
Abstract
Purpose
Ticagrelor is a reversibly binding P2Y12 receptor antagonist for the prevention of atherothrombotic events in patients with acute coronary syndrome. Previous in vitro studies showed that ticagrelor is a substrate and inhibitor of P-glycoprotein (ABCB1). Therefore, we examined the potential interaction between digoxin, a P-glycoprotein substrate, and ticagrelor by evaluating the pharmacokinetics, safety, and tolerability.
Methods
This was a randomized, double-blind, two-period crossover study in healthy volunteers (n = 20). Pharmacokinetic parameters of digoxin and ticagrelor were evaluated following co-administration of ticagrelor 400 mg qd or placebo on days 1–16, and digoxin (0.25 mg bid on day 6 and 0.25 mg qd on days 7–14).
Results
Co-administration of ticagrelor increased the digoxin maximum plasma concentration by 75 %, from 1.8 ng/ml to 3.0 ng/ml (Gmean ratio [GMR] 1.75 [95 % CI, 1.52–2.01]); minimum plasma concentration by 31 %, from 0.5 ng/ml to 0.7 ng/ml (GMR 1.31, 1.13–1.52); and mean area under the curve by 28 %, from 16.8 ng · h/ml to 21.0 ng · h/ml (GMR 1.28, 1.12–1.46), compared with placebo. Renal clearance of digoxin was unaffected by the presence of ticagrelor. Digoxin had no effect on the pharmacokinetics of ticagrelor or its active metabolite, AR-C124910XX. Co-administration of ticagrelor and digoxin was well tolerated.
Conclusions
Collectively, these results indicate that ticagrelor is a weak inhibitor of the P-glycoprotein transporter. Based on these findings, it is recommended that serum concentrations of drugs like digoxin (P-glycoprotein transporter substrates with a narrow therapeutic range) are monitored when initiating or changing ticagrelor therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ticagrelor (Fig. 1a) is a reversibly binding P2Y12 receptor antagonist [1] approved for the prevention of atherothrombotic events in patients with acute coronary syndrome (ACS). During the clinical development program, several studies showed that ticagrelor is rapidly absorbed and exhibits linear and predictable pharmacokinetics over a wide dose range [2–6]. Unlike thienopyridine compounds, ticagrelor does not require metabolic activation for antiplatelet activity [7, 8]. However, ticagrelor is metabolized to the active metabolite, AR-C124910XX (Fig. 1b), by cytochrome P450 3A (CYP3A [9]). AR-C124910XX is formed rapidly [3], and is approximately equipotent to its parent compound with respect to inhibition of platelet aggregation (AstraZeneca, data on file). Ticagrelor is also a substrate for P-glycoprotein (ABCB1; AstraZeneca, data on file), an efflux transporter protein found in the apical membrane of the intestinal mucosa [10] and in the liver and kidney [11].

Chemical structure of a ticagrelor and b its active metabolite AR-C124910XX
Patients with ACS often exhibit multiple comorbidities and may receive a number of drugs concomitantly [12]. Digoxin is a substrate of renal [13] and intestinal P-glycoprotein [14, 15], but not cytochrome P450 enzymes. Therefore, digoxin is a suitable model compound for assessing potential interactions with drugs that interact with P-glycoprotein [16, 17]. In earlier preclinical studies, ticagrelor and AR-C124910XX inhibited the transport of 3H-digoxin in MDR1-MDCK monolayers with an IC50 of 7.8 ± 2.6 μM and 9.9 ± 5.1 μM, respectively [18]. Assuming a steady state maximum plasma concentration (Cmax) for ticagrelor of 1.5 μM [9] and 0.5 μM for AR-C124910XX, corresponding I/IC50 values were approximately 0.19 for ticagrelor and 0.05 for AR-C124910XX. Based on the FDA criteria [16], as the I/IC50 of 0.19 for ticagrelor was ≥ 0.1, an interaction with digoxin was deemed likely and a clinical interaction study necessary. Such an interaction could be clinically relevant for compounds that have a narrow therapeutic range, such as digoxin [19]; the therapeutic range for digoxin is a serum concentration of 0.5–1.0 ng/ml [20, 21]. Furthermore, there is a possibility that digoxin and antiplatelet agents may be co-administered [22, 23]. Current guidelines include recommendations for the use of digoxin in patients with atrial fibrillation, heart failure, or both conditions [24, 25]. Atrial fibrillation is estimated to occur in 2–21 % of patients with ACS [26], and digoxin may be administered to slow a rapid ventricular response in patients with both ACS and atrial fibrillation associated with heart failure [25].
