
Abstract
A 231Pa reference material has been characterized for amount of protactinium. This reference material is primarily intended for calibration of 233Pa tracers produced for 235U–231Pa model age measurements associated with nuclear forensics and nuclear safeguards. Primary measurements for characterization were made by isotope dilution mass spectrometry of a purified 231Pa solution using a 233Pa isotopic spike. The spike was calibrated by allowing multiple aliquots of the 233Pa spike solution to decay to 233U and then measuring the ingrown 233U by isotope dilution mass spectrometry using a certified uranium assay and isotopic standard as a reverse-spike. The new 231Pa reference material will simplify calibration of the 233Pa isotope dilution spikes, provide metrological traceability, and potentially reduce the overall measurement uncertainty of model ages.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Determining the date of the most recent processing step that resulted in separation of a special nuclear material, such as plutonium or enriched uranium, from its daughter products is one of the most diagnostic attributes than can be derived from nuclear forensic analyses. These dates are referred to as model purification ages and are calculated from the measured atomic ratio of parent–daughter nuclide pairs. The radioactive decay of 235U to 231Pa has been used for determining radiometric ages of relatively young (< 200,000 a) uranium-bearing geologic materials since the 1950s [1]. More recently, 231Pa–235U dating has also been utilized to measure model purification ages of uranium materials associated with the nuclear fuel cycle [2,3,4]. Initially, 235U–231Pa dating was performed using α-counting radioactivity measurement methods but the utility of the activity-based measurements was hindered by the limited resolution of the technique. Pickett et al. [5] developed a 231Pa–235U dating method that uses thermal ionization mass spectrometry (TIMS) to achieve faster and more precise measurement results. For this method, the amount of 231Pa in a sample is measured by isotope dilution mass spectrometry (IDMS) using a 233Pa isotopic spike. Regelous et al. [6] demonstrated that 231Pa IDMS measurements can be performed using multi-collector inductively coupled plasma mass spectrometry (MC-ICP-MS) instead of TIMS and noted that the greater ionization efficiency of the MC-ICP-MS method is advantageous when measuring the minute quantity (ng to fg) of ingrown 231Pa that is present in most samples. Despite these advances, the difficulty of preparing and calibrating a 233Pa isotopic IDMS spike has been a significant barrier to routine use of 235U–231Pa dating by the nuclear forensic analytical community.
The two principal methods for obtaining starting material for a 233Pa IDMS spike are separation from 237Np [5, 7] and thermal neutron irradiation of 232Th [7,8,9,10]. The 237Np nuclide has a half-life of (2.144 ± 0.007) 106 a [11], decaying by α particle emission directly to 233Pa with a half-life of only (26.98 ± 0.02) days [12]. 233Pa approaches 99% of the secular equilibrium concentration with 237Np after about 180 days, which approximates the frequency with which a 233Pa spike can be “milked” from a 237Np “cow”. Irradiation of 232Th can produce 233Pa on-demand but requires access to a sufficiently intense thermal neutron flux and appropriate handling facilities for the irradiated material. Regardless of production method, to use 233Pa as an isotopic spike it must be separated from other radionuclides and then calibrated for amount of substance (mol). Once prepared and calibrated, a batch of 233Pa has a useful lifetime of just a few months before there is insufficient material for mass spectrometric analysis.
Calibration of 233Pa has traditionally been performed by reverse-IDMS using geological materials containing uranium that is assumed to be in secular equilibrium with its decay products. This calibration method requires a difficult dissolution of a silicate rock material, is fundamentally dependent on a known or precisely measured uranium concentration, and results in a complicated traceability chain for the measurements. Naperstkow et al. [10] avoided the use of rock standards and reverse IDMS by employing efficiency traced liquid scintillation counting (CNETFootnote 1), 4πβ-γ coincidence, and 4πβ-γ anticoincidence radioactivity measurement methods to calibrate a 233Pa solution with resulting uncertainties between 0.8 and 1% (k = 2). Although this traceable activity-based calibration was performed using sophisticated counting methods that are not routinely used outside of laboratories specializing in radioactivity measurements, an ion chamber can be calibrated for more routine activity-based 233Pa spike characterization. Varga et al. [4] demonstrated an indirect calibration for 233Pa by using γ-counting methods to measure the molality (mol g−1) of 237Np in a solution that was in secular equilibrium with its 233Pa daughter and adding the unseparated neptunium solution directly to analysis samples. This method avoids a laborious 233Pa spike calibration but will typically have a relatively large uncertainty (> 2%, k = 2) due to the limits of neptunium assay measurements. More recently, Treinen et al. [13] demonstrated the use of a characterized solution of high-purity 231Pa as a reverse-spike for accurate and reproducible 233Pa calibrations. This calibration was accomplished with less difficulty than rock standards and with uncertainties that are equivalent to, or lower than, those that can be reasonably obtained using activity measurements.
