
Abstract
Cancer is a complex multistep process involving genetic and epigenetic changes that eventually result in the activation of oncogenic pathways and/or inactivation of tumor suppressor signals. During cancer progression, cancer cells acquire a number of hallmarks that promote tumor growth and invasion. A crucial mechanism by which carcinoma cells enhance their invasive capacity is the dissolution of intercellular adhesions and the acquisition of a more motile mesenchymal phenotype as part of an epithelial-to-mesenchymal transition (EMT). Although many transcription factors can trigger it, the full molecular reprogramming occurring during an EMT is mainly orchestrated by three major groups of transcription factors: the ZEB, Snail and Twist families. Upregulated expression of these EMT-activating transcription factors (EMT-ATFs) promotes tumor invasiveness in cell lines and xenograft mice models and has been associated with poor clinical prognosis in human cancers. Evidence accumulated in the last few years indicates that EMT-ATFs also regulate an expanding set of cancer cell capabilities beyond tumor invasion. Thus, EMT-ATFs have been shown to cooperate in oncogenic transformation, regulate cancer cell stemness, override safeguard programs against cancer like apoptosis and senescence, determine resistance to chemotherapy and promote tumor angiogenesis. This article reviews the expanding portfolio of functions played by EMT-ATFs in cancer progression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer is a multipronged process that requires the acquisition by malignant cells of a number of capabilities—referred as hallmarks of cancer [1]—that ensure their survival and proliferation and, therefore, the growth of the tumor. Cancer cells in a solid tumor can detach from the main mass to invade the surrounding stroma, enter the circulation and eventually metastasize to distant organs. In most cases, invading cancer cells lose their polarity and intercellular adhesions and acquire a more motile phenotype as part of an epithelial-to-mesenchymal transition (EMT) [2–5]. While EMT was identified in the context of embryogenesis almost three decades ago, its underlying molecular mechanisms have only begun to be understood more recently with the discovery of its role in tumor invasiveness.
A hallmark of EMT is the functional loss of the adherens junction protein E-cadherin. Although transcriptional control of E-cadherin during EMT seems to be prevalent, recent studies point to the existence of intricate relationships between epigenetic, transcriptional and translational mechanisms. Although E-cadherin expression is inhibited by a number of transcription factors, only a small set are known to regulate it directly. The main groups of transcription factors that bind to the E-cadherin promoter and directly repress its transcription—which will be referred to hereafter as EMT-activating transcription factors, EMT-ATFs—are the ZEB (ZEB1, ZEB2) and Snail (Snail1, Snail2, Snail3) families of zinc finger proteins and the Twist family of bHLH factors (Twist1, Twist2) [2, 3]. While E12/E47 and Tbx3 also bind to the E-cadherin promoter [6, 7], the ability of Goosecoid and HMGA2 to repress E-cadherin expression and induce an EMT seems to be mediated by other EMT-ATFs [8, 9]. Most of these transcription factors were originally identified as regulators of embryogenesis and cell differentiation and only later recognized for their role in cancer progression. Still, the upstream regulatory signals and downstream targets of EMT-ATFs in cancer largely concur with those during embryogenesis [2, 3].
In addition to their now classical function as promoters of tumor invasiveness, over the last few years EMT-ATFs have gained new relevance in light of their role regulating several other hallmarks of cancer. EMT endows cells not only with greater ability to migrate but also with stem cell characteristics that play a role in tumorigenesis and resistance to chemotherapy. Recent evidence shows that EMT-ATFs also participate in early stages of cancer development cooperating in oncogenic transformation, overriding safeguard programs against cancer like apoptosis and senescence or promoting tumor angiogenesis. The expanding portfolio of functions of EMT-ATFs in cancer set them not only as important diagnostic and prognostic biomarkers but also as potential therapeutic targets. This article reviews the roles of EMT-ATFs in cancer and is organized as follows: The next section summarizes the EMT process in cancer. The following section reviews the main regulatory pathways and mechanisms of action of EMT-ATFs. Later sections describe the regulation of tumor invasiveness and metastasis by EMT-ATFs and overview their newly assigned roles in earlier stages of cancer progression. The final section concludes and presents EMT-ATFs as multifunctional regulators of the hallmarks of cancer.
EMT in cancer
E-cadherin mediates homotypic intercellular adhesion and interacts with intracellular proteins to establish and coordinate the morphology, polarity and function of epithelial cells [10, 11]. The downregulation of E-cadherin is inherent to EMT, but EMT also entails the downregulation of other epithelial specification genes like components of tight and gap junctions, desmosomes, cytokeratins, etc. [2, 3]. In parallel, there is an induction of mesenchymal markers (e.g., N-cadherin, cadherin-11), reorganization of the cytoskeleton (e.g., switch from cytokeratins to vimentin), and the synthesis of extracellular matrix components and metalloproteases [2, 3].
Many signals unleashing an EMT during embryogenesis are also active in cancer. Thus, stimuli such as TGFβ, FGF, EGF, IGF, HGF, PDGF, estrogens, Wnt, Shh, inflammatory cytokines or hypoxia as well as oncogenes like RasV12, ErbB2 or mutant p53 may be involved in EMT during cancer progression (reviewed in [3]). Triggering and maintenance of the mesenchymal state requires cooperation between several of these pathways through autocrine signaling loops [12].
These growth factors, inflammatory, hypoxic and oncogenic signals, along with an increasing number of microRNAs, converge in the induction of EMT-ATFs either at the transcriptional level or by increasing their protein or mRNA stability. E-cadherin expression and function could be downregulated by loss-of-function mutations, but modulation of EMT during embryogenesis and cancer progression mostly involves the participation of EMT-ATFs providing a high degree of functional redundancy and plasticity. E-cadherin has also been found to be silenced by promoter hypermethylation in a number of carcinomas. Some of the signals triggering EMT activate or repress the expression of non-coding microRNAs (miRs), which in turn modulate tumor invasion and metastasis by regulating EMT-ATFs transcripts (see below), targeting E-cadherin itself, or altering the expression of small GTPases or extracellular matrix receptors (reviewed in [13]).
Downregulation of E-cadherin is a critical initial step in EMT, not only because of the disruption of adherens junctions but also because loss of E-cadherin reinforces the EMT process by inducing the expression of Twist1 and ZEB1 in a feed-forward loop [194]. EMT-ATFs do not simply repress E-cadherin but are able to orchestrate the entire EMT program, inhibiting and activating a wide array of epithelial and mesenchymal genes, respectively [14–21].
Expression of EMT markers by primary human tumors correlates with enhanced invasiveness and poorer clinical prognosis. Cancer cells at the tumor invasive edge that have transitioned through an EMT secrete cytokines and proteases that promote angiogenesis, remodel the peritumoral extracellular matrix, and activate non-neoplastic stromal cells. In turn, stromal cells release factors that reinforce the EMT in cancer cells and foster survival, growth, and invasiveness of the tumor, thus creating a reciprocal influence between the tumor and its microenvironment [22, 23]. EMT is required for cancer cells to shift from a collective type of invasion—where cells retain E-cadherin and some intercellular adhesions—to an individual mesenchymal type of invasion, a change that depends on TGFβ signals at the tumor–host interface [24, 25]. While both models of invasion allow cancer cells to reach the lymphatic circulation, optimal hematogenous dissemination only occurs in EMT-mediated individual invasion [22, 25].
In addition to the mesenchymal switch, the molecular reprograming encompassed by the EMT also endows cancer cells with stem-like characteristics [12, 26]. Many of the signals controlling normal stem cell homeostasis are inducers of EMT and seem to contribute to the generation and maintenance of cancer stem cells (CSCs). In an influential article, Brabletz and colleagues [27] proposed a model of tumor progression where only cancer cells at the tumor–host interface, the “migrating CSCs”, undergo EMT and acquire mesenchymal and stem-like characteristics and therefore migratory and self-renewal capacities. CSCs are thought be important in the genesis of primary tumors and metastasis and could also be at the root of tumoral chemoresistance and recurrence [28].
As in embryogenesis, the EMT occurring during cancer progression is a reversible process. At the site of micrometastasis, epigenetic changes and the absence of EMT-inducing signals in the microenvironment lead to the re-expression of particular sets of microRNAs and the downregulation of EMT-ATFs (see below), allowing cancer cells to regain the epithelial characteristics of the primary tumor, through a mesenchymal-to-epithelial transition (MET), and grow to form a secondary tumor [29, 30]. In fact, recent evidence also indicates that EMT and metastatic dissemination of cancer cells may in fact occur from very early in tumor progression [31].
Regulation and mechanisms of action of EMT-ATFs
ZEB1 and ZEB2
The ZEB family comprises zinc finger/homeodomain proteins that are highly conserved across species (see an excellent review in [32]). In higher organisms, the family is constituted by two members: ZEB1 and ZEB2. ZEB factors contain multiple independent domains to interact with other transcriptional regulators (Fig. 1) (reviewed in [32] and [33], see also [34–41]). ZEB proteins bind to CtBP corepressors that in turn recruit histone deacetylases and methyltransferases, polycomb, and coREST [37–43]. Transcriptional repression by ZEB1 is also mediated through recruitment of the histone acetyl transferase Tip60, the SWI/SNF chromatin remodeling ATPase BRG1 and the histone deacetylase SIRT1 [44–46]. Meantime, in addition to CtBP, ZEB2 interacts with the NuRD remodeling and deacetylase repressor complex [47]. On the other hand, ZEB factors can also activate transcription through binding to histone acetyltransferases p300/pCAF ([48–50], reviewed in [33]). ZEB1 and ZEB2 repressor activities are modulated by post-translational modifications. SUMOylation by Pc2 or acetylation by p300/pCAF disrupt ZEB factor binding to CtBP [48–51]. Although its transcriptional significance remains unclear, phosphorylation of ZEB1 varies widely among cell types [52].

Domain structure human EMT-ATFs: human ZEB1 (GenBank reference sequence AAA20602.1), human ZEB2 (AAI27103), human Snail1 (NP_005976.2), human Snail2 (NP_003059.1), human Snail3 (NP_840101.1), human Twist1 (NP_000465.1), and human Twist2 (NP_476527.1)
ZEB1 and ZEB2 trigger an EMT by repression of epithelial markers and activation of mesenchymal ones ([14–16], [20] and reviewed in [33, 53, 54]). ZEB1 and ZEB2 could also regulate EMT through their repressor effect on miRs (see below). ZEB2 mRNA also functions as a competitive endogenous RNA (ceRNA) squelching miRs targeting other transcripts, thus activating their expression [55].
ZEB1 and ZEB2 have overlapping, but still distinct, patterns of expression that result from their regulation by classical signaling pathways and an expanding set of microRNAs (see excellent reviews in [53, 54, 311] and below). In most cell types, TGFβ factors induce both ZEB proteins that in turn modulate TGFβ signaling in opposing ways with ZEB1 synergizing with R-Smads and ZEB2 repressing their activity. In fact, ZEB factors mediate some TGFβ functions during development, cell differentiation, and proliferation [48, 49, 56–58]. ZEB1 and ZEB2 are induced by HIF-1α in hypoxic conditions, inflammatory cytokines, and by ligand-mediated activation (e.g., FGF, IGF-1, PDGFR) of receptor tyrosine kinases [59–66]. ZEB factors are directly activated by downstream signals frequently activated in tumors like Ras-ERK2-Fra1, NFκB, and JAK/STAT3 [61, 67, 68].
Aberrant activation of the canonical Wnt pathway by loss-of-function mutation of APC gene, as occurring in most colorectal carcinomas, induces ZEB1 [69]. ZEB1 expression is also induced by COX2/PGE2, which can activate Wnt signaling by inhibiting GSK3β [70]. The Notch pathway, deregulated in many cancers, also activates ZEB1 expression [71, 72]. In turn, ZEB1 enhances Notch activity by indirectly increasing Jag1 and its coactivators Maml2/Maml3 [72]. ZEB factors are also upregulated by growth and steroid hormones [73–75]. Hippo/YAP signaling and the tumor suppressor Rb/E2F pathway activate and repress ZEB1 transcription, respectively, without affecting ZEB2 levels [76, 77]. SIRT1 is recruited by ZEB1 to repress the E-cadherin promoter but SIRT1 itself also induces ZEB and Snail factors but not Twist [46]. Interestingly, SIRT1 and miR-200a—a microRNA that inhibits ZEB factors (see below)—are involved in a reciprocal negative feedback loop [78]. ZEB1 is repressed at the promoter level by Grainyhead-like 2, a transcription factor tightly co-regulated with E-cadherin [79]. Finally, ZEB1 and ZEB2 are downstream of Snail and Twist factors (see below).
In addition to transcriptional regulation by these upstream signals, ZEB factors expression is also controlled at the mRNA and protein level. They are regulated by a complex network of microRNAs (see below). Furthermore, YB-1, a protein associated with increased invasiveness in breast carcinomas, activates the mRNA of ZEB2 without affecting ZEB1 [80]. ZEB2 is also controlled at the protein level, and its binding to F-box FBXL14 targets it for ubiquitin-proteasome degradation [81].
Snail1, Snail2 and Snail3
The Snail family comprises three members: Snail1 (originally identified as Snail), Snail2 (Slug), and Snail3 (Smuc) (Fig. 1). All three contain a single C-terminal zinc finger cluster that bind to E-boxes in the regulatory regions of target genes. At the N-terminal end, they share a SNAG (Snail-Gfi1) domain that mediates binding to Sin3A/HDAC1/HDAC2 and Ajuba-PRMT5-PRC2 and LSD1-coREST complexes [82–85]. Only Snail2 maintains the central CtBP binding motif present in Drosophila that has also been implicated in recruitment of HDAC and coREST [86]. A recent report shows the recruitment of NCoR and CtBP1 to the SNAG and SLUG domains of Snail2, respectively, with the former being essential for Snail2-mediated EMT [87]. The precise mechanism by which Snail-mediated histone modifications (deacetylation, H3K27 and H4R3 methylation, H3K4 demethylation) cooperate to repress E-cadherin has not been fully elucidated. Snail1 also represses epithelial specification genes, including E-cadherin, by interacting with Smad3/Smad4 [88]. Overexpression of Snail1 decreases binding of AKT1 to the E-cadherin promoter in favor of AKT2 on which Snail1 depends for its repressor activity [89]. Conversely, activation of mesenchymal genes by Snail factors seems to be, at least in part, indirect, via E-cadherin downregulation.
Snail1 and Snail2 transcriptional activities are also modulated by post-transcriptional modifications that alter their protein stability and intracellular localization. Thus, binding of Snail1 to Sin3a is strengthened by PKA and CK2 phosphorylation [90]. In a similar manner, phosphorylation of Snail2 modulates its ability to transcriptionally repress E-cadherin [87]. Phosphorylation of Snail1 by GSK3β functionally inactivates it through both CRM1-dependent export to the cytoplasm and SCF/β-TrCP ubiquitin-mediated proteasome degradation [91]. Interaction of Snail1 with LOXL2 and LOXL3 prevents GSK3β-induced degradation [92], which is, however, fostered by phosphorylation by CK1ε [93]. Snail factors are also targeted for degradation in a GSK3β-independent manner by binding to FBXL14 [81, 94, 95] or, in the case of Snail2, through Mdm2 ubiquitination [222]. In addition, the intracellular localization of Snail factors is also controlled through import and export signals [96, 97]. In addition to GSK3β, phosphorylation of Snail1 by PKD1 also triggers its nuclear export [98]. On the other hand, phosphorylation by PAK1 promotes its nuclear localization, which is also dependent on the breast cancer-associated zinc transporter protein LIV1 [99, 100].
As with other EMT-ATFs, signals regulating Snail proteins in cancer parallel those operating during development. Snail factors are induced by TGFβ, Notch, TNFα, EGF, FGF, Wnt, Shh, SCF/c-kit, hypoxia, and estrogens [3]. Transcriptional activation of Snail1 by TGFβ requires interaction of R-Smads with HMGA2 [101]. In some cancer cell types, induction of Snail1 by TGFβ requires simultaneous oncogenic Ras signaling [102]. On its part, TGFβ-mediated induction of Snail2 depends on the downregulation of KLF4 and FOXA1 that, interestingly, form a double repressor loop with Snail2 [103]. Notch signaling induces Snail1 through direct and indirect mechanisms. In addition to direct activation of the Snail1 promoter, Notch stabilizes Snail1 through HIF-1α-mediated activation of LOX [104]. NFκB signaling by AKT, hypoxia or inflammatory cytokines transcriptionally activates Snail1. Induction of NFκB by inflammatory cytokines also stabilizes Snail1 by inhibiting GSK-3β-mediated ubiquitination [105]. Engagement of receptor tyrosine kinases signaling and COX2/PEG2 also induces expression of Snail1 by inhibition of GSK3β [70]. Snail1 is also able to repress its own transcription [106]. Finally, Snail1 is activated at the translational level by YB-1 [80].
Twist1 and Twist2
Twist1 and Twist2 share a basic/helix-loop-helix (bHLH) domain that mediates their binding to DNA and homo/hetero-dimerization (Fig. 1). At the C-terminal end, there is a “Twist box” that has been implicated in both transcriptional activation and repression (reviewed in [107] and [312]). Regulation of gene expression by Twist factors depends on their binding to other transcriptional regulators, post-translational modifications, and choice of partner for dimerization. Binding of Twist proteins to E-boxes in the promoters of target genes could either activate (e.g., N-cadherin, AKT2 or Gli1) or repress (e.g., E-cadherin) transcription [108, 109]. In some cases, Twist-mediated repression involves binding to other transcription factors and cofactors to inhibit their activity (e.g., Runx2, myogenic bHLH, NFκB, and p300/pCAF). Direct repression of E-cadherin by Twist1 entails recruitment of multiple chromatin remodeling complexes, whose composition and dynamics is currently being uncovered. Twist1 activates transcription of the PRC1 component Bmi1 and binds to polycomb repressor complexes PRC1 and PRC2 at the E-cadherin promoter [310]. Twist1 interaction with components of the NuRD complex is also required for E-cadherin repression [110], a result in line with the finding that recruitment of PRC2 is specified by NuRD histone deacetylation [111]. In addition, it was recently reported that binding of Twist1 to the H4K20 methyltransferase SET8 could simultaneously contribute to repression of E-cadherin and activation of N-cadherin [112].
