
Abstract
In a recent international exercise, 10 international nuclear forensics laboratories successfully performed radiochronometry on three low enriched uranium oxide samples, providing 12 analytical results using three different parent-daughter pairs serving as independent chronometers. The vast majority of the results were consistent with one another and consistent with the known processing history of the materials. In general, for these particular samples, mass spectrometry gave more accurate and more precise analytical results than decay counting measurements. In addition, the concordance of the 235U–231Pa and 234U–230Th chronometers confirmed the validity of the age dating assumptions, increasing confidence in the resulting conclusions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nuclear forensics
Over the past decade, nuclear forensics has developed into an integral part of a robust nuclear security program. It has been specifically identified in the Communiqués, Plans, and Joint Statements of the Nuclear Security Summits in 2010, 2012, 2014, and 2016 as an important focus area for research. The fundamental tenet underlying nuclear forensics is that identifying the origin of nuclear materials found outside of regulatory control can help identify gaps and weaknesses in the physical security and/or safeguards of a particular country or facility, which can then be strengthened in response. In addition, any country or organization that considers thwarting international law or practice by trafficking in such nuclear materials might be deterred by the knowledge that their complicity in such trafficking is likely to be identified [1]. The currently accepted definition of nuclear forensics is “the examination of nuclear or other radioactive materials or of evidence contaminated with radionuclides in the context of international or national law or nuclear security. The analysis of nuclear or other radioactive material seeks to identify what the materials are, how, when, and where the materials were made, and what were their intended uses [2].”
Radiochronometry
At the same time that the policy implications of nuclear forensics have become increasingly mature, laboratories around the world have continued to develop the underlying science, as well as the applications of that science to actual cases. The development of radiochronometry, or “age dating,” and its application to nuclear materials is one area of intense research [3,4,5,6,7,8,9]. Radiometric model ages are determined from measurements of parent and progeny isotopes (typically daughter, but also granddaughter) that accumulate in a material due to decay of the radioactive parent isotope. U- and Pu-series disequilibrium dating are most often used for nuclear forensic investigations. Calculated model ages are based on two fundamental assumptions: (1) the material was completely purified from decay products at the time it was produced, and (2) since the time that it was produced, the material has remained a closed system with neither gain nor loss of parent or progeny except through radioactive decay. The age of nuclear material is an important forensic signature because it can be used to constrain the time of purification or production (a predictive signature) and establish or eliminate potential genetic links among different samples of nuclear materials (a comparative signature). Ideally, the age inferred from the laboratory analysis of a sample (referred to as a “model age”) represents the actual production, processing, or purification age of the nuclear material of interest (referred to as a “sample age”). Model ages may, of course, differ from sample ages due to familiar sources of bias in the measurement process. However, model ages may also differ from sample ages because the sample production history is inconsistent with the model assumptions on which the chronometry determinations are based. Understanding the physical and chemical causes of these inconsistencies and developing experimental and theoretical approaches to address them is an active focus area of nuclear forensics research. Increasing emphasis is being placed on using multiple chronometers to better understand the chemical or physical processes that might “reset” certain chronometers.
The 4th collaborative materials exercise (CMX-4)
The 4th Collaborative Material Exercise (CMX-4), conducted by the Exercise Task Group of the Nuclear Forensics International Technical Working Group (ITWG) [10, 11], was designed to test the resolving power of isotope measurement techniques using low-enriched uranium (LEU) samples of very similar isotopic abundances [12]. However, based upon the outcomes of previous ITWG exercises, information provided by material characteristics other than isotopic abundances, including radiochronometic age, were identified to be of growing interest to the nuclear forensics community. In keeping with ITWG practice, the identity of individual laboratories is obscured through the use of designators or code names. For this exercise, the laboratories were given the name of a famous artist.
Experimental
Sample materials
Three exercise samples (designated as ES-1, ES-2, and ES-3) were generated from two sources of materials and distributed to each laboratory participating in CMX-4. Details regarding the history and preparation of these samples are provided in the introduction to this special section of the Journal [12].
