
Abstract
The Metastatic progression of solid tumors, such as thyroid cancer is a complex process which involves various factors. Current understanding on the role of epithelial–mesenchymal transition (EMT) in thyroid carcinomas suggests that EMT is implicated in the progression from follicular thyroid cancer (FTC) and papillary thyroid cancer (PTC) to poorly differentiated thyroid carcinoma (PDTC) and anaplastic thyroid cancer (ATC). According to the literature, the initiation of the EMT program in thyroid epithelial cells elevates the number of stem cells, which contribute to recurrent and metastatic diseases. The EMT process is orchestrated by a complex network of transcription factors, growth factors, signaling cascades, epigenetic modulations, and the tumor milieu. These factors have been shown to be dysregulated in thyroid carcinomas. Therefore, molecular interferences restoring the expression of tumor suppressors, or thwarting overexpressed oncogenes is a hopeful therapeutic method to improve the treatment of progressive diseases. In this review, we summarize the recent findings on EMT in thyroid cancer focusing on the main role-players and regulators of this process in thyroid tumors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Thyroid cancer is the most prevalent endocrine malignancy and its incidence rates have increased during the past three decades worldwide [1, 2]. Epithelial tumors of thyroid are the most common forms that originate from thyroid follicular cells. These epithelial cancers can be divided into three major pathological forms: papillary thyroid carcinoma (PTC), follicular thyroid carcinoma (FTC), and anaplastic thyroid carcinoma (ATC), which all originate from follicular epithelial cells [3]. The third type of thyroid tumor is known as medullary carcinoma (5–10%), which is derived from C cells of the thyroid gland [4]. Most of the thyroid carcinomas display biologically indolent behavior and have a good prognosis with long-term survival rates (95% at 20 years), although the mortality risk from recurrent or persistent diseases is still high [5]. The conventional treatment for thyroid cancer is thyroidectomy with postoperative radioiodine therapy. However, unresectable recurrence, resistance to radioactive iodine and metastasis in DTC, poorly differentiated thyroid carcinoma (PDTC) and ATC are still the leading causes of death in thyroid cancer [6, 7]. Accordingly, it is critical to elucidate the molecular alterations associated with aggressive behavior of thyroid tumors to develop novel therapeutic strategies to overcome thyroid cancer [6, 8]. Several studies have established that the invasiveness of thyroid cancer is associated with multiphase processes like epithelial–mesenchymal transition (EMT) [3, 9,10,11].
EMT is a morphology-changing program, by which epithelial cells obtain fibroblast-like phenotype by triggering transcriptional events, typical of mesenchymal cells [12]. This program is also implicated in morphogenetic alterations during embryonic development, wound repair, and cancer metastasis [13]. During EMT process, epithelial cells dedifferentiate, start to lose the structural restrictions provided by the tissue architecture and turn from previous pebble shape to fusiform appearance of cells, which is related to cytoskeleton rearrangement [14,15,16]. These cells induce proteolytic digestion of the receptors implicated in cell–cell contact, increase the activity of adhesion molecules that contribute in cell movement and stimulate proteases on the cell surface leading to an incremented digestion of extracellular matrix (ECM) constituents [17]. In this regard, cancer cells switch expression of E-cadherin and cytokeratin, typically found in epithelial cells, to fibronectin, vimentin, or neural cadherin (N-cadherin), which are generally identified in mesenchymal cells. In addition, an elevated activity of matrix metalloproteinases (MMPs), such as MMP-2 and MMP-9 has been previously reported [18,19,20]. The alterations in the expression of E-cadherin and N-cadherin are shown to be associated with resistance to anoikis and increase in invasiveness [16, 21]. Tumor microenvironment is comprised of stromal cells, ECM components, and secreted factors, all of which play a major role in EMT induction and further in tumor metastasis [14, 22]. EMT process may be activated by various conditions, such as tissue hypoxia and fluctuating level of extracellular stimuli, including transforming growth factor β (TGF-β), epithelial growth factor (EGF) family member, and fibroblast growth factor [15, 23].
These stimuli can influence several transcription factors (TFs), such as Snail, Twist, Zeb and others by modulating signal transduction pathways, including Wnt, Notch, mitogen-activated protein kinase (MAPK), and phosphatidylinositol-3-kinase (PI3K) [13, 23]. EMT is also under the control of epigenetic modifications, such as DNA methylation, microRNAs (miRNAs) and long noncoding RNAs (lncRNAs) [24]. Moreover, correlations between some EMT regulating factors and cancer stem cell (CSC) properties have been revealed, because TFs, such as Zeb1 frequently stimulate stemness by repressing stemness-inhibiting miRNAs [25]. Although primary tumor cells and circulating tumor cells (CTCs) show EMT characteristics, they convert back to their epithelial phenotype by a reverse process called mesenchymal to epithelial transition (MET), which causes the induction of cell proliferation and formation of secondary tumors at the distant metastatic site [13, 26]. Many invasive carcinomas, such as thyroid cancer undergo EMT in the invasive front (tumor-stromal boundary) of tumor [27]. A growing body of literature has affirmed that the activation of the EMT program is closely related to thyroid carcinoma progression, which includes extrathyroidal extension, distant metastasis, and increased stemness of cancer cells [10, 28, 29]. Understanding the different aspects of EMT process could aid in unraveling the metastasis in thyroid cancer, and may provide new therapeutic anticancer strategies targeting EMT especially by reversing this transition process. The main goals of this review are to summarize current knowledge on the role of EMT in thyroid cancer focusing on the main role-players and regulators of this process, such as the inducers and signaling pathways, TFs, epigenetic modifications, and microenvironmental factors in thyroid tumors.
EMT regulatory networks in thyroid cancer
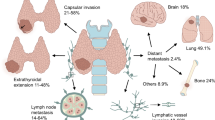
EMT, under physiological and pathological conditions, is controlled by highly evolutionarily conserved regulatory networks [20]. These networks involve two arms: (1) transcriptional control that implicates several TFs, such as Snail, Zeb, Twist, Forkhead box D3 (FOXD3), SOX9, Runx2, and (2) epigenetic mechanisms, such as DNA methylation, lncRNA, and microRNA functions [20, 24]. In addition, various inducers activate several signaling pathways, including TGF-β, Wnt, Notch, and PI3K and MAPK, which converge on the regulation of specific TFs involved in driving EMT in thyroid cancers. Figure 1 depicts several possible regulators and different markers of epithelial and mesenchymal cell types in thyroid cancer EMT.

Schematic representation of EMT modulators in thyroid cancer. Epithelial cells exhibiting apical–basal polarity are held together by cell–cell junctions. These cells express molecules that maintain epithelial phenotype (listed in the pink box). Induction of EMT leads to the expression of genes associated with the mesenchymal state (listed in the green box) which results in cellular changes including the disassembly of epithelial cell–cell junctions and the dissolution of apical–basal cell polarity. The initiation and progression of EMT in thyroid cancer is under the control of multiple regulatory networks, including epigenetic mechanisms, transcription factors, signaling pathways, tumor microenvironmental-associated factors, and specific cellular proteins with various functions in the cells
Signaling pathways
TGF-β signaling
TGF-β family is a key inducer of EMT during cancer metastasis [11]. TGF-β plays two opposing roles in tumorigenesis: it acts as a tumor repressor at the initial phases of tumor formation and stimulates cancer growth and metastasis in later stages [30]. TGF-β signaling functions via stimulation of receptor-regulated SMADs (R-Smads), such as Smad2 and Smad3 or non-Smad signaling pathways. Activated R-Smads bind Smad4, and this complex is transported into the nucleus, where it regulates the transcription of several genes [31]. On the contrary, Smad7 has been shown to antagonize TGF-β/Smad-dependent signaling [32]. The expression of the components of TGF-β/Smad-dependent signaling has been observed in normal thyrocytes, but their expressions have been shown to be significantly higher in PTC tumors as compared with that in normal thyroid tissues [32]. Cerutti et al. reported that Smad7 was upregulated in aggressive undifferentiated thyroid cell lines compared with differentiated ones and that Smad4 was expressed in all tested cell lines, unlike other types of tumors, in which Smad4 expression is deleted [33]. It was supposed that cancer cells become resistant to TGF-β antitumor effects due to mutations in TGF-β receptors and Smads [30]. D’Inzeo et al. detected a mutation in Smad4 gene from node metastases of PTC, and reported that the constant expression of Smad4 in thyroid cells is associated with a significant increase in TGF-β signaling, resulting in mesenchymal conversion and E-cadherin loss, the two important events involved in EMT [34]. Garcia-Rendueles et al. demonstrated TGF-β/Smad suppressing ability against p27 gene in normal thyrocytes that leads to Bax upregulation and apoptosis. However, they further asserted that oncogenic activation stops TGF–β/Smad-dependent p27 suppression in thyroid cancer cells, the phenomenon that causes TGF-β/Smad-dependent growth of thyroid tumors [30]. Moreover, TGF-β has been established to elevate motility and recruitment of immune cells, such as monocytes and macrophages, while prohibiting their antitumor activities. Thus, TGF-β can also assist in the progression of tumorigenesis through secretion of growth factors and cytokines from immune cells [32]. Non-Smad signaling pathways have been identified to be involved in TGF-β-induced invasion of thyroid cancer cells [35, 36]. Baquero et al. clarified the role of TGF-β in promoting EMT process through two independent pathways of activation of MAPK by BRAFV600E and stimulation of Src/FAK signaling [11]. Conversely, Anelli et al. experimentally showed that BRAFV600E expression in thyrocytes results in the upregulation of genes associated with EMT and TGF-β signaling. They additionally suggested the TGF-β and EMT as the most highly differentially induced signaling pathways in BRAFV600E expressing thyrocytes [37].
PI3K/Akt and MAPK/Erk pathways
A number of evidences have shown that the deregulation of PI3K/Akt and MAPK pathways participate in EMT initiation and tumor development in thyroid cancer [38]. MAPK activation is induced by external stimuli (such as growth factors and mitogens) that bind to their cognate cell-surface receptors. Mutations in various components of the MAPK pathway, which can lead to constitutive activation of the pathway, have been found in ~70% of thyroid cancers. RET/PTC rearrangement or RAS or BRAF point mutations are primarily responsible for the initiation and development of thyroid carcinomas. The most frequently alteration in thyroid tumorigenesis is BRAFV600E mutation, which activates MEK-ERK in this pathway [39, 40]. Moreover, it has been revealed that these effects are mediated by overexpression of Snail and loss of E-cadherin, both are hallmarks of EMT [11, 40]. In addition, BRAFV600E expression has been unraveled to increase TGF-β secretion, which in turn can result in an augmented invasion and EMT program in thyroid tumor cells. These effects have been found to be mediated by the activation of two parallel cascades of BRAFV600E/MEK/ERK and Src/FAK signaling [11]. Agrawal et al. by studying the genomic landscape of 496 PTC samples, have found that BRAFV600E-mutated samples more strongly activate the MAPK pathway than RAS-like tumors. This may be due to the insensitivity of BRAFV600E to ERK preventive feedback and responsivity of RAS to this inhibition. However, RAS-like tumors can activate both MAPK and PI3K/AKT pathways by a mechanism distinct from that of BRAFV600E tumors. They also asserted that the RAS-like tumors are a distinct subgroup characterized by a higher differentiation of the tumor [41].
The oncogenes RET/PTC, RAS, and BRAF, by activating MAPK cascade, can induce pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ). TNF-α and IFN-γ are expressed in several types of tumor cells and cells in the tumor microenvironment. They exert numerous pro-tumoral activities in solid tumors. Lv et al. observed that TNF-α and IFN-γ induce EMT and malignant progression in human PTC cells via downregulation of E-cadherin as well as the upregulation of N-cadherin and vimentin [42].
Binding of EGF to its receptor (EGFR) has been reported to induce EMT in breast cancer cells [43]. EGF also plays a crucial role in pathogenesis of ATC and affects the expression of EMT markers, by increasing vimentin and decreasing E-cadherin expressions. A study by Chin et al. revealed a regulatory function for EGF in ATC migration and invasion, which was occurred by modulating the ERK/CREB signaling pathway [44]. Gao et al. showed that the binding of hepatocyte growth factor (HGF) to its receptor, c-Met, could induce the invasion and EMT in thyroid cells as well as the stimulation of metastasis in vivo through the c-Met/PI3K/AKT pathway. They have further proven that HGF-induced EMT was reversed by upregulating E-cadherin and downregulating N-cadherin in cells treated with c-Met and PI3K inhibitors [45]. Various studies have highlighted that upregulation of discoidin domain receptor tyrosine kinase 2 (DDR2), a member of the receptor tyrosine kinase (RTK) family, induces EMT in breast cancer, hepatocellular carcinoma, and head and neck squamous cell carcinoma [46,47,48]. Liang et al. provided evidence to show that DDR2 is upregulated in PTC tissues with local metastasis and that its overexpression induces EMT in PTC cells by stimulating ERK2 and stabilizing Snail1 [49].
The Notch pathway
The Notch pathway plays an oncogenic or tumor suppressor role, depending on the type of malignancy. This pathway involves four receptors (Notch1, 2, 3, and 4) with the Notch1 has been evidenced to participate in the EMT during both normal development and tumorigenesis. The role of abnormal Notch-1 expression has been described in thyroid cancers [50]. Ferretti et al. declared that the expression levels of the Notch pathway components were visibly decreased in undifferentiated tumors in comparison to normal thyroid tissue, and upregulated Notch1 in cancer cells could reestablish the differentiation, diminish cell growth, and induce sodium/iodide symporter expression via a direct act on its promoter [51]. However, Park et al. indicated that the expression of Notch1 receptor in PTCs was meaningfully associated with the presence of clinicopathological complications, such as the large tumor size, capsular invasion, extrathyroidal extension, and node metastasis [52]. A study by Zhang et al. showed that the expression of Notch-Regulated Ankyrin Repeat Protein (NRARP), as a mediator for controlling Notch and Wnt signaling, was highly associated with several EMT markers and poor survival in PTCs [53]. Notch1-induced tumor advancement may involve the modification of the TGF-β signaling pathway. The data obtained from the study of Zhang et al. indicated an elevated expression of TGF-β1, Notch, and p-Smad3 levels in PTC cells [54]. They also affirmed that the activated Notch and TGF-β/Smad3 pathways in tumors could affect their growth and progression.
The Wnt/β-catenin pathway
Wnt ligands are secreted glycoproteins that have been shown to be implicated in the progression of thyroid cancer [55]. The Wnt/β-catenin pathway activation leads to the accumulation of β-catenin in the nucleus where it functions as a coactivator for TFs, such as the T-cell factor (TCF)/lymphoid enhancer factor family [56]. SDC4 is an integrin co-receptor of Wnt and has been reported to considerably affect cell adhesion-promoting MAPK pathway. Chen et al. have demonstrated that SDC4 gene silencing inhibits EMT, and increases cell apoptosis by suppressing the stimulation of Wnt/β-catenin signaling in human PTCs [55]. Thyroid cancer development to PDTC and ATC is associated with constitutive activation of β-catenin and its target genes, which are implicated in carcinogenesis [57, 58]. β-catenin mutations in the most aggressive forms of thyroid cancer and H-RasV12 expression in tumor cells of differentiated thyroid cancer have been shown to induce β-catenin nuclear assembly [59]. An increasing body of evidence supports the role of β-catenin in upregulation of EMT-related genes and an increase in the thyroid tumor cell invasion [57]. CSN6 is a subunit of the COP9 signalosome complex that plays important roles in protein ubiquitination, transcriptional activation, signal transduction, tumorigenesis, and stabilizing β-catenin. Wen et al. showed that the expression of CSN6 was increased in human PTCs and its elevated expression facilitated the EMT in these specimens [60]. Hardin et al. observed that β-catenin silencing could upregulate E-cadherin and reduce the EMT markers and tumor cell invasion [57].