The primary objective of the present study was to compare the pharmacokinetics of digoxin when administered alone and in combination with ticagrelor. Secondary objectives included assessment of the effect of digoxin on the pharmacokinetics of ticagrelor, and evaluation of the safety and tolerability of digoxin and ticagrelor co-administration.
Materials and methods
Study population
The study was conducted in healthy volunteers, who were required to be 18–65 years old with a body mass index (BMI) 18–30 kg/m2; females were to be post-menopausal or surgically sterile. Key exclusion criteria are included in the online supplement.
All volunteers provided written, informed consent prior to study start and the final protocol was approved by an Independent Ethics Committee (Southern Institutional Review Board, Miami, FL, USA; reviewed 18 September 2003). The study was conducted in accordance with the ethical principles established in the Declaration of Helsinki, and was consistent with International Conference on Harmonisation/Good Clinical Practice guidelines, AstraZeneca Bioethics Policy, and regulatory requirements.
Study design and treatment
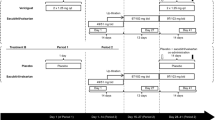
This study (AstraZeneca Trial Identifier: D5130C05265) was a randomized, double-blind, two-period crossover study conducted in a single center (Fig. 2). Volunteers were randomized to one of the two treatment sequences. Both staff and volunteers were blinded to treatment with ticagrelor and placebo, whereas digoxin administration was open label.

Study design
Following randomization, volunteers received the allocated treatment during two 16-day periods, separated by a washout of at least 2 weeks. Ticagrelor at 400 mg once daily (qd) was administered on days 1–16. Digoxin (Lanoxin®, GlaxoSmithKline) at 0.25 mg was given twice daily (bid) on day 6 and at 0.25 mg qd on days 7–14 (Fig. 2). Both ticagrelor and digoxin tablets were administered with 240 ml of room temperature water 1 h before breakfast, and the day 6 evening dose of digoxin was given approximately 12 h after the morning dose. On days 5 and 14, volunteers fasted for 10 h overnight before, and for 4 h after, receiving study medication; water was also prohibited 2 h before and after dose administration.
Key on-study restrictions are included in the online supplement.
Pharmacokinetic assessments
For pharmacokinetic analyses of ticagrelor and AR-C124910XX, blood samples were collected pre-dose on days 1, 4, 5, 13, and 14, and 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 18, and 24 h post-dose on days 5 and 14. Additional samples were collected at 36, 48, and 72 h post-dose on day 14. For digoxin pharmacokinetic analyses, blood samples were collected pre-dose on days 1, 12, 13, and 14 and 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 18, and 24 h post-dose on day 14. Urine samples were also collected pre-dose on day 1 and between 0 and 6 h; 6 and 12 h; and 12 and 24 h post-dose on day 14.
Venous blood (2 ml) was collected into lithium-heparin tubes and immediately placed on ice. Samples were centrifuged at 1,500 g and 4 °C for 10 min and stored frozen at −20 °C until analyzed.
Ticagrelor and AR-C124910XX plasma concentrations were quantified by a validated method using liquid chromatography and tandem mass spectrometry. Lower limits of quantification (LLOQ) were 1 ng/ml for ticagrelor and 2.5 ng/ml for AR-C124910XX [27]. The inter-batch precision and mean accuracy of this method were 3.5–10.7 % and 1.0–12.7 %, respectively, for ticagrelor; corresponding values for AR-C124910XX were 4.7–10.5 % and −3.0–3.2 %, respectively. Digoxin plasma and urine concentrations were measured by PPD Development (Richmond, VA, USA) using validated radioimmunoassays with LLOQs of 0.15 ng/ml (plasma; method ICD 11.1) and 1 ng/ml (urine; method ICD 11.2). Inter-batch precision and mean accuracy were 5.1–10.4 % and −1.5–5.6 %, respectively for the plasma assay; corresponding values for the urine assay were 4.9–13.7 % and 0.65–3.3 %, respectively.
