
Abstract
Congenital heart disease (CHD) is the most prevalent type of birth defect in humans and is the leading non-infectious cause of infant death worldwide. There is a growing body of evidence demonstrating that genetic defects play an important role in the pathogenesis of CHD. However, CHD is a genetically heterogeneous disease and the genetic basis underpinning CHD in an overwhelming majority of patients remains unclear. In this study, the coding exons and splice junction sites of the TBX1 gene, which encodes a T-box homeodomain transcription factor essential for proper cardiovascular morphogenesis, were sequenced in 230 unrelated children with CHD. The available family members of the index patient carrying an identified mutation and 200 unrelated ethnically matched healthy individuals used as controls were subsequently genotyped for TBX1. The functional effect of the TBX1 mutation was predicted by online program MutationTaster and characterized by using a dual-luciferase reporter assay system. As a result, a novel heterozygous TBX1 mutation, p.Q277X, was identified in an index patient with double outlet right ventricle (DORV) and ventricular septal defect (VSD). Genetic analysis of the proband’s available relatives showed that the mutation co-segregated with CHD transmitted in an autosomal dominant pattern with complete penetrance. The nonsense mutation, which was absent in 400 control chromosomes, altered the amino acid that was completely conserved evolutionarily across species and was predicted to be disease-causing by MutationTaster. Biochemical analysis revealed that Q277X-mutant TBX1 lost transcriptional activating function when compared with its wild-type counterpart. This study firstly associates TBX1 loss-of-function mutation with enhanced susceptibility to DORV and VSD in humans, which provides novel insight into the molecular mechanism underlying CHD and suggests potential implications for the development of new preventive and therapeutic strategies for CHD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Congenital heart disease (CHD), a structural defect that arises from abnormal development of the heart or major cardiothoracic blood vessels, is the most prevalent of all congenital malformations in humans, with an estimated prevalence of 1 % in live neonates worldwide [17, 21]. It is the most common cause of infant death caused by birth defects, with approximately 27 % of infants who died of a birth defect having a heart defect. [21]. Clinically, congenital cardiovascular deformities are usually categorized into more than 20 different types with specific anatomic or hemodynamic lesions, including ventricular septal defect (VSD), atrial septal defect, patent ductus arteriosus, endocardial cushion defect, double outlet right ventricle (DORV), tetralogy of Fallot, persistent truncus arteriosus, aortic coarctation, coronary artery anomalies, valvular pulmonary stenosis, pulmonary atresia, abnormal pulmonary venous return, interrupted aortic arch, transposition of the great arteries, and hypoplastic left heart syndrome, of which VSD is the most common form of CHD in children [5, 21, 36, 39, 69, 86]. Distinct kinds of CHDs may occur separately or concomitantly with each other, leading to degraded quality of life, poor exercise tolerance, brain injury, pulmonary hypertension, reduced lung function, impaired muscle function, subclinical hypothyroidism, autonomic nervous dysfunction, aortic aneurysm or dissection, infective endocarditis, thromboembolism, heart failure, arrhythmias, and even cardiac death [1, 3, 6, 8, 14–16, 19, 20, 29, 32–34, 40, 45–48, 51, 55, 57, 59, 60, 62, 66, 67, 87]. Although striking progress in medical treatment of newborns with CHD has contributed to an increasing number of adult survivors, unfortunately, the late morbidity and mortality are still markedly increased in the survivors [49, 68, 70, 71]. Therefore, CHD has conferred a vast economic burden on patients’ families and healthcare systems, and the socioeconomic burden is anticipated to increase in the future as the CHD adults accrue [72]. Despite the high prevalence and important clinical significance, in an overwhelming majority of cases, the etiologies responsible for CHD remain unknown.
Cardiac morphogenesis from a straight tube to a four-chambered heart experiences a complex dynamic biological process that mandates a precise spatial and temporal cooperation of cardiac cell commitment, differentiation, proliferation, and migration, and both environmental and genetic pathogenic factors may disarrange this process of cardiogenesis, resulting in various CHDs [4, 13, 24, 37, 53, 54, 65, 74]. Recently, there is compelling evidence that demonstrates the genetic origin of CHD, and a growing number of mutations in more than 60 genes have been shown to cause CHD [2, 4, 9, 12, 18, 22, 27, 28, 31, 35, 41, 42, 56, 58, 61, 63, 75–78, 82–85]. Nevertheless, these causative genes can only explain the CHDs in a small proportion of patients and in most patients the genetic basis underpinning CHD is still to be revealed.
Chromosome 22q11.2 deletion syndrome (22q11DS), which is caused by a heterozygous multi-gene deletion, is a relatively common genetic disorder, affecting 1 in 4000 live births. CHDs are a prominent part of the 22q11DS phenotype, with an incidence of about 80 % in infants with 22q11DS [44]. Additionally, 22q11DS is found in a small percentage of patients with double outlet right ventricle. The TBX1 gene, a member of the T-box gene family of DNA-binding transcription factors, is mapped to the 22q11.2 and has been identified to be associated with the cardiac phenotype of 22q11.2 DS, including tetralogy of Fallot, truncus arteriosus, and interrupted aortic arch [30, 38, 43, 44]. Interestingly, mutations of the TBX1 gene have been found in some patients featuring 22q11DS who are otherwise devoid of the 22q11.2 deletion [81] and also found in non-syndromic CHD patients [26, 80]. These data strongly suggest TBX1 as an important candidate gene for human CHD.
Materials and Methods
Ethics
This study was performed in conformity to the ethical principles of the revised Declaration of Helsinki (Somerset West, Republic of South Africa, 1996). The study protocol was reviewed and approved by the local institutional ethics committee of Tongji Hospital, Tongji University (the ethical approval number for cases and controls: LL(H)-09-07; the date for the approval: July 27, 2009), and written informed consent was obtained from the parents of each patient and control prior to study.
