
Abstract
Biodiversity loss from disturbances caused by human activities means that species are disappearing at an ever increasing rate. The high number of species that have yet to be described have generated extreme crisis to the taxonomist. Therefore, more than in any other era, effective ways to discover and delimitate species are needed. This paper reviews the historically foremost approaches used to delimit species in Ascomycota, the most speciose phylum of Fungi. These include morphological, biological, and phylogenetic species concepts. We argue that a single property to delineate species boundaries has various defects and each species concept comes with its own advantages and disadvantages. Recently the rate of species discovery has increased because of the advancement of phylogenetic approaches. However, traditional phylogenetic methods with few gene regions lack species-level resolution, and do not allow unambiguous conclusions. We detail the processes that affect gene tree heterogeneity, which acts as barriers to delimiting species boundaries in classical low-rank phylogenies. So far, limited insights were given to the DNA-based methodologies to establish well-supported boundaries among fungal species. In addition to reviewing concepts and methodologies used to delimit species, we present a case study. We applied different species delimitation methods to understand species boundaries in the plant pathogenic and cryptic genus Phyllosticta (Dothideomycetes, Botryosphaeriales). Several DNA-based methods over-split the taxa while in some methods several taxa fall into a single species. These problems can be resolved by using multiple loci and coalescence-based methods. Further, we discuss integrative approaches that are crucial for understanding species boundaries within Ascomycota and provide several examples for ideal and pragmatic approaches of species delimitation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Currently, over 148,000 species of fungi are recognised (Species Fungorum, 2021; http://www.speciesfungorum.org), and an increasing number of new fungal species are added every year with the development of sequence-based methodologies. For example, three fungal classes, 18 orders, 48 families, and 214 genera were published in 2019 (Cheek et al. 2020). Ascomycota is the most speciose phylum of Fungi. Out of the top five orders in which most new species were published in 2019, four (Hypocreales, Pleosporales, Lecanorales, and Capnodiales) belong to Ascomycota. Ascomycota reproduce asexually and sexually, their spores are spread easily by wind, water, soil, or insects and they are well adapted to saprobic, parasitic or mutualistic life modes (Hyde et al. 2019; Naranjo‐Ortiz and Gabaldón 2019).
Why are the Ascomycota so diverse?
Ascomycota are cosmopolitan and can exist in both terrestrial and aquatic habitats. Several classes of Ascomycota produce mycorrhizal associations, while some are adapted to lichen lifestyle and are considered key during the process of terrestrialisation (Naranjo‐Ortiz and Gabaldón 2019). Some groups, such as Saccharomycotina, are adjusted to arctic and highly osmotic habitats, which are typically occupied by prokaryotes (Rodriguez et al. 2009; Zajc et al. 2017; Naranjo‐Ortiz and Gabaldón 2019). Many ascomyceteous classes are important plant and human pathogens (Dean et al. 2012; Cheek et al. 2020). As with most organisms, Ascomycota species richness is highest in the tropics, and they fulfil many vital roles in all ecosystems (Hawksworth 2001; Wu et al. 2019; Maharachchikumbura et al. 2021).
Why is the correct naming of species in Ascomycota important?
Scientific names are standardised (the names are universally accepted and unique for a given organism); they provide information about the similarities and differences between species belonging to the same genus (Winston 1999; Borman and Johnson 2020). We communicate about species of Ascomycota via the use of scientific names. These names provide access to the accrued information on the biology, distribution, ecology, host range, and control of, as well as the risks associated with, fungal species. Erroneous identification can lead to unnecessary control measures and restrictions being applied or, importantly, in no action being taken to control potentially devastating pathogens.
Some Ascomycota species names are descriptive; providing a characteristic or combination of characteristics of the taxon (size of ascomata/conidiomata, the shape of ascus/ascospore, conidial morphology, proportion and the color of the spores). However, numerous paraphyletic and polyphyletic species in Ascomycota independently evolved similar traits to adapt to related environments or geographical locations (Gueidan et al. 2007; Hittinger et al. 2015). Therefore, these different morphological traits are not always evolutionary significant, and these character-based names are less instructive in Ascomycota when compared to plants and animals. Recently, major taxonomic changes have occurred in the taxonomy of Ascomycota. The recent advance of molecular-based data placed asexual fungi, which were previously arranged in the form-phylum Deuteromycota, mainly into the phylum Ascomycota (Bruns et al. 1991).
Some Ascomycota species names are based on geography, derived from the habitat or locality in which the holotype was described. However, in recent history, there have been biological invasions of many non‐native Ascomycota species to different geographical regions and these may have implications for global trade (Fones et al. 2020). Therefore, naming based on distribution is less informative in these ubiquitously distributed Ascomycota species. Furthermore, well known pathogenic genera in Ascomycota, such as Alternaria, Botrytis, Cercospora, Colletotrichum, Fusarium, Mycosphaerella, Pestalotiopsis and Phyllachora were often described based on the host plant from which they were first isolated. However, with the development of sequence data, many authors have questioned this "new host occurrence—new species" concept since many of these species are not highly host specific and found in a range of hosts (Hyde et al. 2009; Quaedvlieg et al. 2011; Groenewald et al. 2013; Aoki et al. 2014; Lawrence et al. 2016; Maharachchikumbura et al. 2016). Also, molecular data have shown that endophytes can dwell in many plants and only produce their sexual structures on certain host plants (Hardoim et al. 2015). The naming of species with a wide range of host is, therefore, less informative in Ascomycota compared to highly host-specific groups such as rusts and smuts. Note that the opposite is also true in some groups of Ascomycota. For example, in the arthropod-associated Laboulbeniales, species such as Arthrorhynchus eucampsipodae and Hesperomyces virescens, once thought to be taxa with wide host ranges, are in reality complexes of species, each with strict host specificity (Haelewaters et al. 2018, 2020a).
Commemorative species names honor a person or event/expedition. It is common to name a fungus in honor of a person who has made a considerable contribution to the taxonomy of the group of organisms involved in naming or more generally to society (e.g., Annabella australiensis, Daldinia hawksworthii, Hysterium barrianum, Malmidea attenboroughii, Phaeosphaeria erikssonii, Phoma cavalliniana, Verticillium lindauianum) (Shoemaker and Babcock 1989; Braun and Dick 2002; Boehm et al. 2009; Pažoutová et al. 2013; Fryar et al. 2019; Guzow-Krzemińska et al. 2019). In some cases, especially in genera with many species, it is often difficult to find descriptive or geography-based names, and in such cases species names may involve random combinations of letters, so-called nonsense species names.
From biodiversity and conservation perspectives, taxonomists, plant pathologists, chemists, ecologists, and landowners need to be able to precisely identify, name, and communicate about the Ascomycota species they encounter day to day. Over the past few decades, different species concepts and delimitation methods have been widely used to study relationships among closely related species, determine species limits, or describe organisms as new species (Lücking et al. 2020; Aime et al. 2021; Crous et al. 2021a, b).
Consequently, the rates of molecular and morphological divergence of species can often be unrelated and mismatched (Stewart et al. 2014). Therefore, various discussions about species concepts and species boundaries have been proposed (Harrington and Rizzo 1999). This article evaluates the most common species concepts and delimitation criteria in the species rank in Ascomycota. We indicate the ways in which a single property can often mislead the species definition. Therefore, we propose a broader, integrative taxonomy or evidence-based approach to understand the species boundaries in Ascomycota.
Species concept/definition vs. species criteria
Whether a particular population or taxonomic group is adequate to be accepted as a fungal species continues to be the main issue in many taxonomic debates (Dupuis et al. 2012; Xu 2020). The lack of a clear difference between the general "species concept" (theoretical basis based on which we describe and name "species") and working or operational criteria (providing in a solid state to resolve whether a particular taxon is or is not a species) is often misunderstood and lacks a clear distinction. However, most of these concepts and criteria are contradictory as they can lead to different conclusions regarding boundaries and the number of species (De Queiroz 2007). We currently use several different species concepts and species criteria in Ascomycota.
The concept of species and the delimitation of species have always been hot topics in fungal taxonomy. Presently biologists define over 30 different species concepts but only a few species concepts are widely used in fungal taxonomy. However, none has been apparent and up to a unified standard, making fungal taxonomy more or less subjective and each with its own advantages and disadvantages. For instance, biochemical traits are very useful in yeast taxonomy; however, morphology leads nowhere, macromorphological characters are helpful to recognise macrofungi but not applicable in microfungi. The morphological, biological, and evolutionary/phylogenetic species concept, which are mainly used in Ascomycota, are summarised here.
The morphological species concept (MSC) in Ascomycota
The norm of “species rank” in the case of MSC is the degree of phenotypic difference (Cronquist 1978; Cracraft 2000). According to this concept, an ascomyceteous species is identifiable by a basic difference in its morphology, and this is what makes a single species clearly different from all other species in Ascomycota. Individuals of the same species are similar to one another in morphology. This concept needs careful morphological examination where the Linnaean system and most traditional methods of classification use the MSC. Traditionally, MSC has been used to establish species in the Ascomycota due to their simpler morphology. Still, MSC is heavily considered as an important scenario in fungal classification than any other organisms. Among the 97,000 fungal species described, ca. 70,000 (72%) were diagnosed by morphological characters or other phenotypic characters, such as growth at different temperatures or water activities (Pitt 1979; Hawksworth et al. 1996; McDonald 2015). Due to its straightforwardness, the MSC was a reasonably satisfactory concept over the last decades. A taxonomist who has to sort numerous collections in space and time and assign them to concrete and preferably clearly delimited taxa may find it more convenient to recognise strictly phenotypic species in these cataloguing activities. However, most biologists and mycologists believe that the morphological species concept is the least satisfactory. It is practical for identification, but it obscures the evolutionary origins of many features.
According to Mayr (1942), morphological species concepts do not share the biological significance he attributes to his species concept (Cracraft 2000). Fungi are well known for their developmental plasticity. Therefore, it is difficult to say what differences in which morphological characteristics are important for distinguishing species and how different two groups of organisms have to be in order for them to be classified as different species (Yang et al. 2017). In general, the application of the MSC to a group of organisms takes experience and practice, and it is often subjective. The morphologically informative characters and what degree a character separates two species depends on the person working on the fungal group.
Many species in the Ascomycota are pleomorphic (Rossman et al. 2015). They have one or several names for the sexual morph associated with one or more asexual morphs (Taylor et al. 2000). As a result of pleomorphism, an organism can be included in different genera. Their morphological characters are very different from each other, and therefore, they are not useful for distinguishing species. Traditionally, many of the teleomorphic Ascomycota species are primarily identified from dried herbarium materials. However, when grown in favourable conditions, these fungi may produce the anamorph (Hawksworth et al. 2013; Wanasinghe et al. 2014; Drenth et al. 2019). This is a major problem in resolving fungal taxonomy. After the One Fungus = One Name (1F1N) practice in the International Code of Nomenclature for algae, fungi and plants (McNeill et al. 2012), fungal species can only have one name regardless of pleomorphic morphs.
When evaluating the taxonomic significance of observed character differences between individuals, it is important to understand how phenotypic variation is generated and regulated in natural populations of the relevant Ascomycota species concerned. Morphological characters often vary on the natural host, culture media, and successive sub-culturing (Ryan et al. 2002; Ansari and Butt 2011; Maharachchikumbura et al. 2014; Senanayake et al. 2020). Therefore, phenotypic characters such as size ranges of spores and fruiting structures can be considerably plastic between sister species.
In the natural selection process, characters resulting in a better-adapted population will be retained during speciation while mal-adapted species will go extinct. This progression plays a crucial role in shaping the development and maintenance of fungal species and therefore, any phenotypic character associated with the fungus growing environment should be helpful in defining species (Harrington and Rizzo 1999; Crowther et al. 2014; Zhang et al. 2017; Thiyagaraja et al. 2021; Mortimer et al. 2021). However, these phenotypic characters are most suitable at the genus, family, and higher taxonomic classifications. Adaptations to particular substrates, temperature conditions, or moisture conditions and competition with other microbes probably plays more prominent roles than morphology in speciation and cohesion. For instance, some Ascomycota species such as Dyfrolomyces produce fully immersed ascomata in the aquatic habitat (D. tiomanensis) and superficial or semi-erumpent ascomata in the terrestrial environment (D. sinensis) (Hyde et al. 2018).
Cryptic species are morphologically indistinguishable. Traditionally several distinct species were identified as a single nominal one due to their morphological similarity. The progress of DNA sequence data and mating type experiments suggests that many Ascomycota species are cryptic, especially those that belong to plant pathogenic genera (e.g., Alternaria, Colletotrichum, Diaporthe, Fusarium, Peronosclerospora). Therefore, many of the previously described diseases and databases based on morpho-taxonomy need to be revisited/updated (Shivas and Cai 2012).
The biological species concept (BSC) in Ascomycota
The current evolutionary synthesis of the BSC was first discussed by Dobzhansky (1935, 1937). Dobzhansky (1937) defined a species as a “stage of the evolutionary process at which the once actually or potentially interbreeding array of forms becomes segregated in two or more separate arrays, which are physiologically incapable of interbreeding.” However, this statement was considered as a definition of the process of speciation, rather than as a definition of a species by Mayr (1940, 1999). As defined by Mayr (1940), “a species consists of a group of populations, which replace each other geographically or ecologically and of which the neighboring ones intergrade or hybridize wherever they are in contact or which are potentially capable of doing so (with one or more of the populations) in those cases where contact is prevented by geographical or ecological barriers.” Mayr (1942) offered a shorter version of the definition of biological species as “groups of actually or potentially interbreeding natural populations, which are reproductively isolated from other such groups.” BSC is based on solitary reproduction criterion, while phenotypic similarity was not considered a criterion (Mayr 1940, 1942). Nonetheless, the history of applying the concept of biological species dates back to 1927 with records on mating tests carried out to delineate fungal species by Shear and Dodge (1927). Shear and Dodge (1927) revealed three species (Neurospora crassa, N. sitophila, and N. tetrasperma) from the morphological species Monilia sitophila based on mating experiments.
To apply biological species definition to a particular taxonomic group, it is required to obtain precise knowledge of geographical distribution and the certainty of the group's reproductive isolation (Mayr 1999). Defining species using BSC is most successful with well-studied, sympatric, sexually out-breeding populations (Worrall 1999). This concept has been criticised due to difficulties that arise when dealing with allopatric populations (Mallet 1995; Worrall 1999). The concept of biological species is more popular with animals than plants and less commonly applied to fungi. There are other limitations associated with BSC when applied to fungi. BSC is inapplicable to fossil fungi as it is not possible to reveal the gene flow and breeding behaviour of fossil remnants. However, understanding gene flow and breeding behaviours in living taxa is challenging and is one of the major drawbacks of BSC (Thorp James and Christopher Rogers 2015).
Asexual reproduction—by producing conidia—is common in the Ascomycota. Dyer and O’Gorman (2011) pointed out that not less than 20% of fungi lack a known sexual phase and exclusively reproduce asexually. BSC entirely define species in terms of sexual reproduction (Mayr 1942; Worrall 1999). Thus, it cannot be applied to asexual (reproducing by fission, fragmentation, budding, or formation of asexual spores) or homothallic fungi, which are capable of self-fertilisation (Worrall 1999). After revealing the existence and expression of sex-related genes, recent studies indicated that fungi known only from their asexual morphs are also likely to reproduce sexually (Han et al. 2003; Kück and Pöggeler 2009; Dyer and O’Gorman 2011). However, BSC cannot be applied even to closely related taxa due to lack of active sexual stage.
