
Abstract
The model age or ‘date of purification’ of a nuclear material is an important nuclear forensic signature. In this study, chemical separation and MC-ICP-MS measurement techniques were developed for 226Ra and 227Ac: grand-daughter nuclides in the 238U and 235U decay chains, respectively. The 230Th–234U, 226Ra–238U, 231Pa–235U, and 227Ac–235U radiochronometers were used to calculate model ages for CRM-U100 standard reference material and two highly-enriched pieces of uranium metal from the International Technical Working Group Round Robin 3 Exercise. Results demonstrate the accuracy of the 226Ra–238U and 227Ac–235U chronometers and provide information about nuclide migration during uranium processing.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nuclear forensic science uses the analysis of nuclear or other radioactive material to reveal information that may provide evidence for nuclear attribution [1, 2]. Because radioactive materials undergo the fundamental process of radioactive decay, paired parent-daughter decay nuclides provide radiochronometers that can be used to determine the model-ages or purification dates of nuclear materials [3–8]. The most commonly used radiochronometer for uranium material is the 230Th–234U chronometer because easily measured quantities of 230Th are produced by 234U decay in a short time [7]. The application of radiochronometers relies on fundamental assumptions that: (1) all daughter nuclides are removed resulting in complete purification of the parent nuclide at the time when the material is produced, time = 0; and (2) the material remains a closed system after production where nuclides are neither added nor removed. While these conditions are often met, there have been multiple examples where daughter nuclides are not completely purified during production [6, 8] or where different chronometers, such as 230Th–234U and 231Pa–235U, yield different or ‘non-concordant’ ages [9, 10].
While non-concordant ages result in difficulty interpreting the model age of a nuclear material, it is important to note that non-concordant ages do not inherently imply that the age determination is incorrect. Assuming that analyses were made accurately, different ages from two chronometers may imply that the assumed conditions for determining an age were not met. For example, if 230Th was efficiently purified from a uranium material at the time of production, but 231Pa was not, the model age determined using the 231Pa–235U chronometer would be anomalously older than the 230Th–234U model age and the actual production age of the material. Such was the case for the International Technical Working Group Round Robin 3 Exercise in 2010 (ITWG RR3) where model ages of highly-enriched uranium (HEU) metal determined by one laboratory from the 231Pa–235U chronometer were much older than model ages determined from the 230Th–234U chronometer [9].
Sturm et al. recently suggested that considering more than one plutonium radiochronometer allowed for additional information about plutonium material to be gained during forensic investigations and prevented erroneous conclusions from being drawn by using only one chronometer [10]. In this work, we apply the same concept to uranium materials and suggest that the measurement of multiple chronometers guides more accurate model age interpretations and also adds to our knowledge of radionuclide migration during the production of uranium materials. For this purpose, we have developed methods to purify 226Ra and 227Ac from uranium matrices and measure concentrations by multiple-collector inductively coupled plasma mass spectrometry (MC-ICP-MS) using isotope dilution in order to combine the 226Ra–238U and 227Ac–235U chronometers with other existing radiochronometers. This study comprehensively uses chronometers in the 238U decay chain (230Th–234U, 226Ra–238U) and 235U decay chain (231Pa–235U, 227Ac–235U).
Experimental
Material investigated was carefully chosen to validate methods developed for 226Ra and 227Ac measurement by MC-ICP-MS and to investigate the behavior of grand-daughter nuclides in uranium metal with a known processing history. The uranium standard CRM-U100 was purified on January 8, 1959 [11], and certified for uranium isotopic composition in 1971 [12]. With a known purification date, CRM-U100 can be used to validate model ages calculated using 226Ra–238U and 227Ac–235U in nuclear era uranium materials. Concordant model-ages between the 230Th–234U, 226Ra–238U, and 227Ac–235U chronometers are expected if the radiochronometry assumptions discussed above are met. A second material used for validation purposes is Table Mountain Latite (TML)—a Pliocene basalt from Sonora, California that has been distributed as a geologic rock standard for uranium-series analyses due to its high concentration of these elements (U ~ 10 ppm, Th ~ 30 ppm) [13, 14]. This basalt is particularly useful because the Pliocene age of TML (>2.6 million years old) ensures that secular equilibrium between 238U–234U and 230Th–226Ra has been established. In secular equilibrium, N 1 λ 1 = N 2 λ 2, where N 1 = number of atoms of the parent isotope and N 2 = number of atoms of the daughter isotope and λ 1 and λ 2 are the decay constants for the parent and daughter isotope. The presumption of secular equilibrium for TML enables a test of the accuracy of our 226Ra concentration measurements. Four ores certified for 226Ra activity by CANMET (Natural Resources Canada; DH-1a, BL4a, UTS-1, UTS-4) were analyzed as an additional accuracy test of 226Ra measurements.
