
Abstract
Drug–drug interactions (DDIs) occur commonly and may lead to severe adverse drug reactions if not handled appropriately. Considerable information to support clinical decision making regarding potential DDIs is available in the literature and through various systems providing electronic decision support for healthcare providers. The challenge for the prescribing physician lies in sorting out the evidence and identifying those drugs for which potential interactions are likely to become clinically manifest. P-glycoprotein (P-gp) is a drug transporting protein that is found in the plasma membranes in cells of barrier and elimination organs, and plays a role in drug absorption and excretion. Increasingly, P-gp has been acknowledged as an important player in potential DDIs and a growing body of information on the role of this transporter in DDIs has become available from research and from the drug approval process. This has led to a clear need for a comprehensive review of P-gp-mediated DDIs with a focus on highlighting the drugs that are likely to lead to clinically relevant DDIs. The objective of this review is to provide information for identifying and interpreting evidence of P-gp-mediated DDIs and to suggest a classification for individual drugs based on both in vitro and in vivo evidence (substrates, inhibitors and inducers). Further, various ways of handling potential DDIs in clinical practice are described and exemplified in relation to drugs interfering with P-gp.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
P-glycoprotein (P-gp) is a drug transporting protein that physicians should be aware of when evaluating potential drug–drug interactions. |
The evidence base for evaluating the role of P-gp in potential drug–drug interactions is varying and interpretation may be complex. |
Potential drug–drug interactions should be identified and appropriate management strategies applied. |
1 Introduction
Adverse drug events (ADEs) are a common cause of hospital admission [1, 2] and display a potential negative impact on patient morbidity and mortality, treatment costs and duration of hospitalization [3, 4]. In this context, interactions between concomitantly used drugs may be a cause of serious ADEs [5, 6]. Formally, a drug interaction is defined as an alteration in a clinically meaningful way of the effect of a drug (object or victim drug) as a result of co-administration of another drug (precipitant or perpetrator drug) [7]. While not all potential drug–drug interactions (DDIs) are clinically significant [8–10], the outcome (therapeutic failure or toxicity) when actual DDIs occur may be severe and sometimes fatal [9]. The clinical challenge lies in sorting out the unimportant interactions and identifying the DDIs that are most likely to be clinically manifest. The evidence base for particular DDIs may be of varying quality and whether or not a particular DDI is clinically meaningful may vary depending on a number of individual patient characteristics such as age, organ function, genetic makeup, comorbidity as well as concurrent medication.
The mechanisms responsible for DDIs may be pharmacodynamic or pharmacokinetic. An example of a pharmacodynamic interaction is excessive bleeding with concurrent use of warfarin, which is a vitamin K antagonist, and low-dose aspirin, where warfarin affects bleeding through decreased production of coagulation factors and aspirin through inhibition of thrombocyte aggregation [7, 11, 12]. Pharmacokinetic interactions can occur due to changes in the absorption, distribution, metabolism and elimination of a drug and may result in higher or lower drug levels leading to potential toxicities or loss of efficacy. These processes may be altered by drugs interfering with drug metabolizing enzymes such as those belonging to the cytochrome P450 system (CYP450), involved in phase I reactions, or uridine diphosphate glucuronosyltransferases (UGTs), involved in phase II reactions. Drugs may also affect the pharmacokinetics of other drugs by interfering with drug transporters such as the p-glycoprotein efflux transporter (P-gp), breast cancer resistance protein (BCRP) or the organic anion transporting polypeptides (OATPs) [13]. Individual drugs may thus be substrates, inhibitors or inducers of specific enzymes or transporters and concurrent administration of, for example, an inhibitor of a transporter or an enzyme may imply an increased concentration of a drug that is a substrate of the same transporter or enzyme and subsequently an increased risk of ADEs. Conversely, administration of an inducer may decrease the concentration of a substrate of a particular enzyme or transporter, and lead to decreased effect of the drug.
The focus of the present review is the P-gp drug transporter in relation to DDIs. Particularly, we aim to synthesize the evidence for classifying various drugs as substrates, inhibitors and inducers of P-gp according to in vitro and in vivo evidence. We searched the literature and drug labels/summaries of product characteristics (SmPCs) to identify human in vitro and in vivo evidence to support the particular P-gp modulatory status (substrate, inhibitor, inducer) of the various drugs. The review consists of the following main parts:
Section 2 is a short introduction to P-gp (gene, protein and transport molecule).
Section 3 includes methodological considerations for compilation of the list of drugs to be evaluated for P-gp modulatory properties and evaluation criteria. The compiled list of drugs was evaluated by means of a two-step procedure evaluating (1) in vitro evidence (human P-gp) and (2) if available, human in vivo studies.
Section 4 consists of five tables of drugs categorized according to therapeutic groups as P-gp modulators (substrates, inhibitors or inducers) for which we found compelling evidence of potential clinical relevance. The remaining drugs, for which we did not find compelling evidence of clinical relevance, are presented in five similar tables in the Electronic Supplementary Material (ESM, Supplementary Tables 1–5). All tables in the main paper and in the ESM provide the specific references forming the basis of the classification of the individual drugs.
Section 5 consists of general considerations regarding clinical management of potential DDIs including elaborations on drugs from the tables in Sect. 4 as well as a discussion of limitations of the suggested classification of drugs as P-gp modulators.
Section 6 consists of conclusions and perspectives.
2 Background
2.1 P-Glycoprotein: Gene, Protein and Transport Molecule
P-gp is a protein encoded for by the ABCB1 (MDR1) gene, which belongs to the group of adenosine triphosphate (ATP)-binding cassette (ABC) genes [14] encoding the widespread ATP-binding cassette transport proteins. At present, 49 members of the ABC superfamily have been identified and grouped into seven subfamilies: ABCA to ABCG [15]. In eukaryotes, all ABC proteins are efflux pumps and play a role in protecting the organism from noxious substances [16].
ABCB1 is located on chromosome 7q21.2 and encodes P-gp which consists of 1276–1280 amino acids and has a molecular mass of around 170 kDa. The topological structure of P-gp consists of two homologous halves, each with six highly hydrophobic transmembrane domains and a nucleotide-binding domain. ATP must bind to both nucleotide-binding domains for activity of P-gp to occur. Moreover, it contains multiple drug-binding sites and is able to simultaneously bind multiple substrates at overlapping binding sites [17].
P-gp exhibits polarized expression and is found in the plasma membranes of cells in barrier and elimination organs. The transporter is expressed on the luminal-facing epithelia of the gut and the bile-facing canaliculi of the liver, where it plays a role in first-pass drug metabolism. It is also found on the luminal membrane in the proximal tubules of the kidney, eliminating substances from the systemic circulation. Further, it is present in the blood-brain barrier and limits the permeability of many drugs into the brain as well as into other specific organs such as the placenta and testis [16].
P-gp substrates display a large diversity in structure including both small molecules (e.g. organic cations, carbohydrates and amino acids) and macromolecules (polysaccharides and proteins) [17–19]. Drugs that are substrates of P-gp may or may not also be inhibitors or inducers of P-gp, and vice versa [17, 20]. A number of drugs that inhibit P-gp, however, have some chemical properties in common, such as aromatic ring structures, a tertiary or secondary amino group and high lipophilicity [17]. Regarding induction of P-gp, this does not occur by the drug binding directly to P-gp but instead by regulation at the transcriptional level by nuclear factors [21]. For instance, the xenobiotic nuclear receptor (PXR) regulates ABCB1 expression and may be activated by, for example, rifampicin [17]; in this context, animal studies demonstrating presence of PXR at the level of the blood–brain barrier might contribute to explaining the decreased efficacy of CNS-acting drugs that are also P-gp substrates [22].
Genetic polymorphisms of clinical importance exist for some of the drug metabolizing enzymes (CYP2C9, CYP2C19 and CYP2D6). These account for a substantial portion of interpersonal variability in drug metabolism [13]. For MDR1, a vast number of commonly occurring single nucleotide polymorphisms have been identified and characterized. However, here results have been inconsistent and no clear association with clinical risk profile has been established. Thus, present evidence does not yield support for recommendations of adjustment in drug dose according to specific ABCB1 sequence variants [16, 23, 24].
2.2 Overlap in Drugs That Interfere with Both P-Glycoprotein (P-gp) and CYP3A4
There is a substantial overlap in drugs that interact with CYP3A4 and P-gp [25]. However, the overlap is by no means complete, and no clear rules seem to exist for determining whether or not an overlap exists [26]. P-gp and CYP3A4 are found in many of the same organs and tissues [25, 27, 28] and seem to function in a complementary fashion to reduce systemic drug exposure: as a drug traverses down the intestinal tract, repeated cycles of P-gp extrusion followed by passive reabsorption facilitates repeated cycles of exposure of the drug to CYP3A4 present in the gut wall and thus to increased metabolism and reduced bioavailability [29]. The structure–activity overlap between CYP3A4 and P-gp may thus complicate a clear determination of the relative contribution of P-gp versus CYP3A4 to a particular DDI. However, various approaches may increase the likelihood of isolating an effect attributable to P-gp. These include use of an appropriate and validated cellular system for in vitro studies, appropriate choice of substrate and inhibitors, and performing in vivo studies when indicated by the results of in vitro studies [30]. Examples of probe drugs are midazolam (CYP3A4 substrate for which P-gp is not a barrier for absorption) and digoxin, dabigatran etexilate or fexofenadine (P-gp substrates that are not metabolized by CYP3A4) [31, 32]. With respect to inhibitors, it is important to note that dual inhibitors of CYP3A4 and P-gp do not per se have the same inhibitory potency towards P-gp and CYP3A4. For example, quinidine and amiodarone are potent P-gp inhibitors (as defined by a >1.5-fold change in fexofenadine or digoxin area under the concentration time curve [AUC]) but weak CYP3A4 inhibitors, whereas itraconazole is a potent inhibitor of both CYP3A4 and P-gp [32].
3 Methodological Considerations
3.1 Compilation of Drugs for Evaluation and Literature Search
First, we compiled a list of drugs to be evaluated for P-gp modulation. The list consisted of (1) drugs mentioned in previous reviews of P-gp: substrates [16, 33–36], inhibitors [16, 33, 36, 37] and inducers [16, 33, 36]; (2) drugs in the list of newly marketed drugs within the last 6 months approved by either the Danish Medicines Agency as of January 2015 [38] and/or the European Medicines Agency (EMA) as of October 2015 [39] and where P-gp modulatory properties were mentioned in the SmPC; and (3) drugs mentioned with P-gp modulatory properties in papers identified in step 1 or references therein. Second, we sought to identify background evidence regarding the respective P-gp modulatory property for each drug in the compiled list. We did this by a reference search of the before-mentioned reviews and/or searched for information in the SmPC from either the Danish Medicines Agency [40], EMA [39] or in the drug label from the United States Food and Drug Administration (FDA) [41]. If approved by the EMA or FDA, we also searched supporting documents available online (e.g. from the EMA, the Scientific Discussion or the European Public Assessment Report (EPAR) and from the FDA, the Clinical Pharmacology and Biopharmaceutics Review(s) [39, 41]. Finally, if necessary we searched PubMed for additional evidence including by use of the following search strings for substrates, inhibitors and inducers, respectively. Substrates and inhibitors: ((“P-Glycoprotein/agonists” [Mesh] OR “P-Glycoprotein/antagonists and inhibitors” [Mesh] OR “P-Glycoprotein/metabolism” [Mesh] OR “P-Glycoprotein/pharmacokinetics” [Mesh] OR “P-Glycoprotein/pharmacology” [Mesh]) AND “P-Glycoprotein” [Mesh]) AND “Suspected Drug” [Mesh] AND “humans” [MeSH terms]. For inducers we searched on combinations of the suspected drug as a Mesh-term combined with “Digoxin” [Mesh], “talinolol” [Supplementary Concept] or “fexofenadine” [Supplementary Concept], respectively.
The evidence base for evaluating the potential for DDIs varies substantially for newer versus older drugs likely due to the increased regulatory focus on discovering potential for DDIs during recent years [42]. Previous reviews and database sources regarding DDIs involving P-gp vary quite substantially both with respect to which drugs are mentioned as substrates, inhibitors or inducers and to what constitutes potential clinical relevance. Explanations for this are likely related to differential target audiences of the data sources (e.g. a referential overview aiming to include all possible evidence as opposed to tools for clinical decision making, which may be more restrictive) as well as different approaches to evaluation and categorization of the available evidence. The scope of the present review is clinical, that is, we aim to provide the clinician with a compilation of the evidence base regarding the P-gp modulatory status for various drugs as well as a classification regarding clinical relevance. This is meant to be used as a supporting document when, for example, performing medication reviews, deciding on a new drug treatment or retrospectively when trying to elucidate a potential mechanistic basis for an observed DDI; that is, it should not be seen as a list of clear-cut contraindications. With this aim in mind, we chose a restrictive approach focusing on maximizing the likelihood of clinical relevance. In terms of classification, this implied disregarding evidence from studies using non-human P-gp and focusing on evidence from in vitro studies of human P-gp and corresponding in vivo studies. The final classification (defined in Sects. 3.2 and 3.3) contained one level for inducers (solely based on in vivo evidence) and two levels for substrates and inhibitors (one based on in vitro evidence and one on in vivo evidence). An initial evidence search and grading of the evidence for all drugs in the compiled list formed the basis for a discussion of the classification of each drug among co-authors and the final classification for each drug was based on a consensus agreement.
