
Abstract
Bio-based production of industrially important chemicals provides an eco-friendly alternative to current petrochemical-based processes. Because of the limited supply of fossil fuel reserves, various technologies utilizing microbial host strains for the sustainable production of platform chemicals from renewable biomass have been developed. Corynebacterium glutamicum is a non-pathogenic industrial microbial species traditionally used for l-glutamate and l-lysine production. It is a promising species for industrial production of bio-based chemicals because of its flexible metabolism that allows the utilization of a broad spectrum of carbon sources and the production of various amino acids. Classical breeding, systems, synthetic biology, and metabolic engineering approaches have been used to improve its applications, ranging from traditional amino-acid production to modern biorefinery systems for production of value-added platform chemicals. This review describes recent advances in the development of genetic engineering tools and techniques for the establishment and optimization of metabolic pathways for bio-based production of major C2–C6 platform chemicals using recombinant C. glutamicum.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Because of the significant increase in the global demand for sustainable production of chemicals and materials from renewable resources instead of fossil fuels, bio-based processes have been developed as an eco-friendly alternative to current petrochemical-based production processes (Jang et al. 2012; Oh et al. 2015a; Joo et al. 2017a). Recently developed bioprocesses allow the utilization of a broad range of biomass feedstocks, such as lignocellulosic hydrolysates, algal residues, and even recalcitrant coal for sustainable production of chemicals with properties similar or superior to those of petrochemical-based products (Chae et al. 2016; Choi et al. 2016; David et al. 2017a; Kind et al. 2014; Lee et al. 2011, 2014a; Sudheer et al. 2016). In bio-based production, the development of efficient microbial cell factories, compatibility with current production processes, optimization of product purification, and availability of renewable substrates are key factors to be considered (Lee et al. 2011). Over the years, systems metabolic engineering, bioinformatics, and biotechnology for engineering microbial cell factories have significantly improved, allowing the production of bio-based platform chemicals and polymers such as succinic acid, 1,4-butanediol, isobutanol, isoprene, poly(2-hydroxybutyrate-co-lactate), and poly(lactate-co-glycolate) (Erickson et al. 2012; Chae et al. 2016; Choi et al. 2016). However, the commercial production of platform chemicals such as butyrate, 3-hydroxypropionic acid, isobutanol, terephthalic acid, and adipic acid still relies on conventional petro-based processes (Lee et al. 2011). Therefore, industrial microbial strains such as Escherichia coli, Saccharomyces cerevisiae, and Corynebacterium glutamicum have been developed for the production of value-added chemicals, polymers, and biofuels through fermentation of renewable biomass (Wendisch et al. 2006a; Buschke et al. 2013; Chae et al. 2016; Choi et al. 2016; David et al. 2017b; Becker and Wittmann 2015; Shin et al. 2016; Yang et al. 2016, 2018). Systems-wide metabolic pathway engineering of these strains has been successfully employed for bio-based production of bio-fuels, bulk and fine chemicals, polymers, feed additives, and high-value compounds for nutritional and pharmaceutical applications (Buschke et al. 2013). However, the use of S. cerevisiae and E. coli as hosts for protein production is limited by the solubility of target compound, as some proteins are retained in the periplasm or aggregate in inclusion bodies (Liu et al. 2012; Liu et al. 2015). Both strains also display strong catabolite repression when mixed pentose and hexose sugars are used as a carbon source, which limits their application in lignocellulosic biorefineries (Buschke et al. 2013). Compared to E. coli and S. cerevisiae, C. glutamicum exhibits low protease activity and is able to secrete properly folded functional protein (Liu et al. 2015). Weak catabolite repression is also observed in C. glutamicum strains engineered for mixed carbon source utilization (Buschke et al. 2013).
C. glutamicum is a gram-positive, non-sporulating bacterium that does not produce endotoxin and, thus, is generally recognized as a safe microorganism. It has been widely used for industrial production of amino acids, such as l-glutamate and l-lysine (Hermann 2003). C. glutamicum has been extensively studied, which has provided substantial insights in its genomics, transcriptomics, central carbon metabolism, systems biology, and physiology that have been used to develop metabolic engineering tools and strategies (Kalinowski et al. 2003; Ikeda and Nakagawa 2003; Wendisch 2003; Sun et al. 2017; Papagianni 2012; Wendisch et al. 2006b; Eggeling and Bott 2005). Genome engineering strategies (Becker et al. 2018; Suzuki and Inui 2013) and expression vector systems (Kirchner and Tauch 2003; Pátek and Nešvera 2013) have been developed and routinely used for broadening its applications in biorefineries for platform chemical production (Woo and Park 2014; Becker and Wittmann 2015; Lee and Kim 2015; Zahoor et al. 2012). Through systematic manipulation of the C. glutamicum genome, the production of natural metabolites, including l-ornithine, l-arginine, l-glutamate, and l-lysine, has been successfully enhanced (Zhang et al. 2017; Park et al. 2014; Schneider et al. 2011; Kind et al. 2010). Improved genetic tools and strategies have enabled successful implementation of heterologous synthetic pathways for the utilization of alternative carbon sources and the production of non-natural products (Heider and Wendisch 2015). For example, heterologous pathway expression using optimized expression vectors and promoters for the production of recombinant proteins, such as endoxylanase, α-amylase, and camelid antibody fragment (VHH), and non-natural metabolites, such as gamma-aminobutyrate, cadaverine, and 5-aminovalerate, by C. glutamicum has been successfully demonstrated (Yim et al. 2016a; Choi et al. 2015; Oh et al. 2015b; Joo et al. 2017b).
Engineering C. glutamicum for sustainable production of chemicals is potentially profitable because of its several unique advantages, including (1) flexible cellular metabolism (Buschke et al. 2013; Gopinath et al. 2012), (2) high stress tolerance to carbon source and target product (Yamamoto et al. 2013; Leßmeier and Wendisch 2015), (3) maintenance of metabolic activity by growth-arrested cells and resistance to fermentation inhibitors (Sakai et al. 2007), and (4) genetic stability because of the lack of a recombination repair system and limited restriction-modification system (Nakamura et al. 2003; Vertès et al. 1993). However, compared to E. coli and S. cerevisiae, relatively few genetic engineering tools and techniques tailored for C. glutamicum are available (Woo and Park 2014; Lee and Kim 2015; Liu et al. 2015). Therefore, comprehensive genetic and physiological studies are needed to maximize its potential as an efficient microbial cell factory for industrial bio-based chemical production. Appropriate expression vector systems for enhanced chemical production in C. glutamicum strains need to be evaluated. Tools and techniques for the introduction and identification of key mutations in C. glutamicum to allow the construction of stable recombinant strains need to be developed. In this review paper, genetic engineering tools and techniques available for metabolic engineering of C. glutamicum for C2–C4 platform chemical production are discussed, and we highlight the strong potential of C. glutamicum as a versatile industrial microbial strain.
Plasmids as synthetic biological circuit for gene expression
In the engineering of recombinant C. glutamicum strains for platform chemical production, the design of efficient biosynthetic pathways and the reinforcement of pathways involved in cell viability maintenance are crucial. For the implementation and evaluation of designed pathways, synthetic biological circuits based on plasmid systems are generally used because of their convenience. Synthetic biological circuits are composed of synthetic DNA parts such as promoters, replication origin, antibiotics as selective markers, 5′ untranslated region, expression cassette, and terminators (Table 1, Fig. 1). Each part can be customized to the specific purpose, such as modulation of the expression level and optimization of designed biosynthetic pathway.

Schematic diagram of plasmid-based synthetic biological circuits for engineering of C. glutamicum to produce biochemicals
Replication origin part for plasmid design in C. glutamicum
For synthetic biological circuit construction, the replication origin is important for plasmid reproduction and maintenance. Several origins of replication have been identified from native plasmids of C. glutamicum, such as the endogenous cryptic medium-copy plasmids (pBL1, pCG1, pGA1) and the broad host-range, low-copy plasmid (pNG2). Using these, C. glutamicum–E. coli shuttle vectors equipped with replication origins for E. coli have been developed to allow assembly of an entire synthetic platform in E. coli and pathway evaluation in C. glutamicum. The expression of key genes in multicopy vectors with replicons from pBL1, pCG1, pGA1, and pNG2 plasmids has enabled enhanced target product synthesis (Nešvera and Pátek 2011; Pátek and Nešvera 2013). This may be owing to higher gene dosage, which results in an increase in desirable flux toward the target product. For instance, the use of a multicopy plasmid with the pCG1 origin significantly enhanced the expression of transketolase biosynthetic operon, resulting in higher aromatic amino acid production (Ikeda et al. 1999). On the other hand, gene dosage reduction may enhance biochemical production in certain cases; for example, when the target product is toxic to the host strain, a low-copy replication origin such as pNG2 may be used. This strategy enhanced isoleucine production without any negative effect on cell growth by modulating the expression of threonine dehydratase from E. coli (Guillouet et al. 1999). Recently, adaptive laboratory evolution was used for the development of high-copy-number plasmid. It was discovered that mutation of the pCG1 replication origin of the pCES208 backbone vector resulted in higher copy numbers. The pCG1 replication origin is composed of three genes, including repA, which encodes plasmid replication initiator, and parAB, which is part of a partitioning system for autonomous plasmid replication. It was identified that a nonsense mutation, TGC to TGA, in the parB locus led to a 10-fold increase in plasmid copy number compared to the wild type. The use of the engineered high-copy plasmid, pHCMS, enhanced endoxylanase production in recombinant C. glutamicum (Choi et al. 2017).
Compatible replication origins for multiple plasmid maintenance in C. glutamicum
Compatibility of replication origins is important in synthetic biological circuit construction because the co-existence of plasmids in the host strain depends on it. The construction of plasmids with compatible replication origins provides an alternative approach to expressing large or multiple genes in a recombinant strain. Current C. glutamicum–E. coli shuttle vectors are limited in terms of the length and/or number of genes that can be inserted because these vectors are already large, with sizes of 4–5 kb, owing to the presence of two replication origins for plasmid maintenance in both C. glutamicum and E. coli (Pátek and Nešvera 2013). Plasmid instability and low transformation efficiency may occur if the final vector size is more than 10 kb (Ohse et al. 1995). Compatible plasmids are useful in establishing pathways that require more than four genes (Anthony et al. 2009). The use of plasmids such as CoryneBrick vectors (pBeB1c-RFP, pBbEB2-c-RFP, pBbEB5RFP) and the pZ8 vector, which have compatible replication origins, pBL1 and pHM1519, may help increase the gene transcription efficiency and enable stronger control of protein expression when different promoters and regulators are used (Harth et al. 2004; Kang et al. 2014). For the implementation of strategies that require the expression of genes in three plasmids, pBL1, pCG1, and pGA1, replication origins should be used as they are compatible and can co-exist in the recombinant strain. Transformation of up to three plasmids was successfully demonstrated using plasmids pVWEx1, pEKEx3, and pECXT99A with compatible pCG1, pBL1, and pGA1 replication origins, respectively (Jorge et al. 2017). For two-plasmid systems in C. glutamicum, the pBL1 replication origin is compatible with the following replication origins: pCG1 (Pátek and Nešvera 2013), pSR1 (Venkova-Canova et al. 2004), pH1519 (Kang et al. 2014), pGA1 (Jorge et al. 2017), and pCC1 (Cho et al. 2017). A recombinant strain harboring two compatible plasmids for the expression of lysine decarboxylase (pVWEx1-ldcC), and putrescine transaminase and γ-aminobutyraldehyde dehydrogenase (pEKEx3-patDA) could effectively produce 5-aminovalerate from cadaverine. Expression of additional genes relevant to starch, xylose, arabinose, and glucosamine utilization in the pECXT99A plasmid enabled 5-aminovalerate production from alternative carbon sources (Jorge et al. 2017).
Promoter part for modulation of gene expression in C. glutamicum
The promoter is an important component of a synthetic biological circuit. This tunable part allows gene transcription regulation and, thus, modulation of gene expression. Promoters are typically classified into two types: inducible and constitutive. Inducible promoters used in recombinant C. glutamicum are either adapted from E. coli systems, such as PlacUV5, PtacM, Ptrp, and ParaBAD, or are native, such as PaceA, PaceB, PgntP, and PgntK. The use of isopropyl β-d-1-thiogalactopyranoside (IPTG) inducible promoters, such as PlacUV5 and PtacM, allows leaky protein expression. As these are not tightly regulated, alternative inducible systems, such as those in which gene transcription is dependent on the concentration of arabinose, have been exploited (Park et al. 2008; Zhang et al. 2012). However, in this system, C. glutamicum requires heterologous expression of l-arabinose permease as cells exhibit low permeability to arabinose (Zhang et al. 2012). Native inducible promoters from acetate (aceA and aceB) and gluconate (gntP and gntK) uptake operons have also been evaluated. However, these promoters are somewhat impractical because for efficient gene transcription regulation, large amounts of acetate and gluconate are required, which is toxic to the host cell (Gerstmeir et al. 2003; Letek et al. 2006). A heat-inducible C. glutamicum–E. coli shuttle vector, pCeHEMG857, was constructed by fusion of two successive lambda OL1 operators to a cryptic CJ1 promoter isolated from Corynebacterium ammoniagenes. This expression vector successfully activated attenuator binding protein (PyrR) expression when the culture temperature was shifted from 30 to 42 °C (Park et al. 2008).