In addition to validation materials, uranium metals ‘A’ and ‘B’ from the ITWG RR3 Exercise were analyzed, referred to here as samples ITWR-RR3-A and ITWG-RR3-B respectively. These metals were cast from two separate batches of HEU scrap material, at the same facility (Y-12 USA), under identical process conditions [9]. They were cuts from two hollow metal logs with slightly different uranium isotope compositions. Results from the round robin exercise showed non-concordant ages between the 230Th–234U and 231Pa–235U chronometers. The 230Th–234U model ages measured corresponded well to the metal casting ages while 231Pa–235U model ages were much older [9].
Enriched spike preparation for isotope dilution analyses
An enriched-228Ra spike was used for isotope dilution Ra concentration measurements. This spike was produced by milking Ra from a Th solution containing 3.25 g of Th, prepared from Th-metal (Ames Laboratory). Complete separation of Ra from the Th-metal solution was accomplished through sorption of Th in 8 M HNO3 to anion exchange columns (BioRad AG1-X8) with varying resin volumes (2–10 ml). Radium was eluted (no sorption in 8 M HNO3), and Th was then washed from the column with 0.1 M HCl and stored. The process was repeated until the thorium concentration in the radium fraction was below ion-counting MC-ICP-MS detection limits (≤2 fg/mL in the test solution). The spike Ra concentration was calibrated by isotope dilution using the NIST SRM4967A 226Ra standard.
The methods for isotope dilution measurement of 227Ac developed here use a 229Th standard to provide 225Ac, which is used as the spike. 229Th decays by alpha decay to 225Ra (t1/2 = 14.9 days) followed by beta decay to 225Ac (t1/2 = 10 days). If a solution of 229Th is older than 3 months, secular equilibrium exists between the grand-daughter 225Ac and parent 229Th. The concentration of 225Ac atoms in the solution can, therefore, be calculated using the half-lives of 229Th and 225Ac and the concentration of 229Th. A 229Th spike solution was prepared at a concentration appropriate for 225Ac isotope dilution analyses, and calibrated by crossing with five aliquots of a Th standard prepared from Ames Th metal. This spike solution has a concentration of 4.542 (14) × 1013 atoms 229Th/g.
A 233Pa spike was prepared for 231Pa analyses following the methods of [8]. The spike was calibrated using multiple aliquot mixtures of 233Pa spike and TML solution. Two separate chemical purification procedures [8 and 15] were used to purify and calibrate this spike, and the concentrations determined from the 233Pa-TML mixtures using both methods were in agreement [15].
Sample preparation and purification
Sample solutions were prepared from powders or small metal pieces by acid dissolution. The uranium oxide (U3O8) standard, CRM-U100, was dissolved in 8 M HNO3 in quartz tubes on a hotplate at 125 °C in two separate aliquots: U100A and U100B (ca. 0.1 g and 0.07 g of CRM-U100, respectively). Pieces of ITWG-RR3-A and ITWG-RR3-B were dissolved individually in concentrated HNO3, directly in Savillex PFA (perfluoroalkoxy) vials. CRM-U100 aliquots were transferred to 30 mL PFA vials and all samples were diluted with Milli-Q H2O to create 2–4 M HNO3 stock solutions. Concentrated HF was added to make the solutions 0.05 M HF. Powdered TML and ores from CANMET were digested in a Milestone Ethos EZ microwave digestion unit using a 8:2 solution of concentrated HNO3:HF. Fluorides created during digestion were decomposed with HClO4 and H3BO3 hotplate evaporation steps, and a final stock sample solution was prepared with 2.5 M HCl + 0.01 M HF.