The 231Pa nuclide has a half-life of (32 670 ± 520) a [11], which is sufficiently long for a batch of purified 231Pa to be prepared as a calibration reference material with a shelf-life of 20 years or even considerably longer. A 231Pa reference material could be utilized to perform reverse-IDMS calibrations in parallel with 233Pa IDMS analyses of unknown samples. This would simplify calibration of 233Pa isotopic tracer, increase the reliability of the IDMS measurements, and reduce uncertainties of 231Pa amount measurements made on unknown samples. The preparation and characterization of such a 231Pa reference material, specifically for calibration of 233Pa isotopic spikes, is described here. This new reference material was produced as part of an ongoing United States Department of Homeland Security program to enhance analytical capabilities for nuclear forensics. Production of the 231Pa reference material and characterization measurements were performed at Lawrence Livermore National Laboratory (LLNL). Activity measurements for verification of the primary characterization value were performed at National Institute of Standards and Technology (NIST), the National Research Council of Canada (NRC), and the National Physical Laboratory in the United Kingdom (NPL).
Experimental
Figure 1 is a schematic of the process for preparation and characterization of the 231Pa reference material. A purified 231Pa master (M) solution and a freshly separated 233Pa spike (S) solution were prepared on consecutive days. Carefully weighed aliquots were taken from both M and S solutions and shipped to NIST, NPL, and NRC for activity measurements. Additional aliquots of the S solution were dispensed for analysis of ingrown 233U by IDMS. A subsample of the 231Pa M solution was then diluted to produce the 231Pa reference material (RM) solution which was dispensed to create individual reference material units. In parallel with unit production, subsamples of RM solution were spiked with the S solution. The protactinium isotopic ratios of the mixed 233Pa–231Pa samples were measured to determinate the molality of 231Pa in the RM solution by IDMS. Radioactivity measurements performed on the M and S solutions were used for verification of IDMS measurement results. Preparation of the protactinium solutions, production of the reference material units, and characterization analyses are described in further detail below. Activity measurements performed for verification of the characterization analyses are described elsewhere [13,14,15] with results summarized here.

231Pa reference material production and characterization process. The “M Solution” is the concentrated 231Pa master solution purified for this project. The “RM Solution” is a stock solution prepared for production of reference material units and the “S Solution” is the 233Pa spike prepared for primary calibration of the RM solution. Heavier arrows in the figure indicate material transfers and analyses performed for the primary calibration for the molality of 231Pa in the RM solution. Lighter arrows indicate material transfer and analyses for verification measurements
Material preparation
An initial clean up step was performed on approximately 20 mg of 231Pa stored at LLNL. This procedure, described in [16], is a precipitation of a hydrous protactinium oxide (Pa2O5·xH2O) from a HF acid solution and repeated rinses of the precipitate. After this step, the protactinium was again dissolved in HF (Seastar Chemicals Inc., Vancouver, Canada)Footnote 2 and a subsample containing 2 mg protactinium was dried first in perchloric acid and then dried two times in concentrated HNO3 (Seastar Chemicals Inc., Vancouver, Canada). An additional separation was then performed on this sample using a silica gel column chemistry technique described in Treinen et al. [17]. A subsample of this purified material (approximately 125 µg of 231Pa) was prepared as the M solution by diluting the 231Pa in a mixed solution of 2 mol L−1 HNO3 and 0.1 mol L−1 HF. Aliquots of the M solution (5 mL) were transferred to Savillex perfluoroalkoxy (PFA) Teflon vials (Minneapolis, MN, USA) and shipped to NPL, NRC, and NIST for massic activity measurements. MC-ICP-MS mass scans of the purified M solution showed a n(227)/n(231Pa) ratio of approximately 1 × 10−7, indicating low levels of 231Pa decay products (227Ac and 227Th). Another mass spectrometric analysis of the 231Pa master solution, performed 6 months after purification, indicated a n(227)/n(231Pa) ratio that was 0.9% higher than expected from ingrowth alone. Based on these data, it is estimated that approximately 85% of the 227 amu contaminant, present immediately after purification, was unsupported 227Th (T1/2 = 18.62 ± 0.09 days [18]) and the remainder was 227Ac (T1/2 = 21.782 ± 0.003 a [19]). This excess material is not detrimental to use of the 231Pa as an IDMS calibration standard, but it does represent a potentially significant source of bias for radioactivity measurements, particularly measurements performed within several months of purification.