Transcription factors
TFs like Zinc finger E-box-binding homeobox (Zeb) 1 (also called TF 8) and Zeb2 (also known as ZFXH1B) are associated with EMT. Zeb1 and Zeb2 are key modulators implicated in the suppression of several crucial regulators of epithelial polarity and thereby are involved in EMT and metastasis processes [61]. Zeb1 upregulation correlates closely with EMT in thyroid cancer cells. It has been demonstrated that Bcl-2-associated athanogene 3 (BAG3) stimulates the nuclear accumulation of β-catenin and augments its transcriptional activity, resulting in the overexpression of Zeb1 in thyroid tumor cells. Overexpressed Zeb1 is then able to directly bind E-box in the E-cadherin promoter, repressing its transcription and leading to the activation of EMT [16]. Twist1 is a basic helix–loop–helix TF capable of straight binding to E-box consensus sites (5-CANNTG-3) to exert its regulatory effects [62, 63]. Twist1 gene amplification stimulates a CSC phenotype, prevents apoptosis, and enhances resistance to chemotherapeutic agents [63,64,65,66]. Elevated Twist1 correlates with poor prognosis and high grade and stage of the tumor [62, 63]. Twist1 has been reported as a major regulator for EMT [67, 68]. Survival rate and motility of thyroid cancer cells is influenced by Twist1 [69]. High expression levels of Twist1 have been observed in ATC resulting in the enhancement of cell migration, invasion, and resistance to apoptosis. Si-RNA-mediated repression of Twist1 decreases these effects [70,71,72]. Twist1 overexpression has also been reported in aggressive follicular carcinomas, which are associated with the loss of E-cadherin expression [73]. Slug (Snail2) is a member of the Snail zinc finger E-box binding family that acts as a transcriptional repressor. Snail family members are involved in a variety of cellular processes related to cell motility and EMT initiation [74]. The increased expression of Snail in mammary cell lines may result in loss of E-cadherin expression and gain of mesenchymal phenotype [75]. It has recently been revealed that Slug is not expressed in normal human thyroid cells, but is highly expressed in thyroid cancer cells and their metastases. Buehler et al. have shown that the Slug expression was significantly higher in the ATC tissues and cell lines than in PTCs and FTCs, suggesting the role of EMT program in the progressive behavior of ATCs [70]. Moreover, Sun et al. confirmed that Slug silencing by siRNA transfection significantly decreases the ability of cell migration and invasion in ATC cells [76]. Wu et al. have described the upregulation of EMT marker, Slug, in WD-FTC and its association with highly aggressive and poorly differentiated phenotype in ATC and FTC tumors [73].
Sex-determining region Y (SRY)-box 9 (Sox9) is another TF that plays different roles in several types of cancers. Sox9 is overexpressed in human PTC cell lines and tissues. Sox9 has been identified to control PTC cell proliferation, migration, invasion, and EMT process through affecting the Wnt/β-catenin pathway [77]. Huang et al. provided evidence to show that knockdown of Sox9 in PTC cells significantly inhibited the expression of downstream target genes of the Wnt pathway, such as β-catenin, cyclin D1, and c-Myc. Their results also showed that knockdown of Sox9 reversed EMT phenotype, by obstructing the expression of N-cadherin and enhancing E-cadherin expression levels [77]. Runx2, runt-related TF2, is a master regulator of embryonic differentiation. Overexpression of Runx2 has been detected in PTCs and thyroid cancer cell lines (KTC-1, TPC-1, WRO, UA-2, and 8305C cells) [78]. Runx2 has been shown to have promoting effects on cancer cell metastasis by stimulating the secretion of MMPs and VEGF and overexpression of EMT-related proteins, including Sox9, Snail2/Slug/SNAI2, and Smad3 [79,80,81].
FOXD3 TF participates in embryonic development and tumorigenesis in many tissues. The upregulation of FOXD3 suppresses metastatic promotion of non-small cell lung cancer and its downregulation in infiltrating ductal carcinomas of breast induces distal metastases [82, 83]. A recent study has indicated that the inhibition of FOXD3 increases the invasiveness and EMT in ATC cell lines and tissue samples. The results of that study also showed that knockdown of FOXD3 could decrease the expression of E-cadherin through the activation of the MAPK/ERK signaling pathway. In addition, the expression of FOXD3 was significantly higher in normal samples compared with tumor samples and was inversely related to p-ERK [84].
TBX1, a member of the T-box family of TFs, is characterized by a conserved and relatively large DNA binding motif known as T-box. Wang et al. declared that TBX1 functions as a tumor inhibitor in thyroid cancers and its expression is downregulated in both PTC tumors and some thyroid cancer cell lines (BCPAP, FTC133, K1, and 8305C). They reported that TBX1 represses EMT via upregulation of E-cadherin and downregulation of N-cadherin, vimentin, and the E-cadherin repressors (Snail, Slug, and Twist) [85].
Specific cellular proteins
E-cadherin
The E-cadherin gene (CDH1) is frequently downregulated by different mechanisms including inherited and somatic mutations, unusual protein processing, hypermethylation of CDH1 promoter, and repressing effects of Zeb1, Zeb2, Snail, E12/E47, and Twist [86, 87]. E-cadherin is expressed in normal thyroid gland and benign thyroid lesions, while its expression diminishes with the progression of differentiated thyroid cancers to undifferentiated ones [35, 88]. The expression profile of PDTC, a rare aggressive malignancy of thyroid follicular cells, exhibits a significant decrease in E-cadherin as compared with paired samples of PTC [35]. Overexpression of BRAFV600E in PTC cells stimulates EMT by enhancing Snail expression and suppressing the E-cadherin levels [87]. Jung et al. also highlighted the loss of E-cadherin expression in majority of ATC patients [89]. In thyroid tumors, the loss of E-cadherin is generally followed by overexpression of several transcriptional factors, such as Snail1, Slug (Snail 2), Zeb, Twist, which are implicated in EMT [16, 17, 70,71,72].
Vimentin
During EMT, the actin and intermediate filament of the cytoskeleton are reorganized, and cells obtain improved cell-matrix contacts, promoting the detachment from nearby cells and triggering migration and invasion. Expression of vimentin, a component of intermediate filaments, is upregulated in mesenchymal cells [20, 90]. Overexpression of vimentin is associated with the induction of EMT in several thyroid cancer cell lines and tissues, which is correlated positively with other mesenchymal markers, including N‐cadherin, Slug, Zeb1, and Snail [9, 16, 91].
Periostin and fibronectin1
Other ECM proteins, such as periostin and fibronectin1 (FN1) are induced during EMT and alter the composition of the ECM, the process that seems to contribute in invasiveness of tumors [28, 92]. Periostin is a secretory protein that acts as a scaffold for assembly of ECM proteins (such as collagen and fibronectin) and plays a role in cell adhesion and ECM organization [92]. Periostin, localized in the ECM, interacts with integrins vß3 and vß5, and thereby stimulates cell adhesion and motility through the modulation of Akt/PKB and FAK-mediated signaling pathways [93, 94]. This protein is highly expressed in many types of human cancers, such as pancreatic, colon, breast, nasopharyngeal, head and neck, gastric, and thyroid malignancies [93,94,95,96,97,98,99]. Periostin has been found to be a marker and inducer of EMT process [93]. Kim et al. affirmed the periostin-mediated induction of the EMT process in prostate cells through loss of E-cadherin under the control of Snail and enhanced p-Akt [93]. Periostin is generally upregulated in PTCs regardless of the presence of a BRAF mutation, and higher levels of periostin mRNA is associated with decreased expression of thyroglobulin and Thyroid Stimulating Hormone Receptor (TSHr), which are differentiation markers. Indeed, periostin overexpression has been shown to be correlated with different clinicopathological characteristics of aggressive PTCs, such as the presence of extrathyroidal extension, distant metastasis, and higher tumor grade and stage. These data suggest that the expression of periostin is related to EMT and tumor aggressiveness [10].
Some immune-histochemical evidences exhibited that the enhanced FN1 expression is seen mainly at tumor invasive front [28]. Sponziello et al. reported the increased levels of FN1 mRNA in classic PTC (vs follicular-variant PTC), in response to BRAFV600E oncogene (vs BRAF-wild type) [28]. Moreover, FN1 expression is higher in PTC patients with lymph node metastasis (LNM) than patients without LNM, and its overexpression is related to larger tumors and an Advanced-stage PTC [100]. Xia et al. found that FN1 sensitivity in identifying overall cervical LNM in PTCs was much higher than ultrasonography evaluation, and its specificity in forecasting LNM in tumors of PTC was similar to ultrasound examination, proposing it as a reliable biomarker for predicting LNM in PTC [100].
S100 proteins
Another family of ECM proteins related to metastatic processes is the S100 proteins, a multigene calcium-binding family that covers more than 20 members. S100 proteins participate in several cellular processes, such as the regulation of protein phosphorylation, enzyme activity, calcium homeostasis, organization of cytoskeletal components, and acting as transcriptional factors. S100 proteins, when are upregulated in the ECM, can disrupt cell–cell adhesion and facilitate the degradation of ECM and metastasis [101, 102]. S100A13, a small S100 calcium-binding protein A13, is characterized by its specificity for diverse forms of cancers. The overexpression of S100A13 increases the risk of relapse in melanoma patients, and invasiveness in lung cancer cell lines [103, 104]. S100A13 has also been found to be enhanced in PTCs compared with normal tissue [105, 106]. Knockdown of S100A13 has been described to reduce the expression of high-mobility group A (HMGA)-1 and Snail and enhances E-cadherin expression in thyroid cells, suggesting that S100A13 may play a key role in EMT and thyroid cancer progression [105].
IQGAP1
IQGAP1, as a scaffold protein widely expressed in human tissues, is involved in various biological processes, such as the cell adhesion, cell cycle, directional migration, transcription, cell division, and extracellular signaling [107, 108]. It has been reported that IQGAP1 is overexpressed in numerous tumors, such as colorectal, gastric, and pancreatic cancers [109,110,111]. In addition, IQGAP1 amplification has been unraveled to be significantly associated with thyroid cancer cell invasion. Su et al. demonstrated that the downregulation of IQGAP1 inhibits cell proliferation and EMT via blocking Wnt/β-catenin signaling in thyroid cancer cells [107].
KIF3a
The onset of EMT process is accompanied by the loss of cellular polarity [112]. Epithelial cell polarity occurs through the uneven distribution of cellular junctions and polarity proteins [113]. Basoapical polarity can change the expression patterns of genes through the interaction of junctions and polarity proteins with remodeling factors of genome [113]. Polarity proteins are a highly conserved set of proteins that form a complex that localizes to the tight junction. These proteins are able to interact with microtubule motor protein KIf3a, leading to control of the polarized trafficking of proteins along microtubules [114]. Recently, several studies have discovered the role of KIF3a in the development of tumorigenesis. Liu et al. described that the exogenous expression of KIF3a in human prostate cancer cell lines transfected by a KIF3a-expressing vector stimulates cell proliferation; however, knockdown of KIF3a decreases the migration and invasion of these cells [115, 116]. Wang et al. provided evidence to describe the significant elevated expression of KIF3a transcript and protein in the tissues and cell lines of human thyroid cancer (FTC133 and TPC1). In addition, they demonstrated that the knockdown of KIF3a inhibits hypoxia-induced EMT of thyroid cancer cells through suppressing the Wnt/β-catenin signaling pathway [117].
BAG3
Once the tumor cells start to metastasize, they need to overcome anoikis, a form of programmed cell death that is initiated by cell detachment from the ECM [20, 118]. BAG3 is an Hsp70 co-chaperone that supports cell survival by retaining the antiapoptotic activity of Bcl-2 family of cell death regulators [119]. It was previously established that BAG3 silencing through inhibition of HSF1 (TF upstream of BAG3 and HSP70) decreases cell motility and ECM adhesion in breast and prostate cancer cells, and primes glioma cells to anoikis [118]. BAG3 has been indicated to control EMT and metastasis in thyroid cancer cells. Meng et al. alluded that BAG3 upregulates Zeb1 through β-catenin activation, which in turn enhances EMT and metastasis of thyroid cancer cells [16].
HMGA proteins
The high-mobility group A (HMGA) proteins are often upregulated in human cancers and have significant roles in the EMT process [120]. HMGA1 controls the transcriptional activity of various genes by changing the chromatin structure (83). The expression of HMGA1 is almost absent in normal cells and adult tissues, but are enhanced in embryonic cells and many cancers, such as breast, pancreas, lung, colon, and thyroid carcinomas [85, 121,122,123,124]. Positive correlation and increased expression levels of HMGA1 protein has been previously described in thyroid cancer cases (83). The downregulation of HMGA1 has shown to decrease the expression of Snail and MMP-2, whereas increases the expression of E-cadherin, which may result in the suppression of proliferation and invasion of thyroid cancer cells [105, 120]. Zhong, et al. pointed out the inducing activity of TGF-β1 on HMGA1 expression through PI3K and ERK signaling in thyroid cancer cells, suggesting a pivotal role of HMGA1 in the progression of thyroid carcinomas [120].
Epigenetic modulators
Epigenetic modifications are dynamic and reversible alterations, affecting chromatin structure for the regulation of gene expression without altering the nucleotide sequence [125]. They include DNA methylation, histone modifications, and noncoding RNA regulations [126]. Increasing evidence has highlighted the critical role of epigenetic modifications in the initiation and maintenance of EMT, and that the reversibility of epigenetic changes can describe the plasticity of EMT for metastatic colonization [127].
