
Abstract
In this work, a temperature-controlled ionic liquid homogeneous liquid phase microextraction (TCIL-HLPME) technique followed by HPLC–UV was applied for preconcentration and determination of diclofenac (DIC) and mefenamic acid (MEF) in urine samples. 1-butyl-3-methylimidazolium hexafluorophosphate ([C4mim][PF6]) was used as the optimum extraction solvent. Experimental design and response surface methodology was used for the optimization process. Firstly, a screening step, using Plackett-Burman design, was carried out to find the significant factors on the extraction efficiency and subsequently, a central composite design (CCD) was employed to find the optimum values of these parameters. The optimal conditions were obtained as extraction solvent volume of 105 µL; sample pH of 2.0, extraction time of 6 min, centrifugation time of 5 min; heating time of 2 min; heating temperature of 50 °C and 20 % of NaCl. Under optimized conditions, the preconcentration factors of 82 and 60 were obtained for DIC and MEF, respectively. The detections limits of 20 and 30 ng mL−1 were achieved for DIC and MEF by the proposed method, respectively. The calibration curves were linear in the range of 40–1000 and 60–1000 ng mL−1 for DIC and MEF, respectively. The intra- and inter-assay precisions (RSD %, n = 3) were in the range of 3.5–4.4 % and 7.3–8.0 % at the concentration level of 100 ng mL−1, respectively. The validated method was successfully applied for the analysis of target analytes in some urine samples.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diclofenac (DIC) and mefenamic acid (MEF) as nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used as analgesic, anti-inflammatory and anti-arthritic agents [1]. Nowadays, determination of pharmaceutical drugs in biological fluids has a great deal of importance in the medical sciences due to the increasing consumption of medicines among human beings. However, determination of the drugs and their metabolites in biological fluids such as plasma and urine is still a very challenging task attributed to the difficulties resulting from their complicated matrices [2]. Therefore, applying of a suitable sample preparation method is an inescapable step before final analysis.
Liquid–liquid extraction (LLE) and solid phase extraction (SPE) are typical sample preparation techniques which have been most widely used in various analytical applications [3–7]. However, both techniques are time consuming and require large volumes of toxic solvents which are not environmental-friendly. Impelled by the need to address these drawbacks, microextraction has gradually appeared as a popular technique for providing many advantages such as quickness and use of negligible volumes of the hazardous organic solvents.
Up to now, different microextraction techniques have been developed which are generally classified into three main groups including liquid phase microextraction (LPME), solid phase microextraction (SPME) and membrane based microextraction techniques [8]. Homogeneous liquid phase microextraction (HLPME) is one of the interesting types of LPME techniques which is based on the complete dissolution of the extraction phase into the sample solution at the first step and then breaking down of the homogenous phase into two separated aqueous-organic phases. This mechanism increases the mass transfer and extraction rate and consequently improves the extraction efficiency.
Until now, different HLPME systems have been reported which are different in the used homogenization and breaking down strategies. These strategies are dictated by the type of organic solvent which is employed as the acceptor phase. For instance, Yamini et al. reported a CHCl3-CH3OH-NaCl three component system; in this system methanol was used as the homogenization agent for the complete dissolution of chloroform into the aqueous sample solution and NaCl was applied to break down the homogenized solution into immiscible aqueous-organic phases [9].
With the emergence of ionic liquids (IL), a big revolution was created in analytical chemistry and sample preparation techniques attributed to the unique properties of these new organic solvents such as low volatility, chemical and thermal stability, and good solubility for both organic and inorganic compounds [10]. In 2009, Zhou and his co-workers have developed a novel, simple and rapid type of HLPME, named temperature-controlled ionic liquid homogeneous liquid phase microextraction (TCIL-HLPME) [11].
It is based on increasing the temperature of the sample solution, which leads to the complete dissolution of IL into the aqueous phase, and breaking down by cooling the solution in an ice-water bath. Then, a centrifugation step is used to collect the cloudy solution as a coherent sedimented phase at the bottom of a test tube. Up to now, this technique has been applied for the determination of various organic and inorganic compounds in different matrices [11–15].
