
Abstract
Purpose
To investigate the underlying mechanism of low bioavailabilities of the water-soluble camptothecin derivatives, belotecan and topotecan.
Methods
The bioavailability of belotecan and topotecan in rats was determined following oral administration of each drug at a dose of 5 mg/kg body weight. The vectorial transport of each drug was measured in Caco-2 and engineered MDCK II cells.
Results
The bioavailability of belotecan (11.4%) and topotecan (32.0%) in rats was increased to 61.5% and 40.8%, respectively, by the preadministration of CsA at a dose of 40 mg/kg. Contrary to the absorptive transport, the secretory transport of these drugs across the Caco-2 cell monolayer was concentration-dependent, saturable, and significantly inhibited by the cis presence of verapamil (a P-gp substrate), MK-571 (an MRP inhibitor), bromosulfophthalein (BSP, an MRP2 inhibitor), fumitremorgin C (FTC, a BCRP inhibitor) and cyclosporine A (CsA, an inhibitor of P-gp and BCRP, and a substrate of P-gp) suggesting the involvement of these transporters, which could be further confirmed in MDCKII/P-gp, MDCKII/MRP2 and MDCKII/BCRP cells.
Conclusion
The involvement of secretory transporters P-gp, MRP2 and BCRP, particularly for belotecan, as well as a low passive permeability, appears to be responsible for the low bioavailability of belotecan and topotecan.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
20-(s)-Camptothecin (CPT), a plant alkaloid isolated from a tree native to China (Camptotheca accuminata), is a novel antitumor agent that exerts its activity exclusively by inhibition of topoisomerase I (1–3). Although, clinical evaluation of CPT was discontinued due to its unpredictably severe toxicity and poor water-solubility, recent development of several semisynthetic CPT analogues including topotecan (Fig. 1A) have succeeded (4,5). Topotecan, a water soluble analogue of CPT, was approved by the FDA for treatment of advanced small cell lung cancers (6). Since then, other CPT analogues are being evaluated in clinical trials. In 2004, a novel water-soluble camptothecin analogue, (20s)-7-(2-isopropylamino)ethylcamptothecin·HCl, also known as belotecan (Fig. 1B), was developed by Chong Kun Dang Pharmaceutical Co. (Seoul, Korea). Belotecan currently is marketed in Korea as Camtobell® for the treatment of ovarian and small cell lung cancers (7,8) based on a series of successful clinical trials (9).

Chemical structure of topotecan (A) and belotecan (B).
CPTs are cell-cycle-specific and are most effective during the S-phase, which is a relatively short phase of the cell cycle. Therefore, prolonged or repetitive exposure to these drugs is needed for efficient killing of malignant cells. Preclinical and clinical studies have shown that prolonged exposure to CPTs, through either continuous intravenous (iv) infusion or repetitive oral (po) administration, results in optimal therapeutic activity (10–12). The oral delivery route is generally preferred over intravenous administration due to increased patient compliance with treatment, decreased vascular access complications, greater convenience and lower costs. However, preclinical studies (13–15) have revealed that the bioavailability of orally administered CPTs is variable and generally low (30∼40%), despite their diverse physicochemical properties.
Multiple transporters including P-gp, multidrug resistance protein2 (MRP2) (16), and breast cancer resistance protein (BCRP) (17–19) are involved in the intestinal efflux of CPTs such as topotecan and irinotecan. Recently, P-gp and MRP2 were reported to be involved in the transport of irinotecan (20), camptothecin (6) and 9-NC (21) in Caco-2 cells and in engineered Madine–Darby canine kidney (MDCK) cells, which overexpress P-gp (MDCKII/P-gp) and MRP2 (MDCKII/MRP2). Most recently, the involvement of MRP2 in the secretory transport of belotecan in Caco-2 cells was demonstrated (22). Therefore, the objective of the present study was to investigate the transport mechanism of belotecan and topotecan in these two cell types as it may relate to the low and variable bioavailability of these orally administered drugs. The involvement of ATP-binding cassette (ABC) transporters, P-gp, MRP2 and BCRP, in the transport of these drugs was preferentially examined.
MATERIALS AND METHODS
Materials
Belotecan and topotecan were kindly provided by Chong Kun Dang Pharm. (Seoul Korea). Fetal bovine serum (FBS) was purchased from Hyclone Laboratories (Logan, UT). Trypsin–EDTA was purchased from Gibco Laboratories (Gaithersburg, MD). Dulbecco’s Modified Eagle’s medium (DMEM), non-essential amino acid solution, l-glutamine, penicillin–streptomycin, Hank’s balanced salt solution (HBSS) and N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), cyclosporin A (CsA) and bromosulfophthalein (BSP) were purchased from Sigma Chemical Co (St. Louis, MO). 3-([{3-(2-[7-chloro-2-quinolinyl]ethyl)phenyl}-{(3-dimethylamino-3-oxopropyl)-thio}-methyl]-thio) propanoic acid (MK-571) was purchased from Cayman chemical (Ann Arbor, MI). Fumitremorgin C (FTC) was purchased from Axxora, LLC (San Diego, CA). All other reagents were analytical grade.
Animals
Male Sprague–Dawley (SD) rats (Dae-Han Biolink, Eumsung, Korea) weighing 260–290 g, were used in the in vivo pharmacokinetic studies. All animal experiments were performed according to the guidelines for Animal Care and Use, Seoul National University.
Cell Culture
A human colonic epithelial cell line, Caco-2 cells, was obtained from the American Type Culture Collection (Rockville, MD). Cells were routinely grown in DMEM containing 10% FBS, 1% non-essential amino acids, 100 U/ml penicillin and 0.1 mg/ml streptomycin at 37°C in an atmosphere of 5% CO2 and 90% relative humidity. For the transport experiments, Caco-2 cells from passage numbers 40 to 55 were seeded on permeable polycarbonate filter inserts (1 cm2, 0.4-μm pore size; Corning Costar Corp., Cambridge, MA) in 12-Transwell plates at a density of 1–1.5 × 105 cells/insert and were grown for 21 days (23). MDCKII/wt cells, MDCKII/P-gp cells, MDCKII/MRP2 cells and MDCKII/BCRP cells were provided by Dr. Borst (The Netherlands Cancer Institute, Amsterdam, The Netherlands). The cells were grown and seeded on filter inserts at a density of 6 × 104 cells/insert in a manner similar to that described for Caco-2 cells.