Gene regulation by Twist factors is also modulated by control of their intracellular localization and the identity of their dimerization partner. Nuclear/cytoplasm shuttling of Twist factors is modulated by integrin-mediated adhesion to the extracellular matrix, post-translational modifications, and partner dimerization [108]. In Drosophila, Twist homodimers activate transcription whereas heterodimers with Daughterless function as repressors. However, in mammalian systems, Twist/E12 heterodimers can both activate and repress transcription [107, 312]. Homo- or heterodimerization of Twist proteins is determined by availability of E12. Likewise, Twist can titrate away E12 from other bHLH proteins thus inhibiting their function. Phosphorylation of the bHLH domain of Twist alters not only dimerization partner choice but also binding affinity for DNA [107].
Understanding the signaling pathways upstream of Twist1 and Twist2 is not as complete as for ZEB and Snail factors. Still, Twist factors are upregulated by classical EMT-inducing pathways during development, inflammation, and cancer, such as TGFβ, Wnt, hypoxia, and ligand-binding activation of receptor tyrosine kinases and inflammatory cytokines receptors [107, 312]. EGF and IL6 induce Twist1 via activation of JAK/STAT signaling and direct binding of STAT3 to the Twist1 promoter [113, 114]. Twist1 and Twist2 form a negative loop with inflammatory cytokines, as Twist factors are transcriptionally induced by NFκB and in turn bind to the TNFα and IL1β promoters blocking NFκB transcriptional activity [115]. As discussed below, upregulation of Twist1 by HIF-1α during hypoxic conditions has significant implications in tumor invasion and angiogenesis [116].
Twist factors are also regulated at the mRNA and protein level as well by their intracellular localization. As with other EMT-ATFs, YB-1 fosters cap-independent translation of Twist1 [80]. Twist1 is also controlled post-translationally by cytoplasmic polyadenylation sites in its 3’UTR [117]. Heterodimerization of Twist1 with E12 and Hand2, fostered by phosphorylation by PKA, stabilizes Twist1 protein that is, on the other hand, targeted for degradation upon binding to FXBL14 [81, 118].
Regulatory networks between EMT-ATFs and microRNAs
In recent years, a plethora of publications have identified miRs regulating each and every step during cancer progression, from cell proliferation, cancer cell stemness, and apoptosis to angiogenesis or tumor invasiveness (reviewed in [13]). miRs regulate invasiveness and metastasis by targeting the transcripts of a large number of genes involved in EMT/MET regulation, including those of EMT-ATFs (Fig. 2).

Regulatory networks between microRNAs and EMT-ATFs of the ZEB (a), Snail (b) and Twist (c) families. For simplification, some relationships have been consolidated or are not shown. Double color identification refers to miRs that have been described as having both pro- and anti-invasion/metastatic roles depending on the context and/or cell system analyzed. See text for discussion and references
Members of the miR-200 family (miR-200a/b/c, miR-141, and miR-429) maintain an epithelial status and prevent EMT through inhibition of ZEB1 and ZEB2 (reviewed in [54], see also [119–121], and other references in [54]). In turn, miR-200 members are transcriptionally repressed by ZEB factors—as well as Snail1—thus forming a double-negative loop that maintain cells in either an epithelial or mesenchymal state [54, 122–124]. Expression of miR-200 and ZEB factors presents an inverse pattern in a number of human cancers. Invading mesenchymal-like cancer cells that extravasate and metastasize eventually need to revert to an epithelial phenotype for the metastatic colony to grow into a secondary tumor. Cancer cells that form macroscopic metastasis have higher levels of miR-200 compared to those that invade but are not able to colonize and, paradoxically to their roles as EMT repressors: overexpression of miR-200 in mouse breast cancer isogenic cell lines fosters lung and liver mestatasis by inhibition of Sec23a, a component of the tumor secretome that blocks metastatic colonization [125–313]. The 5′CpG islands of miR-200 loci are also subjected to dynamic epigenetic regulation [126]. In epithelial cell lines, miR-200 expression is silenced by hypermethylation during TGFβ-induced EMT but reverted by demethylation in MET. Likewise, 5′CpG islands of miR-200 loci are hypermethylated in cancerous cells of primary colorectal carcinomas but remain unmethylated in epithelial cells of the normal colonic mucosa.
miR-200 members mediate the anti-EMT activities of other factors and signaling pathways. For instance, Six1 induces ZEB1 and EMT through transcriptional repression of miR-200 [127]. By contrast, the tumor suppressor p53 inhibits a mesenchymal and stem cell phenotype and/or the invasive capacity of mammary and pancreatic epithelial cells and hepatocarcinoma cell lines by upregulating miR-200—as well as miR-192—to repress ZEB1 and ZEB2 [128–130]. Conversely, oncogenic mutant forms of p53 decrease miR-200 and increase ZEB1 expression [128].
ZEB1 and ZEB2 participate in other miR regulatory networks. ZEB factors repress miR-183 and miR203, which together with miR-200, inhibit the expression of stemness factors Bmi1, Sox2 and KLF4 [131, 132]. ZEB1 and ZEB2 are also targets of miR-205 [13]. In aggressive basal-like breast carcinomas, but not the luminal type, Fra-1 activates miR-221 and miR-222 [133], miRs that have been associated with EMT and tumor invasiveness and are downregulated by EGFR inhibitors. miR-221 and miR-222 downregulate TRPS1, a GATA-like repressor, that in turn inhibits ZEB2 [133]. In contrast, in endothelial cells, ZEB2 is a direct target of miR-221 [134], confirming evidence elsewhere that miR may have different, even opposing, functions depending on the cell type.
Contrary to its inhibitory effect on E-cadherin in breast epithelial cells [135], miR-9 downregulates Snail1 expression in melanoma cells by targeting NFκB, thus inhibiting proliferation and invasiveness [136]. p53 also downregulates Snail1 and Snail2 via induction of miR-34a/b/c. Conversely, Snail1 and Snail2 (and ZEB1) transcriptionally repress miR-34a/b/c, thus forming a double-negative loop similar to the one between ZEB factors and miR-200 [137, 138]. As with miR-200, miR34a inhibits stem cell-related factors Bmi1, CD44, and CD133 [137]. Members of the miR-30 family also target Snail1 [139, 140] and, as with miR-200, overexpression of either miR-30 and miR-34 prevents TGFβ-induced EMT. It is worth noting here that miR-34 also targets several Wnt signaling genes like Wnt1, Wnt3, LEF1 and β-catenin, suggesting that loss of p53 tumor suppressor activity could be accompanied by activation of β-catenin/TCF signaling, which may reinforce EMT and stemness programs [141]. Snail1 is also repressed by let-7d and miR-29b, microRNAs that block EMT and invasiveness in HNSCC and prostate carcinomas, respectively [142, 143].
Snail2 participates in yet another double-negative loop with miRs: miR-1 and miR-200 inhibit Snail2 expression and are repressed by binding of Snail2 to their promoters [144]. Snail2 transcript is also inhibited by miR-203, which is upregulated in primary tumors and hypermethylated and silenced in metastatic cell lines [145].
To date, fewer miRs have been linked to Twist factors. Overexpression of let-7d inhibits Twist1 expression in oral squamous cell carcinomas [142]. Twist1 is also inhibited by miR-29b, miR-580, and miR-214, but it is not clear whether downregulation of Twist1 by miR-29b is mediated by inhibition of Snail1 [117, 143, 146]. In turn, Twist1 activates the expression of miR-10b, miR-199a, and miR-214 genes through direct binding to E-boxes in their promoters [147, 148]. miR-10b is expressed in metastatic breast cancer cells and induces cell motility and migration by indirectly inducing RhoC.
The emergence of all these new regulatory loops between miRs and EMT-ATFs provides evidence of dynamic epigenetic silencing of miR-200 (and likely other miRs) during cancer progression, which endows cancer cells with enhanced plasticity to reversibly switch between EMT and MET during tumor invasiveness and metastasis.
Cross-regulation among EMT-ATFs
Previous sections have highlighted the existence of significant overlap among EMT-ATFs in their regulatory signals, target genes, and mechanisms of action, raising the question of what is the specific contribution of each factor in the regulation of EMT and cancer. Recent evidence indicates that ZEB factors are downstream of the Snail and Twist families in the EMT interactome [149] (Fig. 3). The hierarchical Snail–Twist/ZEB relationship supports earlier evidence of the strongest correlation of ZEB factors, especially ZEB1, with E-cadherin loss and EMT across cancer cell types [150].

Cross-regulation among EMT-ATFs. Dashed lines indicate that different relationships have been reported depending on the cell system analyzed. See text for discussion and references
Snail1 increases ZEB1 protein levels through both transcriptional and post-transcriptional mechanisms [57, 151, 152] (Fig. 3). Snail1 also enhances the stability of Twist1 and triggers nuclear translocation of Ets1, both directly activating the ZEB1 promoter. Additionally, Snail1 not only represses miR-200, thus derepressing ZEB1 mRNA, but also stabilizes ZEB1 protein through still undefined mechanisms. Snail2 also activates ZEB1 by direct binding to its promoter [308].
ZEB2 expression is also regulated by Snail1 through several mechanisms. Repression of miR-200 by Snail1 upregulates ZEB2 mRNA (Fig. 3). In addition, Snail1 induces a natural antisense transcript (NAT) that overlaps with an internal ribosomal entry site in the 5′ UTR of ZEB2, preventing its splicing and increasing ZEB2 [153].
Twist1 mediates some of its EMT-activating effects through Snail2 by binding to its promoter and activating its transcription [154]. Expression of Twist1 and Snail1 are also mutually dependent although in different directions, depending on the cell system. Knock-down of Snail1 downregulates Twist1 protein and mRNA, while knock-down of Twist1 interferes with TGFβ-mediated induction of Snail1 [152]. Alternatively, it has also been reported that Snail1 transcriptionally represses Twist1, which only increases as Snail1 declines in later stages of EMT [155].
Microarray gene expression signatures of human mammary epithelial cells (HMEC) driven towards EMT have confirmed this hierarchical/network relationship among EMT-ATFs. HMEC that have been either treated with TGFβ or subjected to overexpression of Snail1, Twist1, or Goosecoid, or knock-down for E-cadherin, display an overlapping gene signature characterized by the induction of ZEB1, ZEB2, and FOXC2 [149]. In any case, several of the signaling pathways (e.g., TGFβ, Wnt, Notch, TNFα/IL1β, receptor tyrosine kinase, etc.) inducing Snail and Twist factors also activate ZEB1 and ZEB2 expression directly. There is also considerable cross-talk among many of the upstream signals regulating all three EMT-ATF families. For instance, besides regulation of ZEB factors via TGFβ/Snail1-mediated inhibition of miR-200 [152], ZEB1 induction by Notch involves NFκB [71] and growth hormone induces ZEB2 via NAT [75]. TrkB induces EMT through a ZEB1-dependent mechanism that requires the cooperation of Snail1 and Twist1 [63].
Expression and functional redundancy among EMT-ATFs suggests that their specificity could occur at the spatial and temporal level, with Snail1 being required to initiate the EMT and, subsequently, Twist and ZEB factors to consolidate it [57, 152, 155].
EMT-ATFs in EMT and tumor invasiveness
ZEB1 and ZEB2
Expression of ZEB factors drives an EMT by repressing and activating epithelial and mesenchymal specification genes, respectively (reviewed in [33, 53, 54]). ZEB1 and ZEB2 both bind to E-box sequences in the E-cadherin promoter but recruit different set of corepresors: CtBP and SWI/SNF in the case of ZEB1, and CtBP and NuRD by ZEB2. ZEB proteins bind and repress the promoters of other epithelial markers such as P- and R-cadherins, cell polarity markers (Crumbs3, Pals1-associated tight junction protein, lethal giant larvae homologue 2), components of tight junctions (occludin, claudin 7, junctional adhesion molecule 1, zonula occludens protein 3), gap junctions (connexins 26 and 31), and desmosomes (desmoplakin, plakophilin 3) (reviewed in [53] and see also [14–16, 20] Conversely, ZEB proteins activate mesenchymal markers such as vimentin and N-cadherin. Although the mechanism of ZEB1- and ZEB2-mediated induction of mesenchymal genes has not been fully elucidated, in some cases (e.g., vimentin) it involves direct binding of ZEB proteins to the promoter regions of mesenchymal genes and transcriptional activation [156, 157]. ZEB1 and ZEB2 also repress epithelial splicing regulatory proteins-1 and -2 (ESRP-1 and -2) that coordinate the epithelial pattern of alternative splicing of FGFR2 and whose overexpression inhibits EMT [158].
Breakdown of the basement membrane, that separates the epithelial compartment from the surrounding stroma, is considered a key step in the progression from in situ to invasive carcinoma. Interestingly, ZEB1 regulates the expression of several components of the epithelial basement membrane (e.g., α3 chain of laminin 5, α2 chain of collagen IV, γ2 chain of laminin 5), although in a tissue-specific manner [69, 159, 160]. In colorectal carcinomas, the γ2 chain of laminin 5 coexpresses with MT1-MMP in cancer cells at the tumor invasive edge and cleavage of the former by the latter promotes cancer cell migration [69, 161].
As an inhibitor of the epithelial phenotype, ZEB1 is not expressed in normal epithelium, although it is found in isolated fibroblasts and immune cells in the interstitial stroma (Fig. 4). ZEB1 expression is also absent at the center of relatively well-differentiated carcinomas expressing E-cadherin [29, 159]. However, ZEB1 is highly expressed in invading dedifferentiated cancer cells of many tumors including colorectal, breast, liver, endometrial, lung, prostate, and pancreatic carcinomas (Table 1). Like ZEB1, ZEB2 is expressed by stroma cells in epithelial tissues (Fig. 4), but, interestingly, is also detected in normal E-cadherin-positive epithelial cells in several organs [162]. Nevertheless, upregulated expression of ZEB2 at the invasive front has been reported in most carcinomas expressing ZEB1, such as colorectal, breast, gastric, bladder, liver, and pancreatic (Table 1). In most of these tumors, there is also an increase in the number of ZEB1- and ZEB2-positive cells (fibroblasts, macrophages, and endothelial cells) in the peritumoral stroma—often at higher levels than in invading cancer cells—representing both tumoral cells that have undergone an EMT and activated stromal cells (Table 1; e.g., [16, 31, 159, 163–166]). It has been postulated that ZEB-dependent paracrine signaling from the stroma could cooperate in E-cadherin repression in other parts of the tumor [163].

ZEB1 and ZEB2 are expressed in stromal cells of normal colonic mucosa. Immunohistochemistry of colon samples from normal individuals stained by the 3,3′-diaminobenzidine (DAB) method with ZEB1 (H-102; Santa Cruz Biotechnology) and ZEB2 (H-260; Santa Cruz Biotechnology) as described in [69]. Scale bars 25 μm
Expression of ZEB proteins by both cancer and stromal cells at the invasive front of carcinomas translates into increased tumor invasiveness: ZEB proteins promote metastasis in xenograft models [131, 167] and associate with increased aggressiveness and higher metastatic capacity in a wide range of primary human carcinomas (e.g., [65, 70, 131, 164, 168–171]). ZEB factors have been shown to have an independent prognostic value for nodal dissemination, metastasis, response to treatment, and/or survival in multiple carcinomas, inter alia, of the ovary, bladder, colorectal, gastric, pancreas, and hepatocarcinoma [164, 166, 169, 171–176] (see representative entries with superscript letter in Table 1).
Snail1, Snail2 and Snail3
Independently of their inductive effects on ZEB1 and ZEB2, Snail factors also directly regulate epithelial and mesenchymal markers ([21, 177, 178] and references therein). In fact, the emerging consensus in the literature points to Snail1 as the factor responsible for the initiation of EMT in response to inducing signals [152, 155]. In addition to direct binding to the E-cadherin promoter and inhibition of its transcription, Snail factors repress other epithelial markers independently of their effect on E-cadherin—e.g., desmoplakin, adherens junction (claudin-1, -3, -4, -7), tight junctions (occludins), cytokeratins, and Mucin-1. On the other hand, Snail factors activate the expression of mesenchymal-like and pro-invasive genes (e.g., vimentin, fibronectin MMP1, MMP2, MMP7, MT1-MMP) that promote cell migration. While the epithelial and mesenchymal genes controlled by Snail1 and Snail2 overlap to a great extent, there is also evidence of some level of differential regulation in the degree of induction or repression and even in its direction [21]. For some genes, regulation requires cooperation between Snail1 and Snail2 or between Snail1 and Twist1: for example, Snail1 and Snail2 cooperate to transcriptionally repress the vitamin D receptor by which engagement by vitamin D3 promotes cell differentiation which is associated with a good prognosis in colorectal carcinomas [179, 180].
Snail factors are not present in normal epithelial cells but their expression is evidenced, usually higher for Snail1, in cancer cells at the invasive front of carcinomas of the breast, digestive tract, liver, pancreas, ovary ,and lung among others (Table 1). Characterization of Snail3 lags behind, but ESTs have been identified in melanomas and lung carcinomas. In addition to the tumor front, Snail1 is found at areas of inflammation in colorectal carcinomas [181]. Tumor progression correlates with increasing nuclear expression of Snail1, but Snail1 can also be aberrantly detected in the cytoplasm of cancer cells of several carcinomas. Additionally, strong Snail1 staining is found—often at a higher percentage than in tumor cells, when not even exclusively—among fibroblast-like cells, macrophages, and endothelial cells at the peritumoral stroma [181–184].
Expression of Snail factors correlates with malignancy and less differentiated tumors, lymph node invasion, and metastasis, and Snail1, but not always Snail2, is considered an independent prognostic factor of worst evolution and poorer survival in a large number of carcinomas [182, 185–189] (Table 1). Snail1 promotes the recurrence of Her2/neu-induced primary breast tumors in mice, and recurrent human carcinomas tend to display mesenchymal-like characteristics [186]. Thus, Snail1 is spontaneously induced in recurrent breast carcinomas and high levels of Snail1 are an independent predictor for decreased relapse-free survival in breast cancer patients. At least in head and neck carcinomas, only Snail1 nuclear staining, but not cytoplasmic, is associated with a worse clinical outcome [190]. Interestingly, reactivity for Snail1 in stromal but not in cancer cells correlates with distant metastasis and lower survival in early stage colorectal carcinomas [181].