Constitutive promoters, such as Ptrc, PcspB, PgapA, Psod, and Ptuf, have been used to enhance the expression of target genes in C. glutamicum. Constitutive promoters are generally preferred over inducible promoters because they do not require an inducer and optimization of the culture conditions (Yim et al. 2013). The constitutive expression vector, pTRCphb, which expresses key genes for PHB synthesis under control of the trc promoter, has been successfully used to increase production of PHB in C. glutamicum (Liu et al. 2007). Similarly, recombinant C. glutamicum expressing α-amylase from Streptococcus bovis 148 and lysine decarboxylase encoded by cadA from E. coli under the high constitutive promoter HCE was able to produce cadaverine at high concentration (Tateno et al. 2009). However, the introduction of heterologous genes in a recombinant strain often leads to metabolic burden, which can be observed as increased byproduct formation and decreased growth rate. Therefore, optimization of the recruited biosynthetic pathway expression in the recombinant strain is important for balanced target metabolite production and maintenance of cell growth. To this purpose, plasmids equipped with synthetic promoters of various predictable strengths have been developed. Recently, pCES208 vectors harboring synthetic promoters of different strength (PH36 > PH30 > PI64 > PI16 > PL26 > PL10) have been developed for constitutive target gene expression. These plasmids have been employed in recombinant C. glutamicum for enhanced production of cadaverine, 5-aminovalerate, gamma-aminobutyrate, lysine, endoxylanase, α-amylase, camelid antibody fragment (VHH), antibody fragment (scFv), and green fluorescent protein (Oh et al. 2015b; Joo et al. 2017b; Shin et al. 2016; Choi et al. 2015; Yim et al. 2013, 2014, 2016a, 2016b; An et al. 2013).
5′ UTR part for fine tuning of metabolic pathways in C. glutamicum
The UTR of a synthetic biological circuit has a role in translational regulation of the coded genes. Modulation of gene expression to improve biochemical production is possible through modification of the 5′ UTR site via ribosome binding site (RBS) engineering, such as the application of antisense small RNA (asRNA), RBS libraries, and synthetic RBS, and the replacement of the conserved Shine–Dalgarno (SD) sequence. AsRNA is a powerful tool for modulation of protein expression via interference and attenuation of mRNA transcription, RNA cleavage, or gene translation blockage (Fig. 1). In recombinant C. glutamicum, expression of odhA-antisense RNA with overexpression of the odhA gene, encoding the 2-oxoglutarate dehydrogenase complex, enabled enhanced glutamate production (Kim et al. 2009). The sRNAs recently discovered in C. glutamicum could be useful for constructing DNA/RNA parts tailored for C. glutamicum engineering (Mentz et al. 2013). Tunable 5′ UTR libraries with conserved RBS (AGGA) and synthetic riboswitches have been constructed and successfully applied for the synthesis of tunable promoters and a lysine riboswitch (Yim et al. 2013; Zhou and Zeng 2015). Replacement of the conserved SD sequence in the translation initiation region of a target gene has been reported to significantly improve protein expression and enzyme activity. For example, translation efficiency in C. glutamicum significantly increased when the native SD of the tpi gene, encoding triosephosphate isomerase, was replaced with an E. coli SD in a GFP expression vector (Teramoto et al. 2011). For efficient initiation of translation, the predicted putative SD sequence was replaced with a consensus SD sequence in C. glutamicum. High-level expression of nitrile dehydratase from Rhodococcus rhodochrous was achieved by introducing a conserved SD sequence (GAAAGGCGA) and seven random mutations in the translational initiation region (Kang et al. 2014).
Expression cassette part for improvement of protein expression
In synthetic biological circuits, codon optimization, engineered start codons, and addition of a his-tag to the target gene have enabled enhanced protein expression. Codon optimization allows the improvement of protein expression in C. glutamicum by tailoring codon usage and the GC content of genes recruited from heterologous donor strains. Codon optimization of lysine decarboxylase (ldcC) from E. coli and lysine 2-monooxygenase and delta-aminovaleramidase (davBA) from Pseudomonas putida improved cadaverine and 5-aminovalerate production, respectively (Kind et al. 2010; Shin et al. 2016).
Start codon engineering is one of the popular methods to modulate protein expression by regulation of transcription rate. In this approach, rare start codon variants GTG and TTG are changed to ATG. Start codon engineering of genes encoding pyruvate dehydrogenase and isocitrate dehydrogenase resulted in improved lysine production (Becker et al. 2009, 2010). This method has also been applied to enhance putrescine production by fine-tuning the expression of the gene coding for ornithine transcarbamoylase (Schneider et al. 2012). This strategy can also be applied to reduce gene expression, by changing the ATG start codon to GTG, as demonstrated by the reduced expression of isocitrate dehydrogenase for enhanced glycolate production in recombinant C. glutamicum (Zahoor et al. 2014). Target proteins with an N′ terminal His6-tag reportedly demonstrate enhanced expression because of improved stability by the 5′ modification (Cèbe and Geiser 2006). Enhanced 5-aminovalerate production has been demonstrated in recombinant C. glutamicum expressing a codon-optimized davAB operon, encoding lysine 2-monooxygenase and delta-aminovaleramide, through fusion of a His6-tag at the N-terminus of davA (Shin et al. 2016).
Transcription terminators for efficient translational termination in C. glutamicum
Transcription terminator is a region of nucleotide sequence that marks the end of a gene or operon in genomic DNA during transcription. The rrnB T1 terminator from E. coli has commonly been used for construction of expression vectors for C. glutamicum such as pEKEx3, pVWEx1, and pZ8-1 (Lange et al. 2018; Dusch et al. 1999). T7 terminator from E. coli was also used for construction of IPTG-inducible T7 expression vector, pMKEx2 (Kortmann et al. 2015). Palindromic structures that can act as transcription terminators have been found in downstream regions of corynebacterial genes such as thrB, sodA, and nusG (Srivastava and Deb 2002). Two different DNA fragments found downstream of homoserine kinase (thrB) gene were identified as Rho-independent functional transcriptional terminators. These 185- and 127-bp fragments were used for construction of terminator–probe vectors, pULT1 and pULT2 (Mateos et al. 1994). Transcription terminator in Corynebacterium melassecola was found to be located 18 and 88 bp downstream of stop codon in superoxide dismutase (sodA) gene and was used for the construction of shuttle vector, pMM23 (Merkamm and Guyonvarch 2001). Downstream of gene for anti-terminator protein (nusG), a 19-nucleotide inverted repeat that was identified as transcriptional terminator was also used for construction of promoter–probe vector, pULCE0 (Barreiro et al. 2001). Recently, transcriptome sequencing for characterization of small RNAs in C. glutamicum ATCC 13032 have detected 69 small RNAs of Rho-independent terminators (Mentz et al. 2013), which may be used for the construction of expression vectors for C. glutamicum.
Genetic modification in C. glutamicum
The complete genome sequences of C. glutamicum ATCC 13032 (Ikeda and Nakagawa 2003) and C. glutamicum R (Yukawa et al. 2007) laid the foundation for genome-wide analysis of C. glutamicum strains through transcriptomics (Wendisch 2003; Hüser et al. 2005; Inui et al. 2007; Ehira et al. 2009), proteomics (Hermann et al. 2001; Li et al. 2007), metabolomics (Bartek et al. 2008; Woo et al. 2010), and fluxomics (Becker et al. 2007). Comparative analysis of C. glutamicum genomes has provided insights into its native metabolic network by identifying target genes in various strains. The accumulated genome information is used as a detailed guide for designing host strains with beneficial properties by introducing biosynthetic pathways for chemical production and for identifying metabolic bottlenecks in both native and engineered pathways for further strain optimization (Yang and Yang 2017). This strategy has been successfully used for redirecting the carbon flux toward l-lysine, l-glutamate, and l-valine synthesis in C. glutamicum (Kalinowski et al. 2003; Yang and Yang 2017; Bartek et al. 2008).
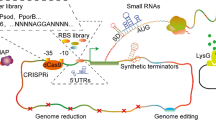
Technologies for gene integration, replacement, and disruption for genome engineering of C. glutamicum have evolved from routinely used homologous recombination-based methods using suicide vector pK19mobsacB and Cre-Lox to the modern RecFACS and clustered regularly interspaced short palindromic repeat (CRISPR) recombination systems (Schäfer et al. 1994; Suzuki et al. 2005; Tsuge et al. 2007; Binder et al. 2013; Jiang et al. 2015; Liu et al. 2017). Genome engineering of C. glutamicum currently relies on random mutagenesis and recombination based on rare double crossover events (Nešvera and Pátek 2011; Park et al. 2014; Schwarzer and Pühler 1991). Suicide vectors have been developed for gene disruption and insertion in the C. glutamicum genome. These are based on either SacB, which hydrolyzes sucrose for levan synthesis leading to sucrose sensitivity, or the Cre-LoxP system, in which Cre recombinase catalyzes specific recombination between two loxP sites (Suzuki et al. 2005; Choi et al. 2015). Other classic methods for gene disruption and insertion are based on integration of a suicide vector into the chromosome, followed by another recombination event for removal of plasmid backbone. A counter-selection step based on a conditionally lethal marker is used for the identification of successful mutants. However, both SacB- and Cre-LoxP-based methods are laborious, time consuming, and inefficient because two rounds of recombination are needed, and frequent spontaneous inactivation of sacB results in false positives (Schäfer et al. 1994).
Recently, RecFACS and CRISPR-based technologies have been developed for simple, rapid, and precise genome editing. In the RecFACS method, heterologous gene integration into the chromosome is based on the multiplex automated genome engineering (MAGE) concept, wherein a set of synthetic single-stranded DNAs is directly introduced in the bacterial chromosome using a phage homologous recombination protein encoded by a different plasmid. Screening of successful mutants is done by rapid and automated fluorescence-activated cell sorting (FACS). An active l-lysine-producing mutant with 12 different amino acid changes in the targeted murE codon was isolated using this strategy (Binder et al. 2013). However, the RecFACS method requires the construction of target-gene specific biosensors and FACS for colony selection, hampering its application in systematic genome engineering.
The CRISPR/Cas9 genome editing system was repurposed for C. glutamicum engineering. CRISPRi technology for gene deletion was developed for C. glutamicum to increase the recombineering efficiency compared to previously developed methods. It involves using deactivated Cas9 (dCas9) for reversible transcription by attaching to RBS to block gene transcription (Fig. 2) (Cleto et al. 2016). For instance, reduced expression of pgi, pck, and pyk encoding glucose-6-phosphate isomerase, phosphoenolpyruvate caboxykinase, and pyruvate kinase, respectively, in C. glutamicum was achieved by using the CRISPR/dCas9 system, resulting in improved l-lysine and l-glutamate titers comparable with those in strains constructed with traditional gene deletion (Cleto et al. 2016).

The CRISPR/Cpf1 system was developed for C. glutamicum because Streptococcus pyogenes Cas9 was toxic to recombinant cells (Jiang et al. 2015). An all-in-one plasmid containing Cpf1, a single-stranded RNA-guided endonuclease from Francisella novicida, CRISPR RNA, and homologous arms enabled large gene deletions and insertions in the C. glutamicum genome. A two-plasmid CRISPR/Cpf1-assisted system using separate FnCpf1 and CRISPR RNA (crRNA) sequences enabled genome editing by codon saturation mutagenesis of γ-glutamyl kinase to relieve l-proline inhibition (Jiang et al. 2015). Another strategy for overcoming S. pyogenes Cas9 toxicity in C. glutamicum is through regulation of Cas9 expression using the P-tac promoter. A two-plasmid system using the developed RNA expression cassette and the CRISPR/Cas9 plasmid mediated ssDNA recombineering based on the RecT phage recombinase system. This enabled the precise introduction of single-nucleotide changes and double-locus editing in C. glutamicum (Liu et al. 2017). Combined use of phage recombinase RecT for target gene editing and CRISPR/Cas9 for counterselection of negative mutants enabled rapid genome engineering and screening of mutant C. glutamicum. This strategy allowed generating and identifying seven different mutants with three genomic deletions for improved gamma-aminobutyric acid production (Cho et al. 2017) (Fig. 2).
Fermentative production of platform chemicals
C. glutamicum is a well-studied non-pathogenic strain that is currently used for industrial production of lysine and glutamate (Zahoor et al. 2012). It is a promising strain for microbial cell factory development because it can utilize a broad spectrum of carbon sources for the production of platform chemicals, materials, and fuels in biorefineries (Becker and Wittmann 2012; Wieschalka et al. 2013; Zahoor et al. 2012) (Table 2). C. glutamicum has a flexible cellular metabolism that has been engineered for application in microalgal, crude glycerol, and lignocellulosic biorefineries (Buschke et al. 2013; Lee et al. 2014a; Meiswinkel et al. 2013; Gopinath et al. 2012). Consolidated bioprocessing of microalgal biomass as a carbon source for the production of succinate has been successfully demonstrated (Lee et al. 2014a). C. glutamicum strains have also been engineered for the utilization of pure and crude glycerol for the production of l-glutamate, l-lysine, l-ornithine, l-arginine, putrescine, succinate, and 1,3-propanediol (Rittmann et al. 2008; Meiswinkel et al. 2013; Litsanov et al. 2013). As for lignocellulosic biorefinery, C. glutamicum strains have been successfully engineered for the production of succinate from xylose (Jo et al. 2017), cadaverine from hydrolyzed dried oat spelts (Buschke et al. 2013), and l-glutamate and l-lysine from rice straw or wheat bran hydrolysate (Gopinath et al. 2011). Engineered C. glutamicum strains have been used for consolidated bioprocessing of hemicellulosic biomass for lysine and xylonic acid production (Yim et al. 2016a; Yim et al. 2017). Growth-arrested cells of C. glutamicum maintain metabolic activity and exhibit high stress tolerance to carbon sources, target products, and fermentation inhibitors, such as organic acids, furans, and phenols, which allows these cells to maintain high production titer, yield, and productivity when laboratory experiments are scaled up for industrial production (Smith et al. 2010; Leßmeier and Wendisch 2015; Sakai et al. 2007). In this section, we will discuss the recent progress in C. glutamicum engineering for the production of representative industrially important C2–C4 chemicals in biorefinery systems (Fig. 3).

