
Abstract
The accurate interpretation of uranium metal 230Th/234U and 231Pa/235U radiochronometry model ages requires an understanding of how uranium parent nuclides and decay progeny (230Th and 231Pa) behave during uranium metal casting. In order to directly measure the spatial distribution of 230Th and 231Pa in uranium metal before and after vacuum induction melting (VIM), Los Alamos National Laboratory identified uranium metal feedstock, characterized the metal feedstock, conducted a controlled casting experiment of an approximately 120 kg uranium metal rod, and characterized the cast metal. This study presents radiochronometry results and quantified 230Th and 231Pa VIM separation factors from bulk uranium.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Radiochronometry, or the science of measuring the time passed since the purification of a radionuclide from its progeny, can be used to produce predictive or comparative model age signatures often used to support nuclear forensic investigations [1, 2, 3, 4]. Model ages are calculated through the precise measurement of progeny/parent nuclide atom ratios in uranium or plutonium sample matrices. Many paired progeny/parent chronometers can be used for radiochronometry–e.g. 230Th/234U, 231Pa/235U, 234U/238Pu, 235U/239Pu, 236U/240Pu, and 241Am/241Pu. Model ages calculated from these radiochronometers correspond to the time passed since the last chemical purification (or production) of a nuclear material assuming that: (1) decay products were completely purified from the parent at the time of production, and (2) the system has remained closed with no loss or addition of parent or daughter since the time of production. These model assumptions are carefully considered when evaluating nuclear forensic data, and although model ages may not correspond to actual purification or production dates if these assumptions are not met, model ages are nonetheless often used to constrain material process history timelines.
Recent studies support this approach, and have shown that the general radiochronometry model assumptions may not be applicable to nuclear materials in many real-world production scenarios [3, 5, 6, 7, 8, 9]. Uranium and plutonium decay progeny may not be completely purified during certain fuel cycle operations resulting in discordant radiochronometric signatures [3, 5, 6, 7, 8, 9]. Differential behavior or chemical fractionation of decay progeny from parent nuclides has been well documented for both uranium metal and uranium hexafluoride (UF6) systems [6, 7, 8, 9]. In uranium metal systems, a unique repeatable pattern of discordance has been documented whereby 231Pa/235U model ages of uranium metals are often significantly older than 230Th/234U model ages of the same materials [6, 9, 10]. Higginson et al. 2022 documented this discordance pattern in five different uranium metals–three depleted uranium metals and two uranium-niobium alloys. The Higginson et al. 2022 study was the first nuclear forensic study to investigate radiochronometry signatures in paired uranium metal feedstock and cast product samples. Data from the paired samples suggested that 231Pa may not be separated during vacuum induction melting (VIM) casting of uranium metal, whereas 230Th is fairly efficiently separated from the bulk uranium product [9]. If 231Pa is not separated during VIM casting, the 231Pa/235U model age of a uranium metal cast using VIM may be a useful nuclear forensic signature that preserves the age of the source uranium used to cast a metal object. While the Higginson et al. 2022 study focused primarily on VIM-cast metals, differential separation of 230Th and 231Pa from a bulk uranium matrix has also been observed during bomb reduction [10] and may occur during other casting operations such as vacuum arc remelt and microwave casting. Within the foundry of the Sigma Division at Los Alamos National Laboratory, there is a continuous stream of work control measurements which indicate that the dose rate from a casting is higher than that of the charge material, with the remnant skull (material remaining in the crucible) being the most radiologically active in the process and the hot top (riser) the most active of the cast material. The decay times, as monitored only for handling purposes (approximately 3 weeks), for particularly radiologically active castings suggest that thorium segregation and subsequent decay is a controlling factor [11]. A summary of the Nuclear Forensics International Technical Working Group Round Robin 3 Exercise by Hanlen in 2011 noted that when UF4 is reduced to uranium metal that reduction generally occurs around 1300 °C at which temperature uranium is molten, while thorium atoms with a melting point of 1750 °C are refractory and flocculate together forming less dense particles that float and become part of the casting slag [10]. In addition, Reilly et al. 2018 suggested that thorium behavior may be more complex during metal casting with thorium transfer to metal slugs and surfaces of cast uranium metal produced from bomb reduction [12].
Discordant radiochronometry ages of uranium metals present challenges during nuclear forensic assessments of material process history. More research is needed to understand the separation of 230Th and 231Pa from uranium during uranium metal production and casting in order to increase confidence during the assessment of radiochronometry signatures for uranium metal found out of regulatory control. Therefore, in this study, we cast depleted uranium metal under controlled laboratory conditions and document the behavior of uranium decay progeny, 230Th and 231Pa, during metal casting. This study, to the best of our knowledge, is the first controlled uranium metal casting experiment designed to quantitatively investigate the spatial distribution of thorium and protactinium in a cast uranium metal. Controlled metal production experiments, such as the one described here, have the potential to provide quantitative thorium-uranium and protactinium-uranium separation factors resulting from VIM casting of uranium and improve the forensic assessment of radiochronometry model ages of unknown seized uranium metals.