In the present work, a simple and quick HLPME method based on application of a temperature-controlled ionic liquid (TCIL-HLPME) was utilized for discrimination of DIC and MEF in urine samples. The effect of different variables on the extraction efficiency of DIC and MEF was investigated using a two-step experimental design and the optimized values were found by response surface methodology (RSM). Finally, TCIL-HLPME was successfully applied for determination of DIC and MEF in some urine samples.
Experimental
Chemicals and reagent
Diclofenac (DIC, pK a = 4.2, log P = 4.5) and mefenamic acid (MEF, pK a = 4.2, log P = 5.1) were kindly donated by the faculty of Pharmacy at Tehran University of Medical Sciences (Tehran, Iran). Sodium chloride, sodium hydroxide, hydrochloric acid and acetic acid were purchased from Merck (Darmstadt, Germany). 1-butyl-3-methylimidazolium hexafluorophosphate ([C4mim][PF6]) and 1-hexyl-3-methylimidazolium hexafluorophosphate ([C6mim][PF6]) were provided from Sigma-Aldrich (Milwaukee, WI, USA). 1-octyl-3-methylimidazolium hexafluorophosphate ([C8mim][PF6]) was obtained from Acros Organics (Geel, Belgium). HPLC grade acetonitrile and methanol were provided from Caledon (Georgetown, Ont., Canada). All other materials used were of analytical reagent grade. Ultrapure water was prepared using a Milli-Q system from Millipore (Bedford, MA, USA).
Instrumentation
Chromatographic separation of MEF and DIC was carried out on a Young Lin HPLC consisting of a YL9100 HPLC pump (Cambridge, England), a six-port two-position Rheodyne HPLC valve (Oak Harbor, Washington, U.S.A.) with a 20 µL sample loop and equipped with a Y19120 HPLC UV–Vis detector. Chromatographic data were recorded and analyzed using Power Stream software (version 3.2). A C18 column (15 cm × 4.6 mm, with particle size of 5 µm) from Capital HPLC Ltd., (Broxburn, United Kingdom) was applied to separate the analytes under isocratic elution conditions. A mixture of 10 mmol L−1 acetate buffer (pH 5.2) and acetonitrile (50:50, v/v) at a flow rate of 1.0 mL min−1 was used as the mobile phase. The injection volume was 25 µL for all of the standards and the samples to ensure full filling of the 20 µL sample loop. Detection was performed at a wavelength of 285 nm.
Biological matrices and standard solution
Urine samples were collected from three young female volunteers who were treated with DIC and MEF and one person who had not consumed these acidic drugs for a long period of time (to construct calibration curves and calculate figures of merit) with respect to human ethical guidelines. In addition, this protocol was approved by an Internal Review Board. Each sample was diluted 1:3 with ultrapure water; adjusted to pH 2.0 by dropwise addition of HCl (100 mM) or NaOH (100 mM) and then filtered through a disposable 0.45 µm cellulose acetate filter prior to analysis.
A stock solution containing 1.0 mg mL−1 of each analyte was prepared in methanol, stored at 4 °C and protected from light. Working standard solutions were prepared by dilution of the stock solution in ultrapure water.
TCIL-HLPME procedure
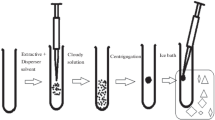
A 5.0 mL aqueous sample solution containing 3.75 mL of water and 1.25 mL of urine sample was spiked with DIC and MEF and its pH was adjusted to 2.0 by dropwise addition of 100 mM HCl or NaOH. The diluted sample was filtered through a disposable 0.45 µm cellulose acetate filter and was placed in a centrifuge tube with a conical bottom. Then, 105 µL of [C4mim][PF6] was added into the solution as extraction solvent. The conical tube was heated in a water bath at 50 °C for 2 min. The used IL was completely dissolved in the solution under these conditions. The tube was cooled in an ice bath for 6 min and the solution became turbid. Then, the solution was centrifuged for 5 min at 3000 rpm. Fine droplets of IL were sedimented at the bottom of the test tube (Fig. 1). The volume of 25 µL of the collected sedimented phase (about 45 µL) was withdrawn using a 50 µL Hamilton microsyringe and injected into the HPLC system for further analysis.