Five separate aliquots of all sample solutions (U100A, U100B, ITWG-RR3-A, ITWG-RR3-B, TML*, and CANMET ores*) were taken from the prepared stock solutions and spiked with appropriate amounts of 228Ra, 229Th (for 225Ac), 229Th (for 230Th), 233Pa and 233U for isotope dilution analysis (*227Ac not measured in TML and ores). Aliquots for 227Ac measurement were spiked with 1.5 mL of 229Th spike providing approximately 100 femtograms of 225Ac in secular equilibrium with 229Th. Spike-sample mixtures were equilibrated in capped Savillex PFA vials on a hotplate at 100 °C overnight before being taken to dryness and re-dissolved in the appropriate acid solutions for chemical separation.
Radium was separated and purified from the bulk uranium and rock matrices by ion-exchange column chromatography developed and modified from methods used by previous studies [16–18]. The column purification procedures chosen were based on sample matrix and varied for nearly pure uranium oxide (CRM-U100), uranium metal (ITWG samples) and geologic matrices (TML and CANMET ores). Radium from U100 was separated from uranium and thorium using two Eichrom UTEVA resin beds (1.8 mL followed by 0.6 mL). Samples were loaded onto and eluted from the resin in 3 M HNO3. Radium was then purified from any Ba impurities using a 1 mL resin bed of Eichrom Sr-Spec resin and 3 M HNO3. The purification of Ra from ITWG-RR3-A and B used the same UTEVA and Eichrom Sr-Spec resin steps described above but included an additional initial purification step on BioRad cation resin (AG50 W-X8) to remove trace contaminants from the metal. The sample was loaded in 1 M HCl and washed with progressively more concentrated HCl in 1 M steps from 1 M to 4 M HCl to remove major cationic species (Ca, Na, Mg etc.). Radium remains on the column and is eluted in 6 M HCl.
Separation of Ra from a rock matrix such as TML or ore material requires a more complex purification procedure than the Ra separation procedure from uranium materials. In this case, the sample is loaded onto a large 10 mL BioRad AG50 W-X8 cation resin bed. Following the washing procedure above, radium is eluted with the rare earth elements in 6 M HCl. After this separation, the sample was dried, reconstituted in 1 M HCl, and loaded onto a 1 mL BioRad AG50 W-X8 resin bed for a second purification of cationic species. The eluted fraction is dried and re-dissolved in 7 M HNO3 for separation from the rare earth elements using a 1 mL Eichrom TRU resin bed. The sample was then dried again, and Ra was separated from Ba through a 1 mL Sr-Spec column (same procedure used above for U100). Prior to analysis, all samples were purified again using a small 0.25 mL BioRad AG50 W-X8 cation resin clean-up column to remove 228Th that is generated from 228Ra by beta-decay (t1/2 = 5.75 years). This final purification is always conducted no more than 1 day prior to measurement of the sample by mass spectrometry.
Actinium is purified from a bulk uranium matrix through a three-stage column process. First, Th is separated from the matrix using a 2 mL anion (BioRad AG-1-X8) resin bed. The sample is loaded in 8 M HNO3, Ac is eluted directly, and Th is left behind sorbed to the resin. The Ac fraction is dried, dried again in concentrated HCl, and then dissolved in 9 M HCl. The sample is then loaded on a 9 M HCl pre-conditioned stacked column arrangement with a 1 mL anion (AG-1-X8) column stacked above a 1 ml Eichrom DGA resin column. In this arrangement, U is sorbed to the upper column, Ac is sorbed to the lower DGA column, and a complete purification of 225Ra is achieved through washing with 9 M HCl. The upper anion column is removed, and Ac is eluted using 0.5 M HCl. It is essential to note that the exact time when samples are loaded onto each column must be recorded during the purification of Ac. Both the parent 229Th and intermediate nuclide 225Ra affect the concentration of 225Ac in the sample during purification. The timing of the separation of these nuclides is, therefore, required for decay corrections explained below for the calculation of 227Ac concentration.
Uranium and thorium purification methods are described in [6]. Protactinium purification from bulk uranium followed procedures presented in [8]. Following purification samples are brought up in 2 % HNO3 for U and Ra analysis and 2 % HNO3 + 0.05 M HF for Th, Pa, and Ac analysis by MC-ICP-MS.