Up to now, evidence for differential roles of Snail1 and Snail2 in cancer progression is scarce. In hepatocarcinomas, only Snail1 but not Snail2 correlates with E-cadherin loss [191], whereas another study reported that Snail2 but not Snail1 is associated with a poorer outcome in breast carcinoma patients [192].
Twist1 and Twist2
Twist factors induce EMT directly and indirectly through their effect on other EMT-ATFs. Thus, Twist1 has been shown to repress E-cadherin by binding to its promoter [19], but also through induction of Snail1 [193] or Snail2 [154]. In HMEC, Twist1 overexpression yields a similar gene signature than Snail1 with repression of E-cadherin and other epithelial specification genes and induction of mesenchymal markers [149]. Several array studies found that overexpression of Twist1 also upregulates the expression of cytoskeletal and extracellular matrix genes involved in cell motility. However, the extent to which induction of mesenchymal genes by Twist is dependent on E-cadherin repression is less clear [194]. Twist1 induces N-cadherin via directly driving its transcription [108] and/or through post-transcriptional mechanisms [195]. Nevertheless, in glioblastoma cells, Twist1 promotes expression of mesenchymal markers without eliciting an E-cadherin/N-cadherin switch [196]. Likewise, Twist1 induces fibronectin in gastric and ovarian cancer cells [195] but not in breast cancer cells [109]. In ovarian carcinoma cells, Twist1 also activates MMP2 and MT1-MMP, and its knock-down reduces cell adhesion to extracellular matrix components in parallel with a decline in several adhesion molecules (e.g., β1 integrin, CD44) [197].
A role for Twist1 in cancer was first evidenced by its ability to promote greater invasiveness and metastasis in xenograft models [19]. Like other EMT-ATFs, Twist1 and Twist2 are absent in normal epithelium but are induced in a number of human carcinomas, including those of the digestive tract, breast, liver, prostate, endometrium, and ovary (Table 1). Interestingly, the Twist1 gene has also been found to be deleted or amplified in some osteosarcomas [198]. Again, mirroring ZEB and Snail factors, Twist1 and Twist2 are not only upregulated among cancer cells at the invasive front of carcinomas but also in stromal cells. Likewise, upregulation of Twist factors is not limited to the nuclei of cancer cells but, in many cases, also in their cytoplasm (e.g., [199–202]).
Twist factors are not only upregulated in human cancers but their reactivity increases during tumor progression from being mostly negative in benign neoplasias to be highly overexpressed in carcinomas of a wide range of tissue origins (Table 1). Knock-down of Twist1 in breast cancer cells inhibits their ability to metastasize in xenograft models but not the formation of primary tumors [19]. Twist factors also correlate with higher tumor grade, invasiveness, and metastasis, being independent prognostic factors for enhanced tumor aggressiveness, tumor recurrence, and poorer patient survival (e.g., [155, 191, 197, 199, 203–207]). Interestingly, in hepatocarcinomas, expression of either Twist1 or Snail1 is associated with shorter survival, and their simultaneous presence has an additive negative effect, suggesting that they somehow play distinct but collaborative roles in cancer progression [191]. In that line, another study in hepatocarcinomas found that Twist1 associates with increased tumor angiogenesis and metastasis but not with downregulation of E-cadherin [205]. Differential expression of Twist and Snail factors in some studies suggests a division of labor among EMT-ATFs. Breast carcinomas with lymph node involvement and poor clinical outcome display high levels of Twist1 and Snail2 while, contrary to other reports, expression of Snail1 declined [192].
Other transcription factors inducing EMT and tumor invasiveness
Like the ZEB, Snail, and Twist factors, E12/E47 binds directly to E-cadherin promoter [6] and induces an EMT by regulating an overlapping—but still distinct—set of epithelial and mesenchymal markers compared to Snail1 and Snail2 [21]. E12/E47 complexes not only promote tumor invasiveness and growth but injection of Madin–Darby Canine Kidney (MDCK) cells overexpressing E47 into nude mice generates tumors with higher invasiveness, growth, and vascularization than those formed by MDCK cells overexpressing Snail1 [177]. T-box transcription factor Tbx3 also binds directly to the E-cadherin promoter and inversely correlates with E-cadherin in human melanomas [7].
The homeobox factor Goosecoid also induces a full EMT by repressing epithelial markers (including E-cadherin) and activating mesenchymal genes [8]. Goosecoid is induced by TGFβ in adult breast epithelial cells and, compared to normal breast tissue, is significantly overexpressed in atypical ductal hyperplasia and ductal breast carcinomas [8].
FOXC2 also induces a mesenchymal phenotype but, departing from the rest of EMT-inducing factors reviewed here, FOXC2 does not alter E-cadherin mRNA levels but rather delocalizes E-cadherin protein from the plasma membrane to the cytoplasm [208]. FOXC2 is induced by TGFβ, and in response to overexpression of Snai1, Twist1, and Goosecoid, and has been suggested to mediate activation of mesenchymal genes by these other EMT-ATFs. FOXC2 is not expressed in normal epithelium but is overexpressed in basal-like breast and esophageal squamous cell carcinomas where it correlates with adverse prognosis [208, 209].
Other roles of EMT-ATFs: beyond EMT and tumor invasiveness
In addition to promoting tumor invasiveness, over the last few years a number of works have demonstrated that EMT-ATFs are also critical during earlier stages of cancer development, collaborating with oncogenes in malignant transformation, inducing tumor formation, contributing to bypass failsafe programs against cancer, and determining resistance to chemotherapy. While these additional cancer hallmarks are not necessarily dependent on EMT, they are jointly regulated with the EMT by EMT-ATFs. While many of these functions are interconnected, they will be addressed here in turn.
EMT, cancer stem cells, and tumorigenesis
The great level of heterogeneity displayed by most human tumors led to the formulation in the 1990s of a hierarchical model of cancer initiation where a small subpopulation of cells in a tumor—referred to as cancer stem cells (CSCs)—retain the capacity for self-renewal and tumor initiation (tumorigenesis) (reviewed in [28]). CSCs have been identified in a number of human cancers including colon, breast, pancreas, ovary, prostate, and brain tumors.
As indicated earlier, Brabletz and coworkers [27] postulated that invading carcinoma cells that have undergone an EMT could function as “migrating CSCs”. Later studies by the Weinberg group and others confirmed that forcing an EMT in non-tumorigenic, immortalized HMECs—by treatment with TGFβ or overexpression of EMT-ATFs—concurrently confers on cells a phenotype similar to breast CSCs (CD44high/CD24low, miR-200low), along with the capacity to form mammospheres, self-renewal, and increasing tumorigenecity in xenotransplants [12, 26, 132, 202]. The ability of miR-200 to suppress colony formation in vitro and tumorigenesis in vivo depends on their repressor effect on Bmi1 and ZEB1 [132].
Signaling pathways that control stem cell homeostasis during embryogenesis and later in adults (e.g., Wnt, Shh, Notch) are active in cancer and trigger—via induction of EMT-ATFs—not only EMT but also stem-like properties [3]. Acquisition and maintenance of stemness by EMT-ATFs could explain the capacity of these factors to induce tumorigenesis and promote recurrence and metastasis.
Isolation of stem-like (CD44high/CD24low) and non-stem-like (CD44low/CD24high) populations from resected primary breast tumors identified high levels of mesenchymal markers (N-cadherin, vimentin, fibronectin) including EMT-ATFs (ZEB2, Snail1, Snail2, Twist1 and Twist2) in the first set but not the second [26]. While ZEB1 did not show up in this study, ZEB1 was found to be uniquely required to maintain the viability and stem-like phenotype of spheres—a property classically linked to stem cells—formed from mouse embryo fibroblasts (MEFs) with targeted deletions of Rb1 and Rb family members [210]. In pancreatic carcinomas, ZEB1 determines stemness in cancer cells by direct repression of several miRs (miR-200, miR-183, miR-203) targeting stemness regulators Bmi1, KLF4, and Sox2 [131]. By contrast, activation of miR-200c by p53 reduces ZEB1 expression and suppresses both EMT and stemness, while downregulation of miR-200c—upon loss of p53 or overexpression of oncogenic p53 mutants—increases ZEB factors and stem cell markers in mammary and pancreatic acinar epithelial cells [128, 130] (Fig. 2).
Similar results have been obtained in other models. Spheres formed out of primary colorectal cancer cells also express high levels of Snail1, whose overexpression upregulates stem cell markers [211]. Another study found that overexpression of Snail1 or Snail2 in ovarian cancer cells derepresses a stemnes gene signature (e.g., Nanog, KLF4, Oct4, Bmi1, Nestin) and increases the number of CSC-like CD44+/C117+ cells, suggesting that CSCs originate from the dedifferentiation of non-stem cancer cells rather than proliferation of existing CSCs [212]. In that line, oncogenic transformation of mammary stem-like cells produces more aggressive tumors than transformation of differentiated mammary epithelial cells and enhances the conversion of non-CSCs into CSCs [213].
The capacity of EMT-ATFs to generate CSCs is important to explain some other EMT-ATFs functions, like enhanced survival, tumor recurrence, and metastasis. The possibility that cancer cells could transition between tumorigenic and non-tumorigenic states [213], and that EMT-ATFs expression could modulate this balance, represents an important avenue in therapy. In the same line, the association between expression of EMT-ATFs by cancer cell lines and primary tumors and resistance to DNA damage and chemotherapy may also be related to EMT-ATFs’ capacity to generate CSCs (see below). It remains to be firmly established whether, in line with the migrating CSC model [27], the joint induction by EMT-ATFs of a proinvasive/metastatic phenotype and a tumorigenic/stemness capacity in cell systems and xenograft models also occurs in human tumors.
EMT-ATFs in resistance to apoptosis and anoikis
EMT allows cancer cells to overcome safeguard mechanisms and become more resistant to signals triggering programmed cell death. Aberrant expression of EMT-ATFs by tumors may thus promote tumorigenesis and tumor growth through increased resistance to apoptosis. EMT triggers pro-survival programs in response to multiple apoptotic stimuli through mechanisms closely linked to the cell cycle. For instance, in an hepatocyte cell line TGFβ triggers an EMT when cells are in G1/S but apoptosis during G2/M [214]. Both Twist1 and Twist2 inhibit myc- and p53-dependent apoptosis by repressing p19ARF [215, 216]. Twist1 also inhibits p53 targets by direct interaction and blocking of the DNA binding domain of p53 [217]. Overexpression of Snail1 induces an EMT at the same time as suppressing TGFβ-induced apoptosis of non-transformed hepatocytes and hepatocarcioma cell lines [218]. Snail1 also protects cells against cell death caused by TNFα or growth factor withdrawal by activating MEK and PI3K signaling [219]. Direct repression by Snail factors of the promoters of p53 and pro-apoptotic target genes protects cells against apoptosis induced by DNA damage in response to genotoxic stress (e.g., BID, PIG8, caspase 6, DFF40) [212, 220]. In hematopoietic progenitors, Snail2 is induced by p53 in response to DNA damage and protects against apoptosis by repressing p53-mediated induction of Puma, an inhibitor of Bcl2 [221]. By contrast, mutant p53 in NSCLC cells results in low Mdm2 and stabilization of Snail2 [222]. In neuroblastomas, overexpression of Snail2 induces Bcl2 [223], and cancer cells with mutant K-Ras that have undergone EMT require Snail2 expression for their survival [224]. In different cell systems, ZEB1 directly inhibits pro-apoptotic TAp73 but also anti-apoptotic ΔNp73 and ΔNp63 [225, 226]. The pro-survival effect of ZEB2 is, however, independent of cell cycle arrest and intercellular adhesion and is mediated through inhibition of cleavage of PARP and pro-caspase 3 and phosphorylation of ATM/ATR substrates [164].
As cancer cells detach from the tumor, start migrating across the stroma, and intravasate into the circulation, they lose most (if not all) of their intercellular contacts and adhesion to the original extracellular matrix. Just like migrating progenitors during development, metastatic cancer cells are able to survive in this new environment, a condition that in normal cells triggers a caspase-dependent apoptosis program known as anoikis or anchorage-dependent cell death (recently reviewed in [227]). Acquisition of resistance to anoikis is therefore a critical cancer cell capability during tumor invasion and metastasis. Downregulation of E-cadherin expression is sufficient to determine resistance to anoikis [14, 194, 228]. ARF induces apoptosis and anoikis, and downregulation of ankyrin G—a binding partner of E-cadherin that concentrates it at sites of cell–cell contact—during EMT promotes the formation of NRAGE–Tbx2 complexes that represses ARF [229]. Several EMT-inducing pathways (e.g., receptor tyrosine kinase, oncogenic Ras, hypoxia) trigger resistance to anoikis through activation of PI3K-AKT and ERK signaling and regulation of Bcl2 family members. Likewise, in different cell systems and conditions, expression of ZEB, Snail, and Twist factors induces anchorage-independent cell growth [14, 64, 194, 230]. In a seminal contribution by Frisch’s group, the capacity of ZEB1 conferring resistance to anoikis was shown to be dependent on the formation of ZEB1/CtBP E-cadherin repressor complexes, in a process reversed by E1a [14]. Snail1 expression promotes resistance to anoikis by activation of the MAPK and PI3K cascades [219]. The tyrosine kinase receptor TrkB, overexpressed in many human cancers, is a known inducer of both EMT and anoikis resistance [63]. Ligand activation of TrkB activates MAPK signaling leading to direct induction of Snail1, Twist1, and ZEB1, but with ZEB1 as the ultimate effector in TrkB-mediated resistance to anoikis. It is of note that TrkB is also a direct target of miR-200 in endometrial and breast carcinoma cells [231], forming yet another miR-regulated regulatory loop, this time for the control of anoikis resistance (Fig. 2a).
EMT-ATFs in regulation of cell cycle, senescence and transformation
Like apoptosis, cell cycle arrest by senescence represents a crucial safeguard mechanism against cancer. In addition to telomerase shortening during replicative senescence, oncogenic transformation and DNA damage can trigger cell cycle arrest and senescence through induction of the p53 and p16INK4a/Rb pathways [232]. Oncogene-induced senescence has been identified in precancerous lesions and needs to be overcome for progression to full tumorigenic status [232, 233]. Evidence accumulated in recent years shows that EMT-ATFs allow cancer cells to bypass senescence thus contributing to the continuous proliferation of immortalized cells. In the same line, an inflammatory environment in the tumor area not only drives cancers cells into an EMT program but also overrides oncogene-induced senescence [234].
ZEB1 transcription, but not ZEB2 or Snail1, is inhibited by the p16INK4a/Rb1 tumor suppressor pathway and in turn represses p15INK4b, p19ARF, and p21CIP/WAF1 ([48], [76], [235]; see [236] for a comprehensive review). Loss of Rb1 is not only involved in tumor initiation but has been reported to also induce EMT [237], and may contribute to overexpression of ZEB1 in proliferating cells and in many primary tumors. MEFs from ZEB1 (+/−) and ZEB1 (−/−) mice undergo premature replicative senescence in a dose-dependent manner compared to wild-type MEFs [235]. Likewise, progenitors in the palate, skeleton, and nervous system in the ZEB1 (−/−) mice display decreased proliferation [235]. Oxidative stress induces senescence through miR-200-mediated inhibition of ZEB1 [224]. As indicated earlier, p53 represses ZEB1 expression through induction of miR-200, [128, 129]. However, while ZEB1 represses ΔNp63 and both isoforms of p73, it does not affect TAp63 or p53 [225].
Evidence for ZEB2 points in both directions—promoting and reverting senescence—perhaps reflecting cellular background differences. ZEB2 induces a G1 arrest in epidermoid and bladder carcinoma cell lines by direct transcriptional repression of cyclin D1 [164, 238]. Overexpression of cyclin D1 uncouples ZEB2-mediated cell cycle arrest from EMT [238]. Contrary to other EMT-ATFs, ZEB2 induces senescence in hepatocarcinoma cells by inhibiting hTERT [239]. However, overexpression of ZEB2 in lung epithelial cells raises the concentration of TGFβ needed to trigger growth arrest [48], and conditional targeted deletion of ZEB2 (−/−) in the developing cerebral cortex decreases proliferation of neural precursor cells [240]. Expression of both ZEB1 and ZEB2 has been shown to abrogate EGFR-induced senescence while their knock-down induces p15INK4b and p16INK4a, thus reactivating the senescence program [241]. In this direction, miR-200 induces senescence in endothelial cells by inhibiting ZEB1 [224]. The regulatory loop between p53/miR-200 and ZEB1/ZEB2 therefore seems to also be involved in control of senescence (Fig. 2a). The ability of ZEB factors to override senescence is closely tied to their activation of EMT: triggering of senescence by knock-down of ZEB1 and ZEB2 (or p53) makes cells insensitive to the EMT-driving effects of TGFβ [241].
In the developing embryo, Snail1 inversely correlates with areas of proliferation and cyclin D2 expression, results confirmed in canine kidney epithelial cells where Snail1 transcriptionally represses cyclin D2 and associates to high levels of p21CIP/WAF1 and G1 arrest [219, 242]. In this line, knock-down of Snail1 drives prostate cancer cells into senescence [243]. Snail2 is also excluded from areas of proliferation in the neural tube of the developing chick [219], and its overexpression in prostate cancer cells represses cyclin D1 [244]. However, other groups have reported opposite results, suggesting that, as in the case of ZEB2, the role of EMT-ATFs in cell cycle arrest and senescence may be cell type-dependent. Thus, in osteosarcoma cells, Snail1 inhibits E12/E47 transcriptional activation of the p21CIP/WAF1 promoter, cooperating in this function with Twist1 [245]. Likewise, in breast cancer cells, Snail2 upregulates cyclin D1 and fosters proliferation by forming a complex with CtBP and transcriptionally repressing UbcH5c, an ubiquitin that targets cyclin D1 [246].