DNA methylation
Aberrant DNA methylation is one of the well characterized epigenetic alterations, which can lead to a number of processes, such as the activation of proto-oncogenes or silencing of tumor suppressor genes [128]. DNA methylation is catalyzed by DNA methyltransferase enzymes that methylate cytosine residues in CpG islands located in gene promoters [127]. Tumor suppressor genes DAPK (death-associated protein kinase), RASSF1A (RAS association domain family 1A), RAPβ2 (retinoic acid receptor beta 2), RAP1GAP (RAP1 GTPase activating protein), TIMP3 (tissue inhibitor of metalloproteinase 3) SLC5A8, and PTEN are epigenetically downregulated in thyroid cancers [128]. The PTEN, a phosphatase that terminates the PI3K/Akt pathway, often exhibits a pattern of promoter hypermethylation with decreasing the differentiation in FTCs [129]. In PTCs, promoter hypermethylation of RASSF1A, DAPK, and TIMP3 genes result in an aggressive phenotype [128]. Thyroid differentiation genes, such as SLC5A5 (NIS), and NKX2-1 also are hypermethylated in undifferentiated thyroid carcinoma [129]. The hypermethylation of E-cadherin gene (CDH1) that is associated with gene silencing has been reported in several human cancers, such as the breast and thyroid tumors [127]. Jensen et al. conducted a study to evaluate the relationship between E-cadherin methylation and its expression in PTC samples as well as the corresponding LNM. They observed that the hypermethylation of CDH1 promoter in PTC samples was correlated with loss of E-cadherin expression, extrathyroidal extension, and lymph node metastases. Loss of epigenetic silencing of CDH1 due to the reduction in the level of gene promoter methylation in LNM was correlated with a gain of E-cadherin expression [27]. TF TBX1 that inhibits EMT and invasiveness has been reported to be inactivated by promoter methylation in PTC tissues and some thyroid cancer cell lines [85].
miRNA
MicroRNAs (miRNAs) are a wide class of naturally occurring, small noncoding, and single- stranded RNA molecules of about 19–25 nucleotides in length. MiRNAs function as posttranscriptional negative regulators of gene expression via either splitting mRNA, or hindering their translation [17, 130]. They are able to regulate majority of cellular processes including cell proliferation, differentiation, adhesion, and programmed cell death [131]. Furthermore, dysregulation of miRNA occurs in several type of cancers, in which the processes of cell migration, invasion, and EMT are intensified [132]. The overexpression and underexpression of specific miRNAs are linked to some cancers, suggesting the oncogenic or tumor suppressor role for these RNAs [133]. Several miRNAs, such as miR-146, miR-221, miR-222 and miR-17-92, and miR-150‐5p are upregulated in thyroid cancers, while some are downregulated, which include miR-200, miR30, and miRNA148a families exclusively in ATC, miR-520a‐3p and miR-144 in PTC, and let-7 family in both ATC and PTC [91, 132, 134,135,136].
Several studies have shown that miR-144 participates in the tumorigenesis of various tumors and acts as a tumor suppressor or an oncogene, depending on the type of tissue. Decreased level of miR-144 is associated with several types of cancers, such as non-small-cell lung cancer, bladder cancer, and colorectal cancer cells [137,138,139]. However, in nasopharyngeal cancer, miR-144 enhances tumor growth, migration, and invasion by suppressing PTEN, acting as an oncogene [61]. MiR-144 has been observed to be meaningfully downregulated in PTC cell lines and tissues, and its expression was associated with larger tumor sizes [134]. Moreover, miR-144 represses Zeb1 and Zeb2 mRNA, and induces a mesenchymal phenotype with inhibition of E-cadherin [61]. Another target of miR‑144 is WW domain-containing transcription regulator 1 (WWTR1) that is highly expressed in PTC tissues with the ability to enhance the cell invasion through EMT [134]. Moreover, miR-144 directly targets E2F8, a typical E2F TF that participates in the regulation of genes required for the progression of cell cycle, to obstruct PTC cell proliferation in vitro and in vivo. E2F8 is generally overexpressed in PTC tissues, and has been shown to cause aggressive clinicopathological characteristic, while its silencing could result in G1-phase arrest by reducing cyclin D1 (CCND1) [140].
MiR-146, miR-146a, and miR-146b family genes are abundantly expressed in human thyroid cancers, especially in ATC, and are under the control of nuclear factor-κB [141]. Indeed, MiR-146b-5p increases the extrathyroidal invasion and stimulates EMT in PTC cells. MiR-146b-5p enhances cell surface Wnt receptor expression and Wnt/β-catenin signaling through downregulation of zinc RING finger 3 (ZNRF3), but on the contrary, overexpression of ZNRF3 can reverse miR-146b-5p effects [142]. MiR146b has been shown to promote EMT of thyroid follicular cells by downregulating PTEN, which may result in the hyperactivation of the PI3K/AKT pathway, increasing Twist expression, and decreasing E-cadherin expression. Ramirez-Moya et al. showed that in spite of the fact that PTEN rescues the miR146b-induced expression of Twist, it fails to rescue reduced level of E-cadherin [143].
Deregulation of miR‐150‐5p has been illustrated to modify the biological and pathological processes of several type of cancers [144]. For example, its overexpression in gastric cancer cells enhances the mutations of DNA repair genes [91, 145]. Upregulation of miR‐150‐5p in PTC cell lines lessens E-cadherin expression, but augments the levels of N‐cadherin, Slug, vimentin, Zeb1, and Snail, suggesting a role in EMT. Indeed, it has been observed that miR‐150‐5p accelerates the EMT switch through the MEK/ERK signal pathway, and controls the BRAFV600E oncogene [91].
MiR-200 family members, including miR-200a, miR-200b, miR-200c, and miR-30 have been recognized as the inhibitors of EMT in ATC. The expressions of these two groups of miRNAs are usually diminished in mesenchymal ATC-derived cell lines. This reduced levels may induce EMT by targeting the Zeb1 and Zeb2 [135]. According to some investigations, the stimulation of the EGF pathway downregulates miR-200s, and enhances the expression of mesenchymal markers [146]. Basal miR-200s level is also regulated by transcription activator p53. It was previously indicated that the presence of a TP53 mutation in ATC could lead to the loss of miR-200 expression [147].
MiR-520 has been reported to serve as a tumor suppressor in several human cancers, such as hepatocellular carcinoma, breast cancer and PTC [136, 148, 149]. MiR-520a-3p is poorly expressed in PTC tissues, where it is associated with the expression of EMT-related biomarkers (including reduced expression of E-cadherin and elevated expression of vimentin). MiR‐520a‐3p has also been claimed to inhibit cancer progression through inactivating the JAK/STAT cascade by negatively regulating JAK1. However, miR‐520a‐3p upregulation or JAK1 knockdown could reduce the expression of JAK1 and EMT biomarkers in PTCs [136].
MiR-199a-5p expression is regulated by tumor specific mechanisms, because it is downregulated in tumors of breast and colorectal tissues, while it is upregulated in gastric cancer [150,151,152]. The expression level of miR-199a-5p is markedly downregulated in PTC tissues and cells. Ma et al. reported that miR-199a-5p overexpression suppresses the progression of PTC by downregulating Snail [153].
LncRNA
LncRNAs are usually referred to RNA transcripts containing more than 200 nucleotides without any protein-coding potential [153]. LncRNAs take part in various physiological and pathological processes, such as carcinogenesis, by changing gene expression through epigenetic regulation, transcriptional control, and posttranscriptional regulation. Accumulating evidence indicates that the lncRNAs participate in tumor progression, thus the aberrant expression of these transcripts are correlated with cancer diagnosis and prognosis [154]. A number of lncRNAs have also been characterized to play important roles in EMT and development of thyroid and several other cancers [57].
BRAF-activated non-protein-coding RNA (BANCR), a 693-bp-long transcript located on chromosome 9, has been found to be related to several human malignancies. Li et al. uncovered high expression levels of BANCR in human melanoma cancer cell lines and tissues, and asserted that the upregulation of BANCR stimulates the cell proliferation through activating ERK1/2 and JNK MAPK cascades [155]. Jiang demonstrated that BANCR expression was downregulated in lung carcinoma cells, and this level was remarkably associated with enhanced cell proliferation and migration of lung carcinomas [156]. A recent study by Wang et al. using thyroid cell lines (including WRO, CAL-62, BCPAP, and FTC-133) indicated that BANCR expression was augmented in tissue samples from PTC patients compared with adjacent normal tissues. They also asserted that the BANCR mediates EMT by downregulating E-cadherin, and upregulating vimentin and N-cadherin. In addition, the results of their study proposed that BANCR overexpression may promote the migration and invasion of PTC cells through the Raf/MEK/ERK pathway [157].
Long intergenic non-protein-coding RNA 673 (LINC00673) has recently been characterized and shown to have oncogenic function [158]. Lu et al. have proven that LINC00673 was overexpressed in non-small cell lung cancer, and that it could sponge miR-150-5p and indirectly regulate the expression of a crucial EMT-TF Zeb1 [158]. Yu et al. reported that LINC00673 was considerably upregulated in tongue squamous cell carcinoma samples, and promoted invasion and metastasis of tumor cells [159]. A recent study by Xia et al. revealed that LINC00673 expression was highly upregulated in thyroid tumor tissues compared with paired adjacent normal tissues, and that high expression of LINC00673 was correlated with larger tumors and LNM. They also showed that LINC00673 might influence EMT, because knocking down of LINC00673 decreased the expression of N-cadherin and vimentin, and increased E-cadherin expression, while its high expression led to the opposite results [160].
LncRNA CASC2 plays a tumor suppressor role, and its downregulation contributes to tumor progression in bladder cancer and renal cell carcinoma [161,162,163]. Zhou et al. examined the expression of LncRNA CASC2 in plasma and tumor tissue samples of patients with PTC in comparison to nodular goiter. They found that lncRNA CASC2 expression was remarkably lower in tumor samples than in nodular goiters. Decreased levels of plasma lncRNA CASC2 was accompanied by LNM of PTC patients. However, results of in vitro functional assays revealed that the overexpression of lncRNA CASC2 inhibits proliferation and migration of PTC cells. In addition, lncRNA CASC2 overexpression inhibited EMT process of PTC by upregulation of E-cadherin and downregulation of Zeb1 and N-cadherin [164].
Taurine upregulated gene 1 (TUG1), an lncRNA located on chromosome 22q12, has been described to be dysregulated in several malignancies. TUG1 is a member of competing endogenous RNAs (ceRNAs), which has been proposed as a novel post-transcriptional regulation, in which lncRNAs interact with miRNAs [165]. Lei et al. reported that the expression level of TUG1 was increased in thyroid cancer tissues. They also described that TUG1 sponged miR-145 and promoted cancer progression and EMT through the miR-145/Zeb1 signaling pathway in thyroid cancer cells [165]. Linc-ROR (regulator of reprogramming) is a lncRNA that plays key controlling roles in interaction with miRNAs, conserving stem cell pluripotency, and activating the EMT [166]. Linc-ROR functions as a sponge to prevent the miRNA-mediated deprivation of the key pluripotency factors Oct4, Sox2, thus linc-ROR may regulate human embryonic stem cell self-renewal and differentiation [167]. Previous studies have demonstrated that miR-145, a known tumor suppressor, obstructs translation of pluripotency TFs during ESC differentiation [166]. Moreover, linc-ROR has a much wider role as a stress-responsive lncRNA, suggesting a mechanistic pathway by which linc-RoR can coordinate cellular responses to their local microenvironment [168]. Dysregulation of linc-ROR has been reported in several malignancies, such as the breast cancer, hepatocellular carcinoma, and endometrial cancer [166, 168,169,170]. Takahashi et al. alluded that in liver cancer cells linc-ROR is regulated by hypoxia and is able to support cell proliferation partially by sponging miR-145 [168]. Zhang et al., by examining linc-ROR expression in PTC tissue specimens and cell line, have found that linc-ROR has higher expression in PTC tissues. Their SiRNA experiments showed that linc-ROR enhanced cell proliferation and invasion and supported the role of ROR as a sponge to downregulate miR-145. They also demonstrated that linc-ROR expression was highly elevated in vitro when the PTC cells were treated with TGF-β, and as a result, the expression of EMT markers was increased and EMT was initiated [171].
Tumor microenvironment factors
Tumors are dependent on their complicated tissue environment that sustains tumor growth, invasion, and metastasis [172]. The tumor stroma consists of a number of cells, such as fibroblasts/myofibroblasts, inflammatory and immune cells, and a vascular system along with noncellular constituents of ECM [173,174,175,176,177]. In the tumor microenvironment, cancer-associated fibroblasts (CAFs) exist in abnormally high numbers, and are implicated in inducing a mesenchymal-like phenotype and enhancing epithelial tumor cell invasion [54]. Some studies have suggested that EMT stimulates the production of CAFs, whereby cancer epithelial cells dedifferentiate to produce mesenchymal cells carrying CAF markers [177, 178]. Zhang et al. clarified that the expression of alpha-smooth muscle actin, a CAF marker, significantly elevated with advanced tumor stages in PTC. Furthermore, they found that cancer cell-derived-TGF-β also influences stromal fibroblasts in a paracrine manner and may lead to tumor development [54].
Immune cells, as significant constituents of tumor stroma, can modulate cancer consequences by either limiting or promoting tumor initiation, development and metastasis [172]. In human PTC, an elevated number of mast cells have been observed to be associated with enhanced EMT, stemness, and invasiveness of tumor [175]. Once activated by thyroid cancer cell-derived conditioned media, mast cells produce high levels of IL-8 that constantly induce EMT in thyroid cancer cells through an IL-8-Akt-Slug axis [12]. Tumor-associated macrophages (TAMs) are one of the main role-players in stroma of various solid tumors and have been shown to promote cancer initiation and malignant progression [179]. Therefore, targeting these cells could result in reducing the number of tumor-initiating cells, overcoming chemotherapy resistance, inhibiting metastatic spread, and increasing antitumor T-cell responses [180]. Qing et al. studied the density of TAMs in benign thyroid lesions and PTC tumors using CD68 immunostaining technique. Their data uncovered that the number of TAMs in PTC tumors was significantly high in comparison to thyroid goiter and follicular adenoma, and their high amount was positively related to LNM. Accordingly, they concluded that TAMs may play an active and key role in the progression of PTC tumors [181]. Another study on diffuse sclerosing variant of PTC has evidenced the accumulation of TAMs in the lymphatic tumor emboli, with the majority (76%) of macrophages being of the M2 phenotype. Results of that study also showed a remarkable association between M2 TAM density and older age and larger tumor size in these patients. In addition, the increase in M2 TAMs has shown to be associated with an increase in lymphatic invasion, size of tumor emboli, and lymph node metastases [182]. Fung et al. found that TAMs can promote PTC metastasis by paracrine secretion of various cytokines, especially CXCL8. Therefore, they suggested that inhibiting CXCL8 can hamper PTC cell invasion in vitro and PTC tumor metastasis in vivo [183].
Accumulating data have confirmed that hypoxia, a critical microenvironmental factor, can activate the EMT process [13]. Hypoxia-inducible factor (HIF)-1 plays a key role in the hypoxia-induced transcription of TFs that include Twist, Snail, Slug, Sip1 (Smad interacting protein 1), and Zeb1 [13, 184]. In thyroid cancer cell lines, hypoxia induces HIF-1α expression and promotes EMT-related changes, which include typical morphologic changes, cadherin shift, and enhanced vimentin expression [3]. Upregulation of HIF-1α, by transfecting this factor, is generally followed by the aforementioned changes without hypoxia, but the suppression of HIF-1α with RNA interference represses EMT. Furthermore, Twist has been demonstrated to have a promoter region containing an HIF1 response element, which can be bound by HIF1 [184]. In thyroid cancer cell lines, Twist has been realized to be controlled by HIF-1α, and suppression of HIF-1α obliterates the hypoxia-induced increase of Twist expression [3]. In patients with PTC, HIF-1α expression was significantly correlated with the presence of LNM, peri-tumoral and extrathyroidal invasion, and larger tumors [185].