Radiochronometers
234U–230Th chronometer
Because the half-life of 234U is relatively short (2.45 × 105 years) compared to the other naturally occurring uranium isotopes, 235U and 238U, the 234U–230Th chronometer is one of the most straightforward chronometers to measure, and certainly the most widely applied [3, 5, 6, 8, 13]. The chronometer becomes increasingly easy to measure as the 234U abundance, and hence the amount of its progeny, 230Th, increases. Because the 234U abundance tends to increase with 235U enrichment for most enrichment technologies, its ease of application increases with increasing enrichment of the sample. Since the samples for CMX-4 were LEU, measuring the 234U–230Th chronometer should have been relatively straightforward,—certainly easier than in natural uranium samples—although not as easy as with highly enriched uranium (HEU) samples.
The two most commonly applied techniques for quantifying the level of 234U and 230Th in uranium samples are mass spectrometry and alpha spectrometry. Due to the relatively long half-lives of 234U and 230Th, mass spectrometry, as an atom counting technique, normally provides lower detection limits and greater precision than alpha spectrometry, which is a decay counting technique. However, an alpha spectrometer is a relatively inexpensive instrument that can be found in many laboratories that do not have the necessary mass spectrometer. Note that both thermal ionization mass spectrometers (TIMS) and inductively coupled plasma mass spectrometers (ICP-MS), which may be equipped with either a single or multiple collector(s), can be used for age dating. The utilization of these techniques and instruments for age dating in CMX-4 are given in Table 1.
Regardless of technique, isotope dilution is typically used for quantification. With this method, quantification is achieved by measuring the isotope of interest (e.g., 234U) relative to a spike or tracer isotope (e.g., 233U). However, in CMX-4, two laboratories used unspiked isotopic analysis in combination with other assay techniques, delayed neutron counting (Cezanne) and external calibration using NIST SRM 4321C (Manet), to calculate the 234U concentration necessary for age dating. For those laboratories that used isotope dilution for U quantification, seven laboratories used 233U spikes, one laboratory used a 232U spike, and another used the NBL U930 standard as 235U spike. For Th quantification, eight laboratories used 229Th, while the remaining three laboratories used 232Th, as isotopic spikes for Th quantification.
All three laboratories that performed alpha spectrometry used chemical separation of the Th from the U, followed by fluoride microprecipitation to create plates for alpha counting. Both TIMS and ICP-MS also typically require chemical separation of Th from the U matrix prior to analysis. This is necessary to improve ionization efficiency (TIMS) and reduce matrix effects, low-mass tailing effects, and memory effects that may result from loading the instrument with high concentrations of U (ICP-MS). However, one of the laboratories used a procedure published by Varga et al. [13], which offers an offline correction for the peak tailing effect and provides accurate Th results using ICP-MS. Purification of a bulk U sample prior to U analysis by TIMS or ICP-MS may not be necessary, depending on the purity of the sample. If the bulk sample is sufficiently pure, then molecular isobaric interferences in the U mass range, for example, 232ThH+, will be insignificant. In addition, tailing to nearby masses and hydride formation usually occur in the range of 10−5–10−6 [14], there will not be a need for correction due to tailing or hydride formation from 232Th on m/z = 233.
For this exercise, all laboratories separated and purified the Th fraction prior to analysis by alpha spectrometry or mass spectrometry. However, only three laboratories purified the U fraction (two from the bulk solution; one from the U fraction after separation from Th prior to analysis by alpha spectrometry). Seven laboratories used a TEVA column to separate the U and Th; three laboratories performed a subsequent purification using another TEVA column. Three laboratories used a multi-step purification starting with an anion exchange column. Cezanne used an anion exchange column followed by a TRU-Spec column, and finished with a cation exchange column. Gauguin used an anion exchange column, followed by a TEVA column, and finished with another anion exchange column. Bondone used back-to-back anion-exchange columns, the first of 120 µm particle size and the second of 25 µm particle size. Monet used a separation procedure based upon a macroporous Lewatit MP5080 ion-exchange resin. All procedures used by the different participants for the 234U–230Th chronometer can be found in Table 2.