Study Participants
A cohort of 230 unrelated children suffered from CHD was enrolled. The available family members of the proband carrying an identified TBX1 mutation were also included. All patients underwent a comprehensive clinical evaluation, including individual and familial histories, medical records, complete physical examination, 12-lead electrocardiogram, and two-dimensional transthoracic echocardiography with color flow Doppler. Cardiac catheterization, angiography, chest X-ray, and cardiac magnetic resonance imaging were performed only if there was a strong clinical indication. Medical records were also reviewed in the case of deceased or unavailable relatives. CHD was confirmed by imaging and/or direct view during cardiac surgery. A total of 200 non-CHD individuals from the same geographic area, who were matched to the CHD patients in ethnicity and gender, were recruited as the controls. After obtaining informed written parental consent, approximately 0.5–2 ml of peripheral venous blood sample was taken from each study participant, and the genomic DNA was extracted from peripheral venous blood leukocytes using a Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions.
Sequencing of TBX1
The coding exons and flanking introns of the TBX1 gene (including isoforms A, B and C) were sequenced in 230 unrelated CHD patients. The available relatives of the index patient carrying an identified TBX1 mutation and 200 unrelated control individuals were subsequently genotyped for TBX1. The referential genomic DNA sequence of TBX1 was derived from nucleotide (Accession No. NC_000022.11), a gene sequence database at the National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov/nucleotide/). With the aid of the online Primer-BLAST program (http://www.ncbi.nlm.nih.gov/tools/primer-blast/), the primer pairs used to amplify the coding regions and splice junction sites of TBX1 by polymerase chain reaction (PCR) were designed as shown in Table 1. The PCR was conducted using HotStar Taq DNA polymerase (Qiagen GmbH, Hilden, Germany) on a Veriti Thermal Cycler (Applied Biosystems, Foster, CA, USA), with standard conditions and concentrations of reagents. Both strands of each PCR product were sequenced with a BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) under an ABI PRISM 3130 XL DNA Analyzer (Applied Biosystems). The DNA sequences were analyzed with the DNA Sequencing Analysis Software v5.1 (Applied Biosystems). Additionally, an identified sequence variation was queried in the single-nucleotide polymorphism (SNP) database at NCBI (http://www.ncbi.nlm.nih.gov/), the human gene mutation (HGM) database (http://www.hgmd.org/), and the 1000 Genome Project (1000 GP) database (http://www.1000genomes.org/) to confirm its novelty.
Multiple Alignments of TBX1 Amino Acid Sequences Among Various Species
Conservation of the amino acid altered by the identified mutation was estimated by aligning human TBX1 to chimpanzee, monkey, dog, mouse, rat, zebrafish, and frog TBX1 using the HomoloGene and Show Multiple Alignment links on the NCBI’s website (http://www.ncbi.nlm.nih.gov/homologene).
Prediction of the Pathogenic Potential of a Novel TBX1 Sequence Variation
The causative potential of a novel TBX1 sequence variation was predicted by MutationTaster (an online program at http://www.mutationtaster.org), which automatically gave a probability for the variation to be either a disease-causing mutation or a benign polymorphism. Of note, here the p value is the probability of the correct prediction, i.e., a p value close to one indicates high accuracy of the prediction.
Plasmids and Site-Directed Mutagenesis
The recombinant expression plasmid TBX1-pcDNA3.1, which contains the full-length cDNA of TBX1 isoform C, was constructed as described previously [80]. For generation of the 4 × T-pGL4.25 luciferase reporter vector (4 × T-luc), four conserved T-half sites “ATTTCACACCT” were oriented head to tail, similar to those reported by Sinha et al. [64], synthesised and subcloned into the KpnI-HindIII sites in the pGL4.25 [luc2CP/minP] plasmid (Promega). The mutant TBX1 expression vector was generated by using a QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA) with a complementary pair of primers and with the wild-type TBX1-pcDNA3.1 used as the template. The mutant TBX1 was sequenced to confirm the desired mutation and to exclude any other sequence variations.
Cell Transfection and Luciferase Assays
The transient cell transfection was performed with Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. COS-7 cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 10 % fetal calf serum (Invitrogen) and seeded in 12-well plates (2 × 105) before transfection. Twenty-four hours after plating, the COS-7 cells at about 75 % confluence were co-transfected with 0.4 μg of wild-type or mutant TBX1-pcDNA3.1, 1.0 μg of 4 × T-luc, and 0.04 μg of pGL4.75 (hRluc/CMV, Promega), a Renilla luciferase reporter plasmid used as an internal control. For co-transfection experiments, 0.2 μg of wild-type TBX1-pcDNA3.1 together with 0.2 μg of mutant TBX1-pcDNA3.1 or 0.2 μg of empty pcDNA3.1 vector was used in the presence of 1.0 μg of 4 × T-luc and 0.04 μg of pGL4.75. The cells were harvested 48 h after transfection. The Firefly and Renilla luciferase activities were measured with the Dual-Glo luciferase assay system (Promega). The activity of the Firefly luciferase was normalized to that of the Renilla luciferase. At least three independent experiments were performed in triplicate for wild-type and mutant TBX1.
Statistical Analysis
Continuous variables are expressed as means ± standard deviations (SD). Student’s unpaired t test was used to compare the continuous variables between two groups. Comparison of the categorical variables between two groups was made by using Pearson’s χ 2 test or Fisher’s exact test when appropriate. A two-tailed p < 0.05 was considered to indicate statistical difference.