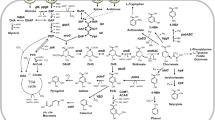
Pathway overview of Corynebacterium glutamicum for amino acids and C2–C5 platform chemical production. The pathways are based on enhanced production of key precursors, dihydroxy-acetone phosphate (dihydroxy-acetone P), glycerate 3-phosphate (3P-glycerate), pyruvate, oxaloacetate, succinate, glyoxylate, and α-ketoglutarate. Production of organic and amino acids with industrially relevant derivatives are indicated in gray. C2 chemicals, ethanol, ethylene glycol, and glycolate, are enclosed in solid square. C3 hydroxy acid chemical, 3-hydroxypropionic acid, is enclosed in dashed square. C3 diols, 1,2-propanediol and 1,3-propanediol, are enclosed in solid round square. C4 alcohols, isobutanol and 2,3-butanediol, are enclosed in dashed round square
C2 chemicals: ethanol, ethylene glycol, and glycolate
Ethanol is widely used as a biofuel and as a solvent in the food and cosmetics industries. Current fermentative production of ethanol relies on starch-based feedstocks using S. cerevisiae as a microbial cell factory. However, its application in emerging lignocellulose-based biorefineries is hampered by its inability to utilize pentose sugars, which are abundant in lignocellulosic biomass. Even in S. cerevisiae engineered for xylose utilization, strong sugar catabolite repression was observed in mixed glucose and xylose culture. S. cerevisiae also displays high sensitivity to fermentation inhibitors, such as organic acid and phenols (Oreb et al. 2012; Lee et al. 2014b; Cao et al. 2014). To establish ethanol production from lignocellulosic biomass, C. glutamicum is preferred because engineered xylose-utilizing strains exhibit minimal sugar catabolite repression. The culture of recombinant strains under oxygen deprivation results in robustly growth-arrested cells, which are resistant to furans, phenols, and acids, fermentation inhibitors usually found in lignocellulosic hydrolysates (Kawaguchi et al. 2006; Sakai et al. 2007). Fermentative ethanol production using C. glutamicum was established through heterologous expression of a pyruvate decarboxylase gene (pdc) from Zymomonas mobilis for the decarboxylation of pyruvic acid to acetaldehyde. A native alcohol dehydrogenase gene (adhB) under lactate dehydrogenase A (ldhA) promoter control was used for acetaldehyde reduction to ethanol. The genes for lactate dehydrogenase (ldhA) and phosphoenolpyruvate carboxylase (ppc) were disrupted to prevent byproduct formation (Fig. 4). Under growth-arrest conditions, 6.9 g/L of ethanol was obtained (Inui et al. 2005). Recently, higher-titer ethanol production by C. glutamicum was achieved by overexpression of pgi, 6-phosphofructokinase (pfkA), glyceraldehyde 3-phosphate dehydrogenase (gapA), pyk, and triosephosphate isomerase (tpi), and heterologous expression of pdc and alcohol dehydrogenase (adhb) from Z. mobilis in a ldhA- and phosphoenolpyruvate carboxylase (ppc)-deficient background strain enabled increased ethanol production rate and yield (Jojima et al. 2015). This recombinant C. glutamicum was capable of producing 119 g/L of ethanol from 245 g/L of glucose, demonstrating a 95% yield. Through the integration of genes for xylose and arabinose utilization, 83 g/L of ethanol, with 90% yield, was achieved in this strain.

Pathway engineering of Corynebacterium glutamicum for production of C2 chemicals; ethanol, ethylene glycol, and glycolate. Ethanol, ethylene glycol, and glycolate are produced via its key precursor: pyruvate, serine, and glyoxylate, respectively. A suggested synthetic pathway for production of glycolate from glycoaldehyde is indicated by broken arrows. Enzymes: pdc, pyruvate decarboxylase; adh, alcohol dehydrogenase; ycdW, glyoxylate reductase; serA, phosphoglycerate dehydrogenase; serC, phosphoserine aminotransferase; serB, phosphoserine phosphatase; AGT, serine decarboxylase; AO, amine oxidase; AGT1, alanine-glyoxylase amino-transferase; mdlC, benzoylformate decarboxylase; yqhD, alcohol dehydrogenase. Abbreviations: 3P-glycerate, glycerate 3-phosphate
Ethylene glycol (EG) is an important precursor for the production of polyethylene terephthalate. It is also a raw material for the production of anti-freezing agent and coolant. To date, natural EG production has been reported only for Caldicellulosiruptor saccharolyticus, which can accumulate only small amounts (Isern et al. 2013). To establish an EG production system and verify the efficiency of constructed pathway, E. coli was initially evaluated as a model host. E. coli was engineered for EG production by direct fermentation of arabinose or xylose (Liu et al. 2013). However, the theoretical yield of the proposed pathway was only 1 mol of EG per mole of pentose. Furthermore, the proposed pentose degradation pathway did not consider the utilization of sucrose and glucose as substrates. To solve these problems, an amino acid-derived synthetic pathway was suggested. Using the serine biosynthesis pathway, 2 mol of EG per mole of glucose is theoretically possible. Therefore, C. glutamicum has been tested as a host for EG production because it is an established amino acid-producing strain capable of high serine accumulation (Stolz et al. 2007; Zhu et al. 2014). To this end, it was firstly engineered for enhanced production of the important precursor serine (Chen et al. 2016a). Then, modules for establishing a synthetic EG production pathway were constructed and transformed into the recombinant strain for the evaluation of EG production (Fig. 4). Firstly, competing pathways for serine degradation to glycine and pyruvate were deleted to increase the intracellular pool of serine in the prophage-free C. glutamicum host strain MB001. An in-frame deletion of the pabABC operon, encoding aminodeoxychorismate synthase and aminodeoxychorismate lyase, resulted in reduced activity of serine hydroxymethyltransferase (glyA), which converts serine to glycine. An additional in-frame substitution in sdaA, encoding serine dehydratase, led to reduced activity of serine dehydratase (sdaA), which converts serine to pyruvate. To drive the metabolic flux toward serine production, feedback-insensitive phosphoglycerate dehydrogenase, phosphoserine aminotransferase, and phosphoserine phosphatase genes coded on the artificial operon serACB under the strong constitutive P1 promoter were introduced into the recombinant strain (Chen et al. 2016a).
An engineered serine-overproducing strain C. glutamicum PABS1, harboring two modules encoding two different biosynthetic pathways, was constructed and evaluated for EG production. Through the pathway on module 1, serine is deaminated into hydroxypyruvate by an alanine-glyoxylase amino-transferase from Arabidopsis thaliana. Hydroxypyruvate is then decarboxylated into glycoaldehyde by a benzoylformate decarboxylase (mdlC) from Pseudomonas putida. Through the pathway module 2, serine is decarboxylated to ethanolamine by a serine decarboxylase from A. thaliana. Ethanolamine is then oxidized to glycoaldehyde by an amine oxidase from Arthrobacter sp., and glycoaldehyde is reduced to EG by alcohol dehydrogenase (yqhD) from E. coli. C. glutamicum PABS1 harboring module 1 yielded 0.7 g/L of EG in shakeflask culture, and the engineered host harboring module 2 produced 1.7 g/L of EG. The strain expressing both modules produced 2.2 g/L of EG in batch fermentation, and accumulation of EG was detected after 6 h of cultivation, with a final concentration of 3.5 g/L at 72 h, resulting in a 0.25 mol/mol glucose yield. Although the final concentration of EG produced using recombinant C. glutamicum was lower than EG production from xylose by recombinant E. coli, the proposed pathway provides a basis for developing efficient microbial strains for EG production using glucose as a carbon source (Chen et al. 2016a).
Glycolate is a simple alpha-hydroxy acid commonly used in the cosmetic industry for skin treatments. Glycolate polymers are used in packaging materials and medical applications. Currently, the production of glycolate relies on high-pressure and high-temperature carbonylation of formaldehyde or enzymatic conversion of glycolonitrile using microbial nitrilases (He et al. 2010; Panova et al. 2007). Even though these methods are well established, the hydrogen cyanide and formaldehyde used in the chemical conversion produce harmful degradation products. A more eco-friendly alternative production process through whole-cell conversion of EG to glycolate by Gluconobacter oxydans has been reported (Wei et al. 2009). However, for sustainable glycolate production in biorefineries, microbial cell factories should be engineered for direct production glycolate from glucose derived from renewable biomass. A biosynthetic pathway of glycolate from glucose has been established using E. coli as a model system. In E. coli, glycolate is a natural metabolite that is synthesized through reduction of the main precursor glyoxylate by endogenous glyoxylate reductase (ycdW), but the glyoxylate shunt is repressed when glucose is used as carbon source. For deregulation of the glyoxylate shunt, isocitrate dehydrogenase kinase/phosphatase (aceK) was expressed to prevent conversion of isocitrate to 2-ketoglutarate. Then, expression of isocitrate lyase (aceA) was employed to direct the flux toward glyoxylate production. Finally, overexpression of glycolate reductase resulted in the production of only 1.4 g/L of glycolate in recombinant E. (Martin et al. 2013). C. glutamicum has been evaluated as a host strain because its glyoxylate shunt remains active in the presence of acetate (Gerstmeir et al. 2003). To direct carbon flux toward glycolate production, isocitrate dehydrogenase expression was repressed by changing the translational start codon from ATG to GTG. To prevent the conversion of glyoxylate to malate, the malate synthase gene (aceB) was deleted (Fig. 4). Finally, heterologous expression of E. coli ycdW in C. glutamicum resulted in the production of 5.3 g/L of glycolate from co-utilization of glucose and acetate. This titer is higher than that achieved by engineered Bacillus subtilis (Kabisch et al. 2013), Kluyveromyces lactis (Koivistoinen et al. 2013), and E. coli (Martin et al. 2013).
C3 hydroxy acids: 3-hydroxypropionic acid (3-HP)
3-HP is a promising platform chemical for the production of a wide range of industrial chemicals, such as acrylic acid, acrylamide, malonic acid, and 1,3 propanediol. E. coli (Chu et al. 2015; Lim et al. 2016; Liu et al. 2016) and Klebsiella pneumoniae (Ashok et al. 2013a, b; Huang et al. 2016) have been extensively engineered for 3-HP production from glucose and glycerol. The pathway for 3-HP production in established strains involves the conversion of glycerol to 3-hydroxypropionaldehyde by diol dehydratase. 3-Hydroxypropionaldehyde is converted to 3-hydroxypropionic acid by diol dehydrogenase. Although 3-HP production is well established in E. coli and K. pneumoniae, 3-HP production in C. glutamicum has been evaluated because it would be safer than using pathogenic K. pneumoniae. Furthermore, C. glutamicum displays weak carbon catabolite repression when used as biocatalyst for the production of organic acids (Zahoor et al. 2012). A 3-HP producing C. glutamicum strain was constructed by improving the pathway for accumulation of the main precursor, glycerol, and introducing a 3-HP biosynthetic pathway (Chen et al. 2016b). Under anaerobic conditions, C. glutamicum naturally accumulates a small amount of glycerol by dephosphorylation and reduction mediated by dihydroxyacetone phosphate (DHAP) phosphatase and (S,S)-butanediol dehydrogenase, encoded by the hdpA and butA genes. To increase glycerol production, codon-optimized gpd and gpp genes, encoding glyceraldehyde 3-phosphate dehydrogenase and glycerol 3-phosphatase from S. cerevisiae, were integrated into the C. glutamicum MB001 strain, resulting in a yield of 0.35 g/g glucose and accumulation of 28 g/L of glycerol. Finally, 3-hydroxypropionaldehyde is converted to 3-HP by heterologous expression of diol dehydrogenase and activator encoded by pduCDEGH from K. pneumoniae and aldehyde dehydrogenase encoded by gabDE209Q/E269Q (Fig. 5). This recombinant C. glutamicum was able to produce 21 ± 1.3 g/L of 3-HP with a yield of 0.27 g/g glucose. As significant amounts of lactate and acetate were produced as byproducts, the ldhA gene was deleted, resulting in a 19.4% increase in 3-HP titer (25.8 ± 71.8 g/L). Furthermore, deletion of pta-ackA and poxB genes, encoding phosphate acetyltransferase, acetate kinase, and pyruvate:quinone oxidase, to block acetate synthesis resulted in a 52.8% reduction of acetate accumulation and an increase in 3-HP production (27.2 ± 1.2 g/L) (Chen et al. 2016b).