Sexual reproduction of fungal species is determined not merely by their compatibility but also by environmental factors (habitat/medium, nutrients, light level and quality, and temperature) and involvement of hormones (Peberdy 1980; Lee et al. 2010; Han et al. 2003; Liu et al. 2016; Wang et al. 2016; Wallen and Perlin 2018). Worrall (1999) indicated the importance of conducting mating tests and recognising barriers to gene flow. Homothallic fungi yield meiospores via self-fertilisation (Reynolds 1993). There are a number of fungi that are nonculturable, and it is challenging to induce mating in vitro, even in some heterothallic fungi (Taylor et al. 2000).
Biological species definition is not valid in the case of interspecific hybridisation of fungi. Hybridisation was previously considered as a rare process between fungal species (James and Rogers 2015). It was recently recognised to occur in fungi in the environment more often than previously thought (Giordano et al. 2018). Interspecific hybridisation between pathogenic fungi may result in hybrid mutualists with increased or decreased virulence or improved host/new niche adaptation (Schardl and Craven 2003). However, the intersterility barriers among closely related species of fungi are stronger than in plants (James and Rogers 2015).
Biological species definition does not infer partial interfertility, which is mainly occurring in in vitro cultivations of fungi (Worrall 1999). Moreover, partial interfertility can be observed in organisms/populations of the same species that have been geographically isolated for an extended period of time (Harrington and Rizzo 1999).
The BSC has been widely applied by organismal biologists during last several decades (James and Rogers 2015). BSC emphasises reproductive isolation, which was advantageous in previous studies to recognise and delimit new fungal species. This concept was applied to a wide range of fungi. Based on mating compatibility Shear and Dodge (1927) revealed that the morphological species Monilia sitophila harbours three biological fungal species. The biological species concept was used to resolve cryptic fungal species within morphological species (Giraud et al. 2008). Pore et al. (1965) applied the criterion intraspecific compatibility to define a new species Arthroderma lenticularum from A. quadrifidum (Pore et al. 1965).
Evolutionary/phylogenetic species concept (PSC) in Ascomycota
This concept foresees a species as a monophyletic group sharing molecular characters derived from a common ancestor, which is the base for the phylogenetic species recognition in Fungi. Compared to the other species recognition methods, PSC performs best because, once progeny evolutionary species have formed from an ancestor, changes in nucleotide sequences occur and can be known before alterations have occurred in mating behaviour or morphology. Speciation of fungi may vary rapidly compared to other organisms, which may explain why traditional characters (i.e., morphology) have frequently proven inadequate for delineating fungal species. Analyses of extant populations of fungi, using either population genetics or phylogenetic analyses, will allow us to obtain a knowledge of the mechanisms of evolution at the species level (Bruns et al. 1991) and should suggest where to look for diagnostic characters in delimiting species. Furthermore, the concept does not have any obvious built-in exclusions or limitations. Another advantage of PSC is that the analysis can be applied to asexual organisms; thus, the sexual and asexual morphs can be enclosed by a single species concept. Apart from that, limitations of morphological or biological or ecological species concept can be eliminated by using PSC. For example, cryptic speciation within the morphologically indistinguishable plant and human pathogenic fungi such as Fusarium oxysporum complex (FOC), Alternaria, Colletotrichum, and Fusarium species complexes, has been successfully resolved by applying PSC (Liu et al. 2016; Achari et al. 2020).
Many fungi are anamorphic and are not known to produce meiospores (Reynolds 1993). Some fungi are homothallic and can produce meiospores without a partner. Besides, some heterothallic fungi cannot be coaxed into mating in cultivation, and many fungi cannot be cultivated; thus, the application of BSR is untenable in many cases. This also applies to sterile fungi, and fungal strains known only from sequence data and that cannot be linked to any physical specimen.
Determination of species boundaries by PSC is presently based on the application of Genealogical Concordance Phylogenetic Species Recognition (GCPSR) based on multi gene phylogeny. The theoretical principles for GCPSR were proposed by Taylor et al. (2000) and Dettman et al. (2003) based on Avise and Ball’s genealogical concordance species concept. By implementing GCSPR, poorly supported non-monophyly in one locus are prohibited from undermining well supported monophyly of another locus. Udayanga et al. (2014) used the GCPSR method to resolve the species limits of the Diaporthe species complex. Manamgoda et al. (2014) applied the same approach to find the species boundaries within Bipolaris. Especially for important plant and human pathogens with cryptic species such as the species complexes that occur in the genera Fusarium, Alternaria and Colletotrichum, delimitation of species boundaries mainly rely on GCPSR (Liu et al. 2016; Achari et al. 2020).
Nevertheless, using GCPSR criteria is somewhat challenging because there are other processes, for instance, incomplete lineage sorting, horizontal gene transfer and population structure, which could cause discordance between gene trees and species trees and hide the true evolutionary connections between closely related taxa (Liu et al. 2016; Achari et al. 2020). Furthermore, the common practice of concatenating DNA sequence data from multiple loci under GCPSR can lead to inaccuracies in species identification (Liu et al. 2016; Achari et al. 2020). Instead, multispecies coalescent models that combine gene tree ambiguity into species recognition may more precisely and accurately define species. Estimation of the speciation process using the Multispecies coalescent model provides a more comprehensive speciation event as it recognises more gene discordant events than GCPSR. Multispecies coalescent model-based species recognition was mainly applied for animal and plant taxa at the beginning, but it has been gradually adopted for resolving species complexes in plant pathogenic fungal groups such as the Alternaria alternata complex by Stewart et al. (2014), for Colletotrichum by Liu et al. (2016) and for FOC by Achari et al. (2020). When compared with the previously proposed model, one advantage of this model is that it allows for the incorporation of understanding from multi gene trees into a single higher level species tree during the delimitation process, excluding the constraint of stipulating a guide tree for showing species associations (Liu et al. 2016; Achari et al. 2020).
The difference within clades also depends on age and thus the time available for speciation. Overall, to decide the limits of the species clades based on phylogeny, results obtained from both Multispecies coalescent and GCPSR should be considered. Clades give support for both the Multispecies coalescent model and GCPSR can be recognised as phylogenetic species in any given population. Furthermore, molecular dating to estimate divergence times within a given population can also be considered as a potential approach to strengthen the species concept determined via phylogeny.
Recently, the dependence of fungal identification on species concepts, delimitation, and recognition approaches have been proposed by Lücking et al. (2020); however, limited insights were pointed out in regard to DNA-based methodologies, although they are further recommended to establish well-supported boundaries among fungal species. DNA-based species delimitation methods are widely available, using distinct strategies including genetic distance (automated barcode gap discovery algorithm, ABGD; and statistical parsimony, SPN), coalescent (generalised mixed Yule coalescent, GMYC; Poisson tree processes, PTP; Bayesian phylogenetics and phylogeography, BPP; and phylogeographic inference using approximate likelihoods, PHRAPL), and genealogical concordance (genealogical concordance phylogenetic species recognition, GCPSR) approaches (e.g., Clement et al. 2000; Rannala and Yang 2003; Hart and Sunday 2007; Powell et al. 2011; Parnmen et al. 2012; Puillandre et al. 2012; Fujisawa and Barraclough 2013; Quaedvlieg et al. 2011; Haelewaters et al. 2018; Bustamante et al. 2019).
Species delimitation via various phylogeny-based methods
The distinction of cryptic lineages in Beauveria bassiana, initially recognised by Rehner et al. (2011), was overcome by integrative analyses where genetic distance (ABGD and SPN) and coalescent (GMYC and BPP) approaches for three markers (Bloc, Rpb1, and Tef1) established a new taxon B. peruviensis (Bustamante et al. 2019). The segregation of B. peruviensis from B. bassiana confirmed that, in addition to phylogenetic analyses, DNA-based methods are optimal to delimit taxa within morphologically defined species. Although the Bloc marker was confirmed as a diagnostic DNA barcode (minimum p-distance = 1.3%); the GMYC, a model that combines diversification between species and coalescence within species (Fujisawa and Barraclough 2013), and the multilocus BPP method, a model that inferences under the multispecies coalescent model with and without introgression, properly recognised B. peruviensis as a separate taxon (Goldstein et al. 2000; Liu et al. 2016; Bustamante et al. 2019). The recognition of B. peruviensis was based on ultrametric trees for single locus with highly significant likelihood ratios for GMYC and based a priori on the concatenated data with high posterior probabilities for BPP (Tables S1, S2, S4; Bustamante et al. 2019). Therefore, the identification of B. peruviensis as a new species was confirmed mainly through phylogenetic analyses and DNA-based algorithms. The use of these methodologies is recommended to delimit species, and subsequently, the achievement of congruent and highly significant results across the methods is likely to prove most useful for framing reliably supported species boundaries that allow the recognition and identification of fungal taxa (Carstens et al. 2013; Bustamante et al. 2019).
The genus Bryoria in the family Parmeliaceae (Myllys et al. 2011). In recent years sequence-based studies advanced the genus, resolved many taxa, and further described cryptic species within the taxa previously identified based on morphology (Boluda et al. 2019). However, there is often a mismatch between the traditionally accepted Bryoria and other Parmeliaceae morphospecies and the use of different gene regions (McMullin et al. 2016; Boluda et al. 2019). This is mainly due to the incomplete lineage sorting, and Boluda et al. (2019) used an integrative taxonomic approach that includes morphology, chemical, molecular, and distributional characters to re-assess species boundaries in Bryoria sect. Implexae. This included standard barcodes ITS, IGS, GAPDH, two new loci (FRBi15 and FRBi16) and microsatellite markers. Sequence datasets were analysed based on various methods that included Bayesian and maximum likelihood phylogenies, phenogram reconstruction, STRUCTURE Bayesian clustering, principal coordinate analysis and haplotype network. ABGD, PTP, GMYC, DISSECT species delimitation analyses, divergence time estimation, and past population demography were carried out. Further detailed morphological examinations and TLC was carried out to understand the species' chemical profile. The morpho-chemical analysis and phylogeny of both FRBi15 and FRBi16 could not conclude the number of putative species, while Poisson tree processes, STRUCTURE, GMYCm, and DISSECT analyses concluded six putative species. The PCoA, Haplotype Network and ABGD methods resulted in the recognition of four putative species. Therefore, these different methods do not support the currently accepted 11 Bryoria sect. Implexae morphospecies and Boluda et al. (2019) proposed to reduce them to four phylogenetic species.
Boluda et al. (2019) further discussed that the sequence data do not reflect the species' evolution but only display the history of the studied loci, which may seldom be distinctive from the species natural history overall. The use of traditional barcodes (ITS, IGS, GAPDH) and microsatellites gave similar topologies. However, the results based on intergenic loci (FRBi15 and FRBi16) were incongruent. These dissimilarities could happen due to numerous reasons such as recombination, hybridisation, or incomplete lineage sorting.
The order Laboulbeniales, comprised of arthropod-associated biotrophic ectoparasites, has been traditionally neglecte by the broader mycological community. These fungi are difficult to study due to their microscopic size, low infection rates, and the fact that they cannot be grown in axenic culture (Haelewaters et al. 2021). One of the most encountered taxa is Hesperomyces virescens, a parasite of ladybirds (Coleoptera, Coccinellidae). Hesperomyces virescens is known from more than 30 ladybird species and has thus far been reported in every continent except Antarctica and Australia (Haelewaters et al. 2014, 2017). It has received considerable attention recently because it is often reported on Harmonia axyridis, an invasive alien species that has been introduced in many countries outside of its native range, with adverse effects on locally native ladybird diversity (Roy et al. 2016; Haelewaters et al. 2017). As a result, researchers have suggested testing H. virescens as a potential biological control agent of H. axyridis (see Haelewaters et al. 2020b). Experimental laboratory work by Cottrell and Riddick (2012) revealed that interspecific transmission of H. virescens occurred at low rates, whereas intraspecific transmission was common. This observation prompted the authors to hypothesize that isolates/strains may exist that only infect closely related ladybird species or even only a single species. Moving forward, Haelewaters et al. (2018) presented evidence from morphometrics, ecology (host associations), and molecular phylogeny to show that H. virescens is a species complex. These authors performed sequence-based species delimitation methods (ABGD, bPTP, GMYC) on ITS and LSU, and concatenated SSU + ITS + LSU datasets and found support for segregation by host.
Even though the majority of species are still being described based on morphology alone, other recent studies in Laboulbeniales have revealed the importance of using molecular data in this order. For example, also Arthrorhynchus eucampsipodae on bat flies and Laboulbenia flagellata on carabid beetles are species complexes (Haelewaters and De Kesel 2020; Haelewaters et al. 2020a), and Haelewaters and Pfister (2019) revealed the existence of position-induced morphological plasticity in taxa of Gloeandromyces, resulting in phylogenetic species with multiple morphotypes depending on the position of the bat fly host.
From multilocus approaches to whole genome approaches
Contemporary methods, including standard barcodes and multilocus sequence analyses, provide more information to better characterise species boundaries (Santos et al. 2017). During the past decade, following the Human Genome Project, advances in genome sequencing technologies have revolutionised fungal systematics and facilitated the phylogenomic era (James et al. 2020). The cost of a draft genome of fungi is decreasing, and more fungal genomes are being deposited to genome sequence databases, including genomes of poorly studied clades such as the class Laboulbeniomycetes (Haelewaters et al. 2020c). Current phylogenies mainly rely on the comparison of several loci of the genome, which has several disadvantages, as mentioned previously. Therefore, established phylogenies need to be revisited, with more aspects of the genomes being compared (Wibberg et al. 2020).
Single-copy genes or single-copy gene families are valuable genetic materials in inferring relationships of unclear lineages across Eukaryotes (Aguileta et al. 2008; Ren et al. 2016). As the number of fungal genomes continues to increase in genomic databases, more robust phylogenies have been reconstructed based on core single-copy genes (Haridas et al. 2020; Kjaerbolling et al. 2020; Vandepol et al. 2020; Shen et al. 2020). Shen et al. (2018) identified 2048 amino acid orthologs from 332 budding yeast species to reconstruct their phylogeny. Recently, Shen et al. (2020) used similar methods to reconstruct a robust phylogeny of the phylum Ascomycota based on 815 BUSCO genes predefined orthologs from Ascomycota in the Benchmarking Universal Single-Copy Orthologs database (Seppey et al. 2019; Shen et al. 2020). These works are helpful in inferring a stable phylogeny at high taxonomic levels without attempting to delineate fungal species.
The state-of-the-art methods to infer a robust species tree in phylogenomics are based on two different strategies; concatenation and coalescence. The examples we have mentioned above adopted the concatenation approach (Shen et al. 2018, 2020), which relies on the entire merged core sequences, single-copy genes. MP method assumed that all individual sequences have no horizontal gene transfer and no paralogs. However, this assumption is unstable due to the evolutionary histories of different genes that can be inconsistent with each other and the species tree (Hahn and Nakhleh 2016). Hence, the coalescent-based method was developed and widely used in inferring species phylogenies (Liu et al. 2009; Mirarab and Warnow 2015; Peter et al. 2018). For example, Peter et al. (2018) built 2018 gene trees based on orthologous groups and further summarised them into a coalescent species tree. Compared with the concatenation-based method, the latter has been proven statistically consistent under vast numbers of gene trees, which can avoid the error induced by incomplete lineage sorting and horizontal gene transfer (Mirarab and Warnow 2015; Chung and Ane 2011). However, this approach can be error prone due to the small number of sites and the large proportion of missing sites in individual gene alignments, especially for deeper nodes (Springer and Gatesy 2016).