Safety and tolerability
Safety and tolerability were evaluated by monitoring the incidence, severity, and causality of adverse events (AEs) during the study, until follow-up. Clinical laboratory parameters (clinical chemistry, hematology, coagulation, urinalysis), 12-lead electrocardiogram (ECG), and vital signs were assessed throughout the study, and a complete physical examination was performed at screening and at the follow-up visit.
Data analyses
Pharmacokinetic parameters (see online supplement) were estimated by non-compartmental methods using WinNonlin Professional (Pharsight Corporation, Mountain View, CA, USA).
Data were analyzed statistically using SAS version 8 (SAS Institute Inc., Cary, NC, USA). Following log-transformation, Cmax, AUCτ and Cmin (digoxin only) were analyzed by analysis of variance; treatment, sequence, and period were fixed terms, and volunteer within sequence was treated as a random effect. Comparisons were expressed as the geometric mean (Gmean) ratio between treatments (ticagrelor plus digoxin versus placebo plus digoxin).
Results
Baseline characteristics and disposition
Twenty healthy volunteers (ten males and ten females) were enrolled and randomized. The mean (range) age was 44 (22–59) years and the mean (range) BMI was 25.9 (21.8–29.5) kg/m2. Sixteen volunteers were classified as ‘other’ race (mainly Hispanic), three were Caucasian, and one was Black.
All 20 volunteers received ticagrelor and 16 completed the study. Of the four volunteers who did not complete the study, two volunteers completed all of the study procedures except the follow-up visit, one was unwilling to continue, and one discontinued because of persistent pruritus (discontinued on day 9). One volunteer missed a ticagrelor dose on day 5, and was excluded from the ticagrelor and AR-C124910XX pharmacokinetic analyses.
Digoxin pharmacokinetic parameters
Digoxin was rapidly absorbed following co-administration with either ticagrelor or placebo (Table 1, Fig. 3). However, plasma levels of digoxin were higher with co-administration of ticagrelor compared with placebo. The median t1/2 of digoxin was slightly prolonged in the presence of ticagrelor (52.1 h) compared with placebo (41.8 h), whereas the median tmax of digoxin remained unchanged (Table 1). Relative increases in AUCτ (28 %), Cmax (75 %), and Cmin (31 %) were observed in the presence of ticagrelor compared with placebo (Table 1). A greater than 50 % increase in these variables occurred in four, twelve, and five volunteers, respectively. The maximum increase observed in these parameters was 2–2.5 fold. In the presence of ticagrelor, digoxin Ae was increased by 24 %, and renal clearance was unchanged, versus placebo (Table 1).

Mean (± SD) plasma concentrations of digoxin, on day 14, in the presence and absence of ticagrelor
Ticagrelor pharmacokinetic parameters
Plasma concentrations of ticagrelor were comparable upon co-administration of digoxin or placebo (Table 2, Fig. 4a). Gmean AUCτ and Cmax values were not altered when co-administered with digoxin, with Gmean ratios for AUCτ and Cmax of 1.06 (90 % confidence interval [CI], 0.97–1.16) and 1.02 (90 % CI, 0.94–1.11), respectively (Table 2). Median tmax for ticagrelor alone was 2.2 h and was comparable (3.2 h) in the presence of digoxin (Table 2).

Mean (± SD) plasma concentrations of a ticagrelor and b AR-C124910XX, in the presence (day 14) and absence (day 5) of digoxin
AR-C124910XX pharmacokinetic parameters
AR-C124910XX plasma concentrations were comparable in the presence and absence of digoxin (Table 2, Fig. 4b). No change in Gmean AUCτ and Cmax values occurred upon co-administration with digoxin, with Gmean ratios for AUCτ and Cmax of 1.11 (90 % CI, 1.01–1.23) and 1.06 (90 % CI, 0.97–1.16), respectively (Table 2). Median tmax values for AR-C124910XX occurred at 3.2 h both in the presence and absence of digoxin (Table 2). Additionally, metabolite:parent ratios for AUCτ and Cmax were unaffected by the presence of digoxin.