Results
Baseline Clinical Characteristics of the Study Population
A cohort of 230 unrelated patients with CHD was clinically investigated in contrast to a total of 200 unrelated non-CHD control individuals (102 males and 98 females, with no family history of CHD in the control individuals). All the patients had confirmed CHD, while the control individuals had no evidence of structural cardiac abnormalities. None of the study participants had established environmental risk factors for CHD, such as maternal illness and drug use in the first trimester of pregnancy, parental smoking, and long-term exposure to chemical toxicants and ionizing radiation. There is no difference in either gender or ethnicity between patient and control groups. The baseline clinical characteristics of the study population are summarized in Table 2.
Identification of a Novel TBX1 Mutation
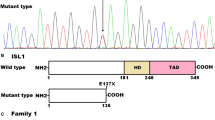
By sequence analysis of the coding exons and exon–intron boundaries of TBX1, a heterozygous sequence variation was identified in 1 of 230 unrelated CHD patients, with a mutational prevalence of roughly 0.43 %. Specifically, a substitution of thymine for cytosine in the first nucleotide of codon 277 (c.829C > T), predicting the transition of glutamine into a premature stop codon at amino acid position 277 (p.Q277X), was identified in an index patient with DORV and a large subpulmonary VSD, who underwent surgical treatment 1 week after birth. The sequence electropherograms showing the identified heterozygous TBX1 variation in contrast to its corresponding control sequence are shown in Fig. 1a. The schematic diagrams of the wild-type TBX1C and mutant TBX1 proteins showing the structural domains and location of the mutation detected in this study are presented in Fig. 1b. The variation was neither observed in 400 control chromosomes nor found in the SNP, HGM, and 1000 GP databases, which were consulted again on November 22, 2014, indicating a novel mutation.

A novel TBX1 mutation associated with congenital heart disease. a Sequence electropherograms showing the TBX1 mutation in contrast to its corresponding control. The arrow indicates the heterozygous nucleotides of C/T in the proband from family 1 (mutant) or the homozygous nucleotides of C/C in the corresponding control individual (wild type). The rectangle means the nucleotides constituting a codon of TBX1. b Schematic diagrams of the wild-type human TBX1 isoform C and mutant human TBX1 protein structures. The mutation associated with congenital heart disease is predicted to produce a truncated protein with only 276 amino acids at the amino-terminus left. NH2, amino-terminus; NLS, nuclear location signal; TAD, transcriptional activation domain; and COOH, carboxyl-terminus. c Pedigree structure of the family with congenital heart disease. Family is designated as family 1. Family members are identified by generations and numbers. Square indicates male family member; circle, female member; closed symbol, affected member; open symbol, unaffected member; symbol with a slash, the deceased member; arrow, proband; ‘‘+’’, carrier of the heterozygous mutation; and ‘‘−’’, non-carrier
Genetic analysis of the proband’s families displayed that the nonsense mutation was present in all affected family members alive, but absent in unaffected family members examined. Analysis of the pedigree demonstrated that the mutation co-segregated with subpulmonary VSD transmitted in an autosomal dominant pattern and with complete penetrance. There are no other anomalies consistent with 22q11 deletion syndrome, and there is no history of speech delay or learning disability in the affected family members. The pedigree structure of the family is shown in Fig. 1c. The phenotypic characteristics and status of TBX1 mutation of the affected family members are listed in Table 3.
Multiple Alignments of TBX1 Protein Sequences Among Various Species
As shown in Fig. 2, a cross-species alignment of multiple TBX1 protein sequences showed that the altered glutamine at amino acid position 277 of human TBX1 was completely conserved evolutionarily, implying that the amino acid is functionally important.

Alignment of multiple TBX1 protein sequences across various species. The altered glutamine at amino acid position 227 of human TBX1 protein is completely conserved evolutionarily among species
Causative Potential of the Identified TBX1 Sequence Variation
The TBX1 sequence variation of c.829C > T was predicted to be disease-causing, with a p value of 1.0000, supporting that Q277X-mutant TBX1 contributes to the occurrence of CHD in these mutation carriers.
Functional Assay of the Q277X-mutant TBX1
As shown in Fig. 3, the Q277X-mutant TBX1 had no transcriptional activity compared with its wild-type counterpart (t = 4.8028, p = 0.0086). When wild-type TBX1 was co-expressed with the same amount of Q277X-mutant TBX1, the induced transcriptional activation was significantly reduced compared with the wild-type TBX1 (t = 4.2947, p = 0.0127).

Transcriptional activation defect of TBX1 resulted from Q277X mutation. Transcriptional activation of the 4 × T-luciferase reporter in cultured COS-7 cells by wild-type TBX1 (WT) or mutant (Q277X), alone or together, showed significantly diminished transcriptional activity by the mutant protein. The activity of the Firefly luciferase was normalized for transfection efficiency to that of Renilla luciferase. The results are shown as the mean and standard deviations of three independent experiments performed in triplicate. ** and * indicate p < 0.01 and p < 0.05, respectively, when compared with wild-type TBX1 (0.4 μg)
Discussion
In the current study, a novel heterozygous mutation in TBX1, p.Q277X, was identified in a family with CHD. The mutant allele was present in all affected family members available but absent in unaffected relatives examined and 400 referral chromosomes from an ethnically matched control population. A cross-species alignment of multiple TBX1 protein sequences showed that the altered amino acid was completely conserved evolutionarily. The p.Q277X variation was predicted to be a disease-causing mutation by MutationTaster, and the functional analysis revealed that the mutant TBX1 had no transcriptional activity. Hence, it is very likely that mutated TBX1 predisposes these mutation carriers to CHD. To the best of our knowledge, this is the first report on the association of TBX1 loss-of-function mutation with enhanced susceptibility to DORV and VSD in humans.