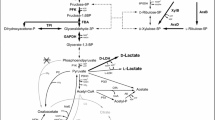
Pathway engineering of Corynebacterium glutamicum for production of C3 chemicals: 3-hydroxypropionic acid, 1,2-propanediol, and 1,3-propanediol. All relevant chemicals are produced via a key precursor, dihydroxy-acetone phosphate. A suggested synthetic pathway for production of 3-HP and 1,3-PD intermediate is indicated by red broken line. Alternative pathway from DHAP to glycerol in C. glutamicum is indicated as black and broken red arrow, which is mediated by endogenous dihydroxyacetone phosphatase (hdpA) and unknown enzyme, respectively (Jojima et al. 2012). Potential alternative pathway, not yet reported in literature, for acetol to S-1,2-propanediol is indicated as violet broken line. Enzymes: gpd, glyceraldehyde 3-phosphate dehydrogenase; gpp, glycerol 3-phosphatase; hdpA, dihydroxyacetone phosphatase; pduCDEGH, diol dehydratase and its activator; yqhD, alcohol dehydrogenase; gabD*, mutant aldehyde dehydrogenase (gabDE209Q/E269); mgs, methyglyoxal synthase; cg_2242, methylglyoxal reductase; fucO, lactaldehyde reductase; gldA, glyceraldehyde dehydrogenase. Abbreviations: dihydroxy-acetone P, dihydroxy-acetone phosphate; 3-HPA, 3-hydroxypropionaldehyde; 3-HP, 3-hydroxypropionic acid; 1,3-PD, 1,3-propanediol; S-1,2-PD, S-1,2-propanediol; R-1,2-PD, R-1,2-propanediol
Production of 3HP from xylose was also demonstrated by integration of the xylAB operon from E. coli, encoding xylose isomerase and xylulokinase, under control of the strong constitutive promoter H36, at the ldhA locus for stable expression of genes relevant for xylose utilization and deletion of lactate byproduct accumulation. Furthermore, arabinose transporter (araE) from C. glutamicum ATCC 31831 under the H36 promoter was integrated into the pyruvate oxidase (poxB) locus. To enhance the flux toward 3-HP production, the phosphotransferase system (PTS) glucose uptake system was replaced with a non-PTS glucose uptake route for decoupling glucose utilization from phosphoenolpyruvate (PEP) synthesis. To this end, the native PEP-dependent PTS was inactivated by deletion of ptsH, encoding the general PTS component HPr (Fig. 5). Then, non-PTS glucose uptake was activated by deletion of iolR (cgp_0196), encoding the transcriptional regulator of the iol regulon (Ikeda et al. 2011) (Fig. 5). A higher titer of 3-HP (36.8 g/L) was achieved; however, the glucose consumption and 3-HP production rates were lower than those of previously constructed strains. To enhance the glucose uptake rate, the strong sod promoter was applied for stronger expression of inositol permease and glucokinase encoded by iolT1 (cgp_0223) and glk (cgp_2399). This resulted in a final production of 38.6 g/L of 3-HP via the non-PTS glucose uptake strategy. The resulting recombinant strain MH15 was capable of simultaneous utilization of glucose and xylose for 3-HP production with a titer and yield of 36.2 g/L and 0.45 g/g of sugar carbon source. Fed-batch fermentation using this strain with glucose and xylose as carbon sources enabled 3-HP production of 54.8 g/L and a yield of 0.49 g/g of sugar. After 72 h, 3.9 g/L of glycerol and 4.3 g/L of glycerol was accumulated in the medium. Fed-batch fermentation using engineered strain MH15 with glucose as a sole carbon source resulted in 62.6 g/L of 3-HP and a yield of 0.51 g/g glucose at 72 h. The only byproduct detected was 3.2 g/L of acetate (Chen et al. 2016b).
C3 diols: 1,2-propanediol (1,2-PD) and 1,3-propanediol (1,3-PDO)
1,2-PD is mainly used in food, cosmetics, and pharmaceutical industries. Currently, the demand for 1,2-PD is accommodated by petroleum-based production processes, which produce toxic intermediates and byproducts. Engineered E. coli, Thermoanaerobacterium thermosaccharolyticum, Clostridium sphenoides, and S. cerevisiae have been proposed as sustainable alternatives for 1,2-PD production (Altaras and Cameron 2000; Altaras et al. 2001; Junget al. 2008). In the pathway for 1,2-PD synthesis in microorganisms, dihydroxyacetone phosphate (DHAP) is the main metabolite from the glycolytic pathway. This is converted to lactaldehyde or acetol, the main precursor for 1,2-PD production (Niimi et al. 2011). Natural production of 1,2-PD from glucose in C. sphenoides and T. thermosaccharolyticum resulted in productions of 2 and 9 g/L, respectively (Tran-Din and Gottschalk 1985; Sanchez-Rivera et al. 1987). However, these strains are not well studied. Therefore, 1,2-PD synthesis in industrial microorganisms such as E. coli, S. cerevisiae, and C. glutamicum has been evaluated. The pathway for R-1,2-propanediol production in E. coli involves the conversion of dihydroxyacetone phosphate to methylglyoxal by overexpression of methyglyoxal synthase (mgs). Methyglyoxal is reduced to acetol by alcohol dehydrogenase (adhI) or l-lactaldehyde by glycerol dehydrogenase (gldA) (Altaras and Cameron 2000). The gldA gene from E. coli exhibits broad substrate specificity, allowing it to convert methyglyoxal and lactaldehyde into R-lactaldehyde and R-1,2-PD, respectively (Misra et al. 1996). Disruption of ldhA in E. coli resulted in a production of up to 4.5 g/L of R-1,2-PD (Altaras and Cameron 2000). In S. cerevisiae, deletion of tpi encoding triosephosphate isomerase and integration of E. coli mgs and gldA enabled it to produce 1.11 g/L of 12-PD in flask culture (Jung et al. 2008). As C. glutamicum naturally produces small amounts of 1,2-PD, its genome was studied for identifying genes encoding 1,2-PD pathway-related enzymes. An NADPH-dependent methylglyoxal reductase gene (cgr_2242) was identified. To increase 1,2-PD production, heterologous expression of mgs for the conversion of dihydroxyacetone phosphate to methylglyoxal was implemented. The resulting strain produced 1.9 g/L of 1,2-propanediol after 96 h of flask culture with high acetol concentration (3.3 g/L) (Niimi et al. 2011). To improve 1,2-propanediol production in recombinant C. glutamicum, heterologous expression of E. coli mgsA, gldA, and yqhD was used. However, heterologous expression of both gldA and yqhD resulted in the production of glycerol as a byproduct as the gene products also reduce DHAP to glycerol; therefore, the hdpA gene was deleted. Further expression of aldehyde dehydrogenase encoded by fucO gene from E. coli for conversion of toxic methylglyoxal to L-lactaldehyde did not improve 1,2-PD production. Additionally, ldh was deleted to improve production under oxygen-deprived conditions. The resulting recombinant C. glutamicum strain was able to produce 4.56 g/L of 1,2-propandiol, with a yield of 0.14 g/g and productivity of 0.09 g/L (Siebert and Wendisch 2015).
1,3-PDO is an important platform chemical used in textile, solvent, food, and pharmaceutical industries. It is used as monomer for the synthesis of polyethers, polyurethanes, and polytrimethylene terephthalate. Commercial production of 1,3-PDO from glucose using recombinant E. coli has been established by DuPont. The recent rapid development of biofuel refineries has generated large amounts of crude glycerol as a byproduct. This provides substantial amounts of substrate for glycerol-based biorefinery, allowing sustainable and economical production of 1,3-PDO. In microorganisms such as Klebsiella, Clostridia, Enterobacter, Citrobacter, and Lactobacilli, 1,3-PDO production from glycerol involves dehydration of glycerol to 3-hydroxypropionaldehyde by glycerol dehydratase and reduction of 3-hydroxypropionaldehyde to 1,3-propanediol by 1,3-propanediol dehydrogenase (Zhang and Xiu 2009; Ren et al. 2016; Gonzalez et al. 2008; Maervoet et al. 2016). Although established strains produce high titers, fermentation processes have low yields of 1,3-propanediol because 40–50% of the glycerol is converted into undesired byproducts, such as formate, acetate, lactate, and 2,3-butanediol, making downstream processes complex and costly (Saxena et al. 2009; Celińska 2010; Kaur et al. 2012). As 1,3-PDO biosynthesis is a reduction process that requires NADH, provision of this cofactor via glycerol oxidation of engineered glutamate-producing strains of C. glutamicum should increase the yield of 1,3-PDO production from glycerol, improving the economic feasibility of glycerol-based biorefinery. C. glutamicum has been evaluated as a host strain for 1,3-PDO production as well as efficient co-factor regeneration by production of glutamate, as strains engineered for glutamate production are able to produce excess NADH during the conversions of glyceraldehyde-3-phosphate to 3-phosphate-glycerate and pyruvate to acetyl-CoA, thus maintaining a high intracellular pool of NADH. Co-production of glutamate and 1,3-PDO is advantageous because oxidative phosphorylation of excess NADH is necessary for high glutamate yield (Huang et al. 2017). However, C. glutamicum cannot utilize glycerol as a sole carbon source; therefore, glycerol assimilation genes pduCDEGH encoding diol dehydratase and its activator from K. pneumoniae and dhaT encoding 1,3-propanediol dehydrogenase were introduced into the recombinant PT01 strain. Additional deletion of 2-oxoglutarate dehydrogenase (odh) was used to drive the flux toward glutamate overproduction (Fig. 5) (Asakura et al. 2007). For the maintenance of cell growth and NADH production, glucose (80 g/L) was added as a co-substrate for cultivation in medium containing 20 g/L glycerol as the carbon source. The engineered C. glutamicum strain OD01 allowed simultaneous production of 1,3-PDO and glutamate, yielding 14.4 g/L of 1,3-PDO and 32.5 g/L of glutamate. Glutamate production in this recombinant strain was better than that in the control strain used, suggesting that 1,3-PDO production benefits glutamate production. The NADH/NAD ratio in the engineered strain was significantly lower, indicating that NADH from glutamate production was utilized for 1,3-PDO production (Huang et al. 2017). The co-produced 1,3-PDO and glutamate were purified by crystallization and distillation.
C4 alcohols: isobutanol and 2,3-butanediol (2,3-BDO)
Isobutanol is currently an attractive bio-based alternative fuel because of its high energy density and low hygroscopic activity compared to ethanol, making it suitable for use in existing pipelines and combustion engines (Smith et al. 2010). Several anaerobic Clostridium species naturally produce isobutanol through a co-enzyme acetyl coA-dependent pathway (Blombach and Eikmanns 2011). In engineered aerobic strains of E. coli and B. subtilis, the pathway for isobutanol production involves conversion of 2-ketoisovalerate to isobutyraldehyde by 2-ketoacid decarboxylase and conversion of isobutyraldehyde to isobutanol by alcohol dehydrogenase (Blombach et al. 2011). High titers of up to 50 g/L of isobutanol were obtained by aerobic fermentation by recombinant E. coli JCL260/pSA55/pSA69 (Baez et al. 2011). However, this process is limited because isobutanol is toxic to the recombinant cells. As isobutanol production involves 2-ketoacid pathways, which are precursors for amino acids, C. glutamicum was evaluated as a host strain. C. glutamicum harbors great potential for application in biorefineries because of its high tolerance to isobutanol compared to E. coli, high 2-ketoisovalerate production, and maintenance of metabolic activity under oxygen deprivation (Smith et al. 2010; Krause et al. 2010).
For isobutanol production in recombinant C. glutamicum, pathway improvement for high intracellular 2-ketoisovalerate accumulation and a recruited isobutanol synthesis pathway were established (Smith et al. 2010). Initial evaluation of isobutanol production was done using a pyruvate carboxylase (pyc) and ldh-deficient host strain for overexpression of acetolactate synthase (alsS) from B. subtilis for pyruvate to acetolactate conversion. Acetohydroxyacid isomeroreductase and dihydroxyacid dehydratase (ilvCD) of C. glutamicum were overexpressed for the conversion of acetolactate to 2-ketoisovalerate. Then, 2-ketoacid decarboxylase (kivD) from L. lactis was introduced for conversion of 2-ketoisovalerate to isobutyraldehyde. Finally, native adhA catalyzed the conversion of isobutyraldehyde to isobutanol (Fig. 6). The resulting recombinant strain was able to produce 4.9 g/L of isobutanol.

Pathway engineering of Corynebacterium glutamicum for production of 2,3-butanediol and isobutanol. The relevant chemicals are produced via key precursor, pyruvate. Endogenous pathway in C. glutamicum is indicated in black arrow. Enzymes: alsS/budB, acetolactate synthase; budA/aldB, acetolactate decarboxylase; butA, acetoin reductase; ilvC, acetohydroxy isomeroreductase; ilvD, dihydroxyacid reductase; kivD, 2-ketoacid decarboxylase; adhA, alcohol dehydrogenase. Abbreviation: DHV, 2,3-dihydroxyisovalerate
For enhanced production of isobutanol, a 2-ketoisovalerate-overproducing strain was constructed. To increase the intracellular 2-ketoisovalerate pool, the pyruvate dehydrogenase complex (aceE), pyruvate:quinone oxidoreductase (pqo), and transaminase B (ilvE) genes were deleted to attenuate competing pathways. Additionally, overexpression of ilvBNCD, encoding acetohydroxyacid synthase, acetohydroxyacid isomeroreductase, and dihydroxyacid dehydratase, was established to drive the flux toward 2-ketoisovalerate. To establish a pathway for isobutanol production, kivD from L. lactis, adh2 from S. cerevisiae, and transhydrogenase (pntAB) from E. coli were introduced. ldhA and malate dehydrogenase (mdh) were deleted to attenuate competing pathways (Fig. 6). The production process was optimized by separating aerobic 2-ketoisovalerate production from oxygen-deprived isobutanol production. Using this strategy, isobutanol production increased up to 13 g/L (Blombach et al. 2011). In another approach, isobutanol production was achieved through in an oxygen-deprived fermentation using a recombinant strain harboring alcohol dehydrogenases from E. coli (adhP) and S. cerevisiae (adh2) and kivD from L. lactis (Yamamoto et al. 2013). A high titer of 73 g/L of isobutanol was achieved by continuous extraction of isobutanol from the reaction mixture using oleyl alcohol.
