
Abstract
In this work, a solidified floating organic drop microextraction was developed based on a vesicular supramolecular solvent consisting of decanoic acid and quaternary ammonium. The method was used for preconcentration of trace amount of cadmium in different rice samples followed by flow-injection analysis–flame atomic absorption spectrometry. Several parameters affecting the extraction efficiency including pH, concentration of 1-(2-pyridylazo)-2-naphthol as the chelating agent, sample and extraction solvent volume, stirring rate, extraction time, salt effect, and interfering ions were investigated and optimized. Under the optimum conditions, a preconcentration factor of 84 was achieved. LOD and LOQ were found to be 0.09 and 0.31 µg L−1, respectively. The calibration curve was linear within the range of 5.0–700 µg L−1 (r2 > 0.9978). Intra- and inter-day precisions (RSD% n = 3) were estimated 2.7 and 3.9% at the concentration of 20 µg L−1, respectively. The accuracy of the method was successfully validated by analysis of an SRM-1643f standard reference material. Relative recoveries were achieved within the range of 93–107% elucidating suitability of the method for determination of cadmium in rice samples.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cadmium has been classified as a human carcinogen by the International Agency for Research on Cancer and by the US National Toxicology Program [1]. Cd(II) is known to damage organs such as kidneys, liver, and lungs, even at very low concentrations [2]. Cd(II) enters the body through different pathways such as daily food uptake, growing plants in contaminated soils, and industrial wastewaters [3]. Thus, determination of Cd(II) in food samples has a great deal of importance [4].
Different instrumental methods such as inductively coupled plasma mass spectrometry [5] or optical emission spectrometry [6], graphite furnace atomic absorption spectrometry [7], flame atomic absorption spectrometry (FAAS) [8], and FAAS coupled with flow-injection analysis (FIA-FAAS) [9,10,11] have been used for determination of Cd(II). Among these, FAAS is a well-known technique because of its good selectivity, low cost of operation and maintenance, rapidness, and simplicity. However, direct determination of Cd(II) in complicated matrices such as food samples is often difficult due to the presence of a vast variety of contaminants and low concentration of the analytes.
To solve this problem, separation and preconcentration procedures are often performed prior to analysis. Preconcentration is a very crucial issue for improvement of the sensitivity and separation is an efficient technique to reduce the interference of the sample matrix. Liquid–liquid extraction (LLE) is one of the classical pretreatment techniques that has been widely employed in analytical chemistry [12]. However, LLE requires large volumes of the organic solvents which are harmful and contaminate the environment [13].
Shortcomings of LLE directed the analytical chemists toward miniaturization of the traditional LLE procedure by reducing the organic solvent-to-aqueous phase ratio, resulting to the development of microextraction techniques such as single drop microextraction [14], homogenous liquid–liquid microextraction [15], cloud point extraction [16], dispersive liquid–liquid microextraction [17], hollow fiber liquid-phase microextraction [18], and solidified floating organic drop microextraction (SFODME) [19]. Therefore, it can be noted that today’s trend in analytical chemistry is seeking for the more effective, simple, environmental-friendly, and green sample preparation methods. Liquid-phase microextraction (LPME) approaches are in accordance with these requirements and have been discussed in several reviews [20,21,22,23,24,25,26,27].
Recently, attention has been paid to the use of alternative solvents, mainly supercritical fluids [28], ionic liquids [29], deep eutectic solvents [30], and supramolecular solvents (SUPRASs) as well as applying of physical energies during the extraction [31, 32]. Utilization of physical energies in dispersive liquid-phase microextraction enhances analyte transfer from the aqueous sample to the extraction medium and reduces the organic solvent consumption by omitting the disperser solvent [33,34,35,36,37,38,39,40].
SUPRASs are water-immiscible liquids made up of large surfactant aggregates dispersed in a continuous phase, usually water [32, 41, 42]. Different parameters influence the formation of larger aggregates such as temperature, electrolyte, pH, and solvent. SUPRASs have two versatile properties that make them suitable for LPME methods. First, they have regions of different polarities that provide a variety of interactions for the analytes. The type of interaction may be tuned by varying the hydrophobic or the polar group of the surfactant and in theory one may design the most appropriate extractant for a specific application. Second, the major feature of SUPRASs is the high concentration of amphiphiles. This characteristic permits to use low volume of the extraction solvent and consequently, achieve to a high preconcentration factor which are essential for the microextraction methods [43,44,45,46,47].
