
Abstract
Protection of normal tissues against toxic effects of ionizing radiation is a critical issue in clinical and environmental radiobiology. Investigations in recent decades have suggested potential targets that are involved in the protection against radiation-induced damages to normal tissues and can be proposed for mitigation of radiation injury. Emerging evidences have been shown to be in contrast to an old dogma in radiation biology; a major amount of reactive oxygen species (ROS) production and cell toxicity occur during some hours to years after exposure to ionizing radiation. This can be attributed to upregulation of inflammatory and fibrosis mediators, epigenetic changes and disruption of the normal metabolism of oxygen. In the current review, we explain the cellular and molecular changes following exposure of normal tissues to ionizing radiation. Furthermore, we review potential targets that can be proposed for protection and mitigation of radiation toxicity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
For many years, it has been known that the use of free radical scavengers immediately before exposure to radiation can alleviate DNA damage and cell death. Further research has led to a large number of experimental studies defining appropriate radioprotectors. Investigations at the United States (US) Army Walter Reed Institute (Washington, DC) during the cold war led to the development of a potent drug with high radioprotection efficiency, named WR2721 or amifostine [1]. This compound is a sulfhydryl containing agents that interact and neutralize free radicals [2]. Afterwards, other agents containing sulfhydryl groups were developed to reduce oxidative stress and radiation toxicity. N-Acetylcysteine and captopril are sulfhydryl agents that have been used for radioprotection in some experimental studies [3]. Although amifostine has been used as a radiation modifier in clinical trials, some studies have reported severe toxicities which led to its use being discontinued [4]. In clinical studies, amifostine has been commonly used for head and neck cancer radiotherapy for reducing xerostomia [5].
To date, several agents have been investigated for use as a radioprotector, with some chemical and natural agents showing effective radioprotection. However, the efficiency of each radioprotector could be limited to some organs/tissues [6]. Moreover, for the protection of normal tissues in radiotherapy, it is crucial to consider possible effects on tumor response [7]. For the protection of a particular organ, we need to choose the best agent, which may be different from those used for other organs [8].
In recent decades, several studies have been conducted to protect and mitigate radiation toxicity using some supplements after exposure to radiation. In addition to clinical applications, radiation mitigators can be used to reduce mortality and normal tissue toxicity after an accidental nuclear or radiological disaster [9]. Radiation mitigators are mostly for the bone marrows and gastrointestinal system injury; however, studies have suggested that mitigators can be used to alleviate radiation toxicity in the lung, heart and kidney [10]. Acute damages to the bone marrows and gastrointestinal system may lead to death some days to weeks following whole-body exposure to radiation. However, in some cases, following local abdomen and chest exposure or non-homogenous whole-body exposure to radiation, the incidence of lung pneumonitis, cardiopulmonary fibrosis or nephropathy may cause death some months to years later [11]. Total body irradiation (TBI) for patients with leukemia may also lead to pneumonitis and nephropathy [12]. Investigations into the development of new agents with high efficiency for mitigation and protection of radiosensitive organs are ongoing. In the current paper, we review recent advances in the mitigation of radiation toxicity and also suggest promising strategies for preventing mortality and side effects that affect the quality of life of exposed people.
Promising targets for radiation protection and mitigation
Emerging evidences indicate that in addition to radiation dose, normal tissue toxicity is highly dependent on hierarchical events that occur in cells after irradiation. At the first level, DNA damage response (DDR) determines cell death or repair of DNA. DDR is initiated some minutes to hours after exposure to radiation [13]. Enhancing DNA repair capacity has been proposed for ameliorating DNA damage and reducing cell death [14]. If damages to the DNA are not completely repaired, oxidized DNA or cell contents from dying cells trigger activation of several signaling pathways that play a pivotal role in the incidence of radiation injury [13]. Cell death through apoptosis, necrosis, autophagy or senescence induces the activation of inflammatory mediators and pro-oxidant enzymes [15,16,17]. It seems that DNA repair enzymes and cell death pathways are activated from the first minutes to hours after exposure to radiation; however, ROS/NO generating enzymes are upregulated after some hours to weeks [18]. The expression of inflammatory and pro-oxidant mediators is time-dependent and tissue-specific. Furthermore, the quality and quantity of their expressions vary for different organs. Thus, it seems that the efficiency of a radiation protector or mitigator is highly dependent on the time of administration as well as irradiated organs [19]. In this review, the potential targets for the amelioration of radiation toxicity in different organs will be discussed. These targets can be proposed for both protection and mitigation of radiation injury for clinical radiotherapy and also for radiological and nuclear disasters.
Targeting cell death after exposure to ionizing radiation
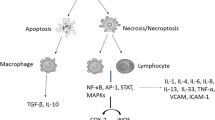
Radiation-induced cell death is the consequence of massive DNA damage and accumulation of unrepaired chromosome breaks. Mitotic catastrophe is a non-immunogenic type of cell death and does not trigger inflammatory or tolerogenic responses. Autophagy also does not play a major role in the side effects of ionizing radiation. Other types of cell death including necrosis, apoptosis and senescence lead to some changes in the response of immune system and reduction/oxidation (redox) reaction that is associated with the appearance of oxidative stress and inflammation [18, 20]. Necrosis usually occurs following massive injury to the membrane, DNA, mitochondria and other organelles within cells. This may be observed some hours after irradiation [21]. Apoptosis and senescence are two important types of cell death that occur following the upregulation and downregulation of some genes that are involved in the progression of death or survival [22]. Modulation of these types of cell death can be suggested as potential strategies for mitigation of radiation injury. This can increase the survival of radiosensitive cells in bone marrow and small intestine thus helps mitigate radiation-induced mortality.
Apoptosis
Apoptosis is the main mechanism of cell death in highly radiosensitive organs such as the bone marrow, gastrointestinal system, testis and skin [23, 24]. High incidence of apoptosis in these organs lead to severe damage to the normal function of organs, leading to the appearance of severe reactions and consequences for exposed people. Apoptosis occurs following DNA damage and increases the release of some cell death signals such as Fas, TNF-α and TGF-β, as well as increased activity of p53 [23]. Each of these mediators via binding to their receptors initiates apoptosis signaling pathways. TGF-β via binding to TGFβR1 or TGFβR2 causes activation of pro-apoptosis Bax via Smad2/3 pathway. Fas binds to Fas ligand (FasL or CD95L); however, TNF-α can bind to TNF receptor (TNFR) or TRAIL. Upregulation of FasL, TNFR or TRAIL cause stimulation of caspase-8 and caspase-10 to induce activation and penetration of Bax into the mitochondria [25]. Upregulation of TNFR and also the production of free radicals after exposure of membrane to ionizing radiation can stimulate the development of ceramide, which suppresses Bcl-2, an inhibitor of pro-apoptosis Bax. Penetration of Bax into the mitochondria leads to the release of cytochrome C. Usually, early apoptosis occurs following damage to mitochondria and release of cytochrome C. This is followed by interaction of cytochrome C with Apaf1 and caspase-9 and the development of apoptosome complex. This complex is able to degrade DNA and other macromolecules such as the membrane, leading to the development of apoptotic bodies [26]. Some mediators such as NF-κB or PI3K can suppress the progression of apoptosis via upregulation of Bcl-2 [27] (Fig. 1).

Apoptosis pathways after exposure to ionizing radiation. Exogenous and endogenous ROS trigger upregulation of apoptosis receptors which cause Bax upregulation and downregulation of Bcl-2. DNA damage and p53 also have key roles in the initiation of apoptosis signaling pathways. Apoptosis clearance by macrophages leads to their activation and is associated with NO generation. NO can interact with Ogg1, a key player in BER pathway, thus suppresses DDR. Neutralization of NO can boost DDR, thus reduces apoptosis. Furthermore, activation of TLR4&5 can increase the expression of NF-κB, leading to inhibition of Bax and upregulation of Bcl-2. P38 has a negative role and its suppression can preserve cell viability
To date, few studies have tried to inhibit apoptosis and improve normal tissue tolerability using selective anti-apoptotic inhibitors. Targeting TGF-β in mice model has been shown to attenuate radiation-induced bone marrow injury. However, there is a possibility that the suppression of TGF-β reduces apoptosis following attenuation of ROS generating enzymes and mitochondrial-derived superoxide suppression [28]. The intestine is another highly radiosensitive organ because of the high incidence of apoptosis in progenitor stem cells. Apoptosis in these cells leads to shortening of villi and crypt. Crypt and villi require permanent turnover by mitosis in intestinal stem cells. In mice intestine, it has been shown that mitochondrial pathway is responsible for apoptosis following exposure to radiation. Inhibition of Bax or PUMA could improve intestinal recovery after exposure to 8 or 18 Gy radiation. Interestingly, PUMA knockdown in mice showed more protection compared to Bax knockdown in mice. Results indicated that PUMA via induction of apoptosis in intestinal progenitor cells plays a main role in radiation-induced intestinal injury in mice [29]. P53 also has a key role in the initiation of apoptosis signaling pathways in the intestine. Although it is a central regulator of DNA damage response, overexpression of p53 has been observed to be associated with increased apoptosis in the crypt [30].
Lymphocytes are one of the most radiosensitive cells. Lymphocytes do not have mitotic activity, while they have a bigger nucleus compared to other cells that may be involved in radiosensitization of lymphocytes. It has been suggested that high expressions of pro-apoptotic genes such as Bax, PUMA and p53 play a key role in the high incidence of apoptosis in lymphocytes. Furthermore, radiation may trigger ROS production by lymphocytes, thus increases the probability of DNA damage and apoptosis [31]. Treatment of lymphocytes with some agents has been shown to reduce the expression of pro-apoptotic genes such as Bax and increase the expression of anti-apoptotic Bcl-2 [32, 33]. Some other strategies have been proposed to mitigate radiation toxicity via inhibition of apoptosis initiation. Enhancing DNA repair capacity is an interesting idea. Furthermore, triggering anti-apoptotic genes such as toll-like receptors (TLRs) or suppression of pro-apoptotic genes such as p38 have shown interesting results. Inhibition of apoptosis is favor for normal tissues sparing, while, in cancer radiotherapy this strategy can reduce the effectiveness of therapy. Thus, apoptosis targeting is an appropriated strategy for radiation mitigation, but not for radiotherapy [34, 35].
Boosting DNA repair for preventing apoptosis
As earlier mentioned, unrepaired DNA damage is responsible for cell death through mitotic catastrophe, senescence and apoptosis. As apoptosis is responsible for the death of highly radiosensitive cells such as the bone marrow and small intestinal progenitor cells, it seems that boosting DNA repair capacity can help prevent cell death through apoptosis. Enoxacin is an agent that has been shown to increase the activity and facilitates faster DNA repair, thus causes more survival of irradiated cells. This agent is able to increase the number of 53BP1 to damaged DNA sites [36]. βarrestin-1 (βarr1) is another key player of DDR that has been linked to active p53 pathway. βarr1 is involved in the regulation of MMD2, a protein that causes degradation of p53. Thus, suppression of βarr1 can stimulate p53 activity and enhance DDR, leading to reduction of radiation toxicity. It has been shown that βarr1 knockdown can increase p53 binding to damaged DNA sites [37].
One of the most important suppressive effects of ionizing radiation on DNA repair mechanisms is nitroacetylation of 8-oxoguanine glycosylase (Ogg1) following the generation of NO by inflammatory cells. Ogg1 is a key enzyme in base excision repair (BER) pathway and is responsible for removing 8-hydroxyguanine (8-oxoG), a product of ROS interaction with DNA. Activation of these enzymes after exposure to ionizing radiation can increase resistance to radiation-induced DNA damage [38]. Furthermore, the acetylation and turnover of Ogg1 by Sirt3 can amplify DNA repair in the mitochondria, thus protects against radiation-induced mitochondrial apoptosis [39].
p53
Although p53 plays a key role in DDR, activation of apoptosis pathway by it can increase radiosensitivity via depletion of stem/progenitor cells. For 2 decades ago, it has shown that suppression of p53 in combination with irradiation can increase the viability of mice C8 cells [40]. Thus, inhibition of p53 may attenuate massive apoptosis to mitigate radiation toxicity in high radiosensitive organs. A study suggested that p53 may have dual role depend on targeted organ. Knockdown of p53 has shown that can increase resistance to radiation-induced hematopoietic system injury, while its inhibition cause more damage to the repopulation of progenitor cells in intestinal epithelial cells [41]. In fact, mechanisms of radiosensitivity in bone marrow and intestine are different. A study showed that although p53 promotes apoptosis in bone marrow progenitor cells, it plays a key role in regulating cell cycle and mitosis in intestine epithelial cells. Targeting p53 can disrupt the regulation of cell cycle checkpoints, thus increase mitotic death of epithelial cells. This shows that p53 targeting increases the radiosensitivity of epithelial cell independent of apoptosis [42]. Pifithrin-mu is an inhibitor of p53 binding to mitochondria. Treatment of mouse thymocytes with pifithrin-mu can increase the viability and reduce apoptosis. Furthermore, administration of Pifithrin-mu has shown that increase survival following whole-body irradiation with lethal doses of ionizing radiation [43]. Sodium orthovanadate is another agent that has shown protect against ionizing radiation through blunting p53 interactions with Bcl-2 and mitochondria. This study suggested that complete radioprotection can be achieved when transcription-dependent and transcription-independent pathways of p53 inhibited [44]. A further study showed that sodium orthovanadate can suppress p53 dependent apoptosis, not p53 independent apoptosis [45].
P53 targeting with sodium orthovanadate also has shown that mitigate lethality of whole-body exposure to radiation. Evaluating different time points for the administration of sodium orthovanadate showed that the best time for mitigation of radiation-induced lethality is immediate after whole-body irradiation. this may indicate that p53 activate apoptosis immediately after exposure of bone marrow stem/progenitor cells to ionizing radiation [46]. Knockdown of p53 can mitigate radiation injury in brain too. In an animal study has shown that inhibition of p53 can attenuate defect in the irradiated brain in mice and help to the development of normal brain [47]. Zinc(II) chelators such as bis(2-pyridylmethyl)amine, tris(2-pyridylmethyl)amine, 1,4,7,10-tetraazacyclododecane, Bispicen, and TPEN are p53 inhibitors that also have shown can protect against radiation-induced apoptosis [48]. P53 has a key role in tumor response to radiation. For the protection of highly radiosensitive organs such as the intestines and bone marrow cells, we need to inhibit p53 without attenuating the responses of cancer cells. For targeting p53, we should consider the expression of p53 in cancer cells. It has been suggested that most solid tumors have mutant p53 [49]. In most conditions, mutant p53 leads to a loss in its function [50]. TP53 genetic test gives a better understanding of the genetic background of tumors for using p53 inhibitors during radiotherapy. In this condition, p53 targeting can be proposed for patients with mutant p53 for protection of normal tissues without adverse effect on tumor response to radiotherapy.