Chemokines, a group of small cytokines, promote the proliferation and migration of tumor cells. Binding of CXC chemokine ligand 12 (CXCL12, also recognized as stromal cell-derived factor 1) to CXC chemokine receptor 4 (CXCR4) increases the migration and invasion in CXCR4-expressing PTCs. In addition, CXCL12/CXCR4 axis induces the EMT program, as evidenced by enhanced N-cadherin and vimentin and reduced E-cadherin expressions [186]. Upregulation of CXCL5 and/or CXCR2 has been reported in some cancer tissues [187]. The activated CXCL5–CXCR2 axis promotes the EMT process in PTC cells. This process is also associated with a downregulation of E-cadherin and an upregulation of N-cadherin and vimentin and Snail in these cells [187]. Exosomes are small (30–150 nm) membranous vesicles secreted by most cells that play a significant role in cell-to-cell communication. These vesicles can modulate tumor microenvironment through transferring their contents, such as DNA, protein, growth factors, miRNAs, and lncRNAs to neighboring or distant cells [188]. Emerging evidence indicates that exosomes and their contents can contribute to the initiation of EMT and cancer progression [189]. One study found that secreted exosomes from thyroid CSC clonal line can induce EMT in the normal thyroid cell lines. It was hypothesized that lncRNAs, especially linc-ROR, transferred via CSC exosomes stimulate EMT and mediate the formation of surrounding tumor microenvironment and distant metastatic niche [189]. Table 1 summarizes different modulators of EMT process in various thyroid cancers.
The relationship between EMT and thyroid-derived CSCs
EMT is a remarkable example of cellular plasticity that implies the capacity of cells to reversible phenotype change [190]. Stemness is defined as the ability of stem cells to replicate in a manner that maintains a reservoir of undifferentiated cells with stem cell identity, yet also generates new differentiated cells [190, 191]. CSCs, as a subset of tumor cells in various type of cancers, are known to be implicated in the development of tumors and resistance to chemotherapy through enhanced levels of ABC (ATP-binding cassette) transporter family and antiapoptotic proteins [19, 192]. Some investigations have declared that the activation of EMT process confer stem cell properties to epithelial cells [190]. It has been clarified that the same factors that control EMT can regulate stemness [193]. A classical EMT TF, Zeb1, induces stemness phenotype by repressing stemness-inhibiting miRNAs (e.g., miR200 family members) [194]. Furthermore, various pieces of evidence have confirmed a controlling activity for the tumor suppressor p53 over EMT-linked stem cell features. Diminished levels of p53 in breast epithelial cells decrease miR-200c, leading to the augmented expression of EMT and stemness-related genes [147]. Stemness-linked signaling pathways, such as Wnt have been discovered to participate in some features of EMT process. The complex of β-catenin/TCF4, for instance, binds directly Zeb1 promoter and stimulates its transcription [195]. The experimental observations in human breast cancer models showed that the expression of mesenchymal genes by carcinoma cells correlates with the expression of many CSC markers. Indeed, epithelial cells that had undergone EMT may produce spheres, soft agar colonies, and in vivo tumors [196]. Several lines of evidence have affirmed the relationship between EMT and an augmented numbers of CSCs in thyroid cancers [197, 198]. Thyroid CSCs are identified by their ability to self-renewal, thyrosphere formation, and expression of stem cell markers, such as Nanog, Sox2, Oct4, aldehyde dehydrogenase (ALDH), stage-specific embryonic antigen 1 (SSEA1), and prominin 1 [29, 192, 199]. Hardin et al. showed that the higher percentage of CSCs exists in ATCs when compared with DTC tumors [197]. They also found that, during TGF-β-induced EMT, PTC cell lines acquire a parallel upregulation of a new EMT inducer Prrx1 (paired-related homeobox protein1) and stem cell-like characteristics, including the overexpression of Sox2 and Oct4, and the formation of thyrospheres [198]. A study on two thyroidectomy specimens of ATC with coexisting DTC presented a strong expression of stem cell markers CD133, CD44, and a neuronal stem cell marker nestin, and decreased expression of E-cadherin in ATC regions. However, the DTCs and nonneoplastic thyroid tissue in both specimens were negative for nestin and positive for E-cadherin, and the expression of CD133 and CD44 were inconstant and generally lower than that of ATCs [200]. Ma et al. reported that SSEA-1 is a specific marker for thyroid CSCs, and that the cells positive for SSEA1 express high levels of stem cell factors Nanog, Sox2, and Oct4. Furthermore, these stem cells displayed evidences of EMT initiation with elevated expression of Snail and vimentin and reduced expression of E-cadherin [29]. Consistently, Heiden et al. reported that the sonic hedgehog pathway plays a significant role in preserving the CSC self-renewal in ATC cells. They showed that Gli1-induced Snail expression increased the number of ALDH+ CSCs and thyrospheres in ATC cell lines [6]. Interestingly, Yasui et al. pointed out that ATC cell lines underwent EMT by overexpressing Snail, and this process was associated with a significant enhancement in the number of thyrospheres. They also discovered that Snail overexpression significantly enhanced spheres formation ability in ALDH− cells, but not in ALDH+ cells [201]. ALDH seems to be a reliable marker for thyroid CSCs and that thyrospheres are originated from thyroid CSCs [6]. Some studies using ATC models have uncovered an association between the stem-like features and chemotherapy resistance. Those data indicated that the multidrug transporters are involved in this refractoriness. Mato et al. were the first to present evidence that links the overexpression of the adult stem marker ABCG2/BCRP and EMT inducer genes in thyroid carcinomas. They identified a cell subpopulation in the PTC cell line (TPC1), which expresses the ABCG2/BCRP gene, and proposed that these cells may contribute in the development of PTC tumors. This cell subpopulation also showed faster migration and higher invasive ability than parental cell line in correlation with overexpression of the BIRC5 gene (an apoptosis inhibitor gene). It is of interest to note that the knocking down of Zeb1 led to the downregulation of ABCG2/BCRP, vimentin, N-cadherin, BIRC5 genes, re-expression of E-cadherin, and decreased cell migration. Their additional analysis of human thyroid carcinoma also showed that the enhanced expression of ABCG2/BCRP, Snail1, Twist1, and Zeb1 genes in PDTCs, ATCs, and PTCs correlates with the advanced forms of the diseases [192].
Concluding remarks and promising targeted therapy in thyroid cancer
Currently, a small number of patients with well-differentiated thyroid carcinoma as well as the most cases with PDTC and ATC that develop recurrent diseases and metastases are resistant to chemotherapy and radiation therapy [7]. Therefore, encouraging trials of novel and effective therapies for these diseases has attracted a lot of attention among researchers. Most of therapy-resistant thyroid cancer cells often exhibit an EMT phenotype. Understanding the molecular and cellular alterations of this transient state and its relationship with the microenvironment components is expected to identify potential targets to help to overcome the tumor recurrence and drug resistant phenotype [202]. Furthermore, in recent years, new treatment approaches have focused on the development of low-molecular-weight compounds targeting EMT-initiating factors and their signaling pathways. Point mutations and/or rearrangement of both serine/threonine kinases, such as tyrosine kinase receptors, such as EGFR, and RET, as well as signaling molecules acting downstream of kinase receptors, such as BRAF or RAS, are known to be involved in the initiation of EMT process. Therefore, considering these important receptors and molecules sheds light on an interesting research area to investigate effective and portent inhibitors for these targets. There are a number of protein kinase inhibitors, such as sorafenib and lenvatinib that have been developed and approved for patients with radioactive iodine-refractory thyroid cancers. In addition, the combination of different kinase inhibitors, such as BRAF inhibitor dabrafenib and the MEK1/2 inhibitor trametinib have been suggested as a therapy for recurrent thyroid cancers [203]. Wnt/β-catenin signaling is also implicated in EMT program of thyroid cancer and can serve as a good choice for thyroid cancer therapy. More interestingly, it has been reported that silencing of β-catenin decreases EMT markers in thyroid cancer [57]. The silencing of the C-Met/PI3K/AKT pathway has also been discovered to reverse EMT and metastasis of thyroid cancer cells [45].
Deregulation of miRNAs is also involved in tumorogenesis and invasiveness of thyroid cancers. Altering the cellular miRNA levels using miRNA mimics or anti-miRNAs appear to be used as adjuvant treatment modalities for thyroid cancer [132]. Restoration of tumor suppressor miRNAs miR-200, miR-30, and miR-144, or the inhibition of oncogenic miR-146, for instance, is expected to reverse the EMT phenotype [132, 134, 141]. Additional attention should also be given to the microenvironment, with a special emphasis on a hypoxic condition, which acts through HIF-1α stabilization and is a hallmark of cancer progression [203]. Combination therapeutic approaches targeting hypoxia-inducible proteins may provide novel therapeutic options [204].
In conclusion, multiple lines of evidences support the involvement of EMT in thyroid tumorigenesis. The clinical significance of the EMT involvement in cancer metastasis, drug resistance, the generation of CSCs, and tumor microenvironment is required to be fully unraveled. Indeed, the development of more detailed and comprehensive experiments is essential to find out different aspects of EMT process and its importance in the progression and metastatic spread of thyroid tumors.
References
G. Pellegriti, F. Frasca, C. Regalbuto, S. Squatrito, R. Vigneri, Worldwide increasing incidence of thyroid cancer: update on epidemiology and risk factors. J. Cancer Epidemiol. 2013, 965212 (2013). https://doi.org/10.1155/2013/965212
S. Rajabi, M.H. Dehghan, R. Dastmalchi, F. Jalali Mashayekhi, S. Salami, M. Hedayati, The roles and role-players in thyroid cancer angiogenesis. Endocr. J. 66(4), 277–293 (2019). https://doi.org/10.1507/endocrj.EJ18-0537
Y.J. Yang, H.J. Na, M.J. Suh, M.J. Ban, H.K. Byeon, W.S. Kim, J.W. Kim, E.C. Choi, H.J. Kwon, J.W. Chang, Y.W. Koh, Hypoxia induces epithelial–mesenchymal transition in follicular thyroid cancer: involvement of regulation of twist by hypoxia inducible factor-1alpha. Yonsei Med. J. 56(6), 1503–1514 (2015). https://doi.org/10.3349/ymj.2015.56.6.1503
S. Rajabi, M. Hedayati, Medullary thyroid cancer: clinical characteristics and new insights into therapeutic strategies targeting tyrosine kinases. Mol. Diagn. Ther. 21(6), 607–620 (2017). https://doi.org/10.1007/s40291-017-0289-5
H. Katoh, K. Yamashita, T. Enomoto, M. Watanabe, Classification and general considerations of thyroid cancer. Ann. Clin. Pathol. 3(1), 1045 (2015)
K.B. Heiden, A.J. Williamson, M.E. Doscas, J. Ye, Y. Wang, D. Liu, M. Xing, R.A. Prinz, X. Xu, The sonic hedgehog signaling pathway maintains the cancer stem cell self-renewal of anaplastic thyroid cancer by inducing snail expression. J. Clin. Endocrinol. Metab. 99(11), E2178–E2187 (2014). https://doi.org/10.1210/jc.2014-1844
Z. Guo, H. Hardin, R.V. Lloyd, Cancer stem-like cells and thyroid cancer. Endocr. Relat. Cancer 21(5), T285–T300 (2014). https://doi.org/10.1530/erc-14-0002
B. Han, H. Cui, L. Kang, X. Zhang, Z. Jin, L. Lu, Z. Fan, Metformin inhibits thyroid cancer cell growth, migration, and EMT through the mTOR pathway. Tumour Biol. 36(8), 6295–6304 (2015). https://doi.org/10.1007/s13277-015-3315-4
V. Vasko, A.V. Espinosa, W. Scouten, H. He, H. Auer, S. Liyanarachchi, A. Larin, V. Savchenko, G.L. Francis, A. de la Chapelle, M. Saji, M.D. Ringel, Gene expression and functional evidence of epithelial-to-mesenchymal transition in papillary thyroid carcinoma invasion. Proc. Natl Acad. Sci. USA 104(8), 2803–2808 (2007). https://doi.org/10.1073/pnas.0610733104
C. Puppin, D. Fabbro, M. Dima, C. Di Loreto, E. Puxeddu, S. Filetti, D. Russo, G. Damante, High periostin expression correlates with aggressiveness in papillary thyroid carcinomas. J. Endocrinol. 197(2), 401–408 (2008). https://doi.org/10.1677/joe-07-0618
P. Baquero, E. Jimenez-Mora, A. Santos, M. Lasa, A. Chiloeches, TGFbeta induces epithelial–mesenchymal transition of thyroid cancer cells by both the BRAF/MEK/ERK and Src/FAK pathways. Mol. Carcinog. 55(11), 1639–1654 (2016). https://doi.org/10.1002/mc.22415