Schematic presentation of TCIL-HLPME procedure
Data analysis and statistical methods
A two-step optimization strategy, including a Placket-Burman screening design and a face-centered central composite design (FCCCD) was employed to optimize the extraction of MEF and DIC. In all cases, design generation and statistical analyses were performed by means of the software package Design Expert version 7.0. The volume of extraction solvent, extraction time, centrifugation time, heating time and temperature, pH and salt effect are the variables that can affect extraction efficiency of MEF and DIC by the proposed extraction procedure.
Calculation of preconcentration factor, extraction recovery and relative recovery
The preconcentration factor (PF) is defined as the ratio between the final analyte concentration in the acceptor phase (C f,a ) and the initial concentration of analyte (C i,s ) in the sample solution. More precisely, it can be defined as the ratio between the slope of the extraction calibration curve (m 2) and direct calibration curve (m 1):
where C f,a was calculated from a calibration graph obtained from direct injection of diclofenac and mefenamic acid standard solutions (5–50 mg L−1) in methanol. The extraction recovery (ER) is defined as the percentage of the number of moles of analyte originally present in the sample (n i,s ) which is extracted into the acceptor phase (n f,a ).
where V f,a and V i,s are the volumes of acceptor phase and sample solution, respectively. Relative recovery (RR %) is acquired from the following equation:
where C found, C real, and C added are the concentration of analyte after addition of known amount of standard into the real sample, the initial concentration of analyte in the real sample, and the concentration of known amount of standard which is spiked into the real sample, respectively.
Results and discussion
Selection of an appropriate extraction solvent is the key step in all liquid phase microextraction techniques. The extraction solvent should be water immiscible, provide high distribution ratio for the target analyte to reach the highest extraction efficiency and be compatible with the used analytical instrument. According to literature, [C4mim][PF6], [C6mim][PF6] and [C8mim][PF6] are the most common ILs used as the extraction solvents. Initial experiments were carried out with these ILs. The results showed much better chromatographic behavior, easier operation as well as proper extraction efficiency and repeatability for [C4mim][PF6]. This observation can be attributed to the lower viscosity of [C4mim][PF6] than two other ILs. Thus, [C4mim][PF6] was selected for subsequent experiments.
Screening design
When a large number of factors affect an analytical system, screening is the first step for the efficient assessment of the factors. Screening design includes examining of the different factors by doing a few experiments to detect the most significant variables. It also provides important information about the movement direction which improves the experimental response. A particular type of such designs is Plackett-Burman design (PBD). The theory and application of PBD has been reviewed elsewhere in more detail [16, 17]. Briefly, PBD is a two-level design (−1, +1) for examining N parameters in K = N + 1 runs. The interactions among factors are completely ignored in this design thus, the main effects are calculated with a reduced number of experiments. The PBD matrix consists of “n” real and “m” dummy factors at two levels (N = n + m). The dummy factors are used to estimate the experimental error which is used in the statistical interpretation.
Considering the literature and our experience, seven variables (n = 7) were used to construct the matrix of PBD design (Table 1). The overall design matrix showed 12 runs to be carried out randomly to eliminate the effects of extraneous or nuisance variables. Therefore, seven factors including volume of extraction solvent, salt %, temperature of the sample solution, pH, extraction time, centrifugation time and heating time were selected and investigated at two levels. To evaluate the work, the sum of peak areas of the target analytes was considered as the experimental response. An analysis of variance (ANOVA) visualized using a Pareto chart was performed on the design to assess the main effects (Fig. 2). In the chart, the bar lengths are proportional to the absolute value of the estimated main effects. The chart also includes a vertical line corresponding to the 95 % confidence interval which statistically judges significant effects. An effect which exceeds this reference line may be considered significant with regard to the response. Furthermore, the positive or negative signs in this chart show whether or not the response is enhanced by changing from the lowest to the highest level set for each factor.