Safety and tolerability
Most AEs reported with co-administration of ticagrelor and digoxin were mild and self-limiting. One volunteer receiving ticagrelor plus digoxin discontinued the study on day 9 due to a treatment-related pruritis that was persistent and required treatment with Benadryl.
Overall, 20 AEs were noted in ten out of 20 volunteers receiving ticagrelor plus digoxin, and 16 AEs were recorded in ten out of 19 volunteers receiving placebo plus digoxin. The most common AEs were an increased tendency to bruise, stomatitis, headache, and arthralgia. Seventeen treatment-related AEs occurred in eight volunteers during ticagrelor plus digoxin administration; 15 treatment-related AEs occurred in nine volunteers during placebo plus digoxin administration.
There were no clinically significant changes in other clinical chemistry parameters, hematology, urinalysis, ECGs, vital signs, or physical findings in either group. However, increases in mean serum uric acid values over time were evident for both treatments; the mean (SD) increase from baseline on day 15 was 30 (64) and 13 (53) μmol/l for the ticagrelor plus digoxin and placebo plus digoxin treatments, respectively.
Discussion
Digoxin is a renal and intestinal P-glycoprotein substrate [13–15], and does not undergo metabolism by cytochrome P450 enzymes. Thus, this drug is a useful model compound with which to assess the effect of co-administered drugs on the exposure to P-glycoprotein substrates [16]. The pharmacokinetics of digoxin in the presence of placebo in our study were comparable to those observed in previous studies [28, 29]. In vitro studies have shown that ticagrelor is a substrate and inhibitor of P-glycoprotein [18]. The present findings confirm an interaction between digoxin and ticagrelor, as predicted by the results of earlier in vitro studies. This study showed that compared with placebo, ticagrelor co-administration increased the Cmax and AUCτ of digoxin by approximately 75 % and 28 %, respectively. Moreover, the renal clearance of digoxin from the plasma was not changed in the presence of ticagrelor versus placebo. Collectively, these findings show that the increased exposure to digoxin observed with co-administration of ticagrelor could be explained by the inhibition of intestinal P-glycoprotein by ticagrelor, thereby reducing the efflux of digoxin into the intestine. As in vitro studies have previously shown the parent compound and active metabolite AR-C124910XX to have similar effects with respect to P-glycoprotein inhibition, both may contribute to the inhibition of intestinal P-glycoprotein observed in the present study. Our results also suggest that interaction of ticagrelor with renal P-glycoprotein is of minimal significance for digoxin. Together with data from previous in vitro studies [18], the present results confirm that ticagrelor is a weak inhibitor of intestinal P-glycoprotein.
The 400 mg qd ticagrelor dose used in the present study is higher than the approved 90 mg bid maintenance dose used clinically [30, 31]. The pharmacokinetics of ticagrelor in the present study were broadly comparable to those observed in previous studies in healthy volunteers [2], and in patients with ACS [6] or atherosclerosis [5]. In our study, digoxin had no effect on the pharmacokinetics of ticagrelor or AR-C124910XX. The lack of effect of digoxin on ticagrelor pharmacokinetic parameters is not surprising, as digoxin does not interact with CYP3A and is not an inhibitor of P-glycoprotein [16].
Patients with ACS often present with cardiovascular comorbidities [26] and may receive multiple drugs concomitantly [12]. Moreover, there are a number of clinical scenarios where digoxin could be co-administered with ticagrelor. For example, current treatment guidelines recommend the use of digoxin monotherapy for heart rate control in patients with permanent atrial fibrillation who are predominantly sedentary [24]. Additionally, digoxin is an appropriate alternative for patients with ACS associated with severe left ventricular dysfunction (LVD) and heart failure [25], and is also recommended for use in symptomatic LVD with and without atrial fibrillation [32, 33]. Indeed, in patients with both heart failure and atrial fibrillation, digoxin is recommended as an adjunctive therapy for heart rate control when monotherapy with β-blockers proves inadequate [25]. Therefore, based on the results of this study, close clinical and laboratory monitoring is recommended in the event of co-administration of digoxin and ticagrelor. As with other narrow therapeutic drugs such as warfarin [34], therapeutic drug monitoring will be useful in maintaining digoxin within the therapeutic range (serum concentration of 0.5–1.0 ng/ml [20]) and avoiding adverse effects associated with higher exposures. Higher serum digoxin concentrations are associated with toxicity. For example, in patients with heart failure and/or atrial fibrillation, toxicity occurred in some patients with digoxin serum concentrations in the range 1.4–2.9 ng/ml, whereas toxicity was consistently observed in patients who had digoxin serum concentrations of 3 ng/ml or greater [35].
