
Abstract
Chemoresistance of cancer cells is a major problem in treating cancer. Knowledge of how cancer cells may die or resist cancer drugs is critical to providing certain strategies to overcome tumour resistance to treatment. Paclitaxel is known as a chemotherapy drug that can suppress the proliferation of cancer cells by inducing cell cycle arrest and induction of mitotic catastrophe. However, today, it is well known that paclitaxel can induce multiple kinds of cell death in cancers. Besides the induction of mitotic catastrophe that occurs during mitosis, paclitaxel has been shown to induce the expression of several pro-apoptosis mediators. It also can modulate the activity of anti-apoptosis mediators. However, certain cell-killing mechanisms such as senescence and autophagy can increase resistance to paclitaxel. This review focuses on the mechanisms of cell death, including apoptosis, mitotic catastrophe, senescence, autophagic cell death, pyroptosis, etc., following paclitaxel treatment. In addition, mechanisms of resistance to cell death due to exposure to paclitaxel and the use of combinations to overcome drug resistance will be discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer therapy is dependent on killing clonogenic malignant cells. A tumour can grow when cancer cells can divide and produce new cells [1]. Usually, cancer cells can escape from the immune system following several mutations. The mutations in cancer cells can change the expression of some antigens to protect them against immune system attacks. Furthermore, cancer cells may undergo further mutations to resist cell death. Mutations in tumour suppressor genes, oncogenes, pro-and anti-apoptosis genes, cell cycle genes, metabolism mediated genes, mitochondria, etc., are the most known abnormal changes that can increase survival and invasion of cancer cells [2,3,4,5]. Anti-cancer agents may kill cancer cells via modulating various mechanisms. Some drugs may induce the activity of the immune system to release pro-apoptosis and necrosis (and also necroptosis) mediators [6]. The stimulation of tumour suppressor genes, generation of free radicals, and the inhibition of antioxidant defence system can induce damage to different organelles and macromolecules in cancer cells, which lead to the induction of various types of cell death such as apoptosis, mitotic catastrophe, autophagy cell death, senescence, pyroptosis, ferroptosis, and others [7, 8].
Apoptosis is known as the most important biomarker of cancer therapy for a wide range of malignancies. The upregulation of pro-apoptosis genes such as Bax, and downregulation of anti-apoptosis genes such as Bcl-2 can predict a better survival for patients with cancer [9, 10]. However, some cancers may show a low level of apoptosis [11]. Autophagy is a process that regulates the metabolism of cells during starvation and stress conditions such as low glucose and oxygen supply [12]. The autophagy process may induce resistance of cancer cells to cell death. However, some autophagy-related proteins (ATG) may be converted to pro-death molecules, leading to the induction of autophagic cell death. The blockade of the autophagy process leads to the inhibition of autophagic cell death [13]. The induction of autophagy in response to anti-cancer drugs may have a prodeath or a prosurvival role. Thus, the inhibition or stimulation of autophagy may cause different consequences in different cancer cell types [14]. Autophagy has an anti-tumour effect in the early stages of the tumour, however, it can facilitate metastasis and invasion in later stages of tumours [15, 16]. Radiotherapy and some chemotherapy drugs may induce autophagy. Furthermore, these modalities that induce the generation of a heavy amount of free radicals can stimulate the induction of other types of cell death such as senescence, pyroptosis, ferroptosis, necrosis, necroptosis, and mitotic catastrophe [17].
To date, a wide range of anti-cancer agents has been approved to induce cell death in cancer cells. Paclitaxel is a herbal-derived agent that can induce cancer-killing following the inhibition of mitosis. However, some studies show it may kill cancer cells by some different mechanisms [18]. In this review, we aim to explain the induction of different mechanisms of cancer-killing by paclitaxel rather than cell cycle arrest.
Paclitaxel
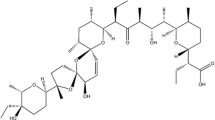
Paclitaxel is a herbal-derived agent with formula C47H51NO14. For the first time, it was extracted from Taxus brevifolia, the bark of the Pacific yew or western yew [19]. Indeed, paclitaxel belongs to a type of anti-cancer agent, which is known as plant alkaloids. This type of chemotherapy drug can be achieved from plants [20]. For the first time, paclitaxel was approved by US Food and Drug Administration (FDA) for the treatment of ovarian cancer [21]. Some years later, FDA approved paclitaxel for breast malignancies [22]. Currently, paclitaxel may be prescribed for some other malignancies such as advanced prostate and breast carcinomas, non-small-cell lung carcinoma (NSCLC), endometrial cancer, bladder cancer, cervical carcinoma and some others [23,24,25,26]. Paclitaxel may be used alone or in the combination with some other chemotherapy drugs to treat early or advanced stages of cancer. Furthermore, combination therapy of cancer with radiation and paclitaxel may be used to treat patients with recurrent malignancies [27].
Several studies have investigated the cellular and molecular mechanisms of the anti-cancer effects of paclitaxel. This has been demonstrated that the main target of paclitaxel is microtubules. Paclitaxel stabilizes microtubules. This effect of paclitaxel prevents the depolymerization of microtubules. Finally, paclitaxel induces mitosis arrest by interference with spindle formation [28]. These modulations by paclitaxel stimulate triggering the spindle assembly checkpoint to induce cell cycle arrest and abnormal mitosis which may lead to cell death [29]. Despite the widespread usage of paclitaxel in cancer therapy, its bioavailability remains a weakness. It has been suggested that the bioavailability of paclitaxel is near 10–30% [30]. Thus, some experiments have investigated some strategies to increase the bioavailability of paclitaxel. Using nanotechnology is an interesting approach to increase the bioavailability of some drugs and adjuvants. To date, some experiments have revealed that using nano-carriers and capsules can be useful to increase the bioavailability of paclitaxel [31]. Lipid nanoparticles have been shown to increase both the absorption and bioavailability of paclitaxel [30]. A study showed that paclitaxel-loaded nanosponges can increase the bioavailability of paclitaxel up to threefold [32]. Micelles and liposomes are other well-known carriers that can be used to increase the bioavailability and absorption of drugs. Micelles and liposomes have been shown to increase the absorption of paclitaxel in the intestine and also augment bioavailability [33,34,35]. Although some chemical structures have been investigated to increase the bioavailability of paclitaxel, it seems that nanoparticles and nanocarriers can overcome this problem more effectively [36, 37]. The structure of paclitaxel and also some well-known carriers are illustrated in the following figure (Fig. 1).

Chemical structure of paclitaxel is shown in A. some carriers to improve the bioavailability of paclitaxel are shown in this figure. B: Liposome; C: Lipid Nanoparticle; D and E: Micelle; F: Polymeric Nanocapsule; G: Nanosponge.
Mechanisms of cell death in cancer
The induction of death in cancer cells is the final purpose of anti-cancer therapy. To achieve this goal, anti-cancer drugs or ionizing radiation may act through different mechanisms. Some drugs such as immunotherapy agents try to change the environment of tumours in favour of anti-cancer therapy. Immunotherapy may induce apoptosis in cancer cells via stimulating natural killer (NK) cells and cytotoxic CD8+ T lymphocytes (CTLs) to release anti-tumour cytokines such as tumour necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) [38]. The immune cells may induce some other types of cell death such as necroptosis. These cells can release some other molecules such as perforin and Granzyme B to induce the perforation and lysis of cancer cells [39]. Chemotherapy agents may kill cancer cells through different mechanisms. Classic chemotherapy drugs such as alkylating agents cause the generation of a heavy amount of free radicals, damage to organelles such as mitochondria, the inhibition of DNA repair capacity, and others. These drugs can induce various types of cell death following damage to DNA and other vital organelles. The induction of mitotic catastrophe, apoptosis, autophagy, pyroptosis, senescence, ferroptosis, necrosis and necroptosis are common following treatment with these drugs. A key weakness of these drugs is the severe toxicity for normal tissues [40,41,42].
Ionizing radiation kills cells including both normal and malignant cells. Thus, radiotherapy is associated with severe cell death in both tumours and normal tissues. However, treatment planning techniques try to deliver a high dose of radiation into the tumour with fewer side effects for the normal organs [43, 44]. Ionizing radiation is a potent clastogenic agent. It mainly acts through the induction of DNA damage in targeted cells. Ionizing radiation can affect the function of other organelles such as mitochondria. It also may suppress the antioxidant defence system. These changes may cause the activation of different mechanisms of cell death including mitotic catastrophe, apoptosis, autophagic cell death, senescence, pyroptosis, ferroptosis, necroptosis, and necrosis [45].
Paclitaxel is a classic chemotherapy drug that induces cell death through different mechanisms. The most known consequence of paclitaxel is mitotic arrest, which may lead to death in cancer cells. However, it has been suggested that paclitaxel may induce cancer-killing through some other mechanisms such as activation of the immune system [46]. The combination of paclitaxel with radiotherapy or immunotherapy may be more effective to eliminate cancer cells, however, normal tissue toxicity should be considered as the major limiting factor [47]. In this section, we describe the induction of various cell death mechanisms following cancer therapy with paclitaxel.
Apoptosis
Apoptosis is one of the main targets of paclitaxel in cancer cells. It seems that paclitaxel induces apoptosis in cancer cells through both extrinsic and intrinsic signalling pathways [48]. The main mechanisms for the induction of apoptosis by paclitaxel are including the generation of ROS, modulation of mitochondria function, modulation of the cell cycle, and also triggering the anti-tumour immunity [49]. In this section, we discuss the mechanisms of apoptosis induction by paclitaxel in the detail.
Paclitaxel may induce apoptosis via inducing anti-tumour immunity
Paclitaxel can affect various types of immune cells. The interactions between paclitaxel and the immune system are critical for both single and combination therapy with some other modalities such as immunotherapy and radiotherapy [50]. The final aim of immunostimulatory agents in cancer therapy is an increase in the proliferation of anti-cancer cells, which leads to an increase in the release of anti-cancer molecules such as perforin, Granzyme B, IFN-γ, TNF-α, and others [51]. It has been suggested that paclitaxel can stimulate macrophages to release anti-tumour cytokines. This is associated with an increase in the proliferation of anti-tumour immune cells including CTLs, NK cells, and also dendritic cells (DCs) [50]. An increase in the production of nitric oxide (NO) by macrophages can stimulate the release of IL-12 following the administration of paclitaxel. This effect of paclitaxel has been observed in mice-bearing tumours [52]. The release of IL-12 by macrophages can attenuate immunosuppression in tumours [53]. Paclitaxel also can suppress the proliferation of Tregs in the tumour [54]. The effect of paclitaxel on tumour-associated macrophages (TAMs) is controversial. It may promote the proliferation and infiltration of TAMs [55]. However, paclitaxel has been shown to induce reprogramming TAMs toward M1 macrophages in some cancers [56]. The consequences of these effects are very different. The suppression of TAMs can boost anti-tumour immunity, while their proliferation following treatment with paclitaxel may suppress immune responses against cancer cells. Paclitaxel also has been shown to interrupt the interactions between CAFs and cancer cells [57].
The interactions between the mentioned immune cells and cancer cells are very vital for eradicating cancer cells and suppressing tumour growth. One of the main effects of immune cells including CD4+ and CD8+ T lymphocytes, and NK cells is the release of pro-apoptosis molecules such as FasL, TRAIL, TNF-α, and IFN-γ [58]. These molecules can bind to their receptors on the surface of malignant cells, leading to the activation of apoptosis mediators such as caspase proteins [59]. The proliferation of CD4+ and CD8+ T lymphocytes, and also NK cells can boost the release of these pro-apoptosis molecules. However, an increase in the infiltration and recruitment of immunosuppressive cells such as Tregs and TAMs can suppress the activity of the mentioned anti-tumour immune cells [60, 61]. As paclitaxel can suppress Tregs and TAMs, and also induce the release of pro-apoptosis molecules by immune cells, it may be interesting to induce apoptosis via modulating anti-tumour immunity [62]. A clinical study confirmed that treatment with paclitaxel can induce the anti-tumour activity of NK cells and CD8+ T lymphocytes in patients with HER2+ breast cancers [63].
Although paclitaxel can induce anti-tumour immunity, the expression and release of some immunosuppressive molecules can induce resistance of malignant cells to paclitaxel. TGF-β is an important immunosuppressive cytokine that can induce epithelial-mesenchymal transition (EMT) in malignant cells, leading to the resistance of cancer cells to anti-cancer drugs such as paclitaxel [64]. A study showed that the inhibition of TGF-β signalling with a TGF-β type I receptor kinase inhibitor can blunt EMT and stemness in breast cancer cells. this was associated with the sensitization of cancer cells to paclitaxel [65]. EDIL3 (EGF like repeats and discoidin domains 3) is an important gene that can induce EMT in cancers through stimulation of TGF-β signalling [66]. It has been reported that EDIL3 plays a key role in drug resistance and EMT in some cancers such as breast, hepatocellular carcinoma, NSCLC, and pancreas malignancies [67,68,69]. An experiment showed plays a key role in EMT and resistance of breast and prostate cancer cells to paclitaxel. The results indicated that a high expression of EDIL3 is associated with EMT in paclitaxel-resistant breast and prostate cancer cells. However, the inhibition of EDIL3 reverted EMT and induced apoptosis following treatment with paclitaxel [70].
Paclitaxel may induce apoptosis via triggering tumour suppressor genes (TSGs)
TSGs play a key role in the suppression of tumour development and growth. Usually, cancers show several mutations in some different TSGs. These mutations can weak the anti-tumour activity of TSGs. Some TSGs such as BRCA1, p53, PTEN, P21, Bax, Fas-associated death domain (FADD) protein, and some others are involved in the induction of apoptosis in both pre-cancer and cancer cells [71]. The mutations in these TSGs can induce resistance to apoptosis and also anti-cancer agents. Some studies have revealed that the activity of TSGs is crucial for inducing apoptosis by paclitaxel. Studies have shown that the presence of BRCA1 plays a key role in the induction of apoptosis in MCF-7 and A549 cancer cells [72, 73]. It has been suggested that paclitaxel can induce apoptosis through BRCA1/JNK and p38 pathway [74]. PTEN and p53 are other TSGs that can be activated by paclitaxel to induce apoptosis in cancer cells. The induction of miR-22 in cancer cells can induce the activity of PTEN and reduce paclitaxel resistance in cancer cells, indicating a key role of miR-22 in the activation of PTEN [75]. Overproduction of ROS following exposure of cancer cells to paclitaxel is another mechanism of PTEN activation by paclitaxel [76]. TP53 is among the most frequent mutated TSGs in malignant cells. A decreased activity of p53 is responsible for escaping cancer cells from apoptosis. Some paclitaxel-resistant cancer cells have low activity of p53 and its downstream proteins such as PUMA [77]. Stimulation of p53 in some cancers may reduce the resistance of cancer cells to paclitaxel-induced apoptosis [78]. However, it seems that this pathway isn’t involved in the paclitaxel-induced apoptosis in all types of cancers [79, 80].
