
Abstract
Two histidines were introduced by site-directed mutagenesis into the structure of Enhanced Green Fluorescent Protein, replacing the serine at position 202 and the glutamine at position 204 for increasing the sensitivity of the protein towards different metal ions by creating possible metal binding sites near the chromophore group. There is no appreciable difference between the absorbance and fluorescence spectra of the two proteins (wild type and the double-histidine mutant) indicating that the mutation does not change the environment of the fluorophore. Fluorescence quenching was measured at different pH (6.5–8) and temperatures (20–45 °C) varying the concentration of metal ions. Under optimal conditions (pH = 7.5, 20 °C) the mutant’s Kd is 16 nM, it binds copper more than 200fold stronger than the wild type EGFP.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Copper is an essential trace element and a catalytic cofactor for many enzymes [1]. Copper ions exist in to oxidation states; +1 and +2 and participates in several red-ox reactions in cells. The concentration of exchangeable copper ions in human blood is in micromolar range (about 8 μM) [2]. Copper imbalance can have an impact on the central nervous system; may lead to vascular dementia, Alzheimer’s and Parkinson’s disease [3]. Overload of cooper ions can cause overproduction of hydroxy radical and other reactive oxygen species (ROS) [4], and on this basis it can induce protease dependent cell death [5]. In our society the wide spread use of copper containing substances—from electro technical industry to agriculture—seriously increased the risk of copper pollution so monitoring the exact quantity of copper in blood, food, and drinking water is an important issue of public health. Besides the classical atom absorption spectroscopy several high-tech detection methods were developed for qualitative and quantitative copper detection, such as evanescent wave infrared absorption spectroscopy [6], electrochemical detection by modified copolymer electrodes [7], and the use of capillary microchips [8]. There are several methods utilizing fluorescent techniques [9–12] as well. These are sensitive and selective methods but it is cumbersome to apply them to determine intracellular copper concentration. The family of fluorescent proteins can be widely applied to detect changes within living cells, because the protein does not have any adverse effect on cellular functions [13]. Copper ions have a quenching effect on the fluorescence of Green Fluorescent Proteins, so it is conceivable to develop GFP as useful sensor even for intracellular applications.
In the past decade green fluorescent protein (GFP) and its descendants became a commonly used tool in cell biology, protein engineering, medicine, etc. GFP was first isolated by O. Shimomura [14] from the jellyfish Aequorea victoria. It was the first green fluorescent protein of which primary structure was described and cloned [15]. The protein has a unique structure with an 11-stranded β- barrel enclosing a chromophore group located on a distorted α-helix in the center of the molecule. Chromophore formation occurs spontaneously through a self-catalyzed intramolecular cyclization of the three amino acids forming the chromophore group [16]. Several experiments showed that the entire structure of the protein is needed for chromophore formation and fluorescence [17].
The wild type GFP has properties which need to be improved, such as low brightness, delay of chromophore formation. Thus the protein was subjected to several mutations in order to improve these characteristics. Wild type GFP was enhanced (EGFP) by modifications concerning the excitation peaks and the brightness. The absorbance peak of the wild type GFP is at 395 and one at 475 nm [18]. EGFP has two mutations (F64L, S65T) in the chromophore region, and its spectral characteristics are improved [19], being among the brightest and most photostable fluorescent proteins [18]. Many improved versions of the GFP are present with enhanced properties such as color-shifted variants with different spectral properties [20] emitting colors from blue to yellow region of the visible spectrum
Several studies aimed to increase the metal binding potential of GFP by examining the protein’s structure and designing several mutations (see Discussion). Cu2+ efficiently decreases the fluorescence intensity of GFP while in the presence of other metal ions (Zn2+, Mn2+) fluorescence intensity is enhanced [21].
In this study we present a mutant of EGFP with increased sensitivity toward copper(II) ions. Using site directed mutagenesis, a metal binding site near the protein’s chromophore group was created by substituting two amino acids for histidines. We show that these mutations do not modify the environment of the chromophore. On the basis of titration with copper ionsdissociation constants were determined at different pH-s and temperatures.