TLRs
TLRs are very critical receptors that mediate inflammatory responses to damage-associated molecular-pattern (DAMP) molecules. These receptors belong to pattern recognition receptors (PRRs) family. Some TLRs including TLR2, TLR4, TLR5, TLR6 and TLR9 which depend on HMGB1 are involved in the progression of inflammatory responses and immune system activation [51]. One of the main effects of TLRs is the induction of anti-apoptotic signaling pathways, which may be involved in the protection of radiosensitive cells to apoptosis [52]. Targeting TLRs using agonists has been shown to protect and mitigate radiation toxicity in normal tissues. In an animal study, Kurkjian et al. showed mitigation of acute radiation syndrome (ARS) following TLR2/6 targeting. Fibroblast-stimulating lipopeptide (FSL-1), an agonist of TLR2/6 administered 24 h after whole-body irradiation with a lethal dose of radiation showed 50% survival for 30 days. However, administration of other TLR agonists did not show any improvement in survival. FSL-1 could induce G-CFU, thus improved hematopoietic activity and increased number of peripheral blood cells [53].
Targeting TLR5 in an in vivo study showed interesting results for both protection and mitigation of radiation injury in highly radiosensitive organs, leading to increased survival. Whole-body exposure of mice to 13 Gy led to 100% mortality, while pre-irradiation treatment with TLR5 agonist caused 87% survival. Interestingly, treatment with TLR5 agonist even 1 h after irradiation led to 70% survival, thereby indicating the potent mitigatory effect of TLR5 agonist. In comparison to amifostine, which is the most common radioprotector, TLR5 agonist showed more efficiency [19]. TLR5 agonists may also help protect epithelial cells from toxic effects of ionizing radiation. Activation of TLR5 by CBLB502 (an optimized type of flagellin) showed protection of mice against radiation-induced dermatitis and mucositis. Interestingly, TLR5 agonist could reduce tumor volume following injection of A549. Similar results have been shown for Entolimod, another agonist of TLR5 [54].
Monophosphoryl lipid A (MPLA) is a TLR4 agonist that has shown radioprotective effect for both in vitro and in vivo models. MLPA triggers translocation of NF-κB and also increases the expression of MyD88, while it reduces the release of inflammatory cytokines. The levels of TNF-α and IL-6 were reduced, while that of IL-2 increased after MLPA administration. MLPA increases the number of nucleated cells and hematopoietic stem cells within the bone marrow. Treatment with MLPA reduced apoptosis in bone marrow cells, which may be due to the activation of NF-κB. Also, MLPA increased IFN-β (TRIF), which may be involved in its radioprotective effect [55]. CBLB502, a TLR5 agonist can also induce the release of IL-6 following NF-κB upregulation, leading to upregulation and activation of STAT-3. These changes suppress apoptosis and preserve progenitor stem cells [56]. Furthermore, TLR5 activation can stimulate the activation of NK cells, which kill cancer cells and inhibit metastasis [57]. It is suggested that upregulation of TLRs can induce maturation of dendritic cells, which activate immune system against cancer cells [58]. This property of TLRs can help to management of normal tissues toxicity by TLRs agonists without negative effect on tumor response to radiotherapy.
The role of TLRs in the development of radiation-induced diseases is complex and needs to be investigated for different organs. Although, TLRs can reduce apoptosis, their upregulation may be involved in the progression of inflammatory-related diseases that may appear in the long term after exposure to ionizing radiation [59]. A study showed that TLR4 knockdown in mice can ameliorate radiation-induced fibrosis in mice lung. Furthermore, TLR4 knockdown led to an improvement in hematopoietic system recovery [60]. In contrast, another study reported that knockdown of both TLR2 and TLR4 can amplify lung fibrosis [61]. Therefore, it seems that stimulation of TLRs is an interesting target for preventing ARS resulting from massive apoptosis in the bone marrow and intestine; however, caution should be observed for other organs.
TLRs agonists have shown interesting results for activation of the immune system against cancer cells. In combination with radiation, TLRs agonists have also shown promising results. However, it needs to be examined for each cancer type [62]. Studies suggested that upregulation of some TLRs like TLR4 is associated with shorter survival for different tumors, while others such as TLR5 upregulation predicts higher survival for lung cancer patients [63]. The main anti-tumor activity of TLRs is mediated through an increase in immune system’s activity like activation of cytotoxic T lymphocytes (CTLs) and triggering immunogenic functions of dendritic cells (DCs) and macrophages [64]. To date, some experimental studies have been conducted to investigate the roles of some TLRs agonists in combination with radiotherapy. Among the mentioned TLRs that can protect normal tissues, TLR7 and TLR9 have been investigated to determine the modulatory effects on tumor response to radiotherapy. CpG oligodeoxynucleotides, which activate TLR9 in DCs, can increase the response of implanted fibrosarcoma tumors in mice, thus increase tumor growth delay and survival in mice [65]. Administration of R848, a TLR7 agonist, in combination with radiotherapy has been shown to increase infiltration of CTLs in mice bearing lymphoma [66]. Similar studies indicated that activation of TLR7 leads to suppression of breast cancers, colorectal carcinoma, fibrosarcoma, colorectal and pancreatic cancers in mice via increasing infiltration of CTLs and NK cells, as well as the release of anti-tumor cytokines such as IFN-γ [67,68,69]. These results show that targeting some TLRs can act as radioprotector for normal tissues and also radiosensitizer for some tumors. For example, TLR5 and TLR7 stimulate the activities of anti-tumor CTLs and NK cells in tumor, while TLR5 suppresses apoptosis and TLR7 triggers the release of IFN-γ and stem cells’ proliferation in hematopoietic system [70]. TLR9 which has anti-tumor activity against fibrosarcoma, can also prevent apoptosis of irradiated cells and preserves the intestines [71].
p38 MAPK
MAPKs include some subfamilies including p38, extracellular signal-regulated kinases 1 and 2 (ERK1/2) as well as c-Jun amino-terminal kinases (JNKs) [72]. Among these subfamilies, p38 targeting has been investigated to ameliorate radiation-induced apoptosis. Inhibition of p38 has been shown to suppress upregulation of pro-apoptotic caspase-3 and PARP, thereby reducing apoptosis in auditory cells [73]. As p38 plays a key role in the promotion of apoptosis following exposure to ionizing radiation, some studies have investigated its targeting for mitigation of radiation toxicity in the bone marrow using p38 inhibition. Suppression of p38 by thioredoxin (TXN) has been shown to mitigate bone marrow hematopoietic stem cell death following exposure to radiation [74]. Inhibition of p38 in combination with granulocyte colony-stimulating factor (G-CSF) administration has also been suggested to improve the recovery of depleted bone marrow cells after exposure to radiation. This combination showed an increase in the number of hematopoietic stem/progenitor cells [75, 76]. Furthermore, inhibition of p38 enhances survival following administration of G-CSF; however, p38 inhibition when administered alone had no significant effect for mitigation of radiation mortality [77] (Fig. 1).
As p38 has a role in the initiation of apoptosis in several cancer cells, its inhibition may protect cancer cells against radiotherapy [78, 79]. Targeting p38 may be a useful candidate for radiation mitigation. However, its targeting during radiotherapy may be inappropriate and needs further elucidation for each cancer type.
Senescence
Senescence is a type of cell death that triggers inflammatory responses [80]. Senescence can occur in different such as fibroblasts, endothelial cells, epithelial cells, astrocytes etc. [81]. It has been suggested that senescence in fibroblasts following exposure to radiation can trigger fibrosis [82]. Clearance of senescent cells can reduce fibrosis and attenuate upregulation of fibrotic markers such as TGF-β [82]. It seems that DNA damage responses (DDRs) after exposure to radiation is responsible for the induction of senescence following upregulation of inflammatory mediators and release of a wide range of cytokines such as IL-1, IL-6, IL-8 and TGF-β [83]. P53 also has a role in senescence [84]. As p53 plays a central role in DDRs following exposure of cells to ionizing radiation, it is predictable that it plays a key role in senescence after irradiation. p53 knockdown has been shown to reduce senescence in irradiated cells [85]. Plasminogen activator inhibitor-1 (PAI-1) is another protein that plays a key role in the development of senescence [86]. It has been suggested that PAI-1 is downstream of TGF-β for inducing senescence [87]. PAI-1 knockdown is associated with increased survival after whole-body irradiation [88]. In fact, PAI-1 expression may predict the severity of radiation toxicity in some radiosensitive organs like intestine [88,89,90]. Chung et al. evaluated the role of PAI-1 in radiation-induced senescence and pulmonary fibrosis. They showed that pre and post-irradiation administration of rPAI-123 (truncated plasminogen activator inhibitor-1) can prevent fibrosis in mice lung. The study concluded that amelioration of radiation-induced fibrosis is mediated through suppression of senescence in type 2 pneumocytes [91]. Similar results have been revealed following inhibition of Bcl-2/xl. A study by Pan et al. has shown that inhibition of Bcl-2/xl by ABT-263 can kill senescent pneumocytes, which cause suppression of pulmonary fibrosis after lung irradiation [92]. In addition to fibrosis, senescence has been observed in other organs such as bone marrow and heart, which can trigger the release of some cytokines such as TGF-β, which may be involved in chronic oxidative stress [93]. It has been suggested that senescence after exposure to radiation can trigger activation of NADPH oxidase enzymes that are associated with increased superoxide and hydrogen peroxide generation [93]. Suppression of senescence can blunt upregulation of NOX enzymes, thus prevents continuous production of free radicals and subsequent consequences such as fibrosis [94]. Furthermore, reduction of senescence has been shown to be associated with the mitigation of radiation toxicity [95]. Role of senescence in cancer response to radiotherapy is complicated and unclear [96] (Fig. 2).

Radiation-induced senescence triggers the activity of pro-oxidant enzymes and fibrosis
Targeting of senescence during radiotherapy and its consequences on tumor response need further examination. However, evidences have shown that inhibition of PAI-1 can induce apoptosis in cancer cells [97]. Genetic knockdown or pharmacological inhibition of PAI-1 has been shown to induce apoptosis in a wide range of cancer cells such as HT-1080 fibrosarcoma, ovarian cancer cells, A549, HCT-116 human colon carcinoma, MDA-MB-231 human breast adenocarcinoma etc.[98, 99]. PAI-1 has a close role with angiogenesis and tumor growth. Thus, its targeting has been proposed to suppress angiogenesis [100]. PAI-1 inhibition can protect endothelial cells against apoptosis and aids the migration of these cells towards fibronectin [101, 102]. Therefore, it seems that targeting PAI-1 can protect some normal tissues such as the lung, while it is able to increase response of some cancers including lung, breast and colon to radiotherapy.
Targeting of inflammation
Inflammation after exposure to a high dose of radiation is involved in several complications caused by radiation. Inflammation is a normal response to massive cell death, which is mediated via some inflammatory mediators such as NF-κB, STATs, COX-2, iNOS etc. [103]. Dermatitis is one of the most common side effects of radiotherapy. Following the Chernobyl accident, some people died because of severe damages and reactions in the skin. Mucositis in the mouth and gastrointestinal system, necrosis in the brain, damage to vessels, pneumonitis and pericarditis are the most common consequences of inflammatory responses to ionizing radiation [104,105,106,107]. Inhibition of inflammatory mediators has been suggested to mitigate several complications after exposure to radiotherapy or an accidental radiation disaster [108]. Furthermore, as inflammation plays a key role in the progression of fibrosis, its inhibition can improve the management of fibrosis in different organs [109].
NF-κB
NF-κB is a central player of innate immune system. It regulates several signaling pathways involved in inflammation, proliferation and apoptosis [34]. The upregulation of NF-κB in highly radiosensitive organs such as bone marrow, testis or small intestine following activation of TLRs has been shown to be associated with protection and mitigation of mortality through suppression of apoptosis and preventing depletion of progenitor cells [57]. However, selective inhibition of NF-κB has been shown to protect bone marrow against toxic effects of ionizing radiation [110]. Furthermore, chronic upregulation of NF-κB plays a key role in chronic inflammation, which is associated with several side effects in normal tissues. Although the upregulation of NF-κB can reduce the incidence of apoptosis in early responding organs such as bone marrow, its suppression has been shown to be associated with the reduction of some side effects of ionizing radiation. An animal study suggested that suppression of NF-κB using the thiol-reactive triterpenoid RTA 408 can protect some radiosensitive organs including gastrointestinal, skin and hematopoietic system. Administration of this agent has shown significant increase in survival [111]. Suppression of NF-κB or upregulation of IκB can induce hematopoiesis and the activity of hematopoietic progenitor cells [110]. Protective effect of NF-κB suppression has also been observed for other organs such as the brain, kidney and gastrointestinal system following administration of different types of NF-κB inhibitors. Interestingly, inhibition of NF-κB showed a reduction of apoptosis in the brain [112]. It has been suggested that some radioprotectors and mitigators such as melatonin, curcumin, resveratrol, soy isoflavones and naringin are able to suppress the expression and nuclear translocation of NF-κB [34, 113, 114]. It seems that the suppression of NF-κB by these agents is a key mechanism for protection or mitigation of radiation injury [115,116,117].