PI3K/Akt signalling pathway
PI3K and Akt are among the key players in tumour resistance to various anti-cancer therapy modalities [81]. PI3K/Akt pathway plays a key role in the upregulation of anti-apoptosis genes such as Bcl-2, transducer and activator of transcription 3 (STAT3), NF-κB, COX-2, and others [82,83,84]. In the normal cells, PI3K can be suppressed by PTEN. However, the mutations in PTEN in malignant cells facilitate upregulation of PI3K and resistance to apoptosis [85, 86]. Several studies have revealed the interactions of paclitaxel with the PI3K/Akt pathway. It seems that paclitaxel may reduce the expression of PI3K. However, emerging studies indicate the PI3K pathway plays a key role in the resistance of cancer cells to paclitaxel [87, 88]. A study showed that paclitaxel attenuates the expression and phosphorylation of PI3K/Akt through the activation of PTEN. The modulation of this pathway led to an increase in apoptosis in the human gastric cancer cells, MGC-803. This effect of paclitaxel was observed in both in vitro and xenograft models [76]. Another study also suggested that paclitaxel induces the regulation of miR-145 in bladder cancer cells, leading to a reduction in survival [89]. MiR-145 can inhibit the expression of Akt, thus inducing apoptosis in cancer cells [90, 91]. Thus, an increase in the regulation of miR-145 may be responsible for the inhibition of PI3K/Akt and the induction of apoptosis following treatment with paclitaxel. Downregulation of the PI3K/Akt pathway can attenuate the expression of anti-apoptosis genes and stimulate the expression of pro-apoptosis mediators. Furthermore, stimulation of epithelial-mesenchymal transition by the PI3K/Akt pathway can suppress apoptosis and induce invasion in cancer cells [87]. A key consequence of Akt inhibition is the induction of apoptosis through Forkhead Box O3 (Foxo3) pathway [92]. Paclitaxel has been shown to stimulate Foxo3 through the inhibition of Akt and stimulation of c-Jun NH2-terminal kinase 1/2 (JNK1/2). Sunters et al. showed that upregulation of JNK1/2 following treatment of MCF-7 breast cancer cells with paclitaxel inhibits Akt, which leads to phosphorylation and nuclear translocation of Foxo3a. These modulatory effects of paclitaxel induce apoptosis in breast cancer cells [93].
Although the activation of PTEN and downregulation of the PI3K/Akt pathway may be involved in the apoptosis of cancer cells, it seems that this pathway has a key role in the cancer resistance to paclitaxel. Thus, targeting the PI3K/Akt pathway may be useful to potentiate cancer-killing by paclitaxel [94]. It has been revealed that the upregulation of SRY (sex determining region Y)-box 2, which is known as SOX2 plays a key role in the paclitaxel resistance via stimulating the PI3K/Akt pathway. Thus the inhibition of this transcription factor may be useful to induce apoptosis in the response to paclitaxel therapy [95, 96]. MiR-145 can inhibit the expression of SOX2. It has been shown that an increase in the regulation of miR-145 can sensitize cancer cells to paclitaxel via downregulation of SOX2 [97]. Furthermore, the release of some growth factors within TME can induce the expression of PI3K by cancer cells. A study showed that CAFs release hepatocyte growth factor (HGF), leading to upregulation and phosphorylation of PI3K/Akt proteins and resistance of A549 lung cancer cells to paclitaxel-induced apoptosis [98]. Some experiments have shown that inhibition of PI3K/Akt can amplify apoptosis induction by paclitaxel [99, 100]. The beneficial effect of Akt inhibition in the combination with paclitaxel has been revealed in a clinical study. In this double-blind placebo-controlled trial study, patients with metastatic triple-negative breast cancer received the AKT inhibitor capivasertib or placebo in the combination with paclitaxel. Results of this study showed a significant increase in overall survival and progression-free survival. Results also indicated that patients with PIK3CA/AKT1/PTEN alterations have a more pronounced response to this combination therapy [101] (Fig. 2).

Mechanisms of paclitaxel-induced apoptosis in cancer cells via targeting the PI3K/Akt pathway. Paclitaxel can suppress PI3K/Akt pathway in some different ways. It induces PTEN via stimulation of ROS and also upregulation of miR-22. Furthermore, upregulation of miR-145 by paclitaxel can inhibit Akt. Paclitaxel also stimulates phosphorylation of Foxo3a, leading to inhibition of Bcl-2
PI3K plays a key role in the upregulation of other anti-apoptosis genes, including Mcl-1 and Bcl-xL [102, 103]. Overexpression of Mcl-1 and Bcl-xL has a key role in the resistance of cancer cells to paclitaxel. Thus, inhibiting these genes can sensitize cancer cells to paclitaxel [104, 105]. The inhibition of Bcl-xL by some small molecules has been shown to increase apoptosis in paclitaxel-treated prostate and pancreas cancer cells [106,107,108]. It has been reported that paclitaxel can also induce phosphorylation of Bcl-xL. This effect of paclitaxel can cause resistance to apoptosis following treatment with paclitaxel. Thus, the inhibition of Bcl-xL and Mcl-1 using their specific inhibitors in combination with paclitaxel can increase the anti-tumour effect of paclitaxel [109, 110]. Obatoclax is a Bcl-2 pan inhibitor that can amplify apoptosis in cancer cells through the inhibition of Mcl-1, Bcl-xL, and Bcl-2 [111]. A synergic effect of paclitaxel and obatoclax has been revealed for urothelial cancer cells [112]. However, another study showed no synergic effect of paclitaxel and obatoclax for ovarian cancer cells [113].
STAT3 phosphorylation
STAT3 is a transcription factor and signal transducer. The aberrant expression of STAT3 can be observed in a wide range of malignancies. The upregulation and phosphorylation of STAT3 are associated with multidrug resistance, invasion, angiogenesis, and metastasis of cancers [114]. The inhibition of the apoptosis pathway is a key mechanism of STAT3 in malignancies. An increase in the expression and phosphorylation of STAT3 can attenuate the anti-tumour activity of paclitaxel. The release of some cytokines and growth factors such as IL-6, IL-10, and IL-22 can induce phosphorylation of STAT3, leading to upregulation of the expression of anti-apoptosis genes such as Bcl-2 and resistance of cancer cells to paclitaxel-induced apoptosis [115,116,117]. Paclitaxel has been shown to downregulate the expression of STAT3 [118]. However, it seems that the inhibition of STAT3 using an inhibitor can augment apoptosis in cancer cells following therapy with paclitaxel [119]. Some studies have shown that the inhibition of STAT3 using miRNAs and siRNAs can increase apoptosis induction of paclitaxel-resistant cancer cells [120,121,122,123].
Endogenous ROS generation
The generation of ROS plays an important role in the damage to cancer cells and suppression of tumour growth. Although ROS may trigger angiogenesis, the induction of new mutations and invasion of cancer cells, a huge production of ROS during cancer therapy can generate unrepairable damages to DNA and other critical organelles [124,125,126,127]. Some anti-cancer drugs such as classic chemotherapy drugs and also radiation can induce the production of a heavy amount of ROS because of mitochondrial dysfunction and upregulation of pro-oxidant enzymes [17]. Paclitaxel has been shown to induce apoptosis in cancer cells via inducing endogenous production of ROS [128]. Furthermore, several studies have revealed that paclitaxel can augment the anti-cancer effects of some other agents via stimulating ROS overproduction in cancer cells.
Paclitaxel can induce apoptosis in a ROS-dependent pathway, which leads to activation of ceramide, p38, JNK, and AMP-activated protein kinase (AMPK) [129, 130]. Ceramide can be produced following interactions of ROS with lipids in the membrane. It can induce some apoptosis signalling such as ER stress, AMPK, and also induce mitochondrial apoptosis [131, 132]. AMPK also can induce p53 activity, which is a potent inducer of apoptosis in cancer cells [133]. It has been revealed that ROS production by paclitaxel can also trigger the activity of PTEN in cancer cells [76]. Paclitaxel has also been shown to enhance the anti-cancer effect of quercetin via the endoplasmic reticulum (ER) stress signalling pathway, which leads to the overproduction of ROS in PC-3 prostate cancer cells [134]. It seems that ROS production in cancer cells following treatment with paclitaxel can induce both extrinsic and intrinsic pathways of apoptosis [135]. Some studies have shown that paclitaxel-loaded nanoparticles such as silver nanoparticles enhance apoptosis in cancers via generating ROS [136,137,138,139].
NF-κB
NF-κB is including 5 subfamilies that regulate a wide range of functions within cells. Usually, cancer cells have a higher expression of NF-κB [140]. The release of some cytokines such as TGF-β, IL-1 and TNF-α by immune cells can trigger the upregulation of NF-κB [141]. Usually, NF-κB can be degraded by the inhibitor of κB (IκB). During stress conditions such as seen following chemotherapy or exposure to ionizing radiation, the release of the mentioned cytokines can induce the degradation of IκB, which lead to the upregulation and nuclear translocation of NF-κB [142]. The interaction of paclitaxel with NF-κB and its signalling pathways need to be more elucidated in various cancer cells. However, it seems that paclitaxel may induce the regulation of NF-κB in some cancer cells such as lung carcinoma [143]. The upregulation of NF-κB in breast cancer cells can induce can increase resistance to paclitaxel via inducing anti-apoptosis genes such as tumour necrosis factor receptor-associated factor 1 (TRAF1), c-inhibitor of apoptosis 2 (c-IAP2), and defender-against cell death (DAD-1). Furthermore, increased activity of superoxide dismutase (SOD) can reduce oxidative damage and apoptosis in cancer cells following exposure to paclitaxel [144]. A study also showed that a low dose of paclitaxel (1 mg/kg) can upregulate NF-κB and induce metastasis in mice-bearing breast tumours. However, higher concentrations of paclitaxel (20–50 mg/kg) can suppress tumour growth and metastasis remarkably [145]. In total, it seems that the inhibition of NF-κB can augment the anti-tumour effect of paclitaxel in some cancer cells [146, 147]. However, paclitaxel may act independently on NF-κB in some cancers, thus, the inhibition of NF-κB may don’t increase apoptosis by paclitaxel [148].
Epigenetics modulation of apoptosis by paclitaxel
Upregulation or downregulation of some miRNAs may be involved in the resistance of cancer cells to paclitaxel. Furthermore, paclitaxel may induce apoptosis via modulating some miRNAs [149]. As earlier mentioned, upregulation of miR-145 by paclitaxel can induce the inhibition of the SOX2/Akt signalling pathway, leading to an increase in the apoptosis of some malignant cells such as bladder cancer cells. By contrast, the upregulation of some miRNAs by paclitaxel can attenuate apoptosis in cancer cells. A study demonstrated an increase in the expression of miR-125b in paclitaxel-resistant cancer cells [150]. The abnormal regulation of some miRNAs in cancer cells can induce resistance of cancer cells to paclitaxel. Downregulation of some miRNAs such as miR-22 and upregulation of some others such as miR-29c can induce resistance of cancer cells to paclitaxel-induced apoptosis [75, 151] (Fig. 3).

A schematic of mechanisms of apoptosis induction in cancer by paclitaxel. Paclitaxel can induce apoptosis via inducing several pathways. Some pathways are illustrated in this figure in summary. ER: Endoplasmic reticulum; PTX: Paclitaxel
Mitotic catastrophe
Mitotic catastrophe is a type of cell death that occurs following massive damage to DNA. It is characterized by the accumulation of unrepaired damages in DNA, aberrant mitosis, and multiple nuclei that are morphologically similar to apoptosis [152]. Although the molecular mechanisms of mitotic catastrophe remained to be illustrated completely, we know that it acts as a tumour suppressor mechanism [153]. Furthermore, mitotic catastrophe can enhance the therapeutic efficiency of anti-cancer modalities such as radiotherapy and chemotherapy. Thus, the stimulation of mitotic catastrophe can be suggested to improve cancer therapy outcomes [154]. This type of cell death mechanism may occur in partnership with some other types of cell death such as necrosis or apoptosis [155]. Mitotic catastrophe occurs independent of p53, thus it may be an appropriate target for cancers with mutated p53 [156].
Paclitaxel has been shown to induce various types of cell death including mitotic catastrophe in cancer cells [157]. Some studies have shown a negative cross-talk of p53 and mitotic catastrophe in cancer cells that were exposed to paclitaxel. Although p53 is essential for the induction of apoptosis and senescence, its activity is associated with cell cycle arrest, DNA repair, and finally the inhibition of mitotic catastrophe in the damaged cells [158]. Thus, antimicrotubule agents including paclitaxel may be effective to kill cancers with mutated p53. A study showed that silencing of the Polo-like kinase family member serum inducible kinase (Snk/Plk2), a target of p53, can amplify mitotic catastrophe following spindle damage. Furthermore, loss of p53 in the human osteosarcoma cell lines (U20S) led to an increase in the induction of mitotic catastrophe both short-term and long-term following treatment with paclitaxel [159]. The combinations of paclitaxel with some other antineoplastic drugs such as oxaliplatin, cetuximab, BZML (a colchicine binding site inhibitor), pazopanib, BPR0L075 (an antimitotic and antivascular agent), and some others have shown that sensitize cancer cells via inducing mitotic catastrophe [160,161,162,163,164]. Furthermore, some natural-derived agents such as fisetin and mulberry water extract have been shown to promote mitotic catastrophe and reduce resistance to paclitaxel [165, 166]. In addition to these drugs, hyperthermia also has been shown to amplify the induction of mitotic catastrophe by paclitaxel [167].