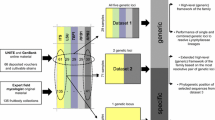
To date, few reports characterise species boundaries of Ascomycota based on genomic analysis (Sepúlveda et al. 2017; Kobmoo et al. 2019; Wibberg et al. 2020; Garcia et al. 2021). However, mycologists have offered useful suggestions for using genome data to define new species in the fungal kingdom. Four criteria were proposed by Matute and Sepulveda (2019) in defining species based on genome sequences: (1) reciprocal monophyly, (2) high concordance among genomic partitions, (3) lower interspecies differentiation than intraspecific differentiation, and (4) low polymorphism. To some extent, these criteria can standardise the use of the concept of genomic species in the genomic era. Xu (2020) proposed that 97% average nucleotide identity for shared house-keeping genes can be used to delineate fungal species. But the number and standard datasets of shared house-keeping genes must be predefined.
Genomic distance is another approach to delineate species, which is not dependent on evolutionary relationships (Gostinčar 2020). Average nucleotide identity (ANI) (Sepúlveda et al. 2017) of the whole genome sequence was introduced to delineate species by comparing two genome sequences. The 95–96% ANI was proposed to be the threshold to characterise species boundaries (Kim et al. 2014). Genomic similarity thresholds to delineate fungal species at phylum, class, order, family, genus, and species levels were evaluated. This method displayed high accuracy in delineating species despite the low discriminative power at higher taxonomic ranks (Gostinčar 2020). Some bacteriologists proposed that core genome phylogenetic analysis and ANI altogether can be used to define bacterial species boundaries (Chan et al. 2012). This combined strategy is not used in fungal taxonomy so far; a case study at the species level is expected in future research.
To our knowledge, there are no universal methods to delimit species boundaries using genome sequences. Here we offer some suggestions. First, we must confirm that a large number of genomes facilitate the development of a stable fungal taxonomy, and genome-based taxonomic approach will play an increasingly important role in fungal species delineation. Genome-based fungal taxonomic studies should be encouraged, and a new proposed classification system should be based on the existing fungal taxonomy foundation and progressively revise it using reliable whole-genome sequences. The standard genome-level marker datasets and criteria for fungal taxonomy should be established by authorities, which will be convenient for pioneering research.
Case study
Genetic markers are not influenced by environmental conditions and, therefore, they are useful to determine the genetic structure of fungal populations and species discrimination. The divergent nature of evolution is reflected in phylogenetic trees. For sister species in a phylogenetic tree, as well as for larger monophyletic groups identified on it, one can always identify characters by which the species included in these groups are mostly similar. More recently, sequence data and phylogenetic analysis techniques have increased unequivocal taxonomic identification of fungal species. However, disagreements among traditional phylogenetic reconstructions may introduce taxonomic questions. Here, by using the genus Phyllosticta, we attempt to understand (1) whether monophyletic clusters of isolates effectively correspond to phylogenetic species, (2) whether different molecular markers result in a different number of phylogenetic species, and (3) whether it is possible to confidently propose a species number based on different concatenation and coalescence methods.
Phyllosticta species are an important group of pathogens causing spots in leaves and stems as well as damage to fruits (Baayen et al. 2002; Glienke et al. 2011; Wikee et al. 2013). Phyllosticta species reduce the photosynthetic ability and cause severe damage to the host by premature fall of leaves and fruits (Glienke et al. 2011). They are also found as endophytes or saprobes from a wide range of host plants (Wikee et al. 2013). The species concept of Phyllosticta has undergone significant changes since its introduction by Persoon (1818). The sexual morph of Phyllosticta is identified as Guignardia. Since Phyllosticta is the oldest and most commonly used name, Guignardia was synonymized under Phyllosticta (Glienke et al. 2011). There are over 3000 specific epithets of Phyllosticta listed in Index Fungorum (2021; http://www.indexfungorum.org/na mes/Names.asp). The taxonomy of the genus has long been problematic due to limited morphological characters and species introduction based only on host associations (Jayawardena et al. 2019). Morphological features of related taxa are always overlapping and change based on host, environment, and culture medium. Currently, six species complexes are recognised in Phyllosticta (Norphanphoun et al. 2020). A natural classification has been established for the genus based on the polyphasic approaches, combining morphological characters and phylogenetic relationships. However, many species of Phyllosticta have overlapping morphologies and recent phylogenetic analysis based on different molecular markers does not always support the delimitation of species (Wang et al. 2020).
A total of 109 strains with sequences for five loci (ITS, LSU, Actin, GAPDH, TEF1) representing 84 species of Phyllosticta in six species complexes were selected for the case study. The species Neofusicoccum mediterraneum (CBS 121718) was used as the outgroup taxon. The ITSx v1.1.2 (Bengtsson-Palme et al. 2013) was used to validate and annotate non-coding sequences (ITS and LSU) and exons and introns of protein-coding regions (Actin, GAPDH, TEF1) were recognised using TBLASTN online.
Phylogenetic analyses
The sequences were aligned using MAFFT v7.475 (Katoh et al. 2019). The best-fit models of DNA evolution for each partition were determined using PartionFinder v. 1.0.1 (Lanfear et al. 2012). Phylogenetic analyses were performed using maximum likelihood (ML), maximum parsimony (MP) and Bayesian approaches with gaps treated as missing data. RAxML-NG v1.0.1 (Kozlov et al. 2019) was used to infer the ML tree with 1000 bootstrap replicates. MP tree was constructed using MPboot v1.1.0 (Hoang et al. 2018) with 1000 ultrafast bootstrap replicates. The Bayesian analysis was conducted using parallel MrBayes v3.2.7 (Huelsenbeck and Ronquist 2001). Two different runs with 50 million generations and four chains were executed, and the initial 25% of sample trees were treated as burn-in. Tracer v1.7.1 (Rambaut et al. 2018) was used to confirm that the MCMC runs reached convergence with all ESS values above 200.
Divergence time estimation
Divergence time was estimated using BEAST v2.6.3 (Bouckaert et al. 2019). The beast control file was set using BEAUti implemented in BEAST software. The evolution model of each partition was in accord with ML tree inference, and the relaxed clock log normal was selected for the analysis. A single calibration point of 103 Mya with a standard deviation of 5 Mya was referred to the genus Phyllosticta using THE TIMESCALE of LIFE (http://timetree.org/). The analyses were conducted with 50 million MCMC iterations and sampling each 5000 generation. Convergence was checked using the same method described previously. The maximum clade credibility tree was inferred in TreeAnnotator v2.6.3 (Bouckaert et al. 2019) after discarding the initial 25% of MCMC trees as burn-in. The result was visualized using ggtree (Yu 2020).
Sequence-based species delimitation methods
We performed species delimitation using seven different methods: Automatic Barcoding Gap Discovery (ABGD) (Puillandre et al. 2012), statistical parsimony network analysis implemented in TCS (Clement et al. 2000), Bayesian Phylogenetics and Phylogeography (BPP) (Yang 2015), an updated version of Bayesian Poisson tree processes (bPTP) (Zhang et al. 2013), multi-rate Poisson tree processes (mPTP) (Kapli et al. 2017), Generalized Mixed Yule Coalescence (GMYC) (Pons et al. 2006), and Species Tree and Classification Estimation, Yarely STACEY (Jones 2017). The web-interface version of ABGD (http://wwwabi.snv.jussieu.fr/public/abgd/) was used to conduct species delimitation with five barcodes as well as the combined dataset without outgroup. The prior maximum divergence of intraspecific diversity (P value) was set from minimum value 0.001 to maximum value 0.1, and the number of steps was set to 50. Kimura-2P model was used to get the matrix of pairwise distances. For TCS analysis, haplotype networks of six datasets were constructed using TCS v1.21 (Clement et al. 2000). Outgroup sequence was removed from all datasets, gaps presenting in alignment were treated as missing data, and the connection limit was set to 95%. BPP v4.3.8 was used to conduct species delimitation using A11 analysis (Flouri et al. 2018). The control file of BPP was prepared according to the A11 example file implemented in BPP. Six phylogenetic trees (5 based marker datasets and 1 based combined dataset) generated using RAxML-NG were submitted to the Bayesian Poisson tree processes (bPTP) (Zhang et al. 2013) webserver (https://species.h-its.org/) to test species limits. The analyses were conducted with 2 million MCMC generations, thinning of 100, and Burn-in of 0.1. Multi-rate Poisson tree processes (mPTP) (Kapli et al. 2017), similar with bPTP, was conducted using five marker phylogenetic trees. The analysis was run two times based on one coalescent rate and multiple coalescent rates, respectively. GMYC analyses (Pons et al. 2006) were conducted using the package splits (Ezard et al. 2017) on the R platform (R Core Team 2021). As input, six ultrametric trees (5 based marker datasets and 1 based combined dataset) were constructed using BEAST with 100 million MCMC iterations, a strict molecular, and a Yule model as prior (Fig. 1).
STACEY (Jones 2017) analysis implemented in BEAST was conducted using the combined datasets. The analysis was performed with 4.5 billion MCMC iterations. The final resulting species tree was subjected to SpeciesDelimitationAnalyzer (Jones et al. 2014) with a burn-in of 10,000, a collapse height of 0.0001 and a simcutoff of 1. The similarity matrix was plotted using R codes supplied by Jones et al. (2014). Defined species of methods were visualised with ITOL online (Letunic and Bork 2019).
As predicted, ABGD, TCS, BPP, PTP, GMYC, and STACEY resulted in different numbers of putative species (Figs. 2, 4; Sup Fig. 1). ABGD approach detects the barcode gap as the first significant gap beyond limit for intraspecific divergence and uses it to partition the sequence alignment data set into candidate species (Puillandre et al. 2012). The percentage of recovered species (PRS) varied with different loci. As a single locus, actin had the highest PRS (90%). The ABGD is fast and straightforward, though it should only be used to grant a preliminary idea of species boundaries (Leavitt et al. 2015). The PTP models could be applied to understand putative species limits on a given rooted phylogenetic tree. Same as ABGD approach, PTP models only could be used to grant an initial idea of species boundaries (Zhang et al. 2013; Leavitt et al. 2015). These methods applied to single-locus gene trees, and bPTPhpp and bPTPml of the Actin and LSU loci had the highest PRS (Sup Fig. 1). Out of all the models, in all loci, mPTPmml gave the lowest PRS (Sup Fig. 1). The Generalized Mixed Yule Coalescent (GMYC) identifies speciation events where branching rates switch from intraspecific (coalescent model) to interspecific (yule model) patterns (Monaghan et al. 2009). Generally, it is firm over a wide range of conditions, including different phylogenies (Leavitt et al. 2015). However, sometimes GMYC approach over-delimitates species because haplotypes of well-supported clades are recognised as independent lineages (Fujisawa and Barraclough 2013; Leavitt et al. 2015). The GMYCm (multiple threshold) and GMYCs (single threshold) model of the combined loci estimated 76 and 86 species, respectively. In this study, the TCS method gave the smallest number of putative species. In the combined analysis, STACEY supported 107 species, which is higher than the actual number of Phyllosticta species. STACEY used multiple loci and is a Bayesian approach under the multispecies coalescent model (Kanz et al. 2015; Chethana et al. 2021). BPP is a Bayesian modelling approach, and it often increased speciation probabilities (Leavitt et al. 2015). In the present study, BPP gave the largest number of putative species (109). Furthermore, the different approaches also showed that as single loci, TEF1 and Actin are the most informative markers. Coalescent-based methods have been commonly used for species delineation in various taxa, and our results agree with that as the GMYC method gave 86 putative species that are most similar to the actual no species studies in this case study (Figs. 2, 4; Sup Fig. 1).

Ascomycota genera discusses in this paper a Beauveria sp. b Colletotrichum sp. c Phyllosticta sp. d Fusarium sp. e Induratia sp. f Hesperomyces sp. Scale bars: a–f = 20 μm

Integrated assessment of results in genus Phyllosticta. Phylogenetic tree based on maximum likelihood inference of combined ITS, LSU, ACT, TEF and GAPDH sequence data, and branches with high bootstrap support values (≥ 70) and high posterior probabilities (≥ 0.70) were shown in boldface. Different symbols represent each complex. Color strips represent species delimitation results from six polyphasic methods, ABGD, bPTP, GMYC, TCS, STACEY and BPP based on combined sequences

The maximum clade credibility tree of species in genus Phyllosticta obtained from a Bayesian approach using BEAST for the ITS, LSU, ACT, TEF and GAPDH sequence data. Bars correspond to the 95% highest posterior density (HPD) intervals. The scale axis shows divergence times as millions of years ago. Geological periods are indicated at the base of the tree. Different symbols represent each complex
Molecular timescales can provide insights into the history of organisms (Samarakoon et al. 2016). Lately, divergence time estimation used to provide additional evidence to stabilize the ranking of species complexes in cryptic genera (Bhunjun et al. 2021). The estimated divergence time for species complexes in Phyllosticta ranged between 39.50 to 62.89 Ma (Fig. 3).

Summary of the number of defined species using 7 methods ABGD, bPTP, GMYC, mPTP, TCS, STACEY, BPP based 5 single locus and combined sequences. The red triangle represents the number of species, and the blue circle represents the percentage of recovered taxonomic species (Number of defined species/number of reported species * 100%)
Limitations of PSC
Naciri and Linder (2015) identified seven processes that affect gene tree heterogeneity and thereby act as barriers when delimiting species boundaries in plants based on fewer gene regions. These processes are fundamental for the Ascomycota in most scenarios. Many of these processes falsify the species relationship among taxa and create fuzzy species boundaries, which is very common in classical low-rank phylogenies (Lumbsch and Leavitt 2011; Naciri and Linder 2015).
Hybridisation is one of the major causes of heterogeneity in gene trees in Fungi. In fungi, hybridisation can occur both by sexual mating and asexual fusion of hyphae or cells (Kohn 2005). Hybridisation can speed up adaptive evolution by transferring adaptive traits among species (Stukenbrock 2016). For instance, hybridisation has been proposed as a significant force in the evolution of phytopathogens (Brasier 2000). For example, the ascomyceteous grass pathogen Zymoseptoria pseudotritici, the Dutch elm disease pathogens Ophiostoma ulmi and O. novo-ulmi, and Brassicaceae pathogen Verticillium longisporum are well studied in hybridising experiments. Above studies revealed that hybridisation is a major force used in the successful spread of plant pathogens and the emergence of new pathogens (Stukenbrock 2016). Hybridisation is also widespread in lichen-forming fungi (Keuler et al. 2020), and therefore, gene trees can usually be different from each other and possibly from the species tree (Naciri and Linder 2015).