The RM solution was prepared from the M solution as a stock material for production of the reference material units. A 4 g subsample of the M solution was transferred to a pre-cleaned and weighed 1 L fluorinated ethylene propylene (FEP) Teflon bottle, diluted with 650 mL of a mixed 2 mol L−1 HNO3 and 0.1 mol L−1 HF solution, and homogenized by hand shaking (gravimetrically determined dilution factor is 187.279 ± 0.047, k = 2). Five (5) subsamples of the 231Pa RM solution were prepared for IDMS analysis by transferring carefully weighed aliquots (0.5–1.0 g) to new pre-cleaned Savillex vials. The potential for weighing bias due to evaporation during dispensing was minimized for these subsamples (and all subsequent spike or sample aliquots for IDMS analyses) by transferring the solution using disposable low-density polyethylene pycnometers (Canus Inc., Ottawa, Canada) with capillary tips. Masses of the subsamples were determined by the difference between the pycnometer mass when filled with solution and after dispensing. At least 3 replicates weighings were performed for all mass measurements. The balance used for this work was a calibrated 5-place XPE 105 electronic balance (Metter-Toledo, Columbus, OH, USA) that was regularly checked for accuracy and linearity using a calibrated weight set.
Following preparation of the IDMS analysis samples, the RM solution was dispensed using a Hamilton MICROLAB 600 dispenser (Reno, NV, USA) outfitted with a new 5 mL dispensing syringe. Intake and dispensing tubing was thoroughly flushed by rinsing with a dilute HNO3 and HF mixed solution followed by deionized water. Prior to dispensing the RM solution, approximately 30 mL of mixed 2 mol L−1 HNO3 and 0.1 mol L−1 HF solution was run through the apparatus (6 dispensing cycles). The uptake tubing was then transferred to the RM solution stock bottle and 30 mL of the solution was dispensed before filling the first reference material unit. A total of 112 231Pa reference material units were prepared with each unit comprised of 5 mL of solution in a pre-cleaned and pre-weighed 30 mL FEP bottle that was previously labelled with a number corresponding to the order in which the bottle was filled. Each unit was tightly capped after receiving the solution and all units were then carefully reweighed. The reference material units were filled in a single effort lasting approximately 1 h. The mean mass of dispensed solution was 5.2978 g with an expanded uncertainty of 0.0022 g (k = 2). Individual unit bottles were packaged by wrapping the top of the bottle and cap with polytetrafluoroethylene (PTFE) Teflon tape which, in turn, was wrapped with vinyl tape to prevent loosening of the cap or unravelling of the PTFE tape. Each bottle was placed in an individual aluminized Mylar pouch along with acid-resistant absorbent pads and the pouch was then heat sealed.
A batch of 233Pa IDMS spike was prepared by separation from a high-purity 237Np solution (65 mg of 237Np in HCl). This separation was performed using an AG MP-1 M anion exchange resin followed by a silica gel separation, as described in [17]. The purification procedure was completed at 22:50 UTC on 27 June 2017. A 1 mol L−1 HNO3 solution is used in the silica gel separation procedure as a rinse prior to eluting the protactinium. MC-ICP-MS mass scans of this rinse solution indicated a n(233Pa)/n(238U) ratio of approximately 0.011 whereas 238U was not observed in the separated 233Pa; thus demonstrating the protactinium was quantitatively separated from uranium, including ingrown 233U. Mass scans of the separated protactinium also indicated a n(233Pa)/n(237Np) atom ratio of > 3 which corresponds to an activity ratio greater than 1.0 × 108. The total integrated quantity of 233Pa that could grow in from this proportion of 237Np, over the course of project activities (300 days), is inconsequential (< 0.01% relative to the amount of 233Pa present after purification). The purified 233Pa was dissolved in a mixed 2 mol L−1 HNO3 and 0.1 mol L−1 HF solution to produce the 233Pa S solution with an estimated massic activity of ≈ 30 kBq g−1 at the time of preparation. On 28 June 2017 aliquots of the S solution were transferred to Savillex Teflon vials and shipped to NIST, NRC, and NPL for activity calibrations. Another 4 aliquots of the 233Pa solution were transferred to vials which were capped, weighed, and stored for later measurement of 233U.