A clinical study suggested that the inhibition of mitotic catastrophe in patients with ovarian cancer following upregulation of inhibitory member of apoptosis-stimulating protein of the p53 family (iASPP) is a mechanism for low response and poor survival of patients that were treated with paclitaxel [168]. Another study showed a key role of forkhead box protein M1 (FOXM1) in the inhibition of mitotic catastrophe and resistance to paclitaxel. FOXM1 is a transcription factor that regulates several biological processes such as DNA damage, cell death, proliferation, differentiation, and others. The upregulation of FOXM1 is associated with tumorigenesis and also the resistance of tumours to therapy. This clinical investigation confirmed a direct relation of FOXM1 overexpression with poor response of ovarian cancer to paclitaxel via inhibiting mitotic catastrophe [169]. Similar results were observed for breast tumour samples [170].
The mitosis slippage is another mechanism that can affect the response of malignant cells to paclitaxel. The spindle assembly checkpoint ensures proper chromosome alignment at the metaphase before chromosome segregation. However, malignant cells may acquire some alternative mechanisms to bypass mitotic arrest before the initiation of cell death. This alternative mechanism in cancer cells is known as mitosis slippage [171, 172]. A long-term cell cycle arrest following treatment with paclitaxel may lead to degradation of cyclin B1 and mitotic slippage. In this condition, cancer cells exit mitosis prematurely, leading to the development of aneuploid cells and resistance to paclitaxel. However, some cells may undergo post-mitotic cell death or cell cycle arrest [173]. In this condition, a combination of paclitaxel with anti-apoptosis inhibitors may be an interesting strategy to overcome paclitaxel resistance in the remained cancer cells [173]. Some recent studies have suggested that targeting anti-apoptosis mediators including myeloid cell leukaemia-1 (Mcl-1) and Bcl-xL can induce cell death and reduce mitosis slippage, which leads to overcoming resistance in paclitaxel-resistant cancer cells [174]. Mcl-1 is a member of the Bcl-2 family that can be synthesised and degraded during mitosis. The upregulation of Mcl-1 in paclitaxel-treated cancer cells can prolong mitosis and suppress cell death after mitosis slippage. However, the inhibition of Mcl-1 has been shown to accelerate post slippage death in paclitaxel-resistant cells [175]. A study by Bennett et al. confirmed the key role of Bcl-xL in mitotic cell death following treatment with paclitaxel. This study confirmed that the inhibition of Bcl-xL sensitizes cancer cells to mitosis inhibitors such as microtubule-binding agents [176].
Autophagy
Autophagy is known as a key regulator of cell metabolism during starvation. Autophagy protects cells against stress situations such as hypoxia, low glucose levels, and oxidative stress. However, this process may cause cancer cell death independent of other types of cell death [177]. The dual role of autophagy in the survival of cancer cells makes it a complicated process in cancer therapy. In some cancers, an increase in autophagy may protect them against chemotherapy and radiotherapy. However, in some others, it may induce autophagic cell death, thus increasing the response of tumours to therapy [178, 179]. Autophagic cell death is a controversial issue. It has been suggested that autophagy may promote cell death in cancers when they lack apoptosis mediators such as Bax or caspase proteins. Autophagic cell death is a type of cell death that can be blocked by the inhibition of the autophagy process. Other types of cell death shouldn’t consider autophagic cell death [13]. In addition to modulation of cell death in cancer cells, autophagy may affect the activity of immune responses during radiotherapy or chemotherapy [180, 181]. Autophagy also may stimulate or inhibit cell death via modulating other cell death pathways such as apoptosis and pyroptosis [182].
Paclitaxel may induce autophagic cell death
As mentioned, paclitaxel may suppress the proliferation of cancers via inducing autophagy. However, it seems that this effect of paclitaxel is highly dependent on the type of cancer and concentration of paclitaxel. An experiment showed that the inhibition of gastric cancer cells by paclitaxel is associated with the appearance of autophagy. This connection was approved by some cell techniques including immunofluorescence of cytoplasm structure, western blot for autophagy-associated proteins, and also 3–(4,5–dimethyl–2–thiazolyl)–2,5–diphenyl–2–H–tetrazolium bromide (MTT) assay [183]. Some studies also have revealed that stimulation of autophagy in the combination with paclitaxel can enhance the cytotoxicity effect of paclitaxel against cancer cells. An in vitro examination showed that treatment of MDA‑MB‑231 human breast cancer cells with 24 µM paclitaxel can induce autophagy without significant inhibition of cancer cells. However, the treatment of cancer cells with a combination of paclitaxel and pristimerin can enhance autophagy and induce a remarkable suppression of cancer cells. This combination could enhance the expression of autophagy mediators including Beclin-1 and light chain 3B (LC3‑II), and also inhibited extracellular signal‑regulated kinase (ERK). The inhibition of autophagy or inducing ERK showed a reduction in cell death following treatment of cells with paclitaxel and pristimerin combination [184].
A study suggested that autophagy has a close relationship with apoptosis in v-Ha-ras-transformed fibroblasts. Exposure of v-Ha-ras-transformed NIH 3T3 cells to paclitaxel was associated with an increase in the expression of autophagy proteins such as GFP-LC3 and LC3. The inhibition of either apoptosis or autophagy in these cells led to an increase in the induction of another mechanism of cell death. This response of cells to paclitaxel indicates that both autophagic cell death and apoptosis cooperate as two partners to suppress the proliferation of these cells in the response to paclitaxel [185]. However, a study suggested that switching from apoptosis to autophagic cell death can increase the resistance of MCF-7 breast cancer cells to paclitaxel [186].
Autophagy attenuates paclitaxel sensitivity in some cancer cells
If autophagy doesn’t lead to autophagic cell death or apoptosis, it can induce resistance of cancer cells to anti-cancer agents including paclitaxel. The upregulation of some autophagy-related genes is responsible for increasing tumour resistance to paclitaxel. A study showed that the upregulation of taurine up-regulated 1 (TUG1) can enhance paclitaxel resistance in ovarian cancer cells via inducing autophagy and inhibiting apoptosis. TUG1 is a long noncoding RNA (lncRNA) and acts as an oncogene [187]. The deletion of TUG1 in ovarian tumours and cancer cells showed a reduction in the formation of autophagosomes and an increase in the induction of apoptosis. Results indicated that autophagy is a mechanism of paclitaxel resistance in ovarian cancer cells [188]. In some types of cancers such as NSCLC and ovarian cancer cells, it seems that autophagy is a mechanism for resistance to paclitaxel-induced apoptosis [189, 190].
Hypoxia is known as a key regulator of tumour resistance to therapy via inducing autophagy. The Warburg effect and glycolysis in cancer cells can upregulate HIF1-α. HIF1-α induces autophagy and resistance to paclitaxel. The selective inhibition of HIF1-α has been shown to suppress autophagy and paclitaxel resistance in HeLa cancer cells via inducing apoptosis [191]. Regulated in development and DNA damage (REDD1) is another regulator of autophagy that increase resistance to paclitaxel. REDD1 can be upregulated during DNA damage, hypoxia, and stress. It induces the expression of some inflammatory mediators such as NF-κB and NLRP3 inflammasome [192]. A study showed that the expression of REDD1 in patients with bladder urothelial carcinoma is associated with poor patient survival. Further experiments also showed that REED1 can induce autophagy in cancer cells and its knockdown is associated with the inhibition of autophagy markers. Analyses showed that REED1 can be inhibited by miR-22, which led to the sensitization of cancer cells to paclitaxel [193].
Autophagy inhibition or stimulation to induce paclitaxel-induced apoptosis
The inhibition or stimulation of autophagy may induce the induction of apoptosis, depending on the type of cancer. Furthermore, the stimulation of autophagy may induce both autophagic cell death and apoptosis in the combination with other adjuvants including paclitaxel. Most studies have shown that the inhibition of autophagy sensitizes cancer cells to paclitaxel. However, some other studies have shown that stimulation of autophagy can enhance the cytotoxicity effect of paclitaxel against breast tumours [194, 195]. The inhibition of the mammalian target of rapamycin (mTOR) is known as the most famous strategy to induce autophagy in cancer cells. A study showed that the inhibition of mTOR suppresses the phosphorylation of Akt/mTOR and inhibits the proliferation of endometrial cancer cells via inducing autophagic cell death and apoptosis in the response to paclitaxel [196].
Inducing autophagy in the response to paclitaxel may be involved in the invasion and metastasis [197]. Thus the targeting of autophagy may be effective to suppress cancer resistance and invasion. It seems that the induction of autophagy in NSCLC is a mechanism for resistance to paclitaxel [189]. An experiment evaluated the role of autophagy inhibition on the paclitaxel resistance and invasive properties of A549 cells. Inhibition of autophagy in NSCLC cells can enhance the generation of ROS and accumulation of mitochondria injury following overproduction of superoxide [198, 199]. Furthermore, the inhibition of autophagy by chloroquine attenuated the phosphorylation of Akt and accumulation of β-catenin, thus reducing metastatic properties of A549 cancer cells [200, 201]. The inhibition of autophagy also triggers ER stress, leading to the induction of apoptosis [202]. Autophagy inhibition using chloroquine has been shown to induce apoptosis in endometrial carcinoma cells too [203]. Obatoclax is a blocker of autophagy flux that may reduce the resistance of some cancer cells to paclitaxel. A study evaluated the effect of a combination of paclitaxel and obatoclax on resistant bladder cancer cells. The results showed that obatoclax can induce the blockade of the autophagic flux in paclitaxel-sensitive bladder cancer cells. However, it couldn’t remarkably inhibit autophagic flux in paclitaxel-resistant bladder cancer cells. A combination of paclitaxel and obatoclax showed a synergic blockade of autophagic flux in paclitaxel-resistant bladder cancer cells. The results also confirmed that blockade of autophagic flux correlates with the induction of apoptosis in bladder cancer cells [112].
Some other studies show the inhibition of autophagy sensitizes cancer cells to paclitaxel via accelerating apoptosis. The paclitaxel sensitization effect of autophagy inhibition has been observed in human SiHa cervical cells, triple-negative breast cancers, renal cell carcinoma, [204,205,206,207]. A clinical study evaluated the effect of autophagy inhibition using hydroxychloroquine in the combination with Gemcitabine/Nab-Paclitaxel in patients with pancreas tumours. this treatment modality resulted in better pathological tumour response and increased overall survival without a significant increase in side effects [208]. Further clinical studies need to illustrate the possible beneficial effects of autophagy inhibitors to overcome paclitaxel resistance in various types of tumours (Fig. 4).

Mechanisms of autophagy modulation by paclitaxel and its effects on the induction or suppression of autophagic cell death and apoptosis. Upregulation of PI3K following downregulation of PTEN or upregulation of some stimulators causes phosphorylation and activation of Akt and mTOR, leading to suppression of autophagic cell death and apoptosis. Paclitaxel can stimulate the activity of PTEN and also inhibit Akt to reactivate apoptosis and autophagic cell death pathways. In some cancer cells, autophagy itself can suppress apoptosis via inhibiting ER stress. However, paclitaxel may reverse this effect of autophagy. Paclitaxel also can downregulate pro-survival mediators in cancer cells via inhibiting REED1
Senescence
Cellular senescence is a type of cell death that may occur following damage to DNA, shortening of telomere, oxidative stress, and also activation of some oncogenes [209]. Senescence has some specific properties such as irreversible cell cycle arrest, dysregulation of the normal metabolism of senescent cells, and an abnormal increase in the β-galactosidase [210]. Senescence may suppress the proliferation of cancer cells. However, senescence-associated secretory phenotype (SASP) may modulate TME in the favour of tumour progression [211]. Due to these properties of cellular senescence, the effect of each anti-cancer modality on the senescence and its consequences on the tumour should be evaluated separately.
Studies have revealed that paclitaxel can induce senescence in cancer cells [212]. However, consequences of senescence can increase resistance to paclitaxel. Senescence may occur dependent or independent of p53. A study showed that paclitaxel can induce premature senescence in NSCLC cells independent of p53 and p21 [213]. However, another experiment showed that transfection of p53 into null p53 NSCLC H358 cells can induce senescence. Interestingly, results indicated that transfection of p53 could increase the resistance of cancer cells to paclitaxel [214]. Another study showed that senescence in ovarian cancer cells is associated with upregulation of p21, p16, and p53 [215]. Some adjuvants have been shown to sensitize cancer cells to paclitaxel via inducing both apoptosis and senescence [216, 217]. However, it seems that SASP can attenuate the induction of apoptosis, thus inducing resistance of cancer cells to paclitaxel [218, 219]. It has been reported that SASP following exposure of cancer cells to paclitaxel is associated with the release of some inflammatory cytokines such as IL-6 and IL-8 that induce migration and resistance of adjacent cells [220]. Similar results were observed in an in vivo model of ovarian tumours. Results of this study also showed that senescence can increase the resistance of neighbouring cells in TME to paclitaxel [221].
Pyroptosis
Pyroptosis is the inflammatory model of cell death that is associated with the activation of the inflammasome and the release of inflammatory cytokines including IL-1 and IL-18. Caspase 1 and gasdermin family proteins including gasdermin E (GSDME) and gasdermin D (GSDMD) play key roles in the activation of inflammasome and execution of pyroptosis [222]. Pyroptosis can be suggested as a target to induce cancer cell death [223]. Pyroptosis may be a mechanism for the sensitization of cancer cells to paclitaxel. A study showed that knockdown of GSDME in MCF-7 breast cancer cells can reduce paclitaxel sensitivity of cancer cells [224]. Paclitaxel has been shown to induce pyroptosis through both caspase 1/GSDMD and caspase 3/ GSDME pathways. A study examined the effect of paclitaxel on pyroptosis via the caspase 3/ GSDME pathway in A549 lung cancer cells. Results of this study indicated that although activation of caspase 3 can induce apoptosis in most cancer cells, however, it mediated cleavage of GSDME in some cancer cells, leading to pyroptosis. Results also showed that the inhibition of pyroptosis via knockdown of GSDME is associated with a reduction in cancer cell death [225]. Another experiment showed that paclitaxel stimulates pyroptosis in advanced nasopharyngeal carcinoma via inducing Caspase-1/GSDMD. Results also confirmed a significant increase in the release of IL-1 and IL-18 up to sixfold. the selective inhibition of GSDMD using siRNA caused the suppression of pyroptosis and increased resistance of cancer cells to paclitaxel [226]. In addition to the direct induction of caspase 1/GSDMD pathway and pyroptosis by paclitaxel, inducing pyroptosis by other adjuvants may be useful to ameliorate paclitaxel-resistance of cancer cells. A study confirmed that activation of caspase 1/GSDMD can sensitize A549 cells to paclitaxel via inducing pyroptosis [227]. Pyroptosis may stimulate the immune system against cancer cells in the tumour. In a murine model of the tumour, the induction of pyroptosis following exposure of the tumour to paclitaxel led to the maturation of dendritic cells and expansion of anti-tumour T lymphocytes [228]. However, the immunologic effects of pyroptosis following administration of paclitaxel need to be more elucidated (Fig. 5).