3.2 Substrates and Inhibitors
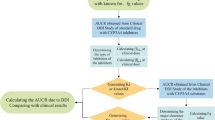
In the 2012 FDA draft guidance on investigation of potential DDIs, it is advised that all newly marketed drugs should be evaluated in vitro to determine whether they are a P-gp substrate and, depending on the therapeutic use (e.g. probability of being prescribed with digoxin, a P-gp substrate with a narrow therapeutic index), evaluation of P-gp inhibitory properties should be considered [32]. Flowcharts for assessment of both P-gp substrate and inhibitor status have previously been published [30] and incorporated by the FDA in their guidance [32]. A similar guidance is provided by EMA [31]. Aiming to construct a classification that would acknowledge and accommodate the differential levels of evidence available for newer versus older drugs, we used the FDA classifications (see Figs. 1, 2) to guide our criteria for evidence needed to obtain the different classifications described below. Overall, the classification was based on two steps for substrates and inhibitors: the first step was based on in vitro evidence and the second step on in vivo evidence.

Decision tree for determining status as P-glycoprotein substrate used for regulatory guidance (modified from [30]). aPreclinical and clinical information should be assessed to determine whether a new molecular entity is a P-gp substrate. Particularly, the relative contribution of the transporter-mediated pathway to the overall clearance of the drug is the primary determinant of whether an inhibitor will have a major effect on the disposition of the new molecular entity; i.e. an in vivo interaction study may not be needed for a drug that has high solubility, high permeability and/or is highly metabolized since it is less likely to be affected by a P-gp inhibitor [30]. DDI drug–drug interaction, P-gp P-glycoprotein

Decision tree for determining status as P-glycoprotein inhibitor used for regulatory guidance (modified from [30]). a[I]1 represents the mean steady-state total (free and unbound) maximum serum concentration (C max) following administration of the highest proposed clinical dose. [I]2 represents concentration of a therapeutic dose of the inhibitor dissolved in 250 mL. IC50 is defined as the concentration of the inhibitor needed to inhibit half the transport rate of the substrate. P-gp P-glycoprotein
In the first step, based on in vitro studies of human P-gp, we categorized evidence of clinical relevance for substrates and inhibitors of P-gp as ‘yes’, ‘no’ or ‘uncertain’. To qualify for categorization ‘yes’ as a substrate, we required evidence for polarized transport from a study using cells overexpressing P-gp in a bidirectional assay with a net efflux above 2. If, with the addition of a known P-gp inhibitor, the positive net efflux was not inhibited by 50% or more or the efflux ratio was not attenuated towards 1, we took this as evidence that other drug transporters were likely to be responsible for the observed polarized net efflux [43]. Acceptable cells for categorization ‘yes’ were (for both substrates and inhibitors) Caco-2 cells (human colon carcinoma cell monolayers) or either of the recombinant epithelial cell lines MDR1-MDCK (Madine-Darby canine kidney cells transfected with the human MDR1 gene) or L-MDR1 cells (porcine kidney epithelial cells, LLC-PK1, transfected with the human MDR1 gene). To qualify for categorization ‘yes’ as an inhibitor, we required evidence for likely clinically relevant inhibition based on calculation of [I]1/IC50 or [I]2/IC50 (as defined in Fig. 2), and if this was not found we classified as ‘no’. If evidence was in various ways insufficient or incomplete (e.g. by being based on other cell lines or by lacking a positive control but otherwise indicating that the drug was a P-gp substrate), we would classify as ‘uncertain’. We also used categorization as ‘uncertain’ if evidence from established cell lines was conflicting (for both substrates and inhibitors). If the drug was marketed within recent years and the label or SmPC explicitly mentioned that the drug had been shown to be an in vitro P-gp substrate or P-gp inhibitor but did not mention specifically which cells had been used, we classified as ‘yes’ assuming that this was likely to be one of the established cell lines (except if mentioned that this was not thought to be clinically relevant in which case we categorized as ‘no’).
In the second step, for drugs classified as ‘yes’ or ‘uncertain’ (as substrates or inhibitors) in the first step, we then classified evidence of clinical relevance as ‘yes’, ‘no’ or ‘uncertain’ based on in vivo evidence. For substrates, we classified evidence of clinical relevance based on changes in the AUC and/or adverse effects taking into account the specific pharmacokinetics of the particular object drug in question such as the therapeutic window (i.e. no global AUC cut-off for substrates). For a drug to qualify for categorization ‘yes’ as an inhibitor, we used an increase in AUC for a model P-gp substrate (digoxin, talinolol, fexofenadine or dabigatran etexilate) of at least 25% as cut-off for assessing clinical relevance [32]. For increases in AUC < 25% we classified as ‘no’, and if results were conflicting, based on a non-model substrate, or if no relevant in vivo evidence was identifiable or otherwise unclear, we categorized as ‘uncertain’.
3.3 Inducers
Since in vitro methods for studying P-gp induction do not currently provide clear evidence, we based classification of inducers solely on in vivo evidence of clinical relevance as ‘yes’, ‘no’ or ‘uncertain’—in the latter case, such as if a drug has been observed to be an inducer of enzymes via nuclear receptors such as PXR, but where no direct in vivo evidence was found [31, 43]. To qualify for classification as ‘yes’, we required a >20% reduction in AUC with digoxin, talinolol, fexofenadine or dabigatran etexilate as substrates [32]. If less was observed, we classified as ‘no’, and if evidence was conflicting or otherwise unclear, or other substrates had been used, we classified as ‘uncertain’. A few drugs were both inhibitors and inducers of P-gp with inhibition being dominant in the acute phase and induction in the chronic phase (rifampicin, ritonavir and tipranavir).
3.4 Notes on Interpretation of Tables
In the main tables (Tables 1, 2, 3, 4, 5), we highlight the drugs for which the evidence regarding the potential for clinically manifest P-gp-mediated DDIs was most compelling; that is, substrates and inhibitors classified as either (in vitro evidence–in vivo evidence): ‘yes’–’yes’, ‘uncertain’–’yes’, or ‘yes’–’uncertain’, and inducers classified as ‘yes’. Drugs with the remaining combinations of in vitro and in vivo classifications are presented in five similar tables in the ESM (Supplementary Tables 1–5). For drugs classified as ‘no’ in the first step based on in vitro evidence (substrates or inhibitors), no evaluation of in vivo evidence was undertaken (assuming that transporters other than P-gp would be responsible for any in vivo evidence).
Before using the results from the present review to guide daily clinical practice, a number of challenges regarding classification and interpretation merit discussion. For instance, an effect attributed to P-gp may in fact be driven by other (known or unknown) drug transporters or drug-metabolizing enzymes and isolating an effect attributable to P-gp may not always be possible. For this reason, if there was evidence qualifying the drug as a P-gp substrate in vitro (classification ‘yes’) and in vivo evidence of a clinically relevant change in AUC, which may be driven by P-gp but also by other enzymes or transporters, we classified as ‘yes’ in the second step with respect to clinical relevance. To acknowledge shared contribution from P-gp and CYP3A4 in vivo, a column denoting whether a drug is a known substrate, inhibitor or inducer of CYP3A4 is included in the tables presented in the main paper. One example of this is the protein kinase inhibitor, dabrafenib, which is an in vitro substrate of human P-gp. Dabrafenib has a high oral bioavailability of 94.5% and a high metabolic clearance. When dabrafenib is co-administered with ketoconazole, which is a potent inhibitor of both CYP3A4 and P-gp, the AUC of dabrafenib increased by 57% [44]. However, given the high oral bioavailability and high metabolic clearance of dabrafenib, this increase in AUC is likely driven by the inhibitory effects on CYP3A4 decreasing the clearance of dabrafenib rather than by the inhibitory effects of P-gp on extent of absorption (cf. Supplementary Table 1, ESM). On the other hand, aliskiren (an antihypertensive drug, cf. Table 2) is an example of a P-gp substrate that is eliminated mainly as an unchanged compound in the faeces and where only approximately 1.4% of the total oral dose is metabolized [45]. Thus, in this case the 4- to 5-fold increase in aliskiren AUC with co-administration of ciclosporin A is likely driven by P-gp as opposed to by CYP3A4.
Cobicistat, a CYP3A4 inhibitor used for pharmacokinetic boosting of HIV-protease inhibitors, is an example of a newly marketed drug where the information available on results of in vitro testing for P-gp and CYP3A4 modulatory properties is more extensive as compared with older drugs. In addition, cobicistat is an example of a drug where the interpretation of P-gp modulatory status is especially complex. In vitro cobicistat has been shown to be both a substrate and an inhibitor of P-gp and CYP3A4 and according to our classification qualifies for classification ‘yes’ in the first step based on in vitro data [46]. With respect to concurrent in vivo administration with digoxin, AUC of digoxin is, however, only increased by 8% with co-administration of cobicistat. Thus, in the second step based on in vivo data, cobicistat classifies as ‘no’ as an inhibitor and is consequently mentioned in Supplementary Table 3 (ESM) [46]. Of note, the manufacturer still recommends monitoring digoxin levels (and also levels of dabigatran etexilate) on co-administration [47]. While cobicistat is a (high-permeability) P-gp substrate in vitro [46], in vivo evidence is sparse. According to the SmPC, concurrent administration with inhibitors or inducers of CYP3A4 (i.e. potentially also P-gp) is warned against. These interactions have not always been studied in vivo (e.g. azole antibiotics) but are based on theoretical extrapolations and the disentangling of the interaction potential may further be complicated by the fact that cobicistat is administered for clinical purposes as a pharmacokinetic enhancer together with the HIV-protease inhibitors darunavir or azatanavir or with the HIV-integrase inhibitor elvitegravir [47, 48]. Thus, the individual effect of P-gp on cobicistat may be hard to isolate, and this is why, based on the current evidence, we have classified evidence of clinical relevance for its status as substrate as ‘uncertain’ (cf. Table 3).
Ketoconazole is an antifungal agent and a dual inhibitor of CYP3A4 and P-gp. However, the clinical relevance of this for potential in vivo DDIs appears to be dose dependant. For instance, when ketoconazole is administered with digoxin (200 mg per day for 4 days), a 9% increase in AUC has been observed [49]. Further, for administration of fexofenadine, an 8% decrease in fexofenadine AUC has been observed with pretreatment with ketoconazole 200 mg daily for 5 days [50], but a 164% increase with coadministration of ketoconazole 400 mg once daily [51] (cf. Table 3). Likewise, for coadministration of ketoconazole 400 mg with dabigatran etexilate, the AUC of the latter increases by approximately 2.5-fold and the combination is contraindicated according to the manufacturer [52].
4 Main Tables of Drugs with P-Glycoprotein-Modulatory Properties Classified According to In Vitro and In Vivo Evidence
5 Clinical Implications
Once a potential DDI has been identified, the next step is to predict the likelihood that this may result in a clinically manifest DDI and decide on the appropriate measures to be taken. These could span from no additional measures (i.e. if the potential DDI is rendered harmless), to additional monitoring (e.g. treatment with closer than usual follow-up with appropriate laboratory tests, physical exams or therapeutic drug monitoring), dose adjustment, staggered administration or in some cases frank contraindication coupled with a search for alternative treatments. Appropriate measures to be taken depend on a number of factors relating to characteristics of both the precipitant and object drugs as well as patient characteristics.
5.1 Considerations Relating to Individual Drug Characteristics
5.1.1 Dose
For P-gp, dose-dependent DDIs may occur. In particular, loperamide, an anti-diarrhoeal agent that reduces motility of the gut by interfering with the opioid receptor in the gut wall [320], is an example of a P-gp substrate for which the clinical relevance of a potential DDI is dose dependent. Sadeque et al. [276] conducted a study in eight healthy volunteers who received a single dose of loperamide 16 mg in the presence of either quinidine 600 mg (P-gp inhibitor) or placebo. The study showed a significant difference in the occurrence of respiratory depression in individuals who were administered quinidine as opposed to placebo. A subsequent study investigated the effects of quinidine and HM30181 (third-generation P-gp inhibitor with a very low bioavailability of 0.3%) on the pharmacokinetics and pharmacodynamics of a single oral dose of loperamide 16 mg [274]. Both inhibitors resulted in a clinically relevant increase in loperamide AUC as compared with loperamide on its own (HM30181 48% increase and quinidine 120% increase), and for concomitant administration of quinidine a marked reduction in pupil size was also seen. Concurrent administration of loperamide 16 mg as a single dose with ketoconazole resulted in a 5-fold increase in the plasma concentration of loperamide but no pharmacodynamic effect as assessed by pupillometry [320]. This may be explained by a differential level of P-gp inhibition in the gut and the CNS, due to differences in the inhibitor concentration at the two sites, implying that a clinically relevant increase in plasma AUC due to administration of an oral P-gp inhibitor may not transfer directly to a clinically manifest CNS affection. Thus, while the transport of loperamide across the blood–brain barrier and into the CNS is increased by P-gp inhibition [274, 321], the clinical relevance of this has been disputed [275]. Evidence to support the presence of a clinically relevant interaction between loperamide and P-gp inhibitors is insufficient when loperamide is taken at the recommended dose, which according to the SmPC is 2 mg at a time for multiple doses [320]. However, the half-life of loperamide is 11 h, which following multiple 2-mg doses may result in drug accumulation, and thus toxicity cannot be excluded in case of repeated administration or abuse [322]. We have therefore chosen to classify loperamide as ‘yes’ with respect to both in vitro and in vivo evidence (substrate, cf. Table 3).