A uranium (U) reverse-spike was prepared for high-precision IDMS measurements of 233U in the decayed 233Pa calibration samples. The starting material for the reverse-spike was CRM 112-A uranium metal assay and isotopic standard [20]. For initial preparation of the uranium solution, a 0.25 g piece CRM 112-A metal was twice cleaned in 8 mol L−1 HNO3 to remove any uranium oxide prior to weighing. The metal was then dried, weighed, and dissolved in 70 mL of a mixed 2 mol L−1 HNO3 and 0.05 mol L−1 HF solution to produce a relatively concentrated master solution. A 2-stage serial dilution of this uranium master solution was performed to create the U reverse-spike with a molality of (111.80 ± 0.14) pmol g−1 (k = 2).
Characterization measurements
The 4 calibration aliquots of the S solution were stored for period of 298 days, after which the samples were reweighed to correct for any change to the solution mass due to evaporation. Individual samples were then split, mixed with the U reverse-spike solution, and measured on the LLNL Nu-plasma HR MC-ICP-MS (Nu Instruments Ltd, Wrexham, UK). Mass spectrometry analysis conditions for these measurements are described in Table 1. The n(233U)/n(238U) isotopic ratios of the spiked samples were measured on 4 May 2018, by which time greater than 99.9% of 233Pa initially present in the S solution had decayed to 233U but less than 0.0003% of the ingrown 233U had decayed to 229Th. Accordingly, with only a small correction for remaining 233Pa, uranium IDMS on these subsamples provides a quantitative value for the amount of 233Pa originally present in the purified solution.
The molality of 231Pa in the RM solution was measured by IDMS using the 233Pa S solution as an isotopic spike. IDMS aliquots of the RM solution were mixed with aliquots of the S solution and a final purification of the mixed solutions was performed using the silica gel method. The n(233Pa)/n(231Pa) isotopic ratio measurements for the IDMS analyses were performed, in duplicate, approximately 7 h after purification. These measurements were made on the LLNL Nu-plasma MC-ICP-MS; details of the analysis procedure are provided in Table 1. The protactinium IDMS analyses were performed 17 days after purification of the 233Pa S solution, so 35% of the 233Pa in the spike had decayed, requiring a substantial decay correction.
Verification measurements
Massic radioactivity analyses for 233Pa in the S solution and 231Pa in the M solutions were incorporated into the reference material production project as metrologically independent verification measurements for the molality of these nuclides. A primary 233Pa massic activity measurement for the S solution was made by NIST using a live-timed 4πβ-γ anti-coincidence counting method (LTAC) and confirmatory measurements were made by γ-spectrometry on efficiency-calibrated HPGe detectors. Details of the methods and data reduction for these analyses are provided in [13]. NRC also performed a primary calibration of S solution using an LTAC method and performed additional activity measurements using a CNET 3H efficiency-traced liquid scintillation counting method and a Triple-to-Double Coincidence Ratio (TDCR) method [14]. Finally, the S solution massic activity was measured by NPL using a 4πβ-γ coincidence counting method (Keightley, personal communication, 7 February 2018).
Massic activity calibrations for 231Pa in the M solution were performed by NRC using two LSC activity measurements methods, CNET and TDCR [14]. Activity measurements of the M solution at NIST included the CNET method and α spectrometry [15]. The NIST measurements were performed in July of 2017, shortly after the samples were received, and additional CNET measurements were made in November of 2017. 233Pa was added to α spectrometry samples as a yield monitor for electrodeposition of the 231Pa sources and the detectors were calibrated for energy and efficiency using U, Am, and Pu electrodeposited sources. NPL did not report any measurements for the 231Pa.
Results
The massic activity values for 231Pa in the M solution and for 233Pa in the S solution that were reported by the radioactivity measurement laboratories are summarized in Table 2. Also shown in the table are values for molality of 233Pa in the S solution and molality of 231Pa in the RM solution that were calculated from the activity data. Fitzgerald et al. [15] noted that both CNET and preliminary γ-spectrometry measurements of the 231Pa master solution showed radioactivity in the samples declining for several weeks after receipt and then increasing again due to ingrowth of 231Pa daughter products. This pattern indicates the presence of an unsupported radioactive contaminant. Corrections were applied to the NIST and NRC counting data for ingrowth of 231Pa decay chain products and an attempt was made by NIST to perform an empirical correction for the contaminant radioactivity. This correction, however, is not likely to be sufficient due the lack of quantitative data about the amount or proportion of nuclide(s) comprising the contaminating material. NRC did not attempt to apply a correction for contaminating nuclides in the 231Pa M solution.