2,3-BDO is an important platform chemical with a wide range of applications, e.g., as plasticizer, fumigant, and antifreezing agent. It is also used as a precursor of important chemicals, such as 1-butanediene and 2-butanone, which are used in synthetic rubber production and as a fuel additive and resin solvent, respectively (Rados et al. 2015). In bacteria, 2,3-BDO is formed from pyruvate via three consecutive steps, starting with the condensation of two pyruvate molecules into α-acetolactate by α-acetolactate synthase. Acetolactate is converted to acetoin by aldehyde decarboxylase (Yang et al. 2015). Finally, acetoin is converted to 2,3-butanediol by 2,3-butanediol dehydrogenase. For microbial production of 2,3-BDO, an engineered K. pneumoniae strain that is capable of producing up to 111.3 g/L of meso-2,3-butanediol, with a productivity of 2.71 g/L·h in fed-batch fermentation, has been well studied (Kim et al. 2014). However, because of safety concerns regarding the use of K. pneumoniae, production of (2R,3R)-2,3-BDO using engineered E. coli strains was evaluated; it was able to produce up to 73.8 g/L (Xu et al. 2014). However, alternative hosts remain to be considered for safe and sustainable production of 2,3 BDO. Therefore, L. lactis, B. subtilis, P. polymyxa, and S. cerevisiae have been engineered; however, the production efficiencies were low or complicated nutrition were required, and thus, these strains were not economically practical (Gaspar et al. 2011; Fu et al. 2014; Xu et al. 2014). C. glutamicum has been studied as an alternative host for 2,3-BDO production because it is a natural 2,3-BDO producer; however, it does not have α-acetolactate decarboxylase (Dickschat et al. 2010). Only a small amount of (2S,3S)-2,3-BDO is produced by conversion of S-acetoin to (2S,3S)-2,3-BDO using native butanediol dehydrogenase (Rados et al. 2015). To improve 2,3-BDO production in C. glutamicum, genes encoding α-acetolactate synthase, α-acetolactate decarboxylase, and butanediol dehydrogenase were heterologously expressed in the host strain to attenuate byproduct formation. Fermentation was established using a two-stage process for aerobic culture of cells for increasing cell density, and oxygen-limited 2,3-BDO production by growth-arrested cells (Rados et al. 2015; Yang et al. 2015).
In one approach, to improve the intracellular pool of the important precursor, pyruvate, and for deletion of acetate, succinate, and lactate byproducts, the E1-subunit of the pyruvate dehydrogenase complex (aceE), mdh, ldhA, and pyruvate:quinone oxidoreductase (pqo) of C. glutamicum were deleted. To improve 2,3-BDO production, α-acetolactate synthase (als), α-acetolactate decarboxylase (aldB), and butanediol dehydrogenase (butA) from L. lactis were introduced into the engineered host (Fig. 6). To optimize conditions for fermentative 2,3-BDO production, a two-stage process was established, wherein the engineered C. glutamicum cells were grown in aerobic conditions using acetate or glucose as a carbon source to achieve high cell density, and then, cells (50 g CDW/L) were collected and used for conversion of glucose to 2,3-BDO under growth-arrest anaerobic conditions. This strategy yielded 6.3 g/L of meso-2,3-BDO (Rados et al. 2015).
In another approach, acetolactate decarboxylase and α-acetolactate synthase (budAB) from K. pneumoniae were introduced into C. glutamicum ATCC 13032 (Fig. 6). The recombinant strain, C. glutamicum SGSC102, produced 18 g/L of 2,3-BDO with byproduct formation of 2.49 g/L acetoin, 8.43 g/L lactate, 2.16 g/L succinate, and 2.36 g/L acetate. 2,3-BDO production for biorefinery by utilization of renewable biomass was demonstrated using this strain in batch cultures with 80 g/L of cassava powder (56.7 g/L glucose and 2.17 g/L fructose) as a carbon source. This resulted in the production of 12.0 g/L of 2,3-BDO (Yang et al. 2015).
Conclusions and outlooks
Recent advances in the development of genetic tools and techniques for metabolic engineering have enabled C. glutamicum to be evolved as an industrial microbial strain beyond traditional amino-acid production, into modern platform chemical production. Combined systems biology and comparative omics analyses have enabled the development and successful application of genetic engineering tools and techniques, such as RecT and CRISPR/Cas9-based genome editing, tunable synthetic promoter-based expression plasmids, and compatible plasmids. These tools have allowed establishing heterologous pathways, thus broadening the product portfolio of recombinant C. glutamicum for efficient production of platform chemicals. C. glutamicum is a promising robust and versatile industrial microbial strain for applications in sustainable platform chemical production through biorefinery.
References
Altaras NE, Cameron DC (2000) Enhanced production of (R)-1,2-propanediol by metabolically engineered Escherichia coli. Biotechnol Prog 16:940–946. https://doi.org/10.1021/bp000076z
Altaras NE, Etzel MR, Cameron DC (2001) Conversion of sugars to 1,2-propanediol by Thermoanaerobacterium thermosaccharolyticum HG-8. Biotechnol Prog 17:52–56. https://doi.org/10.1021/bp000130b
An SJ, Yim SS, Jeong KJ (2013) Development of a secretion system for the production of heterologous proteins in Corynebacterium glutamicum using the Porin B signal peptide. Protein Expr Purif 89(2):251–257. https://doi.org/10.1016/j.pep.2013.04.003
Anthony JR, Anthony LC, Nowroozi F, Kwon G, Newman JD, Keasling JD (2009) Optimization of the mevalonate-based isoprenoid biosynthetic pathway in Escherichia coli for production of the anti-malarial drug precursor amorpha-4,11-diene. Metab Eng 11(1):13–19. https://doi.org/10.1016/j.ymben.2008.07.007
Asakura Y, Kimura E, Usua Y, Kawahara Y, Matsui K, Osumi T, Nakamatsu T (2007) Altered metabolic flux due to deletion of odhA causes L-glutamate overproduction in Corynebacterium glutamicum. Appl Environ Microbiol 73(4):1308–1319. https://doi.org/10.1128/AEM.01867-06
Ashok S, Mohan Raj S, Ko Y, Sankaranarayanan M, Zhou S, Kumar V, Park S (2013a) Effect of puuC overexpression and nitrate addition on glycerol metabolism and anaerobic 3-hydroxypropionic acid production in recombinant Klebsiella pneumoniae ΔglpK ΔdhaT. Metab Eng 15:10–24. https://doi.org/10.1016/j.ymben.2012.09.004
Ashok S, Sankaranarayanan M, Ko Y, Jae KE, Ainala SK, Kumar V, Park S (2013b) Production of 3-hydroxypropionic acid from glycerol by recombinant Klebsiella pneumonia ΔdhaT ΔyqhD which can produce vitamin B12 naturally. Biotechnol Bioeng 110(2):511–524. https://doi.org/10.1002/bit.24726
Baez A, Cho KM, Liao JC (2011) High-flux isobutanol production using engineered Escherichia coli: a bioreactor study with in situ product removal. Appl Microbiol Biotechnol 90:1681–1690. https://doi.org/10.1007/s00253-011-3173-y
Barreiro C, Gonalez-Lavado E, Martin J (2001) Organization and transcriptional analysis of a six-gene cluster around the rplK-rplA operon of Corynebacterium glutamicum encoding the ribosomal proteins L11 and L1. Appl Environ Microbiol 67(5):2183–2190. https://doi.org/10.1128/AEM.67.5.2183-2190.2001
Bartek T, Makus P, Klein B, Lang S, Olidges (2008) Influence of L-isoleucine and pantothenate auxotrophy for L-valine formation in Corynebacterium glutamicum revisited by metabolome analyses. Bioprocess Biosyst Eng 31(3):217–225. https://doi.org/10.1007/s00449-008-0202-z
Becker J, Wittmann C (2012) Bio-based production of chemicals, materials and fuels—Corynebacterium glutamicum as versatile cell factory. Curr Opin Biotechnol 23:631–640. https://doi.org/10.1016/j.copbio.2011.11.012
Becker J, Wittmann C (2015) Advanced biotechnology: metabolically engineered cells for the bio-based production of chemicals and fuels, materials, and health-care products. Angew Chem Int Ed 54(11):3328–3350. https://doi.org/10.1002/anie.201409033
Becker J, Klopprogge C, Herold A, Zelder O, Bolten CJ, Wittmann C (2007) Metabolic flux of L-lysine production in Corynebacterium glutamicum over expression and modification of G6P dehydrogenase. J Biotechnol 132:99–109. https://doi.org/10.1016/j.jbiotec.2007.05.026
Becker J, Klopprogge C, Schroder H, Wittmann C (2009) Metabolic engineering of the tricarboxylic acid cycle for improved lysine production by Corynebacterium glutamicum. Appl Environ Microbiol 75(24):7866–7869. https://doi.org/10.1128/AEM.01942-09
Becker J, Buschke N, Bücker R, Wittmann C (2010) Systems level engineering of Corynebacterium glutamicum—reprogramming translational efficiency for superior production. Eng Life Sci 10(5):430–438. https://doi.org/10.1002/elsc.201000008
Becker J, Gießelmann G, Hoffmann SL, Wittmann C (2018) Corynebacterium glutamicum for sustainable bioproduction: from metabolic physiology to systems metabolic engineering. Adv Biochem Eng Biotechnol 162:217–263. https://doi.org/10.1007/10_2016_21
Binder S, Siedler S, Marienhagen J, Bott M, Eggeling L (2013) Recombineering in Corynebacterium glutamicum combined with optical nanosensors: a general strategy for fast producer strain generation. Nucleic Acid Res 41(12):6360–6369. https://doi.org/10.1093/nar/gkt312
Blombach B, Eikmanns B (2011) Current knowledge on isobutanol production with Escherichia coli, Bacillus subtilis and Corynebacterium glutamicum. Bioeng Bugs 2(6):346–350. https://doi.org/10.4161/bbug.2.6.17845
Blombach B, Riester T, Wieschalka S, Ziert C, Youn JW, Wendisch VF, Eikmanns BJ (2011) Corynebacterium glutamicum tailored for efficient isobutanol production. Appl Environ Microbiol 77:3300–3310. https://doi.org/10.1128/AEM.02972-10
Buschke N, Schäfer R, Becker J, Wittmann C (2013) Metabolic engineering of industrial platform microorganisms for biorefinery applications—optimization of substrate spectrum and process robustness by rational and evolutive strategies. Bioresour Technol 135:544–554. https://doi.org/10.1016/j.biortech.2012.11.047
Cao L, Tang X, Zhang X, Zhang J, Tian X, Wang J, Xiao W (2014) Two-stage transcriptional reprogramming in Saccharomyces cerevisiae for optimizing ethanol production from xylose. Metab Eng 24:150–159. https://doi.org/10.1016/j.ymben.2014.05.001
Cèbe R, Geiser M (2006) Rapid and easy thermodynamic optimization of the 5′-end of mRNA dramatically increases the level of wild type protein expression in Escherichia coli. Protein Expr Purif 45(2):374–380. https://doi.org/10.1016/j.pep.2005.07.007
Celińska E (2010) Debottlenecking the 1,3-propanediol pathway by metabolic engineering. Biotechnol Adv 28:519–530. https://doi.org/10.1016/j.biotechadv.2010.03.003
Chae CG, Kim YJ, Lee SJ, Oh YH, Yang JE, Joo JC, Kang KH, Jang YA, Lee H, Park AR, Song BK, Lee SY, Park SJ (2016) Biosynthesis of poly(2-hydroxybutyrate-co-lactate) and in metabolically engineered Escherichia coli. Biotechnol Bioprocess Eng 21:169–174
Chen Z, Huang J, Wu Y, Liu D (2016a) Metabolic engineering of Corynebacterium glutamicum for the de novo production of ethylene glycol from glucose. Metab Eng 33:12–18. https://doi.org/10.1016/j.ymben.2015.10.013
Chen Z, Huang J, Wu Y, Wu W, Zhang Y, Liu D (2016b) Metabolic engineering of Corynebacterium glutamicum for the production of 3-hydroxypropionic from glucose and xylose. Metab Eng 39:151–158
Cho JS, Choi KR, Prabowo CPS, Shin JH, Yang D, Jang J, Lee SY (2017) CRISPR/Cas9-coupled recombineering for metabolic engineering of Corynebacterium glutamicum. Metab Eng 42:157–167. https://doi.org/10.1016/j.ymben.2017.06.010
Choi JW, Yim SS, Kim MJ, Jeong KJ (2015) Enhanced production of recombinant proteins with Corynebacterium glutamicum by deletion of insertion sequences (IS elements). Microb Cell Factories 14(207):207. https://doi.org/10.1186/s12934-015-0401-7
Choi SY, Park SJ, Kim WJ, Yang JE, Lee H, Shin J, Lee SY (2016) One-step fermentative production of poly(lactate-co-glycolate) from carbohydrates in Escherichia coli. Nat Biotechnol 34:435–440. https://doi.org/10.1038/nbt.3485