In the developing mammalian heart, 6 of the 17 family members, including TBX1, TBX18, and TBX20 of the TBX1 subfamily, and TBX2, TBX3, and TBX5 of the TBX2 subfamily, are expressed and required in a combinatorial fashion in different progenitor pools as well as in different compartments [25]. The human TBX1 gene is located on chromosome 22q11.21, the center of the 22q11DS chromosomal region, which acts in the pharyngeal mesoderm to maintain proliferation of mesenchymal precursor cells for formation of a myocardialized and septated outflow tract, playing a key role in the elongation of the cardiac tube at the anterior pole [10, 25]. To date, three alternatively spliced transcripts, TBX1A (NM_080646.1), TBX1B (NM_005992.1), and TBX1C (NM_080647.1), have been found in humans that differed in their terminal exons. However, analysis of the gene expression levels in human tissues and comparison of the human and mouse genomic sequences showed that TBX1C is the major transcript with the nuclear location signal and the transcriptional activation domain and is highly homologous to mouse Tbx1 [23]. Besides, the apparently pathologic TBX1 mutations identified to date reside on exons 3–8 shared by isoforms A, B, and C or on exon 9C specific to isoform C, with no mutation on exons 9A and 9B specific to isoforms A and B. This is consistent with TBX1C having the primary biological function [52]. Three isoforms of TBX1 proteins share an evolutionarily conserved T-box homeodomain that recognizes and binds to a consensus DNA motif, ATTTCACACCT. The homeodomain is located at amino acid positions 109–302 and is predominantly involved in target DNA binding as well as interaction with other transcription factors [25]. The TBX1 mutation of p.Q277X identified in this study is predicted to generate a truncated protein with amino-terminus along with partial T-box homeodomain left (Fig. 1b) and thus may be anticipated to disable TBX1 by interfering with its binding to target DNA, nuclear distribution, and transcriptional activation.
In order to ascertain the functional consequence of the p.Q277X mutation in TBX1, the major transcript TBX1C was chosen as a representative of TBX1, and introduction of p.Q277X mutation into TBX1C abolished the transcriptional activation of TBX1C. These functional data suggest that haploinsufficiency or dominant-negative effect caused by TBX1 mutation is potentially an alternative pathological mechanism of CHD. Additionally, as the mutation is predicted to lead to a nonsense mutation, it is likely that the mutant mRNA undergoes nonsense-mediated decay, and in that case, it is likely that the mutation results in haploinsufficiency and does not have a dominant-negative effect [50]. However, at present, we cannot assay the mutant protein expression in Q277X-mutant patients due to the inaccessibility to cardiac tissue samples from these patients.
Previous investigations have verified that TBX1 can form complexes with such transcriptionally cooperative partners as NKX2.5 and SRF to synergistically mediate multiple important genes that are expressed in the heart during embryogenesis, including PITX2, FOXA2, FGF8, and FGF10 [25], and loss-of-function mutations in several genes, such as PITX2 and NKX2.5, have been causally linked to CHD including DORV and VSD [4, 11, 73, 76, 83, 84]. Therefore, genetically defective TBX1 may increase the vulnerability to CHD by reducing the expression of such genes essential for cardiovascular genesis.
Association of functionally compromised Tbx1 with increased susceptibility to CHD has been established in experimental animals. In mice, Tbx1 was highly expressed in the mesoderm and endoderm of the pharyngeal arches, and in the outflow tract. Genetic lineage analysis revealed that Tbx1-positive cells of the pharyngeal mesoderm contributed extensively to the outflow tract myocardium, endocardium, and mesenchymal cushions, indicating that Tbx1 plays a key role in an anterior subdomain of the second heart field [25]. Although mice heterozygous for deletion of Tbx1 presented mild anomalies, homozygous Tbx1-null mice died at birth with severe defects in the derivatives of the pharyngeal apparatus, of which cardiac defects included persistent truncus arteriosus, VSD, and mispatterning of the coronaries. Conditional knockout of Tbx1 from the pharyngeal endoderm and mesoderm, respectively, led to a spectrum of cardiovascular defects resembling those of the Tbx1-null mutants. Furthermore, mesodermal re-expression of Tbx1 in a null background corrected most of these defects, highlighting the pivotal role of Tbx1 for proper cardiovascular development [79]. In addition, Tbx1 function in the pharyngeal mesoderm was also required for survival, differentiation, and migration of the neural crest, suggesting a vital role in the development of pharyngeal arch artery [7]. Taken together, these findings support that TBX1 plays a crucial role in human cardiovascular development.
In conclusion, this study firstly associates TBX1 loss-of-function mutation with DORV and VSD, which expands the TBX1 mutation spectrum linked to CHD and provides novel insight into the molecular pathogenesis of CHD, implying the potential implications for genetic counseling and development of new preventive strategies for CHD.