Incongruence is also possible because of incomplete lineage sorting (ILS) (Keuler et al. 2020). The ILS happens when ancestral polymorphisms persist through speciation events, and each ancestral polymorphism can lead to different alleles carried among descendants (Stewart et al. 2014). ILS influences both species delimitation process and species phylogenies and incorrect signals in population relationships (Steenkamp et al. 2018). ILS should be taken into account when reconstructing the phylogeny of related species. For example, single-gene phylogenetic approaches are not useful for ILS between closely related species and inadequate to provide enough evidence to prove that populations relate to a single species (Taylor et al. 2000; Lumbsch and Leavitt 2011). Thus, multiple independent gene regions are essential for species delimitations when incomplete lineage sorting is possible. ILS also may create paraphyletic taxa (Brookfield 2011). The impact of ILS and how it obscures the relationships between sibling species have been well studied in fungi, and some examples include Alternaria alternata sensu lato, the rice blast fungus Magnaporthe oryzae and many lichen-forming Ascomycota (Stewart et al. 2014; Gladieux et al. 2018; Xu 2020).
Genome structure (chromosomes number, polyploidy, and the loci site on the chromosomes corresponding to the centromere) also affects the gene tree heterogeneity (Naciri and Linder 2015). A whole-genome duplication is rare, which creates an organism with double genetic content (Wolfe 2015). Several ascomycetes, especially those in the Saccharomycotina, including the common baker’s yeast Saccharomyces cerevisiae have known ancestral genome duplication events. Chromosomal translocation or the uncommon rearrangement of chromosomes also influences the incongruence in phylogeny, and it may occur more regularly in fungal genomes than earlier noticed (Olarte et al. 2019). For instance, different strains of the entomopathogenic fungus Tolypocladium inflatum exhibit high diversity in production of cyclosporin and other secondary metabolites (Olarte et al. 2019). Gene fusion also plays a significant role in gene structure progression; for example, it can produce more complex proteins (Nevalainen and Peterson 2014; Ntana et al. 2020). During the process, a hybrid gene produced from various genomes or even different gene in the corresponding genome (Naciri and Linder 2015). These genes have different coalescence times; therefore, incongruence in phylogeny can be observed (Fig. 4).
Demographic differences determined the changes in population size and its variation through time (Garza and Williamson 2001; Naciri and Linder 2015; Cissé et al. 2018). It affects the levels of population genetic variation and, therefore, on time to coalescence the effective population size (Naciri and Linder 2015). Migration, bottlenecks, and mating systems are responsible for the demographic changes in fungi (Grünwald et al. 2016).
In addition, selection and phylogeographic structure also affect phylogenetic relationships among taxa to some extent (Naciri and Linder 2015). Phylogeography may decrease genetic diversity between sister species or differing levels of intraspecific diversities within a population, thereby creating difficulties in species delimitation or fuzzy species boundaries. Several studies revealed that selection does not influence the topology of genealogical trees; however, it affects the depth of within-species genealogies (Kanzi et al. 2020).
Therefore, an integrative or polyphasic approach considers a maximum of biological reality. Integrating data from various sources to delimit and describe species has been proposed and used successfully in many lineages of Ascomycota (Lücking et al. 2020). We provided several examples above, as well as a case study using multiple approaches to resolve taxonomic problems.
Additional criteria used to determine species in Ascomycota
Chemotaxonomy
When compared to all other living organisms, species of the Ascomycota produce a large number of secondary metabolites. In chemotaxonomy or chemosystematics, these chemical compounds can be used for the classification and identification of fungi (Frisvad et al. 2008; Helaly et al. 2018). The genus Muscodor (≡ Induratia) was introduced by Worapong et al. (2001) as an endophyte isolated from small limbs of Cinnamomum zeylanicum in Honduras. Hyphal morphologies such as coiling, ropiness, branching patterns, and odour were used as distinguishing characters coupled with phylogenies. The isolate was introduced as a new genus and is typified by Muscodor albus in Xylariaceae. Strobel et al. (2001) and Ezra et al. (2004) evaluated the chemical properties of M. albus and described it as a potential mycofumigant. Subsequently 24 Muscodor species have been described based on mycelia sterilia (MS) mainly using hyphal characteristics and ITS sequence data (e.g. González et al. 2009; Suwannarach et al. 2013; Meshram 2017). The novel taxa identifications have also followed the comparisons of volatile organic compound profiles (e.g. González et al. 2009; Suwannarach et al. 2013; Meshram 2017). Samarakoon et al. (2020) revisited the genus and synonymised Muscodor under Induratia giving priority to the oldest name based on the combination of multi-gene phylogeny, chemical profiling and morphology. Even though, with the presence of only MS, the integration of both multilocus phylogenies and chemical profiling can be used to introduce novel taxa in Indratia/Muscodor. Also, Lambert et al. (2019) introduced Hypomontagnella in Hypoxylaceae based on a combination of morphology, multi-gene phylogeny and chemotaxonomic data. The presence of antifungal polyketides of the sporothriolide type in standardised submerged cultures of Hypomontagnella species is specific to the genus. It is possible to use the combination of phylogeny and chemotaxonomy, even for a MS in this group to introduce novel taxa. Plants, fungi and actinomycetes are particularly good at producing secondary metabolites (Frisvad et al. 2008). Taxonomic groups that are exceptionally talented for production of secondary metabolites are aside from the Xylariales (Becker and Stadler 2021) the invertebrate associated families of the Hypocreales (Zhang et al. 2020) and certain taxa of the Agaricomycetes among the Basidiomycota (Sandargo et al. 2019).
Divergence time estimation
In recent years, molecular dating, or the use of DNA sequences data to estimate divergence times in phylogenetic trees, is rapidly emerging into one of the most useful applications of phylogenetic systematics to determine the species limits of fungi (Berbee and Taylor 1993; Prieto et al. 2013; Beimforde et al. 2014). There have been several analyses of divergence times in the fungal tree of life in the last two decades, but most have yielded contrasting results for the origin of major lineages (Liu et al. 2017; Hyde et al. 2017). There are several studies on the divergence time estimations up to the species level discriminate among reptiles and birds (Guicking et al. 2006; Mays et al. 2015). According to Guicking et al. (2006), phylogeny and divergence time estimation is essential to resolve species boundaries, especially for the species with plastic morphology and geographical distributions. Zhao et al. (2016) conducted a study on Agaricus, which comprises morphologically similar species, and compared divergence time estimations to evaluate up to sections and subgeneric levels. However, there are only a few evolutionary studies for the ascomyceteous species level. Besides, as traditional rank delimitation can be somewhat arbitrary, it is therefore desirable to adopt a standardised, objective, and biologically informative criterion for taxonomic delimitation (Avise and Johns 1999). Hence, several authors have employed divergence time estimates and the study of lineage evolutionary history as criteria to minimise the effects of arbitrary taxonomic systems (Prieto et al. 2013; Beimforde et al. 2014; Pérez-Ortega et al. 2016; Hyde et al. 2017). For example, Qu et al. (2018) estimated the divergence times of Hirsutella in Ophiocordyceps and provided an understanding of the evolution of the phialide structure of the genus. The analysis of the molecular clock calibration based on the fossil record showed that Hirsutella arose from a common ancestor about 102 million years ago (Early Cretaceous, Lower Albian) (Qu et al. 2018). One was generally phialidic, a larger shape, including H. guyana, H. nodulosa, and H. sinensis clades (86.9 Mya, 95% highest posterior density (HPD): 69.1–101.4 Mya). Another main lineage of the phialides was more diversified and smaller than the former, which included H. citriformis and H. thompsonii clades (71.9 Mya, 95% HPD: 41.8–99.6 Mya).
Polyphasic approach for species delimitation in Ascomycota
Each species concept in fungal systematics has pros and cons. In an ideal situation, scientists can combine all species concepts to illustrate limits and the hypothesis of species for any given taxa. However, this is not possible in all cases due to various reasons.
Therefore, species boundaries are often obscured, and only a single method cannot work for delimitation of all fungal species (Leavitt et al. 2011; Haelewaters et al. 2018). Thus, there is debate among mycologists to define species in a biologically meaningful context. In recent years, integrative approaches have become more popular to delimit species in Ascomycota (Araújo et al. 2018; Haelewaters et al. 2018; Sochorová et al. 2019). Furthermore, in some Ascomycota lineages phenotypic methods fail to resolve species boundaries; therefore, it is necessary to have other approaches (Skrede et al. 2017; Boluda et al. 2019). Meanwhile, in some lineages, morphology can successfully delimit species, which can speed up other species delimiting approaches (Bustamante et al. 2019). Further, multiple disciplines help to discriminate species but also their origins. For instance, the integrative approaches connect several species delimitations terms such as reproductive connections, morphology, and evolutionary relationships and integrates them into a monistic concept of species (Liu et al. 2016; Boluda et al 2019). In this section by giving several examples, we explain how integrated methods have improved our knowledge regarding the delimitation of species in the Ascomycota.
In the ideal situation, determining the species limits and understanding the species concept of a given taxon should be interpreted based on a complimentary polyphasic approach including morphological, biological and phylogenetic species concepts. For example, Liu et al. (2016) used MSC, BSC and PSC to understand the species limits of Colletotrichum siamense s. lat., which is a cosmopolitan pathogen that causes serious diseases on many economically important plants. Traditional classification of the C. siamense s. lat. was mainly based on spore morphology, colony characters and host. However, these characters often overlap when it comes to determining the species limits of the genus. Furthermore, larger number of species have been introduced based on a five-locus phylogenetic analysis (ACT, CAL, CHS1, GAPDH, ITS). However, the addition of a larger number of species based only on phylogeny led to significant disagreements regarding the status of C. siamense s. lat, either as a single species or as a species complex. Therefore, Liu et al. (2016) apply a polyphasic methodology that compared morphological characteristics (MSC), mating compatibility test (BSC), PSC based on both single- and concatenated-gene phylogenetic analyses, pairwise homoplasy index test, and coalescent-based species delimitation methods comprising GMYC, PTP and BPP to test their null hypothesis that C. siamense s. lat. is a species complex. Liu et al. (2016) concluded that DNA-based phylogenetic analyses considering both GCPSR and coalescent methods of GMYC, PTP and BPP supported C. siamense s. lat. as a single species rather than a species complex. Additional analyses, i.e. a PHI test, cross fertility and the comparison of ecological characters, strengthened that sympatric speciation, geographic and host plant barriers to gene flow among hypothesised “species” in C. siamense s. lat. have not formed. This study verified that speciation events might be misjudged in fungi if all well-supported clades are accepted as distinct species when using phylogenetic analysis of single-locus or concatenation of multi-loci on small sample size. The polyphasic approach in this study provided a valuable development for species delimitation and can be useful, in principle, to any fungal species that are morphologically indistinguishable. Furthermore, this study emphasised the importance of utilizing a large sample size to robustly estimate species boundaries.
Pragmatic
Species delimitation of mycelia sterilia (MS)
Mycelia sterilia is a group of filamentous fungi that produce only mycelial masses and lacks any spore stages, sclerotia or rhizomorphs, etc. They are morphologically indistinguishable due to lack of macro–micro characters of sexual or asexual morphs (Devanadera 2011). In early classification, MS was considered an informal group known as mitosporic fungi or Deuteromycetes under the class “Agonomycetes” (Carlile et al. 1994). The particular environmental, physiological or biochemical interactions are essential for the reproduction of an organism. The inability of a culture medium to provide optimum nutrients and growing conditions result in the failure of a particular fungal species to produce reproductive spores and spore-bearing structures. Such isolates are termed as MS. However, the group MS does not represent the same fungus and only shares the inability to express diagnostic characters under the conditions provided (Talbot 1971). Also, their sexual or asexual morphs possibly can be discovered in natural environmental conditions. The fungi isolated from different habitats and soil is one of the typical habitats for the high frequency of MS (Devanadera 2011; Nosratabadi et al. 2017).
Traditionally, the identification of MS depended on the comparisons of culture characters (Talbot 1971; Arnold et al. 2001; Naik 2009). Even though the culture-based identification is helpful, it is a limited tool, and morphotypes do not always reflect correct identifications (Guo et al. 2000; Lacap et al. 2003; Naik 2009). Therefore, molecular techniques are considered promising methods for identifying MS (Lacap et al. 2003; Naik 2009; Gnavi et al. 2014; Knapp et al. 2015; Noumeur et al. 2020). Here, we provide an example about the introduction of a genus using MS and later the connection of sexual morph found in the natural habitats based on the polyphasic taxonomic approach.
Species delimitation of dark taxa
There is debate among mycologists about the use of environmental DNA amplified from undescribed, also known as dark taxa, for species delimitation (Hongsanan et al. 2018; Lücking and Hawksworth 2018; Ryberg and Nilsson 2018; Thines et al. 2018; Zamora et al. 2018). Lücking and Hawksworth (2018) proposed a code as “nom. seq.” (nomen sequentiae) for the publication of voucherless, sequence-based names in a consistent manner. Recently, Khan et al. (2020) followed the proposed criteria from the nomenclatural committee and introduced two new species as Archaeorhizomyces victor nom. seq. and A. secundus nom. seq. (Archaeorhizomycetes, Taphrinomycotina, Ascomycota). Species delimitation has been followed by the distinct base pairs comparison of the internal transcribed spacer region ITS1 and ITS2 with similar taxa (Khan et al. 2020). This facilitates the discovery of unknown lineages in the fungal tree in the absence of physical specimens. However, there are various arguments in the scientific community concerning how the dark taxa should be sorted. According to the International Code of Nomenclature for algae, fungi, and plants (ICN), it is necessary to designate a physical specimen as the type species. However, Hawksworth et al. (2016) proposed that it could be possible to permit sequence data to serve as types of names of fungi. Also, use of short sequence reads (ITS1 or ITS2) for species delimitation is often misleading in Ascomycota (Hongsanan et al. 2018).
Species delimitation for pathogenic fungi
Most fungal pathogens in Ascomycota possess cryptic morphology. As we have mentioned early in this study, applying BSC is also somewhat difficult for many pathogenic genera because they do not produce an asexual state in nature. Therefore, the following examples illustrate the recent approaches taken to sort out the species and generic boundaries of cryptic Ascomycota species.
Histoplasma
Whole genomic data provide evidence in identifying cryptic speciation (Wibberg et al. 2020). Histoplasma is a genus of dimorphic human pathogen that causes life threatening chronic lung infections (Hage et al. 2015). In contrast, many aspects regarding the natural history, evolution, systematics and the number of species of this pathogenic genus remain largely unknown due to their cryptic nature. Most species were previously classified as H. capsulatum. Geographic distribution, morphology and clinical symptoms show that this taxon is composed of three distinct groups (Guého et al. 1997). Based on four protein-coding genes (arf, H‐anti, ole and tub1), seven species were proposed by Kasuga et al. (2003). Sepúlveda et al. (2017) reconstructed well-supported species tree using 100-kb sliding-window sequences at the whole-genome level. The result based on the Bayesian concordance analysis suggested that this genus is composed of at least four species (Ané 2010; Sepúlveda et al. 2017). Compared to previous studies, the species tree contradicted proposed relationships that mostly were influenced by incomplete lineage sorting (Teixeira et al. 2016; Qiu et al. 2016).