In this study, the SUPRAS was formed based on coacervation of decanoic acid at the presence of quaternary ammonium (Bu4N+) and used as the extraction phase for SFODME. The method was applied for preconcentration and determination of Cd(II) in rice samples followed by FIA-FAAS. According to the best of our knowledge, there is no report in the literature related to the application of vesicular-based SFODME for determination of metal ions and its combination with FIA-FAAS. Factors affecting the extraction efficiency, such as sample pH, metal-to-chelating agent mole ratio, sample volume, volume of the extraction solvent, ionic strength, stirring rate, extraction time, and potentially interference ions were studied and optimized.
Experimental
Chemicals and solutions
All chemicals were of analytical grade. Tetrabutylammonium hydroxide (40% w/v in water) and decanoic acid (DeA) were purchased from Sigma Aldrich (St. Louis, MO, USA). Ultrapure water was obtained from a Milli-Q water purification system (Millipore, Madrid, Spain). 1-(2-pyridylazo)-2-naphthol (PAN), cadmium nitrate, nitric acid, and sodium hydroxide were purchased from Merck (Darmstadt, Germany). The standard stock solution of cadmium (1000 mg L−1) was prepared by dissolving proper amount of Cd(NO3)2 in 1.0% (v/v) of nitric acid solution. A 0.1 mmol L−1 of PAN solution was prepared by dissolving proper amount in ethanol.
Instrumentation
Determination of Cd(II) was performed by a GBC flame atomic absorption spectrometer model 932 (Victoria, Australia). It was equipped with a cadmium hallow cathode lamp, a deuterium background correction, and air–acetylene burner. The selected parameters were as follows: wavelength of 228.8 nm, lamp current of 5.0 mA, air flow rate of 10 mL min−1, fuel rate of 2.0 mL min−1, sample flow rate of 5.0 mL min−1, and slit width of 0.7 nm. A flow-injection manifold consisting of a Pumpdrive 5001 peristaltic pump (Heidolph, Germany) and a home-made six-port injection valve was coupled to the FAAS system for analysis of the extracts. A digital pH meter, Metrohm 827 (Titrino, Metrohm, Switzerland) was used for pH adjustments. An MR 3200 heater stirrer (Heiddolph, Germany) was used during the extraction procedure.
Vesicular coacervate phase formation
Vesicular-based supramolecular solvent was prepared by mixing 5.15 g of decanoic acid and 3.9 g of tetrabutyl ammonium hydroxide. The mixture was added in 200 mL of distilled water (pH ≈ 7) and stirred at 1200 rpm for 15 min. Finally, phase separation was achieved by centrifugation of the mixture for 5 min at 4000 rpm. The collected vesicular coacervate solvent (about 8 mL) was employed for further experiments. The solvent was less dense than water and was separated at the top of the solution.
SFODME procedure
A schematic representation of the SFODME procedure is shown in Fig. 1. 35 mL of the sample solution (pH ≈ 9) containing appropriate amount of Cd(II) was placed in a 50-mL glass vial. An appropriate amount of PAN was added to the solution. Then, 100 µL of SUPRAS was floated on the surface of the sample solution and stirred at 700 rpm. At the end of extraction time (50 min), the sample vial was placed in an ice bath until the SUPRAS was solidified. The solidified solvent was subsequently transferred into a conical vial and melted. Finally, the solvent was diluted with methanol (1:4) and introduced to the FIA-FAAS system for quantification.

Schematic representation for synthesis of the supramolecular solvent and solidified organic drop microextraction
Sample preparation
Different white rice samples were purchased from a local market in Tehran (Iran). Digestion of the rice samples was done according to literature [48]. The samples were grounded, homogenized and passed through a 850-mm sieve. Then, 0.1 g of each powdered rice sample was placed in a plastic flask and 10 mL of nitric acid (0.5 mol L−1) was added. Finally, each sample was sonicated for 136 min to complete its digestion.