C. Visciano, F. Liotti, N. Prevete, G. Cali, R. Franco, F. Collina, A. de Paulis, G. Marone, M. Santoro, R.M. Melillo, Mast cells induce epithelial-to-mesenchymal transition and stem cell features in human thyroid cancer cells through an IL-8-Akt-Slug pathway. Oncogene 34(40), 5175–5186 (2015). https://doi.org/10.1038/onc.2014.441
J. Jiang, Y.L. Tang, X.H. Liang, EMT: a new vision of hypoxia promoting cancer progression. Cancer Biol. Ther. 11(8), 714–723 (2011). https://doi.org/10.4161/cbt.11.8.15274
A. Voulgari, A. Pintzas, Epithelial–mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim. Biophys. Acta 1796(2), 75–90 (2009). https://doi.org/10.1016/j.bbcan.2009.03.002
J.P. Thiery, H. Acloque, R.Y. Huang, M.A. Nieto, Epithelial–mesenchymal transitions in development and disease. Cell 139(5), 871–890 (2009). https://doi.org/10.1016/j.cell.2009.11.007
X. Meng, D.H. Kong, N. Li, Z.H. Zong, B.Q. Liu, Z.X. Du, Y. Guan, L. Cao, H.Q. Wang, Knockdown of BAG3 induces epithelial–mesenchymal transition in thyroid cancer cells through ZEB1 activation. Cell Death Dis. 5, e1092 (2014). https://doi.org/10.1038/cddis.2014.32
S. Sarkar, G. Horn, K. Moulton, A. Oza, S. Byler, S. Kokolus, M. Longacre, Cancer development, progression, and therapy: an epigenetic overview. Int. J. Mol. Sci. 14(10), 21087–21113 (2013)
S. Al Saleh, L.H. Sharaf, Y.A. Luqmani, Signalling pathways involved in endocrine resistance in breast cancer and associations with epithelial to mesenchymal transition (Review). Int J. Oncol. 38(5), 1197–1217 (2011). https://doi.org/10.3892/ijo.2011.942
J. Huang, H. Li, G. Ren, Epithelial–mesenchymal transition and drug resistance in breast cancer (Review). Int J. Oncol. 47(3), 840–848 (2015). https://doi.org/10.3892/ijo.2015.3084
H. Skovierova, T. Okajcekova, J. Strnadel, E. Vidomanova, E. Halasova, Molecular regulation of epithelial-to-mesenchymal transition in tumorigenesis (Review). Int J. Mol. Med 41(3), 1187–1200 (2018). https://doi.org/10.3892/ijmm.2017.3320
P.W. Derksen, X. Liu, F. Saridin, H. van der Gulden, J. Zevenhoven, B. Evers, J.R. van Beijnum, A.W. Griffioen, J. Vink, P. Krimpenfort, J.L. Peterse, R.D. Cardiff, A. Berns, J. Jonkers, Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell 10(5), 437–449 (2006). https://doi.org/10.1016/j.ccr.2006.09.013
N. Bezdenezhnykh, N. Semesiuk, O. Lykhova, V. Zhylchuk, Y. Kudryavets, Impact of stromal cell components of tumor microenvironment on epithelial–mesenchymal transition in breast cancer cells. Exp. Oncol. 36(2), 72–78 (2014)
J. Heuberger, W. Birchmeier, Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2(2), a002915 (2010). https://doi.org/10.1101/cshperspect.a002915
S. Heerboth, G. Housman, M. Leary, M. Longacre, S. Byler, K. Lapinska, A. Willbanks, S. Sarkar, EMT and tumor metastasis. Clin. Transl. Med 4, 6 (2015). https://doi.org/10.1186/s40169-015-0048-3
S. Wang, S. Huang, Y.L. Sun, Epithelial–mesenchymal transition in pancreatic cancer: a review. Biomed Res. Int. 2017, 2646148 (2017). https://doi.org/10.1155/2017/2646148
G. Christofori, New signals from the invasive front. Nature 441(7092), 444–450 (2006). https://doi.org/10.1038/nature04872
K. Jensen, A. Patel, V. Hoperia, A. Larin, A. Bauer, V. Vasko, Dynamic changes in E-cadherin gene promoter methylation during metastatic progression in papillary thyroid cancer. Exp. Ther. Med. 1(3), 457–462 (2010)
M. Sponziello, F. Rosignolo, M. Celano, V. Maggisano, V. Pecce, R.F. De Rose, G.E. Lombardo, C. Durante, S. Filetti, G. Damante, D. Russo, S. Bulotta, Fibronectin-1 expression is increased in aggressive thyroid cancer and favors the migration and invasion of cancer cells. Mol. Cell Endocrinol. 431, 123–132 (2016). https://doi.org/10.1016/j.mce.2016.05.007
R. Ma, N. Minsky, S.A. Morshed, T.F. Davies, Stemness in human thyroid cancers and derived cell lines: the role of asymmetrically dividing cancer stem cells resistant to chemotherapy. J. Clin. Endocrinol. Metab. 99(3), E400–E409 (2014). https://doi.org/10.1210/jc.2013-3545
A.R. Garcia-Rendueles, J.S. Rodrigues, M.E. Garcia-Rendueles, M. Suarez-Farina, S. Perez-Romero, F. Barreiro, I. Bernabeu, J. Rodriguez-Garcia, L. Fugazzola, T. Sakai, F. Liu, J. Cameselle-Teijeiro, S.B. Bravo, C.V. Alvarez, Rewiring of the apoptotic TGF-beta-SMAD/NFkappaB pathway through an oncogenic function of p27 in human papillary thyroid cancer. Oncogene 36(5), 652–666 (2017). https://doi.org/10.1038/onc.2016.233
J. Zavadil, E.P. Bottinger, TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 24(37), 5764–5774 (2005). https://doi.org/10.1038/sj.onc.1208927
K. Ivanova, I. Manolova, M.M. Ignatova, M. Gulubova, Immunohistochemical expression of TGF-Beta1, SMAD4, SMAD7, TGFbetaRII and CD68-positive TAM densities in papillary thyroid cancer. Open Access Maced. J. Med. Sci. 6(3), 435–441 (2018). https://doi.org/10.3889/oamjms.2018.105
J.M. Cerutti, K.N. Ebina, S.E. Matsuo, L. Martins, R.M. Maciel, E.T. Kimura, Expression of Smad4 and Smad7 in human thyroid follicular carcinoma cell lines. J. Endocrinol. Investig. 26(6), 516–521 (2003). https://doi.org/10.1007/bf03345213
S. D’Inzeo, A. Nicolussi, C.F. Donini, M. Zani, P. Mancini, F. Nardi, A. Coppa, A novel human Smad4 mutation is involved in papillary thyroid carcinoma progression. Endocr. Relat. Cancer 19(1), 39–55 (2012). https://doi.org/10.1530/erc-11-0233
J.A. Knauf, M.A. Sartor, M. Medvedovic, E. Lundsmith, M. Ryder, M. Salzano, Y.E. Nikiforov, T.J. Giordano, R.A. Ghossein, J.A. Fagin, Progression of BRAF-induced thyroid cancer is associated with epithelial–mesenchymal transition requiring concomitant MAP kinase and TGFbeta signaling. Oncogene 30(28), 3153–3162 (2011). https://doi.org/10.1038/onc.2011.44
G. Riesco-Eizaguirre, I. Rodriguez, A. De la Vieja, E. Costamagna, N. Carrasco, M. Nistal, P. Santisteban, The BRAFV600E oncogene induces transforming growth factor beta secretion leading to sodium iodide symporter repression and increased malignancy in thyroid cancer. Cancer Res. 69(21), 8317–8325 (2009). https://doi.org/10.1158/0008-5472.can-09-1248
V. Anelli, J.A. Villefranc, S. Chhangawala, R. Martinez-McFaline, E. Riva, A. Nguyen, A. Verma, R. Bareja, Z. Chen, T. Scognamiglio, O. Elemento, Y. Houvras, Oncogenic BRAF disrupts thyroid morphogenesis and function via twist expression. Elife 6, e20728 (2017). https://doi.org/10.7554/eLife.20728
Q. Zhou, J. Chen, J. Feng, Y. Xu, W. Zheng, J. Wang, SOSTDC1 inhibits follicular thyroid cancer cell proliferation, migration, and EMT via suppressing PI3K/Akt and MAPK/Erk signaling pathways. Mol. Cell. Biochem. 435(1–2), 87–95 (2017). https://doi.org/10.1007/s11010-017-3059-0
M. Zou, E.Y. Baitei, H.A. BinEssa, F.A. Al-Mohanna, R.S. Parhar, R. St-Arnaud, S. Kimura, C. Pritchard, A.S. Alzahrani, A.M. Assiri, B.F. Meyer, Y. Shi, Cyp24a1 attenuation limits progression of Braf(V600E)-induced papillary thyroid cancer cells and sensitizes them to BRAF(V600E) inhibitor PLX4720. Cancer Res. 77(8), 2161–2172 (2017). https://doi.org/10.1158/0008-5472.can-16-2066
I. Palona, H. Namba, N. Mitsutake, D. Starenki, A. Podtcheko, I. Sedliarou, A. Ohtsuru, V. Saenko, Y. Nagayama, K. Umezawa, S. Yamashita, BRAFV600E promotes invasiveness of thyroid cancer cells through nuclear factor kappaB activation. Endocrinology 147(12), 5699–5707 (2006). https://doi.org/10.1210/en.2006-0400
N. Agrawal, R. Akbani, B.A. Aksoy, A. Ally et al. Integrated genomic characterization of papillary thyroid carcinoma. Cell 159(3), 676–690 (2014). https://doi.org/10.1016/j.cell.2014.09.050
N. Lv, Y. Gao, H. Guan, D. Wu, S. Ding, W. Teng, Z. Shan, Inflammatory mediators, tumor necrosis factor-alpha and interferon-gamma, induce EMT in human PTC cell lines. Oncol. Lett. 10(4), 2591–2597 (2015). https://doi.org/10.3892/ol.2015.3518
K.S. Choudhary, N. Rohatgi, S. Halldorsson, E. Briem, T. Gudjonsson, S. Gudmundsson, O. Rolfsson, EGFR signal-network reconstruction demonstrates metabolic crosstalk in EMT. PLoS Comput. Biol. 12(6), e1004924 (2016). https://doi.org/10.1371/journal.pcbi.1004924
L.H. Chin, S.P. Hsu, W.B. Zhong, Y.C. Liang, Involvement of cysteine-rich protein 61 in the epidermal growth factor-induced migration of human anaplastic thyroid cancer cells. Mol. Carcinog. 55(5), 622–632 (2016). https://doi.org/10.1002/mc.22308
W. Gao, J. Han, Overexpression of ING5 inhibits HGF-induced proliferation, invasion and EMT in thyroid cancer cells via regulation of the c-Met/PI3K/Akt signaling pathway. Biomed. Pharmacother. 98, 265–270 (2018). https://doi.org/10.1016/j.biopha.2017.12.045
J. Xu, W. Lu, S. Zhang, C. Zhu, T. Ren, T. Zhu, H. Zhao, Y. Liu, J. Su, Overexpression of DDR2 contributes to cell invasion and migration in head and neck squamous cell carcinoma. Cancer Biol. Ther. 15(5), 612–622 (2014). https://doi.org/10.4161/cbt.28181
K. Zhang, C.A. Corsa, S.M. Ponik, J.L. Prior, D. Piwnica-Worms, K.W. Eliceiri, P.J. Keely, G.D. Longmore, The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat. Cell Biol. 15(6), 677–687 (2013). https://doi.org/10.1038/ncb2743
B. Xie, W. Lin, J. Ye, X. Wang, B. Zhang, S. Xiong, H. Li, G. Tan, DDR2 facilitates hepatocellular carcinoma invasion and metastasis via activating ERK signaling and stabilizing SNAIL1. J. Exp. Clin. Cancer Res. 34, 101 (2015). https://doi.org/10.1186/s13046-015-0218-6
Z. Liang, W.J. Xie, M. Zhao, G.P. Cheng, M.J. Wu, DDR2 facilitates papillary thyroid carcinoma epithelial mesenchymal transition by activating ERK2/Snail1 pathway. Oncol. Lett. 14(6), 8114–8121 (2017). https://doi.org/10.3892/ol.2017.7250
C.W. Jung, J.S. Kong, H. Seol, S. Park, J.S. Koh, S.S. Lee, M.J. Kim, I.J. Choi, J.K. Myung, Expression of activated Notch1 and Hey1 in papillary thyroid carcinoma. Histopathology 70(2), 301–308 (2017). https://doi.org/10.1111/his.13065
E. Ferretti, E. Tosi, A. Po, A. Scipioni, R. Morisi, M.S. Espinola, D. Russo, C. Durante, M. Schlumberger, I. Screpanti, S. Filetti, A. Gulino, Notch signaling is involved in expression of thyrocyte differentiation markers and is down-regulated in thyroid tumors. J. Clin. Endocrinol. Metab. 93(10), 4080–4087 (2008). https://doi.org/10.1210/jc.2008-0528
H.S. Park, C.K. Jung, S.H. Lee, B.J. Chae, D.J. Lim, W.C. Park, B.J. Song, J.S. Kim, S.S. Jung, J.S. Bae, Notch1 receptor as a marker of lymph node metastases in papillary thyroid cancer. Cancer Sci. 103(2), 305–309 (2012). https://doi.org/10.1111/j.1349-7006.2011.02161.x
M. Zhang, Y. Qin, B. Zuo, W. Gong, S. Zhang, Y. Gong, Z. Quan, B. Chu, Overexpression of NOTCH-regulated Ankyrin Repeat Protein is associated with papillary thyroid carcinoma progression. PLoS ONE 12(2), e0167782 (2017). https://doi.org/10.1371/journal.pone.0167782
J. Zhang, Y. Wang, D. Li, S. Jing, Notch and TGF-beta/Smad3 pathways are involved in the interaction between cancer cells and cancer-associated fibroblasts in papillary thyroid carcinoma. Tumour Biol. 35(1), 379–385 (2014). https://doi.org/10.1007/s13277-013-1053-z
L.L. Chen, G.X. Gao, F.X. Shen, X. Chen, X.H. Gong, W.J. Wu, SDC4 gene silencing favors human papillary thyroid carcinoma cell apoptosis and inhibits epithelial mesenchymal transition via Wnt/beta-catenin pathway. Mol. Cells 41(9), 853–867 (2018). https://doi.org/10.14348/molcells.2018.0103
P. Wend, J.D. Holland, U. Ziebold, W. Birchmeier, Wnt signaling in stem and cancer stem cells. Semin. Cell Dev. Biol. 21(8), 855–863 (2010). https://doi.org/10.1016/j.semcdb.2010.09.004
H. Hardin, R. Zhang, H. Helein, D. Buehler, Z. Guo, R.V. Lloyd, The evolving concept of cancer stem-like cells in thyroid cancer and other solid tumors. Lab. Investig. 97(10), 1142–1151 (2017). https://doi.org/10.1038/labinvest.2017.41
K. Ishigaki, H. Namba, M. Nakashima, T. Nakayama, N. Mitsutake, T. Hayashi, S. Maeda, M. Ichinose, T. Kanematsu, S. Yamashita, Aberrant localization of beta-catenin correlates with overexpression of its target gene in human papillary thyroid cancer. J. Clin. Endocrinol. Metab. 87(7), 3433–3440 (2002). https://doi.org/10.1210/jcem.87.7.8648
A. Sastre-Perona, G. Riesco-Eizaguirre, M.A. Zaballos, P. Santisteban, beta-catenin signaling is required for RAS-driven thyroid cancer through PI3K activation. Oncotarget 7(31), 49435–49449 (2016). https://doi.org/10.18632/oncotarget.10356
D. Wen, T. Liao, B. Ma, N. Qu, R.L. Shi, Z.W. Lu, Y.L. Wang, W.J. Wei, Q.H. Ji, Downregulation of CSN 6 attenuates papillary thyroid carcinoma progression by reducing Wnt/β‐catenin signaling and sensitizes cancer cells to FH 535 therapy. Cancer Med. 7(2), 285–296 (2018)
H. Guan, W. Liang, Z. Xie, H. Li, J. Liu, L. Liu, L. Xiu, Y. Li, Down-regulation of miR-144 promotes thyroid cancer cell invasion by targeting ZEB1 and ZEB2. Endocrine 48(2), 566–574 (2015). https://doi.org/10.1007/s12020-014-0326-7
Q. Qin, Y. Xu, T. He, C. Qin, J. Xu, Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 22(1), 90–106 (2012). https://doi.org/10.1038/cr.2011.144
S. Ansieau, A.P. Morel, G. Hinkal, J. Bastid, A. Puisieux, TWISTing an embryonic transcription factor into an oncoprotein. Oncogene 29(22), 3173–3184 (2010). https://doi.org/10.1038/onc.2010.92