Materials and Methods
Bacterial Strains and Plasmids, Site-Directed Mutagenesis
For expression of the EGFP and its mutant, pET15bEGFP vector was used, as described earlier [22]. The same vector was used as a template for site-directed mutagenesis, then for high scale protein production. Site-directed mutagenesis was performed by using the Quickchange Site Directed Mutagenesis kit from Stratagene with specific primers. Mutation was verified by sequencing.
Protein Expression and Purification
Transformation of chemically competent BL21(DE3)STAR cells with the wild type and mutant vectors was carried out by heat-shock. Transformed cells were plated on LB agar containing 100 μg/ml ampicillin, and incubated overnight at 37 °C. Colonies were verified under UV light for fluorescence.
A single green colony was inoculated in 5 ml LB broth (10 g tryptone, 5 g yeast extract, 5 g NaCl, pH 7.5) supplemented with 100 μg/ml ampicillin and incubated for 5 h at 37 °C at 250 rpm. Cells were diluted to 1:100 in 500 ml LB broth with ampicillin and incubated overnight with shaking at 37 °C. Under our condition the system requires no induction. The host cells contain no pLys plasmid and the leaky expression provides satisfactory yield (25–30 mg/l) of EGFP. Cells were collected by centrifugation, resuspended in 10 ml of MOPS buffer pH 7.5 supplemented with 0.25 g/ml lysozyme and freezed at −80 °C for 2 h. To completely disassociate the cellular components, cell suspension was melted and sonicated 5 cycles of 10 s at 70 % amplitude, with 10 s pause between the cycles. This procedure was accomplished on ice to prevent excess heat formation and denaturation of the protein. Soluble proteins were obtained by centrifugation for 15 min at 4 °C, 20000 rpm.
Protein purification was performed by immobilized metal (Ni2+) affinity chromatography (IMAC) using the N-terminal His-tag of the proteins. Purified proteins were dialyzed against 10 mM MOPS buffer pH 7.5 in order to remove the imidazole and other impurities from the solution. 0.5 mg/l cation exchange Amberlite CG50 resin was also added to the dialysis solution to remove residual metal ions from the protein solution. Protein concentration was determined from absorption at 488 nm using ε = 55000 M−1 cm−1 [7, 21].
Sample preparation for electrophoresis was conducted according to Laemmli [23], and the produced proteins separated on a 15 % SDS-PAGE were visualized by Coomassie staining. Absorbance spectrum of the two proteins was determined from 240–600 nm.
Fluorescence Measurements
Fluorescence spectra were recorded with FluoroMax Spex 320 fluorimeter at 20 °C. Fluorescence quenching was measured with the FluoStar Optima fluorescence plate reader using 485 nm filter for excitation and 520 nm filter at emission. Protein samples were diluted into 10 mM MOPS buffer of several pH-s (6.5, 7, 7.5, 8) to 100–340 nM final concentration. pH of buffers was set at 25 C taking into consideration the temperature dependence of MOPS buffer (ΔpKa/ΔT = −0.015 [24]). Different final concentrations of CuSO4 (in the range from 0.001 to 30 μM) were added to the protein solutions, and samples were incubated on water bath at different temperatures (25, 30, 35, 40, 45 °C) for 30 min for protein-metal complex stabilization. Fluorescence intensity was recorded directly, and buffer corrected. All experiments were performed in triplicates using at least to different preparates of each protein (wild type, double-histidine mutant).
Fluorescent quantum yields were determined by the comparative method [25]. For quantum yield measurements concentration of wtEGFP and double-histidine mutant (EGFP S202H/Q204H) in assay buffer and that of fluorescein (Molecular Probes, F1300) in 0.1 M NaOH was adjusted so that all samples have identical absorbances at 480 nm. Then serial dilutions of samples were prepared and the absorbance of the diluted samples was calculated. Fluorescence emission spectra were recorded with excitation at 480 nm, and corrected using the correction factors supplied with the instrument. Baseline corrected fluorescence emission spectra were integrated from 490 to 650 nm and the integrated intensities were plotted against the calculated absorbances. The plots gave straight lines with intercepts = 0 and R2 > 0.999. The gradients of the graphs are proportional to the quantum yield of the different samples. The quantum yield of the fluorescent proteins was calculated by dividing the slope of the test samples by that of fluorescein and by using the known fluorescence quantum yield of fluorescein (0.90) [26].