Neocosmospora vs. Fusarium solani species complex (FSSC)
Formerly Fusarium solani species complex (FSSC) was accepted within the genus Fusarium by many authors (O’Donnell et al. 2008, 2013; Zhang et al. 2006; Nalim et al. 2011; Geiser et al. 2013; Short et al. 2013). Later, the genus was segregated into seven genera, and species in FSSC were transferred to Neocosmospora by Lombard et al. (2015). Their study was based on a multi-locus phylogeny of LSU, ITS, acl1, rpb1, rpb2, α-actin, β-tubulin, calmodulin, histone H3, and tef1-α regions. Nonetheless, Lombard et al. (2015) indicated the need for a comprehensive taxonomic study to describe these genera. The taxonomic concept of Neocosmospora proposed by Lombard et al. (2015) was followed by subsequent authors (Sandoval-Denis and Crous 2018; Sandoval-Denis et al. 2019). Schroers et al. (2016) epitypified Fusarium solani and provided an ex-epitype culture with DNA sequences (NRRL 66304). However, assigning taxa of FSSC to Neocosmospora has been argued with the monophyletic nature of the genus Fusarium that includes FSSC was confirmed by phylogenomic analyses (Geiser et al. 2020; O'Donnell et al. 2020). Geiser et al. (2020) performed phylogenomic calculations of a 55.1 kb, 19- protein-coding gene dataset to assess the monophyly of Fusarium. Besides, 19-locus phylogeny provided a statistically more stable phylogenetic reconstruction, which reaffirmed the very broad species concept of Fusarium was accepted by Geiser et al. (2013). Crous et al. (2021a, b) reanalysed this 19 genes Nectriaceae dataset, and their phylogeny showed that only the concatenated alignment resolved the broad circumscription of Fusarium backbone. Further, they re-analysed the concatenated dataset using different phylogenetic methods and revealed that various Nectriaceae lineages proposed as members of the genus Fusarium by Geiser et al. (2020) have alternate topologies. Crous et al. (2021a, b) emphasised that fusarioid macroconidia character has been gained or lost many times during evolution of Nectriaceae and could not be considered as a generic character, and therefore, the broad circumscription of Fusarium sensu stricto is fuzzy. Furthermore, they noted that the both sexual and asexual morphs of Neocosmospora are distinct, form strongly supported monophyletic groups in phylogeny Neoscosmospora differs drastically in regard to its secondary metabolism from Fusarium sensu stricto, as exemplified by the lack of trichothecene type mycotoxins in the former genus, unlike those in Fusarium sensu stricto.
Conclusion
Species are sets of individuals that, literally, each look different (from the Latin word specere “to look at”). The theory is clear; however, the concept of “species” is a human construct and is not something that we should expect to be able to define a uniform standard for all organisms, including fungi. The above definition is often mysterious in Ascomycota compared to Basidiomycota or to macroorganisms or that can be seen with the naked eye. Ascomycota have a short reproductive generation. For instance, the gap between two generations of asexual reproduction in many species occurs in several days. The fungal population changes rapidly in response to changing environmental conditions, therefore genotypes best-adapted to these changes are selected for. They have a variety of reproduction systems, from random mating to strict clonality. The heterokaryosis is useful to store variability in the form of recessive alleles. These allele frequencies can change, which provides quick adaptation to changing environments. They have various recombination methods such as parasexual recombination, gene conversion and horizontal gene transfer. These circumstances make fungi unique and fascinating among other eukaryotes, and therefore, the above facts need to be considered when integrative approaches are utilized to estimate species boundaries in the Ascomycota.
Based on different methods of prediction, the latest estimated number of fungal species is 11.7–13.2 million (Wu et al. 2019; Hyde et al. 2020). This number was previously lower, from a conservative 1.5 million (Hawksworth 1991) up to 6 million (Taylor et al. 2014), and most recently between 2.2 and 3.8 million (Hawksworth and Lücking 2017). However, some species concepts recognise too many Ascomycota species, whereas some concepts may ignore or understate the number. For example, morphologically defined species can accommodate multiple species based on BSC or PSC. It can be seen that reclassification via the use of the PSC leads to a noticeable rise in the number of species and a more modest remapping of species across previous taxonomic boundaries. These cryptic or sibling species lack distinct morphological characteristics; however, molecular analysis have unveiled these genetically distinct populations and warrant species-level recognition.
The proportion of species descriptions using PSC has been on the rise in the last ten years. However, at present, there is no standard as to which locus/loci should be analysed in PSC (Xu 2020). The identification of different genera has used different genetic datasets. Further, the statistical support and sequence divergence values to designate species in single-gene phylogeny and the combined concatenated tree vary upon the authors. Using these sequence data is marginal among intraspecific and interspecific molecular variation is often unclear in most related sister species (Thines et al. 2018). The addition of further isolates has caused some taxa to become fuzzy because of limited character sampling (Benkert 2011).
References
Achari SR, Kaur J, Dinh Q, Mann R, Sawbridge T, Summerell BA, Edwards J (2020) Phylogenetic relationship between Australian Fusarium oxysporum isolates and resolving the species complex using the multispecies coalescent model. BMC Genom 21:1–20
Aguileta G, Marthey S, Chiapello H, Lebrun MH, Rodolphe F, Fournier E, Gendrault-Jacquemard A, Giraud T (2008) Assessing the performance of single-copy genes for recovering robust phylogenies. Syst Biol 57:613–627
Aime MC, Miller AN, Aoki T, Bensch K, Cai L, Crous PW, Hawksworth DL, Hyde KD, Kirk PM, Lücking R, May TW, Malosso E, Redhead SA, Rossman AY, Stadler M, Thines M, Yurkov AM, Zhang N, Schoch CL (2021) How to publish a new fungal species, or name, version 3.0. IMA Fungus 12:11
Ané C (2010) BUCKy: gene tree/species tree reconciliation with Bayesian concordance analysis. Bioinformatics 26:2910–2911
Ansari MA, Butt TM (2011) Effects of successive subculturing on stability, virulence, conidial yield, germination and shelf-life of entomopathogenic fungi. J App Microbiol 110:1460–1469
Aoki T, O’Donnell K, Geiser DM (2014) Systematics of key phytopathogenic Fusarium species: current status and future challenges. J Gen Plant Pathol 80:189–201
Araújo JP, Evans HC, Kepler R, Hughes DP (2018) Zombie-ant fungi across continents: 15 new species and new combinations within Ophiocordyceps. I. Myrmecophilous hirsutelloid species. Stud Mycol 90:119–160
Arnold AE, Maynard Z, Gilbert GS (2001) Fungal endophytes in dicotyledonous neotropical trees: patterns of abundance and diversity. Mycol Res 105:1502–1507
Avise JC, Johns GC (1999) Proposal for a standardized temporal scheme of biological classification for extant species. PNAS 96:7358–7363
Baayen R, Bonants P, Verkley G, Carroll GC, Van Der Aa HA, de Weerdt M, van Brouwershaven IR, Schutte GC, Maccheroni W Jr, de Blanco CG, Azevedo JL (2002) Nonpathogenic isolates of the citrus black spot fungus, Guignardia citricarpa, identified as a cosmopolitan endophyte of woody plants, G. mangiferae (Phyllosticta capitalensis). Phytopathology 92:464–477
Becker K, Stadler M (2021) Recent progress in biodiversity research on the Xylariales and their secondary metabolism. J Antibiot 74:1–23
Beimforde C, Feldberg K, Nylinder S, Rikkinen J, Tuovila H, Dörfelt H, Gube M, Jackson DJ, Reitner J, Seyfullah LJ, Schmidt AR (2014) Estimating the phanerozoic history of the Ascomycota lineages: combining fossil and molecular data. Mol Phylogenet Evol 78:386–398
Bengtsson-Palme J, Ryberg M, Hartmann M, Branco S, Wang Z, Godhe A, De Wit P, Sánchez-García M, Ebersberger I, de Sousa F, Amend AS, Jumpponen A, Unterseher M, Kristiansson E, Abarenkov K, Bertrand YJK, Sanli K, Eriksson KM, Vik U, Veldre V, Nilsson RH, Bunce M (2013) Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol Evol 4:914–919
Benkert D (2011) Lamprospora bavarica and L. esterlechnerae (Pezizales), zwei neue Arten aus dem Nationalpark Bayerischer Wald (Deutschland, Bayern). Z Mycol 77:149–155
Berbee ML, Taylor JW (1993) Dating the evolutionary radiations of the true fungi. Can J Bot 71:1114–1127
Bhunjun CS, Phukhamsakda C, Jayawardena RS, Jeewon R, Promputtha I, Hyde KD (2021) Investigating species boundaries in Colletotrichum. Fungal Divers 107:107–127
Boehm EWA, Mugambi GK, Miller AN, Huhndorf SM, Marincowitz S, Spatafora JW, Schoch CL (2009) A molecular phylogenetic reappraisal of the Hysteriaceae, Mytilinidiaceae and Gloniaceae (Pleosporomycetidae, Dothideomycetes) with keys to world species. Stud Mycol 64:49–83
Boluda CG, Rico VJ, Divakar PK, Nadyeina O, Myllys L, McMullin RT, Zamora JC, Scheidegger C, Hawksworth DL (2019) Evaluating methodologies for species delimitation: the mismatch between phenotypes and genotypes in lichenized fungi (Bryoria sect. Implexae, Parmeliaceae). Persoonia 42:75–100
Borman AM, Johnson EM (2020) Name changes for fungi of medical importance, 2018 to 2019. J Clin Microbiol 59:e01811-e1820
Bouckaert R, Vaughan TG, Barido-Sottani J, Duchêne S, Fourment M, Gavryushkina A, Heled J, Jones G, Kühnert D, De Maio N, Matschiner M, Mendes FK, Müller NF, Ogilvie HA, du Plessis L, Popinga A, Rambaut A, Rasmussen D, Siveroni I, Suchard MA, Wu C-H, Xie D, Zhang C, Stadler T, Drummond AJ (2019) BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLOS Comput Biol 15(4):e1006650
Brasier C (2000) The rise of the hybrid fungi. Nature 405:134–135
Braun U, Dick MA (2002) Leaf spot diseases of eucalypts in New Zealand caused by Pseudocercospora species. N Z J Sci 32:221–234
Brookfield J (2011) Coalescence: the sharing of ancestry of alleles. eLS. https://doi.org/10.1002/9780470015902.a0001775.pub2
Bruns TD, White TJ, Taylor JW (1991) Fungal molecular systematics. Annu Rev Ecol Syst 22:525–564
Bustamante DE, Oliva M, Leiva S, Mendoza JE, Bobadilla L, Angulo G, Calderon MS (2019) Phylogeny and species delimitations in the entomopathogenic genus Beauveria (Hypocreales, Ascomycota), including the description of B. peruviensis sp. nov. MycoKeys 58:47–68
Carlile M, Watkinson SC, Gooday GW (1994) The fungi, first edit. Academic Press, New York
Carstens BC, Pelletier TA, Reid NM, Satler JD (2013) How to fail at species delimitation. Mol Ecol 22:4369–4383
Chan JZ, Halachev MR, Loman NJ, Constantinidou C, Pallen MJ (2012) Defining bacterial species in the genomic era: insights from the genus Acinetobacter. BMC Microbiol 12:302
Cheek M, Nic Lughadha E, Kirk P, Lindon H, Carretero J, Looney B, Douglas B, Haelewaters D, Gaya E, Llewellyn T, Ainsworth AM (2020) New scientific discoveries: plants and fungi. Plants People Planet 2:371–388
Chethana TKW, Manawasinghe IS, Hurdeal VG, Bhunjun CS, Appadoo MA, Gentekaki E, Raspé O, Promputtha I, Hyde KD (2021) What are fungal species and how to delineate them? Fungal Divers (in press)
Chung Y, Ane C (2011) Comparing two Bayesian methods for gene tree/species tree reconstruction: simulations with incomplete lineage sorting and horizontal gene transfer. Syst Biol 60:261–275
Cissé OH, Ma L, Khil PP, Dekker JP, Kutty G, Bishop L, Liu Y, Deng X, Hauser PM, Pagni M, Hirsch V (2018) Comparative population genomics analysis of the mammalian fungal pathogen Pneumocystis. Mbio 9:e00381-e418
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Cottrell TE, Riddick EW (2012) Limited transmission of the ectoparasitic fungus Hesperomyces virescens between lady beetles. Psyche 2012. Article ID 814378
Cracraft J (2000) Species concepts in theoretical and applied biology: a systematic debate with consequences. Pages 3-14 in species concept and phylogenetic theory: a debate (Q. D. Wheeler, and R. Meier, eds.). Colombia University Press, New York
Cronquist A (1978) Once again, what is a species? In: Knutson LV (ed) Bio systematics in agriculture. Alleheld Osmun, Montclair
Crous PW, Lombard L, Sandoval-Denis M et al (2021a) Fusarium: more than a node or a foot-shaped basal cell. Stud Mycol. https://doi.org/10.1016/j.simyco.2021.100116
Crous PW, Rossman AY, Aime C, Allen C, Burgess T, Groenewald JZ, Castlebury L (2021b) Names of phytopathogenic fungi: a practical guide. Phytopathology. https://doi.org/10.1094/PHYTO-11-20-0512-PER
Crowther TW, Maynard DS, Crowther TR, Peccia J, Smith JR, Bradford MA (2014) Untangling the fungal niche: the trait-based approach. Front Microbiol 5:579
De Queiroz K (2007) Species concepts and species delimitation. Syst Biol 56:879–886
Dean R, Van Kan JA, Pretorius ZA, Hammond-Kosack KE, Di Pietro A, Spanu PD, Rudd JJ, Dickman M, Kahmann R, Ellis J, Foster GD (2012) The top 10 fungal pathogens in molecular plant pathology. Mol Plant Pathol 13:414–430
Dettman JR, Jacobson DJ, Taylor JW (2003) A multilocus genealogical approach to phylogenetic species recognition in the model eukaryote Neurospora. Evolution 57:2703–2720