Pareto chart of the main effects obtained from a Placket-Burman design for DIC and MEF
According to Fig. 2, pH was the most significant factor which presented a negative effect on the extraction efficiency of MEF and DIC by the proposed procedure. The pH of the sample solution has a great importance to keep the ionizable compounds in their completely deprotonated form for efficient extraction. MEF and DIC are acidic analytes which exist in their neutral form at pH values lower than pKa of 4.2, and so they have more tendency to distribute into the extraction solvent. The percentage of salt was the next most important positive significant factor attributed to the salting out effect. It means that, addition of salt into the sample solution leads to the formation of hydrated spheres around the salt ions and thereby decreases the available water for the hydration of acidic analytes [18]. In addition, as presented in Fig. 2, extraction time and volume of the extraction solvent showed positive effects on the extraction efficiency. The extraction time was considered from the moment that the solution containing completely dissolved IL was put into the ice bath for the set interval [9]. Overall, a longer extraction time gives enough opportunity to reach extraction equilibrium. As a general rule, extraction efficiency is increased by increasing the volume of the extraction solvent up to a specified value and after that a decrease is observed due to dilution of the extracted analytes by increasing the collected volume of the extraction phase.
Heating time showed a negative effect whereas both heating temperature and centrifugation time had non-significant positive effects on the extraction efficiency. The heating temperature and time should be selected as enough to dissolve the IL into the sample solution, completely. On the other hand, the complete and fast phase separation of the fine droplets of IL is not achieved in the suspension at short centrifugation times, therefore; it should be set at the value which provides a fast and complete phase separation. Considering the results of the first screening study, two variables including centrifugation time and heating temperature, which were not significant, were eliminated for the next optimization process and fixed at the appropriate values of 5 min and 50 °C, respectively.
Central composite design and optimization
To find the optimized values of the important factors and achieve the maximum extraction efficiency, response surface methodology (RSM) using a central composite design (CCD) was applied. The RSM explains the relationship among the variables and responses, graphically. In this way, the real optimum extraction conditions are obtained because the interactions of parameters and the curvature among experimental variables are considered. CCD, one of the most frequently used RSMs, is a second-order model that uses the following equation for predicting the optimum values [19]:
where Y is the dependent variable (square root of sum of peak area); X i and X j are the independent variables; β i , β ii and β ij represent the regression coefficients for the linear, quadratic and interaction effects of the model and β 0 is the deviation between the observed and predicted responses in the design point. CCD consists of factorial points, star or axial points (α) and center points. Therefore, the total number of design points needed (N) is determined by the following equation:
where f is the number of significant factors from the first screening step, 2f, 2f and C p are the number of the factorial points, axial points and center points, respectively. There are different types of CCD depending on the value of α. One type of CCD is face-centered central composite design (FCCCD) which in α is considered unity.
According to this design, each of the five factors (X 1, X 2, X 3, X 4 and X 5) from the screening step was studied at three levels (Table 2). The design included two replicates of the central point. Hence, according to the Eq. 5, the employed design consisted of 44 randomly performed experiments. A drug-free urine sample was used for the optimization process, and the square root of the sum of the peak areas was assumed as the response to evaluate the extraction efficiency of diclofenac and mefenamic acid.
ANOVA was performed at P = 0.05 to evaluate the significance of the model equation and terms. The p value is the probability of getting a result as extreme or more extreme than the one observed if the proposed null hypothesis is correct. The effect of a factor is considered as significant if its p value is lower than 0.05. As can be seen in Table 2, a model P value lower than 0.0001 implies that the model is significant and the “lack of fit” was not significant. The model was described as follows:
Y = 43.037 − (7.60 × X 1) − (17.96 × X 2) + (4.47 × X 3) − (1.54 × X 4) − (0.24 × X 5) + (9.57 × X 1 × X 2) − (4.27 × X 1 × X 3) + (2.06 X 1 × X 4) − (0.51 × X 1 × X 5) + (5.59 × X 2 × X 3) + (1.58 × X 2 × X 4) + (1.83 × X 2 × X 5) − (3.73 × X 3 × X 4) − (2.22 × X 3 × X 5) − (0.30 × X 4 × X 5) − (9.62 × X 21 ) + (21.72 × X 22 ) + (1.40 × X 23 ) + (0.23 × X 24 ) – (0.18 × X 25 ).