Multi-collector inductively coupled plasma mass spectrometry
Purified U, Th, Pa, Ra, and Ac fractions were analyzed using a Nu Plasma HR MC-ICP-MS at Lawrence Livermore National Laboratory. Samples were introduced in 2 % HNO3 or 2 % HNO3 + HF solutions accordingly. U, Th, and Pa measurements were made according to methods described by [6] and [8]. Static multi-collection routines were written to measure 226Ra and 228Ra simultaneously for Ra and 225Ac and 227Ac simultaneously for Ac on two ion counters (IC0 and IC1). Corrections to the 226Ra/228Ra and 225Ac/227Ac ratios were made using mass bias and relative ion counter efficiency factors calculated from U analyses of the NBL uranium standard U010, which assumes that the instrumental mass bias correction derived from uranium is appropriate for Ra and Ac measurements. NBL uranium standard U005A was analyzed as an unknown for quality control of the uranium mass bias corrections and the cross-calibration of the ion counters. All measurements of U005A agree within the uncertainty envelope of published values for U005A [19]. For both Ra and Ac, data for each sample were collected in one 40-cycle block with a 15 s on-peak integration time for each cycle. All sample signals were corrected for detector baselines measured before analysis at ± half-mass.
Model age determination
Model ages reported here were calculated from the measured 226Ra and 227Ac sample concentrations. Full solutions of the Bateman (U-series decay) equations [20, 21] were used to model 226Ra as well as 227Ac ingrowth from initial 238U, 234U, and 235U concentrations measured directly from the U fractions of samples (following similar procedures as [6] ). This method requires the assumption that at the time of purification, purification was complete and no daughter nuclides of Th, Ra, Pa, or Ac from the decay of uranium were present. The modeled time of ingrowth necessary to produce the measured 226Ra and 227Ac concentrations in the sample is the “model age” of the sample and corresponds to the time that passed between purification of uranium and the time of 226Ra or 227Ac separation and measurement. We report model dates, which correspond to a reference date minus the model age of the sample and, therefore, define the date on which the uranium-bearing sample was originally purified of uranium daughter nuclides, provided that the assumptions of complete purification and subsequent closed-system behavior of the material are met.
Reference dates are determined differently for each chronometer. Because the enriched 228Ra spike is continuously decaying (228Ra t1/2 = 5.75 years), measured 226Ra/228Ra ratios are decay-corrected from the measurement date back to the date of calibration of the 228Ra spike using the following equation:
where t = time between measurement and calibration date. Therefore, model age calculations for Ra use the 228Ra spike calibration date (for this study 26-Apr-13) as the reference date.
Concentrations of 227Ac are determined from a known concentration of 225Ac atoms added to the sample from the 229Th spike with 225Ac in secular equilibrium and the measured 225Ac/227Ac ratio of the sample. However, 225Ac is not the direct daughter of 229Th. As described above, 229Th first decays to 225Ra (t1/2 = 14.5 days), and then 225Ra decays to 225Ac (t1/2 = 10 days). This results in complex behavior of 225Ac during chemical purification of Ac from a U matrix (shown by curve in Fig. 1). Initially, 225Ac atoms represent the concentration predicted from secular equilibrium with 229Th. After the first column purification, 229Th is removed, and 225Ac begins to decay but remains partially supported by decay from 225Ra remaining in the sample:
where t 1 = hours between column 1 (Th purification) and column 2 (Ra purification). In the second column purification, 225Ra is removed from the sample solution leaving 225Ac unsupported. The 225Ac in the sample then decays following simple decay until the time of measurement:
where t 2 = hours between column 2 (Ra purification) and the time of measurement. Using these decay relationships and the timing of purification, the 225Ac atoms at the time of measurement can be calculated. Model age calculations for Ac, are calculated using a reference date that corresponds to the time of measurement. Model ages for the 230Th–234U and 231Pa–235U chronometers were calculated according to [6] and [8] respectively.

225Ac decay from secular equilibrium during chemical purification. At the time of column 1 (black vertical line), 229Th is purified and 225Ac and 225Ra concentrations start to decrease. 225Ac is supported by 225Ra decay until column 2 (black dashed line). At this time, 225Ra is purified and 225Ac decays unsupported until the time of measurement (grey vertical dashed line)
Results and discussion
Concentrations of all measured actinides (238U, 235U, 234U, 230Th, 231Pa, 226Ra, and 227Ac) for samples U100A, U100B, ITWG-RR3-A, ITWG-RR3-B as well as a laboratory blank are reported in Table 1. Uncertainties reported represent a coverage factor of 2, (k = 2) and propagate all known influence factors with the measurement with the exception of the errors associated with the decay constants of 226Ra, 228Ra, 225Ac, and 227Ac. Uncertainty derived from these decay constants represents less than 0.01 % of the total uncertainty. Errors are propagated following standard procedures described by the Joint Committee for Guides in Metrology [22]. Verification 226Ra concentrations for TML and CANMET ores are provided as Supplemental Information. TML 226Ra concentrations agree with those predicted by secular equilibrium, and measured 226Ra concentrations for CANMET ores agree with certified 226Ra activities.