NF-κB plays a key role in the resistance of a wide range of cancer cells to ionizing radiation. It can induce anti-apoptosis genes such as Bcl-2. NF-κB has a higher expression in several cancer types and its inhibition may enhance the death of cancer cells by radiation without adverse effects on normal tissues [34]. Furthermore, NF-κB increases angiogenesis and metastasis via regulation of vascular endothelial growth factor (VEGF), matrix metalloproteinase-3 (MMP-3), signal transducer and activator of transcription 3 (STAT3) etc. [34]. NF-κB upregulation has a link with a wide range of cancers such as hematological malignancies, lung carcinoma, gastrointestinal and breast tumors [118]. Some clinical studies have shown that the suppression of NF-κB is associated with an increase in the survival of patients with multiple myeloma [119, 120]. Targeting NF-κB has been shown to sensitize cancer cells to radiotherapy, thus its inhibition can increase the therapeutic efficiency of radiotherapy with both protection of normal tissues and sensitization of cancer cells [34].
COX-2
COX-2 generates prostaglandins (PGs) through the metabolism of arachidonic acids. PGE2 is one of the most important products of COX-2 which mediates the pathogenesis of several inflammatory responses [121]. The expression of COX-2 is not high in all organs. However, its suppression has been shown to protect and mitigate radiation injury in some organs. In bystander effect, studies have shown that COX-2 plays a key role in DNA damage and cell death after exposure to ionizing radiation [122,123,124,125]. It has been suggested that upregulation of COX-2 is associated with mocusitis; however, it may not be a key player in the inhibition of this process [126]. Overexpression of COX-2 has also been observed in irradiated intestinal mucosa [127].
Inhibition of COX-2 using celecoxib or rofecoxib can alleviate the severity of arthritis and paw edema, and reduces the level of inflammatory cytokines such as TNF-α following exposure to radiation [128, 129]. COX-2 inhibition has also shown protection against radiation-induced skin injury [130]. Targeting COX-2 after irradiation has been shown to mitigate radiation injury in the bone marrow. Inhibition of PGE2 synthesis using meloxicam after irradiation can mitigate mortality. Post-irradiation treatment with meloxicam could also improve hematopoietic system recovery. The mitigation of radiation-induced lethal effect was obtained when meloxicam was administered either 6 or 48 h after exposure to radiation [131]. COX-2 inhibition can also mitigate pulmonary injury after exposure to ionizing radiation [132]. It has been suggested that COX-2 inhibition is involved in the radioprotective effect of some agents such as curcumin [133]. By contrast, some evidences have shown that PGE2 has a role in protecting the intestines and hematopoietic stem cells [134, 135].
COX-2 inhibitors have been shown to sensitize cancer to radiation through suppression of proliferation, angiogenesis and metastasis. COX-2 is a stimulator of VEGF, thus it triggers angiogenesis and tumor growth [136]. COX-2 also induces the expression of anti-apoptosis genes such as Bcl-2, thereby increasing survival following therapy [137]. Due to these reasons, COX-2 inhibitors are known as radiosensitizers [138]. Targeting COX-2 has shown promising results for increasing tumor response to radiotherapy [139]. On the other hand, celecoxib as a selective COX-2 inhibitor can mitigate lung injury. It seems that COX-2 inhibition is interesting for protection and mitigation of radiation-induced lung injury. Other radiosensitive organs will require further examination in clinical studies.
NLRP3 inflammasome
NLRP3 inflammasome is a complex that reacts to stress conditions and pathogens through the release of IL-1 and IL-18. Regulation of this complex has a close relationship with mitochondria injury. Damage to the mitochondria may induce upregulation of NLRP3 inflammasome, thus amplifies inflammatory reactions [140]. Irradiation of macrophages lead to the development of NLRP3 inflammasome complex as well as an increase in pro-inflammatory IL-1 level [141]. This can promote apoptosis in radiosensitive bone marrow cells [142]. Activation of NLRP3 inflammasome has been shown to promote inflammation and fibrosis in some organs such as central nervous system (CNS), gastrointestinal system and lung [143]. In fact, NLRP3 inflammasome may accelerate inflammation and fibrosis following exposure to ionizing radiation [144]. Pyroptosis in bone marrow cells has shown as a key stimulator of NLRP3 inflammasome which acts as a key player in the bone marrow injury and mortality following whole-body irradiation [142]. Activation of NLRP3 inflammasome in immune cells also may play a key role in side effects of other tissues [141]. Inhibition of inflammatory pathways including NLRP3 inflammasome can mitigate radiation toxicity [145]. Melatonin, an anti-inflammatory agent, is able to mitigate radiation-induced mucositis through attenuation of NLRP3 inflammasome pathway [146, 147] (Fig. 3).

Inflammatory responses following exposure to ionizing radiation. Necrosis, necroptosis or autophagy can induce inflammatory responses following the release of DAMPs. NF-κB is a central regulator of inflammatory responses via triggering the release of cytokines and upregulation of COX-2 and iNOS. NF-κB also induces the development of inflammasome. Increased levels of inflammatory cytokines, iNOS and COX-2 amplify oxidative stress and DNA damage, which lead to continuous expression of NF-κB. The positive feedback between inflammatory cytokines, oxidative stress and transcription factors like NF-κB can cause chronic inflammation many years after exposure to radiation. Disruption of this crosstalk can mitigate radiation injury
Experimental studies have confirmed that NLRP3 inflammasome has a role in the pathogenesis of cancer. NLRP3 inflammasome is involved in cancer progression in a wide range of malignancies such as lung, skin, liver, gastrointestinal and prostate cancers [148]. Targeting NLRP3 inflammasome has been proposed for reducing tumor resistance [149, 150]. However, the effect of NLRP3 inflammasome targeting in combination with radiotherapy remains to be elucidated.
Absent in melanoma (AIM)2 inflammasome
AIM2 inflammasome is another type of inflammasome that has a role in cell death through pyroptosis. Pyroptosis is an inflammatory type of cell death that is mediated by inflammasomes [151]. After exposure to clastogenic agents and DNA damage, AIM2 recognizes double-strand breaks (DSBs), then stimulates the activation of caspase-1, the cleavage of gasdermin D (GSDMD) and release of inflammatory cytokines which is associated with membrane perforation and release of cell contents [152,153,154]. Although studies for protection against ionizing radiation via targeting AIM2 inflammasome is very limited, some results are interesting. For the first time, Hu et al. found that mice deficient in AIM2 are more resistant against mortal effects of whole-body exposure to ionizing radiation. Knockdown of AIM2 was shown to reduce pyroptosis in both bone marrow and intestine as well as preserved colony formation of stem/progenitor cells. Results of this study confirmed that AIM2 translocates into the nucleus after irradiation and mediates pyroptosis following activation of caspase-1 [155]. Inhibition of this pathway after irradiation also showed remarkable mitigation of radiation injury in the intestine. After whole-body irradiation, an increase in AIM2 and the cleavage of GSDMD was observed in the intestine, however, inhibition of AIM2 following administration of 5-Androstenediol mitigated intestinal injury and reduced mortality [156]. AIM2-induced pyroptosis is also involved in late effects of ionizing radiation including lung pneumonitis and fibrosis. Suppression of AIM2 can attenuate radiation-induced cell death in lung tissues, which is associated with amelioration of pneumonitis and fibrosis [157].
Heat shock proteins (HSPs)
HSPs are inflammatory mediator proteins that protect cells during stress conditions. It seems that oxidative stress and released cytokines after exposure to stimulus such as ionizing radiation trigger activation of HSPs [158]. HSPs include different subfamilies that may promote or suppress inflammation [159, 160]. HSP70 can prolong G1 phase of cell cycle, leading to the reduction of cell death after exposure to ionizing radiation [161]. In contrast to HSP70, HSP27 is an inflammatory mediator protein. HSP27 has a close relation with NF-κB, thus it can induce inflammation and suppress apoptosis [162]. Inhibition of HSP27 has been shown to reduce inflammation and oxidative stress following lung exposure to radiation [163]. Studies for the mitigation of radiation injury by inhibition of HSPs are very few.
Inhibition of HSP70 is an interesting strategy for tumor sensitization and activation of immune system against cancer [164]. HSP70 has high expression in a wide range of cancer cells, hence improving survival through stimulation of anti-apoptosis genes [165, 166]. Overexpression of HSP70 is associated with malignancy and metastasis in some cancers such as acute myeloid leukemia, hepatocellular carcinoma and cervical cancers [167]. Thus, its activation does not seem to be an appropriate strategy for normal tissue protection during radiotherapy. By contrast, inhibition of HSP27 can be a potential strategy for tumor radiosensitization. HSP27 has a role in the inhibition of apoptosis via inhibition of caspase 9. It can also suppress necrosis in cancer cells, leading to tumor resistance [168]. The expression of HSP27 can predict mortality of patients with lung carcinoma [169]. HSP27 inhibitors can cause both the protection of normal tissues and sensitization of tumor in combination with chemotherapy drugs or radiotherapy [170]. However, targeting HSPs for the mitigation of radiation injury requires further studies.
Hypoxia
Hypoxia is a low oxygen condition within tissues. Hypoxia is a critical issue for tumor response to therapy. However, evidence shows that it also plays a key role in normal tissue injury. Hypoxia stimulates angiogenesis through upregulation of hypoxia-inducible factor (HIF). HIF-1 is the main mediator of VEGF, which is responsible for the development of new vessels. Although angiogenesis causes serious resistance of tumors to radiotherapy, it is a marker of radiation-induced severe injury in normal tissues [171]. Hypoxia occurs when radiation kills endothelial cells, leading to the removal of microvascular [172]. This causes the reduction of vascular density, which leads to oxygen deprivation [173]. Hypoxia and nutrition deprivation following vascular depletion can give rise to necrosis of normal adjacent cells, which lead to inflammation and more amplification in normal cells [174]. For example, in the brain, hypoxia and vascular injury cause reduction of cognition for patients that undergo radiotherapy for brain tumors [173, 175]. It has been shown that hypoxia is a critical issue for late effects of ionizing radiation such as myelopathy, pneumonitis and fibrosis rather than early effects [171, 176, 177]. A radionuclide lung perfusion study showed that hypoxia increases with time after irradiation, which is associated with continuous increase in oxidative stress and the levels of VEGF and TGF-β [178].
HIF-1 plays a key role in hypoxia-induced oxidative injury, inflammation and fibrosis following lung irradiation. It can increase the expression of VEGF, TGF-β and inflammatory cytokines that activate redox reactions and finally facilitates fibrosis and pneumonitis [179]. In addition to the lung, HIF-1 may play a key role in the inflammatory reactions in other organs such as the intestine [180]. In contrast to HIF-1, upregulation of HIF-2 has been shown to protect normal tissues against radiation [181].
Hypoxia is known as a regulator of tumor resistance rather than normal tissue injury [182]. In response to hypoxia, HIF-1 stimulates VEGF and angiogenesis. Furthermore, HIF-1 can increase survival of cancer cells via induction of COX-2 [183]. Thus, targeting hypoxia does not interfere with tumor response to radiotherapy. The effects of hypoxia and HIF-1 on the resistance of a wide range of cancers such as lung, liver, brain, breast, prostate, cervix are well known. Thus, its targeting has been suggested to help overcome the resistance of a wide range of solid tumors and also amelioration of toxicity in adjacent normal tissues during radiotherapy [182].
Targeting fibrosis
Radiation-induced fibrosis can affect most irradiated tissues some months to years after exposure to heavy doses of ionizing radiation. Fibrosis is associated with deposition of collagen which leads to stiffness of tissues. In the muscles, fibrosis can cause stiffness, atrophy and shortening, which limit mobility [184]. This causes problems in the movement of joints and skin breakdown [185]. Fibrosis in the lung may appear the following radiotherapy for chest cancers, leading to pulmonary failure and death [186]. In the heart also, fibrosis increases the risk of heart attack [187]. Studies explaining the cellular and molecular mechanisms of radiation-induced fibrosis are ongoing. However, it is well known that the pivotal role of some cytokines such as TGF-β, IL-4 and IL-13 as well as some other mediators such as renin–angiotensin system and some microRNAs have shown important roles.
Interestingly, fibrosis which leads to tumor stiffness is an important cause of tumor resistance and progression [188]. TGF-β, IL-4 and IL-13 as pro-fibrotic cytokines are the most important suppressors of natural killer (NK) cells and cytotoxic T lymphocytes (CTLs), which cause immune escape of cancer cells [189]. Tumor fibrosis also reduces the penetration of anti-cancer drugs such as chemotherapy and immunotherapy into tumor microenvironment [188]. This effect can reduce the effectiveness of combination modalities such as chemoradiotherapy or immunoradiotherapy. Targeting fibrosis can increase the therapeutic efficiency of radiotherapy through normal tissue protection and also increasing tumor response to radiotherapy.
Pro-fibrotic cytokines
Fibrosis is a late effect of radiotherapy that may appear years after the end of treatment [190]. Fibrosis occurs following deposition of collagen and upregulation of extracellular matrix (ECM) components. Cell death through apoptosis and senescence, oxidative stress and chronic inflammation are key enhancers of fibrosis [191]. Fibrosis is a serious threat for some organs such as lung, skin, heart, intestine and also vessels [192, 193]. Fibrosis in the lung and heart poses a threat to the lives of patients that underwent radiotherapy for chest cancers and may cause death some years after radiotherapy. In the skin and intestine, fibrosis negatively affects the quality of life [194].
TGF-β
TGF-β is the central regulator of collagen deposition and fibrosis development following exposure to ionizing radiation. It is a key regulator of wound healing. However, massive apoptosis and senescence after exposure to high doses of ionizing radiation can cause abnormal upregulation of TGF-β for long times. TGF-β can increase the expression of pro-fibrosis pathways such as Smad2/3 and Rho/Rock pathways, which amplify the expression of ECM. TGF-β is also a potent stimulator of pro-oxidant enzymes such as NADPH oxidase, COX-2 and iNOS, which mediate the generation of ROS, NO and PGE2; which are key players of fibrosis [191]. In response to ionizing radiation, TGF-β has also been shown to induce fibrosis through canonical WNT/β-catenin pathway and PPARγ [17, 195]. Clinical studies have shown that upregulation of TGF-β has a direct relationship with fibrosis in the lung, heart, skin, intestine, bladder, liver etc. [196, 197]. To date, several pharmaceutical agents, antioxidants and radioprotectors have been investigated for mitigating radiation-induced fibrosis through modulation of TGF-β. Cu/Zn superoxide dismutase (SOD) has been shown to reduce radiation-induced fibrosis in the skin through the modulation of TGF-β [198]. Stimulation of SOD has also shown anti-fibrosis effects in the lung [199]. Some agents such as melatonin, curcumin, selenium, MnTE-2-PyP5+, etc., are able to induce SOD after exposure to radiation, thus they may be appropriate candidates for mitigation of fibrosis after exposure to heavy doses of ionizing radiation [200,201,202,203,204]. Experimental studies have shown that some agents such as flaxseed, EUK-207, IPW-5371 and genistein can reduce the expression of TGF-β and mitigate radiation-induced fibrosis [205,206,207].