235U–231Pa chronometer
Even though 235U is much more abundant than 234U in almost all uranium materials, the much longer half-life of 235U (7.04 × 108 years) and the absence of a long-lived spike isotope for Pa has limited routine application of this chronometer until relatively recently. Although radiometric techniques (gamma spectrometry, alpha spectrometry) have been used to quantify 231Pa for age dating in the past [15], the only participant in CMX-4 to apply the 235U–231Pa chronometer (Gauguin) used mass spectrometry, according to the procedure developed by Eppich et al. [4]. The key feature of the method is the use of a 233Pa spike for quantifying 231Pa. Due to the short half-life of 233Pa (26.97 days), no certified 233Pa reference material exists; neither does a certified 231Pa reference material that may be used for a newly prepared 233Pa spike. Rather, the spike must be prepared immediately prior to use and calibrated for 233Pa concentration (atoms of 233Pa g−1), typically using a geologic material as a secular equilibrium standard, for its working-lifetime of approximately 3–4 months.
234U–214Bi chronometer
Age dating U samples using high-resolution gamma spectrometry (HRGS) [16,17,18,19,20] has the advantage of being nondestructive, hence no sample preparation is required. Analogous to the 234U–230Th chronometer the progenies of 234U are used, however nuclides detectable by gamma-spectrometry must be chosen. For practical reasons 214Bi–234U ratio is used based on the secular equilibrium between 214Bi ↔ 226Ra. The method does not require the use of reference materials of known ages. It is most suitable for measuring U samples of higher enrichments and older ages (higher 214Bi levels).
Results and discussion
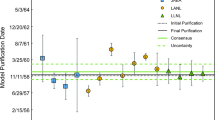
The age dating results for CMX-4 are presented in Table 3 and Fig. 1 (Sample ES-1), Table 4 and Fig. 2 (Sample ES-2), and Table 5 and Fig. 3 (Sample ES-3). All uncertainties are expanded combined standard uncertainties with k = 2. For ES-1 and ES-3, the material processing date is encompassed by a 1-year range (2004), delineated by dashed red lines on the figures, during which the samples were reported to have been manufactured. For ES-2, the material processing date is encompassed by a range extending from the known enrichment date (February 12, 2002) to the known manufacturing date (September 24, 2002), which is the pelletization process. We expect the actual sample material processing date to be the date of conversion from enriched UF6 to UO2, since neither Th nor Pa form volatile fluorides and would, therefore, be expected to deposit out and remain in the cylinder during UF6 release. The conversion date for these samples is unknown, but must have occurred sometime between enrichment and manufacturing of the fuel pellets. For the 234U–214Bi chronometer, an upper limit of ≈ 11 years was estimated uniformly for the three samples from the 214Bi activity corresponding to the detection limit, because of the low enrichment and age of the samples.

Age dating results for ES-1 compared against known sample processing history. The dashed red lines indicate the range of probable processing dates according to the known history of the samples (see text). Results using gamma spectrometry are indicated by black triangles; a blue arrow indicates a method detection limit and extends from the age determined from the detection limit to younger ages. Alpha spectrometry results are indicated by red squares. Mass spectrometry results are indicated by blue diamonds. (Color figure online)

Age dating results for ES-2 compared against known sample processing history. The dashed red line indicates the pellet production date; the dotted red line indicates the uranium enrichment date. Results using gamma spectrometry are indicated by black triangles; a blue arrow indicates a method detection limit and extends from the age determined from the detection limit to younger ages. Alpha spectrometry results are indicated by red squares. Mass spectrometry results are indicated by blue diamonds. (Color figure online)

Age dating results for ES-3 compared against known sample processing history. The dashed red lines indicate the range of probable processing dates according to the known history of the samples (see text). Results using gamma spectrometry are indicated by black triangles; a blue arrow indicates a method detection limit and extends from the age determined from the detection limit to younger ages. Alpha spectrometry results are indicated by red squares. Mass spectrometry results are indicated by blue diamonds. (Color figure online)
Interpretation of results
Upon reviewing the results, the first conclusion is that most of the laboratories measured ages consistent with one another and consistent with the known history of the material. For ES-1 and ES-3, only two of 12 results did not overlap the known history of the material within their stated analytical uncertainty. For ES-2, only three of 12 results did not overlap the known history of the material within their stated analytical uncertainty.