Data Processing, Calculation of Dissociation Constants
The recorded fluorescence intensity values were averaged and processed further depending on the method used for determination of dissociation constants. As first approximation we used the fitting program OneSiteBind of Origin. This program fits a rectangular hyperbola to data points. For this we derived “extent of quenching” by subtracting intensities of copper containing samples (ICu) from the value of samples devoid of copper (I0). It should be noted that this approximation is true under the assumption that the fraction of bound Cu2+ is negligible. In case of strong binding when the dissociation constant is smaller than the protein concentration at low Cu2+ concentration this assumption does not come true we used a version of the familiar equation derived from the quadratic formula [27] where ICu/I0 is the relative intensity, Kd is the dissociation constant, [Cu] and [P] are total concentrations of copper ion and protein, respectively.
Results
In Silico Modeling
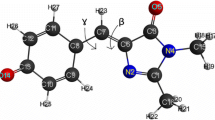
In order to find possible metal binding site in the structure of EGFP, crystal structure of the protein was investigated. Our strategy was to find sites where insertion of histidines would create potential metal binding site. The main criteria were the distance between the chromophore group and the potential locus for mutation. A distance of less than 5 Ǻ [21] is required for a significant effect to occur, the location and distance of the imidazole ring needs to be close to the chromophore group. PyMol [28] was used for visualizing the mutation by in silico changing two amino acids into histidine. Analyzing the 3D crystal structure of EGFP [29] a potential metal binding site was found in the proximity of the chromophore region. We supposed that replacement of serine at position 202 and glutamine at position 204 by histidines would create favorable environment for metal binding. 3D structure of the wild type EGFP (wtEGFP) and the double-histidine mutant (EGFP S202H/Q204H) created by PyMol can be seen on Fig. 1. The β-barrel structure encloses the chromophore group, and the mutated residues are highlighted as sticks.

Structure of wtEGFP (1emm) (a) and molecular model of the double histidine mutant EGFP S202H/Q204H (b). The model was constructed by PyMol [28]. Residues at the positions of the mutations are shown as sticks
Protein Expression and Purification
Protein production and the purity of the sample after purification were verified with SDS-PAGE (Fig. 2). After purification on an immobilized Ni-column, a single band can be observed with a molecular weight of 29 kDa. Absorbance spectra of the two proteins were recorded to compare eventual alterations occurred as a consequence of the mutagenesis. The results (Fig. 3) show that there is no significant difference between the two spectra, thus the mutation does not have any effect on the spectral properties of the EGFP. The figure also illustrates the purity of the samples: the ratio A488/A280 is higher then 1.7 [18], indicating low level of contamination absorbing in the ultraviolet region.

Demonstration of expression and purification of EGFP on SDS-PAGE by Comassie staining. Lane 1: Low molecular weight marker (M3913 Sigma), Lane 2: total cells containing the wtEGFP before cell disruption, Lane 3: total cells containing the EGFP S202H/Q204H before cell disruption, Lane 4: wtEGFP after purification by IMAC (Immobilized Metal Ion Affinity Chromatography), Lane 5: EGFP S202H/Q204H after purification by IMAC

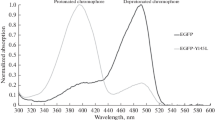
Overlaid normalized absorbance spectra of wtEGFP and the EGFP S202H/Q204H. The maximum at 488 nm corresponds to the chromophore, maximum at 280 nm, to amino acids with aromatic ring
Fluorescence Measurements
Fluorescence spectra of wtEGFP and EGFP S202H/Q204H were recorded as shown in Fig. 4. The spectra were completely identical. Excitation maxima were at 488 nm whereas the maximum of emission was at 507 nm. Interestingly strong emitted light can be detected when excitation wavelength was 280 nm indicating energy transfer from aromatic residue(s), presumably tryptophan to the chromophore.