TGF-β is one of the most potent immune system suppressors in a wide range of malignancies via suppression of CTLs and NK cells [208]. Furthermore, the release of TGF-β will be increased following radiotherapy in both tumor and normal tissues. In this situation, increased level of TGF-β causes immune escape of cancer cells as well as normal tissue toxicity [209]. Hence, inhibition of TGF-β has been suggested to overcome cancer resistance and also normal tissue protection during radiotherapy [210].
IL-4
IL-4 plays a key role in the promotion of fibrosis. IL-4 can induce upregulation of TGF-β, which leads to stimulation of its downstream genes. Knockdown of IL-4 in mice has been shown to attenuate infiltration of macrophages in the lung [211]. It seems that IL-4 is able to upregulate the expression and activity of pro-oxidant enzymes, thus stimulates the production of free radicals and differentiation of myofibroblasts. Duox1 and Duox2 are the most important ROS generating enzymes that upregulate their expression following increased release of IL-4. Suppression of IL-4 and both Duox1 and Duox2 has been shown to be associated with the reduction of radiation-induced fibrosis in the lung. Some radioprotectors such as melatonin and selenium have also shown abilities to reduce infiltration of macrophages that is also associated with the reduction of IL-4 level and inhibition of its downstream signaling pathways.
IL-4 has a high expression in some cancers such as that of the lung, breast, glioma, bladder, pancreas and ovary [212]. In tumor, IL-4 promotes the development of T helper cells type 2 (Th2), while it suppresses proliferation of Th1. These effects lead to attenuation of anti-tumor immunity because of reduction of IFN-γ and IL-2, which are responsible for the proliferation of CTLs and NK cells [213]. In response to radiation, it has been suggested that IL-4 induces proliferation of cancer stem cells (CSCs) in prostate cancer cells [214]. Also, it promotes epithelial-mesenchymal transition in breast cancer cells after irradiation [215]. As suppression of IL-4 can protect lung tissues and inhibit lung and breast cancers, its targeting favours radiotherapy for these malignancies. For other tissues, targeting IL-4 will require further studies to examine its potentials for normal tissue sparing.
IL-13
Similar to IL-4, IL-13 can promote radiation-induced fibrosis. Although the complete mechanisms of the role of IL-13 in the development of radiation toxicity remain to be elucidated, it has been suggested that IL-13 through triggering of IL13Rα2 induces fibrosis [216]. Knockdown of IL-13 in mice has been shown to reduce the severity of lung fibrosis [217]. Interestingly, targeting IL-13 has been suggested for boosting tumor immunity as well as inhibition of IL-13 in some cancer cells including MCA304 sarcoma, 4T1 breast carcinoma and glioma cancer cells [218,219,220]. For mice bearing glioma, inhibition of IL-13 has been shown to increase survival [221]. The combination of radiation and IL-13Rα2-targeted cytotoxin has shown promising results for glioma cancer cells [222, 223]. Studies examining the effects of IL-13 on both tumor and normal tissues are very limited and need to be investigated for each organ and tumor.
Renin–angiotensin system
It is well known that renin–angiotensin system has a key role in the development of late effects of ionizing radiation [224]. Infusion of angiotensin II can augment radiation nephropathy [225]. Furthermore, it has been suggested that angiotensin antagonists can reduce the severity of radiation-induced nephropathy [226, 227]. Total body irradiation can increase the level of renin and the expression of angiotensin some weeks after irradiation, which can cause damages to some organs such as kidney, heart, brain, and lung [227,228,229]. The elevated level of renin–angiotensin can impair the function of kidney and heart, and also stimulates synthase of collagen and development of fibrosis [230, 231]. As the level of renin and angiotensin increases during some weeks to months after exposure to ionizing radiation, it is a potential candidate for mitigation and treatment of radiation-induced side effects. The most important side effects of renin–angiotensin system following exposure to ionizing radiation are renal malfunction and inflammation as well as fibrosis in some organs such as the lung, heart and kidney.
In the kidney, blockade of both angiotensin I and II may be critical in achieving maximum protection against radiation-induced kidney failure [232]. However, a study showed that treatment with an angiotensin II inhibitor can prevent renal failure following whole-body irradiation [233]. In a case report, it was shown that losartan as an angiotensin II inhibitor, can treat renal damage caused by radiation [234]. Comparisons between the different types of angiotensin-converting enzyme inhibitors (ACEI) suggest that captopril is a gold standard ACEI for mitigation of renal nephropathy [235]. Administration of captopril or losartan starting 10 days after whole-body irradiation showed delayed initiation of renal failure [236]. So far, few clinical studies have been conducted to show the mitigatory effect of ACEIs on renal failure after total body irradiation (TBI). A study showed a decline but not significant reduction in renal failure for patients who received hematopoietic stem cells after TBI [237]. However, another study showed a significant improvement in the glomerular filtration rate (GFR) [238]. It has been suggested that the overexpression of heme oxygenase 1 (HO-1) following upregulation of angiotensin II has a role in renal injury caused by ionizing radiation [239]. However, further studies will be required to explain the mechanisms involved in angiotensin II-induced renal function injury.
Inhibition of renin–angiotensin using some agents such as captopril, losartan, enalapril and fosinopril has been shown to mitigate radiation-induced pneumonitis and fibrosis [240, 241]. The combination of renin–angiotensin antagonist captopril with an antioxidant showed more reduction in pneumonitis following lung irradiation, which is an indication that renin–angiotensin can promote inflammation and fibrosis independent of redox system [242]. Delayed treatment with captopril starting from 2 weeks after lung irradiation has also been shown to reduce damages to pulmonary vascular [243]. The use of ACEIs has been suggested to prevent or mitigate radiation-induced pneumonitis in radiotherapy patients [244]. The use of ACEIs for lung cancer radiotherapy patients has shown lower incidence of lung pneumonitis [245]. Similar results were observed in a retrospective study for patients who used ACEIs during radiotherapy [246]. A clinical study also showed promising results for patients who underwent stereotactic body radiotherapy (SBRT) for lung cancer [247]. Lung pneumonitis and renal failure are two important side effects of total body irradiation (TBI) for patients with leukemia. Administration of ACEIs after TBI and hematopoietic stem cell transplantation showed a significant reduction in the incidence of pneumonitis. Captopril showed a reduction in the probability of pneumonitis by 15% [237].
Although the complete mechanisms of renin–angiotensin induced pneumonitis and fibrosis remain to be elucidated, it seems that triggering pro-fibrotic cytokines and mediators by renin–angiotensin has a key role. A study by Molteni et al. confirmed that inhibition of angiotensin II using captopril or enalapril is associated with potent inhibition of TGF-β, COX-2, alpha-actomyosin (α-SMA) thromboxane (TXA2) and PGI2; which are key players in the progression of pulmonary fibrosis [248]. It has been suggested that renin–angiotensin inhibitors can increase the level of heptapeptide angiotensin-(1–7), which causes inhibition of inflammatory mediators such as COX-2 and MAPKs [249]. Furthermore, administration of captopril and other renin–angiotensin inhibitors have been shown to reduce the expression of PAI-1, an important regulator of senescence and fibrosis [250].
Administration of ACEI has also been shown to mitigate radiation toxicity in the bone marrow. Administration of captopril after whole-body irradiation can increase survival, while its administration before irradiation has been shown to reduce survival. Results indicated that administration of captopril only after exposure to radiation can cause bone marrow recovery [251]. Treatment with captopril even after 21 days has been shown to stimulate bone cells’ recovery [252]. It seems that an increase in angiotensin-(1–7) has a role in bone marrow recovery [253].
Angiotensin II has a key role in cancer immunosuppression. It can stimulate the expressions of TGF-β, COX-2, VEGF as well as others which promote tumor growth [254]. Inhibition of angiotensin II has been shown to suppress the growth of colorectal, renal and various types of lung cancer cells [255]. Due to the anti-cancer effects of angiotensin inhibitors on lung cancer as well as its protective effect on normal tissues, the combination of renin–angiotensin system inhibitors with radiotherapy may be applicable for lung cancer patients as shown by promising results from clinical studies [244, 256].
Epigenetic modulators
Epigenetic changes including modifications in the DNA methylation and histone acetylation play a key role in ionizing radiation-induced fibrosis [257]. ROS generation and DNA damage lead to several changes in the expression of miRNAs, DNA methylation and histone acetylation [258, 259]. In this section, we explain some epigenetic targets that are involved in the progression of fibrosis and suggested as potential targets for mitigation of radiation-induced fibrosis.
Bromodomain and extra terminal (BET)
BET proteins are involved in the reading of epigenetic histone modifications. BET proteins act as enhancer of the expression of target genes [260]. These proteins include four subfamilies that regulate the expression of some oncogenes and redox mediators. BET proteins are able to inhibit SOD expression, an important suppressor of fibrosis progression. Furthermore, BET proteins trigger myofibroblast differentiation following stimulation of TGF-β–NOX4 signaling pathway [261]. Silencing BET proteins has been proposed for preventing and also reversing fibrosis [262, 263]. So far, few studies have been conducted to explain the role of BET proteins in radiation-induced fibrosis. BET inhibitor JQ1 showed promising results for the reduction of inflammation and fibrosis following chest irradiation. Results indicated a reduction in pro-fibrosis genes and the differentiation of myofibroblasts [264]. Targeting BET can also regulate the expression of genes involved in fibroblast activation through activation of the diacylglycerol kinase alpha (DGKA). DGKA is an enhancer of alpha smooth muscle actin (α-SMA) and collagen 1A1 (COL1A1). Inhibition of BET proteins can cause reduction of DGKA gene expression, thus mitigates radiation-induced fibrosis [265]. Targeting BET proteins has shown anti-cancer effects for breast, colorectal, glioma, medulloblastoma, prostate and lung cancers [266, 267]. Clinical studies have also shown interesting results [268]. Thus, inhibition of BET may be promising for the amelioration of lung toxicity among patients with lung cancer.
MiR-21
It has been suggested that miR-21 has a close relation with TGF-β [269]. MiR-21 overexpression is associated with upregulation of TGF-β and fibrosis following exposure to ionizing radiation [270]. Upregulation of miR-21 after irradiation can suppress phosphatase and tensin homolog (PTEN) [271], leading to the induction of epithelial to mesenchymal transition (EMT), a key player of fibrosis [272]. MiR-21 overexpression has been shown to be associated with radiation-induced fibrosis [273]. MiR-21 as an inducer of fibroblast senescence causes the release of TGF-β from macrophages [274, 275]. As earlier mentioned, SOD is an important suppressor of radiation-induced fibrosis. One of the most common roles of miR-21 is inhibition of SOD2. TGF-β can induce the expression of miR-21 in fibroblasts, which cause the suppression of SOD and oxidative injury [276]. Downregulation of miR-21 has been suggested for reducing oxidative stress, inflammation and fibrosis following irradiation of the heart [277]. (Table 1).
MiR-21 is known as an important regulator of radioresistance of some malignancies including breast and lung [278]. This could ease the management of radiation-induced fibrosis in the lung without adverse effects on lung or breast cancer responses to radiotherapy [279].
The molecules with unclear selectivity
There are some other mediators that are mainly involved in cellular metabolism and potentiate radiation-induced oxidative stress. Targeting NADPH Oxidase enzymes including NOX1-5 and Duox1&2 have shown that can attenuate radiation injury in some normal cells/tissues. However, their inhibition has shown various consequences on the radiosensitivity of cancer cells [280, 281]. PPAR also is another target that has shown different effects. Some studies suggested that PPAR agonists can protect normal tissues [282]. However, this may increase cancer cells radioresistance [283]. Targeting of these molecules and some other targets such as mitochondria, iNOS, and mTOR need to more accuracy. Suppression of these mediators may reduce ROS and NO levels in tumor, leading to more survival following exposure to ionizing radiation [204, 284, 285] (Fig. 4).

Mechanisms of radiation-induced redox metabolism and its role in the progression of radiation-induced normal tissue injury
Regenerative medicine in the mitigation of ARS
Stem cells responses to ionizing radiation
Stem cells are critical targets for ionizing radiation. The primary reason for hematopoietic and gastrointestinal system is stem cell death, which leads to depletion of progenitor and mature cells during some days after exposure to radiation. Bone marrow is containing two types of multipotent stem cells, including mesenchymal and hematopoietic stem cells [59]. Mesenchymal stem cells are more radioresistant compared to hematopoietic stem cells. Furthermore, mesenchymal stem cells are responsible for the production of fatty cells and osteoblasts that are less critical following exposure to a radiation accident. By contrast, hematopoietic stem cells are responsible for maintain of immune system, red cells and platelets. Hematopoietic stem cells divide into two type progenitor cells, including myeloid and lymphoid cells. Hematopoietic stem cells have very low mitotic activity, while, progenitor cells have higher mitotic index and are more radiosensitive. Lymphoid progenitor cells and also its derivative cells containing lymphocytes have high radiosensitivity. High radiosensitivity of progenitor and mature lymphoid cells leads to fast depletion of lymphocytes in peripheral circulation which may continue for a long time after exposure to a high dose of radiation. Myeloid-derived cells such as neutrophils, platelets, monocytes, basophils and erythrocytes are radioresistant and no immediate reduction is observable after exposure to radiation. However, high radiosensitivity of myeloid progenitor cells lead to remarkable reduction of these cells during some days to weeks. These changes cause severe suppression of immune system.