Choi JW, Yim SS, Jeong KJ (2017) Development of a high-copy-number plasmid via adaptive laboratory evolution of Corynebacterium glutamicum. Appl Microbiol Biotechnol 102:873–883. https://doi.org/10.1007/s00253-017-8653-2
Chu HS, Kim YS, Lee CM, Lee JH, Jung WS, Ahn JH, Song SH, Choi IS, Cho KM (2015) Metabolic engineering of 3-hydroxypropionic acid biosynthesis in Escherichia coli. Biotechnol Bioeng 112:356–364
Cleto S, Jensen JV, Wendisch VF, Lu TK (2016) Corynebacterium glutamicum metabolic engineering with CRISPR interference (CRISPRi). ACS Synth Biol 5:375–385. https://doi.org/10.1021/acssynbio.5b00216
David Y, Baylon MG, Sudheer PDVN, Baritugo KA, Chae CG, Kim YJ, Kim TW, Kim MS, Na JG, Park SJ (2017a) Screening of microorganisms able to degrade low-rank coal in aerobic condition as potential coal biosolubilization mediators from coal to biochemical. Biotechnol Bioprocess Eng 22:178–185. https://doi.org/10.1007/s12257-016-0263-9
David Y, Joo JC, Yang JE, Oh YH, Lee SY, Park SJ (2017b) Biosynthesis of 2-hydroxyacid-containing polyhydroxyalkanoates by employing butyryl-CoA transferases in metabolically engineered Escherichia coli. Biotechnol J 12:1700116. https://doi.org/10.1002/biot.201700116
Dickschat JS, Wickel S, Bolten CJ, Nawrath T, Schulz S, Wittmann C (2010) Pyrazine biosynthesis in Corynebacterium glutamicum. Eur J Org Chem 2010(14):2687–2695. https://doi.org/10.1002/ejoc.201000155
Dusch N, Pühler A, Kalinowski J (1999) Expression of the Corynebacterium glutamicum panD gene encoding L-aspartate-alpha-decarboxylase leads to pantothenate overproduction in Escherichia coli. Appl Environ Microbiol 65(4):1530–1539
Eggeling L, Bott M (2005) Handbook of Corynebacterium glutamicum. Taylor & Francis, Boca Raton
Ehira S, Teramoto H, Inui M, Yukawa H (2009) Regulation of Corynebacterium glutamicum heat shock response by the extracytoplasmic-function sigma factor SigH and transcriptional regulators HspR and HrcA. J Bacteriol 191:2964–2972. https://doi.org/10.1128/JB.00112-09
Eikmanns BJ, Kleinertz E, Liebl W, Sahm H (1991) A family of Corynebacterium glutamicum/Escherichia coli shuttle vectors for cloning, controlled gene expression and promoter probing. Gene 102:93–98. https://doi.org/10.1016/0378-1119(91)90545-M
Erickson B, Nelson JE, Winters P (2012) Perspective on opportunities in industrial biotechnology in renewable chemicals. Biotechnol J 7:176–185. https://doi.org/10.1002/biot.201100069
Fu J, Wang Z, Chen T, Liu W, Shi T, Wang G, Tang Y, Zhao X (2014) NADH plays the vital role for chiral pure D-(−)-2,3-butanediol production in Bacillus subtilis under limited oxygen conditions. Biotechnol Bioeng 111:2126–2131. https://doi.org/10.1002/bit.25265
Gaspar P, Neves AR, Gasson MJ, Shearman CA (2011) Santos H (2011) High yields of 2,3-butanediol and mannitol in Lactococcus lactis through engineering of NAD+ cofactor recycling. Appl Environ Microbiol 77:6826–6835. https://doi.org/10.1128/AEM.05544-11
Gerstmeir R, Wendisch VF, Schnicke S, Ruan H, Farwick M, Reinscheid D, Eikmanns BJ (2003) Acetate metabolism and its regulation in Corynebacterium glutamicum. J Biotechnol 104:99–122. https://doi.org/10.1016/S0168-1656(03)00167-6
Gonzalez R, Murarka A, Dharmadi Y, Yazdani SS (2008) A new model for the anaerobic fermentation of glycerol in enteric bacteria: trunk and auxiliary pathways in Escherichia coli. Metab Eng 10:234–245. https://doi.org/10.1016/j.ymben.2008.05.001
Gopinath V, Meiswinkel TM, Wendisch VF, Nampoothri KM (2011) Amino acid production from rice straw and wheat bran hydrolysates by recombinant pentose-utilizing Corynebacterium glutamicum. Appl Microbiol Biotechnol 92(5):985–996. https://doi.org/10.1007/s00253-011-3478-x
Gopinath V, Murali A, Dhar KS, Nampoothiri KM (2012) Corynebacterium glutamicum as a potent biocatalyst for the bioconversion of pentose sugars to value-added products. Appl Microbiol Biotechnol 93(1):95–106. https://doi.org/10.1007/s00253-011-3686-4
Guillouet S, Rodal AA, An G, Lessard PA, Sinskey AJ (1999) Expression of the Escherichia coli catabolic threonine dehydratase in Corynebacterium glutamicum and its effect on isoleucine production. Appl Environ Microbiol 65:3100–3107
Harth G, Maslesa-Galić S, Horwitz MA (2004) A two-plasmid system for stable, selective-pressure-independent expression of multiple extracellular proteins in mycobacteria. Microbiology 150(7):2143–2151. https://doi.org/10.1099/mic.0.27113-0
He YC, Xu JH, Su JH, Zhou L (2010) Bioproduction of glycolic acid from glycolonitrile with a new bacterial isolate of Alcaligenes sp. ECU0401. Appl Biochem Biotechnol 160:1428–1440. https://doi.org/10.1007/s12010-009-8607-y
Heider SAE, Wendisch VF (2015) Engineering microbial cell factories: metabolic engineering of Corynebacterium glutamicum with a focus on non-natural products. Biotechnol J 10(8):1170–1184. https://doi.org/10.1002/biot.201400590
Hermann T (2003) Industrial production of amino acids by coryneform bacteria. J Biotechnol 104:155–172. https://doi.org/10.1016/S0168-1656(03)00149-4
Hermann T, Pfefferle W, Baumann C, Busker E, Schaffer S, Bott M, Sahm H, Dusch N, Kalinowski J, Puhler A, Bendt AK, Kramer R, Burkovski A (2001) Proteome analysis of Corynebacterium glutamicum. Electrophoresis 22(9):1712–1723. https://doi.org/10.1002/1522-2683(200105)22:9%3C1712::AID-ELPS1712%3E3.0.CO;2-G
Huang Y, Li Z, Ye Q (2016) Transcriptional regulation of genes involved in 3-hydroxypropionic acid production in response to aeration of recombinant Klebsiella pneumoniae. Appl Biochem Biotechnol 178(6):1129–1140. https://doi.org/10.1007/s12010-015-1933-3
Huang J, Wu Y, Wu W, Zhang Y, Liu CZ (2017) Cofactor recycling for co-production of 1,3-propanediol and glutamate by metabolically engineered Corynebacterium glutamicum. Sci Rep 7:42246. https://doi.org/10.1038/srep42246
Hüser AT, Chassagnole C, Lindley ND, Merkamm M, Guyonvarch A, Elisáková V, Pátek M, Kalinowski J, Brune I, Pühler A, Tauch A (2005) Rational design of a Corynebacterium glutamicum pantothenate production strain and its characterization by metabolic flux analysis and genome-wide transcriptional profiling. Appl Environ Microbiol 71(6):3255–3268. https://doi.org/10.1128/AEM.71.6.3255-3268.2005
Ikeda M, Nakagawa S (2003) The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl Microbiol Biotechnol 62:99–109. https://doi.org/10.1007/s00253-003-1328-1
Ikeda M, Okamoto K, Katsumata R (1999) Cloning of the transketolase gene and the effect of its dosage on aromatic amino acid production in Corynebacterium glutamicum. Appl Microbiol Biotechnol 51:201–206. https://doi.org/10.1007/s002530051382
Ikeda M, Mizuno Y, Awane S, Hayashi M, Mitsuhashi S, Takeno S (2011) Identification and application of a different glucose uptake system that functions as an alternative to the phosphotransferase system in Corynebacterium glutamicum. Appl Microbiol Biotechnol 90:1443–1451. https://doi.org/10.1007/s00253-011-3210-x
Inui M, Kawaguchi H, Murakami S, Vertes AA, Yukawa H (2005) Metabolic Engineering of Corynebacterium glutamicum for Fuel Ethanol Production under Oxygen-Deprivation Conditions. J Mol Microbiol Biotechnol 8 (4):243-254. https://doi.org/10.1159/000086705
Inui M, Suda M, Okino S, Nonaka H, Puskas LG, Vertes AA, Yukawa H (2007) Transcriptional profiling of Corynebacterium glutamicum metabolism during organic acid production under oxygen deprivation conditions. Microbiology 153:2491–2504. https://doi.org/10.1099/mic.0.2006/005587-0
Isern NG, Xue J, Rao JV, Cort JR, Ahring BK (2013) Novel monosaccharide fermentation productsin Caldicellulosiruptor saccharolyticus identified using NMR spectroscopy. Biotechnol Biofuels 6:47. https://doi.org/10.1186/1754-6834-6-47
Jakoby M, Ngouoto-Nkili CE, Burkovski A (1999) Construction and application of new Corynebacterium glutamicum vectors. Biotechnol Tech 13:437–441. https://doi.org/10.1023/A:1008968419217
Jang YS, Kim B, Shin JH, Choi YJ, Choi S, Song CW, Lee J, Par HG, Lee SY (2012) Bio-based production of C2-C6 platform chemicals. Biotechnol Bioeng 109(10):2437–2459. https://doi.org/10.1002/bit.24599
Jiang Y, Chen B, Duan C, Sun B, Yang J, Yang S (2015) Multigene editing in the Escherichia coli genome via the CRISPR- Cas9 system. Appl Environ Microbiol 81:2506–2514. https://doi.org/10.1128/AEM.04023-14
Jo S, Yoon J, Lee SM, Um Y, Han SO, Woo HM (2017) Modular pathway engineering of Corynebacterium glutamicum to improve xylose utilization and succinate production. J Biotechnol 258:69–78. https://doi.org/10.1016/j.jbiotec.2017.01.015
Jojima T, Igari T, Gunji W, Suda M, Inui M, Yukawa H (2012) Identification of a HAD superfamily phosphatase, HdpA, involved in 1,3-dihydroxyacetone production during sugar catabolism in Corynebacterium glutamicum. FEBS Lett 586(23):4228–4232. https://doi.org/10.1016/j.febslet.2012.10.028
Jojima T, Noburyu R, Sasaki M, Tajima T, Suda M, Yukawa H, Inui M (2015) Metabolic engineering for improved production of ethanol by Corynebacterium glutamicum. Appl Microbiol Biotechnol 99:1165–1172. https://doi.org/10.1007/s00253-014-6223-4
Joo JC, Khusnutdinova AN, Flick R, Kim T, Bornscheuer UT, Yakunin AF, Mahadevan R (2017a) Alkene hydrogenation activity of enoate reductases for an environmentally benign biosynthesis of adipic acid. Chem Sci 8:1406–1413. https://doi.org/10.1039/C6SC02842J
Joo JC, Oh YH, Yu JH, Hyun SM, Khang TU, Kang KH, Park SJ (2017b) Production of 5-aminovaleric acid in recombinant Corynebacterium glutamicum strains from a Miscanthus hydrolysate solution prepared by a newly developed Miscanthus hydrolysis process. Bioresour Technol 245:1692–1700. https://doi.org/10.1016/j.biortech.2017.05.131
Jung JY, Choi ES, Oh MK (2008) Enhanced production of 1,2-propanediol by tpi1 deletion in Saccharomyces cerevisiae. J Microbiol Biotechnol 18:1797–1802. https://doi.org/10.4014/jmb.0800.010
Jorge JMP, Pérez-García F, Wendisch VF (2017) A new metabolic route for the fermentative production of 5-aminovalerate from glucose and alternative carbon sources. Bioresour Technol 245:1701–1709. https://doi.org/10.1016/J.BIORTECH.2017.04.108
Kabisch J, Pratzka I, Meyer H, Albrecht D, Lalk M, Ehrenreich A, Schweder T (2013) Metabolic engineering of Bacillus subtilis for growth on overflow metabolites. Microb Cell Factories 12:72. https://doi.org/10.1186/1475-2859-12-72
Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A, Tauch A (2003) The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J Biotechnol 104:5–25
Kang MK, Lee J, Um Y, Lee T, Bott M, Park SJ, Woo HM (2014) Synthetic biology platform of CoryneBrick vectors for gene expression in Corynebacterium glutamicum and its application to xylose utilization. Appl Microbiol Biotechnol 98:5991–6002. https://doi.org/10.1007/s00253-014-5714-7
Kaur G, Srivastava AK, Chand S (2012) Advances in biotechnological production of 1,3-propanediol. Biochem Eng J 64:106–118. https://doi.org/10.1016/j.bej.2012.03.002
Kawaguchi H, Vertes AA, Okino S, Inui M, Yukawa H (2006) Engineering of a xylose metabolic pathway in Corynebacterium glutamicum. Appl Environ Microbiol 72:3418–3428. https://doi.org/10.1128/AEM.72.5.3418-3428.2006
Kim J, Hirasawa T, Sato Y, Nagahisa K, Furusawa C, Shimizu H (2009) Effect of odhA overexpression and odhA antisense RNA expression on Tween-40-triggered glutamate production by Corynebacterium glutamicum. Appl Microbiol Biotechnol 81(6):1097–1106. https://doi.org/10.1007/s00253-008-1743-4
Kim BR, Lee SJ, Jeong DU, Yang JM, Oh MK, Lee JW (2014) Redistribution of carbon flux toward 2,3-butanediol production in Klebsiella pneumoniae by metabolic engineering. PLoS One 9(10):e105322. https://doi.org/10.1371/journal.pone.0105322
Kind S, Jeong WK, Schröder H, Wittmann C (2010) Systems-wide metabolic pathway engineering in Corynebacterium glutamicum for bio-based production of diaminopentane. Metab Eng 12(4):341–351. https://doi.org/10.1016/j.ymben.2010.03.005