Fluorescence spectra of wtEGFP (a) and EGFP S202H/Q204H (b). The spectra were recorded at 20 °C in 10 mM MOPS buffer, pH 7.5
It is well known that the fluorescence intensity of EGFP depends on the pH, it falls with decreasing pH with a pKa of 6 [30]. To check further eventual effects of the double histidine mutation on the properties of EGFP S202H/Q204H we measured the pH dependence of fluorescence intensity as well. Again we found no difference (Fig. 5), the pKa of the decrease of fluorescence for both proteins is 6.2. The evidences listed above indicate that mutation at positions 202 and 204 are well tolerated, they do not induce significant structural changes.

Normalized fluorescence intensities of wtEGFP (closed symbols) and EGFP S202H/Q204H (open symbols) at different pH. Data were normalized for the pH = 8 values. Solid line corresponds a titration curve with pKa = 6.2
Cu Titration
To determine the quenching efficiency of copper ion on the fluorescence of EGFP and its double histidine mutant the proteins were incubated with CuSO4. Fluorescence quenching detected for EGFP proteins in the presence of copper has no effect on the position of the emission wavelength maximum as shown in Fig. 6. Spectra show that copper ions efficiently quench fluorescence and the sensitivity of mutant protein considerably increased.

Emission spectra of wtEGFP (a) and EGFP S202H/Q204H (b) in 10 mM MOPS at pH 7.0 in the presence of different concentration of Cu2+. Fluorescent proteins at concentration of 100 nM were incubated with 0.2–30 μM CuSO4 in case of wtEGPF and with 0.01–15 μM CuSO4 in case of EGFP S202H/Q204H for 30 min at 20 °C and spectra were recorded at the same temperature. Excitation wavelength was 470 nm
The same experiment was also performed with fluorescent plate reader with different protein preparations. Fluorescent intensities were processed and analyzed as described in Methods. Figure 7 shows the comparison of the results obtained when the single binding isotherm or the quadratic formula was fit to the same set of data. In the case of wild type EGFP the calculated dissociation constants agree within the experimental error; 4.52 ± 0.77 μM for the hyperbolic equation and 3.92 ± 0.73 μM for the quadratic formula. In the case of double histidines mutant there is a significant difference, the quadratic formula yields more the one order of magnitude lower value: 0.015 ± 0.009 μM vs. 0.26 ± 0.025 μM obtained by the hyberbolic fitting. Kd values for wtEGFP and EGFP S202H/Q204H derived from measurement at different pH and temperature are listed in Table 1 and Table 2, respectively. The values in Table 1 and in Table 2 were obtained by fitting the hyperbolic equation, and quadratic equation, respectively.

Demonstration of the importance of using the correct fitting formula. The same data set was fitted to the hyperbolic equation (a and b) or to the quadratic equation (c and d). The solid lines show the fitted curves. The derived dissociation constants for wtEGFP (a and c) agree well, but in the case of EGFP S202H/Q204H correct values can be obtained only by the quadratic fitting (cf Fig. 6)
To obtain more information about the nature of the putative copper ion binding group(s) we measured quenching at different pH from 6.5 to 8 (Fig. 8). Unfortunately the low intensity values made measurement unreliable below 6.5 but even from this limited set it was evident that the binding weakens as pH drops. This effect is more pronounced as temperature rises. There was no significant difference between wild type and EGFP S202H/Q204H (Fig. 8a and b), although the curves of EGFP S202H/Q204H might indicate somewhat higher pKa for the transition.

pH dependence of the dissociation constants at different temperatures for wtEGFP (a) and for EGFP S202H/Q204H (b)
From the temperature dependence of the binding constants we determined the standard enthalpy of Cu2+ binding using the van’t Hoff plot (Fig. 9). As expected the process is exotherm for both wtEGFP and EGFP S202H/Q204H. At 20 °C and pH = 7.5 the standard enthalpy of binding (ΔH0) is −17.9 and −7.2 kcal/mole for the mutant and wild type EGFP, respectively.