Stem cells in intestine, especially jejunum have high mitotic activity. High self-renewal activity of stem cells in intestine maintains sufficient number of functional cells such as absorptive, epithelial and goblet cells. Functional cells are resistant to ionizing radiation. However, they have low life and then are shed into the lumen during some days. Continuous division of intestine stem cells provide sufficient surface for nutrient and water absorption by crypts and villi. High mitotic activity of intestine stem cells makes them sensitive to ionizing radiation. Damage to intestinal stem cells leads to depletion of goblet cells, shortening of villi and crypt thinning. Furthermore, apoptosis and necrosis of endothelial cells in vessels lead to bleeding and penetration of microorganisms into circulation. The existence of bacteria in circulation and also suppression of immune system can cause severe infection. Reduction of platelets also increase bleeding and more reduction of peripheral cells.
As mentioned earlier, redox interactions and continuous production of ROS and NO play key role in radiation toxicity. In the hematopoietic system and intestine ROS and NO can suppress DNA damage repair and cell cycle progression. Targeting of ROS generating sources such as NOX4, and also mitochondria has shown that reduces damage to hematopoietic stem and progenitor cells and improves hematopoietic function after irradiation. Scavenging of free radicals also can help to repair stem cells and mitigation of radiation injury in intestine [287]. WNT/beta-catenin plays a key role in the proliferation of stem cells and resistance to ionizing radiation [288]. Stimulation of this pathway can increase survival through the induction of repair pathway and suppression of apoptosis [287, 289]. Some other targets such as TLRs, p53 and angiotensin II are promising for stem cells protection, however, there need to studies to explain the role of these targets for each type of stem cells in bone marrow and intestine.
Stem cells therapy for mitigation of ARS
Bone marrow transplantation has been a known strategy for the regeneration of bone marrow function after bone marrow injury such as seen following ARS or total body irradiation for leukemia. Results from Chernobyl accident confirmed successful mitigation of ARS following bone marrow transplantation [290]. It was assumed that hematopoietic stem and progenitor cells are responsible for the regeneration of bone marrow function. However, nowadays it is well known that other stem cells such as mesenchymal, adipose or embryonic stem cells can regenerate radiation-induced stem cell depletion in the bone marrow and gastrointestinal system [291]. To date, some types of stem cells such as mesenchymal and embryonic stem cells have studied in animal studies to mitigate ARS following exposure to lethal doses of ionizing radiation [292,293,294,295]. MSCs have priority for transplantation because their immunoregulatory effects. MSCs are able to attenuate response of T lymphocytes, thus reduces transplant rejection probability [296]. Furthermore, transplantation of endothelial cells has shown that can mitigate ARS, mediated through the regeneration of hematopoietic stem cells, inhibition of apoptosis and also repair of injured vessels [297, 298].
Administration of MSCs to irradiated mice can reduce apoptosis and trigger stemness in both HSCs and stromal stem cells. MSCs are able to upregulate Notch2 signaling, an important regulator of hematopoiesis, thus triggers proliferation of both MSCs and HSCs after exposure to ionizing radiation [299]. MSCs also attenuate inflammation and endogenous free radicals, thus reduce vascular injury [257]. Using MSCs with high expression of high mobility group box 1 (HMGB1) has shown is more effective for mitigation of radiation-induced vascular injury. HMGB1-modified MSCs have higher potential for differentiation toward endothelial cells, thus can repair vessels more effectively [300].
Stem cells therapy in combination with ROS targeting for mitigation of ARS
As some stromal cells such as fibroblasts play a key role in redox reactions after ionizing radiation, inhibition of these targets may help to preserve remaining stem cells and also the success of stem cells transplantation [301]. Furthermore, targeting free radicals by antioxidants or ROS scavenging enzymes may be an interesting idea for better mitigation of ARS following stem cells therapy. A study reported the successful mitigation of ARS using genetic modified MSCs to release extracellular SOD [302]. Similar results observed for modified umbilical cord mesenchymal stromal cells [303]. Activation of ERK/NRF2 pathway is another suggested mechanism that has shown mitigate damage to hematopoietic stem cells [304]. NRF2 triggers the activation of antioxidant defense enzymes such as SOD, thus reduces apoptosis following exposure to ionizing radiation [304]. As SOD has shown that neutralize free radicals produced by stromal cells, activation of this pathway may help to the survival of remaining stem cells after exposure to radiation, or even attenuates damage to transplanted stem cells [305, 306].
Stem cell therapy for mitigation of delayed effect of acute radiation exposure (DEARE)
Although cell therapy can mitigate radiation toxicity in bone marrow and gastrointestinal system, damage to other organs such as kidney and lung need to mitigated through inhibition of other potential targets. Stem cell therapy can reverse the depletion of stem cells in irradiated organs and also attenuate some other side effects. It is reported that MSCs have some properties that help to management of radiation-induced inflammation and fibrosis in late responding tissues such as lung [307]. MSCs can help to restore of radiosensitive cells in the lung, including endothelial cells in vessels and epithelial cells in airways [308]. Furthermore, MSCs have the ability to restore antioxidant defense, thus attenuate oxidative stress [309]. MSCs can release SOD, thus can suppress chronic inflammation, oxidative stress and development of fibrosis in the lung [310, 311]. Clinical usage of stem cells for protection and mitigation of radiation-induced lung fibrosis is in the first steps (NCT02277145).
By contrast to mentioned studies, there is some evidence that stem cell therapy may cause increase progression of lung injury [312]. Furthermore, there are some experimental studies that indicated stem cell transplantation cannot mitigate kidney and lung injury following whole-body exposure to radiation [237, 313]. An experimental study showed that stem cell transplantation following whole-body irradiation with 10 Gy mitigates ARS in hematopoietic and gastrointestinal system. However, a remarkable reduction in survival observed during 15–20 weeks. Results showed a significant increase in BUN level, indicating nephropathy. Treatment with captopril or other renin–angiotensin inhibitors showed a significant reduction in the BUN level and increase of survival [235]. Another study also indicated that bone marrow transplantation after whole-body irradiation with 11 Gy lead to death during 2–4 months because of pneumonitis and kidney failure. However, when rats received renin–angiotensin inhibitors including captopril, enalapril or fosinopril, a significant increase in survival observed, which was associated with amelioration of pneumonitis and nephropathy [314]. A clinical study also confirmed that captopril administration after total body irradiation and bone marrow transplantation increase survival of patients [237].
Importance of time for the onset of radiation mitigation treatment against ARS and DEARE
Timing for initiation of treatment for mitigation of radiation lethality is a vital issue. It is predictable that kinetic of cells and also final consequence of ionizing radiation in each organ play key role in the selection of appropriated mitigators and also timing for supplement. ARS in the hematopoietic and gastrointestinal system is mainly resulting from apoptosis of stem and progenitor cells [315]. Early apoptosis occurs during some hours after exposure to ionizing radiation [316]. Thus, mitigation of ARS in hematopoietic and gastrointestinal system need to suppression of apoptosis at early hours after exposure to radiation. Targeting of p53 and TLRs has confirmed this issue. Inhibition of p53 or treatment with TLRs agonists can mitigate radiation toxicity effectively when treatment initiated during first hours after irradiation [19]. Early apoptosis is p53 dependent and its targeting need to treatment during first minutes to hours [317]. Second wave of radiation-induced toxicity in bone marrow and intestine may be trigger by activation of pro-oxidants and production of free radicals. These free radicals are not produced by ionizing radiation directly, but apoptosis and release of damaged cells contents can trigger ROS production by macrophages, lymphocytes and also mitochondria in some non-immune cells [318]. Neutralization of free radical using ROS/NO scavengers and also mitochondria targeting can suppress further apoptosis and depletion of progenitor cells [319]. Initiation of second wave of ROS production may take some hours to days. Thus, treatment with antioxidants immediately after exposure to radiation may do not cause remarkable improvement in survival and mitigation of ARS [320]. Renin–angiotensin is another known mediator of radiation toxicity in bone marrow. As showed in Fig. 5, it can stimulate upregulation of TGF-β, an important stimulator of apoptosis and redox activation. Targeting of renin–angiotensin system immediately after exposure to radiation to some weeks later can mitigate ARS, probably via inhibition of TGF-β induced apoptosis and chronic oxidative stress.

Suitable timelines for the mitigation of radiation injury in different organs. Each target may need a specific time in each organ. Information for some targets such as COX-2 require further studies. Furthermore, each antioxidant or anti-inflammatory agent may lead to different results, maybe because of their effects on other mechanisms. This timing is based on information adopted from animal studies
The time window for mitigation of DEARE is differ compared to ARS. In the kidney, stem cells have low kinetic and nephropathy can occur during a long time after exposure [321]. Pneumonitis and fibrosis also are late effects of ionizing radiation that may cause death during months to years after exposure to a high dose of ionizing radiation [322]. These side effects are resulting from chronic upregulation of inflammatory and pro-fibrosis cytokines and transcription factors [323]. Oxidative stress following cell death and activation of pro-oxidant enzymes plays a key role in the upregulation of inflammatory and fibrosis mediators. On the other hand, inflammation and fibrosis processes are associated with several changes including ROS and NO production. This process indicates that using ROS/NO scavengers and also some drugs that are able to inhibit inflammation and fibrosis can be used to suppress late effects of ionizing radiation in late responding organs [318]. Mitigation of radiation-induced kidney injury with selenium or angiotensin inhibitors can mitigate nephropathy. Shortening of supplement time showed reduced mitigation of nephropathy [235, 324]. For lung pneumonitis and fibrosis, treatment with antioxidants is necessary during the first 4 weeks. Treatment with flaxseed has shown that treatment during 2–4 weeks is more important compared to previous or later times [325]. Treatment with renin–angiotensin inhibitors or antioxidants including genistein and EUK-207 for longer times can mitigate lung injury. Furthermore, starting treatment as soon as possible may be more useful [326]. Delayed treatment for 4 weeks may abrogate the mitigation of lung injury [327]. Although delayed starting treatment with antioxidants can reduce radiation injury in the lung, it seems that longer time treatment is favor for mitigation of pneumonitis and fibrosis. However, this effect is highly depended on antioxidant or anti-inflammation agent. Another target for mitigation of radiation-induced lung injury is COX-2. A study showed an increased survival when celecoxib administration started at 80 days after chest irradiation [132]. The time effect of treatment with mitigators for lung pneumonitis and injury need further elucidation (Fig. 5).
Experimental models for ARS and DEARE
An important problem for confirmation and approve of a radiation mitigator is extrapolation of animal results to human. This is because it is not possible to test radiation mitigators for human. Thus, it is necessary to use standard animal models to compare results of different mitigator agents. Furthermore, it has been suggested that endpoints in animal models should be related to human, and pharmacodynamics of mitigators should be understood. For evaluating each endpoint, it is necessary to be noted that some genetic differences can affect radiation response in each strain [328].
ARS in hematopoietic and gastrointestinal systems are the most common studies for exploring new radiation mitigators. Mice, canine and nonhuman primate are three types of strains that have been used for investigating hematopoietic system study. The most common mice strains include C57BL/6, BALB/c, C3H/HeN and B6D2F1/J. Radiosensitivity of these strains is varied. Among these strains BALB/c is more radiosensitive. LD50/30 for BALB/c mice is lower than 7 Gy. This may relate to reduced activity of DNA PKcs, a critical DNA repair enzyme in BALB/c strains [328]. NMRI mice are another strain that have studies for several bone marrow experiments with a LD50/30 equal 7.2 Gy [329]. Among larger animals, rhesus macaque is the most common type of nonhuman primate for hematopoietic system model. For gastrointestinal system, C57BL/6, BALB/c were the most common types of strains for investigation of radiation mitigators. However, some other strains such as rats, dogs or non-human primates may be used for this aim [330].
Skin is another important tissue that reacts to ionizing radiation during the early days to several months after irradiation, depending on radiation dose. Rodents are the most common strains for investigating radiation dermatitis. However, skin responses in rodents vary and can be observed at different doses. Furthermore, investigation of dermatitis in some mice may be difficult because of their hairy and dark skins [328]. For skin responses to ionizing radiation, it has been suggested to use pigs instead of mice or rat’s models. This is because of more similar physiological responses and structure of skin in pigs with human [328]. Among large animals, non-human primates and dogs have the most similar immunological responses to human [331].
Lung and kidney are the most important late responding organs, which show different radiosensitivities among various strains. For kidney, rats are in priority among small animal models. Indeed, most researches for nephropathy models can be achieve using rat’s models, while mice show less response for renal following exposure to ionizing radiation [328]. Also, mice strains show different responses to ionizing radiation and recovery, thus it is not easy to compare results of different types of mice to radiation or radiomitigators [332, 333]. WAG/Rij strain rats are one of the most known strains for investigation of kidney diseases including radiation-induced nephropathy [334]. For lung responses, including pneumonitis and fibrosis, non-human primates and dogs have the most similar lung physiology for both pneumonitis and fibrosis. However, mice may show different responses for each of these endpoints [328]. It has been suggested that scientists use two strains of mice for evaluating mitigation of pneumonitis and fibrosis. C57BL/6 mice show extensive fibrosis but very low pneumonitis markers following lung irradiation. On the other hand, C3HeB/FeJ mice show more pneumonitis with no extended fibrosis [328]. Rats may be better candidate compared to mice for lung studies. Sprague–Dawley or WAG/RijCmcr rats show both acute pneumonitis and fibrosis following lung exposure to radiation [205, 241]. Furthermore, the heart, which is a late responding tissue, shows remarkable response following chest irradiation [335]. In this situation, both lung and heart tissues can be investigated for detecting mitigatory effects [336].
Conclusion
In conclusion, ionizing radiation causes several changes in the expressions of pro-inflammatory and pro-fibrotic genes, and also changes the normal metabolism of oxygen. These are as a result of massive DNA damage and cell death, especially through apoptosis, necrosis, necroptosis and senescence. Boosting DNA repair mechanisms and suppression of death mediators such as Bax can help reduce cell death caused by ionizing radiation. Furthermore, the induction of anti-apoptotic pathways has shown promising results. TLRs agonists are interesting for suppression of apoptosis in highly radiosensitive organs such as the bone marrow and intestine. It seems that TLRs can act as protector through modulation of early apoptosis genes. Similar results have also been observed for p53 inhibition. P53, an important regulator of DNA damage repair and apoptosis plays a key role in protection or mitigation of radiation toxicity in hematopoietic system. It seems that p53 has pivotal role in early apoptosis in bone marrow stem cells. Thus, inhibition of p53 immediately or during some early hours after exposure to radiation can increase resistance to ionizing radiation. However, its targeting during some days after exposure to radiation cannot act as a strategy for mitigation of radiation mortality. The roles of these targets in protection or mitigation of radiation injury in late responding tissues remain to be elucidated. However, in these organs such as seen in the lung, senescence and necrosis may be more important compared to apoptosis.