Finally, for classification of some of the CNS drugs as P-gp inhibitors, based on in vitro studies, values of I2/IC50 may be above or below the cut-off of 10 (cf. Fig. 2) depending on the doses recommended for the various therapeutic indications. Thus, for higher doses, in vitro evidence may indicate potential for a clinically relevant DDI whereas this may not be the case for lower doses. In these instances, we have classified these drugs as ‘yes’ with respect to inhibitory status (in vitro evidence) but have mentioned in the table that this may indeed not be clinically relevant for indications with doses in the lower range (cf. Tables 2 and 4).
5.1.2 Therapeutic Index of Object Drug
Potential DDIs are more likely to become clinically manifest in the setting of a drug with a narrow therapeutic index, that is, a short span from occurrence of therapeutic effect to side effects. Examples of P-gp substrates with a narrow therapeutic index are digoxin and dabigatran etexilate. On interpretation of results presented in the tables of this paper, it is therefore important to take into account that while concurrent administration of a given P-gp inhibitor with digoxin may produce AUC changes determined to be clinically relevant for digoxin, this may not be the case for other P-gp substrates with a wider therapeutic index. Thus, results based on narrow therapeutic index substrates such as digoxin should not be over-extrapolated to other P-gp substrates [30, 323].
5.1.3 Potency of the Precipitant Drug in Inhibiting P-gp
Potency of the P-gp inhibitor relative to its unbound plasma levels or estimated intestinal levels after therapeutic doses is another factor influencing recommendations for prescription with P-gp substrates. For example, for edoxaban, a direct oral anticoagulant, dose reduction is recommended with potent P-gp inhibitors such as ciclosporin A and ketoconazole but not with the less potent inhibitor verapamil [162]. Also, for another direct oral anticoagulant, dabigatran etexilate, the impact on the AUC differed quite substantially according to the studied inhibitor in vivo. For clarithromycin, the AUC increased by 19% and C max (peak drug concentration) by 15%, substantially less than with ketoconazole, for example, for which the AUC increased by 138% [150].
5.1.4 Contributions of Other Enzymes or Transporters to the Pharmacokinetics of the Substrate Drug
For many drugs, more than one enzyme or transporter influences the pharmacokinetics of the drug and to a varying extent. In this context, the proportion of a P-gp substrate depending on P-gp transport and metabolization or transport by other enzymes or transporters, respectively, needs to be evaluated together with the modulatory properties of the precipitant drug on the same enzymes and transporters. Examples of this relating to P-gp and CYP3A4 are mentioned in Sect. 3.
5.1.5 Dual Effects Over Time
Some drugs may be both inhibitors and inducers of P-gp depending on the timing. Rifampicin is an example of such a drug. If, for example, rifampicin and digoxin are administered in temporal proximity, the inhibitory effect of rifampicin is clinically manifest whereas the induction effect takes over with time [240]. Likewise, for both fexofenadine and digoxin, the inhibitory properties of ritonavir are predominant in the acute phase and lessen with time as the induction takes over [242, 324].
5.1.6 Route of Administration
Route of administration of the drug might also play a role. If the site of potential interaction is predominantly at the intestinal level affecting absorption, this would not be expected to affect the pharmacokinetics of a drug administered parenterally.
5.1.7 Highly Permeable Drugs
By being an efflux pump present at the apical level of the intestinal membrane, P-gp plays a role in limiting drug absorption. However, the extent to which P-gp limits absorption of a given drug depends also on how much of the drug is absorbed by passive permeability (as well as transport by other transporters). Generally, a P-gp substrate that is highly soluble, highly permeable and/or highly metabolized is less likely to be affected to a clinically relevant extent by a P-gp inhibiting drug [30]. For example, midazolam, which is highly lipophilic, has generally not been thought of as a P-gp substrate, perhaps due to its high intestinal permeability. But there is some evidence that midazolam actually exhibits characteristics of a highly permeable P-gp substrate [325]. Still, in relation to the clinical setting, this would not be of importance with co-administration of a P-gp inducer or P-gp inhibitor since midazolam passes the intestinal membrane anyway due to the high permeability. Midazolam is, however, a CYP3A4 substrate, and in that context interactions would need consideration.
5.1.8 Examples of Drug–Drug Interactions with Complex Interpretation
The new oral hepatitis C drugs are examples where interpretation of an interaction potential may be particularly complex due to multiple drugs administered at the same time. For example, ledipasvir and sofosbuvir are antivirals administered together for the treatment of chronic hepatitis C [220]. Both drugs are in vitro substrates of P-gp, and ledipasvir is an in vitro inhibitor of P-gp. While the inhibitory properties of ledipasvir have not been studied with model substrates such as digoxin or dabigatran etexilate in vivo, it has been shown to cause a more than two-fold increase in sofosbuvir AUC, which is likely driven by the combined effects of P-gp and BCRP modulation [219]. Accordingly, we have classified the inhibitory status of ledipasvir as ‘yes’ with respect to both in vitro and in vivo evidence (cf. Table 3).
Another complex example is the interpretation of the potential for DDIs with ombitasvir, paritaprevir and ritonavir given together (with or without dasabuvir) as part of treatment for chronic hepatitis C. Paritaprevir, ritonavir and dasabuvir all inhibit P-gp in vitro. In vivo, only ritonavir has been administered with digoxin alone, which resulted in an 86% increase in digoxin AUC [243]. When the combination of paritaprevir, ritonavir, ombitasvir (not a P-gp inhibitor in vitro) and dasabuvir are administered together with digoxin in vivo, digoxin AUC increases by 16% compared with digoxin alone [326]. However, when paritaprevir, ritonavir and ombitasvir are administered with digoxin (but without dasabuvir), digoxin AUC increases by 36% compared with digoxin alone [237]. Since the digoxin AUC decreases compared with ritonavir administered alone, a clinically relevant inhibitory potential of dasabuvir and paritaprevir seems unlikely, which is why we have chosen to classify the P-gp inhibitory status of both as ‘yes’ with respect to in vitro evidence and ‘no’ with respect to in vivo evidence (cf. Table 3 and Supplementary Table 3, ESM).
5.2 Clinical Considerations Relating to Patient Characteristics
5.2.1 Organ Impairment and Special Populations
Renal and hepatic impairment are important considerations when evaluating potential DDIs. That is, in patients with impaired hepatic or renal function, the threshold for a DDI becoming clinically manifest is lower if the drug depends highly on that particular organ for elimination. Mirabegron, indicated for treatment of patients with overactive bladder, is an example of a P-gp substrate where the manufacturer recommends caution only in patients with impaired renal or hepatic function. In patients with normal renal or hepatic function, mirabegron can be taken with inhibitors of CYP3A4 and/or P-gp without dose adjustment [298]. Also, for the direct oral anticoagulants (dabigatran etexilate, rivaroxaban, apixaban and edoxaban), dose reduction is recommended in patients with impaired renal function due to a decreased elimination of the drug and thus increased risk of toxicity [138, 150, 162, 177].
Particular consideration should also be taken among special populations such as the elderly and paediatric patients, where individual pharmacokinetics are different compared with healthy adults [32]. When new drugs are approved they will usually not have been tested in an elderly or paediatric population, meaning that knowledge regarding safety and efficacy in these populations depends on post-marketing evidence. The elderly are at increased risk of manifest DDIs for a number of reasons such as age-related pharmacokinetic and pharmacodynamic changes but also because polypharmacy, comorbidities and malnutrition are more prevalent with age [327]. For apixaban, the SmPC states that the AUC was increased by 32% in individuals >65 years old compared with younger individuals (no difference in C max). If the patient also receives acetylsalicylic acid, this additionally increases the risk of bleeding [138]. For a more extensive review of managing DDIs among the elderly we refer to other reviews such as the one by Mallet et al. [327].
For the drug-metabolizing enzyme CYP3A4 there is emerging evidence of a distinct ontogeny of the enzyme, with low activity at birth reaching adult levels during the first years of life [328–331]. For P-gp, evidence is sparser. Nevertheless, there is some human evidence of a pattern with high placental expression levels of P-gp in early pregnancy that decrease with increasing gestational age [332, 333]. Interestingly, paralleling this developmental decrease in placental expression, the P-gp expression of the human blood–brain barrier seems to increase with gestational age [334, 335]. Together this may serve to protect the foetal brain from noxious substances. However, further research is warranted to fully understand whether any difference in developmental P-gp expression in humans plays a role in drug disposition in the foetus and in neonates [336].
5.3 Ways to Handle Drug–Drug Interactions in Clinical Practice
When a DDI with potential clinical relevance has been identified, there are various ways to handle this. At the extremes, there is either no need for additional measures (the potential lack of treatment effect or increased drug effect is rendered harmless) or the medications should not be used concomitantly and the clinician should search for alternative treatments. However, in between those poles various other options for appropriate management exist. These are reviewed and exemplified in the following subsections.
5.3.1 Treatment with Additional Monitoring
Additional monitoring of either the main treatment effect or side effects is a possibility when a potential DDI has been identified. This can be by means of monitoring relevant paraclinical measures or performing additional physical examinations. For a few drugs (e.g. digoxin), an option may be therapeutic drug monitoring where the concentration of the drug is measured in plasma.
Carvedilol is an example of a drug where the evidence regarding the magnitude of the effect on digoxin AUC is heterogeneous. The changes in digoxin AUC with co-administration of carvedilol have been reported to range between a 19% and a 56% increase in various studies [146, 148]. According to the SmPC, the concentration of digoxin is increased by approximately 15% following co-administration with carvedilol, and increased monitoring of digoxin levels at onset, dose adjustment or cessation of carvedilol is recommended [144]. Thus, this is an example of a drug where the interpretation of the potential for a clinical manifest DDI is not straightforward and where increased monitoring may be a solution with respect to clinical management.
5.3.2 Dose Adjustment
Another option is dose adjustment. If an inhibitor or inducer of P-gp is administered with a substrate, then the dose of the substrate may be decreased or increased, respectively. Unfortunately, there are currently no expert dosing guidelines that provide clinicians with information on the dose to use in patients receiving P-gp substrates and concomitant P-gp inhibitors or P-gp inducers.
5.3.3 Staggered Administration
Staggered administration of the object and precipitant drug is another option. As an example, ibrutinib (irreversible Bruton’s tyrosine kinase inhibitor) is an in vitro inhibitor of P-gp with an IC50 of 2.15 µg/mL. At the proposed oral dose of 560 mg and assuming that the drug is taken with 250 mL of water, the concentration in the gut would be 560,000 µg divided by 250 mL and [I2]/IC50 would be 1042 (i.e. much greater than 10). Thus, the potential for a P-gp-mediated DDI cannot be excluded (cf. Fig. 2). However, ibrutinib is predicted to be quickly absorbed (in <2.5 h). Thus, the potential for a clinically relevant P-gp-mediated DDI would be minimized by staggering the dose of ibrutinib and a P-gp substrate by at least 2.5 h [96].
Co-administration of dabigatran etexilate, a direct oral anticoagulant, with the calcium antagonist verapamil is another example of a potential DDI involving P-gp that may be avoided with staggered administration. Härtter et al. have conducted a study to examine the potential for pharmacokinetic and pharmacodynamic interactions between dabigatran etexilate and verapamil [149]. The authors reported a 2.5-fold increase in dabigatran etexilate exposure (as determined by the AUC) when administered 1 h after a single dose of verapamil immediate-release formulation. This was reduced to an approximate 1.8-fold increase in AUC with the extended-release formulation of verapamil. However, administration of dabigatran etexilate 2 h before verapamil did not significantly increase exposure to dabigatran etexilate (<20% increase in AUC). This is explained by absorption of dabigatran etexilate being completed within 2 h [150].
5.3.4 Paused Treatment
Paused treatment may be a solution in those instances where lack of treatment with one of the drugs contributing to the DDI is rendered less harmful. An example of this could be the case of primary prevention with atorvastatin in a patient where a shorter course of a given drug with P-gp inhibitory properties is needed. In this case, paused treatment with atorvastatin during treatment with the perpetrator drug could be a solution. Anticancer drug interactions represent another case in which paused treatment may work. That is, during cancer treatment, perpetrator drugs can be stopped and then resumed following cessation of treatment.
5.4 Discussion of Limitations
We aimed to evaluate, as much as possible, a complete list of all drugs with P-gp modulatory properties. Nevertheless, with the approach described in the Sect. 3, there will inevitably be drugs that are P-gp modulators that have not been included for evaluation as part of this review. Many of these ‘false negatives’ probably represent newer drugs (except for very newly marketed drugs, as mentioned in Subsect. 3.1).