Growth arrest and senescence is also achieved by knocking down Twist1 in immortalized non-malignant prostate cells, while its overexpression inhibits p53-dependent senescence via inhibition of ARF [247]. Repression of p14ARF also results in inhibition of Chk1/2 phosphorylation in response to DNA damage [247]. Twist1 and Twist2 override RasV12-induced senescence of cell lines, and of breast cancer cells in MMTV-Erb2/Neu transgenic mice, by inhibition of p16INK4a and p21CIP/WAF1 [248]. This study also found that Twist factors cooperate with RasV12 in the transformation of MEFs, an effect that was abolished when Ras-induced senescence was inhibited. Twist1 cooperates with N-myc in the transformation of wild-type but not INK4a/ARF (−/−) MEFs, confirming that Twist activity involves inhibition of the ARF/p53 pathway [216]. As in the case of ZEB proteins, Twist1/2-mediated override of senescence is linked to their ability to induce an EMT and promote tumor invasion.
EMT-ATFs and angiogenesis
Snail1 is required for proper vascular development during embryogenesis, and its overexpression in embryonic stem cells induces the formation of VEGFR-2-positive endothelial cells through downregulation of miR-200 [249].
The growth of a tumor from an avascular hyperplasia into a larger mass requires the formation of new vessels through a process known as the angiogenic switch, which involves the production of angiogenic factors and proteases by tumor and stromal cells. Loss of E-cadherin is sufficient to trigger this angiogenic switch. Conditional knock-down of E-cadherin in mouse models of NSCLC promotes the development of tumor vasculature and growth through upregulation of vascular growth factors VEGF-A and VEGF-C and its receptor Flt-4 [250]. Angiogenesis is classically triggered by inflammation and hypoxia, conditions that are also known inducers of EMT [116]. Hypoxic conditions also increase the population of cancer cells with stem-like phenotype [251]. Stabilization of HIF-1α in response to hypoxia or its overexpression promotes invasion and metastasis by directly activating Twist1 and inducing an EMT [116]. Likewise, knock-down of HIF-1α or overexpression of the VHL gene—whose product targets HIF-1α for ubiquitin-mediated degradation—downregulates expression of ZEB1, ZEB2, and Snail1 [60, 252]. Additionally, HIF-1α feeds into upstream EMT-inducing signals (e.g., Notch, NFκB) [104]. Twist1-mediated induction of EMT by HIF-1α is not redundant with Snail1, suggesting that both factors have distinct but complementary roles in the pro-invasive and pro-angiogenic response to hypoxia. In that line, joint expression of HIF-1α, Twist1, and Snail1 in primary HNSCC associates with poorer prognosis [116].
Most EMT-ATFs promote tumor angiogenesis in vivo. ZEB, Snail, and Twist factors are often overexpressed by endothelial cells in the peritumoral stroma. Expression of Twist1 also associates with enhanced tumor microvessel vasculature and VEGF expression in hepatocarcinomas [205]. In mice, injection of lung adenocarcinoma cell lines overexpressing Snail1 or Snail2 generated tumors with enhanced vasculature compared to control cells [177, 253, 254], that in the case of Snail1 is accompanied by higher levels of proangiogenic factors CXCL5 and CXCL8 [254]. Likewise, compared to control cells, xenotransplant of breast cancer cell lines overexpressing Twist1 generate tumors with higher tumor angiogenesis and upregulated expression of several key vascular growth factors and receptors (e.g., VEGF, VEGFR2/KDR, Angiotensin-2, chemokine GRO-α, and CD31) [255, 256]. ZEB2 also promotes angiogenesis by direct transcriptional repression of the anti-angiogenic homeobox GAX factor [134]. However, and contrary to what would be expected from its induction by HIF-1α and its role as promoter of tumor progression, ZEB1 can also function as a negative regulator of angiogenesis in vivo. Xenotransplanted melanoma cells develop larger tumors with more developed vascularization in mice with a haploinsufficient ZEB1 background [257].
EMT-ATFs in oncogenic addiction and resistance to therapy
Activation of oncogenic pathways (or loss/inactivation of tumor suppressor signals) induces pro-survival and pro-growth signals on which tumors can become dependent. This dependency of cancer cells on oncogenes—referred as “oncogenic addiction”—has been exploited in the development of new chemotherapy drugs [258]. The success of antibodies and drugs targeting specific oncogenes in the treatment of a number of solid and hematologic cancers in mice models and humans has helped to reinforce the oncogenic addiction concept.
EMT and EMT-ATFs allow cancer cells to overcome their dependency on the oncogenic signals originally involved in their transformation. Thus, the epithelial status of pancreatic and lung cancer cells with activating mutations of K-Ras determines their dependency on K-Ras for their growth and survival [259]. Cancer cells that are dependent on K-Ras exhibit epithelial characteristics, while those independent of K-Ras have a mesenchymal phenotype. Induction of EMT by TGFβ or ZEB1 overrides K-Ras addiction protecting cancer cells from apoptosis following K-Ras knock-down. Conversely, elimination of ZEB1 in K-Ras-independent cancer cells restores K-Ras dependency [261]. Snail2 is also required for the survival of colorectal cancer cells with mutant K-Ras [260]. These results support EMT as a mechanism for cancer cells to escape from oncogenic addiction and highlight the potential of EMT-ATFs as therapeutic targets.
In the same line, and in parallel with the anti-apoptotic role of EMT described earlier, a wealth of articles have shown an association between EMT and chemotherapy resistance. Cancer cell lines expressing E-cadherin are more sensitive to chemotherapy drugs compared to those displaying a mesenchymal phenotype [261–264]. Resistance to chemotherapy and hormone therapy in patients with breast carcinomas correlates with tumoral expression of stem and mesenchymal markers. Poor response of HNSCC and NSCLC to EGFR inhibitors gefitinib and erlotinib in cell lines, human tumors, and xenograft mice models is associated with expression of EMT and stem-like cell markers [265–268]. Acquisition of resistance to oxaliplatin in colorectal carcinoma cells is also accompanied by expression of mesenchymal markers [261]. Responsiveness to cetuximab (a chimeric mouse–human antibody against EGFR) in Ras wild-type colorectal cancer cells depends on high expression of epithelial markers and low levels of ZEB1, Snail1, and Snail2, and of stem-like phenotype [269]. It is worth noting that the EMT-inducing effect of some drugs is cell-cycle dependent: e.g., in breast cancer cells, doxorubicin induces a mesenchymal phenotype during G1/S and apoptosis in G2/M [270]. The peritumoral stroma also plays an important role in chemoresistance to EGFR inhibitors. Interestingly, in a xenograft model of EGFR-resistant NSCLC, cancer-associated fibroblasts derived out of tumor cells that have undergone an EMT are not only EGFR-resistant but also tumorigenic [268].
The association between EMT and drug resistance is mediated, at least in part, by EMT-ATFs (Table 2). Expression of different EMT-ATFs by cancer cell lines and primary tumors confers tumor cells resistance to chemotherapy and radiotherapy. Resistance to doxorubicin in breast carcinoma cell lines correlates with higher expression of ZEB1 and SIRT1 [271]. In NSCLC cell lines, expression of ZEB1—but not of ZEB2, Snail1, or Snail2—correlates with higher resistance to gefitinib [262]. ZEB1 is also associated with resistance to erlotinib in HNSCC cell lines, and its knock-down increases drug sensitivity with the induction of E-cadherin expression [272]. Interestingly, simultaneous knock-down of ZEB1 and E-cadherin cancels out sensitization to erlotinib by ZEB1 elimination, suggesting that sensitivity to this EGFR inhibitor requires E-cadherin expression. Depletion of ZEB1 also sensitizes pancreatic cancer cell lines to gemcitabine, 5-fluorouracil, and cisplatin, and gemcitabine-resistant clones express higher levels not only of ZEB1 but also of Snail1 and Snail2 [71, 131, 273] (Table 2).
ZEB2 determines resistance to treatment independently of ZEB1, protecting bladder and squamous carcinoma cell lines against DNA damage-inducing agents such as cisplatin or UV radiation [164]. Importantly, patients with ZEB2-negative bladder carcinomas also exhibit better response to radiotherapy [164].
Snail1 is upregulated in NSCLC xenotransplanted tumors resistant to EGFR inhibitors [268] and determines resistance to cisplatin in HNSCC primary tumors and HNSCC and NSCLC cell lines [274, 275] (Table 2). Snail1-related resistance to cisplatin in HNSCC cell lines is mediated by activation of the DNA excision repair protein ERCC1 [275]. Snail1 expression also confers resistance to 5-fluorouracil in breast carcinoma cell lines [276]. In contrast, other studies have found that resistance to gefitinib displayed by some NSCLC cell lines and developed by primary lung adenocarcinomas associated with overexpressed Snail2—but not Snail1, Twist1, or ZEB1—being reversed by Snail2 knock-down through a mechanism involving upregulation of Bim and activation of caspase 9 [277]. In breast carcinoma cell lines refractory to doxorubicin, knock-down of Snail1 and Snail2 have a synergistic effect inducing drug sensitiveness, suggesting that Snail1 and Snail2 determine resistance through non-overlapping mechanisms. Chemotherapy resistance induced by EMT-ATFs is not only tightly linked to the induction of mesenchymal markers but also of stemness. Knock-down of Snail1 and Snail2 in ovarian cancer cell lines increases their sensitivity to cisplatin [278]. As noted earlier, Snail1- and Snail2-induced resistance to paclitaxel and radiation in ovarian cancer cells relates to the repression of pro-apoptotic p53 targets and activation of stemness markers [212]. In malignant mesotheliomas, activation of SCF/c-kit signaling by Snail2 induces multidrug resistance [279].
Twist proteins are also involved in drug resistance. In breast carcinoma cells, Twist1 mediates resistance to paclitaxel via direct transcriptional activation of AKT2 [109]. Twist1 knock-down increases sensitivity of breast cancer cells to doxorubicin—a drug that triggers a p53-dependent DNA damage response—by disrupting p53-Mdm2 association [270]. Independently of p53 and p19ARF pathways, Twist1 and Twist2 block breast cancer cells death by daunorubicin by suppressing daunorubicin-induced phosphorylation of Bcl-2 [280]. In nasopharyngeal, bladder, ovarian, and prostate cancer cell lines, Twist1 increases resistance to paclitaxel and/or vincristine by upregulating Bcl2 and lowering Bax and Bak [281–283].
Concluding remarks
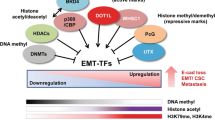
In the span of just a few years, EMT-ATFs have evolved from simple repressors of E-cadherin to inducers of most of the traits that cancer cells need to acquire for successful tumor progresssion (Table 3). Cancer cells can acquire these capabilities through overexpression, gain-of-function mutations, or amplification of oncogenes and/or repression, mutation or deletion of tumor suppressors [1], and mounting evidence indicates that EMT-ATFs critically regulate these cancer hallmarks. From participating only at late stages in cancer (promoting EMT and metastasis), EMT-ATFs are now known to be also involved in the initial phases of tumor development. First, EMT and metastasis may be a much earlier event in cancer progression than previously thought, playing an important role in tumor formation itself [31]. But, more importantly, ZEB, Snail and Twist factors regulate the acquisition of key early cancer hallmarks: EMT-ATFs contribute to overriding cancer safeguard programs (apoptosis, senescence), promoting tumor angiogenesis, cooperating with (or mediating) oncogenic signals (e.g., Ras), and antagonizing tumor suppressor pathways (e.g., p53) (Table 3). In turn, classical tumor repressors like Rb or p53 have recently been shown to regulate EMT and tumor invasiveness [128, 237].
The attention of molecular oncologists is moving from an early focus on the identification of irreversible mechanisms of cancer initiation and progression (e.g., mutations/deletions, amplifications) to the discovery of the dynamics involved in reversible programs of gene regulation in cancer cells (e.g., epigenetic/transcriptional, translational). Human tumors display a great level of heterogeneity, not only among patients but also within different areas and stages in a given tumor. Dynamic regulated expression of miRs and EMT-ATFs grants cancer cells with a great level of functional plasticity [54]. These transcriptional and translational regulatory networks allow cancer cells to reversibly transition between different states as they adapt to the environment during tumor progression, not only between epithelial and mesenchymal phenotypes but also between stemness and differentiation or between proliferation and growth arrest.
The data reviewed here attest to a significant level of overlapping among EMT-ATFs in their pattern of expression, mechanisms of action, target genes and regulation of hallmarks of cancer. Increasing specificity and temporal and spatial hierarchies among EMT-ATFs are progressively being revealed with the identification of an expanding set of upstream regulatory miRs and translational regulators [149, 152, 155]. Similarly, the discovery of new cofactors and chromatin remodeling complexes used by EMT-ATFs to transcriptionally regulate their targets also points to some divergence; even if several EMT-ATFs are coexpressed, availability of these cofactors may dictate the functional capabilities of the EMT-ATFs.
Codification of the hallmarks of cancer by Hanahan and Weinberg [1] has helped researchers not only to systematize their findings and current understanding of cancer but has also contributed to guiding new therapy strategies. Compared to the irreversible effects of mutations and deletions, the dynamic regulation of miRs and EMT-ATFs renders them attractive targets for personalized oncology treatment. Incorporating the analysis of EMT-ATF expression in primary tumors into routine pathology diagnosis could help to prospectively identify resistance to particular chemotherapy. Since chemotherapies targeting a single oncogenic signal or cancer cell trait are not failsafe against resistance and tumor recurrence, a simultaneous approach to several signaling pathways and cancer hallmarks could be more successful. The highly modular structure and complex transcriptional activities of EMT-ATFs and their simultaneous regulation of multiple cancer hallmarks makes them attractive therapeutic targets for translational researchers.
On the other hand, reversibility of the EMT/MET balance and plasticity in its regulation represent adaptive mechanisms developed by cancer cells. Reverting the mesenchymal phenotype of invading cells by blocking the expression and/or function of EMT-ATFs may attenuate the ability of cancer cells to invade and increase their sensitivity to chemotherapy. However, as metastatic cancer cells need to regain an epithelial phenotype to grow into a macroscopic tumor at the site of distant colonization, reverting the mesenchymal status of tumor cells may actually foster metastasis. This is illustrated by the somewhat paradoxical metastatic-enhancing effect of miR-200 [125, 313]. In this regard, if reversible and dynamic regulation of miR and EMT-ATFs during cancer progression offers new avenues for therapy, it also complicates their manipulation. More information of the upstream regulatory networks and mechanisms of transcriptional regulation by EMT-ATFs is therefore needed before interference of their expression/function can be considered in cancer treatment. As far as we know, induction of EMT by EMT-ATFs occurs along with their regulation of other hallmarks (e.g., apoptosis, senescence). It remains to be ascertained how regulation of these hallmarks overlaps at the mechanistic level. To the extent that separate mechanisms are discovered, therapeutic approaches could be more precisely targeted.
In sum, as our understanding on the expanding roles of EMT-ATFs improves, these factors are likely to become not only prognostic and predictive personalized biomarkers in cancer but also important therapeutic targets in the near future.