With respect to efficacy, early high plasma concentrations of digoxin may not give an accurate measurement of the concentration of digoxin at the site of action [36]. However, with chronic digoxin use, steady-state plasma concentrations reach equilibrium with the surrounding tissue concentrations and correlate with specific pharmacologic effects. Therefore, digoxin plasma concentrations measured after the tissue distribution phase may more accurately reflect the likely effect on its efficacy because of the relatively slow equilibration of digoxin within the myocardium. In our study, a 31 % increase in digoxin Cmin was observed in the presence of ticagrelor versus placebo.
The magnitude of the overall increase in digoxin exposure observed in the present study (28 %) is similar to that reported for other P-glycoprotein inhibitors. For example, mibefradil increased digoxin AUC by 31 % [37].
Co-administration of ticagrelor and digoxin was well tolerated. No safety issues were identified during the present study. AEs were generally mild, although one volunteer discontinued from the study due to persistent pruritus that was considered to be treatment related.
In summary, ticagrelor 400 mg qd and digoxin 0.25 mg qd showed a pharmacokinetic interaction. At steady-state, digoxin Cmax, Cmin, and AUCτ were increased by 75 %, 31 %, and 28 %, respectively, in the presence of ticagrelor versus placebo. No effect was seen on the pharmacokinetics and safety of ticagrelor when co-administered with digoxin. These data provide further evidence that ticagrelor is a weak inhibitor of intestinal P-glycoprotein. Based on these findings, it is recommended that serum concentrations of drugs like digoxin (i.e., P-glycoprotein transporter substrates with a narrow therapeutic range) are monitored with initiation of, or any change in, ticagrelor therapy.
References
Husted S, van Giezen JJ (2009) Ticagrelor: the first reversibly binding oral P2Y12 receptor antagonist. Cardiovasc Ther 27:259–274
Butler K, Teng R (2010) Pharmacokinetics, pharmacodynamics, safety and tolerability of multiple ascending doses of ticagrelor in healthy volunteers. Br J Clin Pharmacol 70:65–77
Teng R, Butler K (2010) Pharmacokinetics, pharmacodynamics, tolerability and safety of single ascending doses of ticagrelor, a reversibly binding oral P2Y(12) receptor antagonist, in healthy subjects. Eur J Clin Pharmacol 66:487–496
Teng R, Butler K (2008) AZD6140, the first reversible oral platelet P2Y12 receptor antagonist, has linear pharmacokinetics and provides near complete inhibition of platelet aggregation, with reversibility of effect, in healthy subjects. Can J Clin Pharmacol 15:e426
Husted S, Emanuelsson H, Heptinstall S, Sandset PM, Wickens M, Peters G (2006) Pharmacodynamics, pharmacokinetics, and safety of the oral reversible P2Y12 antagonist AZD6140 with aspirin in patients with atherosclerosis: a double-blind comparison to clopidogrel with aspirin. Eur Heart J 27:1038–1047
Storey RF, Husted S, Harrington RA, Heptinstall S, Wilcox RG, Peters G, Wickens M, Emanuelsson H, Gurbel P, Grande P, Cannon CP (2007) Inhibition of platelet aggregation by AZD6140, a reversible oral P2Y12 receptor antagonist, compared with clopidogrel in patients with acute coronary syndromes. J Am Coll Cardiol 50:1852–1856
Savi P, Combalbert J, Gaich C, Rouchon MC, Maffrand JP, Berger Y, Herbert JM (1994) The antiaggregating activity of clopidogrel is due to a metabolic activation by the hepatic cytochrome P450–1A. Thromb Haemost 72:313–317
Teng R, Oliver S, Hayes MA, Butler K (2010) Absorption, distribution, metabolism, and excretion of ticagrelor in healthy subjects. Drug Metab Dispos 38:1514–1521
Zhou D, Andersson TB, Grimm SW (2011) In vitro evaluation of potential drug-drug interactions with ticagrelor: cytochrome P450 reaction phenotyping, inhibition, induction and differential kinetics. Drug Metab Dispos 39:703–710