Experimental
Identification and Characterization of Depleted Uranium Feedstock
The goal of this work was to cast a depleted uranium rod from a well-characterized uranium feedstock. The feedstock chosen for this study consisted of 402 kg of depleted uranium metal plates (approximately 0.2 wt% 235U) stored at Los Alamos National Laboratory in the Sigma Facility. The feedstock history was not entirely known; however, based on process knowledge at Sigma, the feedstock was likely cast at the Y-12 National Security Complex in the 1970s as a large ingot (~ 2000 kg) using derby metal produced by bomb reduction of UF4 as charge with subsequent warm rolling and shearing to produce manageable plate sizes.
A large rod casting geometry was chosen for this study in order to provide sufficient cooling time to allow for flotation of impurities during casting. This geometry required 14 metal feedstock plates for the desired charge mass of approximately 120 kg. The 14 depleted uranium plates identified for casting were cleaned to remove surface oxide buildup using water, scrubbing, and acetone prior to sampling. Because the plates contained in the drum were likely from the same original casting (based upon markings), or at least from the same well-controlled material stream, the metal was expected to be homogeneous. To characterize the depleted uranium and confirm homogeneity of feedstock composition, samples consisting of 0.25-inch (0.64 cm) drilled chips (sometimes referred to as “pigtails” or turnings) were taken from one location per plate (Fig. 1). With the expectation of homogenous feedstock, one location per plate was deemed sufficient for chemical characterization; however, during sampling documentation was kept recording which plates were from the edge of the rolled ingot based on visual indications as shown by plate 012 (sample ID, S-12-A) in Fig. 1B. Two drill holes were made in approximately the middle of each plate and the collected drilled chips were combined. Two-gram subsamples from each plate were taken for radiochronometry. Exemplar optical microscopy images taken using an Olympus DSX1000 digital microscope of the 2-g radiochronometry subsamples used for destructive analysis are shown in Fig. 1C–E.

Panels A and B–Photographs of 2 of the 14 feedstock plates used in this study. The locations where samples were taken for destructive analysis are visible on each plate. The photo of plate 002 (left) was taken after sectioning on the bandsaw. The bottom edge of plate 012 (S-12-A, right) indicates that this plate was taken from the edge of the rolled ingot. Panels C to E – Optical microscopy images at 25x (left), 60x (middle) and 25x (right) magnification showing examples of feedstock turnings used for destructive analysis. The blue iridescence of the sample surfaces is consistent with surface oxidation of the uranium metal and the samples display a range of shapes and sizes
Uranium Metal Casting
The depleted uranium feedstock plates were quartered on a bandsaw for use as charge material in the rod casting (Fig. 1A). After the feedstock charge was prepared, a depleted uranium rod was cast using VIM at the Los Alamos National Laboratory Sigma Facility. The planned cast rod dimensions were as follows: 6-inch (15.2 cm) outer diameter (OD) × 1.5-inch (3.8 cm) inner diameter (ID) × 12-inch (30.5 cm) length (L). The casting design included a hot top to feed liquid to the solidification front because liquid uranium shrinks upon freezing. Any shrinkage porosity that occurs is designed to be restrained to the hot top, resulting in a sound part without voids. The hot top on the casting had an OD of 7.5-inches (19.2 cm) and an ID of 1.5-inches (3.8 cm). The furnace configuration used for this project is shown in Fig. 2. The feedstock plates were loaded into a graphite crucible sitting on top of the graphite mold. The locations where graphite would contact metal were coated in yttria (yttrium oxide, Y2O3) to limit carbon uptake in the metal during casting. The graphite was instrumented with thermocouples to record the thermal profile during casting. The crucible and mold were stacked atop a graphite pedestal, which was used to place the mold at the correct height within the furnace coils. The entire stack sits inside of the furnace base. The casting preparation also included the addition of insulation, which consisted of graphite felt and graphite foam. The mold was set at the Sigma Facility on November 2, 2021 and casting was conducted on November 3, 2021.

The large rod mold geometry used for this casting. The insulation, crucible, mold, and pedestal sit in the furnace base, and thermocouples used to monitor the thermal profile are also shown
The casting was processed in a single-coil VIM furnace. The metal melted in the crucible while the mold was heated to a specified thermal profile under vacuum conditions. When the metal reached the goal temperature, the stopper rod was raised to allow the molten metal to flow into the mold. At the time of pour, the temperature at the top of the mold was approximately 850 °C, the temperature at the bottom of the mold was approximately 650 °C, and the temperature of the molten metal had reached 1300 °C. The casting remained in the furnace for approximately 18 h to cool. After 18 h, the casting was dropped out of the furnace, moved to a breakout area, removed from the mold, and the outside surface was cleaned by scrubbing with water and acetone. The final cast part weighed 119.40 kg including the central graphite rod. The final completed total casting was 14-inches (35.6 cm) tall with a 1.75-inch-tall (4.4 cm) hot top (Fig. 3).