The integrity of cell monolayers was evaluated prior to the transport experiments by measuring transepithelial electrical resistance (TEER) and/or [14C]mannitol permeability across the monolayers. Cell monolayers were considered intact and suitable for use in transport experiments when TEER values were 300–500 Ω · cm2 for Caco-2 cell monolayers (23) and 130–160 Ω · cm2 for MDCKII cell monolayers. Mannitol transport in the Caco-2 and MDCKII cell monolayers was <0.35% and <2% of the dose/h, respectively.
Measurement of Transepithelial Transport of Belotecan and Topotecan
Transepithelial drug transport across the Caco-2 and MDCKII cell monolayers was conducted in Transwell plates that were placed on an orbital shaker; the plates were shaken at 60 rpm during the transport experiments to minimize the influence of the aqueous boundary layer on transport. Prior to transport experiments, cell monolayers were washed three times with incubation medium (pH 7.4, HBSS containing 25 mM HEPES and 25 mM glucose). After each wash, the plates were incubated in the incubation medium for 30 min at 37°C, and then TEER was measured (23).
For the measurement of apical to basolateral (i.e. absorptive) drug transport, 0.5 ml of incubation medium containing 1–500 μM of either belotecan or topotecan was added to the apical side of the cell monolayer and 1.5 ml of the incubation medium without the drug was added to the basolateral side. The inserts were moved to wells containing fresh incubation medium (1.5 ml) every 20 min for 80 min. At each time point, a 100-μl aliquot of the incubation medium was removed from the basolateral side, and the concentration of the drug in each sample was determined by HPLC.
For the measurement of basolateral to apical (i.e. secretory) drug transport, 1.5 ml of incubation medium containing 1–500 μM of belotecan or topotecan was added to the basolateral side, and 0.5 ml of incubation medium without the drug was added to the apical side. A 0.35 ml aliquot of incubation medium was removed from the apical side every 20 min for 80 min. After removal of each sample, the volume removed (0.35 ml) was replaced with fresh incubation medium. The concentration of the drug in each sample was determined by HPLC.
Inhibitory Effect of P-gp, MRP2 and BCRP Substrates on Belotecan and Topotecan Transport in Caco-2 Cells
The transport of belotecan and topotecan (10 μM) across Caco-2 cell monolayers was investigated in the cis presence of substrates or inhibitors of P-gp [50 μM CsA (24) and 100 μM verapamil (24)], MRP2 [200 μM BSP (25) and 200 μM MK-571 (26)] or BCRP [10 μM FTC (27)]. For the measurement of absorptive transport, each substrate and drug, dissolved in saline, were added to the apical side and the appearance of the drug on the basolateral side was measured every 20 min for 80 min as described above. For secretory transport, each substrate (at the above-mentioned concentration) and the drug (10 μM) were added to the basolateral side and the appearance of the drug on the apical side was measured. To determine the inhibition kinetics, secretory drug transport in the cis presence of increasing concentrations of relevant substrates (verapamil, MK-571 and FTC) was measured.
Pharmacokinetics and the Effect of CsA on the Oral Bioavailability of Belotecan and Topotecan in Rats
The SD rats were fasted overnight prior to the experiments, but were allowed free access to water. Animals were anesthetized with ketamine and then their femoral arteries and veins were cannulated with polyethylene tubing (PE-50; Clay Adams, Parsippany, NJ), and filled with heparinized saline (25 IU/ml) to prevent blood clotting. After recovering from the anesthesia, rats were intravenously (iv) administered either belotecan (n = 3) or topotecan (n = 3) solution in sterile water (2 mg/ml) at a dose of 5 mg/kg body weight. Blood samples (150 μl) were taken from the femoral artery cannula of each rat before drug administration (0 min), and at increments of 2, 7, 15, 30, 60, 120, 240, 480 and 720 min after topotecan administration and at 5, 10, 30, 60, 120, 240, 360, 480 and 1440 min after belotecan administration. The blood volume withdrawn at each time point was replaced with an equal volume of saline to compensate for fluid loss. Plasma was separated by centrifugation and stored at −80°C for later HPLC drug analysis. Oral absorption studies were performed by administering sterile water solutions (2 mg/ml) of belotecan (n = 3) and topotecan (n = 3) at a drug dose of 5 mg/kg. Blood sampling and assay were identical to those for iv administration studies.
The effect of CsA preadministration on the pharmacokinetics and bioavailability of the drugs was also investigated as follows. CsA was dissolved at 40 mg/ml concentration in Cremophor RH40 containing 11.9% (v/v) ethyl alcohol, and the CsA solution was orally (po) administered at a 40 mg/kg CsA-equivalent volume dose (i.e., 1 ml/kg dose) 30 min prior to the iv or oral administration of each drug. Blood sampling and assay were performed as described above.
It was confirmed that the solvent (i.e., 11.9% the ethyl alcohol in Cremophor RH40), under the given dose of 1 ml/kg, did not to influence the oral absorption of belotecan and topotecan (data not shown).
HPLC Analysis of Belotecan and Topotecan
The concentrations of belotecan and topotecan in the samples were determined using modified reverse-phase HPLC (28,29). Briefly, a 50-μl aliquot of the sample was spiked with 50 μl of the internal standard acetonitrile solution, 50 μl acetonitrile and 50 μl of 7% (w/v) perchloric acid, and mixed. The mixture then was centrifuged and 100 μl of supernatant was injected into the HPLC. Tetracycline and belotecan were used as internal standards for belotecan and topotecan, respectively. For the analysis of transport study samples, 100 μl of sample were acidified with 200 μl of phosphoric acid (30%, w/v). The addition of acids (perchloric acid for plasma samples and phosphoric acid for transport samples) was to ensure the conversion of each drug into its respective lactone (29). HPLC was performed using a C18 column (XTerraTM, 250 mm × 4.6 mm, 5 μm, Waters). The mobile phase was a 1:3 (v/v) mixture of acetonitrile and 0.01 M phosphate buffer solution (pH 3.7). The flow rate was maintained at 1 ml/min. A Shimazu RF-535 fluorescence detector was used to detect belotecan (λ ex at 370 nm and λ em at 430 nm) and topotecan (λ ex at 380 nm and λ em at 527 nm).
