
Abstract
Green fluorescent protein (GFP) is frequently utilized for metal ion detection and quantification. To improve the metal binding potential of GFP, three residues (N146, F165, and L201) were substituted to histidines. Each variant responded differently upon interaction with metal ions. More than 80% of N146H, having the most accessible surface area, could bind to immobilized metal ions. However, only F165H exhibited significant differences in quenching by soluble metal ions (22% fluorescence decrease) in comparison with the template protein (12%). These findings can be utilized for designing GFP variants for metal binding and sensor applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The green fluorescent protein variant, GFPuv (Crameri et al. 1996), spontaneously emits green fluorescent light (508 nm) upon excitation by long-wave UV light (399 nm). The three-dimensional structure of GFPuv has been solved, thus allowing its rational engineering for specific applications (Ormö et al. 1996). For instance, several biosensors based on GFPuv have been developed for the determination of metal ions (Prachayasittikul et al. 2000; Richmond et al. 2000; Jensen et al. 2001). The target site of modification is mostly on one side of the GFPuv β-can corresponding to β-sheets 7–10. This region of GFPuv is often regarded as fragile due to structural flexibility and inefficient side chain packing, which both contribute to the irregularly large space between these β-strands (Helms et al. 1999; Seifert et al. 2003). Hence, binding of analytes to an engineered binding patch at this location of GFPuv can cause substantial structural changes that subsequently may alter the fluorescent properties of the GFPuv chromophore.
Recently, we have examined the interactions of chimeric metal binding GFPuv with artificial lipid membranes for future fabrication of membrane-based biosensors (Prachayasittikul et al. 2005a, b). However, for such applications, improvements of metal ion sensitivity of GFPuv are urgently needed. Therefore, several histidine substituted mutants have been designed, constructed and characterized. Positions 146 (on β7), 165 (on β8), and 201 (on β10) were chosen as targets for histidine substitutions due to their accessibility and close proximity to the GFPuv fluorophore. Moreover, determination of accessible surface areas and side chain arrangement of each histidine residue were included in this analysis for elucidating the possibility of protein adsorption via immobilized metal binding interactions. Effects of adding various soluble metal ions to the engineered GFPuv were also studied through fluorescent quenching.
Materials and methods
Bacterial strain and reagents
Escherichia coli strain TG1 (F′ traD36 lacI q Δ(lacZ) M15 proA + B + /supE Δ(hsdM-mcrB)5 (r k -m k -McrB − ) thi Δ(lac-proAB)) was used for the construction and expression of GFPuv in all experiments. pGFPuv (Clontech) was used as expression vector and template for site-directed mutagenesis. All other chemicals were of analytical grade and commercially available.
Construction of sfGFPuv variants
Superfolder (sf) mutations, S30R and Y39N, were initially introduced into GFPuv, by site-specific mutagenesis with mismatching oligonucleotides as primers (Supplementary Table 1), to enhance protein stability (Pédelacq et al. 2005). The product, sfGFPuv, was subsequently subjected to additional mutagenesis for histidine introductions. The amplified products were digested by DpnI to eliminate the template plasmid and the constructed plasmid was transformed into competent E. coli TG1 cells. Colonies exhibiting green fluorescence under UV-light were randomly chosen and sequenced using BigDye-terminators version 3.0 from Applied Biosystems according to the supplier’s instructions.
Protein expression and purification
Cells were cultivated in Luria-Bertani (LB) medium (10 g tryptone/l, 5 g yeast extract/l, and 10 g NaCl/l) supplemented with 100 μg ampicillin/ml at 37°C with vigorous shaking. Protein expression was induced by adding 1 mM IPTG when the cell density (OD600) reached approximately 0.6–0.8, and proteins were expressed overnight at 30°C. The cells were harvested by centrifugation, resuspended in 50 mM phosphate buffer, pH 7.4 and lysed by sonication.