Left–photograph of the as-cast depleted uranium rod after breakout and cleaning. Right – A: Radiograph collected on the exterior of the depleted uranium metal rod; B: Radiograph collected on the interior of the depleted uranium metal rod. Both radiographs were produced from a 5-min exposure of the metal to the imaging film. Radioactivity is shown by an increase in warm colors of each pixel (towards red). An increase in radioactivity near the upper portion (hot top) of the casting is visible primarily in the interior film radiograph. Background radiation/exposure is shown in blue. Radioactivity intensity scales from green (low) to (red) and is qualitative rather than quantitative
Immediately following breakout and cleaning, autoradiography was used to examine the spatial distribution of radioactivity within the cast depleted uranium metal. Two slim photophosphor imaging films (Fujifilm BAS-SR) were used to collect radiographs from the cast metal. One film was taped to the exterior of the cast uranium rod and the second film was placed into the central hole within the cylindrical rod. The films were exposed to the cast uranium rod for five minutes each. After exposure, the films were removed and enclosed in a light-tight cassette, transported to a radiochemistry laboratory, and scanned using an Amersham™ Typhoon™ radiography imaging scanner (Fig. 3).
Sampling for Destructive Analysis
The casting was stored until June 2022 to allow 230Th and 231Pa to ingrow in the freshly cast depleted uranium in sufficient quantities for analysis. Two-gram drill chips, similar to those taken for feedstock characterization, were taken from the cast product for radiochronometry. Samples from the cast depleted uranium rod were taken from 7 spatially different locations representing a gradient from the hot top to the base of the casting (Fig. 4).

Left–two photographs of the casting taken after sampling for destructive analysis. The locations where samples were taken can be seen as holes in the surface of the casting and each location is labeled with an associated sample identification number. The scale bar shown is in units of cm. Middle–a schematic of the casting with dimensions between sampling locations provided in both inches (’’) and cm. Right–three exemplar optical microscopy images of HT1a, 1a, and 3a drilled chips taken for U, Th, and Pa analysis. No notable visual differences were observed between the hot top drilled chips and the bulk casting drilled chips. All samples had a metallic luster and demonstrated a range of minor surface oxidation
Radiochronometry – Radiochemical Purification and Mass Spectrometry
Radiochemistry was used to purify 230Th and 231Pa from the depleted uranium feedstock and cast product drilled chip samples. Approximately 1-g subaliquots of depleted uranium metal from each of 8 feedstock plate samples (S-1-A, S-3-A, S-6-A, S-8-A, S-10-A, S-12-A, S-13-A, and S-14-A) and from each of the 7 cast product samples (HT1a, HT2a, 1a, 2a, 3a, 4a, and 5a) were gravimetrically weighed into Savillex™ PFA jars. The metals were dissolved using 8 M HNO3 in a chemical fume hood on a hotplate at 140 °C. Once in solution, samples were diluted to create 4 M HNO3 + 0.005 M HF primary sample solutions–HF was added to keep 230Th in solution. Serial dilutions of each sample were made using 4 M HNO3 + 0.005 M HF to produce sample solutions with ng uranium / g solution concentrations for uranium assay and isotope composition determination.
For 230Th assay determination, aliquots of each sample primary solution providing 20 to 75 pg of 230Th for analysis were gravimetrically weighed into Savillex™ PFA vials. For all radiochemical purifications (thorium, protactinium, uranium) feedstock samples and cast product samples were processed separately. For each set of samples, a process blank was added. An aliquot of New Brunswick Laboratory certified reference material (CRM) 125-A, previously dissolved at Los Alamos National Laboratory, was also processed through chemistry with the cast product sample set for radiochronometry quality control. All thorium aliquots were spiked with 40 to 75 pg of 229Th from 229Th NFRM Th-1 [13] for isotope dilution mass spectrometry. Thorium was purified from the bulk depleted uranium feedstock using a 3-column anion BioRad® AG 1-X8 anion exchange resin procedure. For the first two separations, samples were loaded in 8 M HNO3, columns were washed with 8 M HNO3 and thorium was eluted with 9 M HCl. For the final column, the thorium samples were reconstituted in 9 M HCl + 0.01 M HF, loaded onto the columns, and collected immediately. Due to the higher uranium matrix mass in the cast product thorium fraction (approximately 250–300 mg of total uranium) the cast product samples were purified using a 3-column procedure that utilized both BioRad® AG 1-X8 and Eichrom® TEVA resins. The first column loaded the sample onto a 2 mL BioRad® AG 1-X8 column in 9 M HCl, whereby uranium was sorbed to the resin and thorium was directly eluted. Thorium fractions were dried and reconstituted in 2 M HNO3 for the second column. The thorium fraction was loaded onto 2 mL Eichrom® TEVA resin beds in 2 M HNO3. Uranium was washed from the columns using 2 M HNO3 and thorium was eluted with 9 M HCl. This column was repeated to remove additional uranium. All purified thorium fractions were dried and reconstituted in 2% HNO3 + 0.005 M HF for analysis by multi-collector inductively-coupled plasma mass spectrometry (MC-ICP-MS).