Abbreviations
- bHLH:
-
Basic helix loop helix
- CSC:
-
Cancer stem cells
- EMT:
-
Epithelial-to-mesenchymal transition
- EMT-ATF:
-
EMT-activating transcription factors
- HNSCC:
-
Head and neck squamous cell carcinoma
- MET:
-
Mesenchymal-to-epithelial transition
- NSCLC:
-
Non-small cell lung carcinoma
References
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119(6):1420–1428
Thiery JP, Acloque H, Huang RY, Nieto MA (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139(5):871–890
Lee K, Nelson CM (2012) New insights into the regulation of epithelial-mesenchymal transition and tissue fibrosis. Int Rev Cell Mol Biol 294:171–221
Nieto MA, Cano A (2012) The epithelial-mesenchymal transition under control: Global programs to regulate epithelial plasticity. Semin Cancer Biol. doi:10.1016/j.semcancer.2012.05.003 (Ahead of print)
Perez-Moreno MA, Locascio A, Rodrigo I, Dhondt G, Portillo F, Nieto MA, Cano A (2001) A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J Biol Chem 276(29):27424–27431
Rodriguez M, Aladowicz E, Lanfrancone L, Goding CR (2008) Tbx3 represses E-cadherin expression and enhances melanoma invasiveness. Cancer Res 68(19):7872–7881
Hartwell KA, Muir B, Reinhardt F, Carpenter AE, Sgroi DC, Weinberg RA (2006) The Spemann organizer gene, Goosecoid, promotes tumor metastasis. Proc Natl Acad Sci USA 103(50):18969–18974
Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin CH, Moustakas A (2006) Transforming growth factor-β employs HMGA2 to elicit epithelial-mesenchymal transition. J Cell Biol 174(2):175–183
Baum B, Georgiou M (2011) Dynamics of adherens junctions in epithelial establishment, maintenance, and remodeling. J Cell Biol 192(6):907–917
Capaldo CT, Macara IG (2007) Depletion of E-cadherin disrupts establishment but not maintenance of cell junctions in Madin-Darby canine kidney epithelial cells. Mol Biol Cell 18(1):189–200
Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, Bell G, Guo W, Rubin J, Richardson AL, Weinberg RA (2011) Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 145(6):926–940
Iorio MV, Croce CM (2012) MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med 4(3):143–159
Grooteclaes ML, Frisch SM (2000) Evidence for a function of CtBP in epithelial gene regulation and anoikis. Oncogene 19(33):3823–3828
Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F (2001) The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell 7(6):1267–1278
Aigner K, Dampier B, Descovich L, Mikula M, Sultan A, Schreiber M, Mikulits W, Brabletz T, Strand D, Obrist P, Sommergruber W, Schweifer N, Wernitznig A, Beug H, Foisner R, Eger A (2007) The transcription factor ZEB1 (δEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 26(49):6979–6988
Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA (2000) The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2(2):76–83
Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, García De Herreros A (2000) The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2(2):84–89
Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA et al (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117(7):927–939
Vandewalle C, Comijn J, De Craene B, Vermassen P, Bruyneel E, Andersen H, Tulchinsky E, Van Roy F, Berx G (2005) SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res 33(20):6566–6578
Moreno-Bueno G, Cubillo E, Sarrio D, Peinado H, Rodriguez-Pinilla SM, Villa S, Bolos V, Jorda M, Fabra A, Portillo F, Palacios J, Cano A (2006) Genetic profiling of epithelial cells expressing E-cadherin repressors reveals a distinct role for Snail, Slug, and E47 factors in epithelial-mesenchymal transition. Cancer Res 66(19):9543–9556
Friedl P, Alexander S (2011) Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147(5):992–1009
Hanahan D, Coussens LM (2012) Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21(3):309–322
Bierie B, Moses HL (2006) Tumour microenvironment: TGFβ: the molecular Jekyll and Hyde of cancer. Nature Rev Cancer 6:506–520
Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E (2009) Localized and reversible TGFβ signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol 11(11):1287–1296
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133(4):704–715
Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T (2005) Opinion: migrating cancer stem cells—an integrated concept of malignant tumour progression. Nat Rev Cancer 5(9):744–749
Magee JA, Piskounova E, Morrison SJ (2012) Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell 21(3):283–296
Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, Knuechel R, Kirchner T (2001) Variable β-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci USA 98(18):10356–10361
Brabletz T (2012) To differentiate or not—routes towards metastasis. Nat Rev Cancer 12(6):425–436
Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, Leach SD, Stanger BZ (2012) EMT and dissemination precede pancreatic tumor formation. Cell 148(1–2):349–361
Gheldof A, Hulpiau P, van Roy F, De Craene B, Berx G (2012) Evolutionary functional analysis and molecular regulation of the ZEB transcription factors. Cell Mol Life Sci 69(15):2527–2541
Sanchez-Tillo E, Siles L, Barrios O, Cuatrecasas M, Vaquero EC, Castells A, Postigo A (2011) Expanding roles of ZEB Factors in tumorigenesis and tumor progression. Am J Cancer Res 1(7):897–912
Postigo AA, Dean DC (1997) ZEB, a vertebrate homolog of Drosophila Zfh-1, is a negative regulator of muscle differentiation. EMBO J 16(13):3935–3943
Postigo AA, Sheppard AM, Dean ML, Mucenski DC (1997) c-Myb and Ets proteins synergize to overcome transcriptional repression by ZEB. EMBO J 16(13):3924–3934
Postigo AA, Dean DC (1999) Independent repressor domains in ZEB regulate muscle and T-cell differentiation. Mol Cell Biol 19(12):7961–7971
Postigo AA, Dean DC (2000) Differential expression and function of members of the zfh-1 family of zinc finger/homeodomain repressors. Proc Natl Acad Sci USA 97(12):6391–6396
Postigo AA, Dean DC (1999) ZEB represses transcription through interaction with the corepressor CtBP. Proc Natl Acad Sci USA 96(12):6683–6688
Postigo AA, Ward E, Skeath JB, Dean DC (1999) zfh-1, the Drosophila homologue of ZEB, is a transcriptional repressor That Regulates somatic myogenesis. Mol Cell Biol 19(12):7255–7263
Furusawa T, Moribe H, Kondoh H, Higashi Y (1999) Identification of CtBP1 and CtBP2 as corepressors of zinc finger-homeodomain factor δEF1. Mol Cell Biol 19(12):8581–8590
van Grunsven LA, Michiels C, Van de Putte T, Nelles L, Wuytens G, Verschueren K, Huylebroeck D (2003) Interaction between Smad-interacting protein-1 and the corepressor C-terminal binding protein is dispensable for transcriptional repression of E-cadherin. J Biol Chem 278(28):26135–26145
Shi Y, Sawada J, Sui G, el Affar B, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y, Shi Y (2003) Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422(6933):735–738
Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA, Zhu P, Garcia-Bassets I, Liu F, Taylor H, Lozach J, Jayes FL, Korach KS, Glass CK, Fu XD, Rosenfeld MG (2007) Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 446(7138):882–887
Hlubek F, Lohberg C, Meiler J, Jung A, Kirchner T, Brabletz T (2001) Tip60 is a cell-type-specific transcriptional regulator. J Biochem 129(4):635–641
Sanchez-Tillo E, Lazaro A, Torrent A, Cuatrecasas M, Vaquero EC, Castells A, Engel P, Postigo A (2010) ZEB1 represses E-cadherin and you induce an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 29(24):3490–3500
Byles V, Zhu L, Lovaas JD, Chmilewski LK, Wang J, Faller DV, Dai Y (2012) SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene. doi:10.1038/onc.2011.612 (Ahead of print)
Verstappen G, van Grunsven LA, Michiels C, Van de Putte T, Souopgui J, Van Damme J, Bellefroid E, Vandekerckhove J, Huylebroeck D (2008) Atypical Mowat-Wilson patient confirms the importance of the novel association between ZFHX1B/SIP1 and NuRD corepressor complex. Hum Mol Genet 17(8):1175–1183
Postigo AA (2003) Opposing functions of ZEB proteins in the regulation of the TGFβ/BMP signaling pathway. EMBO J 22(10):2443–2452
Postigo AA, Depp JL, Taylor JJ, Kroll KL (2003) Regulation of Smad signaling through to differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J 22(10):2453–2462
van Grunsven LA, Taelman V, Michiels C, Opdecamp K, Huylebroeck D, Bellefroid EJ (2006) δEF1 and SIP1 are differentially expressed and have overlapping activities during Xenopus embryogenesis. Dev Dyn 235(6):1491–1500
Long J, Zuo D, Park M (2005) Pc2-mediated sumoylation of Smad-interacting protein 1 attenuates transcriptional repression of E-cadherin. J Biol Chem 280(42):35477–35489
Costantino ME, Stearman RP, Smith GE, Darling DS (2002) Cell-specific phosphorylation of Zfhep transcription factor. Biochem Biophys Res Commun 296:368–373
Vandewalle C, Van Roy F, Berx G (2009) The role of the ZEB family of transcription factors in development and disease. Cell Mol Life Sci 66(5):773–787
Brabletz S, Brabletz T (2010) The ZEB/miR-200 feedback loop—a motor of cellular plasticity in development and cancer? EMBO Rep 11(9):670–677
Karreth FA, Tay Y, Perna D, Ala U, Tan SM, Rust AG, DeNicola G, Webster KA, Weiss D, Perez-Mancera PA, Krauthammer M, Halaban R, Provero P, Adams DJ, Tuveson DA, Pandolfi PP (2011) In vivo identification of tumor-suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell 147(2):382–395
Verschueren K, Remacle JE, Collart C, Kraft H, Baker BS, Tylzanowski P, Nelles L, Wuytens G, Su MT, Bodmer R, Smith JC, Huylebroeck D (1999) SIP1, a novel zinc finger/homeodomain repressor, interacts with Smad proteins and binds to 5′-CACCT sequences in candidate target genes. J Biol Chem 274(29):20489–20498
Shirakihara T, Saitoh M, Miyazono K (2007) Differential regulation of epithelial and mesenchymal markers by δEF1 proteins in epithelial mesenchymal transition induced by TGF-β. Mol Biol Cell 18(9):3533–3544
Nishimura G, Manabe I, Tsushima K, Fujiu K, Oishi Y, Imai Y, Maemura K, Miyagishi M, Higashi Y, Kondoh H, Nagai R (2006) δEF1 mediates TGF-β signaling in vascular smooth muscle cell differentiation. Dev Cell 11(1):93–104
Sheng G, dos Reis M, Stern CD (2003) Churchill, a zinc finger transcriptional activator, regulates the transition between gastrulation and neurulation. Cell 115(5):603–613
Krishnamachary B, Zagzag D, Nagasawa H, Rainey K, Okuyama H, Baek JH, Semenza GL (2006) Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res 66(5):2725–2731
Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H (2007) NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene 26(6):711–724
Chuang MJ, Sun KH, Tang SJ, Deng MW, Wu YH, Sung JS, Cha TL, Sun GH (2008) Tumor-derived tumor necrosis factor-alpha promotes progression and epithelial-mesenchymal transition in renal cell carcinoma cells. Cancer Sci 99(5):905–913
Smit MA, Peeper DS (2011) Zeb1 is required for TrkB-induced epithelial-mesenchymal transition, anoikis resistance and metastasis. Oncogene 30(35):3735–3744
Kong D, Li Y, Wang Z, Banerjee S, Ahmad A, Kim HR, Sarkar FH (2009) miR-200 regulates PDGF-D-mediated epithelial-mesenchymal transition, adhesion, and invasion of prostate cancer cells. Stem Cells 27(8):1712–1721
Graham TR, Zhau HE, Odero-Marah VA, Osunkoya AO, Kimbro KS, Tighiouart M, Liu T, Simons JW, O’Regan RM (2008) Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res 68(7):2479–2488
Lorenzatti G, Huang W, Pal A, Cabanillas AM, Kleer CG (2011) CCN6 (WISP3) decreases ZEB1-mediated EMT and invasion by attenuation of IGF-1 receptor signaling in breast cancer. J Cell Sci 124(10):1752–1758
Shin S, Dimitri CA, Yoon SO, Dowdle W, Blenis J (2010) ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol Cell 38(1):114–127
Xiong H, Hong J, Du W, Lin YW, Ren LL, Wang YC, Su WY, Wang JL, Cui Y, Wang ZH, Fang JY (2012) Roles of STAT3 and ZEB1 proteins in E-cadherin down-regulation and human colorectal cancer epithelial-mesenchymal transition. J Biol Chem 287(8):5819–5832
Sanchez-Tillo E, Barrios O, Siles L, Cuatrecasas M, Castells A, Postigo A (2011) β-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci USA 108(48):19204–19209
Dohadwala M, Yang SC, Luo J, Sharma S, Batra RK, Huang M, Lin Y, Goodglick L, Krysan K, Fishbein MC, Hong L, Lai C, Cameron RB, Gemmill RM, Drabkin HA, Dubinett SM (2006) Cyclooxygenase-2-dependent regulation of E-cadherin: prostaglandin E (2) you induce transcriptional repressors ZEB1 and snail in non-small cell lung cancer. Cancer Res 66(10):5338–5345
Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, Ali S, Abbruzzese JL, Gallick GE, Sarkar FH (2009) Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res 69(6):2400–2407
Brabletz S, Bajdak K, Meidhof S, Burk U, Niedermann G, Firat E, Wellner U, Dimmler A, Faller G, Schubert J, Brabletz T (2011) The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J 30(4):770–782
Chamberlain EM, Sanders MM (1999) Identification of the novel player δEF1 in strogen transcriptional cascades. Mol Cell Biol 19(5):3600–3606
Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB (2002) Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem 277(7):5209–5218
Kumar PA, Kotlyarevska K, Dejkhmaron P, Reddy GR, Lu C, Bhojani MS, Menon RK (2010) Growth hormone (GH)-dependent expression of a natural antisense transcript induces zinc finger E-box-binding homeobox 2 (ZEB2) in the glomerular podocyte: a novel action of GH with implications for the pathogenesis of diabetic nephropathy. J Biol Chem 285(41):31148–31156
Liu Y, Costantino ME, Montoya-Durango D, Higashi Y, Darling DS, Dean DC (2007) The zinc finger transcription factor is linked to cell ZFHX1A proliferation by Rb-E2F1. Biochem J 408(1):79–85
Liu Y, Xin Y, Ye F, Wang W, Lu Q, Kaplan HJ (2010) Dean DC (2010) Taz-tead1 links cell-cell contact to zeb1 expression, proliferation, and dedifferentiation in retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 51(7):3372–3378
Eades G, Yao Y, Yang M, Zhang Y, Chumsri S, Zhou Q (2011) miR-200a regulates SIRT1 expression and epithelial to mesenchymal transition (EMT)-like transformation in mammary epithelial cells. J Biol Chem 286(29):25992–26002
Cieply B, Riley P 4th, Pifer PM, Widmeyer J, Addison JB, Ivanov AV, Denvir J, Frisch SM (2012) Suppression of the epithelial-mesenchymal transition by grainyhead-like-2. Cancer Res 72(9):2440–2453
Evdokimova V, Tognon C, Ng T, Ruzanov P, Melnyk N, Fink D, Sorokin A, Ovchinnikov LP, Davicioni E, Triche TJ, Sorensen PH (2009) Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell 15(5):402–415
Lander R, Nordin K, LaBonne C (2011) The F-box protein Ppa is a common regulator of core EMT factors Twist, Snail, Slug, and Sip1. J Cell Biol 194(1):17–25
Peinado H, Ballestar E, Esteller M, Cano A (2004) Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol 24(1):306–319
Hou Z, Peng H, Ayyanathan K, Yan KP, Langer EM, Longmore GD, Rauscher FJ 3rd (2008) The LIM protein AJUBA recruits protein arginine methyltransferase 5 to mediate SNAIL-dependent transcriptional repression. Mol Cell Biol 28(10):3198–3207
Herranz N, Pasini D, Diaz VM, Franci C, Gutierrez A, Dave N, Escriva M, Hernandez-Muñoz I, Di Croce L, Helin K, Garcia de Herreros A, Peiro S (2008) Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol Cell Biol 28(15):4772–4781
Lin Y, Wu Y, Li J, Dong C, Ye X, Chi YI, Evers BM, Zhou BP (2010) The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J 29(11):1803–1816
Tripathi MK, Misra S, Khedkar SV, Hamilton N, Irvin-Wilson C, Sharan C, Sealy L, Chaudhuri G (2005) Regulation of BRCA2 gene expression by the SLUG repressor protein in human breast cells. J Biol Chem 280(17):17163–17171
Molina-Ortiz P, Villarejo A, MacPherson M, Santos V, Montes A, Souchelnytskyi S, Portillo F, Cano A (2012) Characterization of the SNAG and SLUG domains of Snail2 in the repression of E-cadherin and EMT induction: modulation by serine 4 phosphorylation. PLoS ONE 7(5):e36132
Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL, Crystal RG, de Herreros AG, Moustakas A, Pettersson RF, Fuxe J (2009) A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-β mediated epithelial-mesenchymal transition. Nat Cell Biol 11(8):943–950
Villagrasa P, Diaz VM, Viñas-Castells R, Peiro S, Del Valle-Perez B, Dave N, Rodriguez-Asiain A, Casal JI, Lizcano JM, Duñach M, Garcia de Herreros A (2012) Akt2 interacts with Snail1 in the E-cadherin promoter. Oncogene. doi:10.1038/onc.2011.562 (Ahead of print)