Concentrations from Table 1 are used to calculate model ages for all of the sample materials shown in Table 2. Calculated model ages are subtracted from the appropriate reference dates for each chronometer to provide a model date for the purification of each sample. These model dates as well as the known dates of purification, or the “paper age” of the samples are summarized in Table 2.
U100: validation of methods
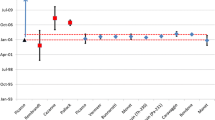
The model dates calculated for two separate aliquots of U100A and U100B are nearly concordant within uncertainty for all chronometers used in this study: 230Th–234U, 226Ra–238U, 231Pa–235U, and 227Ac–235U (Table 2, Fig. 2) (minor discordance between 230Th–234U and 227Ac–235U). However the model dates vary slightly between chronometers. The calculated purification dates from the 230Th–234U chronometer of May 18, 1958 for U100A and April 10, 1958 for U100B are approximately 9 months older than the paper age of the material [11] (Fig. 2). This new model-date is earlier than the date published for CRM-U100 in [6]. This could suggest that uranium purification in January of 1959 was not complete, and that a small amount of 230Th remained in CRM-U100 at the time of purification. Model ages for U100A and B calculated from the 226Ra–238U and 231Pa–235U chronometers agree within uncertainty of the paper age of January 8, 1959 (Fig. 2). These results demonstrate the accuracy (approximately 10 months for a material from 1959) and precision of the 226Ra–238U chronometer developed here for the age-dating of nuclear materials. The 227Ac–235U system provides model dates for U100A and B of July 12, 1960 and April 8, 1960 respectively, which are 15–18 months younger than the known purification date of CRM-U100, though these ages are within uncertainty of ages from the 226Ra–238U and 231Pa–235U chronometers (Fig. 2). A systematic bias between the 238U and 235U decay-series chronometers is suggested, but there are too few results at the present time to be certain of this. Model ages from 227Ac to 235U, despite being slightly younger than the paper age, are relatively accurate given the small quantity of material available for measurement (femtograms of 227Ac). Used together, these model ages represent maximum and minimum ages of purification for CRM-U100 that bracket the age of the material from early 1958 to late 1960 and demonstrate the power of multiple chronometers for determining the age of nuclear material.

Calculated model ages for U100A (symbols with dashed error bars) and U100B (symbols with solid error bars). Model ages show relative concordance between chronometers—with only minor discordance between 230Th–234U and 227Ac–235U. Uncertainties shown represent expanded uncertainties with a coverage factor of 2 (k = 2). Chronometers result in ages that agree well with the paper production ages of CRM-U100 [11]
ITWG round robin 3 exercise uranium metal model ages
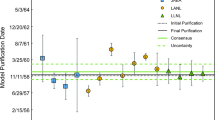
Model ages for the ITWG Round Robin 3 uranium metals from the 230Th–234U, 226Ra–238U, 231Pa–235U, and 227Ac–235U chronometers are significantly discordant and differ by as much as 29 years (231Pa–235U model age 39.9 years relative to 230Th–234U model age of 10.7 years for ITWG-RR3-B) (Table 2; Fig. 3). The 230Th–234U chronometer model dates of August 17, 2003 and May 14, 2004 are 3–5 months younger than the known metal-casting dates of May 22, 2003, and January 14, 2004 for ITWG-RR3 A and B respectively. These ages are outside the uncertainty assigned to the analyses, and while they demonstrate that Th was efficiently segregated from uranium metal during the metal casting at Y-12, these “too young” ages may indicate some post-casting fractionation of Th from U. If not an analytical error, this may be a case where the closed-system assumption of the model age has been violated; perhaps the slightly younger ages reported here reflect aging of the LLNL sample solution from 2010 to 2015 and loss of Th to PFA vial walls [6] in which the sample solutions were stored. 226Ra–238U model dates of August 13, 2001 and April 11, 2002 for ITWG-RR3-A and B respectively, are 20–21 months older than the known casting dates of the metals. These older ages suggest that Ra may not be as efficiently purified as Th during metal casting and minor amounts of excess 226Ra may exist in the uranium metal at the time of casting.