Inflammation and redox mediatory responses are critical targets for mitigation of radiation injury. This is important for both early responding tissues such as bone marrow as well as for late responding tissues such as the lung, kidney, heart, brain and spinal cord. Continuous ROS generation by the mitochondria and pro-oxidant enzymes such as NADPH oxidase promotes apoptosis and senescence in the bone marrow and intestinal progenitor cells. Inhibition of these ROS sources or neutralization of free radicals has shown significant mitigation of radiation mortality. In contrast to TLRs and p53, inhibition of oxidative stress through direct neutralization of free radicals by antioxidants or targeting of mitochondria and pro-oxidant enzymes can mitigate radiation injury even when treatment commences some days after exposure to radiation. Targeting of renin–angiotensin system after irradiation has been shown to mitigate radiation-induced hematopoietic and gastrointestinal system, even when treatment started some weeks after irradiation. Interestingly, suppression of angiotensin before irradiation may not protect these organs. Thus, the cellular and molecular mechanisms of this issue need to be studied.
For late responding organs such as the lung, heart, kidney, brain and gastrointestinal system, chronic inflammation and fibrosis are main concerns. Pro-inflammatory and pro-fibrotic cytokines including IL-1, IL-4, IL-13, TGF-β and TNF-α are important inducers. However, there are some other inflammatory and fibrosis mediators that can affect the release of these cytokines, or may be induced by them. Renin–angiotensin system, mTOR and senescence, hypoxia, epigenetic modulators such as BET and miR-21 are the common enhancers of radiation-induced fibrosis through TGF-β pathway. On the other hand, iNOS, NOX1&2&4, Duox1-2 and COX-2 are important downstream factors of pro-fibrosis cytokines. Targeting these factors can be proposed for the mitigation of radiation injury in these organs. However, their expressions are tissue specific, thus the need for further knowledge about the radiobiological response of each organ is critical for effective mitigation of radiation-induced normal tissue injury.
The timing of treatment with radiation modifiers is a very critical issue. Targeting of early apoptosis can be very effective for protection of radiosensitive organs. However, for an accidental radiological or nuclear disaster it is very difficult to mitigate hematopoietic and gastrointestinal system through this pathway. Thus, some targets such as TLRs and p53 may not be useful for mitigation of radiation injury in real situations. Targeting TLRs is effective before exposure to radiation or some hours after. Thus, TLRs are promising targets for radiation protection, however, mitigation of radiation injury using TLRs agonist acts in a limited time window. The role of TLRs in late responding radiosensitive organs such as the lung is complicated and some studies have shown different results. Although activation of TLRs can suppress apoptosis and ARS for early responding tissues, it is important to note that its role as an inflammatory mediator may cause the severity of late effects of ionizing radiation. In fact, suppression of TLRs may be more promising for late responding organs. Thus, TLRs agonists may be promising radioprotectors in clinical studies, however, their use as radiation mitigators needs further studies. Potent antioxidants and anti-inflammatory agents are interesting for both protection and mitigation via treatment before or after exposure to radiation. As redox interactions start to produce free radicals during some days after exposure to radiation, mitigation of radiation injury using ROS/NO scavengers or redox inhibitors can be done more effectively. In a real situation, treatment of injured people in an accident can be done through the administration of antioxidants starting from some days after the accident. In contrast to TLRs agonists and p53 inhibitors, the time for mitigation of radiation mortality is not very critical. Exposed persons to lethal doses of ionizing radiation may survive if treatments commence even after some days. A more extended time window has been observed for renin–angiotensin inhibitors. Inhibition of renin–angiotensin system can mitigate radiation injury in both early and late responding organs when treatment commence even some weeks after exposure to radiation.
References
Lindegaard JC, Grau C (2000) Has the outlook improved for amifostine as a clinical radioprotector? Radiother Oncol 57(2):113–118
Wasserman T (1999) Radioprotective effects of amifostine. Semin Oncol 20:20
Abt G, Vaghef H, Gebhart E, Dahlgren CV, Hellman B (1997) The role of N-acetylcysteine as a putative radioprotective agent on X-ray-induced DNA damage as evaluated by alkaline single-cell gel electrophoresis. Mutation Res DNA Rep 384(1):55–64
Rades D, Fehlauer F, Bajrovic A, Mahlmann B, Richter E, Alberti W (2004) Serious adverse effects of amifostine during radiotherapy in head and neck cancer patients. Radiother Oncol 70(3):261–264. https://doi.org/10.1016/j.radonc.2003.10.005
Brizel DM, Wasserman TH, Henke M, Strnad V, Rudat V, Monnier A et al (2000) Phase III randomized trial of amifostine as a radioprotector in head and neck cancer. J Clin Oncol 18(19):3339–3345
Gudkov S, Popova N, Bruskov V (2015) Radioprotectors: history, trends and prospects. Biofizika 60(4):801–811
Johnke RM, Sattler JA, Allison RR (2014) Radioprotective agents for radiation therapy: future trends. Future Oncol 10(15):2345–2357
Gudkov AV, Komarova EA (2010) Radioprotection: smart games with death. J Clin Investig 120(7):2270–2273
Deas SD, Huprikar N, Skabelund A (2017) Radiation exposure and lung disease in today's nuclear world. Curr Opin Pulmon Med 23(2):167–172
Citrin D, Cotrim AP, Hyodo F, Baum BJ, Krishna MC, Mitchell JB (2010) Radioprotectors and mitigators of radiation-induced normal tissue injury. Oncologist 15(4):360–371
Delanian S, Lefaix J-L (2007) Current management for late normal tissue injury: radiation-induced fibrosis and necrosis. Semin Radiat Oncol 20:20
Deeg HJ (1990) Delayed complications and long-term effects after bone marrow transplantation. Hematol Oncol Clin 4(3):641–657
Eriksson D, Stigbrand T (2010) Radiation-induced cell death mechanisms. Tumor Biol 31(4):363–372
Riklis E, Emerit I, Setlow R (1996) New approaches to biochemical radioprotection: antioxidants and DNA repair enhancement. Adv Sp Res 18(1–2):51–54
Golden E, Pellicciotta I, Demaria S, Barcellos-Hoff MH, Formenti SC (2012) The convergence of radiation and immunogenic cell death signaling pathways. Front Oncol 2:88
Ashrafizadeh M, Ahmadi Z, Mohammadinejad R, Kaviyani N, Tavakol S (2020) Monoterpenes modulating autophagy: a review study. Basic Clin Pharmacol Toxicol 126:9
Ashrafizadeh M, Ahmadi Z, Kotla NG, Afshar EG, Samarghandian S, Mandegary A et al (2019) Nanoparticles targeting STATs in cancer therapy. Cells 8(10):1158
Holley AK, Miao L, St Clair DK, St Clair WH (2014) Redox-modulated phenomena and radiation therapy: the central role of superoxide dismutases. Antioxid Redox Signal 20(10):1567–1589. https://doi.org/10.1089/ars.2012.5000
Burdelya LG, Krivokrysenko VI, Tallant TC, Strom E, Gleiberman AS, Gupta D et al (2008) An agonist of toll-like receptor 5 has radioprotective activity in mouse and primate models. Science 320(5873):226–230. https://doi.org/10.1126/science.1154986
Ahmadi Z, Roomiani S, Bemani N, Ashrafizadeh M (2019) The targeting of autophagy and endoplasmic reticulum stress mechanisms by honokiol therapy. Rev Clin Med 6(2):66–73
Rainaldi G, Ferrante A, Indovina PL, Santini MT (2003) Induction of apoptosis or necrosis by ionizing radiation is dose-dependent in MG-63 osteosarcoma multicellular spheroids. Anticancer Res 23(3b):2505–2518
Chen Z, Cao K, Xia Y, Li Y, Hou Y, Wang L et al (2019) Cellular senescence in ionizing radiation (Review). Oncol Rep 42(3):883–894. https://doi.org/10.3892/or.2019.7209
Verheij M, Bartelink H (2000) Radiation-induced apoptosis. Cell Tissue Res 301(1):133–142
Ashrafizadeh M, Ahmadi Z (2019) Effects of statins on gut microbiota (microbiome). Rev Clin Med 6(2):55–59
Zhou L, Yuan R, Serggio L (2003) Molecular mechanisms of irradiation-induced apoptosis. Front Biosci J Virtual Lib 8:d9–d19. https://doi.org/10.2741/927
Redza-Dutordoir M, Averill-Bates DA (2016) Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta Mol Cell Res 1863(12):2977–2992. https://doi.org/10.1016/j.bbamcr.2016.09.012
Jung M, Zhang Y, Dimtchev A, Dritschilo A (1998) Impaired regulation of nuclear factor-κB results in apoptosis induced by gamma radiation. Radiat Res 149(6):596–601
Zhang H, Wang Y-A, Meng A, Yan H, Wang X, Niu J et al (2013) Inhibiting TGFβ1 has a protective effect on mouse bone marrow suppression following ionizing radiation exposure in vitro. J Radiat Res 54(4):630–636. https://doi.org/10.1093/jrr/rrs142
Qiu W, Carson-Walter EB, Liu H, Epperly M, Greenberger JS, Zambetti GP et al (2008) PUMA regulates intestinal progenitor cell radiosensitivity and gastrointestinal syndrome. Cell Stem Cell 2(6):576–583. https://doi.org/10.1016/j.stem.2008.03.009
Wang F, Cheng J, Liu D, Sun H, Zhao J, Wang J et al (2014) P53-participated cellular and molecular responses to irradiation are cell differentiation-determined in murine intestinal epithelium. Arch Biochem Biophys 542:21–27. https://doi.org/10.1016/j.abb.2013.11.012
Najafi M, Motevaseli E, Shirazi A, Geraily G, Rezaeyan A, Norouzi F et al (2018) Mechanisms of inflammatory responses to radiation and normal tissues toxicity: clinical implications. Int J Radiat Biol 94(4):335–356. https://doi.org/10.1080/09553002.2018.1440092
Smith TA, Kirkpatrick DR, Smith S, Smith TK, Pearson T, Kailasam A et al (2017) Radioprotective agents to prevent cellular damage due to ionizing radiation. J Transl Med 15(1):232. https://doi.org/10.1186/s12967-017-1338-x
Musa AE, Omyan G, Esmaely F, Shabeeb D (2019) Radioprotective effect of hesperidin: a systematic review. Medicina (Kaunas) 55:7. https://doi.org/10.3390/medicina55070370
Mortezaee K, Najafi M, Farhood B, Ahmadi A, Shabeeb D, Musa AE (2019) NF-kappaB targeting for overcoming tumor resistance and normal tissues toxicity. J Cell Physiol 234(10):17187–17204. https://doi.org/10.1002/jcp.28504
Maier P, Hartmann L, Wenz F, Herskind C (2016) Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int J Mol Sci 17(1):102. https://doi.org/10.3390/ijms17010102
Gioia U, Francia S, Cabrini M, Brambillasca S, Michelini F, Jones-Weinert CW et al (2019) Pharmacological boost of DNA damage response and repair by enhanced biogenesis of DNA damage response RNAs. Sci Rep 9(1):6460. https://doi.org/10.1038/s41598-019-42892-6
Nieto A, Hara MR, Quereda V, Grant W, Saunders V, Xiao K et al (2019) βarrestin-1 regulates DNA repair by acting as an E3-ubiquitin ligase adaptor for 53BP1. Cell Death Differ. https://doi.org/10.1038/s41418-019-0406-6
Ramdzan ZM, Ginjala V, Pinder JB, Chung D, Donovan CM, Kaur S et al (2017) The DNA repair function of CUX1 contributes to radioresistance. Oncotarget 8(12):19021–19038. https://doi.org/10.18632/oncotarget.14875
Cheng Y, Ren X, Gowda AS, Shan Y, Zhang L, Yuan YS et al (2013) Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis 4(7):e731-e. https://doi.org/10.1038/cddis.2013.254
Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV et al (1999) A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285(5434):1733–1737. https://doi.org/10.1126/science.285.5434.1733
Komarova EA, Kondratov RV, Wang K, Christov K, Golovkina TV, Goldblum JR et al (2004) Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic injury, but protects from gastro-intestinal syndrome in mice. Oncogene 23(19):3265–3271. https://doi.org/10.1038/sj.onc.1207494
Kirsch DG, Santiago PM, di Tomaso E, Sullivan JM, Hou WS, Dayton T et al (2010) p53 controls radiation-induced gastrointestinal syndrome in mice independent of apoptosis. Science 327(5965):593–596. https://doi.org/10.1126/science.1166202
Strom E, Sathe S, Komarov PG, Chernova OB, Pavlovska I, Shyshynova I et al (2006) Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol 2(9):474–479. https://doi.org/10.1038/nchembio809