van’t Hoff plots for wtEGFP (a) and EGFP S202H/Q204H (b). Dissociation constants for wtEGFP and EGFP S202H/Q204H were determined by hyperbolic and quadratic fitting, respectively
Discussion
We expressed wtEGFP and a double histidine mutant (EGFP S202H/Q204H) in E. coli system. The spectral properties of the wild type and mutant were identical, showing that mutations exerted no significant change in the immediate surroundings of the chromophore. At the same time the quantum yield of the mutant was somewhat lower than that of the wild type; 0.58 ± 0.022 vs 0.62 ± 0.12. Both values agree well with the previously reported value for EGFP ( 0.60 [31]).
We found however that these mutations made EGFP significantly more sensitive for fluorescence quenching by Cu2+ ions and it is reasonably to conclude that this improvement is due to binding of copper ions to the imidazole moieties of the inserted histidines.
Earlier reports have shown that the red fluorescent protein (DsRed) [32] has intrinsic copper(II) ion binding activity. This binding results in quenching of fluorescence. Interestingly the reported affinities differ widely. Dissociation constants for drFP583 (native DsRed) was reported as 14.8 μM [33] for DsRed-Monomer as 1.7 μM [34] and for DsRed as 0.54 μM [35]. These differences might follow from different experimental conditions and data processing methods, but there was a general observation in these works that under similar conditions GFP showed lower sensitivity for quenching by copper. At the same time Richmond et al [36] have shown that introducing histidines and acidic residue nearby the chromophore GFP can significantly enhance copper binding affinity. In this work a variant of GFP (“10C”, T203Y/S65G/V68L/S72A) was used as the starting point for design, and S147 and Q204 were replaced by histidines and S202 was replaced by an acidic residue. Although no dissociation constants were calculated, Fig. 4 [36] showed that the mutants were quenched more than three orders of magnitude lower metal concentrations than is the wild-type 10C protein. Nevertheless, 50 % quenching of 10C-GFP required higher than 1 mM Cu2+ concentration. This value is dubious because of the presence of hexa-histidine affinity tags (see below).
Recently Hötzer et al. reported that the presence of copper(II) ions leads to a decrease of the fluorescence lifetime (τfl) of several GFP variants. Their meticulous analysis resulted in Kd values for GFP variants T203H, T203V, eYFP, and S65T as 9 μM, 2 μM, 3 μM, and 11 μM, respectively [37]. The investigated proteins all contained hexa-histidine tag; moreover the authors identify the His6-tag as the responsible binding site for Cu2+.
The reported dissociation constants are in good agreement with our results, we got Kd of about 4 μM at pH = 7.5 at 20 °C for the copper(II) binding of wtEGFP containing hexa-histidine tag (Table I). At the same time our mutant EGPF binds copper(II) about 200fold better then wtEGFP at pH = 7.5 and 20 °C. (Table II). Under optimal conditions (10 mM MOPS pH = 7.5, 20 °C) 10 nM Cu+2 induces more then 10 % decrease in intensity, so the limit of detection is well below 1 ppb (1 ppb for Cu2+ is 15.7 nM). To our knowledge it is the lowest reported copper(II) binding constant found for a fluorescent protein. It should be noted that the sites mutated in our work; positions 202 and 204 were also changed by Richmond et al. [36] albeit not two histidines but a histidine and an acidic residue (Asp or Glu) were inserted. This might be the major reason why our mutant is more effective; a His and an Asp or Glu might form an ion pair instead of binding a metal ion. Interestingly, among others positions 202 and 204 were also changed to acidic residues in a work aimed to transform EGFP into an efficient Ca2+ biosensor [38]. These results point to the fact that these positions can be utilized to build metal binding sites.
Although our limited pH dependence studies do not allow identification of the nature of binding residues, they do not contradict the assumption that copper(II) binding of the wild type occurs through the hexa-histidine tag [37]. For EGFP S202H/Q204H it is reasonable to suppose, that the newly introduced histidines serve as binding site for copper. Further studies including X-ray crystallography are planned to clarify this issue.