Also, the evidence base is of varying methodological quality; in general, the P-gp modulatory status of many of the drugs may not always have been investigated under ‘model conditions’. Regarding Caco2 cells, two aspects merit further discussion. First, variation in transporter expression and function of the cells may be seen depending on the exact laboratory conditions that the cells have been maintained under [337]. Second, Caco2 cells exhibit endogenous activity of transporters other than P-gp, for example multidrug resistance-associated proteins (MRPs) or BCRP that may thus have confounded the observed drug transport [338]. To address this issue, Caco2 cells transfected with short interfering RNA (siRNA) to knock down the contribution of other specific transporters may be used [337]. Of note, in the present review we did not make a distinction between transfected versus untransfected Caco2 cells for studies forming the basis of the classification of the individual drugs. However, at least for the underlying references from the scientific literature, all studies that included Caco2 cells were based on untransfected cells.
For the newer drugs, it may also be that specific combinations of drugs have been investigated together because that reflects the intended clinical use (e.g. see the new oral hepatitis C drugs as discussed in Sect. 5). Overall, our chosen classification strategy implies that classifications may change with the emergence of new evidence (e.g. from ‘uncertain’ to ‘yes’ with respect to both in vitro and in vivo evidence). Our chosen classification criteria were decided on with the aim of maximizing objectivity. However, since a consensus agreement was also included in the classification and since model evidence was not always found, this implies a certain degree of subjectivity which should be kept in mind on interpretation of the tables. We therefore provide the references that the classification is based upon for each drug along with the classification (see main tables and supplementary tables) and discuss examples of some of the interpretations of classifications in Sect. 3 (methods) and in Sect. 5 (clinical implications) of the paper.
6 Conclusions and Perspectives
Drug transporters such as P-gp have been increasingly recognized as active players in DDIs. Consequently, the potential for P-gp-mediated DDIs (together with the potential for interactions with other transporters or drug-metabolizing enzymes) is now more systematically investigated as part of the approval of new drugs. This has resulted in an increased amount of information on DDI potential available in the drug labels/SmPCs, including an increase in the complexity of the information. Prescription recommendations for individual drugs on the approved product label may not always reflect data from well characterized DDI studies; for example, a contraindication may be included in the label for various medico-legal reasons rather than due to clear-cut evidence of harm. The real challenge for the prescriber is therefore to navigate the various systems providing electronic decision support (causing potential alert fatigue) and strike the happy mean between an overcautious prescription practice and a practice where DDIs are not considered at all. Many interacting drugs can be safely taken together when appropriate measures are taken. The challenge still lies in narrowing the focus on those DDIs that are likely to be clinically manifest. In this context, more research in those drugs that interfere with more than one transporter or enzyme is needed to aid clinical decision making in this area. Further expert guidelines for dosing adjustments in the setting of concurrent administration of P-gp substrates and inhibitors and inducers, respectively, are needed.
References
Hallas J, Gram LF, Grodum E, Damsbo N, Brøsen K, Haghfelt T, et al. Drug related admissions to medical wards: a population based survey. Br J Clin Pharmacol. 1992;33:61–8.
Fattinger K, Roos M, Vergères P, Holenstein C, Kind B, Masche U, et al. Epidemiology of drug exposure and adverse drug reactions in two swiss departments of internal medicine. Br J Clin Pharmacol. 2000;49:158–67.
Classen DC, Pestotnik SL, Evans RS, Lloyd JF, Burke JP. Adverse drug events in hospitalized patients. Excess length of stay, extra costs, and attributable mortality. JAMA. 1997;277:301–6.
Bates DW, Spell N, Cullen DJ, Burdick E, Laird N, Petersen LA, et al. The costs of adverse drug events in hospitalized patients. Adverse Drug Events Prevention Study Group. JAMA. 1997;277:307–11.
Juurlink DN, Mamdani M, Kopp A, Laupacis A, Redelmeier DA. Drug-drug interactions among elderly patients hospitalized for drug toxicity. JAMA. 2003;289:1652–8.
Pirmohamed M, James S, Meakin S, Green C, Scott AK, Walley TJ, et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. BMJ. 2004;329:15–9.
Preston C, editor. Stockley’s drug interactions. 11th ed. London: Pharmaceutical Press; 2016. pp. 1, 405–407, 942.
Malone DC, Abarca J, Hansten PD, Grizzle AJ, Armstrong EP, Van Bergen RC, et al. Identification of serious drug-drug interactions: results of the partnership to prevent drug–drug interactions. J Am Pharm Assoc. 2003;44:142–51.
Hines LE, Malone DC, Murphy JE. Recommendations for generating, evaluating, and implementing drug–drug interaction evidence. Pharmacotherapy. 2012;32:304–13.
Glintborg B, Andersen SE, Dalhoff K. Drug–drug interactions among recently hospitalised patients–frequent but mostly clinically insignificant. Eur J Clin Pharmacol. 2005;61:675–81.
Delaney JA, Opatrny L, Brophy JM, Suissa S. Drug drug interactions between antithrombotic medications and the risk of gastrointestinal bleeding. Can Med Assoc J. 2007;177:347–51.
Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med. 2002;347:969–74.
Tannenbaum C, Sheehan NL. Understanding and preventing drug–drug and drug–gene interactions. Expert Rev Clin Pharmacol. 2014;7:533–44.
Dassa E, Bouige P. The ABC of ABCS: a phylogenetic and functional classification of ABC systems in living organisms. Res Microbiol. 2001;152:211–29.
Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–66.
Hodges LM, Markova SM, Chinn LW, Gow JM, Kroetz DL, Klein TE, et al. Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein). Pharmacogenet Genom. 2011;21:152–61.
Zhou S-F. Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica. 2008;38:802–32.
Seelig A. A general pattern for substrate recognition by P-glycoprotein. Eur J Biochem. 1998;251:252–61.
Etievant C, Schambel P, Guminski Y, Barret JM, Imbert T, Hill BT. Requirements for P-glycoprotein recognition based on structure-activity relationships in the podophyllotoxin series. Anticancer Drug Des. 1998;13:317–36.
Scala S, Akhmed N, Rao US, Paull K, Lan LB, Dickstein B, et al. P-glycoprotein substrates and antagonists cluster into two distinct groups. Mol Pharmacol. 1997;51:1024–33.
Kuwano M, Oda Y, Izumi H, Yang S-J, Uchiumi T, Iwamoto Y, et al. The role of nuclear Y-box binding protein 1 as a global marker in drug resistance. Mol Cancer Ther. 2004;3:1485–92.
Miller DS, Bauer B, Hartz AMS. Modulation of P-glycoprotein at the blood-brain barrier: opportunities to improve central nervous system pharmacotherapy. Pharmacol Rev. 2008;60:196–209.
Wolking S, Schaeffeler E, Lerche H, Schwab M, Nies AT. Impact of Genetic Polymorphisms of ABCB1 (MDR1, P-Glycoprotein) on Drug Disposition and Potential Clinical Implications: Update of the Literature. Clin Pharmacokinet. 2015;54:709–35.
Marzolini C, Paus E, Buclin T, Kim RB. Polymorphisms in human MDR1 (P-glycoprotein): recent advances and clinical relevance. Clin Pharmacol Ther. 2004;75:13–33.
Wacher VJ, Wu CY, Benet LZ. Overlapping substrate specificities and tissue distribution of cytochrome P450 3A and P-glycoprotein: implications for drug delivery and activity in cancer chemotherapy. Mol Carcinog. 1995;13:129–34.
Kim RB, Wandel C, Leake B, Cvetkovic M, Fromm MF, Dempsey PJ, et al. Interrelationship between substrates and inhibitors of human CYP3A and P-glycoprotein. Pharm Res. 1999;16:408–14.
Christians U. Transport proteins and intestinal metabolism: P-glycoprotein and cytochrome P4503A. Ther Drug Monit. 2004;26:104–6.
Murray GI, Barnes TS, Sewell HF, Ewen SW, Melvin WT, Burke MD. The immunocytochemical localisation and distribution of cytochrome P-450 in normal human hepatic and extrahepatic tissues with a monoclonal antibody to human cytochrome P-450. Br J Clin Pharmacol. 1988;25:465–75.
Benet LZ, Cummins CL. The drug efflux-metabolism alliance: biochemical aspects. Adv Drug Deliv Rev. 2001;50(Suppl 1):S3–11.
Giacomini KM, Huang S-M, Tweedie DJ, Benet LZ, Brouwer KLR, Chu X, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–36.
European Medicines Agency. Guideline on the investigation of drug interactions CPMP/EWP/560/95/Rev. 1 Corr. 2**. 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf.
U.S. Food and Drug Administration. Guidance for industry drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations. Draft Guidance. 2012.
Wessler JD, Grip LT, Mendell J, Giugliano RP. The P-glycoprotein transport system and cardiovascular drugs. J Am Coll Cardiol. 2013;61:2495–502.
Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. 2003;55:3–29.
Fromm MF. The influence of MDR1 polymorphisms on P-glycoprotein expression and function in humans. Adv Drug Deliv Rev. 2002;54:1295–310.
U.S. Food and Drug Administration. Drug Development and drug interactions: table of substrates, inhibitors and inducers. http://www.fda.gov/drugs/developmentapprovalprocess/developmentresources/druginteractionslabeling/ucm093664.htm. Accessed 22 Jan 2015.
Matsson P, Pedersen JM, Norinder U, Bergström CAS, Artursson P. Identification of novel specific and general inhibitors of the three major human ATP-binding cassette transporters P-gp, BCRP and MRP2 among registered drugs. Pharm Res. 2009;26:1816–31.
Danish Medicines Agency (Lægemiddelstyrelsen). Drugs with compassionate reporting requirements. http://laegemiddelstyrelsen.dk/da/bivirkninger/bivirkninger-ved-medicin/medicin-med-skaerpet-indberetningspligt. Accessed 23 Jan 2015.
European Medicines Agency. European Public Assessment Reports. http://www.ema.europa.eu/ema/index.jsp?curl=pages%2Fmedicines%2Flanding%2Fepar_search.jsp&murl=menus%2Fmedicines%2Fmedicines.jsp&mid=WC0b01ac058001d124&searchkwByEnter=false&alreadyLoaded=true&status=Authorised&status=Withdrawn&status=Suspended&status=Ref.
Lægemiddelstyrelsen (Danish Medicines Authority). Summary of Product Characteristics. www.produktresume.dk.
U.S. Food and Drug Administration. FDA approved drug products. https://www.accessdata.fda.gov/scripts/cder/drugsatfda/.
Huang S-M, Strong JM, Zhang L, Reynolds KS, Nallani S, Temple R, et al. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J Clin Pharmacol. 2008;48:662–70.
Zhang L, Zhang YD, Strong JM, Reynolds KS, Huang S-M. A regulatory viewpoint on transporter-based drug interactions. Xenobiotica. 2008;38:709–24.
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 202806Orig1s000 Tafinlar.
European Medicines Agency. Summary of Product Characteristics Rasilez (version dated 07/07/2015).
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 203100 Stribild.
European Medicines Agency. Summary of Product Characteristics Stribild (version dated 05/08/2015).
European Medicines Agency. Summary of Product Characteristics Tybost (version dated 04/06/2015).
Larsen UL, Olesen HL, Nyvold GC, Eriksen J, Jakobsen P, Østergaard M, et al. Human intestinal P-glycoprotein activity estimated by the model substrate digoxin. Scand J Clin Lab Investig. 2007;67:123–34.
Tannergren C, Knutson T, Knutson L, Lennernäs H. The effect of ketoconazole on the in vivo intestinal permeability of fexofenadine using a regional perfusion technique. Br J Clin Pharmacol. 2003;55:182–90.
U.S. Food and Drug Administration. Label Allegra, 60mg capsules (action date 25/07/1996).
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 22-512 Pradaxa.
Hill CR, Jamieson D, Thomas HD, Brown CDA, Boddy AV, Veal GJ. Characterisation of the roles of ABCB1, ABCC1, ABCC2 and ABCG2 in the transport and pharmacokinetics of actinomycin D in vitro and in vivo. Biochem Pharmacol. 2013;85:29–37.
Theis JG, Chan HS, Greenberg ML, Malkin D, Karaskov V, Moncica I, et al. Increased systemic toxicity of sarcoma chemotherapy due to combination with the P-glycoprotein inhibitor cyclosporin. Int J Clin Pharmacol Ther. 1998;36:61–4.
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 201292 Gilotrif.
European Medicines Agency. European Public Assessment Report Giotrif (version dated 16/10/2013).
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 205437Orig1s000 Otezla.
European Medicines Agency. Summary of Product Characteristics Otezla (version dated 08/02/2016).
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 203756Orig1s000 Cometriq.
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 205755 Zykadia.
European Medicines Agency. Summary of Product Characteristics Zykadia (version dated 28/09/2015).
Saeki T, Ueda K, Tanigawara Y, Hori R, Komano T. Human P-glycoprotein transports cyclosporin A and FK506. J Biol Chem. 1993;268:6077–80.
Gupta SK, Bakran A, Johnson RW, Rowland M. Erythromycin enhances the absorption of cyclosporin. Br J Clin Pharmacol. 1988;25:401–2.