Data Analysis
Representative pharmacokinetic parameters of belotecan and topotecan following iv administration of each drug were obtained by noncompartmental analysis of the plasma concentration–time data using WinNonlin version 2.1 (Pharsight, Mountain View, CA). For the plasma samples, the area under the concentration–time curve from time zero to infinity (\(AUC_{0 - \infty } \)) was calculated using standard trapezoidal and extrapolation methods. The bioavailability (F) of belotecan and topotecan after oral administration was calculated using the following equation:
In each transport experiment, the apparent initial transport rate (V, pmol/cm2/min) of the drug was calculated from the initial linear portion of the plot of the total amount of drug transported versus time. Non-linear regression analysis was performed to fit the plot to an equation, as follows:
where V max and K m are the maximum transport rate (pmol/cm2/min) and the Michalis–Menten constant (μM), respectively, and K represents the linear clearance (μl/cm2/min). The intrinsic clearance for the transport (CLint) was obtained from V max /K m.
In the potential inhibition studies, the IC50 values were obtained by fitting the transport inhibition data to an inhibitory sigmoidal model of Hill’s equation (30) using WinNonlin (Pharsight, Mountain View, CA):
where v and Vo are the % inhibition ratio of belotecan or topotecan in the presence and absence of inhibitors, I max is the maximal percent inhibition (%), IC50 is the inhibitor concentration (μM) that causes 50% inhibition of transport, C is the inhibitor concentration (μM), and n is either the curve-fitting coefficient or Hill’s coefficient.
All data are expressed as the mean ± SD for all three experiments. The statistical significance of differences between multiple treatments was evaluated using the Turkey’s test, and a value of P < 0.01 was taken to be statistically significant.
RESULTS
Absorptive and Secretory Transport of Belotecan and Topotecan Across Caco-2 Cell Monolayers
Vectorial transport of belotecan and topotecan was investigated over the initial concentration range of 1∼500 μM. No saturation in flux was observed for absorptive (i.e., apical to basal) transport of belotecan over the concentration range examined (Fig. 2A), suggesting that passive diffusion was the predominant mechanism for absorptive transport. This result was supported by the linearity of the Eadie–Hofstee plot (inset of Fig. 2A). The linear clearance rate of passive diffusion (i.e., CLlinear) of belotecan was calculated to be 0.05 μl/cm2/min (Caco-2 in Table I).

Concentration dependency of belotecan (A) and topotecan (B) for absorptive (i.e., apical to basal, empty circle) and secretory (i.e., basal to apical) transport (filled circle) across Caco-2 cell monolayers. The concentrations of belotecan and topotecan in the transport studies were 1, 5, 10, 20, 50, 100, 300, and 500 μM. The inset shows the Eadie–Hofstee transformation of the transport data for the cell monolayers. Each point represents the mean ± SD for three monolayers.
In contrast to the results observed for belotecan, distinct saturation was observed for the absorptive flux of topotecan over the concentration range examined (1∼500 μM, Fig. 2B), suggesting the involvement of a carrier-mediated mechanism in the absorptive transport of topotecan. Non-linear regression fitting of the data to Eq. 2 yielded 8.57 μM for K m, 2.25 pmol/cm2/min for V max, and 0.08 μl/cm2/min for the linear clearance rate (CLlinear, Caco-2 in Table I). The intrinsic clearance (CLint, i.e., V max/K m) for carrier-mediated absorption was calculated to be 0.26 μl/cm2/min. The CLlinear of belotecan was reduced by 0.6-fold relative to that of topotecan.
The secretory (i.e., basal to apical) fluxes of belotecan and topotecan were saturable over a concentration range of 1 to 500 μM (Fig. 2A, B). Eadie–Hofstee plots demonstrated the presence of both saturable and non-saturable processes in the secretory transport of both drugs (Fig. 2A, B, insets). Non-linear regression fitting of the data to Eq. 2 demonstrated a high-affinity (K m, 10.96 μM) and low-capacity (V max, 6.28 pmol/cm2/min) transport mechanism for belotecan, and a low-affinity (78.27 μM) and high-capacity (29.12 pmol/cm2/min) mechanism for topotecan (Table II). As a result, CLint for the secretory transport of belotecan was 1.5-fold greater than that of topotecan (Caco-2 in Table II). In contrast, CLlinear for belotecan (0.05 μl/cm2/min) was 0.6-fold less than that of topotecan (0.09 μl/cm2/min), consistent with CLlinear for the absorptive transport of these drugs (Caco-2 in Table II).
Effect of Various Substrates or Inhibitors on the Transport of Belotecan and Topotecan Across Caco-2 Cell Monolayers
The involvement of ABC transporters, P-gp, MRP2 and BCRP, in the secretory transport of belotecan and topotecan was examined in Caco-2 cells. The secretory flux of both belotecan and topotecan (10 μM each) was significantly decreased by the cis presence (i.e., basal presence) of cyclosporine A (CsA, 50 μM, a substrate of P-gp, and an inhibitor of P-gp and BCRP (24)) and verapamil (100 μM, a P-gp substrate) (Fig. 3A, B). Consistent with this decrease, the absorptive fluxes of belotecan and topotecan were significantly increased by the cis presence (i.e., apical presence) of CsA (50 μM) and verapamil (100 μM). Therefore, P-gp appeared to be involved in the secretory transport of belotecan and topotecan.

The effect of cis presence of CsA (50 μM), verapamil (100 μM), MK571 (200 μM), BSP (200 μM), FTC (40 μM) on absorptive (i.e., apical to basal) and secretory (i.e., basal to apical) transport of belotecan (A) and topotecan (B) across Caco-2 cell monolayers. Each point represents the mean ± SD for three monolayers. *P < 0.05, transport flux (in the presence of inhibitors) is significantly different from the control (in the absence of inhibitors).