The goodness of fit of the quadratic polynomials is expressed by the coefficient of determination, R 2, which should be at least 0.80 for a good fit of a model [20]. As is observed in Table 2, the coefficient of determination, R 2, is 0.9471 which means that the obtained equation is adequate for correlating experimental results. Figure 3a, b depict the predicted versus actual responses and residuals versus the predicted responses, respectively. As shown, most plots are scattered in close proximity with the line in Fig. 3a, and the residual plots are scattered randomly in Fig. 3b which confirms a good correlation between predicted and actual responses.

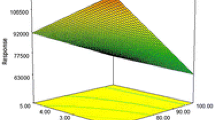
a Predicted vs. actual, b residual vs. predicted and c–f response surfaces of MEF and DIC using FCCCD which illustrates the relationships between the independent variables and the experimental response
After ascertaining the validity of the model, graphs of response surfaces were drawn to find the optimum value for each factor. Dimensions for a quadratic response surface are calculated regarding the number of factors (f) in CCD. Thus, for five factors a six-dimensional response surface should be generated. For ready visualization, three-dimensional (3D) response surfaces are used; in these plots the response model is mapped against two experimental factors while the others are held constant at their optimum. The curvatures of the plots indicate the interaction between the factors. In addition, two-dimensional contour plots which are made on the basis of the model equations were used. These plots display the interaction between the independent variables and assist in determining the optimum operating conditions for the desirable responses.
Figure 3 shows some of the estimated response surfaces with their related contour plots for the extraction of DIC and MEF by the TCIL-HLPME procedure. Figure 3c, d show that concurrently decreasing pH and extraction solvent volume enhances the extraction efficiency. The maximum response was obtained at a pH of 2.0 and a 105.50 µL volume of the extraction solvent. In fact, pH plays the main role in this method to achieve good detection sensitivity of target analytes. The negative effect of pH can be attributed to the fact that DIC and MEF formed ionic structures with increasing of pH, and in these conditions, extraction of the analyte was not effective into the organic phase. Moreover, volume of the IL determines the occurrence of the cloudy state in the sample solution because a portion of it is dissolved in the sample solution. Therefore, the cloudy state is increased with increasing volume of the used IL. But, larger volumes of the IL may lead to the dilution of the extracted analytes and decrease of the determination sensitivity. Enhancement of the extraction efficiency by decreasing of the extraction solvent volume from 300 to 105.50 µL (Fig. 3c) is due to an increase in the concentration of the extracted analytes in the acceptor phase. Figure 3e, f depict the obtained response surface and contour plot for salt versus volume of the extraction solvent. As can be seen, the response was increased by increasing the salt amount indicating its positive effect on the extraction efficiency. The maximum response was obtained when 1.0 g of NaCl was used for 5 mL of the sample volume.
Finally, according to the overall results of the optimization study, the following experimental conditions were chosen: extraction solvent volume, 105 µL; heating temperature, 50 °C; heating time, 2 min; extraction time, 6 min; pH, 2.0; 1.0 g of NaCl in 5 mL of the sample solution; and centrifugation time, 5 min.
Quantitative analysis of diclofenac and mefenamic acid
To evaluate the analytical performance of the TCIL-HLPME method, limit of detection (LOD) and quantification (LOQ), linearity, repeatability (intra-day precision), reproducibility (inter-day precision), preconcentration factor (PF) and extraction recovery (ER %) were investigated under the optimal conditions. The results are presented in Table 3. The reported LOD values of 20 ng mL−1 for DIC and 30 ng mL−1 for MEF were obtained based on practical experiments. For this purpose, concentrations of the target analytes were decreased since a detectable signal-to-noise ratio of 3 (3S/N) was obtained for each of them at their retention times. The LOQ values were considered the low concentrations that linearity of the calibration curves started from. As shown in Table 3, the LOQ values of 40 and 60 ng mL−1 were obtained for DIC and MEF, respectively. The comparative peak areas, calculated based on three replicated extractions at the concentration of 100 ng mL−1 from one urine sample, were applied to calculate the repeatability and were expressed as relative standard deviation percentages (RSD %). Reproducibility was calculated based on the obtained average peak area on each day during three consecutive days. As reported in Table 3, the intra- and inter-day RSD % values were less than 4.4 and 8.0 %, respectively. Figure 4a shows the resulted chromatograms for blank and two replicated extractions of DIC and MEF from a drug-free urine sample indicating the acceptable repeatability of the proposed method. The preconcentration factors were calculated according to Eq. 1 using the slop ratios of extraction and direct calibration curves. Direct calibration curves were constructed by the resultant peak areas of MEF and DIC versus six concentration points of their standard solution in the range of 5–50 mg L−1. The slop values (m2) and determination coefficients (R2) for direct calibration curves were 28.963 and 0.9964 for MEF and 40.34 and 0.9967 for DIC, respectively. Regarding the extraction calibration sensitivities of 3257 (R 2 = 0.9986) for DIC and 1731.5 (R 2 = 0.9912) for MEF, PF values of the proposed method were 82 and 60 for DIC and MEF, respectively. The ER % values of the proposed method were 73.8 % for DIC and 54 % for MEF.