Results and discussion
Composition and binding capability of the vesicular SUPRAS
The vesicular solvent was formed in an aqueous phase containing both protonated (DeA) and deprotonated (De−) decanoic acid at the presence of tetrabutylammonium (Bu4N+). In the aqueous solution, small water-soluble vesicular is formed due to self-assembly of DeA and De−. Vesicle formation near the apparent pKa of acids has been explained on the basis of Vander Waals interactions between the alkyl chains and the formation of unusually strong hydrogen bonds between the deprotonated and protonated carboxylic groups [49]. Moreover, Bu4N+ ions induce the vesicular coacervation by reducing the repulsion between the carboxylate groups in the vesicle and creating electrostatic interaction with De−. As a result, two significant parameters affect the obtaining maximal amount of the vesicle and its stability. The first parameter is the pH of the sample solution. At pH ≈ 7 (pH = pKa), the molar ratio of the alkyl carboxylic acid/carboxylate is around 1.0 in which half of the decanoic acid molecules are neutralized by the hydroxide ions. This permits the formation of aqueous vesicles. Second, molar ratio of Bu4N+/DeA+De− (w/w) in the bulk solution plays an important role on the vesicular coacervation. Our studies showed that an highest formation efficiency of the vesicular aggregates at the molar ratio of 0.5 which was in accordance with the literature [50].
Extraction mechanism of cadmium
Due to existence of polar and apolar groups in the SUPRAS structure, their different interactions with the analytes can improve the extraction efficiency. Figure 2 shows the interactions between the vesicular SUPRAS and the Cd(II)–PAN complex. It seems that three types of interactions are the main extraction driving forces, namely: (1) dispersion forces between the hydrocarbon chains of the amphiphile and the analyte; (2) π-cation interactions between the aromatic rings of Cd(II)–PAN complex and Bu4N+; and (3) hydrogen bonding between the nitrogen and oxygen atoms in the Cd(II)–PAN complex and hydrogen of carboxylic acid. These types of interactions provide good solubilization of Cd(II)–PAN in the SUPRAS and a high extraction efficiency.

a Chemical interactions can influence vesicle formation and its stability, b hydrogen bonding in vesicular formation, and c molecular mechanism of microextraction and different interactions between Cd(II)–PAN complex and the vesicle
Optimization of the extraction conditions
The effect of sample pH
The influence of sample pH on the extraction recovery was investigated within the range of 6–10, whereas the other parameters were kept constant. The results are shown in Fig. 3a. The evaluation of pH is critical in metal extraction procedures, mainly due to its influence on the complex formation and extraction of the hydrophobic complex into the extraction phase. On the other hand, high increasing of pH facilitates hydrolysis of metal ions, which decreases the extraction recovery.

a Effect of pH on the extraction efficiency; extraction conditions: sample solution, 20 mL of 25 µg L−1 Cd(II); SUPRAS amount: 70; 120 µL of PAN (0.1 mM); stirring rate 400 rpm, extraction time, 30 min, b effect of ligand-to-metal mole ratio on the extraction efficiency; sample pH, 9.0; the other conditions are the same as given in a
In the particular case of PAN, it may exist as the three different forms in the solution at various pHs [51,52,53,54]. Acidic solutions lead to protonation of the nitrogen atoms of PAN and consequently, decreasing of their nucleophilicity, whereas an alkaline pH increases the complexation efficiency due to deprotonating of the PAN oxygen and nitrogen heteroatoms (Fig. 2). Moreover, at an alkaline pH, the electrostatic interaction possibility between the nitrogen free pair electrons and Bu4N+ as well as the hydrogen binding between the heteroatoms of Cd(II)–PAN complex and the vesicular coacervate solvent are increased. On the basis of the experimental results, pH of 9.0 was selected for next experiments.
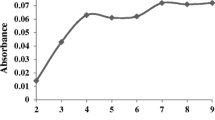
Effect of ligand amount
The molar ratio approach was applied to determine the ligand-to-metal stoichiometric ratio in the Cd(II)–PAN complex. To this aim, different volumes of PAN (0.1 mmol L−1) were added to the sample solutions containing 25 µg L−1 of Cd(II). As it is shown in Fig. 3b, the highest extraction efficiency was obtained when a metal-to-ligand mole ratio of 1:2 was applied and then, reached to a plateau. As a result, considering the existence possibility of other ions in the sample solution, which can complex with PAN, an excess amount of PAN with a molar ratio higher than 1:2 should practically be chosen for the analysis of real samples.