References
Agha H, El Heinady F, El Falaky M, Sobih A (2014) Pulmonary functions before and after pediatric cardiac surgery. Pediatr Cardiol 35:542–549
Al Turki S, Manickaraj AK, Mercer CL, Gerety SS, Hitz MP, Lindsay S, D’Alessandro LC, Swaminathan GJ, Bentham J, Arndt AK, Low J, Breckpot J, Gewillig M, Thienpont B, Abdul-Khaliq H, Harnack C, Hoff K, Kramer HH, Schubert S, Siebert R, Toka O, Cosgrove C, Watkins H, Lucassen AM, O’Kelly IM, Salmon AP, Bu’lock FA, Granados-Riveron J, Setchfield K, Thornborough C, Brook JD, Mulder B, Klaassen S, Bhattacharya S, Devriendt K, Fitzpatrick DF, UK10 K Consortium, Wilson DI, Mital S, Hurles ME (2014) Rare variants in NR2F2 cause congenital heart defects in humans. Am J Hum Genet 94:574–585
Alonso-Gonzalez R, Borgia F, Diller GP, Inuzuka R, Kempny A, Martinez-Naharro A, Tutarel O, Marino P, Wustmann K, Charalambides M, Silva M, Swan L, Dimopoulos K, Gatzoulis MA (2013) Abnormal lung function in adults with congenital heart disease: prevalence, relation to cardiac anatomy, and association with survival. Circulation 127:882–890
Andersen TA, Troelsen Kde L, Larsen LA (2014) Of mice and men: molecular genetics of congenital heart disease. Cell Mol Life Sci 71:1327–1352
Babaoğlu K, Altun G, Binnetoğlu K, Dönmez M, Kayabey Ö, Anık Y (2013) Crossed pulmonary arteries: a report on 20 cases with an emphasis on the clinical features and the genetic and cardiac abnormalities. Pediatr Cardiol 34:1785–1790
Bang JS, Jo S, Kim GB, Kwon BS, Bae EJ, Noh CI, Choi JY (2013) The mental health and quality of life of adult patients with congenital heart disease. Int J Cardiol 170:49–53
Brown CB, Wenning JM, Lu MM, Epstein DJ, Meyers EN, Epstein JA (2004) Cre-mediated excision of Fgf8 in the Tbx1 expression domain reveals a critical role for Fgf8 in cardiovascular development in the mouse. Dev Biol 267:190–202
Cevik A, Kula S, Olgunturk R, Tunaoglu FS, Oguz AD, Saylan B, Cilsal E, Sanli C (2013) Assessment of pulmonary arterial hypertension and vascular resistance by measurements of the pulmonary arterial flow velocity curve in the absence of a measurable tricuspid regurgitant velocity in childhood congenital heart disease. Pediatr Cardiol 34:646–655
Chang SW, Mislankar M, Misra C, Huang N, Dajusta DG, Harrison SM, McBride KL, Baker LA, Garg V (2013) Genetic abnormalities in FOXP1 are associated with congenital heart defects. Hum Mutat 34:1226–1230
Chieffo C, Garvey N, Gong W, Roe B, Zhang G, Silver L, Emanuel BS, Budarf ML (1997) Isolation and characterization of a gene from the DiGeorge chromosomal region homologous to the mouse Tbx1 gene. Genomics 43:267–277
Costa MW, Guo G, Wolstein O, Vale M, Castro ML, Wang L, Otway R, Riek P, Cochrane N, Furtado M, Semsarian C, Weintraub RG, Yeoh T, Hayward C, Keogh A, Macdonald P, Feneley M, Graham RM, Seidman JG, Seidman CE, Rosenthal N, Fatkin D, Harvey RP (2013) Functional characterization of a novel mutation in NKX2-5 associated with congenital heart disease and adult-onset cardiomyopathy. Circ Cardiovasc Genet 6:238–247
Cowan J, Tariq M, Ware SM (2014) Genetic and functional analyses of ZIC3 variants in congenital heart disease. Hum Mutat 35:66–75
Cresci M, Foffa I, Ait-Ali L, Pulignani S, Kemeny A, Gianicolo EA, Andreassi MG (2013) Maternal environmental exposure, infant GSTP1 polymorphism, and risk of isolated congenital heart disease. Pediatr Cardiol 34:281–285
Demir M (2013) The relationship between atrial septal aneurysm and autonomic dysfunction. Exp Clin Cardiol 18:104–106
Dimitropoulos A, McQuillen PS, Sethi V, Moosa A, Chau V, Xu D, Brant R, Azakie A, Campbell A, Barkovich AJ, Poskitt KJ, Miller SP (2013) Brain injury and development in newborns with critical congenital heart disease. Neurology 81:241–248
Dimopoulos K, Wort SJ, Gatzoulis MA (2014) Pulmonary hypertension related to congenital heart disease: a call for action. Eur Heart J 35:691–700
Egbe A, Uppu S, Stroustrup A, Lee S, Ho D, Srivastava S (2014) Incidences and sociodemographics of specific congenital heart diseases in the United States of America: an evaluation of hospital discharge diagnoses. Pediatr Cardiol 35:975–982
Fahed AC, Gelb BD, Seidman JG, Seidman CE (2013) Genetics of congenital heart disease: the glass half empty. Circ Res 112:707–720
Garcia Guerra G, Joffe AR, Robertson CM, Atallah J, Alton G, Sauve RS, Dinu IA, Ross DB, Rebeyka IM, Western Canadian Complex Pediatric Therapies Follow-up Group (2014) Health-related quality of life experienced by children with chromosomal abnormalities and congenital heart defects. Pediatr Cardiol 35:536–541
Gatzoulis MA, Beghetti M, Landzberg MJ, Galiè N (2014) Pulmonary arterial hypertension associated with congenital heart disease: recent advances and future directions. Int J Cardiol 177:340–347
Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER 3rd, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB, American Heart Association Statistics Committee and Stroke Statistics Subcommittee (2014) Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation 129:e28–e292