The crude extract was heated at 70°C for 10 min to denature most endogenous proteins, which were removed by centrifugation. Heat-treated extracts were further purified by salt precipitation from 1.2 M to 2.8 M (NH4)2SO4. The green pellet was dissolved in 50 mM sodium phosphate buffer, pH 7.4 and dialyzed overnight in the same buffer. Protein concentration was determined by the Bradford assay.
Fluorescence measurements
All fluorescence analyses were performed using a fluorometer (Photon Technology International). At first, the excitation and emission spectra of the variants were determined followed by binding analyses to immobilized nickel ions at various pH (6–8) and ionic strengths (0–1 M NaCl). The binding efficiency was calculated as the fluorescence encountered in the eluted fraction normalized for the total fluorescence. The fluorescent quenching by various metal ions was then studied and the quenching of copper ions was studied in more detail by varying metal ion concentration and pH. The fluorescent intensity was recorded directly at the corresponding emission maxima of each mutant. The relative quenching was calculated as the change in fluorescence upon addition of metal ions relative to the non-quenched fluorescence. The protein concentration in the reaction mixtures in all quenching experiments, which were done in at least triplicates, was 20 μg/ml.
Results and discussion
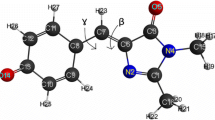
Three histidine substituted mutants (N146H, F165H and L201H) of GFPuv were constructed by site-specific mutagenesis using superfold GFPuv (sfGFPuv) as template (Fig. 1). Substitution to a histidine residue in position 146 led to the most accessible side chain compared to the other two mutants, while the imidazole ring of L201H appeared hidden according to accessible surface area calculations (Table 1). After protein expression and purification, the proteins were scanned to obtain the complete fluorescent spectra (Fig. 2). All variants, except L201H, had a major excitation peak at 399 nm. For F165H, the proportion of anionic fluorophore, which is normally activated by light at 470 nm, was increased compared to sfGFPuv, while the proportion was decreased for N146H. There was only one major peak for L201H, corresponding to the anionic fluorophore. Furthermore, the emission wavelength of L201H was blue-shifted to 500 nm, while the others exhibited emission at 508 nm. It can therefore be concluded that the histidine modifications partly have modified the chemical environment of the GFPuv fluorophore.

The three-dimensional structure of GFPuv (PDB: 1B9C) as visualized by PyMOL version 0.9 (DeLano Scientific LLC). The fluorophore is shown in green. The in silico mutagenesis by introducing histidine residues to positions 146, 165 and 201, are shown in yellow, as is a wild-type histidine at position 148

The fluorescent spectra of the GFPuv variants: L201H (dotted), F165H (dashed), N146H (gray) and sfGFPuv (black). To obtain the excitation spectra, the fluorescent emission was recorded at either 507 or 500 nm, while the emission spectra were recorded with the excitation kept constant at either 399 or 470 nm
The surface accessibility of each introduced histidine residue in sfGFPuv was also examined by immobilized metal affinity chromatography (IMAC). At all tested conditions, the degree of bound protein was in order of N146H, F165H, L201H and sfGFPuv (Fig. 3), which could be directly correlated to the calculated accessible surface area of the introduced histidine residues. All proteins were bound more efficiently with increasing pH as illustrated (Fig. 3a). For instance, at pH 8.0 more than 80% and 50% of N146H and F165H, respectively, were bound to the immobilized nickel ions, whereas only 14% of the wild-type protein could bind to the column. Interestingly, even the most hidden histidine of L201H promoted affinity to a limited extent (35% at pH 8.0), indicating that even partly buried histidines may favor binding in IMAC.

The binding of the engineered sfGFPuv variants was studied using chelating sepharose (GE Healthcare) charged with nickel ions and the binding efficiencies at different pH and ionic strengths were determined. Bound proteins were eluted with imidazole and the fluorescence was determined and normalized for that of the flow-through fraction. (a) pH 6.0, 7.0 and 8.0, (b) 0, 300, 500 and 1,000 mM NaCl. The columns indicate sfGFPuv (black), N146H (gray), F165H (dashed) and L201H (white)
Another important parameter is the ionic strength during binding. There are positive effects on protein binding by increasing salt concentration (Fig. 3b), especially in the range 0–300 mM NaCl. The template protein was most affected by high ionic strengths and the binding efficiency approximately doubled when salt concentration was increased from 0 to 1 M, which probably is due to an increase in the involvement of hydrophobic interactions (Chen et al. 1996).