The secretory transport of belotecan and topotecan was significantly decreased by the cis presence of probenecid (22) and BSP [an MRP2 inhibitor (25)], suggesting the involvement of MRP2 in secretory transport. However, inconsistent with this result, absorptive transport of belotecan was not increased by the cis presence of BSP (200 μM) (Fig. 3A). Moreover, the absorptive transport of topotecan was even decreased (P < 0.05) by the cis presence of MK-571 and BSP (200 μM each, Fig. 3B). Detailed mechanisms for this discrepancy await further investigation.
The cis presence of FTC (100 μM, a BCRP inhibitor) significantly reduced the secretory transport of belotecan and topotecan without affecting absorptive transport (Fig. 3A, B), suggesting the involvement of BCRP, as well as P-gp and MRP2, in the transport of these drugs.
Contribution of P-gp, MRP2 and BCRP to the Secretory Transport of Belotecan and Topotecan Across Caco-2 Cell Monolayers
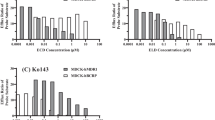
To further elucidate the contribution of P-gp, MRP2 and BCRP to secretory drug transport, the effects of the cis presence of verapamil (1∼500 μM), MK-571 (1∼400 μM) and FTC (0.01∼40 μM) on secretory transport of belotecan and topotecan (10 μM) were examined (Fig. 4A–C, respectively). The 50% inhibition concentration, IC50 (μM) and maximal percent inhibition, I max (%), were calculated using Eq. 3. The IC50 of verapamil for belotecan transport was reduced 0.5-fold compared with topotecan (i.e., 88.9 vs 173.8 μM), while the I max with verapamil was comparable between the two drugs (53.1% vs 48.3%, Table III). These results suggest that belotecan and topotecan share a common secretory transport system, P-gp; moreover, the affinity of belotecan for P-gp was lower than that of topotecan. In contrast, no substantial differences in the values of IC50 and I max for MK-571 (i.e., 129.4 vs 103.0 μM, and 29.2% vs 32.5%, respectively, Table III) and FTC (i.e., 0.6 vs 0.4 μM, and 52.6% vs 59.4%, respectively, Table IV) were observed between belotecan and topotecan, suggesting comparable contributions of MRP and BCRP to secretory transport of these drugs.

The inhibitory effect of verapamil (A), MK-571 (B) and FTC (C) on secretory (i.e., basal to apical) transport of belotecan (filled circle) and topotecan (empty circle) across Caco-2 cell monolayers. The flux of belotecan and topotecan (10 μM each) were measured in the presence of increasing concentration of inhibitors. Each point represents the mean ± SD for three monolayers.
Transport of Belotecan and Topotecan Across the Engineered MDCKII Cell Monolayers
Transporter involvement in the transport of belotecan and topotecan was further investigated using monolayers of polarized MDCKII cells transfected with P-gp (MDCKII/P-gp), MRP2 (MDCKII/MRP2) and BCRP (MDCKII/BCRP) genes. Similar to the results of the Caco-2 cell experiments (Fig. 2A), the absorptive transport of belotecan increased linearly with the concentration of the drug in all MDCKII cells (Fig. 5A, B, C, D), confirming the predominance of passive diffusion to total belotecan absorption. Similar linearity was also observed for absorptive transport of topotecan (Fig. 5A′, B′, C′, D′), suggesting the predominance of passive diffusion in topotecan transport. This finding is inconsistent with the topotecan results in Caco-2 cells (Fig. 2B), which indicated a carrier-mediated transport mechanism for absorptive transport of topotecan. The discrepancy may be attributable to the difference in transporter expression between Caco-2 and MDCK cells. Similar to the Caco-2 cell experiments, however, the passive diffusion clearance (CLlinear) of belotecan was less than that of topotecan (Table I).

Concentration dependency of belotecan (A, B, C, D) and topotecan (A′, B′, C′, D′) transport across MDCKII/wt Cells (A and A′), MDCKII/P-gp cells (B and B′), MDCKII/MRP2 cells (C and C′), and MDCKII/BCRP cells (D and D′), for the absorptive (filled circle) and secretory (empty circle) transport. The concentrations of belotecan and topotecan in the transport studies were 1, 5, 10, 20, 50, 100, 300, and 500 μM. Each point represents the mean ± SD of three monolayers.
In contrast to absorptive transport, secretory transport of belotecan and topotecan in most MDCK cells (Fig. 5) was mediated by saturable and nonsaturable (i.e., linear) processes (Eadie–Hofstee plots, data not shown). The linear portion was smaller than the saturable portion for both drugs, suggesting predominance of carrier-mediated transport over passive diffusion in secretory transport of these drugs. The kinetic parameters for secretory belotecan and topotecan transport are summarized in Table II.
In wild-type MDCKII (MDCKII/wt) cells, the secretory transport of belotecan and topotecan was much greater than that of the corresponding absorptive transport, respectively (Fig. 5A, A′). Non-linear regression fitting of the data to Eq. 2 demonstrated a transport mechanism with high affinity (K m, 73.12 μM) and low capacity (V max, 17.11 pmol/cm2/min) for belotecan, and a passive diffusion (CLlinear, 0.04 μl/cm2/min) plus a transport mechanism with low affinity (K m, 102.39 μM) and high capacity (V max, 30.33 pmol/cm2/min) for topotecan (Table II). The results were comparable to the results obtained in the Caco-2 cell experiments (Table II).
(Involvement of P-gp)
In MDCKII/P-gp cells, secretory transport was saturable for both drugs (Fig. 5B, B′) with lower-affinity (K m, 101.19 μM) and higher-capacity (V max, 59.57 pmol/cm2/min) for belotecan, and higher-affinity (K m, 88.92 μM) and lower-capacity (V max, 38.58 pmol/cm2/min) for topotecan (Table II). The V max for secretory transport of belotecan and topotecan in MDCKII/P-gp cells was 3.2- and 1.1-fold greater, respectively, than that in MDCKII/wt cells. As the result, the intrinsic clearances (CLint) of belotecan and topotecan were greater than those of MDCKII/wt. The CLint of belotecan was larger than that of topotecan (0.58 vs 0.43 μl/cm2/min) due to the higher V max for belotecan.