Analysis of the temperature dependence the dissociation constants (Fig. 9) yielded standard enthalpy for binding for both proteins. As expected both values are negative, the one for the mutant being about 2.5 larger than that of the wild type. Knowledge of these approximate values will be of help in designing the conditions (concentration, temperature) of our planned isothermal titration calorimetric measurements aimed to determine the exact stoichiometry of copper binding.
Finally, our data analysis draws attention to the often neglected fact that fitting the hyperbolic equation describing a binding isotherm is justified only if the concentrations of free ligand can be treated to be equal to their total concentrations or the free ligand concentrations can be precisely determined from experiments. The first assumption is satisfied when the wild type EGFP is titrated with Cu2+ (Fig. 6a) but evidently not in the case of the mutant (Fig. 6b). This problem can be solved if the experimental data are fitted to an explicit mathematical expression presented in terms of the total ligand concentration. For the simplest one-site model, the mathematical expression for describing the equilibrium system is a quadratic equation (see Eq 1; [27]). The correct value of dissociation constant can be obtained only by this way. Both methods of analysis assume a single binding site for copper ions. It might be valid for EGFP but it is evidently cannot be true in the case of EGFP S202H/Q204H since it is unlikely that wild type EGFP binds copper ions by the same residues (Ser202 and Gln204) where the insertion of histidines occurred, i. e. the new binding site eliminates the old one. Therefore the mutant contains at least two different binding sites and it would require a more complicated evaluation method [39]. Fortunately the binding affinities of the two sites differ by about two orders of magnitude so dissociation constant of the stronger sites can be derived with satisfying accuracy by the one-site model too.
References
Tapiero H, Townsend DM, Tew KD (2003) Trace elements in human physiology and pathology. Copper. Biomed Pharmacother 57(9):386–98
Kramer ML et al (2001) Prion protein binds copper within the physiological concentration range. J Biol Chem 276(20):16711–9
Squitti R et al (2011) Copper status in Alzheimer’s disease and other neurodegenerative disorders: genetics, mechanisms, neurophysiology, and therapies. Int J Alzheimers Dis 903–940
Urbanski NK, Beresewicz A (2000) Generation of *OH initiated by interaction of Fe2+ and Cu + with dioxygen; comparison with the Fenton chemistry. Acta Biochim Pol 47(4):951–62
Arnal N, de Alaniz MJT, Marra CA (2012) Cytotoxic effects of copper overload on human-derived lung and liver cells in culture. Biochim Biophys Acta (BBA)—General Subjects 1820(7):931–939
Huang GG, Yang J (2003) Selective detection of copper ions in aqueous solution based on an evanescent wave infrared absorption spectroscopic method. Anal Chem 75(10):2262–9
Lin M et al (2009) Electrochemical detection of copper ion using a modified copolythiophene electrode. Electrochim Acta 54(27):7012–7017
Yang M et al (2010) Detection of copper ion with laser-induced fluorescence in a capillary electrophoresis microchip. Anal Lett 43(18):2883–2891
Liu SR, Wu SP (2011) An NBD-based sensitive and selective fluorescent sensor for copper(II) ion. J Fluoresc 21(4):1599–605
Tang R et al (2012) A rhodamine-based off-on fluorescent chemosensor for selectively sensing Cu(II) in aqueous solution. J Fluoresc 21(1):141–8
Wang Y et al (2012) A novel chemosensor based on rhodamine derivative for colorimetric and fluorometric detection of Cu2+ in aqueous solution. J Fluoresc 22(3):799–805
Yildirim M, Kaya I (2010) Synthesis of a novel fluorescent Schiff base as a possible Cu(II) ion selective sensor. J Fluoresc 20(3):771–7
Tsien RY (1998) The green fluorescent protein. Annu Rev Biochem 67:509–44