Effect of the extraction solvent volume
The volume of the extraction solvent influences the extraction recovery and preconcentration factor of the analyte. Increasing the sample-to-extraction solvent ratio can increase the preconcentration factor. Accordingly, volume of the extraction solvent should be large enough to promote the analyte transport to the acceptor phase. In the present work, the donor-to-acceptor phase ratios were changed within the range of 1000:1–166.7:1, by changing the vesicular solvent volume (20–120 µL), while the sample solution volume was kept constant (20 mL). As it can be seen in Fig. 4a, while the solvent volume was increased from 20 to 100 µL, the absorbance was increased. Further increasing of the extraction solvent volume led to decreasing of the extraction efficiency due to dilution effect. Thus, the optimal volume of 100 µL was selected for further experiments.

a Effect of extraction solvent volume on the extraction efficiency; ligand-to-metal mole ratio, 1:4, the other conditions are the same as given in Fig. 3b. b Effect of sample volume on the extraction efficiency; SUPRAS volume, 100 µL; the other conditions are the same as given in a
Effect of the sample volume
In an LPME procedure, the amount of analyte which is transferred to the extraction solvent is increased by increasing the sample volume leading to the improvement of preconcentration factor and determination sensitivity [55]. However, the speed at which thermodynamic balance is achieved depends on the sample volume-to-acceptor phase ratio. Thus, influence of the sample volume on the extraction efficiency was investigated within the range of 15–55 mL, with a constant Cd(II) concentration of 25 µg L−1. According to the obtained results, shown in Fig. 4b, an optimum sample volume of 35 mL was chosen for the subsequent experiments.
Effect of stirring rate
The sample stirring rate is an essential step in the microextraction methods to reduce the time needed to reach the thermodynamic equilibrium. Moreover, stirring rate had a direct impact on the shape and stability of the droplet. Extractions were examined at different stirring rates within the range of 300–800 rpm (Fig. 5a). While the stirring rate was increased up to 700 rpm, the mass transfer phenomenon was promoted led to an increase in the absorbance signal and the vesicular droplet was also stable. Nevertheless, at higher stirring rates, some problems were appeared in the solvent collection due to dispersion of the vesicular droplet in the sample solution.

a Effect of stirring rate on the extraction efficiency; sample volume, 35 mL; the other conditions are the same, as given in Fig. 4b. b Effect of extraction time on the extraction efficiency; stirring rate, 700 rpm; the other conditions are the same as given in a
Effect of extraction time
An appropriate extraction time guarantees the equilibrium achievement between the aqueous and vesicular phases. The effect of time on the extraction efficiency was examined from 10 to 90 min, whereas the other experimental conditions were kept constant. The results are shown in Fig. 5b. According to the results, the equilibrium between two phases was reached at 50 min.
Salt effect
In general, the addition of NaCl can have three effects on the vesicular-based SFODME procedure including (1) increasing of the ionic strength, decreasing of the analyte solubility and improvement of the extraction efficiency; (2) increasing of the sample viscosity and decreasing of the analyte diffusion rate from the sample solution into the vesicular droplet; and (3) interaction of excess amount of chloride with Bu4N+ and decreasing of its ability for neutralizing the surface charge of the vesicular solvent [56]. In this study, the effect of NaCl concentration was studied within the range of 0–15% (w/v). The results (Fig. 6) showed no meaningful effect on the extraction recovery due to salt addition in the studied range. Hence, further experiments were performed without the salt addition.

Salt effect on the extraction efficiency; extraction time, 50 min; the other conditions are the same, as given in Fig. 5b
Effect of potentially interfering ions
To study whether other ions can interfere during the extraction of Cd(II), the procedure was performed at the presence of cations that often accompany the analyte in the real samples. In these experiments, various amounts of interfering ions were added to 35 mL of the sample solutions containing 20 µg L−1 of Cd(II) and excess amount of PAN. The tolerance limit of ions was fixed as the maximum amount, causing an error not greater than ± 10%. The results shown in Table 1 indicated no obvious influences by the coexisting ions on the recovery of Cd(II) owing to the good tendency of Cd(II) to PAN as well as applying excess amount of the ligand.