(Involvement of MRP2)
In MDCKII/MRP2 cells, the secretory transport of belotecan also was concentration-dependent and saturable (Fig. 5C) with a K m value of 267.93 μM (Table II). The secretory V max for belotecan was increased 6.8-fold, relative to MDCKII/wt cells (Table II), suggesting the involvement of MRP2 in secretory transport of the drug. For the secretory transport of topotecan, higher transport rate compared to absorptive transport was observed, suggesting the involvement of BCRP in the secretion of topotecan. Complete transport saturation was not achieved at even the highest concentration of topotecan (i.e., 500 μM) (Fig. 5C′), suggesting a lower affinity and higher capacity of MRP2 for the transport of topotecan. Non-linear regression fitting of the data to Eq. 2 revealed low-affinity transport of topotecan (K m, >500) cells. The smaller K m value for belotecan compared to that for topotecan in these cells (267.93 vs >500, Table II) suggests that, compared with topotecan, belotecan has a higher affinity for MRP2.
(Involvement of BCRP)
In MDCKII/BCRP cells, the secretory transport of topotecan was concentration dependent and saturable (Fig. 5D′) with the K m value of 213.28 μM (Table II). The secretory V max for topotecan was increased 4-fold, relative to MDCKII/wt cells (Table II). The secretory transport of belotecan was much higher than the absorptive transport of the drug, suggesting the involvement of BCRP in the secretion of belotecan. The transport saturation was not achieved even at the highest concentration of belotecan (i.e., 500 μM) (Fig. 5D), suggesting a lower affinity and higher capacity of BCRP for the transport of belotecan, compared to topotecan (Table II).
In Vivo Pharmacokinetics of Belotecan and Topotecan in Rats
In order to examine the involvement of transporters in the absorption of belotecan and topotecan in vivo, the effect of pretreatment with CsA, a representative inhibitor of P-gp and BCRP (18), on the oral absorption of these drugs was examined in the rat. The mean plasma concentration–time profiles after oral and iv administration of belotecan and topotecan at a dose of 5 mg/kg are shown in Fig. 6 and corresponding pharmacokinetic parameters are summarized in Table IV. The plasma profiles of these drugs followed a biexponential decline after iv administration. Terminal half-lives of belotecan and topotecan were 508 and 93 min, respectively, indicating a slower decline for belotecan compared to topotecan. The oral absorption of belotecan was rapid with a mean C max of 119.8 ng/ml at a t max of 16.7 min, while that of topotecan was somewhat slow with a mean C max of 518.9 ng/ml at a t max of 33.1 min. After achieving C max, the plasma concentrations of these drugs declined in a biexponential fashion. The absolute oral bioavailability (F) of belotecan was calculated (Eq. 1) to be low (11.4%) compared with topotecan (32.0%).

Plasma concentration profiles of belotecan (A, B) and topotecan (C, D) in rats after intravenous (iv) and oral (po) administration of each drug at a dose of 5 mg/kg rat, without (i.e., control, filled circle) and with (i.e., + CsA, empty circle) CsA pretreatment (40 mg/kg, po). Each data point represents the mean ± SE of three (control) or four (+ CsA) rats.
By the 30 min preadministration of CsA at a dose of 40 mg/kg, the absorption of both drugs was increased (Fig. 6 and Table IV). The mean C max of oral belotecan was substantially increased by the CsA pretreatment, while that of topotecan demonstrated a decrease. Nevertheless, the bioavailabilities of both drugs were increased from 11.4 to 61.5 (belotecan, P < 0.01) and from 32.0% to 40.8% (topotecan, insignificant), when calculated based on the control iv \(AUC_{0 - \infty } \) value of each drug, demonstrating a 5.4- (belotecan) and 1.5-fold increase in the absorption of these drugs with CsA pretreatment. The bioavailabilities of belotecan and topotecan were calculated to be 21.0% and 37.0%, respectively, when calculated based on the iv \(AUC_{0 - \infty } \) values under CsA pretreatment conditions. This is due to the significant increase in iv \(AUC_{0 - \infty } \) values under the CsA pretreatment condition (Fig. 6A, C), in which systemic elimination of the drugs might have been inhibited by the presence of CsA in the elimination organs.
Discussion
The bioavailabilities of belotecan and topotecan were low (i.e., 11.4% and 32%, respectively) following oral administration of the drugs to rats at a dose of 5 mg/kg (Table I), consistent with the low and variable (30∼44%) bioavailability of topotecan reported in Phase I clinical trials (13,18). Moreover, a 2.7-fold lower bioavailability of belotecan was observed compared with topotecan. The lower absorption of orally administered belotecan compared with topotecan (Fig. 6), may be due to its lower passive permeability (i.e., CLlinear, Table I) and to the absence of a carrier-mediated absorptive transport mechanism (Fig. 2). Presently, no information regarding the absorptive transport mechanism of topotecan is available. The bioavailabilities of both drugs, however, were increased by the preadministration of CsA (5.4- and 1.5-fold for belotecan and topotecan, respectively, Fig. 6 and Table IV). A similar increase in the bioavailability of topotecan was reported in cancer patients (i.e., from 40.0% to 97.1%) by the coadministration of GF120918, a BCRP and P-gp inhibitor (18), suggesting the involvement of efflux transporters in the absorption of these drugs. Therefore, the present study was to investigate the mechanism of oral absorption of belotecan and topotecan using Caco-2 and engineered MDCKII cells overexpressing P-gp, MRP2 and BCRP.
Despite the involvement of certain efflux transporters during the in vivo absorption of these drugs, the results of the transport experiments in the Caco-2 cell monolayers (Fig. 2 and Table I) suggest that passive diffusion is the primary mechanism of belotecan and topotecan absorption, and the involvement of a carrier-mediated transport mechanism in the absorption is minimal, at best. The absorptive permeability (P app) values, which were calculated by dividing the absorptive flux by the initial drug concentration in the donor compartment, were estimated to be 1.17 ± 0.19 × 10−6 and 3.67 ± 0.02 × 10−6 cm/s for belotecan and topotecan, respectively. These values are well below the suggested threshold of 5 × 10−6 cm/s required for substantial intestinal absorption of drugs (our unpublished data).