Paclitaxel can induce pyroptosis via inducing caspase 3/gasdemine E and inflammasome/caspase 1 pathways. Overproduction of ROS and inhibition of antioxidant defence system play key roles in the initiation of pyroptosis. Upregulation of caspase 3 may cause induction of either apoptosis or pyroptosis
Ferroptosis
Ferroptosis is an iron-dependent mechanism of programmed cell death. This type of cell death was introduced in 2012 [229]. Iron ions can induce the generation of ROS through the Fenton reaction. ROS can interact with phospholipids, leading to the production of lipid peroxidation. Antioxidant defence mediators including glutathione (GSH) and glutathione peroxidase 4 (GPX4) inhibit the production of lipid peroxidation, thus preventing ferroptosis. However, the inhibition of the antioxidant defence system during oxidative stress may lead to ferroptosis [230]. Targeting ferroptosis has been suggested as a strategy for cancer therapy [231]. Paclitaxel may induce cancer cell death via triggering ferroptosis. Furthermore, the stimulation of ferroptosis using other agents may be useful to overcome resistance to paclitaxel. An experiment showed that a low concentration of paclitaxel induces ferroptosis in mutant p53 hypopharyngeal squamous cell carcinoma. Furthermore, treatment of cells with a combination of paclitaxel and RSL3 showed a synergic effect on the induction of ferroptosis. This combination inhibited the activity of GPX4, leading to lipid peroxidation and ferroptosis. Interestingly, results indicated that the status of p53 plays a key role in the promotion of ferroptosis. Upregulation of mutant p53 was associated with an increase in ferroptosis via inhibiting SLC7A11 (xCT) in the treated cancer cells [232]. Another study also confirmed the role of xCT inhibition in the induction of ferroptosis. The inhibition of xCT using sulfasalazine showed that induced ferroptosis in paclitaxel-resistant uterine serous carcinoma. This was associated with an increase in the generation of ROS and upregulation of JNK in paclitaxel-resistant cancer cells, not in paclitaxel-sensitive cells [233]. These results were observed for paclitaxel-tolerant persister cancer cells using different xCT inhibitors [234]. Some other studies have revealed that the induction of ferroptosis enhances the anti-tumour efficiency of paclitaxel, while its inhibition increases resistance to paclitaxel [235,236,237].
Necroptosis
Necroptosis is a type of cell death that occurs independently of caspase proteins. Necroptosis can be induced following activation of the immune system, the release of anti-tumour molecules including TNF-α and FasL, and also upregulation of some receptors such as toll-like receptors (TLRs) [238]. The main mediators of necroptosis are including receptor-interacting protein kinase 1 (RIP1), RIP3, and also RIP3-mixed lineage kinase domain-like protein (MLKL) [239]. Necroptosis is an immunologic type of cell death; thus it can enhance anti-tumour immunity against cancer cells. On the other hand, paclitaxel can stimulate the immune system and release inflammatory cytokines, which may induce necroptosis in cancer cells. However, this effect of paclitaxel needs to be elucidated. Some limited experiments have been performed to show the induction of necroptosis by paclitaxel. A study showed that treatment of paclitaxel induces necroptosis in A549 lung adenocarcinoma cells via phosphorylation of caspase-8 and also activation of RIPK1 and RIPK3. Results of this study indicated that phosphorylation of caspase 8 is associated with resistance of cancer cells to both apoptosis and necroptosis. The dephosphorylation of caspase 8 using a c-Src inhibitor led to a synergic induction of necroptosis by paclitaxel [240]. Another study reported that Fluoxetine (an anti-depression drug with anti-cancer properties) can amplify necroptosis by paclitaxel [241]. Targeting necroptosis in apoptosis-resistant cancer cells is a strategy to kill cancer cells using some specific anti-cancer drugs such as paclitaxel [242]. However, paclitaxel-induced necroptosis may lead to upregulation of some tumour-promoting molecules such as C-X-C-motif chemokine receptor-2 (CXCR2) that need to be considered [243].
Summary and conclusion
To date, a large number of anti-cancer drugs have been developed or discovered. Herbal-derived drugs are among anti-tumour candidates since long years ago. Paclitaxel is a herb-derived agent that has been used to treat some malignancies since a long time ago. Similar to other herbal-derived agents, paclitaxel has low bioavailability and absorption in the intestine. To overcome this problem, scientists have developed some carriers such as micelles, liposomes, nanocapsules, and some others. Evidence shows that these carriers can improve the anti-tumour activity of drugs like paclitaxel. First evaluations indicated that paclitaxel induces cell death in cancers via inhibiting mitosis. However, further experiments during the two last decades show that paclitaxel can induce cancer-killing via modulating several signalling pathways.
Killing all cancer cells with at least normal tissue side effects is an ideal goal in cancer therapy. Knowledge of how cancer cells respond to different types of cell death can illustrate mechanisms of resistance to anti-cancer drugs and also provide new ways to overcome cancer resistance. The first evaluations showed that paclitaxel induces cancer cell death during both mitosis and interphase. Paclitaxel can induce apoptosis via stimulating TSGs such as p53 and PTEN. However, it seems that cancers with mutated p53 are vulnerable to mitotic catastrophe following treatment with paclitaxel. Furthermore, the induction of endogenous ROS generation by paclitaxel can induce both types of cell death. Paclitaxel can stimulate anti-tumour immunity, leading to an increase in the release of pro-apoptosis molecules like FasL and TRAIL, and also some cytokines such as TNF-α and IFN-γ. All these products of anti-tumour immune cells induce apoptosis via binding to their receptors such as Fas, FADD, TNFR, and dead receptors. These molecules can induce extrinsic apoptosis via stimulating caspase 8 and 10.
The inhibition of the PI3K/Akt pathway plays a key role in overcoming apoptosis resistance in cancer cells. paclitaxel can target these proteins via different pathways. Paclitaxel can activate PTEN by ROS overproduction, which leads to the inhibition of the PI3K signalling pathway. Furthermore, overexpression of miR-22 by paclitaxel can induce the activity of PTEN. Paclitaxel also can upregulate miR-145, which directly inhibits Akt activity. The suppression of the PI3K/Akt pathway is associated with the downregulation of Bcl-2. An increase in the Bax/Bcl-2 ratio is associated with the induction of mitochondrial apoptosis. Another pathway of apoptosis induction by paclitaxel is the phosphorylation of Foxo3, which causes upregulation of BIM and downregulation of Bcl-2.
Other mechanisms that can be involved in the anti-tumour activity of paclitaxel are including pyroptosis, ferroptosis, necroptosis, senescence, and also autophagic cell death. It seems that the induction of oxidative stress plays a central role in the induction of these types of cell death. Oxidative stress following treatment with paclitaxel may cause telomere shortening, leading to senescence. An increase in the release of pro-inflammatory cytokines and pro-apoptosis molecules by the immune system can augment the probability of necroptosis following oxidative injury. In addition to ROS generation by mitochondria, ER stress and the inhibition of antioxidant enzymes and peptides such as GSH and GPX4 are involved in oxidative injury and cell death caused by paclitaxel. The inhibition of GPX4 and increased oxidative stress can cause ferroptosis or pyroptosis. Increased release of iron ions is a marker of ferroptosis. However, pyroptosis is associated with upregulation of caspase 1/gasdermine D and caspase 3/gasdermine E.
The induction of cell death or survival by autophagy is a complicated issue during cancer therapy. Although autophagy is known as a mechanism to increase the resistance of cells to stress conditions, some experiments show that it may induce cancer cell death via inducing apoptosis or autophagic cell death. Autophagy may suppress ER stress in cancer cells, leading to resistance to apoptosis and autophagic cell death. However, paclitaxel has been shown to suppress autophagy, leading to ER stress. Paclitaxel also may induce autophagic cell death and apoptosis via suppressing REED1. The combination of paclitaxel with some other adjuvant to suppress autophagy may be interesting to enhance therapy effectiveness.
Emerging evidence from this review shows that paclitaxel can induce killing cancer cells via enhancing apoptosis, mitotic catastrophe, senescence, pyroptosis, and ferroptosis. Furthermore, paclitaxel may induce autophagic cell death in some cancers. However, an increase in autophagy may be involved in the suppression of other types of cell death. It seems that considering the type of cell death or upregulation of survival-related mediators should be noted for each type of cancer. Using some combinations with paclitaxel may enhance tumour control probability.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
References
Papaccio F, Paino F, Regad T, Papaccio G, Desiderio V, Tirino V (2017) Concise review: cancer cells, cancer stem cells, and mesenchymal stem cells: influence in cancer development. Stem Cells Transl Med 6(12):2115–2125
Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M et al (2005) EGFR mutation and resistance of non–small-cell lung cancer to gefitinib. N Engl J Med 352(8):786–792
Shah MA, Schwartz GK (2001) Cell cycle-mediated drug resistance: an emerging concept in cancer therapy. Clin Cancer Res 7(8):2168–2181
Luqmani Y (2005) Mechanisms of drug resistance in cancer chemotherapy. Med Princ Pract 14(Suppl. 1):35–48
Wang H, Feng Z, Wang Y, Zhou R, Yang Z, Xu B (2016) Integrating enzymatic self-assembly and mitochondria targeting for selectively killing cancer cells without acquired drug resistance. J Am Chem Soc 138(49):16046–16055
Mortezaee K, Najafi M (2021) Immune system in cancer radiotherapy: resistance mechanisms and therapy perspectives. Crit Rev Oncol Hematol 157:103180
Fu X, Li M, Tang C, Huang Z, Najafi M (2021) Targeting of cancer cell death mechanisms by resveratrol: a review. Apoptosis 26(11):561–573. https://doi.org/10.1007/s10495-021-01689-7
Yu C, Yang B, Najafi M (2021) Targeting of cancer cell death mechanisms by curcumin: implications to cancer therapy. Basic Clin Pharmacol Toxicol 129(6):397–415. https://doi.org/10.1111/bcpt.13648
Hilska M, Collan YU, Laine VJO, Kössi J, Hirsimäki P, Laato M et al (2005) The significance of tumor markers for proliferation and apoptosis in predicting survival in colorectal cancer. Dis Colon Rectum 48(12):2197–2208
Aaltomaa S, Kärjä V, Lipponen P, Isotalo T, Kankkunen J, Talja M et al (2006) Expression of Ki-67, cyclin D1 and apoptosis markers correlated with survival in prostate cancer patients treated by radical prostatectomy. Anticancer Res 26(6C):4873–4878
Fu D, Lu C, Qu X, Li P, Chen K, Shan L et al (2019) LncRNA TTN-AS1 regulates osteosarcoma cell apoptosis and drug resistance via the miR-134-5p/MBTD1 axis. Aging (Albany NY) 11(19):8374
Mizushima N (2007) Autophagy: process and function. Genes Dev 21(22):2861–2873
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV et al (2012) Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 19(1):107–120. https://doi.org/10.1038/cdd.2011.96
Ramirez JAZ, Romagnoli GG, Kaneno R (2021) Inhibiting autophagy to prevent drug resistance and improve anti-tumor therapy. Life Sci 265:118745
Reyes-Castellanos G, Abdel Hadi N, Carrier A (2022) Autophagy contributes to metabolic reprogramming and therapeutic resistance in pancreatic tumors. Cells 11(3):426
Usman RM, Razzaq F, Akbar A, Farooqui AA, Iftikhar A, Latif A et al (2021) Role and mechanism of autophagy-regulating factors in tumorigenesis and drug resistance. Asia Pac J Clin Oncol 17(3):193–208
Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G et al (2020) ROS in cancer therapy: the bright side of the moon. Exp Mol Med 52(2):192–203