Separate uranium aliquots were taken for uranium assay and uranium isotope composition determination. Aliquots of each sample providing approximately 10 ng of total uranium for assay determination by isotope dilution were weighed gravimetrically into Savillex™ PFA vials. An aliquot of standard reference material (SRM) 960 (also known as CRM 112-A), a uranium metal standard previously dissolved at Los Alamos National Laboratory, was also taken for uranium assay quality control and processed through chemistry with the feedstock samples. Samples were spiked with 1 ng of 233U. For uranium isotope composition measurements, aliquots of each sample providing approximately 30 to 50 ng of total uranium were weighed gravimetrically into Savillex™ PFA vials. All uranium fractions were purified using Eichrom® UTEVA resin. Samples were loaded onto the columns in 3 M HNO3, and sample impurities were rinsed from the resin with 3 M HNO3 followed by 9 M HCl and 5 M HCl. Uranium was then eluted from the columns using 0.1 M HCl. The samples were dried on a hotplate and reconstituted in 2% HNO3 for MC-ICP-MS.
For 231Pa assay determination, aliquots providing single-pg 231Pa quantities of each sample were weighed gravimetrically into Savillex™ PFA vials. An aliquot of New Brunswick Laboratory certified reference material (CRM) 125-A was also processed through chemistry with the cast product sample set for radiochronometry quality control. Samples were spiked with a 233Pa spike for isotope dilution mass spectrometry. The 233Pa spike was produced at Los Alamos National Laboratory by separating the 233Pa from a 237Np stock solution. The produced spike was calibrated twice with the 231Pa nuclear forensic reference material NFRM Pa-1 [14] as it decayed (233Pa half-life = 26.975 ± 0.026 days [15]). A 233Pa spike concentration determined from the average of the two spike calibrations was used for isotope dilution calculations. Protactinium was purified from the bulk depleted uranium matrix using a 3-column procedure. The first column consisted of 2 mL of BioRad® AG 1-X8 anion resin. The samples were loaded onto columns in 9 M HCl with trace concentrated HNO3 and trace saturated H3BO3. Protactinium was sorbed onto the anion resin and the matrix was washed from the resin with 9 M HCl. Protactinium was eluted in 9 M HCl + 0.05 M HF. The samples were dried and reconstituted in 2% HNO3 + trace saturated H3BO3 and loaded onto 2 mL of silica gel resin. The matrix was washed from the resin with 2% HNO3 and protactinium was eluted using 2% HNO3 + 0.05 M HF. The silica gel column was repeated as a final purification to remove ingrown 233U. The purified protactinium fractions in 2% HNO3 + 0.05 M HF were measured by mass spectrometry on the same day as the final purification column to prevent isobaric interference from ingrown 233U on mass 233 during sample analysis.
Purified thorium, uranium, and protactinium sample fractions were analyzed using a Thermo Scientific Neptune Plus MC-ICP-MS. Samples were introduced into the mass spectrometer as solutions (2% HNO3 + 0.005 M HF, 2% HNO3, and 2% HNO3 + 0.05 M HF, respectively) and acid blank solutions were analyzed prior to each sample. For thorium analysis, 229Th, 230Th, and 232Th were measured on Faraday detectors using a static measurement routine. Faraday gain calibrations were performed just prior to the start of the analytical session using an internally supplied voltage. Certified reference material IRMM 074/1 was used to calculate instrumental mass bias corrections.
For uranium isotope composition measurements, all mass bias and detector gain calculations were performed using bracketing standards of New Brunswick Laboratory CRM U010. Retarding Potential Quadrupole energy filters (RPQs) were used to decrease the contribution of low-mass tailing from 238U on 236U. Residual mass tailing effects were assessed by measuring 236U, 234U, and 233U counts at four off-peak masses (− 0.5, − 0.35, + 0.35, + 0.5 amu away from peak center), fitting the points to an exponential curve, and then subtracting the tail contribution from the measured signal. In addition to CRM 125-A that was processed through chemical purification with the samples, IRMM-183 and SRM 960 solutions were also measured during analysis for quality control. For uranium isotope dilution mass spectrometry, 233U and 238U were measured on Faraday detectors and IRMM 074/1 was used to calculate instrumental mass bias corrections.