In contrast to absorptive transport, secretory transport of belotecan and topotecan across Caco-2 cell monolayers was concentration dependent and saturable (Fig. 2), suggesting the significant involvement of a transporter, or of multiple transporters, in the secretion. The secretory transport of belotecan and topotecan was significantly inhibited in the cis presence of typical P-gp substrates (CsA and verapamil), an MRP2 inhibitor and substrate (MK-571 and BSP), and a BCRP inhibitor (FTC), suggesting again the involvement of these transporters in the secretory transport (Fig. 3). This result is consistent with previous reports that suggest the involvement of P-gp (31) and BCRP (32) in the secretory transport of topotecan, and MRP2 in the secretory transport of belotecan (22).
Despite the distinct involvement of efflux transporters, the enhancement of absorptive transport of belotecan and topotecan under the cis presence of high concentration efflux inhibitors was rather small. It may be attributable to still low intracellular concentration of the inhibitors to sufficiently inhibit the efflux of the drug. More significant increase in the absorptive transport of the drugs would be observed for high enough intracellular concentration of the inhibitors.
Since multiple transporters are expressed on Caco-2 cells (33), the interpretation of transport data from the Caco-2 cells is often complicated. Therefore, MDCKII cells overexpressing P-gp, MRP2 or BCRP were utilized to further characterize the vectorial transport of belotecan and topotecan. The increased expression of P-gp, MRP2 (6,20) and BCRP (34) in these cells was confirmed by the enhanced secretory V max values of belotecan and topotecan compared with wild-type MDCKII cells (MDCKII/wt) (Table II). The affinity of belotecan and topotecan for each transporter was estimated from the transport data in these cells. The affinity of belotecan and topotecan for P-gp appeared to be comparable, as the K m values of these drugs in MDCKII/P-gp cells were similar (i.e., 101.19 vs 88.92 μM for belotecan and topotecan, Table II). The affinity of belotecan for MRP2 appeared to be higher than that of topotecan based on the lower K m value for belotecan compared with that for topotecan in MDCKII/MRP2 cells (Table II). The affinity of belotecan for BCRP appeared to be lower than that of topotecan based on the larger K m value for belotecan compared with that for topotecan in MDCKII/BCRP cells (Table II). In addition, based on K m values for P-gp and MRP2, the affinities of belotecan and topotecan for P-gp were higher than the affinities for MRP2 (Table II), consistent with a previous study (35) that reported a high-affinity of camptothecin for P-gp and a low-affinity of CPT-11, a camptothecin analogue, for MRP2. The affinity of topotecan for BCRP was higher than that of belotecan (Table II), consistent with the bioavailability increase in patients based on the treatment of GF120918, a BCRP and P-gp inhibitor (18).
The above results from the engineered MDCK cells further confirm that efflux transporters may limit the bioavailability of these drugs in rats. Moreover, the involvement of efflux transporters seems evident from the increased bioavailability of these drugs under CsA pretreatment (Fig. 6 and Table IV). If this is the case, however, the absorptive transport in Caco-2 cells (i.e., apical to basal flux in Fig. 2) would be expected to increase in more than a linear fashion (i.e., curvilinearly) as the drug concentration apical side increases, possibly due to the saturation of efflux transporters by the drug. However, the absorptive transport was nearly linear over the examined concentration range (Fig. 2). A similar phenomenon was reported for the transport of fexofenadine in the Caco-2 cell model (36), suggesting that such a discrepancy is not a rare case. The discrepancy may indicate that the efflux of the drug from the cell to the apical side across the apical membrane occurs in a linear fashion. It seems to be possible when intracellular drug concentration remains lower than the concentration required to saturate the efflux transport system (i.e., K m), even when apical drugs are administered in high concentrations. It seems reasonable to assume that intracellular concentration of a drug is generally lower compared to its apical concentration as a result of the efflux by the efflux transporters on the apical membrane. Such a gradient in the concentration of a drug does not seem to exist between the inside and the basolateral side of a cell due to the absence of efflux transporters on the basolateral membrane.
In conclusion, the results of the present study suggest that the involvement of P-gp, MRP2 and BCRP in the secretory transport with insignificant carrier-mediated absorptive transport is responsible for the low bioavailability of belotecan and topotecan in rats. Increased bioavailability of these drugs under the CsA administration, which was substantial for belotecan but slight for topotecan, clearly supports this conclusion. The different effect of CsA, with respect to belotecan and topotecan, indicates that the contribution of each efflux transporter to the absorption of each drug may not be identical. Consistent with this, the CLint for the secretory transport of belotecan in Caco-2 cells was 1.5-fold greater than that of topotecan (Table II), suggesting that the lower bioavailability of belotecan, compared with topotecan, is attributable to a greater contribution of efflux transporters to the absorptive transport of belotecan, compared to topotecan.
Abbreviations
- ABC:
-
ATP-binding cassette
- BCRP:
-
breast cancer resistance protein
- BSP:
-
bromosulfophtahlein
- CPT:
-
20-(s)-camptothecin
- CsA:
-
cyclosporine A
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- FTC:
-
fumitremorgin
- HBSS:
-
Hank’s balanced salt solution
- HEPES:
-
N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- MDCK:
-
Mardine–Darby canine kidney
- MK-571:
-
3-([{3-(2-[7-chloro-2-quinolinyl]ethyl)phenyl}-{(3-dimethylamino-3-oxopropyl)-thio}-methyl]-thio) propanoic acid
- MRP2:
-
multidrug resistance protein 2
- P-gp:
-
P-glycoprotein
- SD:
-
Sprague–Dawley
- TEER:
-
transepithelial electrical resistance
References
R. Kim, N. Hirabayashi, M. Nishiyama, K. Jinushi, T. Toge, and K. Okada. Experimental studies on biochemical modulation targeting topoisomerase I and II in human tumor xenografts in nude mice. Int. J. Cancer. 50:760–766 (1992) doi:10.1002/ijc.2910500516.
W. J. Slichenmyer, E. K. Rowinsky, R. C. Donehower, and S. H. Kaufmann. The current status of camptothecin analogues as antitumor agents. J. Natl. Cancer Inst. 85:271–291 (1993) doi:10.1093/jnci/85.4.271.
A. Tanizawa, A. Fujimori, Y. Fujimori, and Y. Pommier. Comparison of topoisomerase I inhibition, DNA damage, and cytotoxicity of camptothecin derivatives presently in clinical trials. J. Natl. Cancer Inst. 86:836–842 (1994) doi:10.1093/jnci/86.11.836.