Gupta SK, Bakran A, Johnson RW, Rowland M. Cyclosporin-erythromycin interaction in renal transplant patients. Br J Clin Pharmacol. 1989;27:475–81.
Fricker G, Drewe J, Huwyler J, Gutmann H, Beglinger C. Relevance of p-glycoprotein for the enteral absorption of cyclosporin A: in vitro–in vivo correlation. Br J Pharmacol. 1996;118:1841–7.
Lown KS, Mayo RR, Leichtman AB, Hsiao HL, Turgeon DK, Schmiedlin-Ren P, et al. Role of intestinal P-glycoprotein (mdr1) in interpatient variation in the oral bioavailability of cyclosporine. Clin Pharmacol Ther. 1997;62:248–60.
Lumen AA, Li L, Li J, Ahmed Z, Meng Z, Owen A, et al. Transport inhibition of digoxin using several common P-gp expressing cell lines is not necessarily reporting only on inhibitor binding to P-gp. PLoS One. 2013;8:e69394.
Dorian P, Strauss M, Cardella C, David T, East S, Ogilvie R. Digoxin-cyclosporine interaction: severe digitalis toxicity after cyclosporine treatment. Clin Invest Med. 1988;11:108–12.
Fenner KS, Troutman MD, Kempshall S, Cook JA, Ware JA, Smith DA, et al. Drug–drug interactions mediated through P-glycoprotein: clinical relevance and in vitro–in vivo correlation using digoxin as a probe drug. Clin Pharmacol Ther. 2009;85:173–81.
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 21-976 Prezista.
U.S. Food and Drug Administration. Label Prezista, oral suspension and tablets for oral use (action date 27/05/2015).
Tidefelt U, Liliemark J, Gruber A, Liliemark E, Sundman-Engberg B, Juliusson G, et al. P-glycoprotein inhibitor valspodar (PSC 833) increases the intracellular concentrations of daunorubicin in vivo in patients with P-glycoprotein-positive acute myeloid leukemia. J Clin Oncol. 2000;18:1837–44.
Wils P, Phung-Ba V, Warnery A, Lechardeur D, Raeissi S, Hidalgo IJ, et al. Polarized transport of docetaxel and vinblastine mediated by P-glycoprotein in human intestinal epithelial cell monolayers. Biochem Pharmacol. 1994;48:1528–30.
Figg WD, Woo S, Zhu W, Chen X, Ajiboye AS, Steinberg SM, et al. A phase I clinical study of high dose ketoconazole plus weekly docetaxel for metastatic castration resistant prostate cancer. J Urol. 2010;183:2219–26.
Malingré MM, Richel DJ, Beijnen JH, Rosing H, Koopman FJ, Ten Bokkel Huinink WW, et al. Coadministration of cyclosporine strongly enhances the oral bioavailability of docetaxel. J Clin Oncol. 2001;19:1160–6.
Morschhauser F, Zinzani PL, Burgess M, Sloots L, Bouafia F, Dumontet C. Phase I/II trial of a P-glycoprotein inhibitor, Zosuquidar. 3HCl trihydrochloride (LY335979), given orally in combination with the CHOP regimen in patients with non-Hodgkin’s lymphoma. Leuk Lymphoma. 2007;48:708–15.
Troutman MD, Thakker DR. Rhodamine 123 requires carrier-mediated influx for its activity as a P-glycoprotein substrate in Caco-2 cells. Pharm Res. 2003;20:1192–9.
Kim J-E, Cho H-J, Kim JS, Shim C-K, Chung S-J, Oak M-H, et al. The limited intestinal absorption via paracellular pathway is responsible for the low oral bioavailability of doxorubicin. Xenobiotica. 2013;43:579–91.
Giaccone G, Linn SC, Welink J, Catimel G, Stieltjes H, van der Vijgh WJ, et al. A dose-finding and pharmacokinetic study of reversal of multidrug resistance with SDZ PSC 833 in combination with doxorubicin in patients with solid tumors. Clin Cancer Res. 1997;3:2005–15.
Bartlett NL, Lum BL, Fisher GA, Brophy NA, Ehsan MN, Halsey J, et al. Phase I trial of doxorubicin with cyclosporine as a modulator of multidrug resistance. J Clin Oncol. 1994;12:835–42.
van der Sandt IC, Blom-Roosemalen MC, de Boer AG, Breimer DD. Specificity of doxorubicin versus rhodamine-123 in assessing P-glycoprotein functionality in the LLC-PK1, LLC-PK1:MDR1 and Caco-2 cell lines. Eur J Pharm Sci. 2000;11:207–14.
Lægemiddelstyrelsen (Danish Medicines Agency). Summary of Product Characteristics Doxorubicin “Teva” (version published online 23/09/2013).
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 203415Orig1s000 Xtandi.
European Medicines Agency. Summary of Product Characteristics Xtandi (version dated 29/10/2015).
European Medicines Agency. Scientific discussion Tarceva (version dated 02/11/2005).
European Medicines Agency. Summary of Product Characteristics Tarceva (version dated 03/02/2014).
Boote DJ, Dennis IF, Twentyman PR, Osborne RJ, Laburte C, Hensel S, et al. Phase I study of etoposide with SDZ PSC 833 as a modulator of multidrug resistance in patients with cancer. J Clin Oncol. 1996;14:610–8.
Lum BL, Kaubisch S, Yahanda AM, Adler KM, Jew L, Ehsan MN, et al. Alteration of etoposide pharmacokinetics and pharmacodynamics by cyclosporine in a phase I trial to modulate multidrug resistance. J Clin Oncol. 1992;10:1635–42.
Guo A, Marinaro W, Hu P, Sinko PJ. Delineating the contribution of secretory transporters in the efflux of etoposide using Madin-Darby canine kidney (MDCK) cells overexpressing P-glycoprotein (Pgp), multidrug resistance-associated protein (MRP1), and canalicular multispecific organic anion. Drug Metab Dispos. 2002;30:457–63.
Lægemiddelstyrelsen (Danish Medicines Agency). Summary of Product Characteristics Etoposid “Accord” (version published online 30/11/2015).
Lamoureux F, Picard N, Boussera B, Sauvage F-L, Marquet P. Sirolimus and everolimus intestinal absorption and interaction with calcineurin inhibitors: a differential effect between cyclosporine and tacrolimus. Fundam Clin Pharmacol. 2012;26:463–72.
European Medicines Agency. European Public Assessment Report Afinitor (version dated 02/09/2009).
European Medicines Agency. Summary of Product Characteristics Afinitor (version dated 27/04/2015).
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 22-334 Afinitor.
European Medicines Agency. Summary of Product Characteristics Iressa (version dated 11/11/2014).
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 205552Orig1s000 Imbruvica.
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 206545Orig1s000 Zydelig.
Giannoudis A, Davies A, Lucas CM, Harris RJ, Pirmohamed M, Clark RE. Effective dasatinib uptake may occur without human organic cation transporter 1 (hOCT1): implications for the treatment of imatinib-resistant chronic myeloid leukemia. Blood. 2008;112:3348–54.
Eadie LN, Hughes TP, White DL. Interaction of the efflux transporters ABCB1 and ABCG2 with imatinib, nilotinib, and dasatinib. Clin Pharmacol Ther. 2014;95:294–306.
Hamada A, Miyano H, Watanabe H, Saito H. Interaction of imatinib mesilate with human P-glycoprotein. J Pharmacol Exp Ther. 2003;307:824–8.
European Medicines Agency. Summary of Product Characteristics Glivec (version dated 11/05/2015).
Lægemiddelstyrelsen (Danish Medicines Agency). Summary of Product Characteristics Irinotecan “Stada” (version published online 15/05/2015).
Bansal T, Mishra G, Jaggi M, Khar RK, Talegaonkar S. Effect of P-glycoprotein inhibitor, verapamil, on oral bioavailability and pharmacokinetics of irinotecan in rats. Eur J Pharm Sci. 2009;36:580–90.
Luo FR, Paranjpe PV, Guo A, Rubin E, Sinko P. Intestinal transport of irinotecan in Caco-2 cells and MDCK II cells overexpressing efflux transporters Pgp, cMOAT, and MRP1. Drug Metab Dispos. 2002;30:763–70.
European Medicines Agency. Summary of Product Characteristics Tyverb (version dated 11/08/2015).
European Medicines Agency. European Public Assessment Report Tyverb (version dated 26/06/2008).
Oka A, Oda M, Saitoh H, Nakayama A, Takada M, Aungst BJ. Secretory transport of methylprednisolone possibly mediated by P-glycoprotein in Caco-2 cells. Biol Pharm Bull. 2002;25:393–6.
Tomita M, Watanabe A, Fujinaga I, Yamakawa T, Hayashi M. Nonlinear absorption of methylprednisolone by absorptive and secretory transporters. Int J Pharm. 2010;387:1–6.
European Medicines Agency. Summary of Product Characteristics Tasigna (version dated 11/11/2015).
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 22-068 Tasigna.
Meerum Terwogt JM, Malingré MM, Beijnen JH, ten Bokkel Huinink WW, Rosing H, Koopman FJ, et al. Coadministration of oral cyclosporin A enables oral therapy with paclitaxel. Clin Cancer Res. 1999;5:3379–84.
Berg SL, Tolcher A, O’Shaughnessy JA, Denicoff AM, Noone M, Ognibene FP, et al. Effect of R-verapamil on the pharmacokinetics of paclitaxel in women with breast cancer. J Clin Oncol. 1995;13:2039–42.
Taub ME, Podila L, Ely D, Almeida I. Functional assessment of multiple P-glycoprotein (P-gp) probe substrates: influence of cell line and modulator concentration on P-gp activity. Drug Metab Dispos. 2005;33:1679–87.
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 230-085 Regorafenib/Stivarga.
European Medicines Agency. Summary of Product Characteristics Stivarga (version dated 09/12/2015).
European Medicines Agency. Summary of Product Characteristics Rapamune (version dated 06/11/2015).
Hebert MF, Fisher RM, Marsh CL, Dressler D, Bekersky I. Effects of rifampin on tacrolimus pharmacokinetics in healthy volunteers. J Clin Pharmacol. 1999;39:91–6.
Hebert MF, Lam AY. Diltiazem increases tacrolimus concentrations. Ann Pharmacother. 1999;33:680–2.
Arima H, Yunomae K, Hirayama F, Uekama K. Contribution of P-glycoprotein to the enhancing effects of dimethyl-beta-cyclodextrin on oral bioavailability of tacrolimus. J Pharmacol Exp Ther. 2001;297:547–55.
Lægemiddelstyrelsen (Danish Medicines Agency). Summary of Product Characteristics Prograf (version published online 08/02/2016).
Lemahieu WPD, Hermann M, Asberg A, Verbeke K, Holdaas H, Vanrenterghem Y, et al. Combined therapy with atorvastatin and calcineurin inhibitors: no interactions with tacrolimus. Am J Transplant. 2005;5:2236–43.
Kishimoto W, Ishiguro N, Ludwig-Schwellinger E, Ebner T, Schaefer O. In vitro predictability of drug-drug interaction likelihood of P-glycoprotein-mediated efflux of dabigatran etexilate based on [I]2/IC50 threshold. Drug Metab Dispos. 2014;42:257–63.
Floren LC, Bekersky I, Benet LZ, Mekki Q, Dressler D, Lee JW, et al. Tacrolimus oral bioavailability doubles with coadministration of ketoconazole. Clin Pharmacol Ther. 1997;62:41–9.
Lin X, Skolnik S, Chen X, Wang J. Attenuation of intestinal absorption by major efflux transporters: quantitative tools and strategies using a Caco-2 model. Drug Metab Dispos. 2011;39:265–74.
Li H, Jin H-E, Kim W, Han Y-H, Kim D-D, Chung S-J, et al. Involvement of P-glycoprotein, multidrug resistance protein 2 and breast cancer resistance protein in the transport of belotecan and topotecan in Caco-2 and MDCKII cells. Pharm Res. 2008;25:2601–12.
Kruijtzer CMF, Beijnen JH, Rosing H, ten Bokkel Huinink WW, Schot M, Jewell RC, et al. Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-glycoprotein inhibitor GF120918. J Clin Oncol. 2002;20:2943–50.
European Medicines Agency. Summary of Product Characteristics Kadcyla (version dated 03/12/2015).
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 125427Orig1s000 Kadcyla.
Samuels BL, Mick R, Vogelzang NJ, Williams SF, Schilsky RL, Safa AR, et al. Modulation of vinblastine resistance with cyclosporine: a phase I study. Clin Pharmacol Ther. 1993;54:421–9.
Bates S, Kang M, Meadows B, Bakke S, Choyke P, Merino M, et al. A Phase I study of infusional vinblastine in combination with the P-glycoprotein antagonist PSC 833 (valspodar). Cancer. 2001;92:1577–90.
Huang RS, Murry DJ, Foster DR. Role of xenobiotic efflux transporters in resistance to vincristine. Biomed Pharmacother. 2008;62:59–64.
Samer CF, Lorenzini KI, Rollason V, Daali Y, Desmeules JA. Applications of CYP450 testing in the clinical setting. Mol Diagn Ther. 2013;17:165–84.