Shimomura O (2005) The discovery of aequorin and green fluorescent protein. J Microsc 217(Pt 1):1–15
Prasher DC et al (1992) Primary structure of the Aequorea victoria green-fluorescent protein. Gene 111(2):229–33
Chalfie M et al (1994) Green fluorescent protein as a marker for gene expression. Science 263(5148):802–5
Zimmer M (2002) Green fluorescent protein (GFP): applications, structure, and related photophysical behavior. Chem Rev 102(3):759–81
Bizzarri R et al (2007) Green fluorescent protein ground states: the influence of a second protonation site near the chromophore. Biochemistry 46(18):5494–504
Heim R, Cubitt AB, Tsien RY (1995) Improved green fluorescence. Nature 373(6516):663–4
Heim R, Tsien RY (1996) Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol 6(2):178–82
Tansila N et al (2008) Metal ion accessibility of histidine-modified superfolder green fluorescent protein expressed in Escherichia coli. Biotechnol Lett 30(8):1391–6
Palfi M, Kovacs E, Miklossy I, Szilagyi L, Abraham B, Lanyi Sz (2009) Engineered Green Fluorescent Protein as a Potential Metal Sensor. Studia Universitatis Babes-Bolyai Chemia (Special Issue): 35–45
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259):680–685
Ellis KJ, Morrison JF (1982) Buffers of constant ionic strength for studying pH-dependent processes. Methods Enzymol 87:405–26
Williams AT, Winfield SA, Miller JN (1983) Relative fluorescence quantum yields using a computer controlled luminescence spectrometer. Analyst 108:1067–107
Brannon JM, Magde D (1978) Absolute quantum yield determination by thermal blooming: fluorescein. J Phys Chem 82(6):705–709
Green NM (1965) A spectrophotometric assay for avidin and biotin based on binding of dyes by avidin. Biochem J 94:23C–24C
Schrodinger LLC (2010) The PyMOL molecular graphics system, Version 1.3r1
Palm GJ et al (1997) The structural basis for spectral variations in green fluorescent protein. Nat Struct Biol 4(5):361–5
Kneen M et al (1998) Green fluorescent protein as a noninvasive intracellular pH indicator. Biophys J 74(3):1591–9
Patterson GH et al (1997) Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys J 73(5):2782–90
Matz MV et al (1999) Fluorescent proteins from nonbioluminescent Anthozoa species. Nat Biotechnol 17(10):969–73
Eli P, Chakrabartty A (2006) Variants of DsRed fluorescent protein: development of a copper sensor. Protein Sci 15(10):2442–7
Rahimi Y et al (2008) Mechanism of copper induced fluorescence quenching of red fluorescent protein, DsRed. Biochem Biophys Res Commun 370(1):57–61
Sumner JP et al (2006) DsRed as a highly sensitive, selective, and reversible fluorescence-based biosensor for both Cu(+) and Cu(2+) ions. Biosens Bioelectron 21(7):1302–8
Richmond TA et al (2000) Engineered metal binding sites on green fluorescence protein. Biochem Biophys Res Commun 268(2):462–5
Hotzer B et al (2011) Determination of copper(II) ion concentration by lifetime measurements of green fluorescent protein. J Fluoresc 21(6):2143–53
Tang S et al (2011) Design and application of a class of sensors to monitor Ca2+ dynamics in high Ca2+ concentration cellular compartments. Proc Natl Acad Sci USA 108(39):16265–70
Wang ZX, Jiang RF (1996) A novel two-site binding equation presented in terms of the total ligand concentration. FEBS Lett 392(3):245–9
Acknowledgement
The work has been funded by the Sectorial Operational Programme Human Resources Development 2007–2013 of the Romanian Ministry of Labour, Family and Social Protection through the Financial Agreement POSDRU/88/1.5/S/60203 and by the University of Sapientia, Department of Bioengineering.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bálint, EÉ., Petres, J., Szabó, M. et al. Fluorescence of a Histidine-Modified Enhanced Green Fluorescent Protein (EGFP) Effectively Quenched by Copper(II) Ions. J Fluoresc 23, 273–281 (2013). https://doi.org/10.1007/s10895-012-1145-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10895-012-1145-y