Model ages for ITWG-RR3-A (left) and ITWG-RR3-B (right) uranium metals. Model ages calculated from the 230Th–234U (black circles) and 231Pa–235U (black squares) chronometers differ significantly for both metals. The older age from the 231Pa–235U chronometer provides evidence for excess 231Pa in the uranium metals at the time of metal casting. If this excess 231Pa is accounted for, new 227Ac–235U corrected model ages can be calculated (white diamonds)—change between uncorrected 227Ac–235U model ages (grey diamonds) and corrected values is shown by the grey dashed line. These corrected model ages agree well with the known casting dates of the metal
Model ages from the 231Pa–235U chronometer, which suggest purification of the uranium metals in 1977 and 1975, are 26–29 years older than the known casting dates of these metals (Table 2, Fig. 3). These much older ages demonstrate that excess 231Pa must have existed during uranium metal casting. Similarly, the 227Ac–235U model ages for ITWG-RR3A and B are January 8, 1987 and October 15, 1986, which again, significantly deviate from the known purification dates (Table 2; Fig. 3). However, because we observe deviation between the 230Th–234U and 231Pa–235U chronometers and relative concordance between 230Th–234U and 226Ra–238U, we can calculate the amount of 231Pa excess that would need to exist at the time of purification in order for the model age of the 231Pa–235U chronometer to agree with the metal casting date [9]. The 231Pa excess required is 2.49 × 1012 per gram primary solution of metal A and 3.42 × 1012 atoms per gram primary solution of metal B. Using these calculated amounts of excess 231Pa at the initial time of casting and re-calculating the 227Ac–235U model ages, the corrected model ages for this system are within 6 months of the known casting date (Fig. 3). These model ages are labeled 227Ac–235Ucorr in Table 1 and Fig. 3 accordingly. These results suggest that the apparent 227Ac excess has been supported by 231Pa excess since the time of metal casting, and that 227Ac was segregated as efficiently as 230Th from U at this time.
Conclusions
We have developed chemical separation and MC-ICP-MS methods to use the grand-daughter chronometers in the decay chains of 238U and 235U age-date materials for nuclear forensic investigations: 226Ra–238U and 227Ac–235U. These methods have been validated by the measurement of model ages calculated for the CRM-U100 reference material where the 230Th–234U, 226Ra–238U, 231Pa–235U, and 227Ac–235U chronometers that agree well with the known purification date of this material. Investigation of uranium metals from the ITWG Round Robin 3 Exercise using these new chronometers demonstrates that during metal casting, 230Th, 226Ra, and 227Ac are efficiently purified from uranium metal, though 226Ra is purified to a lesser extent than 230Th. Protactinium-231 was not completely separated from uranium metal during casting, and this 231Pa excess must be taken into account. None of these model ages are necessarily “incorrect”. Instead, taken together they supply information on relative elemental segregation during U casting, which can be used in the interpretation of unknown samples in a nuclear forensic investigation. Continued use of all four chronometers for nuclear material age-dating has the potential to increase our understanding of radionuclide migration and behavior during uranium processing.
