khajavi M, Inoue K, Lupski JR (2006) Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet 14:1074–1081
Li X, Xu J, Li M (1997) The human delta1261 mutation of the HERG potassium channel results in a truncated protein that contains a subunit interaction domain and decreases the channel expression. J Biol Chem 272:705–708
Paulussen A, Yang P, Pangalos M, Verhasselt P, Marrannes R, Verfaille C, Vandenberk I, Crabbe R, Konings F, Luyten W, Armstrong M (2000) Analysis of the human KCNH2 (HERG) gene: identification and characterization of a novel mutation Y667X associated with long QT syndrome and a non-pathological 9 bp insertion. Hum Mut 15:483
Kupershmidt S, Yang T, Chanthaphaychith S, Wang Z, Towbin JA, Roden DM (2002) Defective human Ether-a-go-go-related gene trafficking linked to an endoplasmic reticulum retention signal in the C terminus. J Biol Chem 277:27442–27448
Gong Q, Keeney DR, Robinson JC, Zhou Z (2004) Defective assembly and trafficking of mutant HERG channels with C-terminal truncations in long QT syndrome. J Mol Cell Cardiol 37:1225–1233
Teng S, Ma L, Dong Y, Lin C, Ye J, Bahring R, Vardanyan V, Yang Y, Lin Z, Pongs O, Hui R (2005) Clinical and electrophysiological characterization of a novel mutation R863X in HERG C-terminus associated with long QT syndrome. J Mol Med 82:189–196
Paulussen AD, Raes A, Jongbloed RJ, Gilissen RA, Wilde AA, Snyders DJ, Smeets HJ, Aerssens J (2005) HERG mutation predicts short QT based on channel kinetics but causes long QT by heterotetrameric trafficking deficiency. Cardiovasc Res 67:467–475
Choe CU, Schulze-Bahr E, Neu A, Xu J, Zhu ZI, Sauter K, Bahring R, Priori S, Guicheney P, Monnig G, Neapolitano C, Heidemann J, Clancy CE, Pongs O, Isbrandt D (2006) C-terminal HERG (LQT2) mutations disrupt IKr channel regulation through 14-3-3ε. Hum Mol Genet 15:2888–2902
Christé G, Thériault O, Chahine M, Millat G, Rodriguez-Lafrasse C, Rousson R, Deschênes I, Ficker E, Chevalier P (2008) A new C-terminal hERG mutation A915fs+47X associated with symptomatic LQT2 and auditory-trigger syncope. Heart Rhythm 5:1577–1586
Nof E, Cordeiro JM, Pérez GJ, Scornik FS, Calloe K, Love B, Burashnikov E, Caceres G, Gunsburg M, Antzelevitch C (2010) A common single nucleotide polymorphism can exacerbate long-QT type 2 syndrome leading to sudden infant death. Circ Cardiovasc Genet 3:199–206
Trudeau MC, Leung LM, Roti ER, Robertson GA (2011) hERG1a N-terminal eag domain-containing polypeptides regulate homomeric hERG1b and heteromeric hERG1a/hERG1b channels: a possible mechanism for long QT syndrome. J Gen Physiol 138:581–592
Uejima H, Lee MP, Cui H, Feinberg AP (2000) Hot-stop PCR: a simple and general assay for linear quantitation of allele ratios. Nat Genet 25:375–376
Kurreeman FA, Schonkeren JJ, Heijmans BT, Toes RE, Huizinga TW (2004) Transcription of the IL10 gene reveals allele-specific regulation at the mRNA level. Hum Mol Genet 13:1755–1762
Paillusson A, Hirschi N, Vallan C, Azzalin CM, Muhlemann O (2005) A GFP-based reporter system to monitor nonsense-mediated mRNA decay. Nucleic Acids Res 33:e54
Lykke-Andersen J, Shu MD, Steitz JA (2000) Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell 103:1121–1131
Gong Q, Stump MR, Dunn AR, Deng V, Zhou Z (2010) Alternative splicing and polyadenylation contribute to the generation of hERG1 C-terminal isoforms. J Biol Chem 285:32233–32241
Acknowledgments
This work was supported in part by NIH grant HL68854.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Science+Business Media LLC
About this protocol
Cite this protocol
Gong, Q., Zhou, Z. (2018). Nonsense-Mediated mRNA Decay of hERG Mutations in Long QT Syndrome. In: Shyng, SL., Valiyaveetil, F., Whorton, M. (eds) Potassium Channels. Methods in Molecular Biology, vol 1684. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-7362-0_4
Download citation
DOI: https://doi.org/10.1007/978-1-4939-7362-0_4
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-7361-3
Online ISBN: 978-1-4939-7362-0
eBook Packages: Springer Protocols