Kind S, Neubauer S, Becker J, Yamamoto M, Volkert M, Von Abendroth G, Zelder O, Wittmann C (2014) From zero to hero—production of bio-based nylon from renewable resources using engineered Corynebacterium glutamicum. Metab Eng 25:113–123. https://doi.org/10.1016/j.ymben.2014.05.007
Kirchner O, Tauch A (2003) Tools for genetic engineering in the amino acid-producing bacterium Corynebacterium glutamicum. J Biotechnol 104:287–299. https://doi.org/10.1016/S0168-1656(03)00148-2
Koivistoinen OM, Kuivanen J, Barth D, Turkia H, Pitkanen JP, Penttila M, Richard P (2013) Glycolic acid production in the engineered yeasts Saccharomyces cerevisiae and Kluyveromyces lactis. Microb Cell Factories 12:82. https://doi.org/10.1186/1475-2859-12-82
Kortmann M, Kuhl V, Klaffl S, Bott M (2015) A chromosomally encoded T7 RNA polymerase-dependent gene expression system for Corynebacterium glutamicum: construction and comparative evaluation at the single-cell level. Microb Biotechnol 8(2):253–365. https://doi.org/10.1111/1751-7915.12236
Kotrba P, Inui M, Yukawa H (2001) The ptsI gene encoding enzyme I of the phosphotransferase system of Corynebacterium glutamicum. Biochem Biophys Res Commun 289:1307–1313. https://doi.org/10.1006/bbrc.2001.6116
Krause FS, Blombach B, Eikmanns BJ (2010) Metabolic engineering of Corynebacterium glutamicum for 2-Ketoisovalerate production. Appl Environ Microbiol 76:8053–8061. https://doi.org/10.1128/AEM.01710-10
Lange J, Muller F, Takors R, Bombach B (2018) Harnessing novel chromosomal integration loci to utilize an organosolv-derived hemicellulose fraction for isobutanol production with engineered Corynebacterium glutamicum. Microb Biotechnol 11(1):257–263. https://doi.org/10.1111/1751-7915.12879
Lee J (2014) Development and characterization of expression vectors for Corynebacterium glutamicum. J Microbiol Biotechnol 24:70–79. https://doi.org/10.4014/jmb.1310.10032
Lee SY, Kim HU (2015) Systems strategies for developing industrial microbial strains. Nat Biotechnol 33:1061–1072. https://doi.org/10.1038/nbt.3365
Lee JW, Kim TY, Jang YS, Choi S, Lee SY (2011) Systems metabolic engineering for chemicals and materials. Trends Biotechnol 29(8):370–378. https://doi.org/10.1016/j.tibtech.2011.04.001
Lee J, Sim SJ, Bott M, Um Y, Oh M, Woo HM (2014a) Succinate production from CO2-grown microalgal biomass as carbon source using engineered Corynebacterium glutamicum through consolidated bioprocessing. Sci Rep 4:5819. https://doi.org/10.1038/srep05819
Lee SM, Jellison T, Alper HS (2014b) Systematic and evolutionary engineering of a xylose isomerase-based pathway in Saccharomyces cerevisiae for efficient conversion yields. Biotechnol Biofuels 7(1):122. https://doi.org/10.1186/s13068-014-0122-x
Leßmeier L, Wendisch VF (2015) Identification of two mutations increasing the methanol tolerance of Corynebacterium glutamicum. BMC Microbiol 15(216):216. https://doi.org/10.1186/s12866-015-0558-6
Letek M, Valbuena N, Ramos A, Ordonez E, Gil JA, Mateos LM (2006) Characterization and use of catabolite-repressed promoters from gluconate genes in Corynebacterium glutamicum. J Bacteriol 188:409–423. https://doi.org/10.1128/JB.188.2.409-423.2006
Li L, Wada M, Yokota A (2007) Cytoplasmic proteome reference map for a glutamic acid-producing Corynebacterium glutamicum ATCC 14067. Proteomics 7(23):4317–4322. https://doi.org/10.1002/pmic.200700269
Lim HG, Noh MH, Jeong JH, Park S, Jung GY (2016) Optimum rebalancing of the 3-hydroxypropionic acid production pathway from glycerol in Esherichia coli. ACS Synth Biol 5(11):1247–1255. https://doi.org/10.1021/acssynbio.5b00303
Litsanov B, Brocker M, Bott M (2013) Glycerol as a substrate for aerobic succinate production in minimal medium with Corynebacterium glutamicum. Microb Biotechnol 6(2):189–195. https://doi.org/10.1111/j.1751-7915.2012.00347.x
Liu Q, Ouyang SP, Kim J, Chen GQ (2007) The impact of PHB accumulation on L-glutamate production by recombinant Corynebacterium glutamicum. J Biotechnol 132(3):273–279. https://doi.org/10.1016/j.jbiotec.2007.03.014
Liu Z, Tyo KEJ, Martínez JL, Petranovic D, Nielsen J (2012) Different expression systems for production of recombinant proteins in Saccharomyces cerevisiae. Biotechnol Bioeng 109:1259–1268. https://doi.org/10.1002/bit.24409
Liu H, Ramos KRM, Valdehuesa KNG, Nisola GM, Lee W, Chung W (2013) Biosynthesis of ethylene glycol in Escherichia coli. Appl Microbiol Biotechnol 97:3409–3417. https://doi.org/10.1007/s00253-012-4618-7
Liu X, Yang Y, Zhang W, Sun Y, Peng F, Jeffrey L, Harvey L, McNeil B, Bai Z (2015) Expression of recombinant protein using Corynebacterium glutamicum: progress, challenges and applications. Crit Rev Biotechnol 36:1–13. https://doi.org/10.3109/07388551.2015.1004519
Liu M, Han X, Xian M, Ding Y, Liu H, Zhao G (2016) Development of a 3-hydroxypropionate resistant Escherichia coli strain. Bioengineered 7(1):21–27. https://doi.org/10.1080/21655979.2015.1122143
Liu X, Zhang W, Zhao Z, Dai X, Yang Y, Bai Z (2017) Protein secretion in Corynebacterium glutamicum. Crit Rev Biotechnol 37:541–551. https://doi.org/10.1080/07388551.2016.1206059
Maervoet VET, Maeseneire SL, De Avci FG, Beauprez J, Soetaert WK (2016) High yield 1,3-propanediol production by rational engineering of the 3 -hydroxypropionaldehyde bottleneck in Citrobacter werkmanii. Microb Cell Factories 15(23):23. https://doi.org/10.1186/s12934-016-0421-y
Martin CH, Dhamankar H, Tseng HC, Sheppard MJ, Reisch CR, Prather KL (2013) A platform pathway for production of 3-hydroxyacids provides a biosynthetic route to 3-hydroxy-gamma-butyrolactone. Nat Commun 4:1414. https://doi.org/10.1038/ncomms2418
Mateos L, Pisabarro A, Patek M, Malumbres M, Guerrero C, Eikmanns B, Sahm H, Martin J (1994) Transcriptional analysis and regulatory signals of the hom-thrB cluster of Brevibacterium lactofermentum. J Bacteriol 176(23):7362–7371. https://doi.org/10.1128/jb.176.23.7362-7371.1994
Meiswinkel TM, Rittmann D, Lindner SN, Wendisch VF (2013) Crude glycerol-based production of amino acids and putrescine by Corynebacterium glutamicum. Bioresour Technol 145:254–258. https://doi.org/10.1016/j.biortech.2013.02.053
Mentz A, Neshat A, Pfeifer-Sancar K, Pühler A, Rückert C, Kalinowski J (2013) Comprehensive discovery and characterization of small RNAs in Corynebacterium glutamicum ATCC 13032. BMC Genomics 14(1):714. https://doi.org/10.1186/1471-2164-14-714
Merkamm M, Guyonvarch A (2001) Cloning of the sodA gene from Corynebacterium melassecola and role of superoxide dismutase in cellular viability. J Bacteriol 183(4):1284–1295. https://doi.org/10.1128/JB.2001.183.4.1284-1295.2001
Misra K, Banerjee AB, Ray S, Ray M (1996) Reduction of methylglyoxal in Escherichia coli K12 by an aldehyde reductase and alcohol dehydrogenase. Mol Cell Biochem 156:117–124. https://doi.org/10.1007/BF00426333
Nakamura Y, Nishio Y, Ikeo K, Gojobori T (2003) The genome stability in Corynebacterium species due to lack of the recombinational repair system. Gene 317:149–155. https://doi.org/10.1016/S0378-1119(03)00653-X
Nešvera J, Pátek M (2011) Tools for genetic manipulations in Corynebacterium glutamicum and their applications. Appl Microbiol Biotechnol 90(5):1641–1654. https://doi.org/10.1007/s00253-011-3272-9
Niimi S, Suzuki N, Inui M, Yukawa H (2011) Metabolic engineering of 1,2-propanediol pathways in Corynebacterium glutamicum. Appl Microbiol Biotechnol 90(5):1721–1729. https://doi.org/10.1007/s00253-011-3190-x
Oh YH, Eom IY, Joo JC, Yu JH, Song BK, Lee SH, Hong SH, Park SJ (2015a) Recent advances in development of biomass pretreatment technologies used in biorefinery for the production of bio-based fuels, chemicals and polymers. Korean J Chem Eng 32:1945–1959. https://doi.org/10.1007/s11814-015-0191-y
Oh YH, Choi JW, Kim EY, Song BK, Jeong KJ, Park K, Kim IK, Woo HM, Lee SH, Park SJ (2015b) Construction of synthetic promoter-based expression cassettes for the production of cadaverine in recombinant Corynebacterium glutamicum. Appl Biochem Biotechnol 176:2065–2075. https://doi.org/10.1007/s12010-015-1701-4
Ohse M, Takahashi K, Kadowaki Y, Kusaoke H (1995) Effects of plasmid DNA sizes and several other factors on transformation of Bacillus subtilis ISW1214 with plasmid DNA by electroporation. Biosci Biotechnol Biochem 59(8):1433–1437. https://doi.org/10.1271/bbb.59.1433
Oreb M, Dietz H, Farwick A, Boles E (2012) Novel strategies to improve co-fermentation of pentoses with D-glucose by recombinant yeast strains in lignocellulosic hydrolysates. Bioengineered 3(6):347–351. https://doi.org/10.4161/bioe.21444
Panova A, Mersingera LI, Liu Q, Foo T, Roe DC, Spillan WL, Sigmund AE, Ben-Bassat A, Wagner LW, DP O‘K, Wu S, Petrillo KL, Payne MS, Breske ST, Gallagher FG, Di Cosimo R (2007) Chemoenzymatic synthesis of glycolic acid. Adv Synth Catal 349:1462–1474. https://doi.org/10.1002/adsc.200700061
Papagianni M (2012) Recent advances in engineering the central carbon metabolism of industrially important bacteria. Microb Cell Factories 11:50. https://doi.org/10.1186/1475-2859-11-50
Park SD, Lee SN, Park IH, Choi JS, Jeong WK, Kim Y, Lee HS (2004) Isolation and characterization of transcriptional elements from Corynebacterium glutamicum. J Microbiol Biotechnol 14:789–795
Park JU, Jo JH, Kim YJ, Chung SS, Lee JH, Lee HH (2008) Construction of heat-inducible expression vector of Corynebacterium glutamicum and C. ammoniagenes: fusion of lambda operator with promoters isolated from C. ammoniagenes. J Microbiol Biotechnol 18:639–647
Park SH, Kim HU, Kim TY, Park JS, Kim SS, Lee SY (2014) Metabolic engineering of Corynebacterium glutamicum for L-arginine production. Nat Commun 5:4618. https://doi.org/10.1038/ncomms5618
Pátek M, Nešvera J (2013) Promoters and plasmid vectors of Corynebacterium glutamicum. In: Tatsumi N, Inui M (eds) Corynebacterium glutamicum: biology and biotechnology. Springer, Berlin, pp 51–88. https://doi.org/10.1007/978-3-642-29857-8_2
Peters-Wendisch PG, Schiel B, Wendisch VF, Katsoulidis E, Möckel B, Sahm H, Eikmanns BJ (2001) Pyruvate carboxylase is a major bottleneck for glutamate and lysine production by Corynebacterium glutamicum. J Mol Microbiol Biotechnol 3:295–300
Rados D, Carvahalo AL, Wieschalka S, Neves AR, Blombach B, Eikmanns BJ, Santos H (2015) Engineering Corynebacterium glutamicum for the production of 2,3-butanediol. Microb Cell Factories 14:171. https://doi.org/10.1186/s12934-015-0362-x
Ravasi P, Peiru S, Gramajo H, Menzella HG (2012) Design and testing of a synthetic biology framework for genetic engineering of Corynebacterium glutamicum. Microb Cell Factories 11:1–11. https://doi.org/10.1186/1475-2859-11-147
Ren C, Wen Z, Xu Y, Jiang W, Gu Y (2016) Clostridia: a flexible microbial platform for the production of alcohols. Curr Opin Chem Biol 35:65–72. https://doi.org/10.1016/j.cbpa.2016.08.024
Rittmann D, Lindner SN, Wendisch VF (2008) Engineering of a glycerol utilization pathway for amino acid production by Corynebacterium glutamicum. Appl Environ Microbiol 74(20):6216–6222. https://doi.org/10.1128/AEM.00963-08
Sakai S, Tsuchida Y, Nakamoto H, Okino S, Ichihashi O, Kawaguchi H, Yukawa H (2007) Effect of lignocellulose-derived inhibitors on growth of and ethanol production by growth-arrested Corynebacterium glutamicum R. Appl Environ Microbiol 73:2349–2353. https://doi.org/10.1128/AEM.02880-06
Sanchez-Rivera F, Cameron DC, Cooney CL (1987) Influence of environmental factors in the production of R-(−)-1,2-propanediol by Clostridium thermosacchrolyticum. Biotechnol Lett 9:449–454. https://doi.org/10.1007/BF01027450
Saxena RK, Anand P, Saran S, Isar J (2009) Microbial production of 1,3-propanediol: recent developments and emerging opportunities. Biotechnol Adv 27:895–913. https://doi.org/10.1016/j.biotechadv.2009.07.003
Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145(1):69–73. https://doi.org/10.1016/0378-1119(94)90324-7