Calaf GM, Ponce-Cusi R, Carrión F (2018) Curcumin and paclitaxel induce cell death in breast cancer cell lines. Oncol Rep 40(4):2381–2388
Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT (1971) Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J Am Chem Soc 93(9):2325–2327
Li D, Fu D, Zhang Y, Ma X, Gao L, Wang X et al (2017) Isolation, purification, and identification of taxol and related taxanes from taxol-producing fungus Aspergillus niger subsp. taxi. J Microbiol Biotechnol 27(8):1379–1385
Menzin AW, King SA, Aikins JK, Mikuta JJ, Rubin SC (1994) Taxol (paclitaxel) was approved by FDA for the treatment of patients with recurrent ovarian cancer. Gynecol Oncol 54(1):103
Cortazar P, Justice R, Johnson J, Sridhara R, Keegan P, Pazdur R (2012) US Food and Drug Administration approval overview in metastatic breast cancer. J Clin Oncol 30(14):1705–1711. https://doi.org/10.1200/JCO.2011.39.2613
De Luca R, Profita G, Cicero G (2019) Nab-paclitaxel in pretreated metastatic breast cancer: evaluation of activity, safety, and quality of life. Onco Targets Ther 12:1621
Kelly WK, Curley T, Slovin S, Heller G, McCaffrey J, Bajorin D et al (2001) Paclitaxel, estramustine phosphate, and carboplatin in patients with advanced prostate cancer. J Clin Oncol 19(1):44–53
Ramalingam S, Belani CP (2004) Paclitaxel for non-small cell lung cancer. Expert Opin Pharmacother 5(8):1771–1780
Park DC, Kim JH, Lew YO, Kim DH, Namkoong SE (2004) Phase II trial of neoadjuvant paclitaxel and cisplatin in uterine cervical cancer. Gynecol Oncol 92(1):59–63
Elstad NL, Fowers KD (2009) OncoGel (ReGel/paclitaxel)—clinical applications for a novel paclitaxel delivery system. Adv Drug Deliv Rev 61(10):785–794
Foland TB, Dentler WL, Suprenant KA, Gupta ML Jr, Himes RH (2005) Paclitaxel-induced microtubule stabilization causes mitotic block and apoptotic-like cell death in a paclitaxel-sensitive strain of Saccharomyces cerevisiae. Yeast (Chichester, England) 22(12):971–978. https://doi.org/10.1002/yea.1284
Priyadarshini K, Keerthi AU (2012) Paclitaxel against cancer: a short review. Med Chem 2(7):139–141
Peltier S, Oger JM, Lagarce F, Couet W, Benoît JP (2006) Enhanced oral paclitaxel bioavailability after administration of paclitaxel-loaded lipid nanocapsules. Pharm Res 23(6):1243–1250. https://doi.org/10.1007/s11095-006-0022-2
Lacoeuille F, Hindre F, Moal F, Roux J, Passirani C, Couturier O et al (2007) In vivo evaluation of lipid nanocapsules as a promising colloidal carrier for paclitaxel. Int J Pharm 344(1–2):143–149. https://doi.org/10.1016/j.ijpharm.2007.06.014
Torne SJ, Ansari KA, Vavia PR, Trotta F, Cavalli R (2010) Enhanced oral paclitaxel bioavailability after administration of paclitaxel-loaded nanosponges. Drug Deliv 17(6):419–425. https://doi.org/10.3109/10717541003777233
Yang FH, Zhang Q, Liang QY, Wang SQ, Zhao BX, Wang YT et al (2015) Bioavailability enhancement of paclitaxel via a novel oral drug delivery system: paclitaxel-loaded glycyrrhizic acid micelles. Molecules 20(3):4337–4356. https://doi.org/10.3390/molecules20034337
Yang T, Feng J, Zhang Q, Wu W, Mo H, Huang L et al (2020) l-Carnitine conjugated chitosan-stearic acid polymeric micelles for improving the oral bioavailability of paclitaxel. Drug Deliv 27(1):575–584. https://doi.org/10.1080/10717544.2020.1748762
Chowdhury N, Singh M (2020) Current development of oral taxane formulations: a review. Crit Rev Ther Drug Carrier Syst 37(3):205–227. https://doi.org/10.1615/CritRevTherDrugCarrierSyst.2020029699
Ma P, Mumper RJ (2013) Paclitaxel nano-delivery systems: a comprehensive review. J Nanomed Nanotechnol 4(2):1000164. https://doi.org/10.4172/2157-7439.1000164
Foote M (2007) Using nanotechnology to improve the characteristics of antineoplastic drugs: improved characteristics of nab-paclitaxel compared with solvent-based paclitaxel. Biotechnol Annu Rev 13:345–357. https://doi.org/10.1016/s1387-2656(07)13012-x
Lee S, Margolin K (2011) Cytokines in cancer immunotherapy. Cancers (Basel) 3(4):3856–3893
Borst J, Ahrends T, Bąbała N, Melief CJ, Kastenmüller W (2018) CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol 18(10):635–647
Mirzaei S, Mohammadi AT, Gholami MH, Hashemi F, Zarrabi A, Zabolian A et al (2021) Nrf2 signaling pathway in cisplatin chemotherapy: potential involvement in organ protection and chemoresistance. Pharmacol Res 167:105575. https://doi.org/10.1016/j.phrs.2021.105575
Najafi M, Mortezaee K, Rahimifard M, Farhood B, Haghi-Aminjan H (2020) The role of curcumin/curcuminoids during gastric cancer chemotherapy: a systematic review of non-clinical study. Life Sci 257:118051. https://doi.org/10.1016/j.lfs.2020.118051
Mortezaee K, Narmani A, Salehi M, Bagheri H, Farhood B, Haghi-Aminjan H et al (2021) Synergic effects of nanoparticles-mediated hyperthermia in radiotherapy/chemotherapy of cancer. Life Sci 269:119020. https://doi.org/10.1016/j.lfs.2021.119020
Farhood B, Mortezaee K, Haghi-Aminjan H, Khanlarkhani N, Salehi E, Nashtaei MS et al (2019) A systematic review of radiation-induced testicular toxicities following radiotherapy for prostate cancer. J Cell Physiol 234(9):14828–14837. https://doi.org/10.1002/jcp.28283
Farhood B, Ashrafizadeh M, Khodamoradi E, Hoseini-Ghahfarokhi M, Afrashi S, Musa AE et al (2020) Targeting of cellular redox metabolism for mitigation of radiation injury. Life Sci 250:117570. https://doi.org/10.1016/j.lfs.2020.117570
Wu Q, Allouch A, Martins I, Brenner C, Modjtahedi N, Deutsch E et al (2017) Modulating both tumor cell death and innate immunity is essential for improving radiation therapy effectiveness. Front Immunol 8:613
Wang T-H, Wang H-S, Soong Y-K (2000) Paclitaxel-induced cell death. Cancer 88(11):2619–2628. https://doi.org/10.1002/1097-0142(20000601)88:11%3c2619::AID-CNCR26%3e3.0.CO;2-J
Choi KH, Jeon JY, Lee Y-E, Kim SW, Kim SY, Yun YJ et al (2019) Synergistic activity of paclitaxel, sorafenib, and radiation therapy in advanced renal cell carcinoma and breast cancer. Transl Oncol 12(2):381–388
Fan W (1999) Possible mechanisms of paclitaxel-induced apoptosis. Biochem Pharmacol 57(11):1215–1221
Nawara HM, Afify SM, Hassan G, Zahra MH, Seno A, Seno M (2021) Paclitaxel-based chemotherapy targeting cancer stem cells from mono-to combination therapy. Biomedicines 9(5):500
Javeed A, Ashraf M, Riaz A, Ghafoor A, Afzal S, Mukhtar MM (2009) Paclitaxel and immune system. Eur J Pharm Sci 38(4):283–290
Papaioannou NE, Beniata OV, Vitsos P, Tsitsilonis O, Samara P (2016) Harnessing the immune system to improve cancer therapy. Ann Transl Med 4(14):261
Mullins DW, Burger CJ, Elgert KD (1999) Paclitaxel enhances macrophage IL-12 production in tumor-bearing hosts through nitric oxide. J Immunol 162(11):6811–6818
Nguyen KG, Vrabel MR, Mantooth SM, Hopkins JJ, Wagner ES, Gabaldon TA et al (2020) Localized interleukin-12 for cancer immunotherapy. Front Immunol. https://doi.org/10.3389/fimmu.2020.575597
Vicari AP, Luu R, Zhang N, Patel S, Makinen SR, Hanson DC et al (2009) Paclitaxel reduces regulatory T cell numbers and inhibitory function and enhances the anti-tumor effects of the TLR9 agonist PF-3512676 in the mouse. Cancer Immunol Immunother 58(4):615–628. https://doi.org/10.1007/s00262-008-0586-2
Shen J, Chen C, Li Z, Hu S (2020) Paclitaxel promotes tumor-infiltrating macrophages in breast cancer. Technol Cancer Res Treat. https://doi.org/10.1177/1533033820945821
Wanderley CW, Colón DF, Luiz JPM, Oliveira FF, Viacava PR, Leite CA et al (2018) Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1 profile in a TLR4-dependent manner. Cancer Res 78(20):5891–5900. https://doi.org/10.1158/0008-5472.can-17-3480
Feng R, Morine Y, Ikemoto T, Imura S, Iwahashi S, Saito Y et al (2018) Nab-paclitaxel interrupts cancer-stromal interaction through C-X-C motif chemokine 10-mediated interleukin-6 downregulation in vitro. Cancer Sci 109(8):2509–2519. https://doi.org/10.1111/cas.13694
Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L et al (2008) Molecular characteristics of immunogenic cancer cell death. Cell Death Differ 15(1):3–12
Lei X, Lei Y, Li J-K, Du W-X, Li R-G, Yang J et al (2020) Immune cells within the tumor microenvironment: biological functions and roles in cancer immunotherapy. Cancer Lett 470:126–133
Jafarzadeh E, Montazeri V, Aliebrahimi S, Sezavar AH, Ghahremani MH, Ostad SN (2022) Combined regimens of cisplatin and metformin in cancer therapy: a systematic review and meta-analysis. Life Sci. https://doi.org/10.1016/j.lfs.2022.120680
Moslehi M, Moazamiyanfar R, Dakkali MS, Rezaei S, Rastegar-Pouyani N, Jafarzadeh E et al (2022) Modulation of the immune system by melatonin; implications for cancer therapy. Int Immunopharmacol 108:108890
Salemme V, Centonze G, Cavallo F, Defilippi P, Conti L (2021) The crosstalk between tumor cells and the immune microenvironment in breast cancer: implications for immunotherapy. Front Oncol. https://doi.org/10.3389/fonc.2021.610303
Muraro E, Comaro E, Talamini R, Turchet E, Miolo G, Scalone S et al (2015) Improved Natural Killer cell activity and retained anti-tumor CD8+ T cell responses contribute to the induction of a pathological complete response in HER2-positive breast cancer patients undergoing neoadjuvant chemotherapy. J Transl Med 13(1):204. https://doi.org/10.1186/s12967-015-0567-0
Bhola N, Arteaga C (2011) PD08-04: inhibition of the TGFb/TGFbR2 pathway prevents enrichment of drug-resistant breast cancer stem cells by anti-cancer chemotherapy. Cancer Res 71(24_Supplement):PD08-4-PD-4. https://doi.org/10.1158/0008-5472.SABCS11-PD08-04
Park SY, Kim MJ, Park SA, Kim JS, Min KN, Kim DK et al (2015) Combinatorial TGF-β attenuation with paclitaxel inhibits the epithelial-to-mesenchymal transition and breast cancer stem-like cells. Oncotarget 6(35):37526–37543. https://doi.org/10.18632/oncotarget.6063
Zhang R, Wei Y-H, Zhao C-Y, Song H-Y, Shen N, Cui X et al (2018) EDIL3 depletion suppress epithelial-mesenchymal transition of lens epithelial cells via transforming growth factor β pathway. Int J Ophthalmol 11(1):18
Jeong D, Ban S, Oh S, Jin Lee S, Yong Park S, Koh YW (2017) Prognostic significance of EDIL3 expression and correlation with mesenchymal phenotype and microvessel density in lung adenocarcinoma. Sci Rep 7(1):8649. https://doi.org/10.1038/s41598-017-08851-9
Jiang SH, Wang Y, Yang JY, Li J, Feng MX, Wang YH et al (2016) Overexpressed EDIL3 predicts poor prognosis and promotes anchorage-independent tumor growth in human pancreatic cancer. Oncotarget 7(4):4226–4240. https://doi.org/10.18632/oncotarget.6772
Xia H, Chen J, Shi M, Gao H, Sekar K, Seshachalam VP et al (2015) EDIL3 is a novel regulator of epithelial-mesenchymal transition controlling early recurrence of hepatocellular carcinoma. J Hepatol 63(4):863–873. https://doi.org/10.1016/j.jhep.2015.05.005
Gasca J, Flores ML, Jiménez-Guerrero R, Sáez ME, Barragán I, Ruíz-Borrego M et al (2020) EDIL3 promotes epithelial–mesenchymal transition and paclitaxel resistance through its interaction with integrin αVβ3 in cancer cells. Cell Death Discov 6(1):86. https://doi.org/10.1038/s41420-020-00322-x
Xu J-H, Hu S-L, Shen G-D, Shen G (2016) Tumor suppressor genes and their underlying interactions in paclitaxel resistance in cancer therapy. Cancer Cell Int 16(1):1–10
Chabalier C, Lamare C, Racca C, Privat M, Valette A, Larminat F (2006) BRCA1 downregulation leads to premature inactivation of spindle checkpoint and confers paclitaxel resistance. Cell Cycle 5(9):1001–1007
Sung M, Giannakakou P (2014) BRCA1 regulates microtubule dynamics and taxane-induced apoptotic cell signaling. Oncogene 33(11):1418–1428
Gilmore PM, McCabe N, Quinn JE, Kennedy RD, Gorski JJ, Andrews HN et al (2004) BRCA1 interacts with and is required for paclitaxel-induced activation of mitogen-activated protein kinase kinase kinase 3. Cancer Res 64(12):4148–4154
Li J, Zhang Y, Zhao J, Kong F, Chen Y (2011) Overexpression of miR-22 reverses paclitaxel-induced chemoresistance through activation of PTEN signaling in p53-mutated colon cancer cells. Mol Cell Biochem 357(1):31–38. https://doi.org/10.1007/s11010-011-0872-8
Wang S-Q, Wang C, Chang L-M, Zhou K-R, Wang J-W, Ke Y et al (2016) Geridonin and paclitaxel act synergistically to inhibit the proliferation of gastric cancer cells through ROS-mediated regulation of the PTEN/PI3K/Akt pathway. Oncotarget 7(45):72990–73002. https://doi.org/10.18632/oncotarget.12166