Devanadera BEA (2011) Molecular identification of Mycelia sterilia by analysis of the internal transcribed spacer (ITS) region DNA sequences and screening for protease and xylanase activities. University Library, University of the Philippines at Los Baños, Los Baños
Dobzhansky T (1935) A critique of the species concept in biology. Philos Sci 2:344–355
Dobzhansky T (1937) Genetics and the origin of species. Columbia University Press, New York
Drenth A, McTaggart AR, Wingfield BD (2019) Fungal clones win the battle, but recombination wins the war. IMA Fungus 10:18
Dupuis JR, Roe AD, Sperling FA (2012) Multi-locus species delimitation in closely related animals and fungi: one marker is not enough. Mol Ecol 18:4422–4436
Dyer PS, O’Gorman CM (2011) A fungal sexual revolution: Aspergillus and Penicillium show the way. Curr Opin Microbiol 14:649–654
Ezard T, Fujisawa T, Barraclough T (2017) splits: SPecies' LImits by Threshold Statistics. R package version 1.0-19/r52. https://R-Forge.R-project.org/projects/splits/
Ezra D, Hess W, Strobel GA (2004) New endophytic isolates of Muscodor albus, a volatile-antibiotic-producing fungus. Microbiol 150:4023–4031
Flouri T, Jiao X, Rannala B, Yang Z (2018) Species Tree Inference with BPP using genomic sequences and the multispecies coalescent. Mol Biol Evol 35:2585–2593
Fones HN, Bebber DP, Chaloner TM, Kay WT, Steinberg G, Gurr SJ (2020) Threats to global food security from emerging fungal and oomycete crop pathogens. Nat Food 1:332–342
Frisvad JC, Andersen B, Thrane U (2008) The use of secondary metabolite profiling in chemotaxonomy of filamentous fungi. Mycol Res 112:231–240
Fryar SC, Haelewaters D, Catcheside DE (2019) Annabella australiensis gen. & sp. nov. (Helotiales, Cordieritidaceae) from South Australian mangroves. Mycol Prog 18:973–981
Fujisawa T, Barraclough TG (2013) Delimiting species using single-locus data and the generalized mixed Yule coalescent approach: a revised method and evaluation on simulated data sets. Syst Biol 62:707–724
Garcia JF, Lawrence DP, Morales-Cruz A, Travadon R, Minio A, Hernandez-Martinez R, Rolshausen PE, Baumgartner K, Cantu D (2021) Phylogenomics of plant-associated Botryosphaeriaceae species. Front Microbiol 12:587
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10:305–318
Geiser DM, Aoki T, Bacon CW, Baker SE, Bhattacharyya MK, Brandt ME, Brown DW, Burgess LW, Chulze S, Coleman JJ, Correll JC (2013) One fungus, one name: defining the genus Fusarium in a scientifically robust way that preserves longstanding use. Phytopathol 103:400–408
Geiser DM, Al-Hatmi A, Aoki T et al (2020) Phylogenomic analysis of a 55.1 kb 19-gene dataset resolves a monophyletic Fusarium that includes the Fusarium solani species complex. Phytopathol. https://doi.org/10.1094/PHYTO-08-20-0330-LE
Giordano L, Sillo F, Garbelotto M, Gonthier P (2018) Mitonuclear interactions may contribute to fitness of fungal hybrids. Sci Rep 8:1–7
Giraud T, Refrégier G, Le Gac M, de Vienne DM, Hood ME (2008) Speciation in fungi. Fungal Genet Biol 45:791–802
Gladieux P, Condon B, Ravel S, Soanes D, Maciel JL, Nhani A, Chen L, Terauchi R, Lebrun MH, Tharreau D, Mitchell T (2018) Gene flow between divergent cereal-and grass-specific lineages of the rice blast fungus Magnaporthe oryzae. Mbio 9(1):e01219-17
Glienke C, Pereira O, Stringari D, Fabris J, Kava-Cordeiro V, GalliTerasawa L, Cunnington J, Shivas RG, Groenewald JZ, Crous PW (2011) Endophytic and pathogenic Phyllosticta species, with reference to those associated with Citrus Black Spot. Persoonia 26:47–56
Gnavi G, Ercole E, Panno L, Vizzini A, Varese GC (2014) Dothideomycetes and Leotiomycetes sterile mycelia isolated from the Italian seagrass Posidonia oceanica based on rDNA data. Springerplus 3:508
Goldstein PZ, Desalle R, Amato G, Vogler AP (2000) Conservation genetics at the species boundary. Conserv Biol 14:120–131
González MC, Anaya AL, Glenn AE, Macías-Rubalcava ML, Hernández-Bautista BE, Hanlin RT (2009) Muscodor yucatanensis, a new endophytic ascomycete from Mexican chakah, Bursera simaruba. Mycotaxon 110:363–372
Gostinčar C (2020) Towards genomic criteria for delineating fungal species. J Fungi 6:246
Groenewald JZ, Nakashima C, Nishikawa J, Shin HD, Park JH, Jama AN, Groenewald M, Braun U, Crous PW (2013) Species concepts in Cercospora: spotting the weeds among the roses. Stud Mycol 75:115–170
Grünwald NJ, McDonald BA, Milgroom MG (2016) Population genomics of fungal and oomycete pathogens. Annu Rev Phytopathol 54:323–346
Guého E, Leclerc MC, de Hoog GS, Dupont B (1997) Molecular taxonomy and epidemiology of Blastomyces and Histoplasma species. Mycoses 40:69–81
Gueidan C, Roux C, Lutzoni F (2007) Using a multigene phylogenetic analysis to assess generic delineation and character evolution in Verrucariaceae (Verrucariales, Ascomycota). Mycol Res 111:1145–1168
Guicking D, Lawson R, Joger U, Wink M (2006) Evolution and phylogeny of the genus Natrix (Serpentes: Colubridae). Biol J Linn Soc 87:127–143
Guo LD, Hyde KD, Liew ECY (2000) Identification of endophytic fungi from Livistona chinensis based on morphology and rDNA sequences. New Phytol 147:617–630
Guzow-Krzemińska B, Flakus A, Kosecka M, Jabłońska A, Rodriguez-Flakus P, Kukwa M (2019) New species and records of lichens from Bolivia. Phytotaxa 397:257–279
Haelewaters D, De Kesel A (2020) Checklist of thallus-forming Laboulbeniomycetes from Belgium and the Netherlands, including Hesperomyces halyziae and Laboulbenia quarantenae spp. nov. MycoKeys 71:23–86
Haelewaters D, Pfister DH (2019) Morphological species of Gloeandromyces (Ascomycota, Laboulbeniales) evaluated using single-locus species delimitation methods. Fungal Syst Evol 3:19–33
Haelewaters D, Comont RF, Zhao SY, Pfister DH (2014) Hesperomyces virescens (Fungi, Ascomycota, Laboulbeniales) attacking Harmonia axyridis (Coleoptera, Coccinellidae) in its native range. Chin Sci Bull 59:528–532
Haelewaters D, Zhao SY, Clusella-Trullas S, Cottrell TE, De Kesel A, Fiedler L, Herz A, Hesketh H, Hui C, Kleespies RG, Losey JE, Minnaar IA, Murray KM, Nedvěd O, Pfliegler WP, Raak-van den Berg CL, Riddick EW, Shapiro-Ilan DI, Smyth RR, Steenberg T, van Wielink PS, Viglášová S, Zhao Z, Ceryngier P, Roy HE (2017) Parasites of Harmonia axyridis: current research and perspectives. Biocontrol 62:355–371
Haelewaters D, De Kesel A, Pfister DH (2018) Integrative taxonomy reveals hidden species within a common fungal parasite of ladybirds. Sci Rep 8:15966
Haelewaters D, Dima B, Abdel-Hafiz BII, Abdel-Wahab MA, Abul-Ezz SR, Acar I, Aguirre-Acosta E, Aime MC, Aldemir S, Ali M, Ayala-Vásquez O, Bakhit MS, Bashir H, Battistin E, Bendiksen E, Castro-Rivera R, Çolak ÖF, De Kesel A, de la Fuente JI, Dizkırıcı A, Hussain S, Jansen GM, Kaygusuz O, Khalid AN, Khan J, Kiyashko AA, Larsson E, Martínez-González CR, Morozova OV, Niazi AR, Noordeloos ME, Pham THG, Popov ES, Psurtseva NV, Schoutteten N, Sher H, Türkekul İ, Verbeken A, Ahmad H, Afshan NS, Christe P, Fiaz M, Glaizot O, Liu J, Majeed J, Markotter W, Nagy A, Nawaz H, Papp V, Péter Á, Pfliegler WP, Qasim T, Riaz M, Sándor AD, Szentiványi T, Voglmayr H, Yousaf N, Krisai-Greilhuber I (2020a) Fungal systematics and evolution: FUSE 6. Sydowia 72:231–356
Haelewaters D, Hiller T, Kemp EA, van Wielink PS, Shapiro-Ilan DI, Aime MC, Nedvěd O, Pfister DH, Cottrell TE (2020b) Mortality of native and invasive ladybirds co-infected by ectoparasitic and entomopathogenic fungi. PeerJ 8:e10110
Haelewaters D, Okrasińska A, Gorczak M, Pfister DH (2020c) Draft genome sequence of the globally distributed cockroach-infecting fungus Herpomyces periplanetae strain D. Haelew. 1187d. Microbiol Res Announc 9:e01458-e1519
Haelewaters D, Blackwell M, Pfister DH (2021) Laboulbeniomycetes: intimate fungal associates of arthropods. Ann Rev Entomol 66:257–276
Hage C, Azar M, Bahr N, Loyd J, Wheat L (2015) Histoplasmosis: up-to-date evidence-based approach to diagnosis and management. Semin Respir Crit Care Med 36:729–745
Hahn MW, Nakhleh L (2016) Irrational exuberance for resolved species trees. Evolution 70:7–17
Han KH, Lee DB, Kim JH, Kim MS, Han KY, Kim WS, Park YS, Kim HB, Han DM (2003) Environmental factors affecting development of Aspergillus nidulans. J Microbiol 41:34–40
Hardoim PR, Van Overbeek LS, Berg G, Pirttilä AM, Compant S, Campisano A, Döring M, Sessitsch A (2015) The hidden world within plants: ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiol Mol Biol Rev 79:293–320
Haridas S, Albert R, Binder M, Bloem J, LaButti K, Salamov A, Andreopoulos B, Baker SE, Barry K, Bills G, Bluhm BH, Cannon C, Castanera R, Culley DE, Daum C, Ezra D, Gonzalez JB, Henrissat B, Kuo A, Liang C, Lipzen A, Lutzoni F, Magnuson J, Mondo SJ, Nolan M, Ohm RA, Pangilinan J, Park HJ, Ramirez L, Alfaro M, Sun H, Tritt A, Yoshinaga Y, Zwiers LH, Turgeon BG, Goodwin SB, Spatafora JW, Crous PW, Grigoriev IV (2020) 101 Dothideomycetes genomes: a test case for predicting lifestyles and emergence of pathogens. Stud Mycol 96:141–153
Harrington TC, Rizzo DM (1999) Defining species in the fungi. In: Worrall JJ (ed) Structure and dynamics of fungal populations. Population and community biology series, vol 25. Springer, Dordrecht
Hart MW, Sunday J (2007) Things fall apart: biological species form unconnected parsimony networks. Biol Lett 3:509–512
Hawksworth DL (1991) The fungal dimension of biodiversity: magnitude, significance, and conservation. Mycol Res 95:641–655
Hawksworth DL (2001) The magnitude of fungal diversity: the 1· 5 million species estimate revisited. Mycol Res 105:1422–1432
Hawksworth DL, Lücking R (2017) Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol Spec 5:1–17
Hawksworth DL, Kirk PM, Sutton B, Pegler D (1996) Ainsworth and Bisby’s dictionary of the fungi, 8th edn. CABI, Wallingford
Hawksworth DL, McNeill J, de Beer W et al (2013) Names of fungal species with the same epithet applied to different morphs: how to treat them. IMA Fungus 4:53–56
Hawksworth DL, Hibbett DS, Kirk PM, Lücking R (2016) (308–310) Proposals to permit DNA sequence data to serve as types of names of fungi. Taxon 65:899–900
Helaly SE, Thongbai B, Stadler M (2018) Diversity of biologically active secondary metabolites from endophytic and saprotrophic fungi of the ascomycete order Xylariales. Nat Prod Rep 35:992–1014
Hittinger CT, Rokas A, Bai FY, Boekhout T, Goncalves P, Jeffries TW, Kominek J, Lachance MA, Libkind D, Rosa CA, Sampaio JP (2015) Genomics and the making of yeast biodiversity. Curr Opin Genet Dev 35:100–109
Hoang DT, Vinh LS, Flouri T, Stamatakis A, von Haeseler A, Minh BQ (2018) MPBoot: fast phylogenetic maximum parsimony tree inference and bootstrap approximation. BMC Evol Biol 18:11
Hongsanan S, Jeewon R, Purahong W, Xie N et al (2018) Can we use environmental DNA as holotypes? Fungal Divers 92:1–30
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Hyde KD, Cai L, McKenzi EHC, Yang YL, Zhang JZ, Prihastuti H (2009) Colletotrichum: a catalogue of confusion. Fungal Divers 39:1–17
Hyde KD, Maharachchikumbura SSN, Hongsanan S et al (2017) The ranking of fungi: a tribute to David L. Hawksworth on his 70th birthday. Fungal Divers 84:1–23
Hyde KD, Chaiwan N, Norphanphoun C, Boonmee S, Camporesi E, Chethana KWT, Dayarathne MC et al (2018) Mycosphere notes 169–224. Mycosphere 9:271–430
Hyde KD, Xu JC, Rapior S, Jeewon R, Lumyong S, Niego AGT, Abeywickrama PD, Aluthmuhandiram JVS, Brahamanage RS, Brooks S, Chaiyasen A, Chethana KWT, Chomnunti P, Chepkirui C, Chuankid B, de Silva NI, Doilom M, Faulds C, Gentekaki E, Gopalan V, Kakumyan P, Harishchandra D, Hemachandran H, Hongsanan S, Karunarathna A, Karunarathna SC, Khan S, Kumla J, Jayawardena RS, Liu JK, Liu N, Luangharn T, Macabeo APG, Marasinghe DS, Meeks D, Mortimer PE, Mueller P, Nadir S, Nataraja KN, Nontachaiyapoom S, O’Brien M, Penkhrue W, Phukhamsakda C, Ramanan US, Rathnayaka AR, Sadaba RB, Sandargo B, Samarakoon BC, Tennakoon DS, Siva R, Sriprom W, Suryanarayanan TS, Sujarit K, Suwannarach N, Suwunwong T, Thongbai B, Thongklang N, Wei DP, Wijesinghe SN, Winiski J, Yan J, Yasanthika E, Stadler M (2019) The amazing potential of fungi: 50 ways we can exploit fungi industrially. Fungal Divers 97:1–136
Hyde KD, Jeewon R, Chen YJ, Bhunjun CS, Calabon MS, Jiang HB, Lin CG, Norphanphoun C, Sysouphanthong P, Pem D, Tibpromma S, Zhang Q, Doilom M, Jayawardena RS, Liu JK, Maharachchikumbura SSN, Phukhamsakda C, Phookamsak R, Al-Sadi AM, Thongklang N, Wang Y, Gafforov Y, Jones EBG, Lumyong S (2020) The numbers of fungi: is the descriptive curve flattening? Fungal Divers 103:219–271
James TY, Stajich JE, Hittinger CT, Rokas A (2020) Toward a fully resolved fungal tree of life. Annu Rev Microbiol 74:291–313
Jayawardena RS, Hyde KD, Jeewon R, Ghobad-Nejhad M, Wanasinghe DN, Liu N, Phillips AJL, Oliveira-Filho JRC, da Silva GA, Gibertoni TB, Abeywikrama P, Carris LM, Chethana KWT, Dissanayake AJ, Hongsanan S, Jayasiri SC, McTaggart AR, Perera RH, Phutthacharoen K, Savchenko KG, Shivas RG, Thongklang N, Dong W, Wei D, Wijayawardena NN, Kang JC (2019) One stop shop II: taxonomic update with molecular phylogeny for important phytopathogenic genera: 26–50. Fungal Divers 94:41–129
Jones G (2017) Algorithmic improvements to species delimitation and phylogeny estimation under the multispecies coalescent. J Math Biol 74:447–467
Jones G, Aydin Z, Oxelman B (2014) DISSECT: an assignment-free Bayesian discovery method for species delimitation under the multispecies coalescent. Bioinformatics 31:991–998