R. P. Hertzberg, M. J. Caranfa, K. G. Holden, D. R. Jakas, G. Gallagher, M. R. Mattern, S. M. Mong, J. O. Bartus, R. K. Johnson, and W. D. Kingsbury. Modification of the hydroxyl lactone ring of camptothecin: inhibition of mammalian topoisomerase I and biological activity. J. Med. Chem. 32:715–720 (1989) doi:10.1021/jm00123a038.
Y. H. Hsiang, L. F. Liu, M. E. Wall, M. C. Wani, A. W. Nicholas, G. Manikumar, S. Kirschenbaum, R. Silber, and M. Potmesil. DNA topoisomerase I-mediated DNA cleavage and cytotoxicity of camptothecin analogues. Cancer Res. 49:4385–4389 (1989).
A. K. Lalloo, F. R. Luo, A. Guo, P. V. Paranjpe, S. H. Lee, V. Vyas, E. Rubin, and P. J. Sinko. Membrane transport of camptothecin: facilitation by human P-glycoprotein (ABCB1) and multidrug resistance protein 2 (ABCC2). BMC Med. 2:16–27 (2004) doi:10.1186/1741-7015-2-16.
S. S. Jew, M. G. Kim, H. J. Kim, E. Y. Rho, H. G. Park, J. K. Kim, H. J. Han, and H. Lee. Synthesis and in vitro cytotoxicity of C(20)(RS)-camptothecin analogues modified at both B (or A) and E ring. Bioorg. Med. Chem. Lett. 8:1797–1800 (1998) doi:10.1016/S0960-894X(98)00317-5.
J. H. Lee, J. M. Lee, J. K. Kim, S. K. Ahn, S. J. Lee, M. Y. Kim, S. S. Jew, J. G. Park, and C. I. Hong. Antitumor activity of 7-[2-(N-isopropylamino)ethyl]-(20s)-camptothecin, CKD-602, as a potent DNA topoisomorase I inhibitor. Arch. Pharm. Res. 21:581–590 (1998).
J. H. Lee, J. M. Lee, K. H. Lim, J. K. Kim, S. K. Ahn, Y. J. Bang, and C. I. Hong. Preclinical and phase I clinical studies with CKD-602, a novel camptothecin derivative. Ann. N. Y. Acad. Sci. 922:324–325 (2000).
G. Del Bino, P. Lassota, and Z. Darzynkiewicz. The S-phase cytotoxicity of camptothecin. Exp. Cell Res. 193:27–35 (1991) doi:10.1016/0014-4827(91)90534-2.
P. J. Houghton, P. J. Cheshire, J. D. Hallman II, L. Lutz, H. S. Friedman, M. K. Danks, and J. A. Houghton. Efficacy of topoisomerase I inhibitors, topotecan and irinotecan, administered at low dose levels in protracted schedules to mice bearing xenografts of human tumors. Cancer Chemother. Pharmacol. 36:393–403 (1995) doi:10.1007/BF00686188.
C. J. Gerrits, H. Burris, J. H. Schellens, J. R. Eckardt, A. S. Planting, M. E. van der Burg, G. I. Rodriguez, W. J. Loos, V. van Beurden, I. Hudson, S. Fields, D. D. Von Hoff, and J. Verweij. Oral topotecan given once or twice daily for ten days: a phase I pharmacology study in adult patients with solid tumors. Clin. Cancer Res. 4:1153–1158 (1998).
J. H. Schellens, G. J. Creemers, J. H. Beijnen, H. Rosing, M. de Boer-Dennert, M. McDonald, B. Davies, and J. Verweij. Bioavailability and pharmacokinetics of oral topotecan: a new topoisomerase I inhibitor. Br. J. Cancer. 73:1268–1271 (1996).
W. C. Zamboni, L. C. Bowman, M. Tan, V. M. Santana, P. J. Houghton, W. H. Meyer, C. B. Pratt, R. L. Heideman, A. J. Gajjar, A. S. Pappo, and C. F. Stewart. Interpatient variability in bioavailability of the intravenous formulation of topotecan given orally to children with recurrent solid tumors. Cancer Chemother. Pharmacol. 43:454–60 (1999) doi:10.1007/s002800050923.
E. Gupta, V. Vyas, F. Ahmed, P. Sinko, T. Cook, and E. Rubin. Pharmacokinetics of orally administered camptothecins. Ann. N. Y. Acad. Sci. 922:195–204 (2000).
E. Gupta, F. Luo, A. Law, S. Ramanathan, V. Vyas, E. Rubin, and P. Sinko. The intestinal absorption of camptothecin, a highly lipophilic drug, across Caco-2 cells is mediated by active transporter(s). Anticancer Res. 20:1013–1016 (2000).
J. H. Schellens, M. Maliepaard, R. J. Scheper, G. L. Scheffer, J. W. Jonker, J. W. Smit, J. H. Beijnen, and A. H. Schinkel. Transport of topoisomerase I inhibitor by the breast cancer resistance protein potential clinical implications. Ann. N. Y. Acad. Sci. 922:188–194 (2000).
C. M. F. Kruijtzer, J. H. Beijnen, H. Rosing, W. W. Bokkel, M. Schot, R. C. Jewell, E. M. Panl, and J. H. M. Schellens. Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-glycoprotein inhibitor GF120918. J. Clin. Oncol. 20:2943–2950 (2002) doi:10.1200/JCO.2002.12.116.
A. Gupta, Y. Dai, R. R. Vethanayagam, M. F. Herbert, K. E. Thummel, J. D. Unadkat, D. D. Ross, and Q. Mao. Cyclosporin A, tacrolimus and sirolimus are potent inhibitors of the human breast cancer resistance protein (ABCG2) and reverse resistance to mitoxantrone and topotecan. Cancer Chemother. Pharmacol. 58:374–383 (2006) doi:10.1007/s00280-005-0173-6.
F. R. Luo, P. V. Paranjpe, A. Guo, E. Rubin, and P. Sinko. Intestinal transport of irinotecon in Caco-2 cells and MDCKII cells overexpressing efflux transporters PGP, cMOAT, and MRP1. Drug Metab. Dispos. 30:763–770 (2002) doi:10.1124/dmd.30.7.763.