Protactinium measurements were made using a static multi-ion counting routine. There are no certified isotopic protactinium reference materials available so uranium certified reference material CRM U010 was used to calculate mass bias and detector gains. Low-mass tailing from 238U and 235U on 236U and 234U was corrected as described above for uranium analyses. Certified reference material U005-A was measured for quality control of the mass bias and gain corrections applied to the protactinium sample fractions. An internal LANL thorium standard, LATH-1, was used to monitor for potential hydride interference (232Th1H) on 233Pa. No hydride corrections at mass 233—potentially resulting from the natural 232Th chemistry blanks—were necessary for this analysis. Certified reference material CRM 125-A that was processed through chemistry was measured with the samples for Pa quality control.
Results and Discussion
A photograph of the depleted uranium cast rod produced in this study and radiographs collected after casting on the exterior and interior of the uranium rod are provided in Fig. 3. Increased activity near the hot top of the casting was observed through autoradiography (Fig. 3B). Due to the limited range that radiation can traverse through solids to generate an autoradiography image (ranging from approximately tens of µm for alpha particles to several cm of sample volume for gamma radiation [16]), autoradiography results cannot be used to determine whether the observed excess activity near the hot top persists through the volume of the hot top or is constrained to the surface. Dimensions of the depleted uranium cast rod, sampling locations, and exemplar optical microscope images of the casting samples are provided in Fig. 4. Samples for destructive analysis were taken from 7 different sampling locations. Two samples were taken from the hot top of the casting: HT1a and HT2a. Five samples were taken from the cast rod from top to bottom: 1a, 2a, 3a, 4a, and 5a (Fig. 4).
Uranium assay and isotope composition data as well as measured 230Th and 231Pa concentrations are provided in Tables 1, 2, and 3, respectively. Uranium isotope compositions measured in the feedstock and casting samples are highly consistent and, in general, agree between samples within analytical uncertainty (Table 1). The samples have 234U/238U between 0.000007390 ± 0.000000064 and 0.000007464 ± 0.000000048 and 235U/238U between 0.0020178 ± 0.0000029 and 0.0020241 ± 0.0000029. One sample of the feedstock uranium plates, S-12-A, has a slightly elevated 235U/238U relative to the other samples which may represent very minor laboratory contamination during sampling in the foundry or during radiochemistry. All samples have detectable 236U consistent with feedstock produced from enrichment tails resulting from enriching uranium that included a component of recycled reprocessed reactor fuel. The measured uranium isotope compositions of the feedstock are consistent with the known history of the depleted uranium and the uranium composition of the feedstock was not altered by VIM casting (Table 1). Uranium concentrations of the feedstock and casting samples ranged from 0.9919 ± 0.0010 to 0.99933 ± 0.00095 g of uranium per gram of metal; therefore, the feedstock and casting were comprised of very pure (> 99%) depleted uranium. Measured uranium isotope compositions for quality control standards CRM 125-A and SRM 960 agreed with the certified uranium compositions for these standards. The measured uranium concentration of SRM 960 also agreed with the certified uranium assay of this standard (also known as CRM 112-A). Process blanks handled with the samples during uranium purification and analysis contained between 5 and 9 pg of total U; therefore, laboratory blanks were insignificant relative to the 10 to 50 ng uranium aliquot sizes.
Measured 230Th concentrations are consistent to within less than 1% variation between the different feedstock plates used as the charge for VIM casting. The average concentration of 230Th in the 8 feedstock plates analyzed was 0.7369 ± 0.0012 ng 230Th per gram of depleted uranium metal (Table 2). Thorium-230 concentrations in the casting ranged from 0.13942 ± 0.00025 (sample 2a) to 0.16565 ± 0.00032 (sample 5a) ng 230Th per gram of cast metal (Table 2). Process blank 230Th concentrations were below the limit of detection on a Faraday detector. The concentrations of 230Th in the cast metal represent between 18.5 and 22.4% of the original average 230Th present in the feedstock. Therefore, VIM casting removed between 77.6 and 81.6% of the 230Th (Table 2). Assuming 1) a charge mass of 120 kg, 2) an average 230Th concentration in the feedstock of 0.7369 ± 0.0012 ng 230Th per gram of depleted uranium metal, and 3) a range of VIM casting 230Th removal from 77.6 to 81.6%, of the approximately 88 mg of 230Th present in the uranium metal feedstock, approximately 69 to 72 mg was removed during the casting process. Interestingly, very little spatial variation in 230Th concentration was observed with height in the casting. Slightly higher 230Th concentrations are shown in the hot top (HT1a and HT2a) and base of the casting (5a) relative to the center height of the casting (3a); however, this variation is only on the order of ~ 2%.