The results for the IDMS calibration of the 233Pa S solution are summarized in Fig. 2 and details are provided Table 3. The 8 replicate measurement results agree within uncertainties and have an observed variability of only 0.07% relative standard deviation. The mean measured 233Pa molality for a reference date of 27 June 2017 22:50 UTC is 0.14690 pmol g−1 with an expanded uncertainty of 0.00035 pmol g−1 (0.24% relative, k = 2). This value is consistent with 233Pa molality values derived from primary and confirmatory massic activities calibrations performed at the radioactivity laboratories.

Measurement data for molality of 233Pa in the S solution. All values are for a reference time of 27 June 2017 22:50 UTC. Error bars for data points are expanded uncertainties (U = k uc) with a coverage (k) factor of 2. A mean value for the IDMS data (solid line) and an expanded uncertainty interval for the mean value (dashed lines) are included in the figure
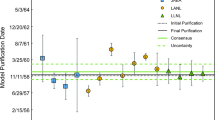
The molality of 233Pa in the S solution was a key variable for the protactinium IDMS measurements made to determine the molality of 231Pa in the RM solution. The 231Pa IDMS results are shown in Fig. 3 with details provided in Table 4. These results are also highly repeatable and have an observed variability of only 0.07% relative standard deviation. The mean molality of 231Pa in the RM solution is 27.55 pmol g−1 with an expanded uncertainty of 0.12 pmol g−1 (0.35% relative, k = 2). The activity-based molality value calculated from alpha spectrometry measurements (Table 2) is essentially indistinguishable from the IDMS data. Activity-based molality values calculated from the LSC-based measurement methods range from 3 to 8% higher than the IDMS value.

Molality of 231Pa in the RM solution. All values are for a reference time of 27 June 2017 22:50 UTC. Error bars for data points are expanded uncertainties (U = k uc) with a coverage factor (k) of 2. A mean value for the IDMS data (solid line) and an expanded uncertainty (k = 2) interval for this mean value (dashed lines) are included in the figure
The measured masses of RM solution dispensed to the 231Pa reference material units have a maximum difference of only 3.1 mg (excluding a single outlier unit) which is only 0.06% of the 5.3 g of solution dispensed to each unit. The 0.35% relative expanded uncertainty for the molality of 231Pa in the RM solution is much greater than the relative difference in solution masses. As a result, the variability in the mass of RM solution dispensed to individual reference material units corresponds to trivial differences in the calculated amount of 231Pa in each unit and this variability does not significantly increase the uncertainty for the amount 231Pa. Accordingly, a single value and uncertainty for amount 231Pa can be applied to every unit in the production run. This value, of (145.96 ± 0.51) pmol (k = 2), was calculated using the mean buoyancy-corrected mass of solution aliquots dispensed to the units and uncertainty components for the range of observed masses and for a correction applied to compensate for solution evaporation during dispensing. The uncertainty for the 231Pa amount value for the reference material units was estimated in accordance with international guidance for measurement uncertainty [25, 26] and includes all significant sources of uncertainty. A detailed uncertainty budget for the calculated amount of 231Pa is provided in Table 5.
Discussion
The goal of this project was to produce a 231Pa reference material characterized for amount of 231Pa. Although this material is not being provided as a Certified Reference Material (CRM) per the definition in [27], the production project was planned and executed to satisfy the quality requirements for CRMs as outlined in international guidance documents [28, 29]. General requirements for a high-quality analytical reference material include stability, homogeneity, and metrological traceability. Additionally, characterized attribute values should have GUM-compliant uncertainties [24] and independent verification of the attribute values should be performed, when possible.
Stability of the 231Pa reference material units is an important consideration. Protactinium typically occurs in solution as the pentavalent cation, Pa(V), and has long been known to form colloids that adsorb onto container walls [30]. This tendency could result in highly variable characteristics for a production run of a protactinium reference material. To mitigate the potential for this source of variability, the 231Pa RM solution was prepared as a mixture of 2 mol L−1 HNO3 and 0.1 mol L−1 HF. Sub μg g−1 concentrations of Pa(V) are reasonably stable in HNO3 solutions of greater than 1.0 mol L−1 and Pa(V) is highly soluble in HF solutions, even in relatively dilute solutions (e.g. 3.9 g L−1 in 0.05 mol L−1 HF) [31]. Furthermore, the authors have observed that protactinium in a 2 mol L−1 HNO3 and 0.1 mol L−1 HF mixed solution shows no apparent change in molality of protactinium for durations of greater than 1 year. Although it is anticipated that the 231Pa will remain in solution, it is also expected that the solution in the reference material units will change in molality due to evaporative losses of the aqueous solution from the container. To ensure that the 231Pa reference material will remain fit-for-purpose over the long-term, the units are characterized for amount of 231Pa and each unit is contained in a weighed Teflon bottle (bottle mass values to be provided with units). With these values, a user can utilize the RM solution as received, dilute the solution in the unit container with an appropriate acid solution, or perform a quantitative transfer of the 231Pa.