To identify the strongest fluorescent metal ion quencher, all protein variants were mixed with 50 μM of Cu2+, Co2+, Mn2+, Ni2+, and Zn2+ and the quenching was measured (Fig. 4). The addition of the metal ions caused no wavelength shift of fluorescent excitation and emission peaks (data not shown). Cu2+ was the only metal which was able to efficiently decrease the fluorescent activity of all proteins, whereas there were no significant effects from Co2+, Mn2+, and Ni2+. This is in good agreement with previous studies on copper binding to DsRed, a red fluorescent protein (Sumner et al. 2006; Eli and Chakrabartty 2006). Interestingly, the fluorescence of F165H was enhanced in the presence of Zn2+ or Mn2+ ions.

The fluorescent quenching of the sfGFPuv variants by five metal ions (50 μM final concentration of Cu2+, Co2+, Mn2+, Ni2+, Zn2+). The columns represent sfGFPuv (black), N146H (gray), F165H (dashed) and L201H (white)
The quenching effect of Cu2+ was further investigated at low metal ion concentrations. sfGFPuv and the histidine substituted variants were quenched at different Cu2+ concentrations (1–50 μM) and their fluorescent reduction was monitored (Fig. 5). Clearly, F165H was the most sensitive variant to copper quenching. The percentages of maximum quenching (at 50 μM metal concentration) for F165H, N146H, L201H, and sfGFPuv were 22%, 15%, 13%, and 12%, respectively. Even though the order of metal sensitivity does not positively correlate with the calculated accessible surface area and binding capacity towards immobilized metal ions, it indicates that the locations of introduced histidines are important in the interaction with metal ions. If a residue is more solvent-exposed, as in N146H, the metal ion binding would be facilitated, but the metal ions would not be able to come in close proximity to the fluorophore for the quenching effect to occur (N146H is positioned 10.15 Å from GFPuv fluorophore). The introduced histidine residue of L201H is buried inside the β-barrel, and this fact offers a plausible explanation for the decreased efficiency in binding metal ions. Furthermore, it is not in close enough proximity to the GFPuv fluorophore (9.94 Å), and is therefore not much more quenched than the template protein. The side chain of F165H is situated more close to the fluorophore (4.93 Å); bound copper ions can therefore readily quench its fluorescent emission. Additionally, in the vicinity of F165H modification, there is a native histidine at position 148 (3.65 Å to the H165 residue and 3.22 Å to the fluorophore) which can cooperatively function as a metal binding partner to increase the metal sensitivity. These results confirm that the metal quenching process is highly concentration and distance dependent and also suggest that both structural orientation and distance of corresponding side chain towards the GFPuv fluorophore should be taken into consideration when designing a highly sensitive fluorescent protein-based biosensor for metal ions. In the presence of 200 μM CuSO4, the fluorescent emission of each protein was measured at its maximum excitation wavelength in the pH range 5.5–9.5. The metal ion sensitivity for all proteins was clearly more pronounced at pH 5.5 than at pH 9.5 (Fig. 6). Both F165H and L201H exhibited a more pronounced pH-dependent metal quenching compared to the other two proteins, especially at the lower pH values.