Analytical performance
To evaluate practical applicability of the proposed SFODME, linearity, relative standard deviations (RSD%), limits of detection (LOD), and quantification (LOQ) and preconcentration factors (PF) were investigated under the optimized conditions. LOD (3 Sb/m) and LOQ (10 Sb/m), where Sb is the standard deviation of ten replicates of the blank signal and m is the slope of the extraction calibration curve, were 0.09 and 0.31 µg L−1, respectively. In addition, practical LOD of 2.0 µg L−1 and LOQ of 5.0 µg L−1 were obtained. For this aim, the concentration of the analyte was decreased, since the obtained response created a detectable signal-to-noise ratio of 3 (3 S/N). LOQ was considered the lowest concentration that the linearity of the calibration curve was started.
Calibration curve, Abs = 0.0015C (µg L1−) + 0.0628, was linear within the concentration range of 5–700 µg L−1 with the regression coefficient (R2) higher than 0.9976. The intra- and inter-day precisions (RSD% n = 3), at the concentration of 20 µg L−1 of Cd(II), were found to be 2.7 and 3.9%, respectively. Preconcentration factor (PF) was calculated as the slope ratio of the extraction calibration curve to the direct calibration curve and was estimated 84.
Furthermore, the accuracy of the method was validated by analyzing a standard reference material (SRM-1643f) from the National Institute of Standard and Technology (NIST). The presented Cd(II) in SRM was 5.83 ± 0.13 µg L−1. The amount of Cd(II) found by the proposed method was 5.6 ± 0.4 µg L−1 (n = 3) which was in good agreement with the certified value.
A comparison between the proposed method with the other works in literature is provided in Table 2. The SFODME method has generally low LOD, LOQ, and RSD%, high PF, and a wide dynamic linear range that are comparable with the other studies. Moreover, SUPRASs have different polarity regions that provide several types of interactions with the analytes and consequently, a mixed mechanism for their solubilization. As a result, the analytes with different polarities can be extracted simultaneously and efficiently.
Analysis of rice samples
The proposed SFODME method was applied for determination of Cd(II) in different rice samples. Each sample was digested according to the procedure, as described in sect. “Sample preparation”, and analyzed in triplicates. Results are shown in Table 3.
Furthermore, each sample was spiked with Cd(II) standard at the concentrations of 20 µg L−1 and the procedure was used to investigate the relative recovery (RR%) using the following equation:
where Cfound is the concentration of Cd(II) after the addition of known amount of standard to the sample solution, Creal is the initial concentration of Cd(II) which existed in the sample, and Cadded is the certain concentration of Cd(II) standard solution which is added to the sample solution. As shown in Table 3, RR% and RSD% values were achieved within the range of 93–107 and 2.0–3.6%, respectively, confirming capability of the SFODME/FIA-FAAS method for preconcentration of Cd(II) in rice samples at the trace levels.
Conclusion
A solidified floating organic drop microextraction procedure based on vesicular supramolecular solvent was developed for extraction of cadmium from rice samples followed by FIA-FAAS. The supramolecular solvent was formed by coacervation of decanoic acid at the presence of Bu4N+, as the environmental-friendly reagents, in a simple way without any special device and complicated operation. Because of existence of amphiphilic nanostructures in SUPRASs, they can be considered as the multifunctional solvents for efficient extraction of a wide variety of compounds. Owing this characteristic of SUPRASs, extraction of Cd(II)–PAN complex was performed based on different interactions with the vesicular aggregates mainly hydrophobic, hydrogen bonding, and π-cation interactions. Moreover, low volatility and inflammability of SUPRASs render them as very attractive candidates to replace with the toxic organic solvents for the extraction purposes. The proposed method was environmentally friendly, simple, low cost, precise, reproducible, selective, sensitive, and linear over a broad concentration range and provided a high preconcentration factor. Moreover, it was better or comparable with the other reported methods and, thus, can be used as a suitable alternative for extraction of trace amount of Cd(II) from rice samples.