a The resultant chromatograms for the blank and two replicated extractions from a drug-free urine sample (urine 1) at the concentrations of 100 ng mL−1 of DIC (1) and 200 ng mL−1 of MEF (2) and b the non-spiked (a) and spiked (b) chromatograms of a urine sample (urine 4) which was taken from a 30-year-old female who was treated with DIC
The results provided by TCIL-HLPME for extraction and determination of MEF and DIC were compared with corresponding literature data obtained by other methods (Table 4). Regarding the complexity of urine matrix compared with simple matrices such as deionized water reported in Table 4, utilization of the low volumes of urine samples which is a considerable advantage for analysis of biological fluids, utilization of less expensive and more general determination instrument (HPLC–UV) in comparison with expensive and not available ones in routine analytical labs such as LC–MS/MS, the proposed method provided a wide linearity range, good recoveries and acceptable LOD, repeatability and reproducibility values. Moreover, TCIL-HLPME is a very simple, quick and inexpensive microextraction technique which can easily be used for the aims of field analysis. More important, a microliter volume of a green organic solvent is used in this technique instead of milliliter volumes of the toxic organic solvents which improves the safety. In addition, lower detection limits and higher determination sensitivities can be achieved by applying larger volumes of urine samples during extraction procedure as well as usage of more powerful analytical instruments such as LC–MS/MS. Overall, considering the mentioned advantages, it seems that TCIL-HLPME can be proposed as a good alternative for the existing sample preparation methods to analyse DIC and MEF in urine samples.
Analysis of real samples
Finally, the proposed method was applied for the determination of DIC and MEF in four urine samples which were taken from four female volunteers. The results are summarized in Table 5. Figure 4a shows the chromatograms obtained for a blank and two replicated extractions from a drug-free urine sample (urine 1) at the concentrations of 100 ng mL−1 of DIC and 200 ng mL−1 of MEF, respectively. Figure 4b presents the (1) non-spiked and (2) spiked chromatograms obtained from a urine sample (urine 4) which was taken from a 30-year-old female who was treated with DIC. The obtained chromatograms indicated the suitable applicability of TCIL-HLPME technique for the determination of low levels of DIC and MEF in urine samples due to providing acceptable repeatability and clean separations.
To examine the accuracy of the proposed method, all urine samples were spiked with different concentrations of DIC and MEF proportional to the founded concentrations in each sample and the relative recovery (RR %) values were calculated according to Eq. 3. The spiked recoveries were good in the range of 85.4–95.4 %. RSD % values for the determination of the target analytes were obtained less than 6.6 %. Overall, the results demonstrated a good performance and accuracy of the presented method for the determination of DIC and MEF in urine samples.
Conclusion
In the present study, a simple, quick, environmentally friendly and easy-to-use microextraction technique based on temperature-controlled ionic liquid homogeneous liquid phase microextraction followed by HPLC–UV was developed for the extraction and determination of mefenamic acid and diclofenac from urine samples. Placket-Burman design and response surface methodology based on face-centered central composite design was used to optimize the effective parameters on the performance of TCIL-HLPME. The experimental results indicated lower limits of detection, good linearity and repeatability for the proposed method which will make it a competitive alternative to routine methods in the future.