One previous hypothesis for 230Th removal during uranium metal casting was buoyancy of the less-dense 230Th relative to the bulk uranium metal during the molten stage of the casting [6, 10]. This would result in a vertical gradient or separation of 230Th to the hot top of the casting. The data presented here suggest that 230Th is partially removed during VIM casting, but if the 230Th is concentrated in the hot top through floatation, it is not homogenously distributed within the hot top because our 2 discrete hot top samples did not detect the presence of excess accumulated 230Th. Another possible explanation for the lowered 230Th concentration in the cast product compared to the feedstock could be 230Th accumulation in the crucible, dross, or skull during casting [12]. While buoyancy driven segregation is possible, this effect would tend to be small since the driving forces are low and the melt is far from quiesscent in either the crucible or the casting. Another likely mechanism is pre-wetting of the surface as is observed in Al-In [17] and related phenomena observed in other monotectic systems. Total segregation would be highly dependent on fluid residence time, which would be highly exagerated in the crucible and result in a weak vertical gradient effect in the casting. Unfortunately, samples were not taken from the crucible, dross, or skull in this study, but should be targets of future research.
The measured 231Pa concentrations in the 8 feedstock plate samples ranged from 93.2 ± 1.0 to 94.2 ± 1.0 pg 231Pa per gram of metal demonstrating a homogenous 231Pa distribution in the feedstock used for VIM casting (Table 3). The chemical homogeneity observed in both 230Th and 231Pa concentrations in the feedstock plates is consistent with the expected history of the feedstock which included casting of a large ingot and subsequent warm rolling. In contrast to 230Th, 231Pa concentrations in the depleted uranium metal were relatively unchanged by the VIM casting process (Table 3). Measured 231Pa concentrations in the casting samples ranged from 92.0 ± 0.6 to 93.5 ± 0.6 pg 231Pa per gram of metal, which represents retention of between 98.1 and 99.8% of the 231Pa from the average feedstock after VIM casting (Table 3). Process blank 231Pa concentrations were below the instrument limit of detection.
The observation in this study that the 231Pa concentration of the feedstock uranium metal is relatively unchanged during VIM casting is consistent with data from another paired feedstock and VIM uranium metal casting sample published by Higginson et al. 2022 [9]. Therefore, repeated observations of 231Pa during uranium production by VIM suggest that the 231Pa decay product of 235U is essentially unpurified from uranium metal during VIM casting. Retention of the feedstock 231Pa signature through casting, therefore, may consitute a powerful nuclear forensic signature that can provide provenance assessment teams with a predictive signature, and potential high-fidelity model age, of the source feedstock that was used to produce a uranium metal.
To further explore the effect of VIM casting on uranium model ages, all measured uranium, thorium, and protactinium concentration data are provided in units of atoms per gram uranium metal in Table 4. The concentrations provided in Table 4 can be combined with the model age equation (Eq. 1) and reference dates to calculate model ages and model purification dates for each sample using the 230Th/234U and 231Pa/235U radiochronometers.
In the equation above, t is equivalent to the model age or time since last chemical purification, λD and λP represent the decay constants of the daughter and parent nuclides, respectively, and ND/NP is the measured daughter/parent atom ratio. Model ages were calculated using half-lives from [18, 19, 20]. The calculated 230Th/234U and 231Pa/235U model ages and model purification dates for both feedstock and casting samples are provided in Table 5 and are shown graphically in Fig. 5.

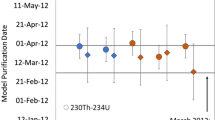
Calculated 230Th/234U (circles) and 231Pa/235U (squares) model purification dates for depleted uranium feedstock and casting samples. Model purification dates are presented in MM/DD/YY format. Uncertainties are smaller than symbols. The controlled VIM casting date is shown as a black dashed line. Preservation of the 231Pa/235U model purification age of the uranium through VIM casting is highlighted with a yellow shaded region for model purification dates from 1973–1974. The calculated 230Th/234U model purification dates of the casting samples (light blue circles) are biased older than the experimentally controlled VIM casting date of November 3, 2021
Calculated 230Th/234U and 231Pa/235U model ages demonstrate discordant uranium model ages for both the feedstock and casting. As discussed in the introduction to this work, discordant 230Th/234U and 231Pa/235U—with 231Pa/235U model ages biased older than 230Th/234U model ages—are often observed during nuclear forensic investigations of uranium metals [6, 9, 10]. Therefore, data presented from this controlled uranium metal casting experiment are consistent with historically measured uranium metal radiochronometric data.