Kanz B, von Brackel W, Cezanne R, Eichler M, Hohmann ML, Teuber D, Printzen C (2015) DNA barcodes for the distinction of reindeer lichens: a case study using Cladonia rangiferina and C. stygia. Herzogia 28:445–464
Kanzi AM, Trollip C, Wingfield MJ, Barnes I, Van der Nest MA, Wingfield BD (2020) Phylogenomic incongruence in Ceratocystis: a clue to speciation? BMC Genom 21:362
Kapli P, Lutteropp S, Zhang J, Kobert K, Pavlidis P, Stamatakis A, Flouri T (2017) Multi-rate Poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics 33:1630–1638
Kasuga T, White TJ, Koenig G, Mcewen J, Restrepo A, Castañeda E, Da Silva LC, Heins-Vaccari EM, De Freitas RS, Zancopé-Oliveira RM, Qin Z (2003) Phylogeography of the fungal pathogen Histoplasma capsulatum. Mol Ecol 12:3383–3401
Katoh K, Rozewicki J, Yamada KD (2019) MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 20:1160–1166
Keuler R, Garretson A, Saunders T, Erickson RJ, Andre NS, Grewe F, Smith H, Lumbsch HT, Huang JP, Clair LL, Leavitt SD (2020) Genome-scale data reveal the role of hybridization in lichen-forming fungi. Sci Rep 10:1497
Khan FK, Kluting K, Tångrot J, Urbina H, Ammunet T, Sahraei SE, Rydén M, Ryberg M, Rosling A (2020) Naming the untouchable –environmental sequences and niche partitioning as taxonomical evidence in fungi. IMA Fungus 11:23
Kim M, Oh H-S, Park S-C, Chun J (2014) Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol 64:346–351
Kjaerbolling I, Vesth T, Frisvad JC, Nybo JL, Theobald S, Kildgaard S, Petersen TI, Kuo A, Sato A, Lyhne EK, Kogle ME, Wiebenga A, Kun RS, Lubbers RJM, Makela MR, Barry K, Chovatia M, Clum A, Daum C, Haridas S, He G, LaButti K, Lipzen A, Mondo S, Pangilinan J, Riley R, Salamov A, Simmons BA, Magnuson JK, Henrissat B, Mortensen UH, Larsen TO, de Vries RP, Grigoriev IV, Machida M, Baker SE, Andersen MR (2020) A comparative genomics study of 23 Aspergillus species from section Flavi. Nat Commun 11:1106
Knapp DG, Kovács GM, Zajta E, Groenewald JZ, Crous PW (2015) Dark septate endophytic pleosporalean genera from semiarid areas. Persoonia 35:87–100
Kobmoo N, Mongkolsamrit S, Arnamnart N, Luangsa-Ard JJ, Giraud T (2019) Population genomics revealed cryptic species within host-specific zombie-ant fungi (Ophiocordyceps unilateralis). Mol Phylogenet Evol 140:106580
Kohn LM (2005) Mechanisms of fungal speciation. Annu Rev Phytopathol 43:279–308
Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A (2019) RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35:4453–4455
Kück U, Pöggeler S (2009) Cryptic sex in fungi. Fungal Biol Rev 23:86–90
Lacap DC, Hyde KD, Liew ECY (2003) An evaluation of the fungal ‘morphotype’ concept based on ribosomal DNA sequences. Fungal Divers 12:53–66
Lambert C, Wendt L, Hladki AI et al (2019) Hypomontagnella (Hypoxylaceae): a new genus segregated from Hypoxylon by a polyphasic taxonomic approach. Mycol Prog 18:187–201
Lanfear R, Calcott B, Ho SYW, Guindon S (2012) PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol 29:1695–1701
Lawrence DP, Rotondo F, Gannibal PB (2016) Biodiversity and taxonomy of the pleomorphic genus Alternaria. Mycol Prog 15:1–22
Leavitt SD, Johnson L, St. Clair LL (2011) Species delimitation and evolution in morphologically and chemically diverse communities of the lichen-forming genus Xanthoparmelia (Parmeliaceae, Ascomycota) in western North America. Am J Bot 98:175–188
Leavitt SD, Moreau CS, Lumbsch HT (2015) The dynamic discipline of species delimitation: progress toward effectively recognizing species boundaries in natural populations. In: Recent advances in lichenology. Springer, New Delhi
Lee SC, Ni M, Li W, Shertz C, Heitman J (2010) The evolution of sex: a perspective from the fungal kingdom. Microbiol Mol Biol Rev 74:298–340
Letunic I, Bork P (2019) Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47(W1):W256–W259
Liu L, Yu L, Pearl DK, Edwards SV (2009) Estimating species phylogenies using coalescence times among sequences. Syst Biol 58:468–477
Liu F, Wang M, Damm U, Crous PW, Cai L (2016) Species boundaries in plant pathogenic fungi: a Colletotrichum case study. BMC Evol Biol 16:1–4
Liu J-K, Hyde KD, Jeewon R et al (2017) Ranking higher taxa using divergence times: a case study in Dothideomycetes. Fungal Divers 84:75–99
Lombard L, van der Merwe NA, Groenewald JZ, Crous PW (2015) Generic concepts in Nectriaceae. Stud Mycol 80:189–245
Lücking R, Hawksworth DL (2018) Formal description of sequence-based voucherless Fungi: promises and pitfalls, and how to resolve them. IMA Fungus 9:143–165
Lücking R, Aime MC, Robbertse B et al (2020) Unambiguous identification of fungi: where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 11:14
Lumbsch HT, Leavitt SD (2011) Goodbye morphology? A paradigm shift in the delimitation of species in lichenized fungi. Fungal Divers 50:59–72
Maharachchikumbura SSN, Guo LD, Chukeatirote E, Hyde KD (2014) Improving the backbone tree for the genus Pestalotiopsis; addition of P. steyaertii and P. magna sp. nov. Mycol Prog 13:617–624
Maharachchikumbura SSN, Larignon P, Hyde KD, Al-Sadi AM, Liu ZY (2016) Characterization of Neopestalotiopsis, Pestalotiopsis and Truncatella species associated with grapevine trunk diseases in France. Phytopathol Mediterr 55:380–390
Maharachchikumbura SSN, Wanasinghe DN, Cheewangkoon R, Al-Sadi AM (2021) Uncovering the hidden taxonomic diversity of fungi in Oman. Fungal Divers 106:229–268
Mallet J (1995) A species definition for the modern synthesis. Trends Ecol Evol 10:294–299
Manamgoda DS, Rossman AY, Castlebury LA, Crous PW, Madrid H, Chukeatirote E, Hyde KD (2014) The genus Bipolaris. Stud Mycol 79:221–288
Matute DR, Sepulveda VE (2019) Fungal species boundaries in the genomics era. Fungal Genet Biol 131:103249
Mayr E (1940) Speciation phenomena in birds. Am Nat 74(752):249–278
Mayr E (1942) Systematics and the origin of species. Columbia University Press, New York
Mayr E (1999) Systematics and the origin of species, from the viewpoint of a zoologist. Harvard University Press, Cambridge
Mays HL, McKay BD, Tietze DT et al (2015) A multilocus molecular phylogeny for the avian genus Liocichla (Passeriformes: Leiothrichidae: Liocichla). Avian Res 6:17
McDonald J (2015) Morphological and molecular systematics of Resupinatus (Basidiomycota). The University of Western Ontario, London
McMullin RT, Lendemer JC, Braid HE, Newmaster SG (2016) Molecular insights into the lichen genus Alectoria (Parmeliaceae) in North America. Botany 94:165–175
McNeill J, Barrie FR, Buck WR, Demoulin V, Greuter W, Hawksworth DL, Herendeen PS, Knapp S, Marhold K, Prado J, Prud’homme van Reine WF, Smith GF, Wiersema JH, Turland NJ (2012) International code of nomenclature for algae, fungi, and plants (Melbourne Code). Koeltz Scientific Books, Königstein
Meshram V (2017) Muscodor camphora, a new endophytic species from Cinnamomum camphora. Mycosphere 8:568–582
Mirarab S, Warnow T (2015) ASTRAL-II: coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 31:i44–i52
Monaghan MT, Wild R, Elliot M, Fujisawa T, Balke M, Inward DJ, Lees DC, Ranaivosolo R, Eggleton P, Barraclough TG, Vogler AP (2009) Accelerated species inventory on Madagascar using coalescent-based models of species delineation. Syst Biol 58:298–311
Mortimer PE, Jeewon R, Xu J-C, Lumyong S, Wanasinghe DN (2021) Morpho-phylo taxonomy of novel dothideomycetous fungi associated with dead woody twigs in Yunnan province China. Front Microbiol 12:654683
Myllys L, Velmala S, Holien H, Halonen P, Li-Song WA, Goward T (2011) Phylogeny of the genus Bryoria. Lichenologist 43:617–638
Naciri Y, Linder HP (2015) Species delimitation and relationships: the dance of the seven veils. Taxon 64:3–16
Naik BS (2009) Taxonomic placement for mycelia sterilia in endophytic fungal research: a molecular approach. Curr Sci 97:1276–1277
Nalim FA, Samuels GJ, Wijesundera RL, Geiser DM (2011) New species from the Fusarium solani species complex derived from perithecia and soil in the Old World tropics. Mycologia 103:1302–1330
Naranjo-Ortiz MA, Gabaldón T (2019) Fungal evolution: major ecological adaptations and evolutionary transitions. Biol Rev 94:1443–1476
Nevalainen H, Peterson R (2014) Making recombinant proteins in filamentous fungi-are we expecting too much? Front Microbiol 5:75
Norphanphoun C, Hongsanan S, Gentekaki E, Chen YJ, Kuo CH, Hyde KD (2020) Differentiation of species complexes in Phyllosticta enables better species resolution. Mycosphere 11:2542–2628
Nosratabadi M, Kordbacheh P, Kachuei R, Safara M, Rezaie S, Afshari MA, Jafari H (2017) Isolation and identification of non-pathogenic and pathogenic fungi from the soil of Greater Tunb, Abu-Musa and Sirri Islands, Persian Gulf, Iran. J Appl Biotechnol Rep 4:713–718
Noumeur SR, Teponno RB, Helaly SE et al (2020) Diketopiperazines from Batnamyces globulariicola, gen. & sp. nov.(Chaetomiaceae), a fungus associated with roots of the medicinal plant Globularia alypum in Algeria. Mycol Prog 19:589–603
Ntana F, Mortensen UH, Sarazin C, Figge R (2020) Aspergillus: a powerful protein production platform. Catalysts 10:1064
O’Donnell K, Sutton DA, Fothergill A, McCarthy D, Rinaldi MG, Brandt ME, Zhang N, Geiser DM (2008) Molecular phylogenetic diversity; multilocus haplotype nomenclature, and in vitro antifungal resistance within the Fusarium solani species complex. J Clin Microbiol 46:2477–2490
O’Donnell K, Rooney AP, Proctor RH, Brown DW, McCormick SP, Ward TJ, Frandsen RJ, Lysøe E, Rehner SA, Aoki T, Robert VA et al (2013) Phylogenetic analyses of RPB1 and RPB2 support a middle Cretaceous origin for a clade comprising all agriculturally and medically important fusaria. Fungal Genet Biol 52:20–31
O’Donnell K, Al-Hatmi AM, Aoki T, Brankovics B, Cano-Lira JF, Coleman JJ, de Hoog GS, Di Pietro A, Frandsen RJN, Geiser DM et al (2020) No to Neocosmospora: phylogenomic and practical reasons for continued inclusion of the Fusarium solani species complex in the genus Fusarium. mSphere 5:e810–20
Olarte RA, Menke J, Zhang Y, Sullivan S, Slot JC, Huang Y, Badalamenti JP, Quandt AC, Spatafora JW, Bushley KE (2019) Chromosome rearrangements shape the diversification of secondary metabolism in the cyclosporin producing fungus Tolypocladium inflatum. BMC Genom 20:1–23
Parnmen S, Rangsiruji A, Mongkolsuk P, Boonpragob K, Nutakki A, Lumbsch HT (2012) Using phylogenetic and coalescent methods to understand the species diversity in the Cladia aggregata complex (Ascomycota, Lecanorales). PLoS ONE 7:e52245
Pažoutová S, Follert S, Bitzer J, Keck M, Surup F, Šrůtka P, Holuša J, Stadler M (2013) A new endophytic insect-associated Daldinia species, recognised from a comparison of secondary metabolite profiles and molecular phylogeny. Fungal Divers 60:107–123
Peberdy JF (1980) Sexual reproduction in fungi. In: Developmental microbiology. Springer, Boston
Pérez-Ortega S, Garrido-Benavent I, Grube M, Olmo R, de los Ríos A (2016) Hidden diversity of marine borderline lichens and a new order of fungi: Collemopsidiales (Dothideomyceta). Fungal Divers 80:285–300
Persoon CH (1818) Traite sur les champignons comestibles, contenant l’undication des especes nuisible precede d’une introduction a l’historie des Champignons–Paris
Peter J, De Chiara M, Friedrich A, Yue JX, Pflieger D, Bergstrom A, Sigwalt A, Barre B, Freel K, Llored A, Cruaud C, Labadie K, Aury JM, Istace B, Lebrigand K, Barbry P, Engelen S, Lemainque A, Wincker P, Liti G, Schacherer J (2018) Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 556:339–344
Pitt JI (1979) The genus Penicillium and its teleomorphic states Eupenicillium and Talaromyces. Academic Press, London
Pizarro D, Divakar PD, Grewe F et al (2018) Phylogenomic analysis of 2556 single-copy protein-coding genes resolves most evolutionary relationships for the major clades in the most diverse group of lichen-forming fungi. Fungal Divers 92:31–41
Pons J, Barraclough TG, Gomez-Zurita J, Cardoso A, Duran DP, Hazell S, Kamoun S, Sumlin WD, Vogler AP (2006) Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst Biol 55:595–609
Pore RS, Tsao GC, Plunkett OA (1965) A new species of Arthroderma established according to biological species concepts. Mycologia 57:969–973
Powell JR, Monaghan MT, Öpik M, Rillig MC (2011) Evolutionary criteria outperform operational approaches in producing ecologically relevant fungal species inventories. Mol Ecol 20:655–666
Prieto M, Baloch E, Tehler A, Wedin M (2013) Mazaedium evolution in the Ascomycota (Fungi) and the classification of mazaediate groups of formerly unclear relationship. Cladistics 29:296–308
Puillandre N, Lambert A, Brouillet S, Achaz G (2012) ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol Ecol 21:1864–1877
Qiu H, Cai G, Luo J, Bhattacharya D, Zhang N (2016) Extensive horizontal gene transfers between plant pathogenic fungi. BMC Biol 14:41
Qu J, Zhou Y, Yu J, Zhang J, Han Y, Zou X (2018) Estimated divergence times of Hirsutella (asexual morphs) in Ophiocordyceps provides insight into evolution of phialide structure. BMC Evol Biol 18:111
Quaedvlieg W, Kema GHJ, Groenewald JZ et al (2011) Zymoseptoria gen. nov.: a new genus to accommodate Septoria-like species occurring on graminicolous hosts. Persoonia 26:57–69
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904
Rannala B, Yang Z (2003) Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics 164:1645–1656
Rehner SA, Minnis AM, Sung GH, Luangsa-ard JJ, Devotto L, Humber RA (2011) Phylogeny and systematics of the anamorphic, entomopathogenic genus Beauveria. Mycologia 103:1055–1073