X. Sha, and X. Fang. Transport characteristics of 9-nitrocamptothecin in the human intestinal cell line Caco-2 and everted gut sacs. Int. J. Pharm. 272:161–171 (2004) doi:10.1016/j.ijpharm.2003.12.023.
E. M. Namkoong, I. W. Kim, D. D. Kim, S. J. Chung, and C. K. Shim. Effect of probenecid on the biliary excretion of belotecan. Arch. Pharm. Res. 30:1482–1488 (2007).
H. Li, S. J. Chung, H. S. Kim, J. W. Lee, and C. K. Shim. The transport of a reversible proton pump antagonist, 5,6-dimethyl-2-(4-fluorophenylamino)-4-(1-methyl-1,2,3,4-tetrahydroisoquinoline-2-yl) pyrimidine hydrochloride (YH1885), across Caco-2 cell monolayers. Drug Metab. Dispos. 29:54–59 (2001).
M. H. Silbermann, A. W. Boersma, A. L. Janssen, R. J. Scheper, H. Herweijer, and K. Nooter. Effects of cyclosporin A and verapamil on the intracellular daunorubicin accumulation in Chinese hamster ovary cells with increasing levels of drug-resistance. Int. J. Cancer. 44:722–726 (1989) doi:10.1002/ijc.2910440428.
M. Horikawa, Y. Kato, C. A. Tyson, and Y. Sugiyama. The potential for an interaction between MRP2 (ABCC2) and various therapeutic agents: probenecid as a candidate inhibitor of the biliary excretion of irinotecan metabolites. Drug Metab. Pharmacokinet. 17:23–33 (2002) doi:10.2133/dmpk.17.23.
J. Kőnig, A. T. Nies, Y. Cui, I. Leier, and D. Keppler. Conjugate export pumps of the multidrug resistance protein (MRP) family: localization, substrate specificity, and MRP2-mediated drug resistance. Biochim. Biophys. Acta. 1461:377–394 (1999) doi:10.1016/S0005-2736(99)00169-8.
S. K. Rabindran, H. He, M. Singh, E. Brown, K. I. Collins, T. Annable, and L. M. Greenberger. Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin C. Cancer Res. 58:5850–5858 (1998).
J. Y. Cho, H. B. Seo, K. S. Yu, K. S. Bae, S. Y. Yi, I. J. Jang, and S. G. Shin. Simple and sensitive determination of the new antitumor drug CKD-602 in human plasma by liquid chromatography. J. Chromatogr. B. 784:25–31 (2003) doi:10.1016/S1570-0232(02)00750-X.
A. M. Vali, B. Shafaghi, and S. Dadashzadeh. Simple and sensitive high performance liquid chromatographic method for the simultaneous quantitation of the lactone and carboxylate forms of topotecan in human plasma. J. Chromatogr. B. 818:205–212 (2005) doi:10.1016/j.jchromb.2004.12.027.
X. Zhou, X. Yang, P. Wanf, R. A. Coburn, and M. E. Morris. Effect of dihydropyridiens and pyridines on multidrug resistance mediated by breast cancer resistance protein: in vitro and in vivo studies. Drug Metab. Dispos. 33:1220–1228 (2005) doi:10.1124/dmd.104.003558.
C. B. Hendricks, E. K. Rowinsky, L. B. Grochow, R. C. Donehower, and S. H. Kaufmann. Effect of P-glycoprotein expression on the accumulation and cytotoxicity of topotecan (SK&F 104864), a new camptothecin analogue. Cancer Res. 52:2268–2278 (1992).
J. W. Jonker, J. W. Smit, R. F. Brinkhuis, M. Maliepaard, J. H. Beijnen, J. H. M. Schellens, and A. H. Schinkel. Role of breast cancer resistance protein in the bioavailability and fetal penetration of topotecan. J. Natl. Cancer Inst. 92:1651–1656 (2000) doi:10.1093/jnci/92.20.1651.
J. Taipalesuu, H. Tornblom, G. Lindberg, C. Einarsson, F. Sjoqvst, H. Melhus, P. Garberg, B. Sjostrom, B. Lundgren, and P. Artursson. Correlation of gene expression of ten drug efflux proteins of the ATP-binding cassette transporter family in normal human jejunum and in human intestinal epithelial Caco-2cell monolayers. J. Pharmacol. Exp. Ther. 299:164–170 (2001).
G. Merino, A. I. Alvarez, M. M. Pulido, A. J. Molina, A. H. Schinkel, and J. G. Prieto. Breast Cancer Resistance Protein (BCRP/ABCG2) transports fluoroquinolone antibiotics and affects their oral availability, pharmacokinetics and milk secretion. Drug Metab. Dispos. 34:690–693 (2006) doi:10.1124/dmd.105.008219.
X. Y. Chu, Y. Kato, and Y. Sugiyama. Possible involvement of P-glycoprotein in biliary excretion of CPT-11 in rats. Drug Metab. Dispos. 27:440–441 (1999).
N. Petri, C. Tannergreen, D. Rungstad, and H. Lennernäs. Transport characteristics of fexofenadine in the Caco-2 cell model. Pharm. Res. 21:1398–1404 (2004) doi:10.1023/B:PHAM.0000036913.90332.b1.
Acknowledgments
We sincerely thank Dr. Borst at The Netherlands Cancer Institute (Amsterdam, The Netherlands) for providing MDCKII/wt, MDCKII/P-gp, MDCKII/MRP2, and MDCKII/BCRP cells. This work was supported by the Korea Science and Engineering Foundation (KOSEF) through the National Research Lab. Program funded by the Ministry of Science and Technology (No.R0A2006000102900).
Author information
Authors and Affiliations
Corresponding author
Additional information
Hong Li and Hyo-Eon Jin have contributed equally to this work.
Rights and permissions
About this article
Cite this article
Li, H., Jin, HE., Kim, W. et al. Involvement of P-glycoprotein, Multidrug Resistance Protein 2 and Breast Cancer Resistance Protein in the Transport of Belotecan and Topotecan in Caco-2 and MDCKII Cells. Pharm Res 25, 2601–2612 (2008). https://doi.org/10.1007/s11095-008-9678-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-008-9678-0