Calculated 230Th/234U model purification dates for the depleted uranium feedstock samples range from October 4, 1985 ± 127 days to May 9, 1986 ± 90 days, whereas 231Pa/235U model purification dates are older and range from December 1, 1972 ± 211 days to July 9, 1973 ± 199 days (Table 5). Based on interpretation from previous studies [9], the 231Pa/235U model purification dates in 1973–1974 may more accurately reflect the timing of uranium metal production by bomb reduction (derby production). It is possible that the later 230Th/234U model purification dates are more closely related to the timing of the feedstock ingot casting and warm rolling potentially in 1985–1986, though we note that this interpretation is somewhat speculative because the exact history of the depleted uranium feedstock is unknown. Measured 230Th/234U and 231Pa/235U model ages of CRM 125-A agree with the certified model purification date of CRM 125-A (August 19, 1994 ± 116 days) and provide quality control of reported radiochronometry data (Table 5).
Had the VIM casting at Sigma Facility completely purified the 230Th and 231Pa decay progeny of bulk uranium, an expected model purification date of November 3, 2021 would have been measured for uranium casting samples. However, neither the 230Th/234U or 231Pa/235U model purification dates of the cast uranium metal correspond to this exact controlled VIM casting date (Table 5). Calculated 230Th/234U model purification dates range from June 17, 2014 ± 28 days to November 19, 2015 ± 22 days and are biased 6.0 to 7.4 years older than the known casting date of the uranium metal. This model purification date bias from the known casting date is the result of only 77.6 to 81.6% purification of 230Th during VIM casting. Calculated 231Pa/235U model purification dates of the cast uranium metal are similar to the model purification dates of the feedstock depleted uranium and range from October 9, 1973 ± 126 days to July 23, 1974 ± 120 days. These measured post-VIM 231Pa/235U model purification dates demonstrate signature preservation of the original feedstock 231Pa/235U model ages to within an approximately 1-year timeline and are likely useful uranium metal nuclear forensic signatures for source attribution (Fig. 5).
Conclusions
A single-coil VIM furnace was used to cast an approximately 120 kg uranium rod on a known date to investigate the separation of thorium and protactinium from uranium during uranium metal casting, and quantify the behavior of the 230Th/234U and 231Pa/235U radiochronometry systems. The goal of this controlled study was to increase the nuclear forensic community’s understanding of the behavior of nuclides used for radiochronometry to improve model age data interpretation to support nuclear forensic investigations. Quantification of 230Th and 231Pa in the feedstock and cast rod produced in this study demonstrate that VIM casting purified 77.6 to 81.6% of the original 230Th present in the feedstock, whereas only 0.2 to 1.9% of the feedstock 231Pa was purified from the uranium during casting. This differential separation of 230Th and 231Pa during metal casting is consistent with the previously observed discordance between 230Th/234U and 231Pa/235U radiochronometric model ages of uranium metals [6, 9, 10]. Measured 230Th/234U model purification dates of the cast metal rod were biased approximately 6.0 to 7.4 years older than the controlled VIM casting date of November 3, 2021. Measured 231Pa/235U model purification dates of the casting samples were similar to the feedstock 231Pa/235U model purification dates (1973 – 1974) demonstrating feedstock model age signature preservation through casting. More research is needed to investigate the reproducibility of thorium and protactinium separation from bulk uranium during VIM casting. In particular, investigation of thorium and protactinium signatures of uranium metals cast at different scales and geometries for elucidation of separation mechanism is a natural extension of this work. Mass balance studies to confirm the location of the separated thorium are also needed. Results presented here quantify thorium and protactinium behavior during uranium metal production and help increase confidence assigned to provenance assessment during nuclear forensic investigations.