MacPherson MR, Molina P, Souchelnytskyi S, Wernstedt C, Martin-Pérez J, Portillo F, Cano A (2010) Phosphorylation of serine 11 and serine 92 as new positive regulators of human Snail1 function: potential involvement of casein kinase-2 and the cAMP-activated kinase protein kinase A. Mol Biol Cell 21(2):244–253
Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC (2004) Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol 6(10):931–940
Peinado H, Iglesias-de Del Carmen, la Cruz M, Olmeda D, Csiszar K, Fong KS, Vega S, Nieto MA, Cano A, Portillo F (2005) A molecular role for lysyl oxidase-like 2 enzyme in snail regulation and tumor progression. EMBO J 24(19):3446–3458
Xu Y, Lee SH, Kim HS, Kim NH, Piao S, Park SH, Jung YS, Yook JI, Park BJ, Ha NC (2010) Role of CK1 in GSK3β-mediated phosphorylation and degradation of snail. Oncogene 29(21):3124–3133
Vernon AE, LaBonne C (2006) Slug stability is dynamically regulated during neural crest development by the F-box protein Ppa. Development 133(17):3359–3370
Viñas-Castells R, Beltran M, Valls G, Gomez I, Garcia JM, Montserrat-Sentis B, Baulida J, Bonilla F, de Herreros AG, Diaz VM (2010) The hypoxia-controlled FBXL14 ubiquitin ligase targets SNAIL1 for proteasome degradation. J Biol Chem 285(6):3794–3805
Dominguez D, Montserrat-Sentis B, Virgos-Soler A, Guaita S, Grueso J, Porta M, Puig I, Baulida J, Franci C, Garcia de Herreros A (2003) Phosphorylation regulates the subcellular location and activity of the snail transcriptional repressor. Mol Cell Biol 23(14):5078–5089
Mingot JM, Vega S, Maestro B, Sanz JM, Nieto MA (2009) Characterization of Snail nuclear import pathways as representatives of C2H2 zinc finger transcription factors. J Cell Sci 122(9):1452–1460
Du C, Zhang C, Hassan S, Biswas MH, Balaji KC (2010) Protein kinase D1 suppresses epithelial-to-mesenchymal transition through phosphorylation of snail. Cancer Res 20:7810–7819
Yamashita S, Miyagi C, Fukada T, Kagara N, Che YS, Hirano T (2004) Zinc transporter LIVI controls epithelial-mesenchymal transition in zebrafish gastrula organizer. Nature 429(6989):298–302
Yang Z, Rayala S, Nguyen D, Vadlamudi RK, Chen S, Kumar R (2005) Pak1 phosphorylation of snail, a master regulator of epithelial-to-mesenchyme transition, modulates snail’s subcellular localization and functions. Cancer Res 65(8):3179–3184
Thuault S, Tan EJ, Peinado H, Cano A, Heldin CH, Moustakas A (2008) HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J Biol Chem 283(48):33437–33446
Horiguchi K, Shirakihara T, Nakano A, Imamura T, Miyazono K, Saitoh M (2009) Role of Ras signaling in the induction of snail by Transforming Growth Factor-β. J Biol Chem 284(1):245–253
Liu YN, Abou-Kheir W, Yin JJ, Fang L, Hynes P, Casey O, Hu D, Wan Y, Seng V, Sheppard-Tillman H, Martin P, Kelly K (2012) Critical and reciprocal regulation of KLF4 and SLUG in transforming growth factor β-initiated prostate cancer epithelial-mesenchymal transition. Mol Cell Biol 32(5):941–953
Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U (2008) Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci USA 105(17):6392–6397
Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP (2009) Stabilization of snail by NFkB is required for inflammation-induced cell migration and invasion. Cancer Cell 15(5):416–428
Peiro S, Escriva M, Puig I, Barbera MJ, Dave N, Herranz N, Larriba MJ, Takkunen M, Franci C, Muñoz A, Virtanen I, Baulida J, García de Herreros A (2006) Snail1 transcriptional repressor binds to its own promoter and controls its expression. Nucleic Acids Res 34(7):2077–2084
Franco HL, Casanovas J, Rodríguez-Medina JR, Cadilla CL (2011) Redundant or separate entities? Roles of Twist1 and Twist2 as molecular switches during gene transcription. Nucleic Acids Res 39(4):1177–1186
Alexander NR, Tran NL, Rekapally H, Summers CE, Glackin C, Heimark RL (2006) N-cadherin gene expression in prostate carcinoma is modulated by integrin-dependent nuclear translocation of Twist1. Cancer Res 66(7):3365–3369
Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH (2007) Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res 67(5):1979–1987
Fu J, Qin L, He T, Qin J, Hong J, Wong J, Liao L, Xu J (2011) The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res 21(2):275–289
Reynolds N, Salmon-Divon M, Dvinge H, Hynes-Allen A, Balasooriya G, Leaford D, Behrens A, Bertone P, Hendrich B (2012) NuRD-mediated deacetylation of H3K27 facilitates recruitment of Polycomb Repressive Complex 2 to direct gene repression. EMBO J 31(3):593–605
Yang F, Sun L, Li Q, Han X, Lei L, Zhang H, Shang Y (2012) SET8 promotes epithelial-mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J 31(1):110–123
Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, Abbruzzese JL, Hortobagyi GN, Hung MC (2007) Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res 67(19):9066–9076
Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola D, Mansour M, Xu LM, Costanzo C, Cheng JQ, Wang LH (2008) Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J Biol Chem 283(21):14665–14673
Sosic D, Richardson JA, Yu K, Ornitz DM, Olson EN (2003) Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity. Cell 112(2):169–180
Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, Teng SC, Wu KJ (2008) Direct regulation of TWIST by HIF-1α promotes metastasis. Nat Cell Biol 10(3):295–305
Nairismägi ML, Vislovukh A, Meng Q, Kratassiouk G, Beldiman C, Petretich M, Groisman R, Füchtbauer EM, Harel-Bellan A, Groisman I (2012) Translational control of TWIST1 expression in MCF-10A cell lines recapitulating breast cancer progression. Oncogene. doi:10.1038/onc.2011.650 (Ahead of print)
Firulli BA, Krawchuk D, Centonze VE, Vargesson N, Virshup DM, Conway SJ, Cserjesi P, Laufer E, Firulli AB (2005) Altered Twist1 and Hand2 dimerization is associated with Saethre-Chotzen syndrome and limb abnormalities. Nat Genet 37(4):373–381
Christoffersen NR, Silahtaroglu A, Orom UA, Kauppinen S, Lund AH (2007) miR-200b mediates post-transcriptional repression of ZFHX1B. RNA 13(8):1172–1178
Park SM, Gaur AB, Lengyel E, Peter ME (2008) The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 22(7):894–907
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10(5):593–601
Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T (2008) A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep 9(6):582–589
Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ (2008) A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res 68(19):7846–7854
Mizuguchi Y, Specht S, Lunz JG 3rd, Isse K, Corbitt N, Takizawa T, Demetris AJ (2012) Cooperation of p300 and PCAF in the control of microRNA 200c/141 transcription and epithelial characteristics. PLoS ONE 7(2):e32449
Dykxhoorn DM, Wu Y, Xie H, Yu F, Lal A, Petrocca F, Martinvalet D, Song E, Lim B, Lieberman J (2009) miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS ONE 4(9):e7181
Davalos V, Moutinho C, Villanueva A, Boque R, Silva P, Carneiro F, Esteller M (2012) Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene 31(16):2062–2074
Ono H, Imoto I, Kozaki K, Tsuda H, Matsui T, Kurasawa Y, Muramatsu T, Sugihara K, Inazawa J (2012) SIX1 promotes epithelial-mesenchymal transition in colorectal cancer through ZEB1 activation. Oncogene. doi:10.1038/onc.2011.646 (Ahead of print)
Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW, Yu WH, Rehman SK, Hsu JL, Lee HH, Liu M, Chen CT, Yu D, Hung MC (2011) p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol 13(3):317–323
Kim T, Veronese A, Pichiorri F, Lee TJ, Jeon YJ, Volinia S, Pineau P, Marchio A, Palatini J, Suh SS, Alder H, Liu CG, Dejean A, Croce CM (2011) p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med 208(5):875–883
Pinho AV, Rooman I, Real FX (2011) p53-dependent regulation of growth, epithelial-mesenchymal transition and stemness in Normal pancreatic epithelial cells. Cell Cycle 10(8):1312–1321
Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, zur Hausen A, Brunton VG, Morton J, Sansom O, Schüler J, Stemmler MP, Herzberger C, Hopt U, Keck T, Brabletz S, Brabletz T (2009) The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol 11(12):1487–1495
Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E, Dirbas FM, Somlo G, Pera RA, Lao K, Clarke MF (2009) Downregulation of miRNA-200c links breast cancer stem cells with Normal stem cells. Cell 138(3):592–603
Stinson S, Lackner MR, Adai AT, Yu N, Kim HJ, O’Brien C, Spoerke J, Jhunjhunwala S, Boyd Z, Januario T, Newman RJ, Yue P, Bourgon R, Modrusan Z, Stern HM, Warming S, de Sauvage FJ, Amler L, Yeh RF, Dornan D (2011) TRPS1 targeting by miR-221/222 promotes the epithelial-to-mesenchymal transition in breast cancer. Sci Signal 4(177):ra41
Chen Y, Banda M, Speyer CL, Smith JS, Rabson AB, Gorski DH (2010) Regulation of the expression and activity of the antiangiogenic homeobox gene GAX/MEOX2 by ZEB2 and microRNA-221. Mol Cell Biol 30(15):3902–3913
Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S, Westermann F, Speleman F, Vandesompele J, Weinberg RA (2010) miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol 12(3):247–256
Liu S, Kumar SM, Lu H, Liu A, Yang R, Pushparajan A, Guo W, Xu X (2012) MicroRNA-9 up-regulates E-cadherin through inhibition of NF-κB1-Snail1 pathway in melanoma. J Pathol 226(1):61–72
Siemens H, Jackstadt R, Hünten S, Kaller M, Menssen A, Götz U, Hermeking H (2011) miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 10(24):4256–4271
Kim NH, Kim HS, Li XY, Lee I, Choi HS, Kang SE, Cha SY, Ryu JK, Yoon D, Fearon ER, Rowe RG, Lee S, Maher CA, Weiss SJ, Yook JI (2011) A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J Cell Biol 195(3):417–433
Kumarswamy R, Mudduluru G, Ceppi P, Muppala S, Kozlowski M, Niklinski J, Papotti M, Allgayer H (2012) MicroRNA-30a inhibits epithelial-to-mesenchymal transition by targeting Snai1 and is downregulated in non-small cell lung cancer. Int J Cancer 130(9):2044–2053
Zhang J, Zhang H, Liu J, Tu X, Zang Y, Zhu J, Chen J, Dong L, Zhang J (2012) miR-30 inhibits TGF-β1-induced epithelial-to-mesenchymal transition in hepatocyte by targeting Snail1. Biochem Biophys Res Commun 417(3):1100–1105
Cha YH, Kim NH, Park C, Lee I, Kim HS, Yook JI (2012) miRNA-34 p53 tumor suppressor intrinsically links Wnt signaling. Cell Cycle 11(7):1273–1281
Chang CJ, Hsu CC, Chang CH, Tsai LL, Chang YC, Lu SW, Yu CH, Huang HS, Wang JJ, Tsai CH, Chou MY, Yu CC, Hu FW (2011) Let-7d functions as novel regulator of epithelial-mesenchymal transition and chemoresistant property in oral cancer. Oncol Rep 26(4):1003–1010
Ru P, Steele R, Newhall P, Phillips NJ, Toth K, Ray RB (2012) MicroRNA-29b suppresses prostate cancer metastasis by regulating epithelial-mesenchymal transition signaling. Mol Cancer Ther 11(5):1166–1173
Liu YN, Yin JJ, Abou-Kheir W, Hynes PG, Casey OM, Fang L, Yi M, Stephens RM, Seng V, Sheppard-Tillman H, Martin P, Kelly K (2012) MiR-1 and miR-200 inhibit EMT via Slug-dependent and tumorigenesis via Slug-independent mechanisms. Oncogene. doi:10.1038/onc.2012.58 (Ahead of print)
Zhang Z, Zhang B, Li W, Fu L, Fu L, Zhu Z, Dong JT (2011) Epigenetic Silencing of miR-203 Upregulates SNAI2 and Contributes to the Invasiveness of Malignant Breast Cancer Cells. Genes Cancer 2(8):782–791
Li B, Han Q, Zhu Y, Yu Y, Wang J, Jiang X (2012) Downregulated miR-214 contributes to intrahepatic cholangiocarcinoma metastasis by targeting Twist. FEBS J 279(13):2393–2398
Ma L, Teruya-Feldstein J, Weinberg RA (2007) Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 449(7163):682–688
Lee YB, Bantounas I, Lee DY, Phylactou L, Caldwell MA, Uney JB (2009) Twist-1 regulates the miR-199a/214 cluster during development. Nucleic Acids Res 37(1):123–128
Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, Hollier BG, Ram PT, Lander ES, Rosen JM, Weinberg RA, Mani SA (2010) Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci USA 31(35):15449–15454
Reinhold WC, Reimers MA, Lorenzi P, Ho J, Shankavaram UT, Ziegler MS, Bussey KJ, Nishizuka S, Ikediobi O, Pommier YG, Weinstein JN (2010) Multifactorial regulation of E-cadherin expression: an integrative study. Mol Cancer Ther 9(1):1–16
Guaita S, Puig I, Franci C, Garrido M, Dominguez D, Batlle E, Sancho E, Dedhar S, De Herreros AG, Baulida J (2002) Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J Biol Chem 277(42):39209–39216
Dave N, Guaita-Esteruelas S, Gutarra S, Frias A, Beltran M, Peiro S, de Herreros AG (2011) Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J Biol Chem 286(14):12024–12032
Beltran M, Puig I, Peña C, Garcia JM, Alvarez AB, Peña R, Bonilla F, de Herreros AG (2008) A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev 22(6):756–769
Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J (2011) Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res 71(1):245–254
Tran DD, Corsa CA, Biswas H, Aft RL, Longmore GD (2011) Temporal and spatial cooperation of Snail1 and Twist1 during epithelial-mesenchymal transition predicts for human breast cancer recurrence. Mol Cancer Res 9(12):1644–1657
Bindels S, Mestdagt M, Vandewalle C, Jacobs N, Volders L, Noël A, van Roy F, Berx G, Foidart JM, Gilles C (2006) Regulation of vimentin by SIP1 in human epithelial breast tumor cells. Oncogene 25(36):4975–4985
Nam EH, Lee Y, Park YK, Lee JW, Kim S (2012) ZEB2 upregulates integrin α5 expression through cooperation with Sp1 to induce invasion during epithelial-mesenchymal transition of human cancer cells. Carcinogenesis 33(3):563–571
Horiguchi K, Sakamoto K, Koinuma D, Semba K, Inoue A, Inoue S, Fujii H, Yamaguchi A, Miyazawa K, Miyazono K, Saitoh M (2012) TGF-β drives epithelial-mesenchymal transition through δEF1-mediated downregulation of ESRP. Oncogene 31(26):3190–3201
Spaderna S, Schmalhofer O, Hlubek F, Berx G, Eger A, Merkel S, Jung A, Kirchner T, Brabletz T (2006) A transient, EMT-linked loss of basement membranes indicates metastasis and poor survival in colorectal cancer. Gastroenterology 131(3):830–840
Drake JM, Barnes JM, Madsen JM, Domann FE, Stipp CS, Henry MD (2010) ZEB1 coordinately regulates laminin-332 and β4 integrin expression altering the invasive phenotype of prostate cancer cells. J Biol Chem 285(44):33940–33948
Hlubek F, Spaderna S, Jung A, Kirchner T, Brabletz T (2004) β-catenin activates a coordinated expression of the proinvasive factors laminin-5 γ2 chain and MT1-MMP in colorectal carcinomas. Int J Cancer 108(2):321–326
Oztas E, Avci ME, Ozcan A, Sayan AE, Tulchinsky E, Yagci T (2010) Novel monoclonal antibodies detect Smad-interacting protein 1 (SIP1) in the cytoplasm of human tumor cells from multiple tissue arrays. Exp Mol Pathol 89:182–189
Gemmill RM, Roche J, Potiron VA, Nasarre P, Mitas M, Coldren CD, Helfrich BA, Garrett-Mayer E, Bunn PA, Drabkin HA (2011) ZEB1-responsive genes in non-small cell lung cancer. Cancer Lett 300(1):66–78
Sayan AE, Griffiths TR, Pal R, Browne GJ, Ruddick A, Yagci T, Edwards R, Mayer NJ, Qazi M, Goyal S, Fernandez S, Straatman K, Jones GD, Bowman KJ, Colquhoun A, Mellon JK, Kriajevska M, Tulchinsky E (2009) SIP1 protein protects cells from DNA damage-induced apoptosis and has independent prognostic value in bladder cancer. Proc Natl Acad Sci USA 106(35):14884–14889
Soini Y, Tuhkanen H, Sironen R, Virtanen I, Kataja V, Auvinen P, Mannermaa A, Kosma VM (2011) Transcription factors ZEB1, twist and snai1 in breast carcinoma. BMC Cancer 11:73
Cai MY, Luo RZ, Chen JW, Pei XQ, Lu JB, Hou JH, Yun JP (2012) Overexpression of ZEB2 in Peritumoral Liver Tissue Correlates with Favorable Survival after Curative Resection of Hepatocellular Carcinoma. PLoS ONE 7:e32838
Spaderna S, Schmalhofer Or Wahlbuhl M, Dimmler A, Bauer K, Sultan A, Hlubek F, Jung A, Strand D, Eger A, Kirchner T, Behrens J, Brabletz T (2008) The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res 68(2):537–544
Spoelstra NS, Manning NG, Higashi Y, Darling D, Singh M, Shroyer KR, Broaddus RR, Horwitz KB, Richer JK (2006) The transcription factor ZEB1 is aberrantly expresse in aggressive uterine in cancers. Cancer Res 66(7):3893–3902
Elloul S, Elstrand MB, Nesland JM, Trope CG, Kvalheim G, Goldberg I, Reich R, Davidson B (2005) Snail, Slug, and Smad-interacting protein 1 as novel parameters of disease aggressiveness in metastatic ovarian and breast carcinoma. Cancer 103(8):1631–1643
Singh M, Spoelstra NS, Jean A, Howe E, Torkko KC, Clark RH, Darling DS, Shroyer KR, Horwitz KB, Broaddus RR, Richer JK (2008) ZEB1 expression in type I vs type II endometrial Cancers: a marker of aggressive disease. Mod Pathol 21:912–923