References
IAEA Nuclear Security Series No. 2, 2006. Nuclear Forensics Support. http://wwwpub.iaea.org/MTCD/publications/PDF/Pub1241_web.pdf
Mayer K, Wallenius M, Varga Z (2013) Nuclear forensic science: correlating measurable material parameters to the history of nuclear material. Chem Rev 113:884–900
Varga Z, Suryani G (2007) Production date determination of uranium-oxide materials by inductively coupled plasma mass spectrometry. Anal Chim Acta 599:16–23
Williams RW, Gaffney AM, Kristo MJ, Hutcheon ID (2009) 230Th–234U model-ages of some uranium standard reference materials. In: INMM 50th annual meeting conference papers
Varga Z, Wallenius M, Mayer K (2010) Age determination of uranium samples by inductively coupled plasma mass spectrometry using direct measurement and spectral deconvolution. J Anal At Spectrom 25:1958–1962
Willams RW, Gaffney AM (2011) 230Th–234U model ages of some uranium standard reference materials. Proc Radiochim Acta 1:31–35
Varga Z, Nicholl A, Wallenius M, Mayer K (2012) Development and validation of a methodology for uranium radiochronometry reference material preparation. Anal Chim Acta 718:25–31
Eppich GE, Williams RW, Gaffney AM, Schorzman KC (2013) 235U–231Pa age dating of uranium materials for nuclear forensic investigations. J Anal At Spectrom 28:666–674
Nuclear Forensics International Technical Working Group (ITWG) Round Robin 3 Exercise after action and lessons learned report. Report #PNNL-20079. Coordinator: Hanlen R (2011) Pacific Northwest National Laboratory, Richland, Washington
Sturm M, Richter S, Aregbe Y, Wellum R, Mialle S, Mayer K, Prohaska T (2014) Evaluation of chronometers in plutonium age determination for nuclear forensics: what if the ‘Pu/U clocks’ do not match? J Radioanal Nucl Chem 302:399–411
Petit GS (1960) Preparation of uranium isotopic standards for the National Bureau of Standards. Union Carbide Nuclear Company, Oak Ridge, Tennessee. Oak Ridge National Laboratory, Report #KL-8, Department of Energy/K25 Archives
Garner EL, Machlan LA, Shields WR (1971) Standard reference materials: uranium isotopic standard reference materials. National Bureau of Standards US Special Publication 260-27
Williams RW, Collerson KD, Gill JB, Daniel C (1992) High Th/U ratios in subcontinental lithospheric mantle: mass spectrometric measurement of Th isotopes in Gaussberg lamproites. Earth Planet Sci Lett 111:257–268
Sims KWW, Gill JB, Dosseto A, Hoffman DL, Lundstrom CC, Williams RW, Ball L, Tollstrup D, Turner S, Prytulak J, Glessner JJG, Standish JJ, Elliot T (2008) An inter-laboratory assessment of the thorium isotopic composition of synthetic and rock reference materials. Geostand Anal Res 32:65–91
Regelous M, Turner SP, Elliott TR, Rostami K, Hawkesworth CJ (2004) Measurement of femtogram quantities of protactinium in silicate rock samples by multicollector inductively coupled plasma mass spectrometry. Anal Chem 76:3584–3589
Volpe AM, Olivares JA, Murrell MT (1991) Determination of radium isotope ratios and abundances in geologic samples by thermal ionization mass spectrometry. Anal Chem 63:913–916
Chabaux F, Ben Othman D, Birck JL (1994) A new Ra-Ba chromatographic separation and its application to Ra mass-spectrometric measurement in volcanic rocks. Chem Geol 114:191–197
Koornneef JM, Stracke A, Aciego S, Reubi O, Bourdon B (2010) A new method for U-Th-Pa-Ra separation and accurate measurement of 234U–230Th–231Pa–226Ra disequilibria in volcanic rocks by MC-ICPMS. Chem Geol 277:30–41
Richter S, Goldberg SA (2003) Improved techniques for high accuracy isotope ratio measurements of nuclear materials using thermal ionization mass spectrometry. Int J Mass Spec 229:181–197
Ivanovich M, Harmon RS (1992) Uranium series disequilibria: applications to earth, marine and environmental sciences. Clarendon Press, Oxford
Bateman H (1910) Solution of a system of differential equations occurring in the theory of radioactive transformations. Proc Camb Philos Soc 15:423
JCGM (2008) Evaluation of measurement data—Guide to the expression of uncertainty in measurement. Joint Committee for Guides in Metrology. JCGM 100
Acknowledgments
Research herein was supported by the National Nuclear Security Administration’s Nuclear Noncompliance Verification Program (Office of Nuclear Verification; NA-243) and a Lawrence Livermore National Laboratory Postdoctoral Appointment provided to T. Kayzar. The authors would like to thank Kerri C. Schorzman for laboratory support. This work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344 LLNL-JRNL-669502.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kayzar, T.M., Williams, R.W. Developing 226Ra and 227Ac age-dating techniques for nuclear forensics to gain insight from concordant and non-concordant radiochronometers. J Radioanal Nucl Chem 307, 2061–2068 (2016). https://doi.org/10.1007/s10967-015-4435-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-015-4435-4