Rebello S, Compain S, Feng A, Hariry S, Dieterich H-A, Jarugula V. Effect of cyclosporine on the pharmacokinetics of aliskiren in healthy subjects. J Clin Pharmacol. 2011;51:1549–60.
Vaidyanathan S, Camenisch G, Schuetz H, Reynolds C, Yeh C-M, Bizot M-N, et al. Pharmacokinetics of the oral direct renin inhibitor aliskiren in combination with digoxin, atorvastatin, and ketoconazole in healthy subjects: the role of P-glycoprotein in the disposition of aliskiren. J Clin Pharmacol. 2008;48:1323–38.
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 22081 Letairis.
European Medicines Agency. Summary of Product Characteristics Volibris (version dated 12/01/2016).
Robinson K, Johnston A, Walker S, Mulrow JP, McKenna WJ, Holt DW. The digoxin-amiodarone interaction. Cardiovasc Drugs Ther. 1989;3:25–8.
European Medicines Agency. Summary of Product Characteristics Eliquis (version dated 15/04/2016).
Zhang D, He K, Herbst JJ, Kolb J, Shou W, Wang L, et al. Characterization of efflux transporters involved in distribution and disposition of apixaban. Drug Metab Dispos. 2013;41:827–35.
Wu X, Whitfield LR, Stewart BH. Atorvastatin transport in the Caco-2 cell model: contributions of P-glycoprotein and the proton-monocarboxylic acid co-transporter. Pharm Res. 2000;17:209–15.
Andrén L, Andreasson A, Eggertsen R. Interaction between a commercially available St. John’s wort product (Movina) and atorvastatin in patients with hypercholesterolemia. Eur J Clin Pharmacol. 2007;63:913–6.
Hermann M, Asberg A, Christensen H, Holdaas H, Hartmann A, Reubsaet JLE. Substantially elevated levels of atorvastatin and metabolites in cyclosporine-treated renal transplant recipients. Clin Pharmacol Ther. 2004;76:388–91.
Pham PA, la Porte CJL, Lee LS, van Heeswijk R, Sabo JP, Elgadi MM, et al. Differential effects of tipranavir plus ritonavir on atorvastatin or rosuvastatin pharmacokinetics in healthy volunteers. Antimicrob Agents Chemother. 2009;53:4385–92.
Lægemiddelstyrelsen (Danish Medicines Agency). Summary of Product Characteristics Carvedilol “Sandoz” (version published online 21/12/2015).
Bachmakov I, Werner U, Endress B, Auge D, Fromm MF. Characterization of beta-adrenoceptor antagonists as substrates and inhibitors of the drug transporter P-glycoprotein. Fundam Clin Pharmacol. 2006;20:273–82.
Baris N, Kalkan S, Güneri S, Bozdemir V, Guven H. Influence of carvedilol on serum digoxin levels in heart failure: is there any gender difference? Eur J Clin Pharmacol. 2006;62:535–8.
Oldham HG, Clarke SE. In vitro identification of the human cytochrome P450 enzymes involved in the metabolism of R(+)- and S(−)-carvedilol. Drug Metab Dispos. 1997;25:970–7.
De Mey C, Brendel E, Enterling D. Carvedilol increases the systemic bioavailability of oral digoxin. Br J Clin Pharmacol. 1990;29:486–90.
Härtter S, Sennewald R, Nehmiz G, Reilly P. Oral bioavailability of dabigatran etexilate (Pradaxa®) after co-medication with verapamil in healthy subjects. Br J Clin Pharmacol. 2013;75:1053–62.
European Medicines Agency. Summary of Product Characteristics Pradaxa (version dated 09/12/2015).
Ishiguro N, Kishimoto W, Volz A, Ludwig-Schwellinger E, Ebner T, Schaefer O. Impact of endogenous esterase activity on in vitro p-glycoprotein profiling of dabigatran etexilate in Caco-2 monolayers. Drug Metab Dispos. 2014;42:250–6.
Doering W. Quinidine–digoxin interaction: pharmacokinetics, underlying mechanism and clinical implications. N Engl J Med. 1979;301:400–4.
Mordel A, Halkin H, Zulty L, Almog S, Ezra D. Quinidine enhances digitalis toxicity at therapeutic serum digoxin levels. Clin Pharmacol Ther. 1993;53:457–62.
Woodland C, Ito S, Koren G. A model for the prediction of digoxin-drug interactions at the renal tubular cell level. Ther Drug Monit. 1998;20:134–8.
Pedersen KE, Christiansen BD, Klitgaard NA, Nielsen-Kudsk F. Effect of quinidine on digoxin bioavailability. Eur J Clin Pharmacol. 1983;24:41–7.
Shimizu M, Uno T, Sugawara K, Tateishi T. Effects of itraconazole and diltiazem on the pharmacokinetics of fexofenadine, a substrate of P-glycoprotein. Br J Clin Pharmacol. 2006;61:538–44.
Mahgoub AA, El-Medany AH, Abdulatif AS. A comparison between the effects of diltiazem and isosorbide dinitrate on digoxin pharmacodynamics and kinetics in the treatment of patients with chronic ischemic heart failure. Saudi Med J. 2002;23:725–31.
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics review(s) NDA 22425 Multaq.
Vallakati A, Chandra PA, Pednekar M, Frankel R, Shani J. Dronedarone-induced digoxin toxicity: new drug, new interactions. Am J Ther. 2013;20:e717–9.
Mendell J, Zahir H, Matsushima N, Noveck R, Lee F, Chen S, et al. Drug-drug interaction studies of cardiovascular drugs involving P-glycoprotein, an efflux transporter, on the pharmacokinetics of edoxaban, an oral factor Xa inhibitor. Am J Cardiovasc Drugs. 2013;13:331–42.
Mikkaichi T, Yoshigae Y, Masumoto H, Imaoka T, Rozehnal V, Fischer T, et al. Edoxaban transport via P-glycoprotein is a key factor for the drug’s disposition. Drug Metab Dispos. 2014;42:520–8.
European Medicines Agency. Summary of Product Characteristics Lixiana (version dated 18/11/2015).
Kamiyama E, Nakai D, Mikkaichi T, Okudaira N, Okazaki O. Interaction of angiotensin II type 1 receptor blockers with P-gp substrates in Caco-2 cells and hMDR1-expressing membranes. Life Sci. 2010;86:52–8.
Lægemiddelstyrelsen (Danish Medicines Agency). Summary of Product Characteristics Trandate (version published online 15/6/2015).
Incecayir T, Tsume Y, Amidon GL. Comparison of the permeability of metoprolol and labetalol in rat, mouse, and Caco-2 cells: use as a reference standard for BCS classification. Mol Pharm. 2013;10:958–66.
Thiel-Demby VE, Humphreys JE, St John Williams LA, Ellens HM, Shah N, Ayrton AD, et al. Biopharmaceutics classification system: validation and learnings of an in vitro permeability assay. Mol Pharm. 2009;6:11–8.
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 203858Orig1s000 lomitapide.
European Medicines Agency. European Public Assessment Report Lojuxta (version dated 18/08/2015).
Soldner A, Benet LZ, Mutschler E, Christians U. Active transport of the angiotensin-II antagonist losartan and its main metabolite EXP 3174 across MDCK-MDR1 and caco-2 cell monolayers. Br J Pharmacol. 2000;129:1235–43.
Belz GG, Doering W, Munkes R, Matthews J. Interaction between digoxin and calcium antagonists and antiarrhythmic drugs. Clin Pharmacol Ther. 1983;33:410–7.
Bachmakov I, Rekersbrink S, Hofmann U, Eichelbaum M, Fromm MF. Characterisation of (R/S)-propafenone and its metabolites as substrates and inhibitors of P-glycoprotein. Naunyn Schmiedebergs Arch Pharmacol. 2005;371:195–201.
Jerling M. Clinical pharmacokinetics of ranolazine. Clin Pharmacokinet. 2006;45:469–91.
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 21-256 Ranexa.
European Medicines Agency. Summary of Product Characteristics Adempas (version dated 08/02/2016).
Rickert V, Haefeli WE, Weiss J. Pharmacokinetic interaction profile of riociguat, a new soluble guanylate cyclase stimulator, in vitro. Pulm Pharmacol Ther. 2014;28:130–7.
European Medicines Agency. European Public Assessment Report Xarelto (version dated 09/11/2008).
European Medicines Agency. Summary of Product Characteristics Xarelto (version dated 17/07/2015).
Mueck W, Kubitza D, Becka M. Co-administration of rivaroxaban with drugs that share its elimination pathways: pharmacokinetic effects in healthy subjects. Br J Clin Pharmacol. 2013;76:455–66.
Gnoth MJ, Buetehorn U, Muenster U, Schwarz T, Sandmann S. In vitro and in vivo P-glycoprotein transport characteristics of rivaroxaban. J Pharmacol Exp Ther. 2011;338:372–80.
Hochman JH, Pudvah N, Qiu J, Yamazaki M, Tang C, Lin JH, et al. Interactions of human P-glycoprotein with simvastatin, simvastatin acid, and atorvastatin. Pharm Res. 2004;21:1686–91.
Neuvonen PJ, Kantola T, Kivistö KT. Simvastatin but not pravastatin is very susceptible to interaction with the CYP3A4 inhibitor itraconazole. Clin Pharmacol Ther. 1998;63:332–41.
Kantola T, Kivistö KT, Neuvonen PJ. Erythromycin and verapamil considerably increase serum simvastatin and simvastatin acid concentrations. Clin Pharmacol Ther. 1998;64:177–82.
Westphal K, Weinbrenner A, Zschiesche M, Franke G, Knoke M, Oertel R, et al. Induction of P-glycoprotein by rifampin increases intestinal secretion of talinolol in human beings: a new type of drug/drug interaction. Clin Pharmacol Ther. 2000;68:345–55.
Wetterich U, Spahn-Langguth H, Mutschler E, Terhaag B, Rösch W, Langguth P. Evidence for intestinal secretion as an additional clearance pathway of talinolol enantiomers: concentration- and dose-dependent absorption in vitro and in vivo. Pharm Res. 1996;13:514–22.
Teng R, Butler K. A pharmacokinetic interaction study of ticagrelor and digoxin in healthy volunteers. Eur J Clin Pharmacol. 2013;69:1801–8.
European Medicines Agency. Summary of Product Characteristics Brilique (version dated 11/08/2015).
Teng R, Mitchell P, Butler K. Effect of rifampicin on the pharmacokinetics and pharmacodynamics of ticagrelor in healthy subjects. Eur J Clin Pharmacol. 2013;69:877–83.
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 22433 Brilinta.
European Medicines Agency. European Public Assessment Report Samsca (version dated 18/08/2009).
Shoaf SE, Ohzone Y, Ninomiya S, Furukawa M, Bricmont P, Kashiyama E, et al. In vitro P-glycoprotein interactions and steady-state pharmacokinetic interactions between tolvaptan and digoxin in healthy subjects. J Clin Pharmacol. 2011;51:761–9.
Pauli-Magnus C, von Richter O, Burk O, Ziegler A, Mettang T, Eichelbaum M, et al. Characterization of the major metabolites of verapamil as substrates and inhibitors of P-glycoprotein. J Pharmacol Exp Ther. 2000;293:376–82.
Klein HO, Lang R, Weiss E, Di Segni E, Libhaber C, Guerrero J, et al. The influence of verapamil on serum digoxin concentration. Circulation. 1982;65:998–1003.
Rodin SM, Johnson BF, Wilson J, Ritchie P, Johnson J. Comparative effects of verapamil and isradipine on steady-state digoxin kinetics. Clin Pharmacol Ther. 1988;43:668–72.
Shaik N, Giri N, Pan G, Elmquist WF. P-glycoprotein-mediated active efflux of the anti-HIV1 nucleoside abacavir limits cellular accumulation and brain distribution. Drug Metab Dispos. 2007;35:2076–85.
European Medicines Agency. European Public Assessment Report Triumeq (version dated 15/10/2014).
Hughes J, Crowe A. Inhibition of P-glycoprotein-mediated efflux of digoxin and its metabolites by macrolide antibiotics. J Pharmacol Sci. 2010;113:315–24.
Eberl S, Renner B, Neubert A, Reisig M, Bachmakov I, König J, et al. Role of p-glycoprotein inhibition for drug interactions: evidence from in vitro and pharmacoepidemiological studies. Clin Pharmacokinet. 2007;46:1039–49.
Bouquié R, Deslandes G, Renaud C, Dailly E, Haloun A, Jolliet P. Colchicine-induced rhabdomyolysis in a heart/lung transplant patient with concurrent use of cyclosporin, pravastatin, and azithromycin. J Clin Rheumatol. 2011;17:28–30.
Rengelshausen J, Göggelmann C, Burhenne J, Riedel K-D, Ludwig J, Weiss J, et al. Contribution of increased oral bioavailability and reduced nonglomerular renal clearance of digoxin to the digoxin-clarithromycin interaction. Br J Clin Pharmacol. 2003;56:32–8.
European Medicines Agency. Summary of Product Characteristics Daklinza (version dated 05/11/2015).
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 206843Orig1s000 Daklinza.
European Medicines Agency. Summary of Product Characteristics Triumeq (version dated 09/11/2015).