Goldmuntz E, Clark BJ, Mitchell LE, Jawad AF, Cuneo BF, Reed L, McDonald-McGinn D, Chien P, Feuer J, Zackai EH, Emanuel BS, Driscoll DA (1998) Frequency of 22q11 deletions in patients with conotruncal defects. J Am Coll Cardiol 32:492–498
Gong W, Gottlieb S, Collins J, Blescia A, Dietz H, Goldmuntz E, McDonald-McGinn DM, Zackai EH, Emanuel BS, Driscoll DA, Budarf ML (2001) Mutation analysis of TBX1 in non-deleted patients with features of DGS/VCFS or isolated cardiovascular defects. J Med Genet 38:e45
Gorini F, Chiappa E, Gargani L, Picano E (2014) Potential effects of environmental chemical contamination in congenital heart disease. Pediatr Cardiol 35:559–568
Greulich F, Rudat C, Kispert A (2011) Mechanisms of T-box gene function in the developing heart. Cardiovasc Res 91:212–222
Griffin HR, Töpf A, Glen E, Zweier C, Stuart AG, Parsons J, Peart I, Deanfield J, O’Sullivan J, Rauch A, Scambler P, Burn J, Cordell HJ, Keavney B, Goodship JA (2010) Systematic survey of variants in TBX1 in non-syndromic tetralogy of Fallot identifies a novel 57 base pair deletion that reduces transcriptional activity but finds no evidence for association with common variants. Heart 96:1651–1655
Huang W, Meng H, Qiao Y, Pang S, Chen D, Yan B (2013) Two novel and functional DNA sequence variants within an upstream enhancer of the human NKX2-5 gene in ventricular septal defects. Gene 524:152–155
Huang RT, Xue S, Xu YJ, Zhou M, Yang YQ (2014) Somatic GATA5 mutations in sporadic tetralogy of Fallot. Int J Mol Med 33:1227–1235
Idorn L, Jensen AS, Juul K, Reimers JI, Johansson PI, Sørensen KE, Ostrowski SR, Søndergaard L (2013) Thromboembolic complications in Fontan patients: population-based prevalence and exploration of the etiology. Pediatr Cardiol 34:262–272
Jerome LA, Papaioannou VE (2001) DiGeorge syndrome phenotype in mice mutant for the T-box gene, TBX1. Nat Genet 27:286–291
Jiang JQ, Li RG, Wang J, Liu XY, Xu YJ, Fang WY, Chen XZ, Zhang W, Wang XZ, Yang YQ (2013) Prevalence and spectrum of GATA5 mutations associated with congenital heart disease. Int J Cardiol 165:570–573
Klitsie LM, Roest AA, Blom NA, ten Harkel AD (2014) Ventricular performance after surgery for a congenital heart defect as assessed using advanced echocardiography: from doppler flow to 3D echocardiography and speckle-tracking strain imaging. Pediatr Cardiol 35:3–15
Knöchelmann A, Geyer S, Grosser U (2014) Maternal understanding of infective endocarditis after hospitalization: assessing the knowledge of mothers of children with congenital heart disease and the practical implications. Pediatr Cardiol 35:223–231
Kröönström LA, Johansson L, Zetterström AK, Dellborg M, Eriksson P, Cider Å (2014) Muscle function in adults with congenital heart disease. Int J Cardiol 170:358–363
Lahm H, Deutsch MA, Dreßen M, Doppler S, Werner A, Hörer J, Cleuziou J, Schreiber C, Böhm J, Laugwitz KL, Lange R, Krane M (2013) Mutational analysis of the human MESP1 gene in patients with congenital heart disease reveals a highly variable sequence in exon 1. Eur J Med Genet 56:591–598
Laux D, Bertail C, Bajolle F, Houyel L, Boudjemline Y, Bonnet D (2014) Anomalous left coronary artery connected to the pulmonary artery associated with other cardiac defects: a difficult joint diagnosis. Pediatr Cardiol 35:1198–1205
Lee LJ, Lupo PJ (2013) Maternal smoking during pregnancy and the risk of congenital heart defects in offspring: a systematic review and metaanalysis. Pediatr Cardiol 34:398–407
Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, Bradley A, Baldini A (2001) Tbx1 haploinsufficiency in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 410:97–101
Mandal S, Tadros SS, Soni S, Madan S (2014) Single coronary artery anomaly: classification and evaluation using multidetector computed tomography and magnetic resonance angiography. Pediatr Cardiol 35:441–449
Martínez-Quintana E, Rodríguez-González F, Nieto-Lago V (2013) Subclinical hypothyroidism in grown-up congenital heart disease patients. Pediatr Cardiol 34:912–917
McCulley DJ, Black BL (2012) Transcription factor pathways and congenital heart disease. Curr Top Dev Biol 100:253–277
McElhinney DB, Geiger E, Blinder J, Benson DW, Goldmuntz E (2003) NKX2.5 mutations in patients with congenital heart disease. J Am Coll Cardio 42:1650–1655
Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier R, DemayMB Russell RG, Factor S, Tokooya K, Jore BS, Lopez M, Pandita RK, Lia M, Carrion D, Xu H, Schorle H, Kobler JB, Scambler P, Wynshaw-Boris A, Skoultchi AI, Morrow BE, Kucherlapati R (2001) TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 104:619–629
Momma K (2010) Cardiovascular anomalies associated with chromosome 22q11.2 deletion syndrome. Am J Cardiol 105:1617–1624
Moutafi AC, Manis G, Dellos C, Tousoulis D, Davos CH (2014) Cardiac autonomic nervous activity in adults with coarctation of the aorta late after repair. Int J Cardiol 173:566–568
Mueller GC, Sarikouch S, Beerbaum P, Hager A, Dubowy KO, Peters B, Mir TS (2013) Health-related quality of life compared with cardiopulmonary exercise testing at the midterm follow-up visit after tetralogy of Fallot repair: a study of the German competence network for congenital heart defects. Pediatr Cardiol 34:1081–1087