J.M. Schmidt, E. Panzilius, H.S. Bartsch, M. Irmler, J. Beckers, V. Kari, J.R. Linnemann, D. Dragoi, B. Hirschi, U.J. Kloos, S. Sass, F. Theis, S. Kahlert, S.A. Johnsen, K. Sotlar, C.H. Scheel, Stem-cell-like properties and epithelial plasticity arise as stable traits after transient Twist1 activation. Cell Rep. 10(2), 131–139 (2015). https://doi.org/10.1016/j.celrep.2014.12.032
R. Maestro, A.P. Dei Tos, Y. Hamamori, S. Krasnokutsky, V. Sartorelli, L. Kedes, C. Doglioni, D.H. Beach, G.J. Hannon, Twist is a potential oncogene that inhibits apoptosis. Genes Dev. 13(17), 2207–2217 (1999). https://doi.org/10.1101/gad.13.17.2207
X. Wang, M.T. Ling, X.Y. Guan, S.W. Tsao, H.W. Cheung, D.T. Lee, Y.C. Wong, Identification of a novel function of TWIST, a bHLH protein, in the development of acquired taxol resistance in human cancer cells. Oncogene 23(2), 474–482 (2004). https://doi.org/10.1038/sj.onc.1207128
H. Zheng, Y. Kang, Multilayer control of the EMT master regulators. Oncogene 33(14), 1755–1763 (2014). https://doi.org/10.1038/onc.2013.128
E. Sanchez-Tillo, Y. Liu, O. de Barrios, L. Siles, L. Fanlo, M. Cuatrecasas, D.S. Darling, D.C. Dean, A. Castells, A. Postigo, EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell. Mol. Life Sci. 69(20), 3429–3456 (2012). https://doi.org/10.1007/s00018-012-1122-2
G. Di Maro, F.M. Orlandella, T.C. Bencivenga, P. Salerno, C. Ugolini, F. Basolo, R. Maestro, G. Salvatore, Identification of targets of Twist1 transcription factor in thyroid cancer cells. J. Clin. Endocrinol. Metab. 99(9), E1617–E1626 (2014). https://doi.org/10.1210/jc.2013-3799
D. Buehler, H. Hardin, W. Shan, C. Montemayor-Garcia, P.S. Rush, S. Asioli, H. Chen, R.V. Lloyd, Expression of epithelial–mesenchymal transition regulators SNAI2 and TWIST1 in thyroid carcinomas. Mod. Pathol. 26(1), 54–61 (2013). https://doi.org/10.1038/modpathol.2012.137
P. Salerno, G. Garcia-Rostan, S. Piccinin, T.C. Bencivenga, G. Di Maro, C. Doglioni, F. Basolo, R. Maestro, A. Fusco, M. Santoro, G. Salvatore, TWIST1 plays a pleiotropic role in determining the anaplastic thyroid cancer phenotype. J. Clin. Endocrinol. Metab. 96(5), E772–E781 (2011). https://doi.org/10.1210/jc.2010-1182
R.G. Hardy, C. Vicente-Duenas, I. Gonzalez-Herrero, C. Anderson, T. Flores, S. Hughes, C. Tselepis, J.A. Ross, I. Sanchez-Garcia, Snail family transcription factors are implicated in thyroid carcinogenesis. Am. J. Pathol. 171(3), 1037–1046 (2007). https://doi.org/10.2353/ajpath.2007.061211
J. Wu, Y. Zhang, R. Cheng, W. Gong, T. Ding, Q. Zhai, Y. Wang, B. Meng, B. Sun, Expression of epithelial–mesenchymal transition regulators TWIST, SLUG and SNAIL in follicular thyroid tumours may relate to widely invasive, poorly differentiated and distant metastasis. Histopathology 74(5), 780–791 (2019). https://doi.org/10.1111/his.13778
H. Son, A. Moon, Epithelial–mesenchymal Transition and Cell Invasion. Toxicol. Res 26(4), 245–252 (2010). https://doi.org/10.5487/tr.2010.26.4.245
A. Cano, M.A. Perez-Moreno, I. Rodrigo, A. Locascio, M.J. Blanco, M.G. del Barrio, F. Portillo, M.A. Nieto, The transcription factor snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2(2), 76–83 (2000). https://doi.org/10.1038/35000025
W. Sun, S. Yang, Q. Ma, S. Zheng, J. Wang, D. Chen, K. Zhang, SLUG promotes invasion and metastasis of anaplastic thyroid cancer cells through repression of E-cadherin. Int. J. Clin. Exp. Pathol. 9(8), 8373–8379 (2016)
J. Huang, L. Guo, Knockdown of SOX9 inhibits the proliferation, invasion, and EMT in thyroid cancer cells. Oncol. Res. 25(2), 167–176 (2017). https://doi.org/10.3727/096504016x14732772150307
D.F. Niu, T. Kondo, T. Nakazawa, N. Oishi, T. Kawasaki, K. Mochizuki, T. Yamane, R. Katoh, Transcription factor Runx2 is a regulator of epithelial–mesenchymal transition and invasion in thyroid carcinomas. Lab. Investig. 92(8), 1181–1190 (2012). https://doi.org/10.1038/labinvest.2012.84
J. Akech, J.J. Wixted, K. Bedard, M. van der Deen, S. Hussain, T.A. Guise, A.J. van Wijnen, J.L. Stein, L.R. Languino, D.C. Altieri, J. Pratap, E. Keller, G.S. Stein, J.B. Lian, Runx2 association with progression of prostate cancer in patients: mechanisms mediating bone osteolysis and osteoblastic metastatic lesions. Oncogene 29(6), 811–821 (2010). https://doi.org/10.1038/onc.2009.389
S.K. Baniwal, O. Khalid, Y. Gabet, R.R. Shah, D.J. Purcell, D. Mav, A.E. Kohn-Gabet, Y. Shi, G.A. Coetzee, B. Frenkel, Runx2 transcriptome of prostate cancer cells: insights into invasiveness and bone metastasis. Mol. Cancer 9, 258 (2010). https://doi.org/10.1186/1476-4598-9-258
H.F. Yuen, W.K. Kwok, K.K. Chan, C.W. Chua, Y.P. Chan, Y.Y. Chu, Y.C. Wong, X. Wang, K.W. Chan, TWIST modulates prostate cancer cell-mediated bone cell activity and is upregulated by osteogenic induction. Carcinogenesis 29(8), 1509–1518 (2008). https://doi.org/10.1093/carcin/bgn105
J.H. Yan, C.L. Zhao, L.B. Ding, X. Zhou, FOXD3 suppresses tumor growth and angiogenesis in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 466(1), 111–116 (2015). https://doi.org/10.1016/j.bbrc.2015.08.116
H. Zhao, D. Chen, J. Wang, Y. Yin, Q. Gao, Y. Zhang, Downregulation of the transcription factor, FoxD3, is associated with lymph node metastases in invasive ductal carcinomas of the breast. Int. J. Clin. Exp. Pathol. 7(2), 670–676 (2014)
H. Yin, T. Meng, L. Zhou, F. Zhao, X. Li, Y. Li, M. Hu, H. Chen, D. Song, FOXD3 regulates anaplastic thyroid cancer progression. Oncotarget 8(20), 33644–33651 (2017). https://doi.org/10.18632/oncotarget.16853
N. Wang, Y. Li, J. Wei, J. Pu, R. Liu, Q. Yang, H. Guan, B. Shi, P. Hou, M. Ji, TBX1 functions as a tumor suppressor in thyroid cancer through inhibiting the activities of the PI3K/AKT and MAPK/ERK pathways. Thyroid 29(3), 378–394 (2019). https://doi.org/10.1089/thy.2018.0312
G. Berx, F. van Roy, Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb. Perspect. Biol. 1(6), a003129 (2009). https://doi.org/10.1101/cshperspect.a003129
P. Baquero, I. Sánchez-Hernández, E. Jiménez-Mora, J.L. Orgaz, B. Jiménez, A. Chiloeches, V600EBRAF promotes invasiveness of thyroid cancer cells by decreasing E-cadherin expression through a Snail-dependent mechanism. Cancer Lett. 335(1), 232–241 (2013). https://doi.org/10.1016/j.canlet.2013.02.033
S.M. Wiseman, O.L. Griffith, S. Deen, A. Rajput, H. Masoudi, B. Gilks, L. Goldstein, A. Gown, S.J. Jones, Identification of molecular markers altered during transformation of differentiated into anaplastic thyroid carcinoma. Arch. Surg. 142(8), 717–729 (2007). https://doi.org/10.1001/archsurg.142.8.717.
C.W. Jung, K.H. Han, H. Seol, S. Park, J.S. Koh, S.S. Lee, M.J. Kim, I.J. Choi, J.K. Myung, Expression of cancer stem cell markers and epithelial–mesenchymal transition-related factors in anaplastic thyroid carcinoma. Int. J. Clin. Exp. Pathol. 8(1), 560–568 (2015)
B.T. Beaty, J. Condeelis, Digging a little deeper: the stages of invadopodium formation and maturation. Eur. J. Cell Biol. 93(10–12), 438–444 (2014). https://doi.org/10.1016/j.ejcb.2014.07.003
R. Yan, T. Yang, H. Zhai, Z. Zhou, L. Gao, Y. Li, MicroRNA-150-5p affects cell proliferation, apoptosis, and EMT by regulation of the BRAF(V600E) mutation in papillary thyroid cancer cells. J. Cell. Biochem. 119(11), 8763–8772 (2018). https://doi.org/10.1002/jcb.27108
A. Kudo, I. Kii, Periostin function in communication with extracellular matrices. J. Cell Commun. Signal. 12(1), 301–308 (2018). https://doi.org/10.1007/s12079-017-0422-6
L. Morra, H. Moch, Periostin expression and epithelial–mesenchymal transition in cancer: a review and an update. Virchows Arch. 459(5), 465–475 (2011). https://doi.org/10.1007/s00428-011-1151-5
C. Puppin, N. Passon, F. Frasca, R. Vigneri, F. Tomay, S. Tomaciello, G. Damante, In thyroid cancer cell lines expression of periostin gene is controlled by p73 and is not related to epigenetic marks of active transcription. Cell. Oncol. 34(2), 131–140 (2011). https://doi.org/10.1007/s13402-011-0009-9
H. Zhong, X. Li, J. Zhang, X. Wu, Overexpression of periostin is positively associated with gastric cancer metastasis through promoting tumor metastasis and invasion. J. Cell. Biochem. 120(6), 9927–9935 (2019). https://doi.org/10.1002/jcb.28275
B. Yu, K. Wu, X. Wang, J. Zhang, L. Wang, Y. Jiang, X. Zhu, W. Chen, M. Yan, Periostin secreted by cancer-associated fibroblasts promotes cancer stemness in head and neck cancer by activating protein tyrosine kinase 7. Cell Death Dis. 9(11), 1082 (2018). https://doi.org/10.1038/s41419-018-1116-6
Y. Liu, F. Li, F. Gao, L. Xing, P. Qin, X. Liang, J. Zhang, X. Qiao, L. Lin, Q. Zhao, L. Du, Role of microenvironmental periostin in pancreatic cancer progression. Oncotarget 8(52), 89552–89565 (2017). https://doi.org/10.18632/oncotarget.11533
I. Vardaki, S. Ceder, D. Rutishauser, G. Baltatzis, T. Foukakis, T. Panaretakis, Periostin is identified as a putative metastatic marker in breast cancer-derived exosomes. Oncotarget 7(46), 74966–74978 (2016). https://doi.org/10.18632/oncotarget.11663
Z.M. Xiao, X.Y. Wang, A.M. Wang, Periostin induces chemoresistance in colon cancer cells through activation of the PI3K/Akt/survivin pathway. Biotechnol. Appl. Biochem. 62(3), 401–406 (2015). https://doi.org/10.1002/bab.1193
S. Xia, C. Wang, E.L. Postma, Y. Yang, X. Ni, W. Zhan, Fibronectin 1 promotes migration and invasion of papillary thyroid cancer and predicts papillary thyroid cancer lymph node metastasis. Onco Targets Ther. 10, 1743–1755 (2017). https://doi.org/10.2147/ott.s122009
G.S. Mack, A. Marshall, Lost in migration. Nat. Biotechnol. 28(3), 214–229 (2010). https://doi.org/10.1038/nbt0310-214
Y.F. Ji, H. Huang, F. Jiang, R.Z. Ni, M.B. Xiao, S100 family signaling network and related proteins in pancreatic cancer (Review). Int J. Mol. Med. 33(4), 769–776 (2014). https://doi.org/10.3892/ijmm.2014.1633
A. Azimi, M. Pernemalm, M. Frostvik Stolt, J. Hansson, J. Lehtio, S. Egyhazi Brage, C. Hertzman Johansson, Proteomics analysis of melanoma metastases: association between S100A13 expression and chemotherapy resistance. Br. J. Cancer 110(10), 2489–2495 (2014). https://doi.org/10.1038/bjc.2014.169
A. Pierce, N. Barron, R. Linehan, E. Ryan, L. O'Driscoll, C. Daly, M. Clynes, Identification of a novel, functional role for S100A13 in invasive lung cancer cell lines. Eur. J. Cancer 44(1), 151–159 (2008). https://doi.org/10.1016/j.ejca.2007.10.017
J. Zhong, C. Liu, Y.J. Chen, Q.H. Zhang, J. Yang, X. Kang, S.R. Chen, G.B. Wen, X.Y. Zu, R.X. Cao, The association between S100A13 and HMGA1 in the modulation of thyroid cancer proliferation and invasion. J. Transl. Med. 14, 80 (2016). https://doi.org/10.1186/s12967-016-0824-x
J. Martinez-Aguilar, R. Clifton-Bligh, M.P. Molloy, A multiplexed, targeted mass spectrometry assay of the S100 protein family uncovers the isoform-specific expression in thyroid tumours. BMC Cancer 15, 199 (2015). https://doi.org/10.1186/s12885-015-1217-x
D. Su, Y. Liu, T. Song, Knockdown of IQGAP1 inhibits proliferation and epithelial–mesenchymal transition by Wnt/beta-catenin pathway in thyroid cancer. Onco Targets Ther. 10, 1549–1559 (2017). https://doi.org/10.2147/ott.s128564
X.X. Wang, K. Wang, X.Z. Li, L.Q. Zhai, C.X. Qu, Y. Zhao, Z.R. Liu, H.Z. Wang, Q.J. An, L.W. Jing, X.H. Wang, Targeted knockdown of IQGAP1 inhibits the progression of esophageal squamous cell carcinoma in vitro and in vivo. PLoS ONE 9(5), e96501 (2014). https://doi.org/10.1371/journal.pone.0096501
X.X. Wang, X.Z. Li, L.Q. Zhai, Z.R. Liu, X.J. Chen, Y. Pei, Overexpression of IQGAP1 in human pancreatic cancer. Hepatobiliary Pancreat. Dis. Int. 12(5), 540–545 (2013)
H. Hayashi, K. Nabeshima, M. Aoki, M. Hamasaki, S. Enatsu, Y. Yamauchi, Y. Yamashita, H. Iwasaki, Overexpression of IQGAP1 in advanced colorectal cancer correlates with poor prognosis-critical role in tumor invasion. Int. J. Cancer 126(11), 2563–2574 (2010). https://doi.org/10.1002/ijc.24987
A. Walch, S. Seidl, C. Hermannstadter, S. Rauser, J. Deplazes, R. Langer, C.H. von Weyhern, M. Sarbia, R. Busch, M. Feith, S. Gillen, H. Hofler, B. Luber, Combined analysis of Rac1, IQGAP1, Tiam1 and E-cadherin expression in gastric cancer. Mod. Pathol. 21(5), 544–552 (2008). https://doi.org/10.1038/modpathol.2008.3
M.W. Klymkowsky, P. Savagner, Epithelial–mesenchymal transition: a cancer researcher's conceptual friend and foe. Am. J. Pathol. 174(5), 1588–1593 (2009). https://doi.org/10.2353/ajpath.2009.080545
D. Bazzoun, S. Lelievre, R. Talhouk, Polarity proteins as regulators of cell junction complexes: implications for breast cancer. Pharmacol. Ther. 138(3), 418–427 (2013). https://doi.org/10.1016/j.pharmthera.2013.02.004
S. Fan, T.W. Hurd, C.J. Liu, S.W. Straight, T. Weimbs, E.A. Hurd, S.E. Domino, B. Margolis, Polarity proteins control ciliogenesis via kinesin motor interactions. Curr. Biol. 14(16), 1451–1461 (2004). https://doi.org/10.1016/j.cub.2004.08.025
Y.-Y. Shan, S.-L. Li, KIF3a inhibits TGF-β1-induced epithelial–mesenchymal transition in lung cancer cells. Int. J. Clin. Exp. Med. 9(2), 2528–2534 (2016).