Kurnik D, Wood AJJ, Wilkinson GR. The erythromycin breath test reflects P-glycoprotein function independently of cytochrome P450 3A activity. Clin Pharmacol Ther. 2006;80:228–34.
Schuetz EG, Yasuda K, Arimori K, Schuetz JD. Human MDR1 and mouse mdr1a P-glycoprotein alter the cellular retention and disposition of erythromycin, but not of retinoic acid or benzo(a)pyrene. Arch Biochem Biophys. 1998;350:340–7.
European Medicines Agency. Summary of Product Characteristics Telzir (version dated 12/01/2016).
European Medicines Agency. Scientific discussion Telzir (version dated 05/12/2005).
Polli JW, Jarrett JL, Studenberg SD, Humphreys JE, Dennis SW, Brouwer KR, et al. Role of P-glycoprotein on the CNS disposition of amprenavir (141W94), an HIV protease inhibitor. Pharm Res. 1999;16:1206–12.
U.S. Food and Drug Administration. Label Crixivan, capsules (action date 27/03/2015).
Rouquayrol M, Gaucher B, Roche D, Greiner J, Vierling P. Transepithelial transport of prodrugs of the HIV protease inhibitors saquinavir, indinavir, and nelfinavir across Caco-2 cell monolayers. Pharm Res. 2002;19:1704–12.
Jalava KM, Partanen J, Neuvonen PJ. Itraconazole decreases renal clearance of digoxin. Ther Drug Monit. 1997;19:609–13.
Lægemiddelstyrelsen (Danish Medicines Agency). Summary of Product Characteristics Itraconazol “Ratiopharm” (version published online 02/11/2015).
Wang E, Lew K, Casciano CN, Clement RP, Johnson WW. Interaction of common azole antifungals with P glycoprotein. Antimicrob Agents Chemother. 2002;46:160–5.
Griffin J, Fletcher N, Clemence R, Blanchflower S, Brayden DJ. Selamectin is a potent substrate and inhibitor of human and canine P-glycoprotein. J Vet Pharmacol Ther. 2005;28:257–65.
Schinkel AH, Wagenaar E, Mol CA, van Deemter L. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest. 1996;97:2517–24.
U.S. Food and Drug Administration. Label Stromectol, tablets (action date 15/12/2009).
Profit L, Eagling VA, Back DJ. Modulation of P-glycoprotein function in human lymphocytes and Caco-2 cell monolayers by HIV-1 protease inhibitors. AIDS. 1999;13:1623–7.
Davit B, Reynolds K, Yuan R, Ajayi F, Conner D, Fadiran E, et al. FDA evaluations using in vitro metabolism to predict and interpret in vivo metabolic drug–drug interactions: impact on labeling. J Clin Pharmacol. 1999;39:899–910.
Simpson K, Jarvis B. Fexofenadine: a review of its use in the management of seasonal allergic rhinitis and chronic idiopathic urticaria. Drugs. 2000;59:301–21.
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 205834 Harvoni.
European Medicines Agency. Summary of Product Characteristics Harvoni (version dated 23/02/2016).
Ito T, Yano I, Tanaka K, Inui KI. Transport of quinolone antibacterial drugs by human P-glycoprotein expressed in a kidney epithelial cell line, LLC-PK1. J Pharmacol Exp Ther. 1997;282:955–60.
Lægemiddelstyrelsen (Danish Medicines Agency). Summary of Product Characteristics Levofloxacin “Amneal” (version published online 02/03/2015).
Vishnuvardhan D, Moltke LL, Richert C, Greenblatt DJ. Lopinavir: acute exposure inhibits P-glycoprotein; extended exposure induces P-glycoprotein. AIDS. 2003;17:1092–4.
van Heeswijk RPG, Bourbeau M, Campbell P, Seguin I, Chauhan BM, Foster BC, et al. Time-dependent interaction between lopinavir/ritonavir and fexofenadine. J Clin Pharmacol. 2006;46:758–67.
European Medicines Agency. Summary of Product Characteristics Kaletra (version dated 01/04/2016).
European Medicines Agency. Summary of Product Characteristics Celsentri (version dated 08/01/2016).
European Medicines Agency. European Public Assessment Report–scientific discussion Celsentri (version dated 18/12/2007).
Lægemiddelstyrelsen (Danish Medicines Agency). Summary of Product Characteristics Lariam (version published online 19/12/2014).
Pham YT, Régina A, Farinotti R, Couraud P, Wainer IW, Roux F, et al. Interactions of racemic mefloquine and its enantiomers with P-glycoprotein in an immortalised rat brain capillary endothelial cell line, GPNT. Biochim Biophys Acta. 2000;1524:212–9.
Kirby BJ, Collier AC, Kharasch ED, Whittington D, Thummel KE, Unadkat JD. Complex drug interactions of the HIV protease inhibitors 3: effect of simultaneous or staggered dosing of digoxin and ritonavir, nelfinavir, rifampin, or bupropion. Drug Metab Dispos. 2012;40:610–6.
Kim RB, Fromm MF, Wandel C, Leake B, Wood AJ, Roden DM, et al. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J Clin Invest. 1998;101:289–94.
U.S. Food and Drug Administration. Label Viracept, tablets for oral use and powder for oral use (action date 27/03/2015).
European Medicines Agency. Summary of Product Characteristics Viracept (version dated 10/06/2014).
European Medicines Agency. European Public Assessment Report Farydak (version dated 11/09/2015).
European Medicines Agency. Summary of Product Characteristics Farydak (version dated 11/09/2015).
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 206619Orig1s000 Viekirax Pak.
European Medicines Agency. Summary of Product Characteristics Viekirax (version dated 30/11/2015).
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) 203469Orig1s000 Iclusig.
European Medicines Agency. Summary of Product Characteristics Iclusig (version dated 11/01/2016).
Reitman ML, Chu X, Cai X, Yabut J, Venkatasubramanian R, Zajic S, et al. Rifampin’s acute inhibitory and chronic inductive drug interactions: experimental and model-based approaches to drug–drug interaction trial design. Clin Pharmacol Ther. 2011;89:234–42.
Hamman MA, Bruce MA, Haehner-Daniels BD, Hall SD. The effect of rifampin administration on the disposition of fexofenadine. Clin Pharmacol Ther. 2001;69:114–21.
European Medicines Agency. Summary of Product Characteristics Norvir (version dated 23/11/2015).
Ding R, Tayrouz Y, Riedel K-D, Burhenne J, Weiss J, Mikus G, et al. Substantial pharmacokinetic interaction between digoxin and ritonavir in healthy volunteers. Clin Pharmacol Ther. 2004;76:73–84.
Alsenz J, Steffen H, Alex R. Active apical secretory efflux of the HIV protease inhibitors saquinavir and ritonavir in Caco-2 cell monolayers. Pharm Res. 1998;15:423–8.
Khaliq Y, Gallicano K, Venance S, Kravcik S, Cameron DW. Effect of ketoconazole on ritonavir and saquinavir concentrations in plasma and cerebrospinal fluid from patients infected with human immunodeficiency virus. Clin Pharmacol Ther. 2000;68:637–46.
Corallo CE, Rogers IR. Roxithromycin-induced digoxin toxicity. Med J Aust. 1996;165:433–4.
European Medicines Agency. Summary of Product Characteristics Invirase (version dated 24/02/2016).
Smalley J, Marino AM, Xin B, Olah T, Balimane PV. Development of a quantitative LC-MS/MS analytical method coupled with turbulent flow chromatography for digoxin for the in vitro P-gp inhibition assay. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;854:260–7.
European Medicines Agency. European Public Assessment Report Olysio (version dated 04/06/2014).
European Medicines Agency. Summary of Product Characteristics Olysio (version dated 25/09/2015).
U.S. Food and Drug Administration. Label Zagam, tablets (action date 4/04/2003).
de Lange EC, Marchand S, van den Berg D, van der Sandt IC, de Boer AG, Delon A, et al. In vitro and in vivo investigations on fluoroquinolones; effects of the P-glycoprotein efflux transporter on brain distribution of sparfloxacin. Eur J Pharm Sci. 2000;12:85–93.
Cormet-Boyaka E, Huneau JF, Mordrelle A, Boyaka PN, Carbon C, Rubinstein E, et al. Secretion of sparfloxacin from the human intestinal Caco-2 cell line is altered by P-glycoprotein inhibitors. Antimicrob Agents Chemother. 1998;42:2607–11.
European Medicines Agency. Summary of Product Characteristics Aptivus (version dated 10/12/2015).
Giessmann T, May K, Modess C, Wegner D, Hecker U, Zschiesche M, et al. Carbamazepine regulates intestinal P-glycoprotein and multidrug resistance protein MRP2 and influences disposition of talinolol in humans. Clin Pharmacol Ther. 2004;76:192–200.
Akamine Y, Miura M, Yasui-Furukori N, Kojima M, Uno T. Carbamazepine differentially affects the pharmacokinetics of fexofenadine enantiomers. Br J Clin Pharmacol. 2012;73:478–81.
Yamada S, Yasui-Furukori N, Akamine Y, Kaneko S, Uno T. Effects of the P-glycoprotein inducer carbamazepine on fexofenadine pharmacokinetics. Ther Drug Monit. 2009;31:764–8.
Zhang C, Zuo Z, Kwan P, Baum L. In vitro transport profile of carbamazepine, oxcarbazepine, eslicarbazepine acetate, and their active metabolites by human P-glycoprotein. Epilepsia. 2011;52:1894–904.
El Ela AA, Härtter S, Schmitt U, Hiemke C, Spahn-Langguth H, Langguth P. Identification of P-glycoprotein substrates and inhibitors among psychoactive compounds–implications for pharmacokinetics of selected substrates. J Pharm Pharmacol. 2004;56:967–75.
Saruwatari J, Yasui-Furukori N, Niioka T, Akamine Y, Takashima A, Kaneko S, et al. Different effects of the selective serotonin reuptake inhibitors fluvoxamine, paroxetine, and sertraline on the pharmacokinetics of fexofenadine in healthy volunteers. J Clin Psychopharmacol. 2012;32:195–9.
Weiss J, Dormann S-MG, Martin-Facklam M, Kerpen CJ, Ketabi-Kiyanvash N, Haefeli WE. Inhibition of P-glycoprotein by newer antidepressants. J Pharmacol Exp Ther. 2003;305:197–204.
Iwaki K, Sakaeda T, Kakumoto M, Nakamura T, Komoto C, Okamura N, et al. Haloperidol is an inhibitor but not substrate for MDR1/P-glycoprotein. J Pharm Pharmacol. 2006;58:1617–22.
Yasui-Furukori N, Saito M, Niioka T, Inoue Y, Sato Y, Kaneko S. Effect of itraconazole on pharmacokinetics of paroxetine: the role of gut transporters. Ther Drug Monit. 2007;29:45–8.
Rameis H. The importance of prospective planning of pharmacokinetic trials. Considerations of studies on the phenytoin–digoxin-(P–D) and phenytoin–digitoxin-(P–DT) interaction. Int J Clin Pharmacol Ther Toxicol. 1992;30:528–9.
Collett A, Higgs NB, Sims E, Rowland M, Warhurst G. Modulation of the permeability of H2 receptor antagonists cimetidine and ranitidine by P-glycoprotein in rat intestine and the human colonic cell line Caco-2. J Pharmacol Exp Ther. 1999;288:171–8.
Lægemiddelstyrelsen (Danish Medicines Agency). Summary of Product Characteristics Domperidon “Alternova” (version published online 14/12/2015).
Yoshizato T, Kotegawa T, Imai H, Tsutsumi K, Imanaga J, Ohyama T, et al. Itraconazole and domperidone: a placebo-controlled drug interaction study. Eur J Clin Pharmacol. 2012;68:1287–94.
Faassen F, Vogel G, Spanings H, Vromans H. Caco-2 permeability, P-glycoprotein transport ratios and brain penetration of heterocyclic drugs. Int J Pharm. 2003;263:113–22.
Pauli-Magnus C, Rekersbrink S, Klotz U, Fromm MF. Interaction of omeprazole, lansoprazole and pantoprazole with P-glycoprotein. Naunyn Schmiedebergs Arch Pharmacol. 2001;364:551–7.
Lægemiddelstyrelsen (Danish Medicines Agency). Summary of Product Characteristics Lansoprazol “Stada” (version published online 11/01/2016).
Wandel C, Kim R, Wood M, Wood A. Interaction of morphine, fentanyl, sufentanil, alfentanil, and loperamide with the efflux drug transporter P-glycoprotein. Anesthesiology. 2002;96:913–20.
Zhou Y, Sridhar R, Shan L, Sha W, Gu X, Sukumar S. Loperamide, an FDA-approved antidiarrhea drug, effectively reverses the resistance of multidrug resistant MCF-7/MDR1 human breast cancer cells to doxorubicin-induced cytotoxicity. Cancer Invest. 2012;30:119–25.
Crowe A, Wong P. Potential roles of P-gp and calcium channels in loperamide and diphenoxylate transport. Toxicol Appl Pharmacol. 2003;193:127–37.