Schneider J, Niermann K, Wendisch VF (2011) Production of the amino acids L-glutamate, L-lysine, L-ornithine and L-arginine from arabinose by recombinant Corynebacterium glutamicum. J Biotechnol 154:191–198. https://doi.org/10.1016/j.jbiotec.2010.07.009
Schneider J, Eberhardt D, Wendisch VF (2012) Improving putrescine production by Corynebacterium glutamicum by fine-tuning ornithine transcarbamoylase activity using a plasmid addiction system. Appl Microbiol Biotechnol 95(1):169–178. https://doi.org/10.1007/s00253-012-3956-9
Schwarzer A, Pühler A (1991) Manipulation of Corynebacterium glutamicum by gene disruption and replacement. Nat Biotechnol 9(1):84–87. https://doi.org/10.1038/nbt0191-84
Shin JH, Park SH, Oh YH, Choi JW, Lee MH, Cho JS, Jeong KJ, Joo JC, Yu J, Park SJ, Lee SY (2016) Metabolic engineering of Corynebacterium glutamicum for enhanced production of 5-aminovaleric acid. Microb Cell Factories 15:174. https://doi.org/10.1186/s12934-016-0566-8
Siebert D, Wendisch VF (2015) Metabolic pathway engineering for production of 1,2-propanediol and 1-propanol by Corynebacterium glutamicum. Biotechnol Biofuels 8:91. https://doi.org/10.1186/s13068-015-0269-0
Smith KM, Cho KM, Liao JC (2010) Engineering Corynebacterium glutamicum for isobutanol production. Appl Microbiol Biotechnol 87:1045–1055. https://doi.org/10.1007/s00253-010-2522-6
Srivastava P, Deb JK (2002) Construction of fusion vectors of Corynebacteria: expression of glutathione-S-transferase fusion protein in Corynebacterium acetoacidophilum ATCC 21476. FEMS Microbiol Lett 212:209–216. https://doi.org/10.1111/j.1574-6968.2002.tb11268.x
Stansen C, Uy D, Delaunay S, Eggeling L, Goergen JL, Wendisch VF (2005) Characterization of a Corynebacterium glutamicum lactate utilization operon induced during temperature-triggered glutamate production. Appl Environ Microbiol 71:5920–5928. https://doi.org/10.1128/AEM.71.10.5920-5928.2005
Stolz M, Peters-Wendisch P, Etterich H, Gerharz T, Faurie R, Sahm H, Fersterra H, Eggeling L (2007) Reduced folate supply as a key to enhanced L-serine production by Corynebacterium glutamicum. Appl Environ Microbiol 73:750–755. https://doi.org/10.1128/AEM.02208-06
Sudheer PDVN, David Y, Chae CG, Kim YJ, Baylon MG, Baritugo KA, Kim TW, Kim MS, Na JG, Park SJ (2016) Advances in biological treatment of coal for synthetic natural gas and chemicals. Korean J Chem Eng 33:2788–2801. https://doi.org/10.1007/s11814-016-0225-0
Sun Y, Guo W, Wang F, Zhan C, Yang Y, Liu X, Bai Z (2017) Transcriptome analysis of Corynebacterium glutamicum in the process of recombinant protein expression in bioreactors. PLoS One 12(4):e0174824. https://doi.org/10.1371/journal.pone.0174824
Suzuki N, Inui M (2013) Genome engineering of Corynebacterium glutamicum. In: Tatsumi N, Inui M (eds) Corynebacterium glutamicum: biology and biotechnology. Springer, Berlin, pp 89–105. https://doi.org/10.1007/978-3-642-29857-8_3
Suzuki N, Nonaka H, Tsuge Y, Inui M, Yukawa H (2005) New multiple-deletion method for the Corynebacterium glutamicum genome, using a mutant lox sequence. Appl Environ Microbiol 71:8472–8480. https://doi.org/10.1128/AEM.71.12.8472-8480.2005
Tateno T, Okada Y, Tsuchidate T, Tanaka T, Fukuda H, Kondo A (2009) Direct production of cadaverine from soluble starch using Corynebacterium glutamicum coexpressing alpha-amylase and lysine decarboxylase. Appl Microbiol Biotechnol 82:115–121. https://doi.org/10.1007/s00253-008-1751-4
Teramoto H, Watanabe K, Suzuki N, Inui M, Yukawa H (2011) High yield secretion of heterologous proteins in Corynebacterium glutamicum using its own tat-type signal sequence. Appl Microbiol Biotechnol 91(3):677–687. https://doi.org/10.1007/s00253-011-3281-8
Tran-Din K, Gottschalk G (1985) Formation of D(−)-1,2-propanedioland D(−)-lactate from glucose by Clostridium sphenoides underphosphate limitation. Arch Microbiol 142:87–92. https://doi.org/10.1007/s00253-011-3281-8
Tsuchiya M, Morinaga Y (1988) Genetic control systems of Escherichia coli can confer inducible expression of cloned genes in coryneform bacteria. Nat Biotechnol 6:428–430. https://doi.org/10.1038/nbt0488-428
Tsuge Y, Suzuki N, Inui M, Yukawa H (2007) Random segment deletion based on IS31831 and Cre/loxP excision system in Corynebacterium glutamicum. Appl Microbiol Biotechnol 74:1333–1341. https://doi.org/10.1007/s00253-006-0788-5
Venkova-Canova T, Pátek M, Nesvera J (2004) Characterization of the cryptic plasmid pCC1 from Corynebacterium callunae and its use for vector construction. Plasmid 51(1):54–60. https://doi.org/10.1016/j.plasmid.2003.09.002
Vertès AA, Inui M, Kobayashi M, Kurusu Y, Yukawa H (1993) Presence of mrr- and mcr-like restriction systems in coryneform bacteria. Res Microbiol 144:181–185. https://doi.org/10.1016/0923-2508(93)90043-2
Wei G, Yang X, Gan T, Zhou W, Lin J, Wei D (2009) High cell density fermentation of Gluconobacter oxydans DSM 2003 for glycolic acid production. J Ind Microbiol Biotechnol 36:1029–1034. https://doi.org/10.1007/s10295-009-0584-1
Wendisch VF (2003) Genome-wide expression analysis in Corynebacterium glutamicum using DNA microarrays. J Biotechnol 104:273–285. https://doi.org/10.1016/S0168-1656(03)00147-0
Wendisch VF, Bott M, Eikmanns BJ (2006a) Metabolic engineering of Escherichia coli and Corynebacterium glutamicum for biotechnological production of organic acids and amino acids. Curr Opin Microbiol 9:268–274. https://doi.org/10.1016/j.mib.2006.03.001
Wendisch VF, Bott M, Kalinowski J, Oldiges M, Wiechert W (2006b) Emerging Corynebacterium glutamicum systems biology. J Biotechnol 124:74–92. https://doi.org/10.1016/j.jbiotec.2005.12.002
Wieschalka S, Blombach B, Bott M, Eikmanns BJ (2013) Bio-based production of organic acids with Corynebacterium glutamicum. Microb Biotechnol 6:87–102. https://doi.org/10.1111/1751-7915.12013
Woo HM, Park JB (2014) Recent progress in development of synthetic biology platforms and metabolic engineering of Corynebacterium glutamicum. J Biotechnol 180:43–51. https://doi.org/10.1016/j.jbiotec.2014.03.003
Woo HM, Noack S, Seibold G, Willbold S, Eikmanns B, Bott M (2010) Link between phosphate starvation and glycogen metabolism in Corynebacterium glutamicum, revealed by metabolomics. Appl Environ Microbiol 76:6910–6919. https://doi.org/10.1128/AEM.01375-10
Xu Y, Chu H, Gao C, Tao F, Zhou Z, Li K, Li L, Ma C, Xu P (2014) Systematic metabolic engineering of Escherichia coli for high-yield production of fuel biochemical 2,3-butanediol. Metab Eng 23:22–33. https://doi.org/10.1016/j.ymben.2014.02.004
Yamamoto S, Suda M, Niimi S, Inui M, Yukawa H (2013) Strain optimization for efficient isobutanol production using Corynebacterium glutamicum under oxygen deprivation. Biotechnol Bioeng 110(11):2938–2948. https://doi.org/10.1002/bit.24961
Yang J, Yang S (2017) Comparative analysis of Corynebacterium glutamicum genomes: a new perspective for the industrial production of amino acids. BMC Genomics 18:940. https://doi.org/10.1186/s12864-016-3255-4
Yang J, Kim B, Kim H, Kweon Y, Lee S, Lee J (2015) Industrial production of 2,3-butanediol from the engineered Corynebacterium glutamicum. Appl Biochem Biotechnol 176(8):2303–2313. https://doi.org/10.1007/s12010-015-1719-7
Yang JE, Kim JW, Oh YH, Choi SY, Lee H, Park AR, Shin J, Park SJ, Lee SY (2016) Biosynthesis of poly(2-hydroxyisovalerate-co-lactate) by metabolically engineered Escherichia coli. Biotechnol J 11:1572–1585. https://doi.org/10.1002/biot.201600420
Yang JE, Park SJ, Kim WJ, Kim HJ, Kim BJ, Lee H, Shin J, Lee SY (2018) One-step fermentative production of aromatic polyesters from glucose by metabolically engineered Escherichia coli strains. Nat Commun 9(79):79. https://doi.org/10.1038/s41467-017-02498-w
Yim SS, An SJ, Kang M, Lee J, Jeong KJ (2013) Isolation of fully synthetic promoters for high-level gene expression in Corynebacterium glutamicum. Biotechnol Bioeng 110(11):2959–2969. https://doi.org/10.1002/bit.24954
Yim SS, An SJ, Choi JW, Ryu AJ, Jeong KJ (2014) High-level secretory production of recombinant single-chain variable fragment (scFv) in Corynebacterium glutamicum. Appl Microbiol Biotechnol 98:273–284
Yim SS, Choi JW, Lee SH, Jeong KJ (2016a) Modular optimization of a hemicellulose-utilizing pathway in Corynebacterium glutamicum for consolidated bioprocessing of hemicellulosic biomass. ACS Synth Biol 5:334–343. https://doi.org/10.1021/acssynbio.5b00228
Yim SS, Choi JW, Lee RJ, Lee YJ, Lee SH, Kim SY, Jeong KJ (2016b) Development of a new platform for secretory production of recombinant proteins in Corynebacterium glutamicum. Biotechnol Bioeng 113(1):163–172. https://doi.org/10.1002/bit.25692
Yim SS, Choi JW, Lee SH, Jeon EJ, Chung WJ, Jeong KJ (2017) Engineering of Corynebacterium glutamicum for consolidated conversion of hemicellulosic biomass into xylonic acid. Biotechnol J 12(11):1700040. https://doi.org/10.1002/biot.201700040
Yukawa H, Omumasaba CA, Nonaka H, Kos P, Okai N, Suzuki N, Suda M, Tsuge Y, Watanabe J, Ikeda Y, Vertes AA, Inui M (2007) Comparative analysis of the Corynebacterium glutamicum group and complete genome sequence of strain R. Microbiology 153:1042–1058. https://doi.org/10.1099/mic.0.2006/003657-0
Zahoor A, Lindner SN, Wendisch VF (2012) Metabolic engineering of Corynebacterium glutamicum aimed at alternative carbon sources and new products. Comput Struct Biotechnol J 3:e201210004. https://doi.org/10.5936/csbj.201210004
Zahoor A, Otten A, Wendisch VF (2014) Metabolic engineering of Corynebacterium glutamicum for glycolate production. J Biotechnol 192:366–375. https://doi.org/10.1016/j.jbiotec.2013.12.020
Zhang Q, Xiu Z (2009) Metabolic pathway analysis of glycerol metabolism in Klebsiella pneumoniae incorporating oxygen regulatory system. Biotechnol Prog 25:103–115. https://doi.org/10.1002/btpr.70
Zhang Y, Shang X, Lai S, Zhang G, Liang Y, Wen T (2012) Development and application of an arabinose-inducible expression system by facilitating inducer uptake in Corynebacterium glutamicum. Appl Environ Microbiol 78:5831–5838. https://doi.org/10.1128/AEM.01147-12
Zhang B, Yu M, Zhou Y, Li Y, Ye BC (2017) Systematic pathway engineering of Corynebacterium glutamicum S9114 for L-ornithine production. Microb Cell Factories 16(158):158. https://doi.org/10.1186/s12934-017-0776-8
Zhou LB, Zeng AP (2015) Engineering a lysine-ON riboswitch for metabolic control of lysine production in Corynebacterium glutamicum. ACS Synth Biol 4(12):1335–1340. https://doi.org/10.1021/acssynbio.5b00075
Zhu Q, Zhang X, Luo Y, Guo W, Xu G, Shi J, Xu Z (2014) L-serine over-production with minimization of by-product synthesis by engineered Corynebacterium glutamicum. Appl Microbiol Biotechnol 99:1665–1673. https://doi.org/10.1007/s00253-014-6243-0
Acknowledgements
This work was supported by the Mid-career Researcher Program through the National Research Foundation (NRF) of Korea funded by the Ministry of Science and ICT (MSIT) (NRF-2016R1A2B4008707), a basic research grant from the KRIBB, and the Lignin Biorefinery from MSIT through the NRF of Korea (NRF-2017M1A2A2087634).
Availability of data and material
Please contact corresponding author for any data requests.
Funding
Funding sources are declared in acknowledgement section.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interest
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable. This article does not contain any studies with human participants or animals performed by any of the authors.
Consent for publication
Not applicable. Our manuscript does not contain any individual person’s data in any form.
Rights and permissions
About this article

Cite this article
Baritugo, KA., Kim, H.T., David, Y. et al. Metabolic engineering of Corynebacterium glutamicum for fermentative production of chemicals in biorefinery. Appl Microbiol Biotechnol 102, 3915–3937 (2018). https://doi.org/10.1007/s00253-018-8896-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-8896-6