The second conclusion is that these age-dating results were useful in comparing the three samples (ES-1, ES-2, and ES-3) with one another, one of the key points of the exercise. When combined with other signatures, including the similarity of the U isotopic composition between ES-1 and ES-3, these age-dating results supported the conclusion that ES-1 and ES-3 might have had a similar processing history. Again, when combined with other signatures, these age-dating results also supported the conclusion that ES-2 was a different material from ES-1 to ES-3, sharing neither isotopic similarity nor processing history.
The third conclusion is that mass spectrometric methods of measuring these chronometers provided more accurate and precise results than counting methods offered for these samples. Picasso reported that the level of enrichment in these samples was too low to measure more than an upper limit on the age using gamma spectrometry and the 214Bi–234U chronometer. In addition, those laboratories that used alpha spectrometry to perform chronometry provided results that tended to be either less accurate or less precise than results provided by laboratories that used mass spectrometry. In particular, for ES-2, none of the alpha spectrometry results agree with the stated sample history or with one another. Following the conclusion of the exercise, Rembrandt (who conducted only a single U/Th column separation to generate their Th alpha sources) further purified their Th alpha sources. The repurified sources yielded comparable ages to mass spectrometry. This finding clearly demonstrates the need for multiple U/Th separation steps to adequately remove 234U from the 230Th spectral region.
The final conclusion, drawn from Gauguin’s results, is that the consistency of the 234U–230Th and 235U–231Pa results validated the age-dating assumption, namely that there was a real purification event that removed Th and Pa from the U quantitatively. It is highly unlikely that there would be a process that would partially purify both Th and Pa, but in a proportion that retained the concordancy of the two chronometers. This is a different conclusion than that of ITWG round robin 3, in which, due to the complex production history of the HEU metal samples, these two chronometers differed remarkably from one another [7, 21, 22]. Knowing the true material processing date (assumed to be the conversion date) would be helpful in a real investigation in identifying potential material sources and excluding others.
Conclusions
In the CMX-4 exercise, 10 international nuclear forensics laboratories successfully performed radiochronometry on the three CMX-4 samples (ES-1, ES-2, and ES-3), providing 12 analytical results using three different parent-daughter pairs serving as independent chronometers (234U–230Th, 235U–231Pa, 234U–214Bi). The vast majority of the results were consistent with one another and consistent with the known processing history of the materials. In general, for these particular samples, mass spectrometry gave more accurate and more precise analytical results than decay counting measurements. In addition, the concordancy of the 235U–231Pa chronometer with the 234U–230Th confirmed the validity of the age dating assumption, increasing confidence in the nuclear forensic conclusions, and in the model age in particular. When combined with other analytical results, age dating helped confirm a relationship between ES-1 and ES-3 and a lack of relationship between ES-2 and ES-1 and ES-3.
References
Allison G (2008) Nuclear deterrence in the Age of Nuclear Terrorism. Tech Rev 14(1):68–73
IAEA (2015) Nuclear forensics in support of investigations, iaea nuclear security series no. 2-G (Rev. 1). International Atomic Energy Agency, Vienna
LaMont SP, Hall G (2005) Uranium age determination by measuring the 230Th/234U ratio. J Radioanal Nucl Chem 264(2):423–427
Eppich GR, Williams RW, Gaffney AM, Schorzman KC (2013) 235U–231Pa age dating of uranium materials for nuclear forensic investigations. J Anal Atom Spectrom 28:666–674
Gaffney AM, Hubert A, Kinman WS, Magara M, Okubo A (2015) Round-robin 230Th–234U age dating of bulk uranium for nuclear forensics. J Radioanal Nucl Chem. https://doi.org/10.1007/s10967-015-4334-8
Williams RW, Gaffney AM (2011) 230Th–234U model ages of some uranium standard reference materials. Proc Radiochim Acta 1:31–35