von Richter O, Burk O, Fromm MF, Thon KP, Eichelbaum M, Kivisto KT (2004) Cytochrome P450 3A4 and P-glycoprotein expression in human small intestinal enterocytes and hepatocytes: a comparative analysis in paired tissue specimens. Clin Pharmacol Ther 75:172–183
Glaeser H (2011) Importance of P-glycoprotein for drug-drug interactions. Handb Exp Pharmacol 201:285–297
Taneva E, Bogdanova V, Shtereva N (2004) Acute coronary syndrome, comorbidity, and mortality in geriatric patients. Ann N Y Acad Sci 1019:106–110
Tanigawara Y, Okamura N, Hirai M, Yasuhara M, Ueda K, Kioka N, Komano T, Hori R (1992) Transport of digoxin by human P-glycoprotein expressed in a porcine kidney epithelial cell line (LLC-PK1). J Pharmacol Exp Ther 263:840–845
de Lannoy I, Silverman M (1992) The MDR1 gene product, P–glycoprotein, mediates the transport of the cardiac glycoside, digoxin. Biochem Biophys Res Commun 189:551–557
Drescher S, Glaeser H, Murdter T, Hitzl M, Eichelbaum M, Fromm MF (2003) P–glycoprotein-mediated intestinal and biliary digoxin transport in humans. Clin Pharmacol Ther 73:223–231
Food and Drug Administration. Guidance for industry: Drug interaction studies – study design, data analysis, implications for dosing, and labeling (Feb 2012) http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf. Accessed 11 October 2012
Greiner B, Eichelbaum M, Fritz P, Kreichgauer HP, von Richter O, Zundler J, Kroemer HK (1999) The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest 104:147–153
Food and Drug Administration. New Drug Application #22433, Appendix 1: clinical pharmacology and biopharmaceutics review (2010) http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/022433Orig1s000ClinPharmR.pdf. Accessed 30 January 2013
Englund G, Hallberg P, Artursson P, Michaelsson K, Melhus H (2004) Association between the number of coadministered P-glycoprotein inhibitors and serum digoxin levels in patients on therapeutic drug monitoring. BMC Med 2:8
Morris SA, Hatcher HF, Reddy DK (2006) Digoxin therapy for heart failure: an update. Am Fam Physician 74:613–618
Rathore SS, Curtis JP, Wang Y, Bristow MR, Krumholz HM (2003) Association of serum digoxin concentration and outcomes in patients with heart failure. JAMA 289:871–878
Mahmud A, Bennett K, Okechukwu I, Feely J (2007) National underuse of anti-thrombotic therapy in chronic atrial fibrillation identified from digoxin prescribing. Br J Clin Pharmacol 64:706–709
Lip GY, Huber K, Andreotti F, Arnesen H, Airaksinen KJ, Cuisset T, Kirchhof P, Marin F (2010) Management of antithrombotic therapy in atrial fibrillation patients presenting with acute coronary syndrome and/or undergoing percutaneous coronary intervention/ stenting. Thromb Haemost 103:13–28
NICE. Clinical guideline 36: Atrial Fibrillation—the management of atrial fibrillation. http://www.nice.org.uk/nicemedia/live/10982/30052/30052.pdf Accessed 16 May 2012
Camm AJ, Kirchhof P, Lip GY, Schotten U, Savelieva I, Ernst S, Van Gelder IC, Al-Attar N, Hindricks G, Prendergast B, Heidbuchel H, Alfieri O, Angelini A, Atar D, Colonna P, De Caterina R, De Sutter J, Goette A, Gorenek B, Heldal M, Hohloser SH, Kolh P, Le Heuzey JY, Ponikowski P, Rutten FH (2010) Guidelines for the management of atrial fibrillation: the Task Force for the Management of Atrial Fibrillation of the European Society of Cardiology (ESC). Eur Heart J 31:2369–2429
Schmitt J, Duray G, Gersh BJ, Hohnloser SH (2009) Atrial fibrillation in acute myocardial infarction: a systematic review of the incidence, clinical features and prognostic implications. Eur Heart J 30:1038–1045
Sillén H, Cook M, Davis P (2010) Determination of ticagrelor and two metabolites in plasma samples by liquid chromatography and mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 878:2299–2306