Kim T-E, Lee H, Lim KS, Lee S, Yoon S-H, Park K-M, et al. Effects of HM30181, a P-glycoprotein inhibitor, on the pharmacokinetics and pharmacodynamics of loperamide in healthy volunteers. Br J Clin Pharmacol. 2014;78:556–64.
Vandenbossche J, Huisman M, Xu Y, Sanderson-Bongiovanni D, Soons P. Loperamide and P-glycoprotein inhibition: assessment of the clinical relevance. J Pharm Pharmacol. 2010;62:401–12.
Sadeque AJ, Wandel C, He H, Shah S, Wood AJ. Increased drug delivery to the brain by P-glycoprotein inhibition. Clin Pharmacol Ther. 2000;68:231–7.
European Medicines Agency. European Public Assessment Report Moventig (version dated 17/12/2014).
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 204760 Movantik.
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 204042Orig1s000 Invokana.
European Medicines Agency. Summary of Product Characteristics Vokanamet (version dated 29/01/2016).
European Medicines Agency. European Public Assessment Report Cerdelga (version dated 13/02/2015).
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 205494 Cerdelga.
Niemi M, Backman JT, Neuvonen M, Neuvonen PJ, Kivistö KT. Effects of rifampin on the pharmacokinetics and pharmacodynamics of glyburide and glipizide. Clin Pharmacol Ther. 2001;69:400–6.
Golstein PE, Boom A, van Geffel J, Jacobs P, Masereel B, Beauwens R. P-glycoprotein inhibition by glibenclamide and related compounds. Pflugers Arch. 1999;437:652–60.
Lilja JJ, Niemi M, Fredrikson H, Neuvonen PJ. Effects of clarithromycin and grapefruit juice on the pharmacokinetics of glibenclamide. Br J Clin Pharmacol. 2007;63:732–40.
European Medicines Agency. European Public Assessment Report Onglyza (version dated 29/03/2011).
European Medicines Agency. Summary of Product Characteristics Onglyza (version dated 23/11/2015).
Lægemiddelstyrelsen (Danish Medicines Agency). Summary of Product Characteristics Revitelle (version published online 04/01/2016).
Church MK. Safety and efficacy of bilastine: a new H(1)-antihistamine for the treatment of allergic rhinoconjunctivitis and urticaria. Expert Opin Drug Saf. 2011;10:779–93.
Crowe A, Wright C. The impact of P-glycoprotein mediated efflux on absorption of 11 sedating and less-sedating antihistamines using Caco-2 monolayers. Xenobiotica. 2012;42:538–49.
Polli JW, Baughman TM, Humphreys JE, Jordan KH, Mote AL, Salisbury JA, et al. P-glycoprotein influences the brain concentrations of cetirizine (Zyrtec), a second-generation non-sedating antihistamine. J Pharm Sci. 2003;92:2082–9.
Conen S, Theunissen EL, Vermeeren A, van Ruitenbeek P, Stiers P, Mehta MA, et al. The role of P-glycoprotein in CNS antihistamine effects. Psychopharmacology. 2013;229:9–19.
Desrayaud S, Guntz P, Scherrmann JM, Lemaire M. Effect of the P-glycoprotein inhibitor, SDZ PSC 833, on the blood and brain pharmacokinetics of colchicine. Life Sci. 1997;61:153–63.
Tröger U, Lins H, Scherrmann J-M, Wallesch C-W, Bode-Böger SM. Tetraparesis associated with colchicine is probably due to inhibition by verapamil of the P-glycoprotein efflux pump in the blood-brain barrier. BMJ. 2005;331:613.
Eleftheriou G, Bacis G, Fiocchi R, Sebastiano R. Colchicine-induced toxicity in a heart transplant patient with chronic renal failure. Clin Toxicol. 2008;46:827–30.
Dahan A, Sabit H, Amidon GL. Multiple efflux pumps are involved in the transepithelial transport of colchicine: combined effect of p-glycoprotein and multidrug resistance-associated protein 2 leads to decreased intestinal absorption throughout the entire small intestine. Drug Metab Dispos. 2009;37:2028–36.
Cvetkovic M, Leake B, Fromm MF, Wilkinson GR, Kim RB. OATP and P-glycoprotein transporters mediate the cellular uptake and excretion of fexofenadine. Drug Metab Dispos. 1999;27:866–71.
European Medicines Agency. European Public Assessment Report Betmiga (version dated 15/01/2013).
Takusagawa S, Ushigome F, Nemoto H, Takahashi Y, Li Q, Kerbusch V, et al. Intestinal absorption mechanism of mirabegron, a potent and selective β3-adrenoceptor agonist: involvement of human efflux and/or influx transport systems. Mol Pharm. 2013;10:1783–94.
Kharasch ED, Hoffer C, Whittington D, Sheffels P. Role of P-glycoprotein in the intestinal absorption and clinical effects of morphine. Clin Pharmacol Ther. 2003;74:543–54.
Fromm MF, Eckhardt K, Li S, Schänzle G, Hofmann U, Mikus G, et al. Loss of analgesic effect of morphine due to coadministration of rifampin. Pain. 1997;72:261–7.
Heiskanen T, Backman JT, Neuvonen M, Kontinen VK, Neuvonen PJ, Kalso E. Itraconazole, a potent inhibitor of P-glycoprotein, moderately increases plasma concentrations of oral morphine. Acta Anaesthesiol Scand. 2008;52:1319–26.
Skarke C, Jarrar M, Erb K, Schmidt H, Geisslinger G, Lötsch J. Respiratory and miotic effects of morphine in healthy volunteers when P-glycoprotein is blocked by quinidine. Clin Pharmacol Ther. 2003;74:303–11.
Schinkel AH, Wagenaar E, van Deemter L, Mol CA, Borst P. Absence of the mdr1a P-Glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J Clin Invest. 1995;96:1698–705.
Crowe A. The influence of P-glycoprotein on morphine transport in Caco-2 cells. Comparison with paclitaxel. Eur J Pharmacol. 2002;440:7–16.
European Medicines Agency. Summary of Product Characteristics Vargatef (version dated 27/05/2015).
U.S. Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review(s) NDA 205832 Ofev.
Machavaram KK, Gundu J, Yamsani MR. Effect of ketoconazole and rifampicin on the pharmacokinetics of ranitidine in healthy human volunteers: a possible role of P-glycoprotein. Drug Metabol Drug Interact. 2006;22:47–65.
Bourdet DL, Pollack GM, Thakker DR. Intestinal absorptive transport of the hydrophilic cation ranitidine: a kinetic modeling approach to elucidate the role of uptake and efflux transporters and paracellular vs. transcellular transport in Caco-2 cells. Pharm Res. 2006;23:1178–87.
Bourdet DL, Thakker DR. Saturable absorptive transport of the hydrophilic organic cation ranitidine in Caco-2 cells: role of pH-dependent organic cation uptake system and P-glycoprotein. Pharm Res. 2006;23:1165–77.
Schwarz UI, Hanso H, Oertel R, Miehlke S, Kuhlisch E, Glaeser H, et al. Induction of intestinal P-glycoprotein by St John’s wort reduces the oral bioavailability of talinolol. Clin Pharmacol Ther. 2007;81:669–78.
Mueller SC, Uehleke B, Woehling H, Petzsch M, Majcher-Peszynska J, Hehl E-M, et al. Effect of St John’s wort dose and preparations on the pharmacokinetics of digoxin. Clin Pharmacol Ther. 2004;75:546–57.
Tian R, Koyabu N, Morimoto S, Shoyama Y, Ohtani H, Sawada Y. Functional induction and de-induction of P-glycoprotein by St. John’s wort and its ingredients in a human colon adenocarcinoma cell line. Drug Metab Dispos. 2005;33:547–54.
Lægemiddelstyrelsen (Danish Medicines Agency). Summary of Product Characteristics Kinin “DAK” (version published online 08/02/2016).
Aronson JK, Carver JG. Interaction of digoxin with quinine. Lancet. 1981;1:1418.
Hedman A, Angelin B, Arvidsson A, Dahlqvist R, Nilsson B. Interactions in the renal and biliary elimination of digoxin: stereoselective difference between quinine and quinidine. Clin Pharmacol Ther. 1990;47:20–6.
Hsiao P, Bui T, Ho RJY, Unadkat JD. In vitro-to-in vivo prediction of P-glycoprotein-based drug interactions at the human and rodent blood-brain barrier. Drug Metab Dispos. 2008;36:481–4.
Hu Y, Sieck DE, Hsu WH. Why are second-generation H1-antihistamines minimally sedating? Eur J Pharmacol. 2015;765:100–6.
Bagal S, Bungay P. Restricting CNS penetration of drugs to minimise adverse events: role of drug transporters. Drug Discov Today Technol. 2014;12:e79–85.
Sundhedsstyrelsen (Danish Health and Medicines Authority). Summary of Product Characteristics Imodium “McNeil” (version published online 04/27/15).
Kreisl WC, Liow J-S, Kimura N, Seneca N, Zoghbi SS, Morse CL, et al. P-glycoprotein function at the blood-brain barrier in humans can be quantified with the substrate radiotracer 11C-N-desmethyl-loperamide. J Nucl Med. 2010;51:559.
Eggleston W, Clark KH, Marraffa JM. Loperamide abuse associated with cardiac dysrhythmia and death. Ann Emerg Med. 2016. doi:10.1016/j.annemergmed.2016.03.047.
Shi JG, Zhang Y, Yeleswaram S. The relevance of assessment of intestinal P-gp inhibition using digoxin as an in vivo probe substrate. Nat Rev Drug Discov. 2011;10:75.
Kharasch ED, Bedynek PS, Walker A, Whittington D, Hoffer C. Mechanism of ritonavir changes in methadone pharmacokinetics and pharmacodynamics: II. Ritonavir effects on CYP3A and P-glycoprotein activities. Clin Pharmacol Ther. 2008;84:506–12.
Tolle-Sander S, Rautio J, Wring S, Polli JW, Polli JE. Midazolam exhibits characteristics of a highly permeable P-glycoprotein substrate. Pharm Res. 2003;20:757–64.
European Medicines Agency. Summary of Product Characteristics Exviera (version dated 10/02/2016).
Mallet L, Spinewine A, Huang A. The challenge of managing drug interactions in elderly people. Lancet. 2007;370:185–91.
Ince I, Knibbe CAJ, Danhof M, de Wildt SN. Developmental changes in the expression and function of cytochrome P450 3A isoforms: evidence from in vitro and in vivo investigations. Clin Pharmacokinet. 2013;52:333–45.
Lacroix D, Sonnier M, Moncion A, Cheron G, Cresteil T. Expression of CYP3A in the human liver–evidence that the shift between CYP3A7 and CYP3A4 occurs immediately after birth. Eur J Biochem. 1997;247:625–34.
Fanni D, Ambu R, Gerosa C, Nemolato S, Castagnola M, Van Eyken P, et al. Cytochrome P450 genetic polymorphism in neonatal drug metabolism: role and practical consequences towards a new drug culture in neonatology. Int J Immunopathol Pharmacol. 2014;27:5–13.
Hines RN. Ontogeny of human hepatic cytochromes P450. J Biochem Mol Toxicol. 2007;21:169–75.
Mathias AA, Hitti J, Unadkat JD. P-glycoprotein and breast cancer resistance protein expression in human placentae of various gestational ages. Am J Physiol Regul Integr Comp Physiol. 2005;289:R963–9.
Sun M, Kingdom J, Baczyk D, Lye SJ, Matthews SG, Gibb W. Expression of the multidrug resistance P-glycoprotein, (ABCB1 glycoprotein) in the human placenta decreases with advancing gestation. Placenta. 2006;27:602–9.
Daood M, Tsai C, Ahdab-Barmada M, Watchko JF. ABC transporter (P-gp/ABCB1, MRP1/ABCC1, BCRP/ABCG2) expression in the developing human CNS. Neuropediatrics. 2008;39:211–8.
Virgintino D, Errede M, Girolamo F, Capobianco C, Robertson D, Vimercati A, et al. Fetal blood-brain barrier P-glycoprotein contributes to brain protection during human development. J Neuropathol Exp Neurol. 2008;67:50–61.
Lam J, Koren G. P-glycoprotein in the developing human brain: a review of the effects of ontogeny on the safety of opioids in neonates. Ther Drug Monit. 2014;36:699–705.
Artursson P, Matsson P, Karlgren M. In vitro characterization of interactions with drug transporting proteins. In: Sugiyama Y, Steffansen B, editors. Transporters in drug development, AAPS advances in the pharmaceutical sciences series 7. New York: Springer Science + Business Media; 2013.
Ahlin G, Hilgendorf C, Karlsson J, Szigyarto CA-K, Uhlen M, Artursson P. endogenous gene and protein expression of drug-transporting proteins in cell lines routinely used in drug discovery programs. Drug Metab Dispos. 2009;37:2275–83.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study received no specific funding.
Conflict of interest
Drs Lund, Petersen and Dalhoff have no conflicts of interest to declare.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lund, M., Petersen, T.S. & Dalhoff, K.P. Clinical Implications of P-Glycoprotein Modulation in Drug–Drug Interactions. Drugs 77, 859–883 (2017). https://doi.org/10.1007/s40265-017-0729-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-017-0729-x