Okugawa Y, Toiyama Y, Tanaka K, Matsusita K, Fujikawa H, Saigusa S, Ohi M, Inoue Y, Mohri Y, Uchida K, Kusunoki M (2012) Clinical significance of zinc finger E-box binding homeobox 1 (ZEB1) in human gastric cancer. J Surg Oncol 106(3):280–285
Yoshihara K, Tajima A, Komata D, Yamamoto T, Kodama S, Fujiwara H, Suzuki M, Onishi Y, Hatae M, Sueyoshi K, Fujiwara H, Kudo Y, Inoue I, Tanaka K (2009) Gene expression profiling of advanced-stage serous ovarian cancers distinguishes novel subclasses and implicates ZEB2 in tumor progression and prognosis. Cancer Sci 100(8):1421–1428
Kahlert C, Lahes S, Radhakrishnan P, Dutta S, Mogler C, Herpel E, Brand K, Steinert G, Schneider M, Mollenhauer M, Reissfelder C, Klupp F, Fritzmann J, Wunder C, Benner A, Kloor M, Huth C, Contin P, Ulrich A, Koch M, Weitz J (2011) Overexpression of ZEB2 at the invasion front of colorectal cancer is an independent prognostic marker and regulates tumor invasion in vitro. Clin Cancer Res 17(24):7654–7663
Zhou YM, Cao L, Li B, Zhang RX, Sui CJ, Yin ZF, Yang JM (2012) Clinicopathological Significance of ZEB1 Protein in Patients with Hepatocellular Carcinoma. Ann Surg Oncol 19(5):1700–1706
Dai YH, Tang YP, Zhu HY, Lv L, Chu Y, Zhou YQ, Huo JR (2012) ZEB2 Promotes the Metastasis of Gastric Cancer and Modulates Epithelial Mesenchymal Transition of Gastric Cancer Cells. Dig Dis Sci 57(5):1253–1260
Kurahara H, Takao S, Maemura K, Mataki Y, Kuwahata T, Maeda K, Ding Q, Sakoda M, Iino S, Ishigami S, Ueno S, Shinchi H, Natsugoe S (2012) Epithelial-mesenchymal transition and mesenchymal-epithelial transition via regulation of ZEB-1 and ZEB-2 expression in pancreatic cancer. J Surg Oncol 105(7):655–661
Peinado H, Marin F, Cubillo E, Stark HJ, Fusenig N, Nieto MA, Cano A (2004) Snail and E47 repressors of E-cadherin induce distinct invasive and angiogenic properties in vivo. J Cell Sci 117(13):2827–2839
Bolós V, Peinado H, Pérez-Moreno MA, Fraga MF, Esteller M, Cano A (2003) The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci 116(Pt 3):499–511
Palmer HG, Larriba MJ, García JM, Ordoñez-Morán P, Peña C, Peiro S, Puig I, Rodriguez R, de la Fuente R, Bernad A, Pollán M, Bonilla F, Gamallo C, de Herreros AG, Muñoz A (2004) The transcription factor SNAIL represses vitamin D receptor expression and responsiveness in human colon cancer. Nat Med 10(9):917–919
Larriba MJ, Martín-Villar E, García JM, Pereira F, Peña C, de Herreros AG, Bonilla F, Muñoz A (2009) Snail2 cooperates with Snail1 in the repression of vitamin D receptor in colon cancer. Carcinogenesis 30(8):1459–1468
Franci C, Gallen M, Alameda F, Baro T, Iglesias M, Virtanen I, Garcia de Herreros A (2009) Snail1 protein in the stroma as a new putative prognosis marker for colon tumours. PLoS ONE 4(5):e5595
Côme C, Magnino F, Bibeau F, De Barbara Santa P, Becker KF, Theillet C, Savagner P (2006) Snail and slug play distinct roles during breast carcinoma progression. Clin Cancer Res 12(18):5395–5402
Tuhkanen H, Soini Y, Kosma VM, Anttila M, Sironen R, Hämäläinen K, Kukkonen L, Virtanen I, Mannermaa A (2009) Nuclear expression of Snail1 in borderline and malignant epithelial ovarian tumours is associated with tumour progression. BMC Cancer 9:289
Bezdekova M, Brychtova S, Sedlakova E, Langova K, Brychta T, Belej K (2012) Analysis of snail-1, E-cadherin and claudin-1 expression in colorectal adenomas and carcinomas. Int J Mol Sci 13(2):1632–1643
Sugimachi K, Tanaka S, Kameyama T, Taguchi K, Aishima S, Shimada M, Sugimachi K, Tsuneyoshi M (2003) Transcriptional repressor snail and progression of human hepatocellular carcinoma. Clin Cancer Res 9(7):2657–2664
Moody SE, Perez D, Pan TC, Sarkisian CJ, Portocarrero CP, Sterner CJ, Notorfrancesco KL, Cardiff RD, Chodosh LA (2005) The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell 8(3):197–209
Miyoshi A, Kitajima Y, Kido S, Shimonishi T, Matsuyama S, Kitahara K, Miyazaki K (2005) Snail accelerates cancer invasion by upregulating MMP expression and is associated with poor prognosis of hepatocellular carcinoma. Br J Cancer 92(2):252–258
Uchikado Y, Natsugoe S, Okumura H, Setoyama T, Matsumoto M, Ishigami S, Aiko S (2005) Slug Expression in the E-cadherin preserved tumors is related to prognosis in patients with esophageal squamous cell carcinoma. Clin Cancer Res 11(3):1174–1180
Blechschmidt K, Sassen S, Schmalfeldt B, Schuster T, Höfler H, Becker KF (2008) The E-cadherin repressor Snail is associated with lower overall survival of ovarian cancer patients. Br J Cancer 98(2):489–495
Luo WR, Li SY, Cai LM, Yao KT (2012) High Expression of Nuclear Snail, but not Cytoplasmic Staining. Predicts Poor Survival in Nasopharyngeal Carcinoma. Ann Surg Oncol. doi:10.1245/s10434-012-2347-x (Ahead of print)
Yang MH, Chen CL, Chau GY, Chiou SH, Su CW, Chou TY, Peng WL, Wu JC (2009) Comprehensive analysis of the independent effect of twist and snail in promoting metastasis of hepatocellular carcinoma. Hepatology 50(5):1464–1474
Martin TA, Goyal A, Watkins G, Jiang WG (2005) Expression of the transcription factors snail, slug, and twist and their clinical significance in human breast cancer. Ann Surg Oncol 12(6):488–496
Smit MA, Geiger TR, Song JY, Gitelman I, Peeper DS (2009) A Twist-Snail Axis Critical for TrkB-Induced Epithelial-Mesenchymal Transition-Like Transformation, Anoikis Resistance, and Metastasis. Mol Cell Biol 29(13):3722–3737
Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA (2008) Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res 68(10):3645–3654
Yang Z, Zhang X, Gang H, Li X, Li Z, Wang T, Han J, Luo T, Wen F, Wu X (2007) Up-regulation of gastric cancer cell invasion by Twist is accompanied by N-cadherin and fibronectin expression. Biochem Biophys Res Commun 358(3):925–930
Mikheeva SA, Mikheev AM, Petit A, Beyer R, Oxford RG, Khorasani L, Maxwell JP, Glackin CA, Wakimoto H, Gonzalez-Herrero I, Sanchez-Garcia I, Silber JR, Horner PJ, Rostomily RC (2010) TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Mol Cancer 9:194
Terauchi M, Kajiyama H, Yamashita M, Kato M, Tsukamoto H, Umezu T, Hosono S, Yamamoto E, Shibata K, Ino K, Nawa A, Nagasaka T, Kikkawa F (2007) Possible involvement of TWIST in enhanced peritoneal metastasis of epithelial ovarian carcinoma. Clin Exp Metastasis 24(5):329–339
Entz-Werle N, Stoetzel C, Berard-Marec P, Kalifa C, Brugiere L, Pacquement H, Schmitt C, Tabone MD, Gentet JC, Quillet R, Oudet P, Lutz P, Babin-Boilletot A, Gaub MP, Perrin-Schmitt F (2005) Frequent genomic abnormalities at TWIST in human pediatric osteosarcomas. Int J Cancer 117(3):349–355
Song LB, Liao WT, Mai HQ, Zhang HZ, Zhang L, Li MZ, Hou JH, Fu LW, Huang WL, Zeng YX, Zeng MS (2006) The clinical significance of twist expression in nasopharyngeal carcinoma. Cancer Lett 242(2):258–265
Yuen HF, Chan YP, Wong ML, Kwok WK, Chan KK, Lee PY, Srivastava G, Law SY, Wong YC, Wang X, Chan KW (2007) Upregulation of Twist in oesophageal squamous cell carcinoma is associated with neoplastic transformation and distant metastasis. J Clin Pathol 60(5):510–514
Zhang Z, Xie D, Li X, Wong YC, Xin D, Guan XY, Chua CW, Leung SC, Na Y, Wang X (2007) Significance of TWIST expression and its association with E-cadherin in bladder cancer. Hum Pathol 38(4):598–606
Fang X, Cai Y, Liu J, Wang Z, Wu Q, Zhang Z, Yang CJ, Yuan L, Ouyang G (2011) Twist2 contributes to breast cancer progression by promoting an epithelial-mesenchymal transition and cancer stem-like cell self-renewal. Oncogene 30(47):4707–4720
Kajiyama H, Hosono S, Terauchi M, Shibata K, Ino K, Yamamoto E, Nomura S, Nawa A, Kikkawa F (2006) Twist Expression Predicts Poor Clinical Outcome of Patients with Clear Cell Carcinoma of the Ovary. Oncology 71(5–6):394–401
Hosono S, Kajiyama H, Terauchi M, Shibata K, Ino K, Nawa A, Kikkawa F (2007) Expression of Twist increases the risk for recurrence and for poor survival in epithelial ovarian carcinoma patients. Br J Cancer 96(2):314–320
Niu RF, Zhang L, Xi GM, Wei XY, Yang Y, Shi YR, Hao XS (2007) Up-regulation of Twist induces angiogenesis and correlates with metastasis in hepatocellular carcinoma. J Exp Clin Cancer Res 26(3):385–394
Song Z, Xue-Yan G, Shuang H, Yu C, Fu-Lin G, Fei-Hu B, Shi-Ren S, Xu-Feng W, Jie D, Dai-Ming F (2007) Expression and significance of TWIST basic helix-loop-helix protein over-expression in gastric cancer. Pathology 39(5):470–475
Sasaki K, Natsugoe S, Ishigami S, Matsumoto M, Okumura H, Setoyama T, Uchikado Y, Kita Y, Tamotsu K, Sakamoto A, Owaki T, Aikou T (2009) Significance of Twist expression and its association with E-cadherin in esophageal squamous cell carcinoma. J Exp Clin Cancer Res 28:158
Mani SA, Yang J, Brooks M, Schwaninger G, Zhou A, Miura N, Kutok JL, Hartwell K, Richardson AL, Weinberg RA (2007) Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal- like breast cancers. Proc Natl Acad Sci USA 104:10069–10074
Nishida N, Mimori K, Yokobori T, Sudo T, Tanaka F, Shibata K, Ishii H, Doki Y, Mori M (2011) FOXC2 is a novel prognostic factor in human esophageal squamous cell carcinoma. Ann Surg Oncol 18(2):535–542
Liu Y, Clem B, Zuba-Surma EK, El-Naggar S, Telang S, Jenson AB, Wang Y, Shao H, Ratajczak MZ, Chesney J, Dean DC (2009) Mouse fibroblasts Lacking RB1 function form spheres and undergo reprogramming to a cancer stem cell phenotype. Cell Stem Cell 4(4):336–347
Hwang WL, Yang MH, Tsai ML, Lan HY, Su SH, Chang SC, Teng HW, Yang SH, Lan YT, Chiou SH, Wang HW (2011) SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology 141(1):279–291
Kurrey NK, Jalgaonkar SP, Joglekar AV, Ghanate AD, Chaskar PD, Doiphode RY, Bapat SA (2009) Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 7:2059–2068
Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, Arendt LM, Kuperwasser C, Bierie B, Weinberg RA (2011) Normal and neoplastic cells can spontaneously convert nonstem to a stem-like state. Proc Natl Acad Sci USA 108(9):7950–7955
Yang Y, Pan X, Lei W, Wang J, Song J (2006) Transforming Growth Factor-β1 induces epithelial-to-mesenchymal transition and apoptosis via a cell cycle-dependent mechanism. Oncogene 25(55):7235–7244
Maestro R, Dei Tos AP, Hamamori Y, Krasnokutsky S, Sartorelli V, Kedes L, Doglioni C, Beach DH, Hannon GJ (1999) Twist is a potential oncogene that inhibits apoptosis. Genes Dev. 13:2207–2217
Valsesia-Wittmann S, Magdeleine M, Dupasquier S, Garin E, Jallas AC, Combaret V, Krause A, Leissner P, Puisieux A (2004) Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. Cancer Cell 6(6):625–630
Shiota M, Izumi H, Onitsuka T, Miyamoto N, Kashiwagi E, Kidani A, Yokomizo A, Naito S, Kohno K (2008) Twist promotes tumor cell growth through YB-1 expression. Cancer Res 68(1):98–105
Franco DL, Mainez J, Vega S, Sancho P, Murillo MM, de Frutos CA, Del Castillo G, López-Blau C, Fabregat I, Nieto MA (2010) Snail1 suppresses TGF-β-induced apoptosis and is sufficient to trigger EMT in hepatocytes. J Cell Sci 123(20):3467–3477
Vega S, Morales AS, Ocana OH, Valdes F, Fabregat I, Nieto MA (2004) Snail blocks the cell cycle and confers resistance to cell death. Genes Dev 18(10):1131–1143
Kajita M, McClinic KN, Wade PA (2004) Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress. Mol Cell Biol 24(17):7559–7566
Wu WS, Heinrichs S, Xu D, Garrison SP, Zambetti GP, Adams JM, Look AT (2005) Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing Puma. Cell 123(4):641–653
Wang SP, Wang WL, Chang YL, Wu CT, Chao YC, Kao SH, Yuan A, Lin CW, Yang SC, Chan WK, Li KC, Hong TM, Yang PC (2009) p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat Cell Biol 11(6):694–704
Vitali R, Mancini C, Cesi V, Tanno B, Mancuso M, Bossi G, Zhang Y, Martinez RV, Calabretta B, Dominici C, Raschella G (2008) Slug (SNAI2) down-regulation by RNA interference facilitates apoptosis and inhibits invasive growth in neuroblastoma preclinical models. Clin Cancer Res 14(14):4622–4630
Magenta A, Cencioni C, Fasanaro P, Zaccagnini G, Greco S, Sarra-Ferraris G, Antonini A, Martelli F, Capogrossi MC (2011) miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ 18(10):1628–1639
Fontemaggi G, Gurtner A, Damalas A, Costanzo A, Higashi Y, Sacchi A, Strano S, Piaggio G, Blandino G (2005) δEF1 repressor controls selectively p53 family members during differentiation. Oncogene 24(49):7273–7280
Bui T, Sequeira J, Wen TC, Sola A, Higashi Y, Kondoh H, Genetta T (2009) ZEB1 Links p63 and p73 in a Novel Neuronal Survival Pathway Rapidly Induced in Response to Cortical Ischemia. PLoS ONE 4:e4373
Taddei ML, Giannoni E, Fiaschi T, Chiarugi P (2012) Anoikis: an emerging hallmark in health and diseases. J Pathol 226(2):380–393
Derksen PW, Liu X, Saridin F, van der Gulden H, Zevenhoven J, Evers B, van Beijnum JR, Griffioen AW, Vink J, Krimpenfort P, Peterse JL, Cardiff RD, Berns A, Jonkers J (2006) Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell 10(5):437–449
Kumar S, Park SH, Cieply B, Schupp J, Killiam E, Zhang F, Rimm DL, Frisch SM (2011) A pathway for the control of anoikis sensitivity by E-cadherin and epithelial-to-mesenchymal transition. Mol Cell Biol 31(19):4036–4051
Takeyama Y, Sato M, Horio M, Hase T, Yoshida K, Yokoyama T, Nakashima H, Hashimoto N, Sekido Y, Gazdar AF, Minna JD, Kondo M, Hasegawa Y (2010) Knockdown of ZEB1, a master epithelial-to-mesenchymal transition (EMT) gene, suppresses anchorage-independent cell growth of lung cancer cells. Cancer Lett 296(2):216–224
Howe EN, Cochrane DR, Richer JK (2011) Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res 13(2):R45
Collado M, Serrano M (2010) Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 10(1):51–57
Lanigan F, Geraghty JG, Bracken AP (2011) Transcriptional regulation of cellular senescence. Oncogene 30(26):2901–2911
Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS (2008) Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133(6):1019–1031
Liu Y, El-Naggar S, Darling DS, Higashi Y, Dean DC (2008) Epithelial-mesenchymal ZEB1 links and cellular senescence transition. Development 135(3):579–588
Browne G, Sayan AE, Tulchinsky E (2010) ZEB proteins link cell motility with cell cycle control and cell survival in cancer. Cell Cycle 9(5):886–891
Arima Y, Inoue Y, Shibata T, Hayashi H, Nagano O, Saya H, Taya Y (2008) Rb depletion results in deregulation of E-cadherin and induction of cellular phenotypic changes that are characteristic of the epithelial-to-mesenchymal transition. Cancer Res 68(13):5104–5112
Mejlvang J, Kriajevska M, Vandewalle C, Chernova T, Sayan AE, Berx G, Mellon JK, Tulchinsky E (2007) Direct repression of cyclin D1 by SIP1 attenuates cell cycle progression in cells undergoing an epithelial mesenchymal transition. Mol Cell Biol 18(11):4615–4624
Ozturk N, Erdal E, Mumcuoglu M, Akcali KC, Yalcin O, Senturk S, Arslan-Ergul A, Gur B, Yulug I, Cetin-Atalay R, Yakicier C, Yagci T, Tez M, Ozturk M (2006) Reprogramming of replicative senescence in hepatocellular carcinoma-derived cells. Proc Natl Acad Sci USA 103(7):2178–2183
Miquelajauregui A, Van de Putte T, Polyakov A, Nityanandam A, Boppana S, Seuntjens E, Karabinos A, Higashi Y, Huylebroeck D, Tarabykin V (2007) Smad-interacting protein-1 (Zfhx1b) Acts of Wnt signaling upstream in the mouse hippocampus and controls STI formation. Proc Natl Acad Sci USA 104(31):12919–12924
Ohashi S, Natsuizaka M, Wong GS, Michaylira CZ, Grugan KD, Stairs DB, Kalabis J, Vega ME, Kalman RA, Nakagawa M, Klein-Szanto AJ, Herlyn M, Diehl JA, Rustgi AK, Nakagawa H (2010) Epidermal growth factor receptor and mutant p53 expand an esophageal cellular subpopulation capable of epithelial-to-mesenchymal transition through ZEB transcription factors. Cancer Res 70:4174–4184
de Frutos CA, Vega S, Manzanares M, Flores JM, Huertas H, Martínez-Frías ML, Nieto MA (2007) Snail1 is a transcriptional effector of FGFR3 signaling during chondrogenesis and achondroplasias. Dev Cell 13(6):872–883
Emadi Baygi M, Soheili ZS, Schmitz I, Sameie S, Schulz WA (2010) Snail regulates cell survival and inhibits cellular senescence in human metastatic prostate cancer cell lines. Cell Biol Toxicol 26(6):553–567
Liu J, Uygur B, Zhang Z, Shao L, Romero D, Vary C, Ding Q, Wu WS (2010) Slug inhibits proliferation of human prostate cancer cells via downregulation of cyclin D1 expression. Prostate 70(16):1768–1777
Takahashi E, Funato N, Higashihori N, Hata Y, Gridley T, Nakamura M (2004) Snail regulates p21(WAF/CIP1) expression in cooperation with E2A and Twist. Biochem Biophys Res Commun 325:1136–1144