Kayzar TM, Williams RW (2015) Developing Radium-226 and Actinium-227 age-dating techniques for nuclear forensics to gain insight from concordant and non-concordant radiochronometers. J Radioanal Nucl Chem 307(3):2061–2068
Wallenius M, Morgenstern A, Apostolidis A, Mayer K (2002) Determination of the age of highly enriched uranium. Anal Bioanal Chem 374:379–384
Varga Z, Mayer K, Bonamici CE, Hubert A, Hutcheon I, Kinman W, Kristo M, Pointurier F, Spencer K, Stanley F, Steiner R, Tandon L, Williams R (2015) Validation of reference materials for uranium radiochronometry in the frame of nuclear forensic investigations. Appl Rad Isotopes 102:81–86
Niemeyer S, Koch L (2003) The nuclear smuggling International Technical Working Group: Making a difference in combating illicit trafficking. In: Proceedings from the conference on advances in destructive and non-destructive analysis for environmental monitoring and nuclear forensics, Karlsruhe, Germany, October 21–23. International Atomic Energy Agency, Vienna
Smith DK, Biro T, Chartier B, Mayer K, Niemeyer S, Thompson P (2007) Recent activities of the nuclear smuggling international technical working group to thwart illicit trafficking. In: Proceedings of the iaea conference on illicit nuclear trafficking, Edinburgh, Scotland, November 19–22, 2007. International Atomic Energy Agency, Vienna
Schwantes JM, Marsden O, Reilly D (2018) Fourth collaborative materials exercise of the nuclear forensics international technical working group. J Radioanal Nucl Chem 311:1441–1452
Varga Z, Wallenius M, Mayer K, Hrnecek E (2011) Alternative method for the production date determination of impure uranium ore concentrate samples. J Radioanal Nucl Chem 290:485–492
Deschamps P, Doucelance R, Ghaleb B, Michelot J-L (2003) Further investigation on optimized tail correction and high-precision measurement of uranium isotopic ratios using multi-collector ICP-MS. Chem Geol 201:141–160
Morgenstern A, Apostolidis C, Mayer K (2002) Age determination of highly enriched uranium: separation and Analysis of 231Pa. Anal Chem 74:5513–5516
Mayer K, Wallenius M, Varga Z (2013) Nuclear forensic science: correlating measurable material parameters to the history of nuclear material. Chem Rev 113:884–900
Nguyen CT (2005) Age-dating of highly enriched uranium by gamma-spectrometry. Nucl Instrum Method B 229:103–110
Nguyen CT, Zsigrai J (2006) Gamma-spectrometric uranium age-dating using intrinsic efficiency calibration. Nucl Instrum Method B 243:187–192
Nguyen CT, Zsigrai J (2006) Basic characterization of highly enriched uranium by gamma spectrometry. Nucl Instrum Method B 246:417–424
Nguyen CT, Zsigrai J, Lakosi L (2014) Uranium age dating by gamma spectrometry. In: Advances in nuclear forensics: countering the evolving threat of nuclear and other radioactive material. summary of an international conference, IAEA, Vienna, Austria, 7–10 July 2014, Proc. Series 2015, CD-ROM, IAEA-CN-218/013
Hanlen R. (2011) Round robin 3 exercise after action and lessons learned report, PNNL-20079. Pacific Northwest National Laboratory, Richland, Washington. http://www.nf-itwg.org/sites/default/files/pdfs/Round_Robin_3_Final_Report.pdf
Kristo MJ, Tumey SJ (2013) The state of nuclear forensics. Nucl Instrum Method B 294:656–661
Acknowledgements
All participants acknowledge the Nuclear Forensics International Technical Working Group, in particular, the co-chairs of the Exercise Task Group, Jon Schwantes and Olivia Marsden, for organizing a successful international exercise. Some of this work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344 with funding from the U.S. Federal Bureau of Investigation. This work was supported by the Nuclear Safety Research program through the Korea Foundation of Nuclear Safety (KOFONS), granted financial resource from the Nuclear Safety and Security Commission (NSSC), Republic of Korea (No. 1405020).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kristo, M.J., Williams, R., Gaffney, A.M. et al. The application of radiochronometry during the 4th collaborative materials exercise of the nuclear forensics international technical working group (ITWG). J Radioanal Nucl Chem 315, 425–434 (2018). https://doi.org/10.1007/s10967-017-5680-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-017-5680-5