Mulkey SB, Swearingen CJ, Melguizo MS, Schmitz ML, Ou X, Ramakrishnaiah RH, Glasier CM, Bradley Schaefer G, Bhutta AT (2013) Multi-tiered analysis of brain injury in neonates with congenital heart disease. Pediatr Cardiol 34:1772–1784
Mulkey SB, Swearingen CJ, Melguizo MS, Reeves RN, Rowell JA, Gibson N, Holland G, Bhutta AT, Kaiser JR (2014) Academic proficiency in children after early congenital heart disease surgery. Pediatr Cardiol 35:344–352
Müller J, Engelhardt A, Fratz S, Eicken A, Ewert P, Hager A (2014) Improved exercise performance and quality of life after percutaneous pulmonary valve implantation. Int J Cardiol 173:388–392
Nicholson P, Yepiskoposyan H, Metze S, Zamudio Orozco R, Kleinschmidt N, Mühlemann O (2010) Nonsense-mediated mRNA decay in human cells: mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell Mol Life Sci 67:677–700
O’Byrne ML, Mercer-Rosa L, Ingall E, McBride MG, Paridon S, Goldmuntz E (2013) Habitual exercise correlates with exercise performance in patients with conotruncal abnormalities. Pediatr Cardiol 34:853–860
Ogata T, Niihori T, Tanaka N, Kawai M, Nagashima T, Funayama R, Nakayama K, Nakashima S, Kato F, Fukami M, Aoki Y, Matsubara Y (2014) TBX1 mutation identified by exome sequencing in a Japanese family with 22q11.2 deletion syndrome-like craniofacial features and hypocalcemia. PLoS One 9:e91598
Passarella G, Trifirò G, Gasparetto M, Moreolo GS, Milanesi O (2013) Disorders in glucidic metabolism and congenital heart diseases: detection and prevention. Pediatr Cardiol 34:931–937
Patel SS, Burns TL (2013) Nongenetic risk factors and congenital heart defects. Pediatr Cardiol 34:1535–1555
Perry JC (2012) Sudden cardiac death and malignant arrhythmias: the scope of the problem in adult congenital heart patients. Pediatr Cardiol 33:484–490
Pierpont ME, Basson CT, Benson DW Jr, Gelb BD, Giglia TM, Goldmuntz E, McGee G, Sable CA, Srivastava D, Webb CL, American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young (2007) Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation 115:3015–3038
Priromprintr B, Rhodes J, Silka MJ, Batra AS (2014) Prevalence of arrhythmias during exercise stress testing in patients with congenital heart disease and severe right ventricular conduit dysfunction. Am J Cardiol 114:468–472
Qu XK, Qiu XB, Yuan F, Wang J, Zhao CM, Liu XY, Zhang XL, Li RG, Xu YJ, Hou XM, Fang WY, Liu X, Yang YQ (2014) A novel NKX2.5 loss-of-function mutation associated with congenital bicuspid aortic valve. Am J Cardiol 114:1891–1895
Rushani D, Kaufman JS, Ionescu-Ittu R, Mackie AS, Pilote L, Therrien J, Marelli AJ (2013) Infective endocarditis in children with congenital heart disease: cumulative incidence and predictors. Circulation 128:1412–1419
Sakata M, Hayabuchi Y, Inoue M, Onishi T, Kagami S (2013) Left atrial volume change throughout the cardiac cycle in children with congenital heart disease associated with increased pulmonary blood flow: evaluation using a novel left atrium-tracking method. Pediatr Cardiol 34:105–111
Sanchez-Castro M, Gordon CT, Petit F, Nord AS, Callier P, Andrieux J, Guérin P, Pichon O, David A, Abadie V, Bonnet D, Visel A, Pennacchio LA, Amiel J, Lyonnet S, Le Caignec C (2013) Congenital heart defects in patients with deletions upstream of SOX9. Hum Mutat 34:1628–1631
Schuck R, Abd El Rahman MY, Rentzsch A, Hui W, Weng Y, Alexi-Meskishvili V, Lange PE, Berger F, Abdul-Khaliq H (2014) Altered right ventricular function in the long-term follow-up evaluation of patients after delayed aortic reimplantation of the anomalous left coronary artery from the pulmonary artery. Pediatr Cardiol 35:530–535
Shi LM, Tao JW, Qiu XB, Wang J, Yuan F, Xu L, Liu H, Li RG, Xu YJ, Qian Wang Q, Zheng HZ, Li X, Wang XZ, Qu XK, Yang YQ (2014) GATA5 loss-of-function mutations associated with congenital bicuspid aortic valve. Int J Mol Med 33:1219–1226
Sinha S, Abraham S, Gronostajski RM, Campbell CE (2000) Differential DNA binding and transcription modulation by three T-box proteins, T, TBX1 and TBX2. Gene 15:15–29
Starikov R, Bohrer J, Goh W, Kuwahara M, Chien EK, Lopes V, Coustan D (2013) Hemoglobin A1c in pregestational diabetic gravidas and the risk of congenital heart disease in the fetus. Pediatr Cardiol 34:1716–1722
Tabib A, Khorgami MR, Meraji M, Omidi N, Mirmesdagh Y (2014) Accuracy of Doppler-derived indices in predicting pulmonary vascular resistance in children with pulmonary hypertension secondary to congenital heart disease with left-to-right shunting. Pediatr Cardiol 35:521–529
Tripathi A, Black GB, Park YM, Jerrell JM (2014) Factors associated with the occurrence and treatment of supraventricular tachycardia in a pediatric congenital heart disease cohort. Pediatr Cardiol 35:368–373
Tutarel O, Kempny A, Alonso-Gonzalez R, Jabbour R, Li W, Uebing A, Dimopoulos K, Swan L, Gatzoulis MA, Diller GP (2014) Congenital heart disease beyond the age of 60: emergence of a new population with high resource utilization, high morbidity, and high mortality. Eur Heart J 35:725–732