It is difficult to envision a mechanism that would produce unit-to-unit heterogeneity in the 231Pa reference material production run. The separated protactinium used to prepare the RM solution was essentially monoisotopic and mass spectrometric analysis of the material did not indicate the presence of significant quantities of parent elements for protactinium nuclides, such as uranium or neptunium. Considering the purity of the starting material and the fact that 231Pa is the only isotope of protactinium with a half-life greater than 27 d, it can be stated, with a high degree of confidence, that the characterized reference material units are monoisotopic and therefore isotopically homogeneous.
To minimize the potential for temporal changes to the molality of 231Pa in the RM solution, preparation of the solution, preparation of the IDMS characterization samples, and production of the reference material units were performed during a single work day with all 112 units dispensed over a relatively short period (1 h). Variability that could be caused by differential protactinium contamination was minimized by performing preparation and production work in a class 10 000 clean facility, using only new precleaned labware for all stages of the project, and using high-purity reagents for preparation, production, and analysis (double distilled acids, 18.2 MΩ cm water). Finally, variability in the amount of material transferred to the units was constrained by use of a high-precision automated dispenser and verified by carefully weighing of all unit bottles before and after dispensing.
Metrological traceability [27] for amount of 231Pa was maintained for the two main stages of the calibration process. First, the molality of 233Pa in the spike solution was measured by a primary IDMS analysis method [32, 33] which took advantage of the relatively rapid decay of 233Pa to the long-lived 233U nuclide (Fig. 4a). This allowed for a certified uranium assay and isotopic reference material (CRM 112-A) to be used as an isotopic spike. All masses for the analyses were measured on a calibrated balance that was verified against calibrated weight sets during project activities. Mass spectrometric measurements were corrected for instrumental bias using replicate measurement of an appropriate uranium isotopic reference material (CRM U010). A value for the molality of 233Pa in the S solution, at the time of the 231Pa IDMS analyses, was calculated by using the evaluated half-life for 233Pa to perform the appropriate decay correction from the time of purification. For the second stage of the calibration process (Fig. 4b), the traceable molality of 233Pa in the S solution was utilized for IDMS analyses of 231Pa in the RM solution. All masses for the 231Pa IDMS sample preparation were also measured on a calibrated balance that was verified using calibrated check weights. An isotopic standard for protactinium is not feasible, so mass spectrometric measurements were calibrated using the CRM U010 isotopic reference material. Although the MC-ICP-MS mass fractionation behavior of uranium and protactinium are likely to differ, these differences tend to be small for neighboring elements [34] and an estimated uncertainty component for potential bias in the mass spectrometry results was included in the budget for the attribute value. Finally, the average mass for the 231Pa RM solution distributed to each unit was determined from multiple replicate weighings of each sample unit on a calibrated balance.

Schematic of the traceability chain for amount of 231Pa. The diagram indicates the direct relationship between measurements necessary for calibration of the 233Pa S solution (a) and the 231Pa reference material (b)
The molality of 233Pa in the S solution, as measured by IDMS, was independently verified by activity-based analyses performed at NIST, NRC, and NPL, providing a high level of confidence in the calibration of this material. Values for molality of 231Pa in the RM solution calculated from LSC-based massic activity measurement on the M solution are, however, higher than those measured by IDMS. Observations from mass spectrometry on the 231Pa M solution and the activity measurements themselves provide an explanation for this discrepancy. Mass spectrometric scans of the 231Pa Master solution following the final purification procedure (prior to shipment for activity measurements) showed a small, but measurable, proportion of nuclides at 227 amu, probably 227Th and 227Ac daughter products. The presence of unsupported radioactive contaminants was also observed in LSC and γ spectrometry measurements made at NIST shortly after the samples were received. Calibration methods that measure total activity (i.e. LSC based methods CNET and TDCR) must be corrected for radioactive contaminants and for ingrowth of decay chain products from the time of purification. Due to limited data available to the radioactivity laboratories, developing a correction for the unsupported daughter products was not performed or could only be poorly constrained. As expected, this contamination resulted in elevated massic activity values from the LSC-based measurement methods used at NIST and NRC. The α spectrometry measurements performed at NIST had the advantage that only alpha particle energies characteristic of 231Pa decay were measured, so these results were not affected by the presence of small proportions of unsupported decay-chain products. The calculated value for the 231Pa molality of the RM solution based on the α spectrometry measurements is essentially identical to the IDMS results, providing independent verification of the characterized value.