Quenching of the sfGFPuv variants by various concentrations of Cu2+, which has the highest quenching effect of the metal ions, at pH 6.0. The curves represent sfGFPuv (black), N146H (gray), F165H (dashed) and L201H (dotted)

The effect of pH (from pH 5.5–9.5) on the quenching by Cu2+ (200 μM). Plots for the quenching are (a) sfGFPuv, (b) N146H, (c) F165H and (d) L201H
Conclusions
Variants of sfGFPuv carrying a single histidine substitution have been constructed and characterized. These substitutions influence the equilibrium of both the neutral and anionic states of the GFPuv fluorophore as evident by changes in their excitation profiles. The variants behave differently in the binding towards immobilized nickel ions and fluorescent quenching induced by soluble metals due to differences in surface accessibility. The location and distance of the imidazole ring need to be very close to the GFPuv fluorophore (<5 Å) in order to effectively quench the fluorescence. This is essential for generating GFPuv quenching variants able to bind not only copper ions but also heavy metals like cadmium and mercury using specific peptide sequences (Mejare and Bulow 2001). The development of sfGFPuv variants can thereby meet practical biosensor applications for monitoring metal ions in soluble or immobilized forms.
References
Ahmed S, Gromiha M, Fawareh H et al (2004) ASAview: Database and tool for solvent accessibility representation. BMC Bioinformatics 5:51–55
Chen WY, Wu CF, Liu CC (1996) Interactions of imidazole and proteins with immobilized Cu(II) ions: Effects of structure, salt concentration, and pH in affinity and binding capacity. J Colloid Interface Sci 180:135–143
Crameri A, Whitehorn EA, Tate E et al (1996) Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat Biotechnol 14:315–19
Eli MA, Chakrabartty A (2006) Variants of DsRed fluorescent protein: Development of a copper sensor. Protein Sci 15:2442–2447
Helms V, Straatsma TP, McCammon JA (1999) Internal dynamics of green fluorescent protein. J Phys Chem 103:3263–3269
Jensen KK, Martini L, Schwartz TW (2001) Enhanced fluorescence response energy transfer between spectral variants of green fluorescent protein through zinc-site engineering. Biochemistry 40:938–945
Mejare M, Bulow L (2001) Metal-binding proteins and peptides in bioremediation and phytoremediation of heavy metals. Trends Biotechnol 19:67–73
Ormö M, Cubitt AB, Kallio K et al (1996) Crystal structure of the Aequorea victoria green fluorescent protein. Science 273:1392–1395
Pédelacq JD, Cabantous S, Tran T et al (2005) Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol 24:79–88
Prachayasittikul V, Isarankura-Na-Ayudhya C, Galla HJ (2004) Binding of chimeric metal-binding green fluorescent protein to lipid monolayer. Eur Biophys J 33:522–534
Prachayasittikul V, Isarankura-Na-Ayudhya C, Tantimongcolwat T et al (2005a) Nanoscale orientation and lateral organization of chimeric metal-binding green fluorescent protein on lipid membrane determined by epifluorescence and atomic force microscopy. Biochem Biophys Res Commun 326:298–306
Prachayasittikul V, Isarankura-Na-Ayudhya C, Hilterhaus L et al (2005b) Interaction analysis of chimeric metal-binding green fluorescent protein and artificial solid-supported lipid membrane by quartz crystal microbalance and atomic force microscopy. Biochem Biophys Res Commun 327:174–182
Richmond TA, Takahashi TT, Shimkhada R et al (2000) Engineered metal binding sites on green fluorescence protein. Biochem. Biophys Res Commun 268:462–465
Seifert MHJ, Georgescu J, Ksiazek D et al (2003) Backbone dynamics of green fluorescent protein and the effect of histidine 148 substitution. Biochemistry 42:2500–2512
Sumner JP, Westerberg NM, Stoddard AK et al (2006) DsRed as a highly sensitive, selective, and reversible fluorescence-based biosensor for both Cu1+ and Cu2+ ions. Biosen Bioelec 21:1302–1308
Acknowledgements
NT is grateful for the Royal Golden Jubilee (RGJ) Ph.D. scholarship under V.P. supervision from the Thailand Research Fund (TRF). This project was also supported by the National Science Research Foundation (VR) and an annual budget grant of Mahidol University (Project No. 02012053-0003).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Tansila, N., Becker, K., Na-Ayudhya, C.I. et al. Metal ion accessibility of histidine-modified superfolder green fluorescent protein expressed in Escherichia coli . Biotechnol Lett 30, 1391–1396 (2008). https://doi.org/10.1007/s10529-008-9692-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-008-9692-7