Liu Q, Sui R, Li R, Miao J, Liu J (2015) Biological characteristics of Taxol-resistant ovarian cancer cells and reversal of Taxol resistance by adenovirus expressing p53. Mol Med Rep 11(2):1292–1297
Guntur VP, Waldrep JC, Guo JJ, Selting K, Dhand R (2010) Increasing p53 protein sensitizes non-small cell lung cancer to paclitaxel and cisplatin in vitro. Anticancer Res 30(9):3557–3564
Vikhanskaya F, Vignati S, Beccaglia P, Ottoboni C, Russo P, D’Incalci M et al (1998) Inactivation of p53 in a human ovarian cancer cell line increases the sensitivity to paclitaxel by inducing G2/M arrest and apoptosis. Exp Cell Res 241(1):96–101
Debernardis D, Siré EG, De Feudis P, Vikhanskaya F, Valenti M, Russo P et al (1997) p53 status does not affect sensitivity of human ovarian cancer cell lines to paclitaxel. Cancer Res 57(5):870–874
Liu R, Chen Y, Liu G, Li C, Song Y, Cao Z et al (2020) PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis 11(9):1–12
Hutti JE, Pfefferle AD, Russell SC, Sircar M, Perou CM, Baldwin AS (2012) Oncogenic PI3K mutations lead to NF-κB-dependent cytokine expression following growth factor deprivation. Cancer Res 72(13):3260–3269. https://doi.org/10.1158/0008-5472.CAN-11-4141
Spangle JM, Roberts TM, Zhao JJ (2017) The emerging role of PI3K/AKT-mediated epigenetic regulation in cancer. Biochim Biophys Acta Rev Cancer 1868(1):123–131. https://doi.org/10.1016/j.bbcan.2017.03.002
Butler DE, Marlein C, Walker HF, Frame FM, Mann VM, Simms MS et al (2017) Inhibition of the PI3K/AKT/mTOR pathway activates autophagy and compensatory Ras/Raf/MEK/ERK signalling in prostate cancer. Oncotarget 8(34):56698–56713. https://doi.org/10.18632/oncotarget.18082
Gu L, Zhu N, Findley HW, Zhou M (2004) Loss of PTEN expression induces NF-kB via PI3K/Akt pathway involving resistance to chemotherapy in acute lymphoblastic leukemia cell lines. Blood 104(11):4438
Álvarez-Garcia V, Tawil Y, Wise HM, Leslie NR (2019) Mechanisms of PTEN loss in cancer: it’s all about diversity. Semin Cancer Biol 59:66–79. https://doi.org/10.1016/j.semcancer.2019.02.001
Du F, Wu X, Liu Y, Wang T, Qi X, Mao Y et al (2013) Acquisition of paclitaxel resistance via PI3K-dependent epithelial-mesenchymal transition in A2780 human ovarian cancer cells. Oncol Rep 30(3):1113–1118. https://doi.org/10.3892/or.2013.2567
Chen D, Lin X, Zhang C, Liu Z, Chen Z, Li Z et al (2018) Dual PI3K/mTOR inhibitor BEZ235 as a promising therapeutic strategy against paclitaxel-resistant gastric cancer via targeting PI3K/Akt/mTOR pathway. Cell Death Dis 9(2):1–11
Papadopoulos EI, Scorilas A (2015) Cisplatin and paclitaxel alter the expression pattern of miR-143/145 and miR-183/96/182 clusters in T24 bladder cancer cells. Clin Transl Sci 8(6):668–675. https://doi.org/10.1111/cts.12323
Xin Z, Tong Z, Tan J, Liu C (2021) MicroRNA-145-5p aggravates cell apoptosis and oxidative stress in tongue squamous cell carcinoma. Exp Ther Med 21(4):373. https://doi.org/10.3892/etm.2021.9804
Wang J, Sun Z, Yan S, Gao F (2019) Effect of miR-145 on gastric cancer cells. Mol Med Rep 19(5):3403–3410. https://doi.org/10.3892/mmr.2019.10015
Das TP, Suman S, Alatassi H, Ankem MK, Damodaran C (2016) Inhibition of AKT promotes FOXO3a-dependent apoptosis in prostate cancer. Cell Death Dis 7(2):e2111. https://doi.org/10.1038/cddis.2015.403
Sunters A, Madureira PA, Pomeranz KM, Aubert M, Brosens JJ, Cook SJ et al (2006) Paclitaxel-induced nuclear translocation of FOXO3a in breast cancer cells is mediated by c-Jun NH2-terminal kinase and Akt. Cancer Res 66(1):212–220
Kim S-H, Juhnn Y-S, Song Y-S (2007) Akt involvement in paclitaxel chemoresistance of human ovarian cancer cells. Ann N Y Acad Sci 1095(1):82–89. https://doi.org/10.1196/annals.1397.012
Li Y, Chen K, Li L, Li R, Zhang J, Ren W (2015) Overexpression of SOX2 is involved in paclitaxel resistance of ovarian cancer via the PI3K/Akt pathway. Tumor Biol 36(12):9823–9828. https://doi.org/10.1007/s13277-015-3561-5
Li D, Zhao L-N, Zheng X-L, Lin P, Lin F, Li Y et al (2014) Sox2 is involved in paclitaxel resistance of the prostate cancer cell line PC-3 via the PI3K/Akt pathway. Mol Med Rep 10(6):3169–3176. https://doi.org/10.3892/mmr.2014.2630
Ozen M, Karatas OF, Gulluoglu S, Bayrak OF, Sevli S, Guzel E et al (2015) Overexpression of miR-145–5p inhibits proliferation of prostate cancer cells and reduces SOX2 expression. Cancer Investig 33(6):251–258
Ying L, Zhu Z, Xu Z, He T, Li E, Guo Z et al (2015) Cancer associated fibroblast-derived hepatocyte growth factor inhibits the paclitaxel-induced apoptosis of lung cancer A549 cells by up-regulating the PI3K/Akt and GRP78 signaling on a microfluidic platform. PLoS ONE 10(6):e0129593. https://doi.org/10.1371/journal.pone.0129593
Kim K-J, Kim J-W, Sung JH, Suh KJ, Lee JY, Kim SH et al (2020) PI3K-targeting strategy using alpelisib to enhance the antitumor effect of paclitaxel in human gastric cancer. Sci Rep 10(1):12308. https://doi.org/10.1038/s41598-020-68998-w
Lin Y-H, Chen BY-H, Lai W-T, Wu S-F, Guh J-H, Cheng A-L et al (2015) The Akt inhibitor MK-2206 enhances the cytotoxicity of paclitaxel (Taxol) and cisplatin in ovarian cancer cells. Naunyn-Schmiedeberg’s Arch Pharmacol 388(1):19–31. https://doi.org/10.1007/s00210-014-1032-y
Schmid P, Abraham J, Chan S, Wheatley D, Brunt AM, Nemsadze G et al (2020) Capivasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer: the PAKT trial. J Clin Oncol 38(5):423–433. https://doi.org/10.1200/jco.19.00368
Choudhary GS, Al-harbi S, Mazumder S, Hill BT, Smith MR, Bodo J et al (2015) MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis 6(1):e1593. https://doi.org/10.1038/cddis.2014.525
Chan G, Nogalski MT, Bentz GL, Smith MS, Parmater A, Yurochko AD (2010) PI3K-dependent upregulation of Mcl-1 by human cytomegalovirus is mediated by epidermal growth factor receptor and inhibits apoptosis in short-lived monocytes. J Immunol 184(6):3213–3222. https://doi.org/10.4049/jimmunol.0903025
Song T, Chai G, Liu Y, Xie M, Chen Q, Yu X et al (2015) Mechanism of synergy of BH3 mimetics and paclitaxel in chronic myeloid leukemia cells: Mcl-1 inhibition. Eur J Pharm Sci 70:64–71. https://doi.org/10.1016/j.ejps.2015.01.003
Song L, Coppola D, Livingston S, Cress WD, Haura EB (2005) Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol Ther 4(3):267–276
Yamanaka K, Miyake H, Zangemeister-wittke U, Jansen B, Gleave M (2004) Novel bispecific antisense oligonucleotides inhibiting both Bcl-2 and Bcl-xL expression induce apoptosis and enhance chemosensitivity in human androgen-independent prostate cancer cells. Cancer Res 64(7_Supplement):677
Kasai S, Sasaki T, Watanabe A, Nishiya M, Yasuhira S, Shibazaki M et al (2017) Bcl-2/Bcl-xL inhibitor ABT-737 sensitizes pancreatic ductal adenocarcinoma to paclitaxel-induced cell death. Oncol Lett 14(1):903–908. https://doi.org/10.3892/ol.2017.6211
Parrondo R, de Las PA, Reiner T, Perez-Stable C (2013) ABT-737, a small molecule Bcl-2/Bcl-xL antagonist, increases antimitotic-mediated apoptosis in human prostate cancer cells. PeerJ 1:e144
Basu A, Haldar S (2003) Identification of a novel Bcl-xL phosphorylation site regulating the sensitivity of taxol- or 2-methoxyestradiol-induced apoptosis. FEBS Lett 538(1):41–47. https://doi.org/10.1016/S0014-5793(03)00131-5
Flores ML, Castilla C, Ávila R, Ruiz-Borrego M, Sáez C, Japón MA (2012) Paclitaxel sensitivity of breast cancer cells requires efficient mitotic arrest and disruption of Bcl-xL/Bak interaction. Breast Cancer Res Treat 133(3):917–928. https://doi.org/10.1007/s10549-011-1864-9
Or C-HR, Huang C-W, Chang C-C, Lai Y-C, Chen Y-J, Chang C-C (2020) Obatoclax, a Pan-BCL-2 inhibitor, downregulates survivin to induce apoptosis in human colorectal carcinoma cells via suppressing WNT/β-catenin signaling. Int J Mol Sci 21(5):1773
Jiménez-Guerrero R, Gasca J, Flores ML, Pérez-Valderrama B, Tejera-Parrado C, Medina R et al (2018) Obatoclax and paclitaxel synergistically induce apoptosis and overcome paclitaxel resistance in urothelial cancer cells. Cancers (Basel) 10(12):490
Stamelos VA, Fisher N, Bamrah H, Voisey C, Price JC, Farrell WE et al (2016) The BH3 mimetic obatoclax accumulates in lysosomes and causes their alkalinization. PLoS ONE 11(3):e0150696. https://doi.org/10.1371/journal.pone.0150696
Siveen KS, Sikka S, Surana R, Dai X, Zhang J, Kumar AP et al (2014) Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta Rev Cancer 1845(2):136–154
Wang S, Yao Y, Yao M, Fu P, Wang W (2018) Interleukin-22 promotes triple negative breast cancer cells migration and paclitaxel resistance through JAK-STAT3/MAPKs/AKT signaling pathways. Biochem Biophys Res Commun 503(3):1605–1609. https://doi.org/10.1016/j.bbrc.2018.07.088
Yang C, He L, He P, Liu Y, Wang W, He Y et al (2015) Increased drug resistance in breast cancer by tumor-associated macrophages through IL-10/STAT3/bcl-2 signaling pathway. Med Oncol 32(2):14
Wang L, Zhang F, Cui JY, Chen L, Chen YT, Liu BW (2018) CAFs enhance paclitaxel resistance by inducing EMT through the IL-6/JAK2/STAT3 pathway. Oncol Rep 39(5):2081–2090. https://doi.org/10.3892/or.2018.6311
Zhang X, Wu X, Zhang F, Mo S, Lu Y, Wei W et al (2017) Paclitaxel induces apoptosis of esophageal squamous cell carcinoma cells by downregulating STAT3 phosphorylation at Ser727. Oncol Rep 37(4):2237–2244. https://doi.org/10.3892/or.2017.5503
Liu H, Tekle C, Chen Y-W, Kristian A, Zhao Y, Zhou M et al (2011) B7–H3 silencing increases paclitaxel sensitivity by abrogating Jak2/Stat3 phosphorylation. Mol Cancer Ther 10(6):960–971
Sun C-C, Li S-J, Zhang F, Zhang Y-D, Zuo Z-Y, Xi Y-Y et al (2016) The novel miR-9600 suppresses tumor progression and promotes paclitaxel sensitivity in non–small-cell lung cancer through altering STAT3 expression. Mol Ther Nucleic Acids 5:e387. https://doi.org/10.1038/mtna.2016.96
Gao J, Shao Z, Yan M, Fu T, Zhang L, Yan Y (2018) Targeted regulationof STAT3 by miR-29a in mediating Taxol resistance of nasopharyngeal carcinoma cell line CNE-1. Cancer Biomark 22:641–648. https://doi.org/10.3233/CBM-170964
Su W-P, Cheng F-Y, Shieh D-B, Yeh C-S, Su W-C (2012) PLGA nanoparticles codeliver paclitaxel and Stat3 siRNA to overcome cellular resistance in lung cancer cells. Int J Nanomed 7:4269–4283. https://doi.org/10.2147/IJN.S33666
Fan Z, Cui H, Yu H, Ji Q, Kang L, Han B et al (2016) MiR-125a promotes paclitaxel sensitivity in cervical cancer through altering STAT3 expression. Oncogenesis 5(2):e197. https://doi.org/10.1038/oncsis.2016.1
Höll M, Koziel R, Schäfer G, Pircher H, Pauck A, Hermann M et al (2016) ROS signaling by NADPH oxidase 5 modulates the proliferation and survival of prostate carcinoma cells. Mol Carcinog 55(1):27–39
Fukai T, Ushio-Fukai M (2020) Cross-talk between NADPH oxidase and mitochondria: role in ROS signaling and angiogenesis. Cells 9(8):1849
Ushio-Fukai M (2007) VEGF signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal 9(6):731–739
Kim J, Kim J, Bae J-S (2016) ROS homeostasis and metabolism: a critical liaison for cancer therapy. Exp Mol Med 48(11):e269
Lin H-L, Liu T-Y, Chau G-Y, Lui W-Y, Chi C-W (2000) Comparison of 2-methoxyestradiol-induced, docetaxel-induced, and paclitaxel-induced apoptosis in hepatoma cells and its correlation with reactive oxygen species. Cancer 89(5):983–994. https://doi.org/10.1002/1097-0142(20000901)89:5%3c983::AID-CNCR7%3e3.0.CO;2-G
Sun H, Yu T, Li J (2011) Co-administration of perifosine with paclitaxel synergistically induces apoptosis in ovarian cancer cells: more than just AKT inhibition. Cancer Lett 310(1):118–128. https://doi.org/10.1016/j.canlet.2011.06.010
Liu W, Gu J, Qi J, Zeng XN, Ji J, Chen ZZ et al (2015) Lentinan exerts synergistic apoptotic effects with paclitaxel in A549 cells via activating ROS-TXNIP-NLRP 3 inflammasome. J Cell Mol Med 19(8):1949–1955
Young MM, Kester M, Wang H-G (2013) Sphingolipids: regulators of crosstalk between apoptosis and autophagy. J Lipid Res 54(1):5–19. https://doi.org/10.1194/jlr.R031278
Huang W-C, Chen C-L, Lin Y-S, Lin C-F (2011) Apoptotic sphingolipid ceramide in cancer therapy. J Lipids 2011:565316. https://doi.org/10.1155/2011/565316
Adamovich Y, Adler J, Meltser V, Reuven N, Shaul Y (2014) AMPK couples p73 with p53 in cell fate decision. Cell Death Differ 21(9):1451–1459. https://doi.org/10.1038/cdd.2014.60