References
N.I.o.E.H. Sciences, Report on carcinogens. US Department of Health and Human Services, Public Health Service, National Toxicology Program (2005)
W.-S. Zhong, T. Ren, L.-J. Zhao, J. Food Drug Anal. 24, 46 (2016)
C.B. Ojeda, F.S. Rojas, Anal. Bioanal. Chem. 394, 759 (2009)
A.C. Davis, P. Wu, X. Zhang, X. Hou, B.T. Jones, Appl. Spectrosc. Rev. 41, 35 (2006)
S. Chen, Q. Guo, L. Liu, Food Anal. Method. 10, 740 (2017)
M.V.B. Krishna, G. Venkateswarlu, D. Karunasagar, Anal. Methods 9, 2031 (2017)
M. Pirsaheb, N. Fattahi, Anal. Methods 7, 6266 (2015)
M. Shirani, A. Akbari, M. Hassani, Anal. Methods 7, 6012 (2015)
R. Zare-Dorabei, S. Boroun, M. Noroozifar, Talanta 178, 722 (2018)
ÇA. Şahin, İ Tokgöz, S. Bektaş, J. Hazard. Mater. 181, 359 (2010)
A. Anthemidis, V. Kazantzi, V. Samanidou, A. Kabir, K.G. Furton, Talanta 156, 64 (2016)
N. Yoshikuni, T. Baba, N. Tsunoda, K. Oguma, Talanta 66, 40 (2005)
G. Leng, W.-J. Chen, W.-B. Xu, Y. Wang, Food Anal. Methods 10, 3071 (2017)
M.A. Jeannot, F.F. Cantwell, Anal. Chem. 68, 2236 (1996)
L. Fotouhi, S. Seidi, F. Shahsavari, J. Iran. Chem. Soc. 13, 1289 (2016)
T. Li, J. Yang, J. Iran. Chem. Soc. 12, 367 (2015)
S. Seidi, J. Iran. Chem. Soc. 14, 1159 (2017)
Y. Mao, H. Chen, J. Han, Y. Wang, X. Tang, L. Ni, L. Wang, J. Iran. Chem. Soc. 13, 403 (2016)
A. Alahabadi, A. Rastegar, A. Esrafili, Z. Rezai, A.H. Bandegharaei, M. Farzadkia, J. Iran. Chem. Soc. 14, 1725 (2017)
V. Sharifi, A. Abbasi, A. Nosrati, J. Food Drug Anal. 24, 264 (2016)
F.R. Mansour, M.A. Khairy, J. Chromatogr. B 1061, 382 (2017)
I. de la Calle, F. Pena-Pereira, I. Lavilla, C. Bendicho, Anal. Chim. Acta 936, 12 (2016)
A. Szreniawa-Sztajnert, B. Zabiegała, J. Namieśnik, Trends Anal, Chem 49, 45 (2013)
M. Tobiszewski, J. Namieśnik, Curr. Opin. Green Sustain. Chem. 5, 1 (2017)
M. Alexovič, B. Horstkotte, P. Solich, J. Sabo, Anal, Chim. Acta 906, 22 (2016)
M. Alexovič, B. Horstkotte, P. Solich, J. Sabo, Anal, Chim. Acta 907, 18 (2016)
M. Alexovič, B. Horstkotte, I. Šrámková, P. Solich, J. Sabo, Trends Anal. Chem. 86, 39 (2017)
N.L. Rozzi, R.K. Singh, Compr. Rev. Food Sci. Food Saf. 1, 33 (2002)
G.F.B. Cruz, R.J. Cassella, Anal. Methods 7, 6848 (2015)
L. Alavi, S. Seidi, A. Jabbri, T. Baheri, New J. Chem. 14, 7038 (2017)
B. Ebrahimpour, Y. Yamini, S. Seidi, F. Rezaei, Anal. Methods 6, 2936 (2014)
M. Moradi, Y. Yamini, B. Ebrahimpour, J. Iran. Chem. Soc. 11, 1087 (2014)
S.-P. Chu, W.-C. Tseng, P.-H. Kong, C.-K. Huang, J.-H. Chen, P.-S. Chen,S.-D. Huang, Food Chem. 185, 377 (2015)
P.-S. Chen, W.-Y. Haung, S.-D. Huang, J. Chromatogr. B 955, 116 (2014)
J. Regueiro, M. Llompart, C. Garcia-Jares, J.C. Garcia-Monteagudo, R. Cela, J. Chromatogr. A 1190, 27 (2008)
E. Yiantzi, E. Psillakis, K. Tyrovola, N. Kalogerakis, Talanta 80, 2057 (2010)