Morita A, Yamamoto S, Wang B, Tanaka K, Suzuki N, Aoki S et al (2010) Sodium orthovanadate inhibits p53-mediated apoptosis. Cancer Res 70(1):257–265. https://doi.org/10.1158/0008-5472.can-08-3771
Morita A, Zhu J, Suzuki N, Enomoto A, Matsumoto Y, Tomita M et al (2006) Sodium orthovanadate suppresses DNA damage-induced caspase activation and apoptosis by inactivating p53. Cell Death Differ 13(3):499–511. https://doi.org/10.1038/sj.cdd.4401768
Wang B, Tanaka K, Morita A, Ninomiya Y, Maruyama K, Fujita K et al (2013) Sodium orthovanadate (vanadate), a potent mitigator of radiation-induced damage to the hematopoietic system in mice. J Radiat Res 54(4):620–629. https://doi.org/10.1093/jrr/rrs140
de Guzman AE, Ahmed M, Li Y-Q, Wong CS, Nieman BJ (2019) p53 loss mitigates early volume deficits in the brains of irradiated young mice. Int J Radiat Oncol Biol Phys 103(2):511–520. https://doi.org/10.1016/j.ijrobp.2018.09.014
Morita A, Ariyasu S, Ohya S, Takahashi I, Wang B, Tanaka K et al (2013) Evaluation of zinc (II) chelators for inhibiting p53-mediated apoptosis. Oncotarget 4(12):2439–2450. https://doi.org/10.18632/oncotarget.1535
Muller PAJ, Vousden KH (2013) p53 mutations in cancer. Nat Cell Biol 15(1):2–8. https://doi.org/10.1038/ncb2641
Mantovani F, Collavin L, Del Sal G (2019) Mutant p53 as a guardian of the cancer cell. Cell Death Differ 26(2):199–212. https://doi.org/10.1038/s41418-018-0246-9
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11(5):373
Shcheblyakov DV, Logunov DY, Tukhvatulin AI, Shmarov MM, Naroditsky BS, Gintsburg AL (2010) Toll-like receptors (TLRs): the role in tumor progression. Acta Nat 2(3):21–29
Kurkjian CJ, Guo H, Montgomery ND, Cheng N, Yuan H, Merrill JR et al (2017) The toll–like Receptor 2/6 agonist, FSL–1 lipopeptide, therapeutically mitigates acute radiation syndrome. Sci Rep 7(1):17355
Burdelya LG, Gleiberman AS, Toshkov I, Aygun-Sunar S, Bapardekar M, Manderscheid-Kern P et al (2012) Toll-like receptor 5 agonist protects mice from dermatitis and oral mucositis caused by local radiation: implications for head-and-neck cancer radiotherapy. Int J Radiat Oncol Biol Phys 83(1):228–234
Guo J, Chen Y, Lei X, Xu Y, Liu Z, Cai J et al (2017) Monophosphoryl lipid a attenuates radiation injury through TLR4 activation. Oncotarget 8(49):86031
Ashrafizadeh M, Ahmadi Z, Farkhondeh T, Samarghandian S (2019) Modulatory effects of statins on the autophagy: a therapeutic perspective. J Cell Physiol 20:20
Burdelya LG, Brackett CM, Kojouharov B, Gitlin II, Leonova KI, Gleiberman AS et al (2013) Central role of liver in anticancer and radioprotective activities of Toll-like receptor 5 agonist. Proc Natl Acad Sci 110(20):E1857–E1866
Roses RE, Xu M, Koski GK, Czerniecki BJ (2008) Radiation therapy and Toll-like receptor signaling: implications for the treatment of cancer. Oncogene 27(2):200
Ashrafizadeh M, Ahmadi Z, Mohamamdinejad R, Farkhondeh T, Samarghandian S (2019) Curcumin activates the Nrf2 pathway and induces cellular protection against oxidative injury. Curr Mol Med 20:20
Epperly M, Rhieu B, Cao S, Goff J, Shields D, Franicola D et al (2014) Reduced radiation pulmonary fibrosis in toll-like receptor-4 (TLR4) deletion recombinant negative mice. Int J Radiat Oncol Biol Phys 90(1):S150. https://doi.org/10.1016/j.ijrobp.2014.05.625
Paun A, Fox J, Balloy V, Chignard M, Qureshi ST, Haston CK (2010) Combined Tlr2 and Tlr4 deficiency increases radiation-induced pulmonary fibrosis in mice. Int J Radiat Oncol Biol Phys 77(4):1198–1205. https://doi.org/10.1016/j.ijrobp.2009.12.065
Urban-Wojciuk Z, Khan MM, Oyler BL, Fåhraeus R, Marek-Trzonkowska N, Nita-Lazar A et al (2019) The role of TLRs in anti-cancer immunity and tumor rejection. Front Immunol 10:2388. https://doi.org/10.3389/fimmu.2019.02388
Gu J, Liu Y, Xie B, Ye P, Huang J, Lu Z (2018) Roles of toll-like receptors: From inflammation to lung cancer progression. Biomed Rep 8(2):126–132. https://doi.org/10.3892/br.2017.1034
Huang L, Xu H, Peng G (2018) TLR-mediated metabolic reprogramming in the tumor microenvironment: potential novel strategies for cancer immunotherapy. Cell Mol Immunol 15(5):428–437. https://doi.org/10.1038/cmi.2018.4
Mason KA, Ariga H, Neal R, Valdecanas D, Hunter N, Krieg AM et al (2005) Targeting toll-like receptor 9 with CpG oligodeoxynucleotides enhances tumor response to fractionated radiotherapy. Clin Cancer Res 11(1):361–369
Dovedi SJ, Melis MH, Wilkinson RW, Adlard AL, Stratford IJ, Honeychurch J et al (2013) Systemic delivery of a TLR7 agonist in combination with radiation primes durable antitumor immune responses in mouse models of lymphoma. Blood 121(2):251–259. https://doi.org/10.1182/blood-2012-05-432393
Dewan MZ, Vanpouille-Box C, Kawashima N, DiNapoli S, Babb JS, Formenti SC et al (2012) Synergy of topical toll-like receptor 7 agonist with radiation and low-dose cyclophosphamide in a mouse model of cutaneous breast cancer. Clin Cancer Res 18(24):6668–6678. https://doi.org/10.1158/1078-0432.ccr-12-0984
Adlard AL, Dovedi SJ, Telfer BA, Koga-Yamakawa E, Pollard C, Honeychurch J et al (2014) A novel systemically administered Toll-like receptor 7 agonist potentiates the effect of ionizing radiation in murine solid tumor models. Int J Cancer 135(4):820–829. https://doi.org/10.1002/ijc.28711
Nicolay NH, Schölch S, Rauber C, Lopez Perez R, Debus J, Huber PE (2016) The combination of ionizing radiation and toll-like receptor 7/8 agonists creates local and abscopal tumor immune responses in vivo. Int J Radiat Oncol Biol Phys 96(2):E561. https://doi.org/10.1016/j.ijrobp.2016.06.2033
Liu Z, Lei X, Li X, Cai J, Gao F, Yang Y (2018) Toll-like receptors and radiation protection. Eur Rev Med Pharmacol Sci 22(1):31–39
Saha S, Bhanja P, Liu L, Alfieri AA, Yu D, Kandimalla ER et al (2012) TLR9 agonist protects mice from radiation-induced gastrointestinal syndrome. PLoS ONE 7(1):e29357-e. https://doi.org/10.1371/journal.pone.0029357
Cargnello M, Roux PP (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 75(1):50–83. https://doi.org/10.1128/MMBR.00031-10
Shin YS, Hwang HS, Kang SU, Chang JW, Oh Y-T, Kim C-H (2014) Inhibition of p38 mitogen-activated protein kinase ameliorates radiation-induced ototoxicity in zebrafish and cochlea-derived cell lines. NeuroToxicology 40:111–122. https://doi.org/10.1016/j.neuro.2013.12.006
Sundaramoorthy P, Wang Q, Zheng Z, Jiao Y, Chen BJ, Doan PL et al (2017) Thioredoxin mitigates radiation-induced hematopoietic stem cell injury in mice. Stem Cell Res Ther 8(1):263. https://doi.org/10.1186/s13287-017-0711-2
Li D, Wang Y, Wu H, Lu L, Wang X, Zhang J et al (2013) The effects of p38 MAPK inhibition combined with G-CSF administration on the hematoimmune system in mice with irradiation injury. PLoS ONE 8(4):e62921-e. https://doi.org/10.1371/journal.pone.0062921
Wang Y, Liu L, Zhou D (2011) Inhibition of p38 MAPK attenuates ionizing radiation-induced hematopoietic cell senescence and residual bone marrow injury. Radiat Res 176(6):743–752. https://doi.org/10.1667/rr2727.1
Li D, Wang Y, Wu H, Lu L, Zhang H, Chang J et al (2011) Mitigation of ionizing radiation-induced bone marrow suppression by p38 inhibition and G-CSF administration. J Radiat Res 52(6):712–716. https://doi.org/10.1269/jrr.11007
Kang YH, Lee SJ (2008) Role of p38 MAPK and JNK in enhanced cervical cancer cell killing by the combination of arsenic trioxide and ionizing radiation. Oncol Rep 20(3):637–643
Munshi A, Ramesh R (2013) Mitogen-activated protein kinases and their role in radiation response. Genes Cancer 4(9–10):401–408. https://doi.org/10.1177/1947601913485414
Lasry A, Ben-Neriah Y (2015) Senescence-associated inflammatory responses: aging and cancer perspectives. Trends Immunol 36(4):217–228
He S, Sharpless NE (2017) Senescence in health and disease. Cell 169(6):1000–1011
Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ et al (2017) Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8:14532
Nguyen HQ, To NH, Zadigue P, Kerbrat S, De La Taille A, Le Gouvello S et al (2018) Ionizing radiation-induced cellular senescence promotes tissue fibrosis after radiotherapy. A review. Crit Rev Oncol Hematol 129:13–26. https://doi.org/10.1016/j.critrevonc.2018.06.012
Qian Y, Chen X (2013) Senescence regulation by the p53 protein family. Methods Mol Biol (Clifton, NJ) 965:37–61. https://doi.org/10.1007/978-1-62703-239-1_3
Poleszczuk J, Krzywon A, Forys U, Widel M (2015) Connecting radiation-induced bystander effects and senescence to improve radiation response prediction. Radiat Res 183(5):571–577
Vaughan DE, Rai R, Khan SS, Eren M, Ghosh AK (2017) Plasminogen activator inhibitor-1 is a marker and a mediator of senescence. Arterioscler Thromb Vasc Biol 37(8):1446–1452. https://doi.org/10.1161/ATVBAHA.117.309451
Kortlever RM, Nijwening JH, Bernards R (2008) Transforming growth factor-beta requires its target plasminogen activator inhibitor-1 for cytostatic activity. J Biol Chem 283(36):24308–24313. https://doi.org/10.1074/jbc.M803341200
Milliat F, Sabourin J-C, Tarlet G, Holler V, Deutsch E, Buard V et al (2008) Essential role of plasminogen activator inhibitor type-1 in radiation enteropathy. Am J Pathol 172(3):691–701. https://doi.org/10.2353/ajpath.2008.070930
Abderrahmani R, Francois A, Buard V, Tarlet G, Blirando K, Hneino M et al (2012) PAI-1-dependent endothelial cell death determines severity of radiation-induced intestinal injury. PLoS ONE 7(4):e35740. https://doi.org/10.1371/journal.pone.0035740
Abderrahmani R, Francois A, Buard V, Benderitter M, Sabourin JC, Crandall DL et al (2009) Effects of pharmacological inhibition and genetic deficiency of plasminogen activator inhibitor-1 in radiation-induced intestinal injury. Int J Radiat Oncol Biol Phys 74(3):942–948. https://doi.org/10.1016/j.ijrobp.2009.01.077
Chung EJ, McKay-Corkum G, Chung S, White A, Scroggins BT, Mitchell JB et al (2016) Truncated plasminogen activator inhibitor-1 protein protects from pulmonary fibrosis mediated by irradiation in a murine model. Int J Radiat Oncol Biol Phys 94(5):1163–1172. https://doi.org/10.1016/j.ijrobp.2015.11.044
Pan J, Li D, Xu Y, Zhang J, Wang Y, Chen M et al (2017) Inhibition of Bcl-2/xl with ABT-263 selectively kills senescent type II pneumocytes and reverses persistent pulmonary fibrosis induced by ionizing radiation in mice. Int J Radiat Oncol Biol Phys 99(2):353–361. https://doi.org/10.1016/j.ijrobp.2017.02.216
Wang Y, Boerma M, Zhou D (2016) Ionizing radiation-induced endothelial cell senescence and cardiovascular diseases. Radiat Res 186(2):153–161
Choi SH, Kim M, Lee HJ, Kim EH, Kim CH, Lee YJ (2016) Effects of NOX1 on fibroblastic changes of endothelial cells in radiationinduced pulmonary fibrosis. Mol Med Rep 13(5):4135–4142. https://doi.org/10.3892/mmr.2016.5090
Sivananthan A, Shields D, Fisher R, Hou W, Zhang X, Franicola D et al (2018) Continuous one year oral administration of the radiation mitigator, MMS350, after total-body irradiation, restores bone marrow stromal cell proliferative capacity and reduces senescence in fanconi anemia (Fanca−/−) mice. Radiat Res 191(2):139–153
Sabin RJ, Anderson RM (2011) Cellular senescence—its role in cancer and the response to ionizing radiation. Genome Integrity 2(1):7. https://doi.org/10.1186/2041-9414-2-7
Placencio VR, DeClerck YA (2015) Plasminogen activator inhibitor-1 in cancer: rationale and insight for future therapeutic testing. Can Res 75(15):2969–2974. https://doi.org/10.1158/0008-5472.CAN-15-0876
Fang H, Placencio VR, DeClerck YA (2012) Protumorigenic activity of plasminogen activator inhibitor-1 through an antiapoptotic function. J Natl Cancer Inst 104(19):1470–1484. https://doi.org/10.1093/jnci/djs377
Mashiko S, Kitatani K, Toyoshima M, Ichimura A, Dan T, Usui T et al (2015) Inhibition of plasminogen activator inhibitor-1 is a potential therapeutic strategy in ovarian cancer. Cancer Biol Ther 16(2):253–260. https://doi.org/10.1080/15384047.2014.1001271
Gomes-Giacoia E, Miyake M, Goodison S, Rosser CJ (2013) Targeting plasminogen activator inhibitor-1 inhibits angiogenesis and tumor growth in a human cancer xenograft model. Mol Cancer Ther 12(12):2697–2708. https://doi.org/10.1158/1535-7163.mct-13-0500