References
M. Starek, J. Krzek, Talanta 77, 925 (2009)
M.R. Moradiya, K.P. Solanki, P.A. Shah, K.G. Patel, V.T. Thakkar, T.R. Gandhi, J. Pharam. Sci. Tech. 67, 164 (2013)
M. Mirzaei, H. Aghabozorg, H. Eshtiagh-Hosseini, J. Iran. Chem. Soc. 8, 580 (2011)
M. Shamsipur, N. Fattahi, M. Sadeghi, M. Pirsaheb, J. Iran. Chem. Soc. 11, 249 (2014)
P. Kubica, H. Garraud, J. Szpunar, R. Lobinski, J. Chromatogr. A 1417, 30 (2015)
S. Song, E.N. Ediage, A. Wu, S. De Saeger, J. Chromatogr. A 1292, 111 (2013)
R.G. Melcher, P.L. Morabito, Anal. Chem. 62, 2183 (1990)
Y. Yamini, S. Seidi, M. Rezazadeh, Anal. Chim. Acta 814, 1 (2014)
H. Ebrahimzadeh, Y. Yamini, F. Kamarei, S. Shariati, Anal. Chim. Acta 594, 93 (2007)
F. Geng, J. Liu, L.Q. Zheng, L. Yu, Z. Li, G.Z. Li, C.H. Tung, J. Chem. Eng. Data 55, 147 (2010)
Q. Zhou, H. Bai, G. Xie, J. Xiao, J. Chromatogr. A 1177, 43 (2008)
M.R. Jamali, Y. Assadi, R.R. Kozani, F. Shemirani, Anal. Chim. Acta 594, 93 (2007)
C. Yao, W.R. Pitner, J.L. Anderson, Anal. Chem. 81, 5054 (2009)
L. He, Y. Luo, H. Xie, C. Wang, X. Jiang, K. Lu, Anal. Chim. Acta 665, 52 (2009)
Q.X. Zhou, H.H. Bai, G.H. Xie, J.P. Xie, J. Chromatogr. A 1177, 43 (2008)
D.B. Hibbert, J. Chromatogr. B 910, 2 (2012)
M.P. Callao, Trends Anal. Chem. 62, 86 (2014)
H. Farahani, M.R. Ganjali, R. Dinarvand, P. Norouzi, J. Agric. Food Chem. 57, 2633 (2009)
G.E.P. Box, K.B. Wilson, J. Roy. Statist. Soc. Ser. B 13, 1 (1951)
A.M. Joglekar, A.T. May, Cereal Food World 32, 857 (1987)
S.H. Davarani, A. Pourahadi, S. Nojavan, M.H. Banitaba, M. Nasivi, Aghdam. Anal. Chim. Acta 722, 55 (2012)
P.L. Kole, J. Millership, J.C. Mcelnay, J. Pharm. Biomed. Anal. 54, 701 (2011)
F. Rezaei, Y. Yamini, M. Moradi, B. Ebrahimpour, Talanta 105, 173 (2013)
A.R.M. Silva, F.C.M. Portugal, J.M.F. Nogueira, J. Chromatogr. A 1209, 10 (2008)
S. Kamaruzaman, M.M. Sanagi, S. Endud, W.A.W. Ibrahim, N. Yahaya, J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 940, 59 (2013)
M.R. Rouini, A. Asadipour, Y. Hoseinzadeh-Ardakani, F. Asadi, J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 800, 189 (2004)
B. Barfi, A. Asghari, M. Rajabi, A.G. Moghadam, N. Mirkhani, F. Ahmadi, J. Pharm. Biomed. Anal. 111, 297 (2015)
L. Fotouhi, S. Seidi, Y. Yamini, E. Hosseini, Anal. Methods 7, 2848 (2015)
Acknowledgments
The authors express their gratitude for financial support from Alzahra University. Thanks also goes to Dr. Zahra Talebpour for providing the HPLC instrument.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fotouhi, L., Seidi, S. & Shahsavari, F. Optimization of temperature-controlled ionic liquid homogenous liquid phase microextraction followed by high performance liquid chromatography for analysis of diclofenac and mefenamic acid in urine sample. J IRAN CHEM SOC 13, 1289–1299 (2016). https://doi.org/10.1007/s13738-016-0843-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-016-0843-9