P.-P. Zhang, Z.-G. Shi, Q.-W. Yu, Y.-Q. Feng, Talanta 83, 1711 (2011)
M. Alexovič, V. Andruch, I.S. Balogh, J. Šandrejová, Anal. Methods 5, 2497 (2013)
M. Alexovič, M. Wieczorek, J. Kozak, P. Kościelniak, I.S. Balogh, V. Andruch, Talanta 133, 127 (2015)
Z. Lin, J. Li, X. Zhang, M. Qiu, Z. Huang, Y. Rao, J. Chromatogr. B 1046, 177 (2017)
W. Hinze, Ann. Chim. 77, 167 (1987)
E. Pramauro, E. Pelezetti, Surfactants in Analytical Chemistry: Applications of Organized Amphiphilic Media. (Elsevier, Amsterdam, 1996), p. 179
A. Ballesteros-Gómez, S. Rubio, D. Pérez-Bendito, J. Chromatogr. A 1216, 530 (2009)
A. Ballesteros-Gómez, M.D. Sicilia, S. Rubio, Anal. Chim. Acta 677, 108 (2010)
M. Cantero, S. Rubio, D. Pérez-Bendito, J. Chromatogr. A 1046, 147 (2004)
R. Carabias-Martınez, E. Rodrıguez-Gonzalo, B. Moreno-Cordero, J. Pérez-Pavón, E.F. Garcıa-Pinto,Laespada, J. Chromatogr. A 902, 251 (2000)
I. Casero, D. Sicilia, S. Rubio, D. Pérez-Bendito, Anal. Chem. 71, 4519 (1999)
J.F. Paula, R.E. Froes-Silva, V.S. Ciminelli, Microchem. J. 104, 12 (2012)
C.L. Apel, D.W. Deamer, M.N. Mautner, Biochim. Biophys. Acta 1559, 1 (2002)
F.-J. Ruiz, S. Rubio, D. Pérez-Bendito, Anal. Chem. 78, 7229 (2006)
T. Asadollahi, A.M. Shabani, S. Dadfarnia, J. Ghasemi, Curr. Anal. Chem. 8, 373 (2012)
S. Khan, M. Soylak, T.G. Kazi, Biol. Trace Elem. Res. 156, 49 (2013)
F. Shah, T.G. Kazi, H.I. Afridi, M.B. Arain, J.A. Baig, J. Hazard. Mater. 192, 1132 (2011)
S.R. Yousefi, S.J. Ahmadi, Microchim. Acta 172, 75 (2011)
M.A. Farajzadeh, D. Djozan, P. Khorram, Anal. Chim. Acta 713, 70 (2012)
F. Rezaei, Y. Yamini, M. Moradi, J. Chromatogr. A 1327, 155 (2014)
M. Tuzen, S. Sahiner, B. Hazer, Food Chem. 210, 115 (2016)
R. Galbeiro, S. Garcia, I. Gaubeur, J. Trace Elem. Med. Biol. 28, 160 (2014)
M.I. ul Hoque, D.A. Chowdhury, R. Holze, A.-N. Chowdhury, M.S. Azam, J. Environ. Chem. Eng. 3, 831 (2015)
F. Omidi, M. Behbahani, M.K. Bojdi, S.J. Shahtaheri, J. Magn. Magn. Mater. 395, 213 (2015)
G. Xiang, S. Wen, X. Wu, X. Jiang, L. He, Y. Liu, Food Chem. 132, 532 (2012)
M. Chamsaz, A. Atarodi, M. Eftekhari, S. Asadpour, M. Adibi, J. Adv. Res. 4, 35 (2013)
Acknowledgements
The authors gratefully acknowledge the financial support for this project by K. N. Toosi University of Technology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Seidi, S., Alavi, L. & Jabbari, A. Trace determination of cadmium in rice samples using solidified floating organic drop microextraction based on vesicular supramolecular solvent followed by flow-injection analysis–flame atomic absorption spectrometry. J IRAN CHEM SOC 15, 2083–2092 (2018). https://doi.org/10.1007/s13738-018-1401-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-018-1401-4