Conclusion
The molality of 233Pa in the S solution prepared for calibration of 231Pa in the RM solution was precisely measured using a primary analysis method (i.e. IDMS) and independently verified by massic activity measurements at 3 metrology laboratories. This uniquely well characterized 233Pa material enabled high-precision primary measurements of the molality of 231Pa in the RM solution used to prepare units of the 231Pa reference material. The molality of 231Pa was independently verified by α spectrometry massic activity measurements performed at NIST. Individual units were carefully prepared from the RM solution resulting in a metrologically traceable reference material that is suitable for calibration of 233Pa spikes that are produced by analytical laboratories on an as-needed basis. The relative expanded uncertainty for amount of 231Pa in each reference material unit is 0.35% which should allow for reliable 233Pa spike calibrations and similarly well constrained measurements of 231Pa for determination of 235U–231Pa model ages.
Notes
The acronym CNET refers to the 3H efficiency-traced liquid scintillation counting methodology developed by Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas (CIEMAT) and the National Institute of Standards and Technology (NIST). This method is also referred to as the CIEMAT/NIST method.
Certain commercial equipment, instruments, software, or materials are identified in this paper to foster understanding. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
References
Cheng H, Edwards RL, Murrell MT, Benjamin TM (1998) Uranium-thorium-protactinium dating systematics. Geochim Cosmochim Acta 62:3437–3452
Morgenstern A, Apostolidis C, Mayer K (2002) Age determination of highly enriched uranium: separation and analysis of 231Pa. Anal Chem 74:5513–5516
Eppich GR, Williams RW, Gaffney AM, Schorzman KC (2013) 235U–231Pa age dating of uranium materials for nuclear forensic investigations. J Anal At Spectrom 28:666–674
Varga Z, Nicholl A, Wallenius M, Mayer K (2018) Measurement of the 231Pa/235U ratio for the age determination of uranium materials. J Radioanal Nucl Chem 318:1565–1571
Pickett DA, Murrell MT, Williams RW (1994) Determination of femtogram quantities of protactinium in geologic samples by thermal ionization mass spectrometry. Anal Chem 66:1044–1049
Regelous M, Turner SP, Elliot TR, Rostami K, Hawkesworth CJ (2004) Measurement of femtogram quantities of protactinium in silicate rock samples by multicollector inductively coupled plasma mass spectrometry. Anal Chem 76:3584–3589
Sill CW (1966) Preparation of protactinium-233 tracer. Anal Chem 38(11):1458–1463
Bourdon B, Joron J-L, Allège CL (1999) A method for 231Pa analysis by thermal ionization mass spectrometry in silicate rocks. Chem Geol 157:147–151
Keegan E, Stopic A, Griffiths G (2014) Protactinium-231 (231Pa) measurement for isotope chronometry in nuclear forensics. IAEA-CN-218-12. https://conferences.iaea.org/indico/event/16/contributions/7228/attachments/3117/3738/Liz_Keegan_Full_Paper_Int_conf_on_Advances_in_NF_Vienna_July_2014.pdf
Naperstkow Z, Moore K, Szames D, Varlow C, Armstrong AF, Galea R (2018) Production and standardization of an on-demand protactinium-233 tracer. J Radioanal Nucl Chem 318:703–709
BIPM (2011) Monograph 5: Table of radionuclides, 6-A: 22:242. http://www.nucleide.org/DDEP_WG/DDEPdata.htm. Accessed 11 March 2019
BIPM (2010) Monograph 5: table of radionuclides, 5-A: 22:244. http://www.nucleide.org/DDEP_WG/DDEPdata.htm. Accessed 11 March 2019
Fitzgerald R, Pibida L (2018) Primary standardization of the massic activity of a 233Pa solution. J Radioanal Nucl Chem 318:149–155
Galea R (2018) Summary of results from the NRC standardization of LLNL supplied 233Pa and 231Pa. Internal Report, National Research Council Canada, Ottawa, Canada
Fitzgerald R, Colle R, LaRosa J, Nour S, Laureano-Perez L, Pidiba L (2018) Massic activity of a protactinium-233 solution determined by two methods. (Abstract 361) MARC XI, April 8–13, 2018, Kailua-Kona, Hawaii. http://www.marcconference.org/wp-content/uploads/marcxi_BookofAbstracts_20180410.pdf