Taylor A, Beerahee A, Citerone D, Davy M, Fitzpatrick K, Lopez-Gil A, Stocchi F (1999) The effect of steady-state ropinirole on plasma concentrations of digoxin in patients with Parkinson’s disease. Br J Clin Pharmacol 47:219–222
Schwartz JI, Agrawal NG, Wehling M, Musser BJ, Gumbs CP, Michiels N, De SM, Wagner JA (2008) Evaluation of the pharmacokinetics of digoxin in healthy subjects receiving etoricoxib. Br J Clin Pharmacol 66:811–817
Brilique, summary of product characteristics (2010) http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001241/WC500100494.pdf. Accessed 16 February 2012
BrilintaTM US full prescribing information (July 2011) http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/022433s000lbl.pdf. Accessed16 February 2012
Fuster V, Ryden LE, Cannom DS, Crijns HJ, Curtis AB, Ellenbogen KA, Halperin JL, Le Heuzey JY, Kay GN, Lowe JE, Olsson SB, Prystowsky EN, Tamargo JL, Wann S, Smith SC Jr, Jacobs AK, Adams CD, Anderson JL, Antman EM, Halperin JL, Hunt SA, Nishimura R, Ornato JP, Page RL, Riegel B, Priori SG, Blanc JJ, Budaj A, Camm AJ, Dean V, Deckers JW, Despres C, Dickstein K, Lekakis J, McGregor K, Metra M, Morais J, Osterspey A, Tamargo JL, Zamorano JL (2006) ACC/AHA/ESC 2006 Guidelines for the Management of Patients with Atrial Fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 Guidelines for the Management of Patients With Atrial Fibrillation): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation 114:e257–e354
Dickstein K, Cohen-Solal A, Filippatos G, McMurray JJ, Ponikowski P, Poole-Wilson PA, Strömberg A, van Veldhuisen DJ, Atar D, Hoes AW, Keren A, Mebazaa A, Nieminen M, Priori SG, Swedberg K, ESC Committee for Practice Guidelines (CPG) (2008) ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the diagnosis and treatment of acute and chronic heart failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur J Heart Fail 10:933–989
Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G (2008) Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-based Clinical Practice Guidelines (8th edition). Chest 133:160S–198S
Miura T, Kojima R, Sugiura Y, Mizutani M, Takatsu F, Suzuki Y (2000) Effect of aging on the incidence of digoxin toxicity. Ann Pharmacother 34:427–432
Digoxin Tablets [prescribing information]. West-ward Pharmaceutical Corp. Eatontown, NJ. July 2010. http://druginserts.com/lib/rx/meds/digoxin-18/. Accessed 16 February 2012
Siepmann M, Kleinbloesem C, Kirch W (1995) The interaction of the calcium antagonist RO 40–5967 with digoxin. Br J Clin Pharmacol 39:491–496
Acknowledgments
The authors wish to thank Kenneth Lasseter (lead investigator) and the team at the clinical pharmacology unit for conducting the study. Staff at York Bioanalytical Solutions (York, UK) and PPD Development (Richmond, VA, USA) are acknowledged for their contribution to the pharmacokinetic sample analyses. Statistical support was provided by Stephanie Dunbar, PhD. Editorial support in the preparation of this manuscript was provided by David Evans, PhD (Medical Writer, Gardiner-Caldwell Communications).
Funding sources
Editorial support was funded by AstraZeneca.
Authors’ contributions
Both authors participated in research design, performed the data analysis, and wrote or contributed to the writing of the manuscript.
Conflicts of interest
This study was funded by AstraZeneca. The authors are employees of AstraZeneca.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 14 kb)
Rights and permissions
About this article
Cite this article
Teng, R., Butler, K. A pharmacokinetic interaction study of ticagrelor and digoxin in healthy volunteers. Eur J Clin Pharmacol 69, 1801–1808 (2013). https://doi.org/10.1007/s00228-013-1543-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-013-1543-3