Uysal F, Bostan OM, Semizel E, Signak IS, Asut E, Cil E (2014) Congenital anomalies of coronary arteries in children: the evaluation of 22 patients. Pediatr Cardiol 35:778–784
Valente AM, Gauvreau K, Assenza GE, Babu-Narayan SV, Evans SP, Gatzoulis M, Groenink M, Inuzuka R, Kilner PJ, Koyak Z, Landzberg MJ, Mulder B, Powell AJ, Wald R, Geva T (2013) Rationale and design of an International Multicenter Registry of patients with repaired tetralogy of Fallot to define risk factors for late adverse outcomes: the INDICATOR cohort. Pediatr Cardiol 34:95–104
van der Bom T, Zomer AC, Zwinderman AH, Meijboom FJ, Bouma BJ, Mulder BJ (2011) The changing epidemiology of congenital heart disease. Nat Rev Cardiol 8:50–60
Verheugt CL, Uiterwaal CS, van der Velde ET, Meijboom FJ, Pieper PG, Sieswerda GT, Plokker HW, Grobbee DE, Mulder BJ (2010) The emerging burden of hospital admissions of adults with congenital heart disease. Heart 96:872–878
Wang J, Xin YF, Xu WJ, Liu ZM, Qiu XB, Qu XK, Xu L, Li X, Yang YQ (2013) Prevalence and spectrum of PITX2c mutations associated with congenital heart disease. DNA Cell Biol 32:708–716
Wang C, Zhou K, Xie L, Li Y, Zhan Y, Qiao L, Qin C, Liu R, Hua Y (2014) Maternal medication use, fetal 3435 C > T polymorphism of the ABCB1 gene, and risk of isolated septal defects in a Han Chinese population. Pediatr Cardiol 35:1132–1141
Wei D, Bao H, Zhou N, Zheng GF, Liu XY, Yang YQ (2013) GATA5 loss-of-function mutation responsible for the congenital ventriculoseptal defect. Pediatr Cardiol 34:504–511
Wei D, Gong XH, Qiu G, Wang J, Yang YQ (2014) Novel PITX2c loss-of-function mutations associated with complex congenital heart disease. Int J Mol Med 33:1201–1208
Werner P, Paluru P, Simpson AM, Latney B, Iyer R, Brodeur GM, Goldmuntz E (2014) Mutations in NTRK3 suggest a novel signaling pathway in human congenital heart disease. Hum Mutat 35:1459–1468
Xiang R, Fan LL, Huang H, Cao BB, Li XP, Peng DQ, Xia K (2014) A novel mutation of GATA4 (K319E) is responsible for familial atrial septal defect and pulmonary valve stenosis. Gene 534:320–323
Xu H, Morishima M, Wylie JN, Schwartz RJ, Bruneau BG, Lindsay EA, Baldini A (2004) Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development 131:3217–3227
Xu YJ, Chen S, Zhang J, Fang SH, Guo QQ, Wang J, Fu QH, Li F, Xu R, Sun K (2014) Novel TBX1 loss-of-function mutation causes isolated conotruncal heart defects in Chinese patients without 22q11.2 deletion. BMC Med Genet 15:78
Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, Kamatani N, Momma K, Takao A, Nakazawa M, Shimizu N, Matsuoka R (2003) Role of TBX1 in human del22q11.2 syndrome. Lancet 362:1366–1373
Yang YQ, Gharibeh L, Li RG, Xin YF, Wang J, Liu ZM, Qiu XB, Xu YJ, Xu L, Qu XK, Liu X, Fang WY, Huang RT, Xue S, Nemer G (2013) GATA4 loss-of-function mutations underlie familial tetralogy of Fallot. Hum Mutat 34:1662–1671
Yuan F, Zhao L, Wang J, Zhang W, Li X, Qiu XB, Li RG, Xu YJ, Xu L, Qu XK, Fang WY, Yang YQ (2013) PITX2c loss-of-function mutations responsible for congenital atrial septal defects. Int J Med Sci 10:1422–1429
Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, Carriero NJ, Cheung YH, Deanfield J, DePalma S, Fakhro KA, Glessner J, Hakonarson H, Italia MJ, Kaltman JR, Kaski J, Kim R, Kline JK, Lee T, Leipzig J, Lopez A, Mane SM, Mitchell LE, Newburger JW, Parfenov M, Pe’er I, Porter G, Roberts AE, Sachidanandam R, Sanders SJ, Seiden HS, State MW, Subramanian S, Tikhonova IR, Wang W, Warburton D, White PS, Williams IA, Zhao H, Seidman JG, Brueckner M, Chung WK, Gelb BD, Goldmuntz E, Seidman CE, Lifton RP (2013) De novo mutations in histone-modifying genes in congenital heart disease. Nature 498:220–223
Zhao L, Ni SH, Liu XY, Wei D, Yuan F, Xu L, Xin-Li Li RG, Qu XK, Xu YJ, Fang WY, Yang YQ, Qiu XB (2014) Prevalence and spectrum of Nkx2.6 mutations in patients with congenital heart disease. Eur J Med Genet 57:579–586
Zheng J, Song H, Jiang S, Li T (2013) Congenital atresia of the left main coronary artery with noncompaction of the ventricular myocardium in an asymptomatic young child. Pediatr Cardiol 34:1998–2002
Zomer AC, Vaartjes I, van der Velde ET, de Jong HM, Konings TC, Wagenaar LJ, Heesen WF, Eerens F, Baur LH, Grobbee DE, Mulder BJ (2013) Heart failure admissions in adults with congenital heart disease; risk factors and prognosis. Int J Cardiol 168:2487–2493
Acknowledgements
The authors are really thankful to the participants for their dedication to the study. This work was supported in part by grants from the National Natural Science Fund of China (81270161) and the key program for Basic Research of Shanghai, China (14JC1405500).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Yun Pan and Zha-Gen Wang have contributed equally to the work.
Rights and permissions
About this article

Cite this article
Pan, Y., Wang, ZG., Liu, XY. et al. A Novel TBX1 Loss-of-Function Mutation Associated with Congenital Heart Disease. Pediatr Cardiol 36, 1400–1410 (2015). https://doi.org/10.1007/s00246-015-1173-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-015-1173-x