Mittal MK, Singh K, Misra S, Chaudhuri G (2011) SLUG-induced elevation of D1 cyclin in breast cancer cells through the inhibition of its ubiquitination. J Biol Chem 286(1):469–479
Kwok WK, Ling MT, Yuen HF, Wong YC, Wang X (2007) Role of p14ARF in TWIST-mediated senescence in prostate epithelial cells. Carcinogenesis 28(12):2467–2475
Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S, Maestro R, Voeltzel T, Selmi A, Valsesia-Wittmann S, Caron de Fromentel C, Puisieux A (2008) Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 14(1):79–89
Gill JG, Langer EM, Lindsley RC, Cai M, Murphy TL, Murphy KM (2012) Snail promotes the cell-autonomous generation of Flk1(+) endothelial cells through the repression of the microRNA-200 family. Stem Cells Dev 21(2):167–176
Ceteci F, Ceteci S, Karreman C, Kramer BW, Asan E, Götz R, Rapp UR (2007) Disruption of tumor cell adhesion promotes angiogenic switch and progression to micrometastasis in RAF-driven murine lung cancer. Cancer Cell 12(2):145–159
Heddleston JM, Li Z, Lathia JD, Bao S, Hjelmeland AB, Rich JN (2010) Hypoxia inducible factors in cancer stem cells. Br J Cancer 102(5):789–795
Evans AJ, Russell RC, Roche O, Burry TN, Fish JE, Chow VW, Kim WY, Saravanan A, Maynard MA, Gervais ML, Sufan RI, Roberts AM, Wilson LA, Betten M, Vandewalle C, Berx G, Marsden PA, Irwin MS, Teh BT, Jewett MA, Ohh M (2007) VHL promotes E2 box-dependent E-cadherin transcription by HIF-mediated regulation of SIP1 and snail. Mol Cell Biol 27(1):157–169
Shih JY, Tsai MF, Chang TH, Chang YL, Yuan A, Yu CJ, Lin SB, Liou GY, Lee ML, Chen JJ, Hong TM, Yang SC, Su JL, Lee YC, Yang PC (2005) Transcription repressor slug promotes carcinoma invasion and predicts outcome of patients with lung adenocarcinoma. Clin Cancer Res 11(22):8070–8078
Yanagawa J, Walser TC, Zhu LX, Hong L, Fishbein MC, Mah V, Chia D, Goodglick L, Elashoff DA, Luo J, Magyar CE, Dohadwala M, Lee JM, St John MA, Strieter RM, Sharma S, Dubinett SM (2009) Snail promotes CXCR2 ligand-dependent tumor progression in non-small cell lung carcinoma. Clin Cancer Res 15(22):6820–6829
Mironchik Y, Winnard PT Jr, Vesuna F, Kato Y, Wildes F, Pathak AP, Kominsky S, Artemov D, Bhujwalla Z, Van Diest P, Burger H, Glackin C, Raman V (2005) Twist overexpression induces in vivo angiogenesis and correlates with chromosomal instability in breast cancer. Cancer Res 65(23):10801–10809
Hu L, Roth JM, Brooks P, Ibrahim S, Karpatkin S (2008) Twist is required for thrombin-induced tumor angiogenesis and growth. Cancer Res 68(11):4296–4302
Inuzuka T, Tsuda M, Tanaka S, Kawaguchi H, Higashi Y, Ohba Y (2009) Integral role of transcription factor 8 in the negative regulation of tumor angiogenesis. Cancer Res 69(4):1678–1684
Luo J, Solimini NL, Elledge SJ (2009) Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136(5):823–837
Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, Settleman J (2009) A gene expression signature associated with ‘K-Ras addiction’ Reveals Regulators of EMT and tumor cell survival. Cancer Cell 15(6):489–500
Wang Y, Ngo VN, Marani M, Yang Y, Wright G, Staudt LM, Downward J (2010) Critical role for transcriptional repressor Snail2 in transformation by oncogenic RAS in colorectal carcinoma cells. Oncogene 29(33):4658–4670
Yang AD, Fan F, Camp ER, van Buren G, Liu W, Somcio R, Gray MJ, Cheng H, Hoff PM, Ellis LM (2006) Chronic oxaliplatin resistance you induce epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res 12(14):4147–4153
Witta SE, Gemmill RM, Hirsch FR, Coldren CD, Hedman K, Ravdel L, Helfrich B, Dziadziuszko R, Chan DC, Sugita M, Chan Z, Baron A, Franklin W, Drabkin HA, Girard L, Gazdar AF, Minna JD, Bunn PA Jr (2006) Restoring E-cadherin expression Increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res 66(2):944–950
Fuchs BC, Fujii T, Dorfman JD, Goodwin JM, Zhu AX, Lanuti M, Tanabe KK (2008) Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res 68(7):2391–2399
Li Y, VandenBoom TG 2nd, Kong D, Wang Z, Ali S, Philip PA, Sarkar FH (2009) Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res 69:6704–6712
Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N, Iwata KK, Gibson N, Haley JD (2005) Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res 65(20):9455–9462
Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, Pham TQ, Soriano R, Stinson J, Seshagiri S, Modrusan Z, Lin CY, O’Neill V, Amler LC (2005) Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res 11(24):8686–8698
Frederick BA, Helfrich BA, Coldren CD, Zheng D, Chan D, Bunn PA Jr, Raben D (2007) Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol Cancer Ther 6(6):1683–1691
Mink SR, Vashistha S, Zhang W, Hodge A, Agus DB, Jain A (2010) Cancer-associated fibroblasts derived from EGFR-TKI-resistant tumors reverse EGFR pathway inhibition by EGFR-TKIs. Mol Cancer Res 8(6):809–820
Oliveras-Ferraros C, Vazquez-Martin A, Cufí S, Queralt B, Báez L, Guardeño R, Hernández-Yagüe X, Martin-Castillo B, Brunet J, Menendez JA (2011) Stem cell property epithelial-to-mesenchymal transition is a core transcriptional network for predicting cetuximab (Erbitux™) efficacy in KRAS wild-type tumor cells. J Cell Biochem 112(1):10–29
Li QQ, Xu JD, Wang WJ, Cao XX, Chen Q, Tang F, Chen ZQ, Liu XP, Xu ZD (2009) Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin Cancer Res 15(8):2657–2665
Tryndyak VP, Beland FA, Pogribny IP (2010) E-cadherin transcriptional down-regulation by epigenetic and microRNA-200 family alterations is related to mesenchymal and drug-resistant phenotypes in human breast cancer cells. Int J Cancer 126(11):2575–2583
Haddad Y, Choi W, McConkey DJ (2009) δEF1 controls the epithelial to mesenchymal transition phenotype and resistance to the epidermal growth factor receptor inhibitor erlotinib in human head and neck squamous carcinoma cell lines. Clin Cancer Res 15(2):532–542
Arumugam T, Ramachandran V, Fournier KF, Wang M, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey DJ, Choi W (2009) Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res 69(14):5820–5828
Zhuo W, Wang Y, Zhuo X, Zhang Y, Ao X, Chen Z (2008) Knockdown of Snail, a novel zinc finger transcription factor, via RNA interference increases A549 cell sensitivity to cisplatin via JNK/mitochondrial pathway. Lung Cancer 62(1):8–14
Hsu DS, Lan HY, Huang CH, Tai SK, Chang SY, Tsai TL, Chang CC, Tzeng CH, Wu KJ, Kao JY, Yang MH (2010) Regulation of excision repair cross-complementation group 1 by Snail contributes to cisplatin resistance in head and neck cancer. Clin Cancer Res 16(18):4561–4571
Zhang W, Feng M, Zheng G, Chen Y, Wang X, Pen B, Yin J, Yu Y, He Z (2012) Chemoresistance to 5-fluorouracil induces epithelial-mesenchymal transition via up-regulation of Snail in MCF7 human breast cancer cells. Biochem Biophys Res Commun 417(2):679–685
Chang TH, Tsai MF, Su KY, Wu SG, Huang CP, Yu SL, Yu YL, Lan CC, Yang CH, Lin SB, Wu CP, Shih JY, Yang PC (2011) Slug confers resistance to the epidermal growth factor receptor tyrosine kinase inhibitor. Am J Respir Crit Care Med 183(8):1071–1079
Haslehurst AM, Koti M, Dharsee M, Nuin P, Evans K, Geraci J, Childs T, Chen J, Li J, Weberpals J, Davey SK, Squire J, Park PC, Feilotter H (2012) EMT snail and slug transcription factors directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 12:91
Catalano A, Rodilossi S, Rippo MR, Caprari P, Procopio A (2004) Induction of stem cell factor/c-Kit/slug signal transduction in multidrug-resistant malignant mesothelioma cells. J Biol Chem 279(45):46706–46714
Pham CG, Bubici C, Zazzeroni F, Knabb JR, Papa S, Kuntzen C, Franzoso G (2007) Upregulation of Twist-1 by NF-kappaB blocks cytotoxicity induced by chemotherapeutic drugs. Mol Cell Biol 27(11):3920–3935
Wang X, Ling MT, Guan XY, Tsao SW, Cheung HW, Lee DT, Wong YC (2004) Identification of a novel function of TWIST, a bHLH protein, in the development of acquired taxol resistance in human cancer cells. Oncogene 23(2):474–482
Kwok WK, Ling MT, Lee TW, Lau TC, Zhou C, Zhang X, Chua CW, Chan KW, Chan FL, Glackin C, Wong YC, Wang X (2005) Up-regulation of TWIST in prostate cancer and its implication as a therapeutic target. Cancer Res 65(12):5153–5162
Zhang X, Wang Q, Ling MT, Wong YC, Leung SC, Wang X (2007) Anti-apoptotic role of TWIST and its association with Akt pathway in mediating taxol resistance in nasopharyngeal carcinoma cells. Int J Cancer 120(9):1891–1898
McConkey DJ, Choi W, Marquis L, Martin F, Williams MB, Shah J, Svatek R, Das A, Adam L, Kamat A, Siefker-Radtke A, Dinney C (2009) Role of epithelial-to-mesenchymal transition (EMT) in drug sensitivity and metastasis in bladder cancer. Cancer Metastasis Rev 28(3–4):335–344
Kenney PA, Wszolek MF, Rieger-Christ KM, Neto BS, Gould JJ, Harty NJ, Mosquera JM, Zeheb R, Loda M, Darling DS, Libertino JA, Summerhayes IC (2011) Novel ZEB1 expression in bladder tumorigenesis. BJU Int 107(4):656–663
Choi W, Shah JB, Tran M, Svatek R, Marquis L, Lee IL, Yu D, Adam L, Wen S, Shen Y, Dinney C, McConkey DJ, Siefker-Radtke A (2012) p63 expression defines a lethal subset of muscle-invasive bladder cancers. PLoS ONE 7(1):e30206
Graham TR, Yacoub R, Taliaferro-Smith L, Osunkoya AO, Odero-Marah VA, Liu T, Kimbro KS, Sharma D, O’Regan RM (2010) Reciprocal regulation of ZEB1 and AR in triple negative breast cancer cells. Breast Cancer Res Treat 123(1):139–147
Ohashi S, Natsuizaka M, Naganuma S, Kagawa S, Kimura S, Itoh H, Kalman RA, Nakagawa M, Darling DS, Basu D, Gimotty PA, Klein-Szanto AJ, Diehl JA, Herlyn M, Nakagawa H (2011) A NOTCH3-mediated squamous cell differentiation program limits expansion of EMT-competent cells that express the ZEB transcription factors. Cancer Res 71(21):6836–6847
Dohadwala M, Wang G, Heinrich E, Luo J, Lau O, Shih H, Munaim Q, Lee G, Hong L, Lai C, Abemayor E, Fishbein MC, Elashoff DA, Dubinett SM, St John MA (2010) The role of ZEB1 in the inflammation-induced promotion of EMT in HNSCC. Otolaryngol Head Neck Surg 142(5):753–759
Rosivatz E, Becker I, Specht K, Fricke E, Luber B, Busch R, Hofler H, Becker KF (2002) Differential expression of the epithelial-mesenchymal transition regulators snail, SIP1, and twist in gastric cancer. Am J Pathol 161(5):1881–1891
Maeda G, Chiba T, Okazaki M, Satoh T, Taya Y, Aoba T, Kato K, Kawashiri S, Imai K (2005) Expression of SIP1 in oral squamous cell carcinomas: implications for E-cadherin expression and tumor progression. Int J Oncol 27(6):1535–1541
Kojc N, Zidar N, Gale N, Poljak M, Fujs Komlos K, Cardesa A, Höfler H, Becker KF (2009) Transcription factors Snail, Slug, Twist, and SIP1 in spindle cell carcinoma of the head and neck. Virchows Arch 454:549–555
Acun T, Oztas E, Yagci T, Yakicier MC (2011) SIP1 is downregulated in hepatocellular carcinoma by promoter hypermethylation. BMC Cancer 11:223
Imamichi Y, Konig A, Gress T, Menke A (2007) Collagen type I-induced Smad- interacting protein 1 expression downregulates E-cadherin in pancreatic cancer. Oncogene 26(16):2381–2385
Blanco MJ, Moreno-Bueno G, Sarrio D, Locascio A, Cano A, Palacios J, Nieto MA (2002) Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene 21(20):3241–3246
Geradts J, de Herreros AG, Su Z, Burchette J, Broadwater G, Bachelder RE (2011) Nuclear Snail1 and nuclear ZEB1 protein expression in invasive and intraductal human breast carcinomas. Hum Pathol 42(8):1125–1131
Roy HK, Smyrk TC, Koetsier J, Victor TA, Wali RK (2005) The transcriptional repressor SNAIL is overexpressed in human colon cancer. Dig Dis Sci 50(1):42–46
Zidar N, Gale N, Kojc N, Volavsek M, Cardesa A, Alos L, Höfler H, Blechschmidt K, Becker KF (2008) Cadherin-catenin complex and transcription factor Snail-1 in spindle cell carcinoma of the head and neck. Virchows Arch 453(3):267–274
Hotz B, Arndt M, Dullat S, Bhargava S, Buhr HJ, Hotz HG (2007) Epithelial to mesenchymal transition: expression of the regulators snail, slug, and twist in pancreatic cancer. Clin Cancer Res 13(16):4769–4776
Castro Alves C, Rosivatz E, Schott C, Hollweck R, Becker I, Sarbia M, Carneiro F, Becker KF (2007) Slug is overexpressed in gastric carcinomas and may act synergistically with SIP1 and Snail in the down-regulation of E-cadherin. J Pathol 211(5):507–515
Katoh M, Katoh M (2003) Identification and characterization of human SNAIL3 (SNAI3) gene in silico. Int J Mol Med 11(3):383–388
Valdes-Mora F, Gómez del Pulgar T, Bandres E, Cejas P, Ramirez de Molina A, Perez-Palacios R, Gallego-Ortega D, Garcia-Cabezas MA, Casado E, Larrauri J, Nistal M, Gonzalez-Baron M, Garcia-Foncillas J, Lacal JC (2009) TWIST1 overexpression is associated with nodal invasion and male sex in primary colorectal cancer. Ann Surg Oncol 16(1):78–87
Lee TK, Poon RT, Yuen AP, Ling MT, Kwok WK, Wang XH, Wong YC, Guan XY, Man K, Chau KL, Fan ST (2006) Twist overexpression correlates with hepatocellular carcinoma metastasis through induction of epithelial-mesenchymal transition. Clin Cancer Res 12(18):5369–5376
Ohuchida K, Mizumoto K, Ohhashi S, Yamaguchi H, Konomi H, Nagai E, Yamaguchi K, Tsuneyoshi M, Tanaka M (2007) Twist, a novel oncogene, is upregulated in pancreatic cancer: clinical implication of Twist expression in pancreatic juice. Int J Cancer 120(8):1634–1640
Gasparotto D, Polesel J, Marzotto A, Colladel R, Piccinin S, Modena P, Grizzo A, Sulfaro S, Serraino D, Barzan L, Doglioni C, Maestro R (2011) Overexpression of TWIST2 correlates with poor prognosis in head and neck squamous cell carcinomas. Oncotarget 2(12):1165–1175
Li Y, Wang W, Wang W, Yang R, Wang T, Su T, Weng D, Tao T, Li W, Ma D, Wang S (2012) Correlation of TWIST2 up-regulation and epithelial-mesenchymal transition during tumorigenesis and progression of cervical carcinoma. Gynecol Oncol 124(1):112–118
Shiota M, Izumi H, Onitsuka T, Miyamoto N, Kashiwagi E, Kidani A, Hirano G, Takahashi M, Naito S, Kohno K (2008) Twist and p53 reciprocally regulate target genes via direct interaction. Oncogene 27(42):5543–5553
Wels C, Joshi S, Koefinger P, Bergler H, Schaider H (2011) Transcriptional activation of ZEB1 by Slug leads to cooperative regulation of the epithelial-mesenchymal transition-like phenotype in melanoma. J Invest Dermatol 131(9):1877–1885
Yuen HF, Chua CW, Chan YP, Wong YC, Wang X, Chan KW (2007) Significance of TWIST and E-cadherin expression in the metastatic progression of prostatic cancer. Histopathology 50(5):648–658
Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY, Yang WH, Huang CH, Kao SY, Tzeng CH, Tai SK, Chang SY, Lee OK, Wu KJ (2010) Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol 12(10):982–992
Hill L, Browne G, Tulchinsky E (2012) ZEB/miR-200 at the crossroads of signal transduction in cancer. Int J Cancer. doi:10.1002/ijc.27708 (Ahead of print)
Qin Q, Xu Y, He T, Qin C, Xu J (2012) Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res 22(1):90–106
Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celia-Terrassa T, Mercatali L, Khan Z, Goodarzi H, Hua Y, Wei Y, Hu G, Garcia BA, Ragoussis J, Amadori D, Harris AL, Kang Y (2011) Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med 17(9):1101–1108
Acknowledgments
We apologize to those researchers whose relevant work was cited only indirectly through reviews because of space limitations. Experimental work was conducted by E.S.T., Y.L., O.d.B., L.S. and L.F.. M.C. and A.C. identified tissue samples and/or advised in the interpretation of immunostaining. A.P. wrote the article and all authors contributed to its critical revision. Comments on the manuscript by E.C. Vaquero are also greatly appreciated. Tissue samples for Fig. 4 were obtained from IDIBAPS’ Tumor Bank. Work in A.P.’s laboratory was funded by grants from Olga Torres Foundation, AVON Cosmetics SAU, Spanish Association Against Cancer (AECC), Spanish Ministry of Economy and Competitiveness (formerly of Science and Innovation, BFU2007-60302, BFU2010-15163), La Caixa Foundation, and the European Commission. E.S.T.’s salary is funded by CIBERehd. O.d.B. and L.S. are recipients of PhD scholarships from the Spanish Ministry of Education, Culture and Sports (FPU Program). L.S.’s salary was previously funded by AECC.
Author information
Authors and Affiliations
Corresponding author
Additional information
E. Sánchez-Tilló, Y. Liu, O. de Barrios and L. Siles contributed equally.
Rights and permissions
About this article
Cite this article
Sánchez-Tilló, E., Liu, Y., de Barrios, O. et al. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell. Mol. Life Sci. 69, 3429–3456 (2012). https://doi.org/10.1007/s00018-012-1122-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-012-1122-2