Wilson RE (2014) Retrieval and purification of the an aged 231Pa source from its decay daughters. Radiochim Acta 102(6):505–511
Treinen KC, Gaffney AM, Rolison JM, Samperton KM, McHugh KC, Miller ML, Williams RW (2018) Improved protactinium spike calibration method applied to 231Pa–235U age-dating of certified reference materials for nuclear forensics. J Radioanal Nucl Chem 318:209–219
NNDC, 2011, Nuclear Wallet Cards. National Nuclear Date Center, Brookhaven National Laboratory, www.nndc.bnl.gov. Accessed 11 March 2019
BIPM, 2004, Monograph 5: Table of Radionuclides, 2-A: 151:242. http://www.nucleide.org/DDEP_WG/DDEPdata.htm. Accessed 11 March 2019
CRM 112-A (2010) Certificate of analysis CRM 112-A uranium (normal) metal assay isotopic standard. NBL Program Office, Argonne, IL. https://science.energy.gov/nbl/certified-reference-materials/prices-and-certificates/
Richter S, Goldberg SA (2003) Improved techniques for high accuracy isotope ratio measurements of nuclear materials using thermal ionization mass spectrometry. Int J Mass Specctrom 229:181–197
CRM U010, 2008, Certificate of Analysis CRM U010 uranium isotopic standard. NBL Program Office, Argonne, IL. https://science.energy.gov/nbl/certified-reference-materials/prices-and-certificates/
CRM U005-A, 2008, Certificate of Analysis CRM U005-A uranium isotopic standard. NBL Program Office, Argonne, IL. https://science.energy.gov/nbl/certified-reference-materials/prices-and-certificates/uranium-and-thorium-crms/
CRM 129-A, 2003, Certificate of Analysis CRM 129-A uranium oxide (U3O8) assay and isotopic standard, NBL Program Office, Argonne, IL. https://science.energy.gov/nbl/certified-reference-materials/prices-and-certificates/uranium-and-thorium-crms/
JCGM-Joint Committee for Guides in Metrology, 2008, Evaluation of measurement data – Guide to the expression of uncertainty in measurement. JCGM 100, 2008 (E/F)
Taylor BN, Kuyatt CE (1994) Guideline for evaluating and expressing the uncertainty of NIST measurement results. National Institute of Standards and Technology, Gaithersburg, MD, Technical Note 1297
JCGM-Joint Committee for Guides in Metrology (2012) International vocabulary of metrology-basic and general concepts and terms (VIM). JCGM 200:2012
ISO, 2017, International Organization for Standardization, Reference materials - Guidance for characterization and assessment of homogeneity and stability. ISO GUIDE 35:2017 (E)
ISO, 2016, International Organization for Standardization, General requirements for the competence of reference material producers. ISO 17034:2016 (E)
Kirby, H. W., 1959, The Radiochemistry of Protactinium. National Academy of Sciences National Research Council, Nuclear Series, (NAS-NS 3016)
Morss LR, Edelstein N, Fuger J, Katz JJ (2011) The chemistry of the actinide and transactinide elements. Springer, Dordrecht
De Bièvre P, Peiser HS (1993) Basic equations and uncertainties in isotope-dilution mass spectrometry for traceability to SI of values obtained by this primary method. Fresenius J Anal Chem 359:523–525
Milton MJT, Quinn TJ (2001) Primary methods for the measurement of amount of substance. Metrologia 38:289–296
Albarède F, Telouk P, Blichert-Toft J, Boyet M, Agraneir A, Nelson B (2004) Precise and accurate isotopic measurements using multiple-collector ICPMS. Geochim Cosmochim Acta 68:2725–2744
Acknowledgements
Roger Henderson at LLNL performed the initial purification of the 231Pa that was used as the starting material for this project. His expertise and assistance is gratefully acknowledged. Funding for project activities at LLNL and at NIST was provided by the United Stated Department of Homeland Security. Analyses performed at NRC and NPL were internally funded.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Human and animal rights
This article does not contain any studies with human or animal subjects performed by the any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Essex, R.M., Williams, R.W., Treinen, K.C. et al. Preparation and calibration of a 231Pa reference material. J Radioanal Nucl Chem 322, 1593–1604 (2019). https://doi.org/10.1007/s10967-019-06711-6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-019-06711-6