Zhang X, Huang J, Yu C, Xiang L, Li L, Shi D et al (2020) Quercetin enhanced paclitaxel therapeutic effects towards PC-3 prostate cancer through ER stress induction and ROS production. Onco Targets Ther 13:513–523. https://doi.org/10.2147/OTT.S228453
Zhao Y, Zeng X, Tang H, Ye D, Liu J (2019) Combination of metformin and paclitaxel suppresses proliferation and induces apoptosis of human prostate cancer cells via oxidative stress and targeting the mitochondria-dependent pathway. Oncol Lett 17(5):4277–4284. https://doi.org/10.3892/ol.2019.10119
Subramaniam Y, Subban K, Chelliah J (2021) A novel synergistic anticancer effect of fungal cholestanol glucoside and paclitaxel: apoptosis induced by an intrinsic pathway through ROS generation in cervical cancer cell line (HeLa). Toxicol In Vitro 72:105079. https://doi.org/10.1016/j.tiv.2021.105079
Li Y, Guo M, Lin Z, Zhao M, Xiao M, Wang C et al (2016) Polyethylenimine-functionalized silver nanoparticle-based co-delivery of paclitaxel to induce HepG2 cell apoptosis. Int J Nanomed 11:6693–6702. https://doi.org/10.2147/IJN.S122666
Zou J, Zhu B, Li Y (2020) Functionalization of silver nanoparticles loaded with paclitaxel-induced A549 cells apoptosis through ROS-mediated signaling pathways. Curr Top Med Chem 20(2):89–98. https://doi.org/10.2174/1568026619666191019102219
Li X, Lu X, Xu H, Zhu Z, Yin H, Qian X et al (2012) Paclitaxel/tetrandrine coloaded nanoparticles effectively promote the apoptosis of gastric cancer cells based on “Oxidation Therapy.” Mol Pharm 9(2):222–229. https://doi.org/10.1021/mp2002736
Chandler NM, Canete JJ, Callery MP (2004) Increased expression of NF-κB subunits in human pancreatic cancer cells1, 2. J Surg Res 118(1):9–14
Naugler WE, Karin M (2008) NF-κB and cancer—identifying targets and mechanisms. Curr Opin Genet Dev 18(1):19–26
Magné N, Toillon R-A, Bottero V, Didelot C, Van Houtte P, Gérard J-P et al (2006) NF-κB modulation and ionizing radiation: mechanisms and future directions for cancer treatment. Cancer Lett 231(2):158–168
Collins TS, Lee L-F, Ting JPY (2000) Paclitaxel up-regulates interleukin-8 synthesis in human lung carcinoma through an NF-κB- and AP-1-dependent mechanism. Cancer Immunol Immunother 49(2):78–84. https://doi.org/10.1007/s002620050605
Patel NM, Nozaki S, Shortle NH, Bhat-Nakshatri P, Newton TR, Rice S et al (2000) Paclitaxel sensitivity of breast cancer cells with constitutively active NF-κB is enhanced by IκBα super-repressor and parthenolide. Oncogene 19(36):4159–4169
Li Q, Ma Z, Liu Y, Kan X, Wang C, Su B et al (2016) Low doses of paclitaxel enhance liver metastasis of breast cancer cells in the mouse model. FEBS J 283(15):2836–2852
Oyaizu H, Adachi Y, Okumura T, Okigaki M, Oyaizu N, Taketani S et al (2001) Proteasome inhibitor 1 enhances paclitaxel-induced apoptosis in human lung adenocarcinoma cell line. Oncol Rep 8(4):825–829. https://doi.org/10.3892/or.8.4.825
Liu GH, Wang SR, Wang B, Kong BH (2006) Inhibition of nuclear factor-kappaB by an antioxidant enhances paclitaxel sensitivity in ovarian carcinoma cell line. Int J Gynecol Cancer 16(5):1777–1782. https://doi.org/10.1111/j.1525-1438.2006.00652.x
Bellarosa D, Binaschi M, Maggi CA, Goso C (2005) Sabarubicin-(MEN 10755) and paclitaxel show different kinetics in nuclear factor-kappaB (NF-kB) activation: effect of parthenolide on their cytotoxicity. Anticancer Res 25(3B):2119
Yan H, Wang S, Yu H, Zhu J, Chen C (2013) Molecular pathways and functional analysis of miRNA expression associated with paclitaxel-induced apoptosis in hepatocellular carcinoma cells. Pharmacology 92(3–4):167–174
Zhou M, Liu Z, Zhao Y, Ding Y, Liu H, Xi Y et al (2010) MicroRNA-125b confers the resistance of breast cancer cells to paclitaxel through suppression of pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) expression. J Biol Chem 285(28):21496–21507. https://doi.org/10.1074/jbc.M109.083337
Huang L, Hu C, Chao H, Wang R, Lu H, Li H et al (2019) miR-29c regulates resistance to paclitaxel in nasopharyngeal cancer by targeting ITGB1. Exp Cell Res 378(1):1–10. https://doi.org/10.1016/j.yexcr.2019.02.012
Portugal J, Mansilla S, Bataller M (2010) Mechanisms of drug-induced mitotic catastrophe in cancer cells. Curr Pharm Des 16(1):69–78. https://doi.org/10.2174/138161210789941801
Mc Gee MM (2015) Targeting the mitotic catastrophe signaling pathway in cancer. Mediat Inflamm 2015:146282. https://doi.org/10.1155/2015/146282
Denisenko TV, Sorokina IV, Gogvadze V, Zhivotovsky B (2016) Mitotic catastrophe and cancer drug resistance: a link that must to be broken. Drug Resist Updat 24:1–12. https://doi.org/10.1016/j.drup.2015.11.002
Sia J, Szmyd R, Hau E, Gee HE (2020) Molecular mechanisms of radiation-induced cancer cell death: a primer. Front Cell Dev Biol. https://doi.org/10.3389/fcell.2020.00041
Fragkos M, Beard P (2011) Mitotic catastrophe occurs in the absence of apoptosis in p53-null cells with a defective G1 checkpoint. PLoS ONE 6(8):e22946. https://doi.org/10.1371/journal.pone.0022946
Khing TM, Choi WS, Kim DM, Po WW, Thein W, Shin CY et al (2021) The effect of paclitaxel on apoptosis, autophagy and mitotic catastrophe in AGS cells. Sci Rep 11(1):23490. https://doi.org/10.1038/s41598-021-02503-9
Khodamoradi E, Hoseini-Ghahfarokhi M, Amini P, Motevaseli E, Shabeeb D, Musa AE et al (2020) Targets for protection and mitigation of radiation injury. Cell Mol Life Sci 77(16):3129–3159. https://doi.org/10.1007/s00018-020-03479-x
Burns TF, Fei P, Scata KA, Dicker DT, El-Deiry WS (2003) Silencing of the novel p53 target gene Snk/Plk2 leads to mitotic catastrophe in paclitaxel (taxol)-exposed cells. Mol Cell Biol 23(16):5556–5571. https://doi.org/10.1128/MCB.23.16.5556-5571.2003
Lin Y-W, Raj EN, Liao W-S, Lin J, Liu K-K, Chen T-H et al (2017) Co-delivery of paclitaxel and cetuximab by nanodiamond enhances mitotic catastrophe and tumor inhibition. Sci Rep 7(1):9814. https://doi.org/10.1038/s41598-017-09983-8
Isham CR, Bossou AR, Negron V, Fisher KE, Kumar R, Marlow L et al (2013) Pazopanib enhances paclitaxel-induced mitotic catastrophe in anaplastic thyroid cancer. Sci Transl Med. https://doi.org/10.1126/scitranslmed.3004358
Bai Z, Gao M, Zhang H, Guan Q, Xu J, Li Y et al (2017) BZML, a novel colchicine binding site inhibitor, overcomes multidrug resistance in A549/Taxol cells by inhibiting P-gp function and inducing mitotic catastrophe. Cancer Lett 402:81–92. https://doi.org/10.1016/j.canlet.2017.05.016
Soares AS, Costa VM, Diniz C, Fresco P (2014) Combination of Cl-IB-MECA with paclitaxel is a highly effective cytotoxic therapy causing mTOR-dependent autophagy and mitotic catastrophe on human melanoma cells. J Cancer Res Clin Oncol 140(6):921–935. https://doi.org/10.1007/s00432-014-1645-z
Wang X, Wu E, Wu J, Wang T-L, Hsieh H-P, Liu X (2013) An antimitotic and antivascular agent BPR0L075 overcomes multidrug resistance and induces mitotic catastrophe in paclitaxel-resistant ovarian cancer cells. PLoS ONE 8(6):e65686. https://doi.org/10.1371/journal.pone.0065686
Chen N-C, Chyau C-C, Lee Y-J, Tseng H-C, Chou F-P (2016) Promotion of mitotic catastrophe via activation of PTEN by paclitaxel with supplement of mulberry water extract in bladder cancer cells. Sci Rep 6(1):20417. https://doi.org/10.1038/srep20417
Klimaszewska-Wisniewska A, Halas-Wisniewska M, Tadrowski T, Gagat M, Grzanka D, Grzanka A (2016) Paclitaxel and the dietary flavonoid fisetin: a synergistic combination that induces mitotic catastrophe and autophagic cell death in A549 non-small cell lung cancer cells. Cancer Cell Int 16(1):10. https://doi.org/10.1186/s12935-016-0288-3
Michalakis J, Georgatos SD, Romanos J, Koutala H, Georgoulias V, Tsiftsis D et al (2005) Micromolar taxol, with or without hyperthermia, induces mitotic catastrophe and cell necrosis in HeLa cells. Cancer Chemother Pharmacol 56(6):615–622. https://doi.org/10.1007/s00280-005-1002-7
Jiang L, Siu MK, Wong OG, Tam K-F, Lu X, Lam EW et al (2011) iASPP and chemoresistance in ovarian cancers: effects on paclitaxel-mediated mitotic catastrophe. Clin Cancer Res 17(21):6924–6933
Zhao F, Siu MKY, Jiang L, Tam KF, Ngan HYS, Le XF et al (2014) Overexpression of forkhead box protein M1 (FOXM1) in ovarian cancer correlates with poor patient survival and contributes to paclitaxel resistance. PLoS ONE 9(11):e113478. https://doi.org/10.1371/journal.pone.0113478
Khongkow P, Gomes AR, Gong C, Man EPS, Tsang JWH, Zhao F et al (2016) Paclitaxel targets FOXM1 to regulate KIF20A in mitotic catastrophe and breast cancer paclitaxel resistance. Oncogene 35(8):990–1002. https://doi.org/10.1038/onc.2015.152
Burgess A, Rasouli M, Rogers S (2014) Stressing mitosis to death. Front Oncol 4:140. https://doi.org/10.3389/fonc.2014.00140
Chan KS, Koh CG, Li HY (2012) Mitosis-targeted anti-cancer therapies: where they stand. Cell Death Dis 3(10):e411. https://doi.org/10.1038/cddis.2012.148
Henriques AC, Ribeiro D, Pedrosa J, Sarmento B, Silva PMA, Bousbaa H (2019) Mitosis inhibitors in anticancer therapy: when blocking the exit becomes a solution. Cancer Lett 440–441:64–81. https://doi.org/10.1016/j.canlet.2018.10.005
Cheng B, Crasta K (2017) Consequences of mitotic slippage for antimicrotubule drug therapy. Endocr Relat Cancer 24(9):T97-t106. https://doi.org/10.1530/erc-17-0147
Sloss O, Topham C, Diez M, Taylor S (2016) Mcl-1 dynamics influence mitotic slippage and death in mitosis. Oncotarget 7(5):5176
Bennett A, Sloss O, Topham C, Nelson L, Tighe A, Taylor SS (2016) Inhibition of Bcl-xL sensitizes cells to mitotic blockers, but not mitotic drivers. Open Biol 6(8):160134
Lin Y, Jiang M, Chen W, Zhao T, Wei Y (2019) Cancer and ER stress: mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed Pharmacother 118:109249
Linder B, Kögel D (2019) Autophagy in cancer cell death. Biology 8(4):82. https://doi.org/10.3390/biology8040082
Lin L, Baehrecke EH (2015) Autophagy, cell death, and cancer. Mol Cell Oncol 2(3):e985913. https://doi.org/10.4161/23723556.2014.985913
Zhong Z, Sanchez-Lopez E, Karin M (2016) Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell 166(2):288–298
Gerada C, Ryan KM (2020) Autophagy, the innate immune response and cancer. Mol Oncol 14(9):1913–1929
Kong Y, Feng Z, Chen A, Qi Q, Han M, Wang S et al (2019) The natural flavonoid galangin elicits apoptosis, pyroptosis, and autophagy in glioblastoma. Front Oncol 9:942
Yu Y-F, Hu P-C, Wang Y, Xu X-L, Rushworth GM, Zhang Z et al (2017) Paclitaxel induces autophagy in gastric cancer BGC823 cells. Ultrastruct Pathol 41(4):284–290. https://doi.org/10.1080/01913123.2017.1334019
Lee Y, Na J, Lee MS, Cha EY, Sul JY, Park JB et al (2018) Combination of pristimerin and paclitaxel additively induces autophagy in human breast cancer cells via ERK1/2 regulation. Mol Med Rep 18(5):4281–4288. https://doi.org/10.3892/mmr.2018.9488
Eum K-H, Lee M (2011) Crosstalk between autophagy and apoptosis in the regulation of paclitaxel-induced cell death in v-Ha-ras-transformed fibroblasts. Mol Cell Biochem 348(1):61–68. https://doi.org/10.1007/s11010-010-0638-8
Ajabnoor GMA, Crook T, Coley HM (2012) Paclitaxel resistance is associated with switch from apoptotic to autophagic cell death in MCF-7 breast cancer cells. Cell Death Dis 3(1):e260. https://doi.org/10.1038/cddis.2011.139
Ghaforui-Fard S, Vafaee R, Taheri M (2019) Taurine-upregulated gene 1: a functional long noncoding RNA in tumorigenesis. J Cell Physiol 234(10):17100–17112. https://doi.org/10.1002/jcp.28464
Gu L, Li Q, Liu H, Lu X, Zhu M (2020) Long noncoding RNA TUG1 promotes autophagy-associated paclitaxel resistance by sponging miR-29b-3p in ovarian cancer cells. Onco Targets Ther 13:2007–2019. https://doi.org/10.2147/OTT.S240434
Chen K, Shi W (2016) Autophagy regulates resistance of non-small cell lung cancer cells to paclitaxel. Tumor Biol 37(8):10539–10544. https://doi.org/10.1007/s13277-016-4929-x
Zhang S-F, Wang X-Y, Fu Z-Q, Peng Q-H, Zhang J-Y, Ye F et al (2015) TXNDC17 promotes paclitaxel resistance via inducing autophagy in ovarian cancer. Autophagy 11(2):225–238
Peng X, Gong F, Chen Y, Jiang Y, Liu J, Yu M et al (2014) Autophagy promotes paclitaxel resistance of cervical cancer cells: involvement of Warburg effect activated hypoxia-induced factor 1-α-mediated signaling. Cell Death Dis 5(8):e1367. https://doi.org/10.1038/cddis.2014.297