Z. Liu, R.E. Rebowe, Z. Wang, Y. Li, Z. Wang, J.S. DePaolo, J. Guo, C. Qian, W. Liu, KIF3a promotes proliferation and invasion via Wnt signaling in advanced prostate cancer. Mol. Cancer Res. 12(4), 491–503 (2014)
S.-C. Wang, Z. Hu, D.-S. Chai, C.-B. Chen, Z.-Y. Wang, L. Wang, Knockdown of KIF3a inhibits hypoxia-induced epithelial-to-mesenchymal transition via suppression of the Wnt/β-catenin pathway in thyroid cancer. Int. J. Clin. Exp. Pathol. 9(2), 1014–1021 (2016)
P. Antonietti, B. Linder, S. Hehlgans, I.C. Mildenberger, M.C. Burger, S. Fulda, J.P. Steinbach, F. Gessler, F. Rodel, M. Mittelbronn, D. Kogel, Interference with the HSF1/HSP70/BAG3 pathway primes glioma cells to matrix detachment and BH3 mimetic-induced apoptosis. Mol. Cancer Ther. 16(1), 156–168 (2017). https://doi.org/10.1158/1535-7163.mct-16-0262
C.K. Das, B. Linder, F. Bonn, F. Rothweiler, I. Dikic, M. Michaelis, J. Cinatl, M. Mandal, D. Kogel, BAG3 overexpression and cytoprotective autophagy mediate apoptosis resistance in chemoresistant breast cancer cells. Neoplasia 20(3), 263–279 (2018). https://doi.org/10.1016/j.neo.2018.01.001
J. Zhong, C. Liu, Q.H. Zhang, L. Chen, Y.Y. Shen, Y.J. Chen, X. Zeng, X.Y. Zu, R.X. Cao, TGF-beta1 induces HMGA1 expression: the role of HMGA1 in thyroid cancer proliferation and invasion. Int. J. Oncol. 50(5), 1567–1578 (2017). https://doi.org/10.3892/ijo.2017.3958
S.N. Shah, L. Cope, W. Poh, A. Belton, S. Roy, C.C. Talbot Jr., S. Sukumar, D.L. Huso, L.M. Resar, HMGA1: a master regulator of tumor progression in triple-negative breast cancer cells. PLoS ONE 8(5), e63419 (2013). https://doi.org/10.1371/journal.pone.0063419
N. Abe, T. Watanabe, T. Masaki, T. Mori, M. Sugiyama, H. Uchimura, Y. Fujioka, G. Chiappetta, A. Fusco, Y. Atomi, Pancreatic duct cell carcinomas express high levels of high mobility group I(Y) proteins. Cancer Res. 60(12), 3117–3122 (2000)
B. Meyer, S. Loeschke, A. Schultze, T. Weigel, M. Sandkamp, T. Goldmann, E. Vollmer, J. Bullerdiek, HMGA2 overexpression in non-small cell lung cancer. Mol. Carcinog. 46(7), 503–511 (2007). https://doi.org/10.1002/mc.20235
A. Belton, A. Gabrovsky, Y.K. Bae, R. Reeves, C. Iacobuzio-Donahue, D.L. Huso, L.M. Resar, HMGA1 induces intestinal polyposis in transgenic mice and drives tumor progression and stem cell properties in colon cancer cells. PLoS ONE 7(1), e30034 (2012). https://doi.org/10.1371/journal.pone.0030034
J. Roche, R.M. Gemmill, H.A. Drabkin, Epigenetic regulation of the epithelial to mesenchymal transition in lung cancer. Cancers 9(7) (2017). https://doi.org/10.3390/cancers9070072
J.W. Wei, K. Huang, C. Yang, C.S. Kang, Non-coding RNAs as regulators in epigenetics (Review). Oncol. Rep. 37(1), 3–9 (2017). https://doi.org/10.3892/or.2016.5236
J.Y. Lee, G. Kong, Roles and epigenetic regulation of epithelial–mesenchymal transition and its transcription factors in cancer initiation and progression. Cell. Mol. Life Sci. 73(24), 4643–4660 (2016). https://doi.org/10.1007/s00018-016-2313-z
T. Sasanakietkul, T.D. Murtha, M. Javid, R. Korah, T. Carling, Epigenetic modifications in poorly differentiated and anaplastic thyroid cancer. Mol. Cell. Endocrinol. 469, 23–37 (2018). https://doi.org/10.1016/j.mce.2017.05.022
D. Vu-Phan, R.J. Koenig, Genetics and epigenetics of sporadic thyroid cancer. Mol. Cell. Endocrinol. 386(1–2), 55–66 (2014). https://doi.org/10.1016/j.mce.2013.07.030
D.P. Bartel, MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116(2), 281–297 (2004)
T. Wang, H. Xu, M. Qi, S. Yan, X. Tian, miRNA dysregulation and the risk of metastasis and invasion in papillary thyroid cancer: a systematic review and meta-analysis. Oncotarget 9(4), 5473–5479 (2018). https://doi.org/10.18632/oncotarget.16681
C.S. Fuziwara, E.T. Kimura, MicroRNA deregulation in anaplastic thyroid cancer biology. Int. J. Endocrinol. 2014, 743450 (2014). https://doi.org/10.1155/2014/743450
P. Xu, Y. Zhu, B. Sun, Z. Xiao, Colorectal cancer characterization and therapeutic target prediction based on microRNA expression profile. Sci. Rep. 6, 20616 (2016). https://doi.org/10.1038/srep20616
W. Sun, X. Lan, Z. Wang, W. Dong, L. He, T. Zhang, P. Zhang, H. Zhang, MicroRNA-144 inhibits proliferation by targeting WW domain-containing transcription regulator protein 1 in papillary thyroid cancer. Oncol. Lett. 15(1), 1007–1013 (2018). https://doi.org/10.3892/ol.2017.7440
F. Marini, E. Luzi, M.L. Brandi, MicroRNA role in thyroid cancer development. J. Thyroid Res. 2011, 407123 (2011). https://doi.org/10.4061/2011/407123
C.L. Bi, Y.Q. Zhang, B. Li, M. Guo, Y.L. Fu, MicroRNA-520a-3p suppresses epithelial–mesenchymal transition, invasion, and migration of papillary thyroid carcinoma cells via the JAK1-mediated JAK/STAT signaling pathway. J. Cell. Physiol. 234(4), 4054–4067 (2019). https://doi.org/10.1002/jcp.27199
R. Xiao, C. Li, B. Chai, miRNA-144 suppresses proliferation and migration of colorectal cancer cells through GSPT1. Biomed. Pharmacother. 74, 138–144 (2015). https://doi.org/10.1016/j.biopha.2015.08.006
S. Chen, P. Li, J. Li, Y. Wang, Y. Du, X. Chen, W. Zang, H. Wang, H. Chu, G. Zhao, G. Zhang, MiR-144 inhibits proliferation and induces apoptosis and autophagy in lung cancer cells by targeting TIGAR. Cell. Physiol. Biochem. 35(3), 997–1007 (2015). https://doi.org/10.1159/000369755
Y. Guo, L. Ying, Y. Tian, P. Yang, Y. Zhu, Z. Wang, F. Qiu, J. Lin, miR-144 downregulation increases bladder cancer cell proliferation by targeting EZH2 and regulating Wnt signaling. FEBS J. 280(18), 4531–4538 (2013). https://doi.org/10.1111/febs.12417
J. Sun, R. Shi, S. Zhao, X. Li, S. Lu, H. Bu, X. Ma, C. Su, E2F8, a direct target of miR-144, promotes papillary thyroid cancer progression via regulating cell cycle. J. Exp. Clin. Cancer Res. 36(1), 40 (2017). https://doi.org/10.1186/s13046-017-0504-6
F. Pacifico, E. Crescenzi, S. Mellone, A. Iannetti, N. Porrino, D. Liguoro, F. Moscato, M. Grieco, S. Formisano, A. Leonardi, Nuclear factor-{kappa}B contributes to anaplastic thyroid carcinomas through up-regulation of miR-146a. J. Clin. Endocrinol. Metab. 95(3), 1421–1430 (2010). https://doi.org/10.1210/jc.2009-1128
X. Deng, B. Wu, K. Xiao, J. Kang, J. Xie, X. Zhang, Y. Fan, MiR-146b-5p promotes metastasis and induces epithelial–mesenchymal transition in thyroid cancer by targeting ZNRF3. Cell. Physiol. Biochem. 35(1), 71–82 (2015). https://doi.org/10.1159/000369676
J. Ramírez-Moya, L. Wert-Lamas, P. Santisteban, MicroRNA-146b promotes PI3K/AKT pathway hyperactivation and thyroid cancer progression by targeting PTEN. Oncogene 37(25), 3369–3383 (2018). https://doi.org/10.1038/s41388-017-0088-9
F. An, M. Xia, Q. Zhan, X. Wu, F. Wu, Sa1797 body fluid miRNA profile of cholangiocarcinoma identifies miR-150-5p as a tumor suppressor gene involved in tumor invasion and metastasis. Gastroenterology 150(4), S368–S369 (2016)
J.C. Santos, M.T. Brianti, V.R. Almeida, M.M. Ortega, W. Fischer, R. Haas, A. Matheu, M.L. Ribeiro, Helicobacter pylori infection modulates the expression of miRNAs associated with DNA mismatch repair pathway. Mol. Carcinog. 56(4), 1372–1379 (2017). https://doi.org/10.1002/mc.22590
L. Xue, D. Su, D. Li, W. Gao, R. Yuan, W. Pang, MiR-200 regulates epithelial–mesenchymal transition in anaplastic thyroid cancer via EGF/EGFR signaling. Cell Biochem. Biophys. 72(1), 185–190 (2015). https://doi.org/10.1007/s12013-014-0435-1
C.J. Chang, C.H. Chao, W. Xia, J.Y. Yang, Y. Xiong, C.W. Li, W.H. Yu, S.K. Rehman, J.L. Hsu, H.H. Lee, M. Liu, C.T. Chen, D. Yu, M.C. Hung, p53 regulates epithelial–mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell Biol. 13(3), 317–323 (2011). https://doi.org/10.1038/ncb2173
Y.Y. Park, S.B. Kim, H.D. Han, B.H. Sohn, J.H. Kim, J. Liang, Y. Lu, C. Rodriguez-Aguayo, G. Lopez-Berestein, G.B. Mills, A.K. Sood, J.S. Lee, Tat-activating regulatory DNA-binding protein regulates glycolysis in hepatocellular carcinoma by regulating the platelet isoform of phosphofructokinase through microRNA 520. Hepatology 58(1), 182–191 (2013). https://doi.org/10.1002/hep.26310
I. Keklikoglou, C. Koerner, C. Schmidt, J.D. Zhang, D. Heckmann, A. Shavinskaya, H. Allgayer, B. Guckel, T. Fehm, A. Schneeweiss, O. Sahin, S. Wiemann, U. Tschulena, MicroRNA-520/373 family functions as a tumor suppressor in estrogen receptor negative breast cancer by targeting NF-kappaB and TGF-beta signaling pathways. Oncogene 31(37), 4150–4163 (2012). https://doi.org/10.1038/onc.2011.571
X.J. He, Y.Y. Ma, S. Yu, X.T. Jiang, Y.D. Lu, L. Tao, H.P. Wang, Z.M. Hu, H.Q. Tao, Up-regulated miR-199a-5p in gastric cancer functions as an oncogene and targets klotho. BMC Cancer 14, 218 (2014). https://doi.org/10.1186/1471-2407-14-218
J. Chen, V.Y. Shin, M.T. Siu, J.C. Ho, I. Cheuk, A. Kwong, miR-199a-5p confers tumor-suppressive role in triple-negative breast cancer. BMC Cancer 16(1), 887 (2016). https://doi.org/10.1186/s12885-016-2916-7
Y. Hu, J. Liu, B. Jiang, J. Chen, Z. Fu, F. Bai, J. Jiang, Z. Tang, MiR-199a-5p loss up-regulated DDR1 aggravated colorectal cancer by activating epithelial-to-mesenchymal transition related signaling. Dig. Dis. Sci. 59(9), 2163–2172 (2014). https://doi.org/10.1007/s10620-014-3136-0
S. Ma, W. Jia, S. Ni, miR-199a-5p inhibits the progression of papillary thyroid carcinoma by targeting SNAI1. Biochem. Biophys. Res. Commun. 497(1), 181–186 (2018). https://doi.org/10.1016/j.bbrc.2018.02.051
G. Su, Q. He, J. Wang, Clinical values of long non-coding RNAs in bladder cancer: a systematic review. Front. Physiol. 9, 652 (2018). https://doi.org/10.3389/fphys.2018.00652
R. Li, L. Zhang, L. Jia, Y. Duan, Y. Li, L. Bao, N. Sha, Long non-coding RNA BANCR promotes proliferation in malignant melanoma by regulating MAPK pathway activation. PLoS ONE 9(6), e100893 (2014). https://doi.org/10.1371/journal.pone.0100893
W. Jiang, D. Zhang, B. Xu, Z. Wu, S. Liu, L. Zhang, Y. Tian, X. Han, D. Tian, Long non-coding RNA BANCR promotes proliferation and migration of lung carcinoma via MAPK pathways. Biomed. Pharmacother. 69, 90–95 (2015). https://doi.org/10.1016/j.biopha.2014.11.027
Y. Wang, J. Gu, X. Lin, W. Yan, W. Yang, G. Wu, lncRNA BANCR promotes EMT in PTC via the Raf/MEK/ERK signaling pathway. Oncol. Lett. 15(4), 5865–5870 (2018). https://doi.org/10.3892/ol.2018.8017
W. Lu, H. Zhang, Y. Niu, Y. Wu, W. Sun, H. Li, J. Kong, K. Ding, H.M. Shen, H. Wu, D. Xia, Y. Wu, Long non-coding RNA linc00673 regulated non-small cell lung cancer proliferation, migration, invasion and epithelial mesenchymal transition by sponging miR-150-5p. Mol. Cancer 16(1), 118 (2017). https://doi.org/10.1186/s12943-017-0685-9
J. Yu, Y. Liu, Z. Gong, S. Zhang, C. Guo, X. Li, Y. Tang, L. Yang, Y. He, F. Wei, Y. Wang, Q. Liao, W. Zhang, X. Li, Y. Li, G. Li, W. Xiong, Z. Zeng, Overexpression long non-coding RNA LINC00673 is associated with poor prognosis and promotes invasion and metastasis in tongue squamous cell carcinoma. Oncotarget 8(10), 16621–16632 (2017). https://doi.org/10.18632/oncotarget.14200
E. Xia, A. Bhandari, Y. Shen, X. Zhou, O. Wang, lncRNA LINC00673 induces proliferation, metastasis and epithelial–mesenchymal transition in thyroid carcinoma via Kruppel-like factor 2. Int. J. Oncol. 53(5), 1927–1938 (2018). https://doi.org/10.3892/ijo.2018.4524