Ren R, Sun Y, Zhao Y, Geiser D, Ma H, Zhou X (2016) Phylogenetic resolution of deep eukaryotic and fungal relationships using highly conserved low-copy nuclear genes. Genome Biol Evol 8:2683–2701
Reynolds DR (1993) The fungal holomorph: An overview. In: Reynolds DR, Taylor JW (eds) The fungal holomorph: mitotic, meiotic and pleomorphic speciation in fungal systematics. CAB International, Wallingford, pp 15–25
Rodriguez RJ, White JF Jr, Arnold AE, Redman AR (2009) Fungal endophytes: diversity and functional roles. New Phytol 182:314–330
Rossman AY, Crous PW, Hyde KD, Hawksworth DL, Aptroot A, Bezerra JL, Bhat JD, Boehm E, Braun U, Boonmee S, Camporesi E (2015) Recommended names for pleomorphic genera in Dothideomycetes. IMA Fungus 6:507–523
Roy HE, Brown PMJ, Adriaens T, Berkvens N, Borges I, Clusella-Trullas S, Comont RF, De Clercq P, Eschen R, Estoup A, Evans EW, Facon B, Gardiner MM, Gil A, Grez AA, Guillemaud T, Haelewaters D, Herz A, Honek A, Howe AG, Hui C, Hutchison WD, Kenis M, Koch RL, Kulfan J, Lawson Handley L, Lombaert E, Loomans A, Losey J, Lukashuk AO, Maes D, Magro A, Murray KM, San Martin G, Martinkova Z, Minnaar I, Nedvěd O, Orlova-Bienkowskaja MJ, Rabitsch W, Ravn HP, Rondoni G, Rorke SL, Ryndevich SK, Saethre M-G, Sloggett JJ, Soares AO, Stals R, Tinsley MC, Vandereycken A, van Wielink P, Viglášová S, Zach P, Zaviezo T, Zhao Z (2016) The harlequin ladybird, Harmonia axyridis: global perspectives on invasion history and ecology. Biol Invasions 18:997–1044
Ryan M, Bridge P, Smith D, Jeffries P (2002) Phenotypic degeneration occurs during sector formation in Metarhizium anisopliae. J Appl Microbiol 93:163–168
Ryberg M, Nilsson RH (2018) New light on names and naming of dark taxa. MycoKeys 30:31–39
Samarakoon MC, Hyde KD, Promputtha I, Hongsanan S et al (2016) Evolution of Xylariomycetidae (Ascomycota: Sordariomycetes). Mycosphere 7:1746–1761
Samarakoon MC, Thongbai B, Hyde KD et al (2020) Elucidation of the life cycle of the endophytic genus Muscodor and its transfer to Induratia in Induratiaceae fam. nov., based on a polyphasic taxonomic approach. Fungal Divers 101:177–210
Sandargo B, Chepkirui C, Cheng T, Chaverra-Munoz L, Thongbai B, Stadler M, Hüttel S (2019) Biological and chemical diversity go hand in hand: Basidomycota as source of new pharmaceuticals and agrochemicals. Biotechnol Adv 37(6):107344
Sandoval-Denis M, Crous PW (2018) Removing chaos from confusion: assigning names to common human and animal pathogens in Neocosmospora. Persoonia 41:109–129
Sandoval-Denis M, Lombard L, Crous PW (2019) Back to the roots: a reappraisal of 1238 Neocosmospora. Persoonia 43:90–185
Santos L, Alves A, Alves R (2017) Evaluating multi-locus phylogenies for species boundaries determination in the genus Diaporthe. PeerJ 5:3120
Schardl CL, Craven KD (2003) Interspecific hybridization in plant-associated fungi and oomycetes: a review. Mol Ecol 12:2861–2873
Schroers HJ, Samuels GJ, Zhang N, Short DP, Juba J, Geiser DM (2016) Epitypification of Fusisporium (Fusarium) solani and its assignment to a common phylogenetic species in the Fusarium solani species complex. Mycologia 108:806–819
Senanayake IC, Rathnayake AR, Marasinghe DS, Calabon MS, Gentekaki E, Lee HB, Hurdeal VG, Pem D, Dissanayake LS, Wijesinghe SN, Bundhun D, Nguyen TT, Goonasekara ID, Abeywickrama PD, Bhunjun CS, Jayawardena RS, Wanasinghe DN, Jeewon R, Bhat DJ, Xiang MM (2020) Morphological approaches in studying fungi: collection, examination, isolation, sporulation and preservation. Mycosphere 11:2678–2754
Seppey M, Manni M, Zdobnov EM (2019) BUSCO: assessing genome assembly and annotation completeness. Methods Mol Biol 1962:227–245
Sepúlveda VE, Márquez R, Turissini DA, Goldman WE, Matute DR (2017) Genome sequences reveal cryptic speciation in the human pathogen Histoplasma capsulatum. Mbio 8(6):e01339-e11317
Shear CL, Dodge BO (1927) Life histories and heterothallism of the red bread-mold fungi of the Monilia sitophila group. US Government Printing Office, pp 1019–1042
Shen XX, Opulente DA, Kominek J, Zhou X, Steenwyk JL, Buh KV, Haase MAB, Wisecaver JH, Wang M, Doering DT, Boudouris JT, Schneider RM, Langdon QK, Ohkuma M, Endoh R, Takashima M, Manabe RI, Cadez N, Libkind D, Rosa CA, DeVirgilio J, Hulfachor AB, Groenewald M, Kurtzman CP, Hittinger CT, Rokas A (2018) Tempo and mode of genome evolution in the budding yeast Subphylum. Cell 175:1533-1545.e20
Shen XX, Steenwyk JL, LaBella AL, Opulente DA, Zhou X, Kominek J, Li Y, Groenewald M, Hittinger CT, Rokas A (2020) Genome-scale phylogeny and contrasting modes of genome evolution in the fungal phylum Ascomycota. Sci Adv 6(45):eabd0079
Shivas RG, Cai L (2012) Cryptic fungal species unmasked. Microbiol Aust 33:36–37
Shoemaker RA, Babcock CE (1989) Phaeosphaeria. Can J Bot 67:1500–1599
Short DPG, O’Donnell K, Thrane U, Nielsen KF, Zhang N, Juba JH, Geiser DM (2013) Phylogenetic relationships among members of the Fusarium solani species complex in human infections and the descriptions of F. keratoplasticum sp. nov. and F. petroliphilum stat. nov. Fungal Genet Biol 53:59–70
Skrede I, Carlsen T, Schumacher T (2017) A synopsis of the saddle fungi (Helvella: Ascomycota) in Europe–species delimitation, taxonomy and typification. Persoonia 39:201–253
Sochorová Z, Döbbeler P, Sochor M, van Rooy J (2019) Octospora conidiophora (Pyronemataceae)—a new species from South Africa and the first report of anamorph in bryophilous Pezizales. MycoKeys 54:49–76
Springer MS, Gatesy J (2016) The gene tree delusion. Mol Phylogenet Evol 94:1–33
Steenkamp ET, Wingfield MJ, McTaggart AR, Wingfield BD (2018) Fungal species and their boundaries matter–definitions, mechanisms and practical implications. Fungal Biol Rev 32:104–116
Stewart JE, Timmer LW, Lawrence CB, Pryor BM, Peever TL (2014) Discord between morphological and phylogenetic species boundaries: incomplete lineage sorting and recombination results in fuzzy species boundaries in an asexual fungal pathogen. BMC Evol Biol 14:1–4
Strobel GA, Dirkse E, Sears J, Markworth C (2001) Volatile antimicrobials from Muscodor albus, a novel endophytic fungus. Microbiology 147:2943–2950
Stukenbrock EH (2016) The role of hybridization in the evolution and emergence of new fungal plant pathogens. Phytopathology 106:104–112
Suwannarach N, Kumla J, Bussaban B et al (2013) Molecular and morphological evidence support four new species in the genus Muscodor from northern Thailand. Ann Microbiol 63:1341–1351
Talbot PHB (1971) Fungi with a sterile mycelium or conidia: Eumycota subdivision Deuteromycotina. In: Principles of fungal taxonomy. Macmillan Education UK, London
Taylor JW, Jacobson DJ, Kroken S, Kasuga T, Geiser DM, Hibbett DS, Fisher MC (2000) Phylogenetic species recognition and species concepts in fungi. Fungal Genet Biol 31:21–32
Taylor DL, Hollingsworth TN, McFarland JW, Lennon NJ, Nusbaum C, Ruess RW (2014) A first comprehensive census of fungi in soil reveals both hyperdiversity and fine-scale niche partitioning. Ecol Monogr 84:3–20
Teixeira MD, Patané JS, Taylor ML, Gómez BL, Theodoro RC, de Hoog S, Engelthaler DM, Zancopé-Oliveira RM, Felipe MS, Barker BM (2016) Worldwide phylogenetic distributions and population dynamics of the genus Histoplasma. PLoS Negl Trop Dis 10(6):e0004732
Thines M, Crous PW, Aime MC, Aoki T, Cai L, Hyde KD, Miller AN, Zhang N, Stadler M (2018) Ten reasons why a sequence-based nomenclature is not useful for fungi anytime soon. IMA Fungus 9:177–183
Thiyagaraja V, Lücking R, Ertz D, Karunarathna SC, Wanasinghe DN, Lumyong S, Hyde KD (2021) The evolution of life modes in Stictidaceae, with three novel taxa. J Fungi 7:105
Thorp James H, Christopher Rogers D (eds) (2015) Thorp and Covich’s freshwater invertebrates. Elsevier, London
Udayanga D, Castlebury LA, Rossman AY, Chukeatirote E, Hyde KD (2014) Insights into the genus Diaporthe: phylogenetic species delimitation in the D. eres species complex. Fungal Divers 67:203–229
Vandepol N, Liber J, Desirò A, Na H, Kennedy M, Barry K, Grigoriev IV, Miller AN, O’Donnell K, Stajich JE, Bonito G (2020) Resolving the Mortierellaceae phylogeny through synthesis of multi-gene phylogenetics and phylogenomics. Fungal Divers 104:267–289
Wallen RM, Perlin MH (2018) An overview of the function and maintenance of sexual reproduction in dikaryotic fungi. Front Microbiol 9:503
Wanasinghe DN, Jones EG, Camporesi E, Boonmee S, Ariyawansa HA, Wijayawardene NN, Mortimer PE, Xu J, Yang JB, Hyde KD (2014) An exciting novel member of Lentitheciaceae in Italy from Clematis vitalba. Cryptogam Mycol 35:323–337
Wang Z, Li N, Li J, Dunlap JC, Trail F, Townsend JP (2016) The fast-evolving phy-2 gene modulates sexual development in response to light in the model fungus Neurospora crassa. Mbio 7:e02148
Wang M, Liu B, Ruan R, Zeng Y, Luo J, Li H (2020) Genomic sequencing of Phyllosticta citriasiana provides insight into its conservation and diversification with two closely related Phyllosticta species associated with Citrus. Front Microbial 10:2979
Wibberg D, Stadler M, Lambert C, Bunk B, Spröer C, Rückert C, Kalinowski J, Cox RJ, Kuhnert E (2020) High quality genome sequences of thirteen Hypoxylaceae (Ascomycota) strengthen the phylogenetic family backbone and enable the discovery of new taxa. Fungal Divers 106:7–28
Wikee S, Lombard L, Crous PW, Nakashima C, Motohashi K, Chukeatirote E, Alias SA, McKenzie EHC, Hyde KD (2013) Phyllosticta capitalensis, a widespread endophyte of plants. Fungal Divers 60:91–105
Winston JE (1999) Describing species: practical taxonomic procedure for biologists. Columbia University Press, New York
Wolfe KH (2015) Origin of the yeast whole-genome duplication. PLoS Biol 13(8):e1002221
Worapong J, Strobel G, Ford E et al (2001) Muscodor albus anam. gen. et sp nov., an endophyte from Cinnamomum zeylanicum. Mycotaxon 79:67–79
Worrall JJ (ed) (1999) Structure and dynamics of fungal populations, vol 25. Springer, Berlin
Wu B, Hussain M, Zhang W, Stadler M, Liu X, Xiang M (2019) Current insights into fungal species diversity and perspective on naming the environmental DNA sequences of fungi. Mycology 10:127–140
Xu J (2020) Fungal species concepts in the genomics era. Genome 63:459–468
Yang Z (2015) The BPP program for species tree estimation and species delimitation. Curr Zool 61:854–865
Yang J, Maharachchikumbura SSN, Liu JK, Hyde KD, Jones EG, Al-Sadi AM, Liu ZY (2017) Pseudostanjehughesia aquitropica gen. et sp. nov. and Sporidesmium sensu lato species from freshwater habitats. Mycol Prog 17:591–616
Yu G (2020) Using ggtree to visualize data on tree-like structures. Curr Protoc Bioinform 69:96
Zajc J, Zalar P, Gunde-Cimerman N (2017) Yeasts in hypersaline habitats. Yeasts in natural ecosystems: diversity 2017. Springer, Cham, pp 293–329
Zamora JC, Svensson M, Kirschner R, Olariaga I, Ryman S, Parra LA, Geml J, Rosling A, Adamčík S, Ahti T, Aime MC et al (2018) Considerations and consequences of allowing DNA sequence data as types of fungal taxa. IMA Fungus 9:167–175
Zhang N, O’Donnell K, Sutton DA, Nalim FA, Summerbell RC, Padhye AA, Geiser DM (2006) Members of the Fusarium solani species complex that cause infections in both humans and plants are common in the environment. J Clin Microbiol 44:2186–2190
Zhang J, Kapli P, Pavlidis P, Stamatakis A (2013) A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29:2869–2876
Zhang H, Dong W, Hyde KD, Maharachchikumbura SSN, Hongsanan S, Bhat DJ, Al-Sadi AM, Zhang D (2017) Towards a natural classification of Annulatascaceae-like taxa: introducing Atractosporales ord. nov. and six new families. Fungal Divers 85:75–110
Zhang L, Fasoyin OE, Molnár I, Xu Y (2020) Secondary metabolites from hypocrealean entomopathogenic fungi: novel bioactive compounds. Nat Prod Rep 37(9):1181–1206
Zhao R-L, Zhou J-L, Chen J et al (2016) Towards standardizing taxonomic ranks using divergence times—a case study for reconstruction of the Agaricus taxonomic system. Fungal Divers 78:239–292
Acknowledgements
Danny Haelewaters is supported by the Research Foundation – Flanders (junior postdoctoral fellowship 1206620N). Dhanushka Wanasinghe thanks the CAS President’s International Fellowship Initiative (PIFI) for funding his postdoctoral research (number 2021FYB0005), the Postdoctoral Fund from Human Resources and Social Security Bureau of Yunnan Province.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Handling Editor: Chayanard Phukhamsakda.
Supplementary Information
Below is the link to the electronic supplementary material.
13225_2021_486_MOESM1_ESM.pdf
Supplementary file1 (PDF 721 kb) Supplementary Fig. 1. Color strips represent the species delimitation results from 5 methods, ABGD, bPTP, GMYC, mPTP, TCS based 5 single loci and combined sequences, respectively. Different symbols represent each complex. White means missing data. The number of defined species from different methods, number of reported species and percentages of recovered taxonomic species (Number of defined species/number of reported species * 100%) were displayed on the top.
Rights and permissions
About this article

Cite this article
Maharachchikumbura, S.S.N., Chen, Y., Ariyawansa, H.A. et al. Integrative approaches for species delimitation in Ascomycota. Fungal Diversity 109, 155–179 (2021). https://doi.org/10.1007/s13225-021-00486-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13225-021-00486-6