Pastor F, Dumas K, Barthélémy M-A, Regazzetti C, Druelle N, Peraldi P et al (2017) Implication of REDD1 in the activation of inflammatory pathways. Sci Rep 7(1):7023. https://doi.org/10.1038/s41598-017-07182-z
Zeng Q, Liu J, Cao P, Li J, Liu X, Fan X et al (2018) Inhibition of REDD1 sensitizes bladder urothelial carcinoma to paclitaxel by inhibiting autophagy. Clin Cancer Res 24(2):445–459
Zou C-F, Jia L, Jin H, Yao M, Zhao N, Huan J et al (2011) Re-expression of ARHI (DIRAS3) induces autophagy in breast cancer cells and enhances the inhibitory effect of paclitaxel. BMC Cancer 11(1):22. https://doi.org/10.1186/1471-2407-11-22
Xu S, Wang P, Zhang J, Wu H, Sui S, Zhang J et al (2019) Ai-lncRNA EGOT enhancing autophagy sensitizes paclitaxel cytotoxicity via upregulation of ITPR1 expression by RNA-RNA and RNA-protein interactions in human cancer. Mol Cancer 18(1):89. https://doi.org/10.1186/s12943-019-1017-z
Wang H, Li D, Li X, Ou X, Liu S, Zhang Y et al (2016) Mammalian target of rapamycin inhibitor RAD001 sensitizes endometrial cancer cells to paclitaxel-induced apoptosis via the induction of autophagy. Oncol Lett 12(6):5029–5035. https://doi.org/10.3892/ol.2016.5338
Zamora A, Alves M, Chollet C, Therville N, Fougeray T, Tatin F et al (2019) Paclitaxel induces lymphatic endothelial cells autophagy to promote metastasis. Cell Death Dis 10(12):956. https://doi.org/10.1038/s41419-019-2181-1
Zhan Y, Wang K, Li Q, Zou Y, Chen B, Gong Q et al (2018) The novel autophagy inhibitor alpha-hederin promoted paclitaxel cytotoxicity by increasing reactive oxygen species accumulation in non-small cell lung cancer cells. Int J Mol Sci 19(10):3221
Wang K, Liu X, Liu Q, Ho Ih, Wei X, Yin T et al (2020) Hederagenin potentiated cisplatin- and paclitaxel-mediated cytotoxicity by impairing autophagy in lung cancer cells. Cell Death Dis 11(8):611. https://doi.org/10.1038/s41419-020-02880-5
Datta S, Choudhury D, Das A, Mukherjee DD, Dasgupta M, Bandopadhyay S et al (2019) Autophagy inhibition with chloroquine reverts paclitaxel resistance and attenuates metastatic potential in human nonsmall lung adenocarcinoma A549 cells via ROS mediated modulation of β-catenin pathway. Apoptosis 24(5):414–433. https://doi.org/10.1007/s10495-019-01526-y
Xi G, Hu X, Wu B, Jiang H, Young CYF, Pang Y et al (2011) Autophagy inhibition promotes paclitaxel-induced apoptosis in cancer cells. Cancer Lett 307(2):141–148. https://doi.org/10.1016/j.canlet.2011.03.026
Xu L, Liu J-H, Zhang J, Zhang N, Wang Z-H (2015) Blockade of autophagy aggravates endoplasmic reticulum stress and improves paclitaxel cytotoxicity in human cervical cancer cells. Cancer Res Treat 47(2):313–321. https://doi.org/10.4143/crt.2013.222
Liu S, Li X (2015) Autophagy inhibition enhances sensitivity of endometrial carcinoma cells to paclitaxel. Int J Oncol 46(6):2399–2408. https://doi.org/10.3892/ijo.2015.2937
Zou S, Du X, Lin H, Wang P, Li M (2018) Paclitaxel inhibits the progression of cervical cancer by inhibiting autophagy via lncRNARP11-381N20. 2. Eur Rev Med Pharmacol Sci 22(10):3010–3017
Wang R-X, Xu X-E, Huang L, Chen S, Shao Z-M (2019) eEF2 kinase mediated autophagy as a potential therapeutic target for paclitaxel-resistant triple-negative breast cancer. Ann Transl Med 7(23):783. https://doi.org/10.21037/atm.2019.11.39
Wen J, Yeo S, Wang C, Chen S, Sun S, Haas MA et al (2015) Autophagy inhibition re-sensitizes pulse stimulation-selected paclitaxel-resistant triple negative breast cancer cells to chemotherapy-induced apoptosis. Breast Cancer Res Treat 149(3):619–629. https://doi.org/10.1007/s10549-015-3283-9
Zhang Q, Si S, Schoen S, Chen J, Jin X-B, Wu G (2013) Suppression of autophagy enhances preferential toxicity of paclitaxel to folliculin-deficient renal cancer cells. J Exp Clin Cancer Res 32(1):99. https://doi.org/10.1186/1756-9966-32-99
Zeh HJ, Bahary N, Boone BA, Singhi AD, Miller-Ocuin JL, Normolle DP et al (2020) A randomized phase II preoperative study of autophagy inhibition with high-dose hydroxychloroquine and gemcitabine/nab-paclitaxel in pancreatic cancer patients. Clin Cancer Res 26(13):3126–3134
Wyld L, Bellantuono I, Tchkonia T, Morgan J, Turner O, Foss F et al (2020) Senescence and cancer: a review of clinical implications of senescence and senotherapies. Cancers 12(8):2134. https://doi.org/10.3390/cancers12082134
Wang B, Kohli J, Demaria M (2020) Senescent cells in cancer therapy: friends or foes? Trends Cancer 6(10):838–857. https://doi.org/10.1016/j.trecan.2020.05.004
Coppé JP, Desprez PY, Krtolica A, Campisi J (2010) The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5:99–118. https://doi.org/10.1146/annurev-pathol-121808-102144
Franco MS, Roque MC, Oliveira MC (2019) Short and long-term effects of the exposure of breast cancer cell lines to different ratios of free or co-encapsulated liposomal paclitaxel and doxorubicin. Pharmaceutics 11(4):178
Chen JY-F, Hwang C-C, Chen W-Y, Lee J-C, Fu T-F, Fang K et al (2010) Additive effects of C2-ceramide on paclitaxel-induced premature senescence of human lung cancer cells. Life Sci 87(11):350–357. https://doi.org/10.1016/j.lfs.2010.06.017
Ling YH, Zou Y, Perez-Soler R (2000) Induction of senescence-like phenotype and loss of paclitaxel sensitivity after wild-type p53 gene transfection of p53-null human non-small cell lung cancer H358 cells. Anticancer Res 20(2a):693–702
Uruski P, Sepetowska A, Konieczna C, Pakuła M, Wyrwa M, Tussupkaliyev A et al (2021) Primary high-grade serous ovarian cancer cells are sensitive to senescence induced by carboplatin and paclitaxel in vitro. Cell Mol Biol Lett 26(1):44. https://doi.org/10.1186/s11658-021-00287-4
Zhou J, Jiang Y-Y, Wang H-P, Chen H, Wu Y-C, Wang L et al (2020) Natural compound Tan-I enhances the efficacy of paclitaxel chemotherapy in ovarian cancer. Ann Transl Med 8(12):752. https://doi.org/10.21037/atm-20-4072
Chou Y-S, Yen C-C, Chen W-M, Lin Y-C, Wen Y-S, Ke W-T et al (2016) Cytotoxic mechanism of PLK1 inhibitor GSK461364 against osteosarcoma: mitotic arrest, apoptosis, cellular senescence, and synergistic effect with paclitaxel. Int J Oncol 48(3):1187–1194. https://doi.org/10.3892/ijo.2016.3352
Schmidt S, Schneider L, Essmann F, Cirstea IC, Kuck F, Kletke A et al (2010) The centrosomal protein TACC3 controls paclitaxel sensitivity by modulating a premature senescence program. Oncogene 29(46):6184–6192. https://doi.org/10.1038/onc.2010.354
Mohiuddin M, Kasahara K (2021) The mechanisms of the growth inhibitory effects of paclitaxel on gefitinib-resistant non-small cell lung cancer cells. Cancer Genomics Proteomics 18(5):661–673
Prencipe M, Fitzpatrick P, Gorman S, Mosetto M, Klinger R, Furlong F et al (2009) Cellular senescence induced by aberrant MAD2 levels impacts on paclitaxel responsiveness in vitro. Br J Cancer 101(11):1900–1908. https://doi.org/10.1038/sj.bjc.6605419
Weiner-Gorzel K, Dempsey E, Milewska M, McGoldrick A, Toh V, Walsh A et al (2015) Overexpression of the microRNA miR-433 promotes resistance to paclitaxel through the induction of cellular senescence in ovarian cancer cells. Cancer Med 4(5):745–758
Tan Y, Chen Q, Li X, Zeng Z, Xiong W, Li G et al (2021) Pyroptosis: a new paradigm of cell death for fighting against cancer. J Exp Clin Cancer Res 40(1):153. https://doi.org/10.1186/s13046-021-01959-x
Wang Y-Y, Liu X-L, Zhao R (2019) Induction of pyroptosis and its implications in cancer management. Front Oncol 9:971
Shi Y, Ren J, Liang C, Wang F, Li W, Li X (2019) GSDME influences sensitivity of breast cancer MCF-7 cells to paclitaxel by regulating cell pyroptosis. Chin J Cancer Biother pp 146–151
Zhang C-c, Li C-g, Wang Y-f, Xu L-h, He X-h, Zeng Q-z et al (2019) Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis 24(3):312–325. https://doi.org/10.1007/s10495-019-01515-1
Wang X, Li H, Li W, Xie J, Wang F, Peng X et al (2020) The role of caspase-1/GSDMD-mediated pyroptosis in Taxol-induced cell death and a Taxol-resistant phenotype in nasopharyngeal carcinoma regulated by autophagy. Cell Biol Toxicol 36(5):437–457. https://doi.org/10.1007/s10565-020-09514-8
Cheng Z, Li Z, Gu L, Li L, Gao Q, Zhang X et al (2020) Ophiopogonin B-inducing pyroptosis through caspase-1/gsdmd pathway contributes to alleviation of paclitaxel resistance in lung cancer cells. J Cancer 13(2):715–727. https://doi.org/10.7150/jca.66432
Xiao Y, Zhang T, Ma X, Yang QC, Yang LL, Yang SC et al (2021) Microenvironment-responsive prodrug-induced pyroptosis boosts cancer immunotherapy. Adv Sci. 8(24):e2101840. https://doi.org/10.1002/advs.202101840
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149(5):1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Wu Y, Yu C, Luo M, Cen C, Qiu J, Zhang S et al (2020) Ferroptosis in cancer treatment: another way to Rome. Front Oncol. https://doi.org/10.3389/fonc.2020.571127
Jiang M, Qiao M, Zhao C, Deng J, Li X, Zhou C (2020) Targeting ferroptosis for cancer therapy: exploring novel strategies from its mechanisms and role in cancers. Transl Lung Cancer Res 9(4):1569–1584. https://doi.org/10.21037/tlcr-20-341
Ye J, Jiang X, Dong Z, Hu S, Xiao M (2019) Low-concentration PTX and RSL3 inhibits tumor cell growth synergistically by inducing ferroptosis in mutant p53 hypopharyngeal squamous carcinoma. Cancer Manag Res 11:9783–9792. https://doi.org/10.2147/CMAR.S217944
Sugiyama A, Ohta T, Obata M, Takahashi K, Seino M, Nagase S (2020) xCT inhibitor sulfasalazine depletes paclitaxel-resistant tumor cells through ferroptosis in uterine serous carcinoma. Oncol Lett 20(3):2689–2700. https://doi.org/10.3892/ol.2020.11813
You JH, Lee J, Roh J-L (2021) PGRMC1-dependent lipophagy promotes ferroptosis in paclitaxel-tolerant persister cancer cells. J Exp Clin Cancer Res 40(1):350. https://doi.org/10.1186/s13046-021-02168-2
Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D et al (2020) CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer 19(1):43. https://doi.org/10.1186/s12943-020-01168-8
Wei D, Ke Y-Q, Duan P, Zhou L, Wang C-Y, Cao P (2021) MicroRNA-302a-3p induces ferroptosis of non-small cell lung cancer cells via targeting ferroportin. Free Radic Res 55(7):821–830. https://doi.org/10.1080/10715762.2021.1947503
Qiu Y, Yu Q, Ji M, Zhang Z, Kang L, Fu Y et al (2021) Activation ferroptosis enhanced the therapy sensitivity of TNBC to paclitaxel via NCOA4 mediated ferritinophagy. Res Sq. PPR: PPR310530. https://doi.org/10.21203/rs.3.rs-360631/v1
Su Z, Yang Z, Xu Y, Chen Y, Yu Q (2015) Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer 14:48. https://doi.org/10.1186/s12943-015-0321-5
Su Z, Yang Z, Xie L, DeWitt JP, Chen Y (2016) Cancer therapy in the necroptosis era. Cell Death Differ 23(5):748–756. https://doi.org/10.1038/cdd.2016.8
Diao Y, Ma X, Min W, Lin S, Kang H, Dai Z et al (2016) Dasatinib promotes paclitaxel-induced necroptosis in lung adenocarcinoma with phosphorylated caspase-8 by c-Src. Cancer Lett 379(1):12–23. https://doi.org/10.1016/j.canlet.2016.05.003
Khing TM, Po WW, Sohn UD (2019) Fluoxetine enhances anti-tumor activity of paclitaxel in gastric adenocarcinoma cells by triggering apoptosis and necroptosis. Anticancer Res 39(11):6155. https://doi.org/10.21873/anticanres.13823
Jang MS, Lee SJ, Kang NS, Kim E (2011) Cooperative phosphorylation of FADD by Aur-A and Plk1 in response to taxol triggers both apoptotic and necrotic cell death. Cancer Res 71(23):7207–7215. https://doi.org/10.1158/0008-5472.can-11-0760
Ando Y, Ohuchida K, Otsubo Y, Kibe S, Takesue S, Abe T et al (2020) Necroptosis in pancreatic cancer promotes cancer cell migration and invasion by release of CXCL5. PLoS ONE 15(1):e0228015. https://doi.org/10.1371/journal.pone.0228015
Funding
No funding was obtained for this study.
Author information
Authors and Affiliations
Contributions
All authors were involved in the preparing first draft. The scientific edition was performed by MN. All authors wrote and approved the article.
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical approval
This article does not contain human or animal studies performed by any of the authors.
Informed consent
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhao, S., Tang, Y., Wang, R. et al. Mechanisms of cancer cell death induction by paclitaxel: an updated review. Apoptosis 27, 647–667 (2022). https://doi.org/10.1007/s10495-022-01750-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-022-01750-z