Y. Dong, W. Wu, Downregulation of lncRNA CASC2 promotes the postoperative local recurrence of early oral squamous cell carcinoma. Eur. Arch. Otorhinolaryngol. 276(2), 605–610 (2019). https://doi.org/10.1007/s00405-018-5209-8
Z. Pei, X. Du, Y. Song, L. Fan, F. Li, Y. Gao, R. Wu, Y. Chen, W. Li, H. Zhou, Y. Yang, J. Zeng, Down-regulation of lncRNA CASC2 promotes cell proliferation and metastasis of bladder cancer by activation of the Wnt/beta-catenin signaling pathway. Oncotarget 8(11), 18145–18153 (2017). https://doi.org/10.18632/oncotarget.15210
Y. Cao, R. Xu, X. Xu, Y. Zhou, L. Cui, X. He, Downregulation of lncRNA CASC2 by microRNA-21 increases the proliferation and migration of renal cell carcinoma cells. Mol. Med. Rep. 14(1), 1019–1025 (2016). https://doi.org/10.3892/mmr.2016.5337
T. Zhou, M. Zhong, S. Zhang, Z. Wang, R. Xie, C. Xiong, Y. Lv, W. Chen, J. Yu, LncRNA CASC2 expression is down- regulated in papillary thyroid cancer and promotes cell invasion by affecting EMT pathway. Cancer Biomark. 23(2), 185–191 (2018). https://doi.org/10.3233/cbm-181198
H. Lei, Y. Gao, X. Xu, LncRNA TUG1 influences papillary thyroid cancer cell proliferation, migration and EMT formation through targeting miR-145. Acta Biochim. Biophys. Sin. 49(7), 588–597 (2017). https://doi.org/10.1093/abbs/gmx047
Y. Pan, C. Li, J. Chen, K. Zhang, X. Chu, R. Wang, L. Chen, The emerging roles of long noncoding RNA ROR (lincRNA-ROR) and its possible mechanisms in human cancers. Cell Physiol. Biochem. 40(1–2), 219–229 (2016). https://doi.org/10.1159/000452539
A.P. Hutchins, D. Pei, Transposable elements at the center of the crossroads between embryogenesis, embryonic stem cells, reprogramming, and long non-coding RNAs. Sci. Bull. 60(20), 1722–1733 (2015). https://doi.org/10.1007/s11434-015-0905-x
K. Takahashi, I.K. Yan, H. Haga, T. Patel, Modulation of hypoxia-signaling pathways by extracellular linc-RoR. J. Cell Sci. 127(Pt 7), 1585–1594 (2014). https://doi.org/10.1242/jcs.141069
Y.M. Chen, Y. Liu, H.Y. Wei, K.Z. Lv, P. Fu, Linc-ROR induces epithelial–mesenchymal transition and contributes to drug resistance and invasion of breast cancer cells. Tumour Biol. 37(8), 10861–10870 (2016). https://doi.org/10.1007/s13277-016-4909-1
X.Y. Xu, J. Zhang, Y.H. Qi, M. Kong, S.A. Liu, J.J. Hu, Linc-ROR promotes endometrial cell proliferation by activating the PI3K-Akt pathway. Eur. Rev. Med. Pharmacol. Sci. 22(8), 2218–2225 (2018). https://doi.org/10.26355/eurrev_201804_14807
R. Zhang, H. Hardin, W. Huang, D. Buehler, R.V. Lloyd, R.N.A. Long Non-coding, Linc-ROR Is upregulated in papillary thyroid carcinoma. Endocr. Pathol. 29(1), 1–8 (2018). https://doi.org/10.1007/s12022-017-9507-2
D.F. Quail, J.A. Joyce, Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19(11), 1423–1437 (2013). https://doi.org/10.1038/nm.3394
M. Ryder, R.A. Ghossein, J.C. Ricarte-Filho, J.A. Knauf, J.A. Fagin, Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr. Relat. Cancer 15(4), 1069–1074 (2008). https://doi.org/10.1677/erc-08-0036
J.D. French, G.R. Kotnis, S. Said, C.D. Raeburn, R.C. McIntyre Jr., J.P. Klopper, B.R. Haugen, Programmed death-1+ T cells and regulatory T cells are enriched in tumor-involved lymph nodes and associated with aggressive features in papillary thyroid cancer. J. Clin. Endocrinol. Metab. 97(6), E934–E943 (2012). https://doi.org/10.1210/jc.2011-3428
R.M. Melillo, V. Guarino, E. Avilla, M.R. Galdiero, F. Liotti, N. Prevete, F.W. Rossi, F. Basolo, C. Ugolini, A. de Paulis, M. Santoro, G. Marone, Mast cells have a protumorigenic role in human thyroid cancer. Oncogene 29(47), 6203–6215 (2010). https://doi.org/10.1038/onc.2010.348
H. Li, X. Fan, J. Houghton, Tumor microenvironment: the role of the tumor stroma in cancer. J. Cell. Biochem. 101(4), 805–815 (2007). https://doi.org/10.1002/jcb.21159
R.O. Hynes, The extracellular matrix: not just pretty fibrils. Science 326(5957), 1216–1219 (2009). https://doi.org/10.1126/science.1176009
O.W. Petersen, H.L. Nielsen, T. Gudjonsson, R. Villadsen, F. Rank, E. Niebuhr, M.J. Bissell, L. Ronnov-Jessen, Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am. J. Pathol. 162(2), 391–402 (2003). https://doi.org/10.1016/s0002-9440(10)63834-5
R.Z. Panni, D.C. Linehan, D.G. DeNardo, Targeting tumor-infiltrating macrophages to combat cancer. Immunotherapy 5(10), 1075–1087 (2013). https://doi.org/10.2217/imt.13.102
J.B. Mitchem, D.J. Brennan, B.L. Knolhoff, B.A. Belt, Y. Zhu, D.E. Sanford, L. Belaygorod, D. Carpenter, L. Collins, D. Piwnica-Worms, S. Hewitt, G.M. Udupi, W.M. Gallagher, C. Wegner, B.L. West, A. Wang-Gillam, P. Goedegebuure, D.C. Linehan, D.G. DeNardo, Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 73(3), 1128–1141 (2013). https://doi.org/10.1158/0008-5472.can-12-2731
W. Qing, W.Y. Fang, L. Ye, L.Y. Shen, X.F. Zhang, X.C. Fei, X. Chen, W.Q. Wang, X.Y. Li, J.C. Xiao, G. Ning, Density of tumor-associated macrophages correlates with lymph node metastasis in papillary thyroid carcinoma. Thyroid 22(9), 905–910 (2012). https://doi.org/10.1089/thy.2011.0452
W.C. Chang, J.Y. Chen, C.H. Lee, A.H. Yang, Expression of decoy receptor 3 in diffuse sclerosing variant of papillary thyroid carcinoma: correlation with M2 macrophage differentiation and lymphatic invasion. Thyroid 23(6), 720–726 (2013). https://doi.org/10.1089/thy.2012.0261
W. Fang, L. Ye, L. Shen, J. Cai, F. Huang, Q. Wei, X. Fei, X. Chen, H. Guan, W. Wang, X. Li, G. Ning, Tumor-associated macrophages promote the metastatic potential of thyroid papillary cancer by releasing CXCL8. Carcinogenesis 35(8), 1780–1787 (2014). https://doi.org/10.1093/carcin/bgu060
C.D. Yeo, N. Kang, S.Y. Choi, B.N. Kim, C.K. Park, J.W. Kim, Y.K. Kim, S.J. Kim, The role of hypoxia on the acquisition of epithelial–mesenchymal transition and cancer stemness: a possible link to epigenetic regulation. Korean J. Intern. Med. 32(4), 589–599 (2017). https://doi.org/10.3904/kjim.2016.302
O. Koperek, E. Akin, R. Asari, B. Niederle, N. Neuhold, Expression of hypoxia-inducible factor 1 alpha in papillary thyroid carcinoma is associated with desmoplastic stromal reaction and lymph node metastasis. Virchows Arch. 463(6), 795–802 (2013). https://doi.org/10.1007/s00428-013-1484-3
Y. Lin, Q. Ma, L. Li, H. Wang, The CXCL12-CXCR4 axis promotes migration, invasiveness, and EMT in human papillary thyroid carcinoma B-CPAP cells via NF-kappaB signaling. Biochem. Cell Biol. 96(5), 619–626 (2018). https://doi.org/10.1139/bcb-2017-0074
D. Cui, Y. Zhao, J. Xu, Activated CXCL5-CXCR2 axis promotes the migration, invasion and EMT of papillary thyroid carcinoma cells via modulation of beta-catenin pathway. Biochimie 148, 1–11 (2018). https://doi.org/10.1016/j.biochi.2018.02.009
C. Thery, L. Zitvogel, S. Amigorena, Exosomes: composition, biogenesis and function. Nat. Rev. Immunol. 2(8), 569–579 (2002). https://doi.org/10.1038/nri855
H. Hardin, H. Helein, K. Meyer, S. Robertson, R. Zhang, W. Zhong, R.V. Lloyd, Thyroid cancer stem-like cell exosomes: regulation of EMT via transfer of lncRNAs. Lab. Investig 98(9), 1133–1142 (2018). https://doi.org/10.1038/s41374-018-0065-0
I. Fabregat, A. Malfettone, J. Soukupova, New insights into the crossroads between EMT and stemness in the context of cancer. J. Clin. Med. 5(3) (2016). https://doi.org/10.3390/jcm5030037
S. Mukherjee, J. Kong, D.J. Brat, Cancer stem cell division: when the rules of asymmetry are broken. Stem Cells Dev. 24(4), 405–416 (2015). https://doi.org/10.1089/scd.2014.0442
E. Mato, C. Gonzalez, A. Moral, J.I. Perez, O. Bell, E. Lerma, A. de Leiva, ABCG2/BCRP gene expression is related to epithelial–mesenchymal transition inducer genes in a papillary thyroid carcinoma cell line (TPC-1). J. Mol. Endocrinol. 52(3), 289–300 (2014). https://doi.org/10.1530/jme-14-0051
N.K. Kurrey, S.P. Jalgaonkar, A.V. Joglekar, A.D. Ghanate, P.D. Chaskar, R.Y. Doiphode, S.A. Bapat, Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 27(9), 2059–2068 (2009). https://doi.org/10.1002/stem.154
X. Ye, W.L. Tam, T. Shibue, Y. Kaygusuz, F. Reinhardt, E. Ng Eaton, R.A. Weinberg, Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 525(7568), 256–260 (2015). https://doi.org/10.1038/nature14897
E. Sanchez-Tillo, O. de Barrios, L. Siles, M. Cuatrecasas, A. Castells, A. Postigo, beta-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc. Natl Acad. Sci. USA 108(48), 19204–19209 (2011). https://doi.org/10.1073/pnas.1108977108
S.A. Mani, W. Guo, M.J. Liao, E.N. Eaton, A. Ayyanan, A.Y. Zhou, M. Brooks, F. Reinhard, C.C. Zhang, M. Shipitsin, L.L. Campbell, K. Polyak, C. Brisken, J. Yang, R.A. Weinberg, The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell 133(4), 704–715 (2008). https://doi.org/10.1016/j.cell.2008.03.027
H. Hardin, X.M. Yu, A.D. Harrison, C. Larrain, R. Zhang, J. Chen, H. Chen, R.V. Lloyd, Generation of novel thyroid cancer stem-like cell clones: effects of resveratrol and valproic acid. Am. J. Pathol. 186(6), 1662–1673 (2016). https://doi.org/10.1016/j.ajpath.2016.02.003
H. Hardin, Z. Guo, W. Shan, C. Montemayor-Garcia, S. Asioli, X.M. Yu, A.D. Harrison, H. Chen, R.V. Lloyd, The roles of the epithelial–mesenchymal transition marker PRRX1 and miR-146b-5p in papillary thyroid carcinoma progression. Am. J. Pathol. 184(8), 2342–2354 (2014). https://doi.org/10.1016/j.ajpath.2014.04.011
W. Li, A.N. Reeb, W.A. Sewell, G. Elhomsy, R.Y. Lin, Phenotypic characterization of metastatic anaplastic thyroid cancer stem cells. PLoS ONE 8(5), e65095 (2013). https://doi.org/10.1371/journal.pone.0065095
J. Liu, R.E. Brown, Immunohistochemical detection of epithelialmesenchymal transition associated with stemness phenotype in anaplastic thyroid carcinoma. Int. J. Clin. Exp. Pathol. 3(8), 755–762 (2010)
K. Yasui, M. Shimamura, N. Mitsutake, Y. Nagayama, SNAIL induces epithelial-to-mesenchymal transition and cancer stem cell-like properties in aldehyde dehydroghenase-negative thyroid cancer cells. Thyroid 23(8), 989–996 (2013). https://doi.org/10.1089/thy.2012.0319
L. Lan, Y. Luo, D. Cui, B.Y. Shi, W. Deng, L.L. Huo, H.L. Chen, G.Y. Zhang, L.L. Deng, Epithelial–mesenchymal transition triggers cancer stem cell generation in human thyroid cancer cells. Int. J. Oncol. 43(1), 113–120 (2013). https://doi.org/10.3892/ijo.2013.1913
A. Antonelli, C. La Motta, Novel therapeutic clues in thyroid carcinomas: the role of targeting cancer stem cells. Med. Res. Rev. 37(6), 1299–1317 (2017). https://doi.org/10.1002/med.21448
A. Klaus, O. Fathi, T.W. Tatjana, N. Bruno, K. Oskar, Expression of hypoxia-associated protein HIF-1alpha in follicular thyroid cancer is associated with distant metastasis. Pathol. Oncol. Res. 24(2), 289–296 (2018). https://doi.org/10.1007/s12253-017-0232-4
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shakib, H., Rajabi, S., Dehghan, M.H. et al. Epithelial-to-mesenchymal transition in thyroid cancer: a comprehensive review. Endocrine 66, 435–455 (2019). https://doi.org/10.1007/s12020-019-02030-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-019-02030-8