References
Kristo MJ, Gaffney AM, Marks N, Knight K, Cassata WS, Hutcheon ID (2016) Nuclear forensic science: analysis of nuclear material out of regulatory control. Annu Rev Earth Planet Sci 44:555–579
Kristo MJ, Williams R, Gaffney AM, Kayzar-Boggs TM, Schorzman KC, Lagerkvist P, Vesterlund A, Rameback H, Nelwamondo AN, Kotze D, Song K, Lim SH, Han S, Lee C, Okubo A, Maloubier D, Cardona D, Samuleev P, Dimayuga I, Varga Z, Wallenius M, Mayer K, Loi E, Keegan E, Harrison J, Thiruvoth S, Stanley FE, Spencer KJ, Tandon L (2018) The application of radiochronometry during the 4th collaborative materials exercise of the nuclear forensics international technical working group (ITWG). J Radioanal Nucl Chem 315:425–434
Gaffney AM, Wimpenny JBN, Parsons-Davis T, Williams RW, Torres RA, Chung BW (2018) A case study in plutonium radiochronometry using multiple systems. J Radioanal Nucl Chem 318:287–295
Varga Z, Krajko J, Penkin M, Novak M, Eke Z, Wallenius M, Mayer K (2017) Identification of uranium signatures relevant for nuclear safeguards and forensics. J Radioanal Nucl Chem 312:639–654
Sturm M, Richter S, Aregbe Y, Wellum R, Maille S, Mayer K, Prohaska T (2014) Evaluation of chronometers in plutonium age determination for nuclear forensics: What if the ‘Pu/U clocks’ do not match? J Radioanal Nucl Chem 302:399–411
Kayzar TM, Williams RW (2016) Developing Ra-226 and Ac-227 age-dating techniques for nuclear forensics to gain insight from concordant and non-concordant radiochronometers. J Radioanal Nucl Chem 307:2061–2068
Rolison JM, Williams RW (2018) Application of the Ra-226-Th230-U-234 and Ac-227-Pa-231-U-235 radiochronometers to UF6 cylinders. J Radioanal Nucl Chem 317:897–905
Harrison LN, Gaffney AM (2021) Th-230-U-234 and Pa-231-U-235 radiochronometry of hydrolyzed uranium hexafluoride gas. J Radioanal Nucl Chem 329:1513–1521
Higginson MA, Kayzar-Boggs TM, Chen CY, Cross STJ, Denton JS, Dunne JA, Edwards MA, Eng C, Gaffney AM, Gilligan CRD, Morris MN, Rolison JM, Sanborn ME, Wende AM (2022) Establishing discordance as a radiochronometric signature for nuclear forensic investigations: a multi-laboratory intercomparison exercise. J Radioanal Nuclear Chem. https://doi.org/10.1007/s10967-022-08428-5
Hanlen R (2011) Round Robin 3 Exercise After Action and Lessons Learned Report. PNNL-20079. Pacific Northwest National Laboratory, U.S. Department of Energy, Richland, WA, p 78
Bliss J (2016) Uranium Foundry Dosimetry Study. November 15, 2016, Presentation to the Sigma Division. Los Alamos National Laboratory, unpublished.
Reilly DR, Athon MT, Corbey JF, Leavy II, McCoy KM, Schwantes JM (2018) Trace element migration during UF4 bomb reduction: Implications to metal fuel production, worker health and safety, and nuclear forensics. J Nucl Mat 510:156–162
Essex RM, Mann JL, Williams RW, Kinman WS, Hubert A, Bennett ME, Gourgiotis A (2018) A new thorium-229 reference material. Appl Radiat Isot 134:23–31
Essex RM, Williams RW, Treinen KC, Collé R, Fitzgerald R, Galea R, Keightley J, LaRosa J, Laureano-Pérez L, Nour S, Pibida L (2019) Preparation and calibration of a 231Pa reference material. J Radioanal Nucl Chem 322(3):1593–1604
Usman K, MacMahon TD (2000) Determination of the half-life of 233Pa. Appl Radiat Isot 52:585–589
Parsons-Davis T, Knight K, Fitzgerald M, Stone G, Caldeira L, Ramon C, Kristo M (2018) Application of modern autoradiography to nuclear forensic analysis. Forensic Sci Int 286:223–232
Kaban I, Curiotto S, Chatain D, Hoyer W (2010) Surfaces, interfaces and phase transitions in Al-In monotectic alloys. Acta Mater 58(9):3406–3414
Cheng H, Lawrence Edwards R, Shen C, Polyak VJ, Asmerom Y, Woodhead J, Hellstrom J, Wang Y, Kong X, Spotl C, Wang X, Alexander EC (2013) Improvements in 230Th dating, 230Th and 234U half-life values, and U-Th measurements by multi-collector inductively coupled plasma mass spectrometry. Earth and Planet Sci Lett 371–372:82–91
Jaffey AH, Flynn KF, Glendenin LE, Bentley WC, Essling AM (1971) Precision measurement of half-lives and specific activities of 235U and 238U. Phys Rev C 4:1889
Robert J, Miranda CF, Muxart R (1968) Mesure de la periode du protactinium-231 par microcalorimetrie. Radiochim Acta 11(2):104–108
Acknowledgements
This work was funded by the National Nuclear Security Administration Office of Defense Nuclear Nonproliferation’s Nonproliferation Research and Development Program (NA-22). We have no conflicts of interest to disclose. This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government, nor any agency thereof, nor any of their employees, nor any of their contractors, subcontractors, or their employees, make any warranty, express or implied, or assume any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represent that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government, any agency thereof, or any of their contractors or subcontractors. The views and opinions expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof. LA-UR-23-20097.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kayzar-Boggs, T.M., Luitjohan, K.E., Imhoff, S.D. et al. Results from a controlled depleted uranium metal casting experiment designed to investigate nuclear Forensic Radiochronometry Signatures. J Radioanal Nucl Chem 332, 1695–1706 (2023). https://doi.org/10.1007/s10967-023-08881-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-023-08881-w