Isogai C, Laug WE, Shimada H, Declerck PJ, Stins MF, Durden DL et al (2001) Plasminogen activator inhibitor-1 promotes angiogenesis by stimulating endothelial cell migration toward fibronectin. Cancer Res 61(14):5587–5594
Bajou K, Peng H, Laug WE, Maillard C, Noel A, Foidart JM et al (2008) Plasminogen activator inhibitor-1 protects endothelial cells from FasL-mediated apoptosis. Cancer Cell 14(4):324–334. https://doi.org/10.1016/j.ccr.2008.08.012
Ashrafizadeh M, Ahmadi Z, Kotla N, Afshar E, Samarghandian S, Mandegary A et al (2019) Nanoparticles targeting STATs in cancer therapy. Cells 8:10
Galvano A, Novo G, Roselli M, Giordano A, Russo A (2019) Cardiovascular damage induced by radiotherapy. Cardiovascular complications in cancer therapy. Springer, Berlin, pp 21–31
Salem A, Mistry H, Backen A, Hodgson C, Koh P, Dean E et al (2018) Cell death, inflammation, tumor burden, and proliferation blood biomarkers predict lung cancer radiotherapy response and correlate with tumor volume and proliferation imaging. Clin Lung Cancer 19(3):239 e7–248 e7
Hager A, Meissner F, Riechardt AI, Bonaventura T, Löwen J, Heufelder J et al (2019) Breakdown of the blood-eye barrier in choroidal melanoma after proton beam radiotherapy. Graefe's Arch Clin Exp Ophthalmol 257(10):2323–2328
Schaue D, Micewicz ED, Ratikan JA, Xie MW, Cheng G, McBride WH (2015) Radiation and inflammation. Semin Radiat Oncol 20:20
Dörr H, Meineke V (2011) Acute radiation syndrome caused by accidental radiation exposure-therapeutic principles. BMC Med 9(1):126
Yarnold J, Brotons M-CV (2010) Pathogenetic mechanisms in radiation fibrosis. Radiother Oncol 97(1):149–161
Poulos MG, Ramalingam P, Gutkin MC, Kleppe M, Ginsberg M, Crowley MJP et al (2016) Endothelial-specific inhibition of NF-κB enhances functional haematopoiesis. Nat Commun 7(1):13829. https://doi.org/10.1038/ncomms13829
Alexeev V, Lash E, Aguillard A, Corsini L, Bitterman A, Ward K et al (2014) Radiation protection of the gastrointestinal tract and growth inhibition of prostate cancer xenografts by a single compound. Mol Cancer Ther 13(12):2968–2977. https://doi.org/10.1158/1535-7163.mct-14-0354
Daroczi B, Kari G, Ren Q, Dicker AP, Rodeck U (2009) Nuclear factor kappaB inhibitors alleviate and the proteasome inhibitor PS-341 exacerbates radiation toxicity in zebrafish embryos. Mol Cancer Ther 8(9):2625–2634. https://doi.org/10.1158/1535-7163.mct-09-0198
Farhood B, Aliasgharzadeh A, Amini P, Rezaeyan A, Tavassoli A, Motevaseli E et al (2019) Mitigation of radiation-induced lung pneumonitis and fibrosis using metformin and melatonin: a histopathological study. Medicina (Kaunas) 55:8. https://doi.org/10.3390/medicina55080417
Azmoonfar R, Amini P, Yahyapour R, Rezaeyan A, Tavassoli A, Motevaseli E et al (2019) Mitigation of radiation-induced pneumonitis and lung fibrosis using alpha-lipoic acid and resveratrol. Antiinflamm Antiallergy Agents Med Chem. https://doi.org/10.2174/1871523018666190319144020
Machado ND, Fernández MA, Díaz DD (2019) Recent strategies in resveratrol delivery systems. ChemPlusChem 84(7):951–973
Ahmadi Z, Mohammadinejad R, Ashrafizadeh M (2019) Drug delivery systems for resveratrol, a non-flavonoid polyphenol: emerging evidence in last decades. J Drug Deliv Sci Technol 20:20
Ahmadi Z, Ashrafizadeh M (2019) Melatonin as a potential modulator of Nrf2. Fundam Clin Pharmacol 20:20
Baud V, Karin M (2009) Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov 8(1):33–40. https://doi.org/10.1038/nrd2781
Hideshima T, Chauhan D, Schlossman R, Richardson P, Anderson KC (2001) The role of tumor necrosis factor alpha in the pathophysiology of human multiple myeloma: therapeutic applications. Oncogene 20(33):4519–4527. https://doi.org/10.1038/sj.onc.1204623
Mitsiades N, Mitsiades CS, Richardson PG, Poulaki V, Tai YT, Chauhan D et al (2003) The proteasome inhibitor PS-341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: therapeutic applications. Blood 101(6):2377–2380. https://doi.org/10.1182/blood-2002-06-1768
Fernandes JV, Cobucci RNO, Jatobá CAN, de Medeiros Fernandes TAA, de Azevedo JWV, de Araújo JMG (2015) The role of the mediators of inflammation in cancer development. Pathol Oncol Res 21(3):527–534
Zhao Y, de Toledo SM, Hu G, Hei TK, Azzam EI (2014) Connexins and cyclooxygenase-2 crosstalk in the expression of radiation-induced bystander effects. Br J Cancer 111(1):125–131. https://doi.org/10.1038/bjc.2014.276
Chai Y, Calaf GM, Zhou H, Ghandhi SA, Elliston CD, Wen G et al (2013) Radiation induced COX-2 expression and mutagenesis at non-targeted lung tissues of gpt delta transgenic mice. Br J Cancer 108(1):91–98. https://doi.org/10.1038/bjc.2012.498
Kobayashi A, Konishi T (2018) Radiation quality effects alteration in COX-2 pathway to trigger radiation-induced bystander response in A549 lung carcinoma cells. J Radiat Res 59(6):754–759. https://doi.org/10.1093/jrr/rry065
Chai Y, Lam RK, Calaf GM, Zhou H, Amundson S, Hei TK (2013) Radiation-induced non-targeted response in vivo: role of the TGFbeta-TGFBR1-COX-2 signalling pathway. Br J Cancer 108(5):1106–1112. https://doi.org/10.1038/bjc.2013.53
Sonis ST, O'Donnell KE, Popat R, Bragdon C, Phelan S, Cocks D et al (2004) The relationship between mucosal cyclooxygenase-2 (COX-2) expression and experimental radiation-induced mucositis. Oral Oncol 40(2):170–176
Yeoh AS, Gibson RJ, Yeoh EE, Bowen JM, Stringer AM, Giam KA et al (2007) A novel animal model to investigate fractionated radiotherapy-induced alimentary mucositis: the role of apoptosis, p53, nuclear factor-kappaB, COX-1, and COX-2. Mol Cancer Ther 6(8):2319–2327. https://doi.org/10.1158/1535-7163.mct-07-0113
Khayyal MT, El-Ghazaly MA, El-Hazek RM, Nada AS (2009) The effects of celecoxib, a COX-2 selective inhibitor, on acute inflammation induced in irradiated rats. Inflammopharmacology 17(5):255–266. https://doi.org/10.1007/s10787-009-0014-z
Pinheiro RM, Calixto JB (2002) Effect of the selective COX-2 inhibitors, celecoxib and rofecoxib in rat acute models of inflammation. Inflamm Res 51(12):603–610
Liang L, Hu D, Liu W, Williams JP, Okunieff P, Ding I (2003) Celecoxib reduces skin damage after radiation: selective reduction of chemokine and receptor mRNA expression in irradiated skin but not in irradiated mammary tumor. Am J Clin Oncol 26(4):S114–S121. https://doi.org/10.1097/01.coc.0000074149.95710.40
Hoggatt J, Singh P, Stilger KN, Plett PA, Sampson CH, Chua HL et al (2013) Recovery from hematopoietic injury by modulating prostaglandin E(2) signaling post-irradiation. Blood Cells Mol Dis 50(3):147–153. https://doi.org/10.1016/j.bcmd.2012.11.006
Hunter NR, Valdecanas D, Liao Z, Milas L, Thames HD, Mason KA (2013) Mitigation and treatment of radiation-induced thoracic injury with a cyclooxygenase-2 inhibitor, celecoxib. Int J Radiat Oncol Biol Phys 85(2):472–476. https://doi.org/10.1016/j.ijrobp.2012.04.025
Cho YJ, Yi CO, Jeon BT, Jeong YY, Kang GM, Lee JE et al (2013) Curcumin attenuates radiation-induced inflammation and fibrosis in rat lungs. Korean J Physiol Pharmacol 17(4):267–274. https://doi.org/10.4196/kjpp.2013.17.4.267
Hanson WR, Thomas C (1983) 16, 16-dimethyl prostaglandin E2 increases survival of murine intestinal stem cells when given before photon radiation. Radiat Res 96(2):393–398
Hanson WR, Ainsworth EJ (1985) 16,16-Dimethyl prostaglandin E2 induces radioprotection in murine intestinal and hematopoietic stem cells. Radiat Res 103(2):196–203
Toomey DP, Murphy JF, Conlon KC (2009) COX-2, VEGF and tumour angiogenesis. Surgeon 7(3):174–180. https://doi.org/10.1016/s1479-666x(09)80042-5
Singh B, Cook KR, Vincent L, Hall CS, Berry JA, Multani AS et al (2008) Cyclooxygenase-2 induces genomic instability, BCL2 expression, doxorubicin resistance, and altered cancer-initiating cell phenotype in MCF7 breast cancer cells. J Surg Res 147(2):240–246. https://doi.org/10.1016/j.jss.2008.02.026
Sminia P, Kuipers G, Geldof A, Lafleur V, Slotman B (2005) COX-2 inhibitors act as radiosensitizer in tumor treatment. Biomed Pharmacother 59:S272–S275. https://doi.org/10.1016/S0753-3322(05)80044-7
Choy H, Milas L (2003) Enhancing radiotherapy with cyclooxygenase-2 enzyme inhibitors: a rational advance? J Natl Cancer Inst 95(19):1440–1452
West AP, Shadel GS (2017) Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol 17(6):363
Perrone MS, Missiroli S, Previati M, Fiorica F, Pinton P, Carlotta G (2017) Radiation induces IL-1b production and promotes activation of NLRP3 inflammasome. Int J Radiat Oncol Biol Phys 99(2):E613–E614. https://doi.org/10.1016/j.ijrobp.2017.06.2078
Liu Y-G, Chen J-K, Zhang Z-T, Ma X-J, Chen Y-C, Du X-M et al (2017) NLRP3 inflammasome activation mediates radiation-induced pyroptosis in bone marrow-derived macrophages. Cell Death Dis 8(2):e2579-e. https://doi.org/10.1038/cddis.2016.460
Wei J, Wang H, Wang H, Wang B, Meng L, Xin Y et al (2019) The role of NLRP3 inflammasome activation in radiation damage. Biomed Pharmacother 118:109217. https://doi.org/10.1016/j.biopha.2019.109217
Sohn S-H, Lee JM, Park S, Yoo H, Kang JW, Shin D et al (2015) The inflammasome accelerates radiation-induced lung inflammation and fibrosis in mice. Environ Toxicol Pharmacol 39(2):917–926. https://doi.org/10.1016/j.etap.2015.02.019
Chatterjee S, Pietrofesa RA, Park K, Tao J-Q, Carabe-Fernandez A, Berman AT et al (2019) LGM2605 reduces space radiation-induced NLRP3 inflammasome activation and damage in in vitro lung vascular networks. Int J Mol Sci 20(1):176. https://doi.org/10.3390/ijms20010176
Ortiz F, Acuña-Castroviejo D, Doerrier C, Dayoub JC, López LC, Venegas C et al (2015) Melatonin blunts the mitochondrial/NLRP 3 connection and protects against radiation-induced oral mucositis. J Pineal Res 58(1):34–49
Fernández-Gil B, Moneim AEA, Ortiz F, Shen Y-Q, Soto-Mercado V, Mendivil-Perez M et al (2017) Melatonin protects rats from radiotherapy-induced small intestine toxicity. PLoS ONE 12(4):e0174474
Moossavi M, Parsamanesh N, Bahrami A, Atkin SL, Sahebkar A (2018) Role of the NLRP3 inflammasome in cancer. Mol Cancer 17(1):158. https://doi.org/10.1186/s12943-018-0900-3
Guo B, Fu S, Zhang J, Liu B, Li Z (2016) Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci Rep 6(1):36107. https://doi.org/10.1038/srep36107
Xu S, Li X, Liu Y, Xia Y, Chang R, Zhang C (2019) Inflammasome inhibitors: promising therapeutic approaches against cancer. J Hematol Oncol 12(1):64. https://doi.org/10.1186/s13045-019-0755-0
Shi J, Gao W, Shao F (2017) Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci 42(4):245–254
Man SM, Kanneganti TD (2015) Regulation of inflammasome activation. Immunol Rev 265(1):6–21
Rathinam VA, Vanaja SK, Fitzgerald KA (2012) Regulation of inflammasome signaling. Nat Immunol 13(4):333
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H et al (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526(7575):660–665
Hu B, Jin C, Li H-B, Tong J, Ouyang X, Cetinbas NM et al (2016) The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 354(6313):765–768. https://doi.org/10.1126/science.aaf7532
Wu T, Liu W, Fan T, Zhong H, Zhou H, Guo W et al (2020) 5-Androstenediol prevents radiation injury in mice by promoting NF-κB signaling and inhibiting AIM2 inflammasome activation. Biomed Pharmacother 121:109597. https://doi.org/10.1016/j.biopha.2019.109597
Gao J, Peng S, Shan X, Deng G, Shen L, Sun J et al (2019) Inhibition of AIM2 inflammasome-mediated pyroptosis by Andrographolide contributes to amelioration of radiation-induced lung inflammation and fibrosis. Cell Death Dis 10(12):957. https://doi.org/10.1038/s41419-019-2195-8
Kalmar B, Greensmith L (2009) Induction of heat shock proteins for protection against oxidative stress. Adv Drug Deliv Rev 61(4):310–318
Yenari MA, Liu J, Zheng Z, Vexler ZS, Lee JE, Giffard RG (2005) Antiapoptotic and anti-inflammatory mechanisms of heat-shock protein protection. Ann N Y Acad Sci 1053(1):74–83
Tsan M-F, Gao B (2004) Heat shock protein and innate immunity. Cell Mol Immunol 1(4):274–279
Lee SJ, Choi SA, Lee KH, Chung HY, Kim TH, Cho CK et al (2001) Role of inducible heat shock protein 70 in radiation-induced cell death. Cell Stress Chaperones 6(3):273–281. https://doi.org/10.1379/1466-1268(2001)006%3c0273:roihsp%3e2.0.co;2
Salari S, Seibert T, Chen Y-X, Hu T, Shi C, Zhao X et al (2013) Extracellular HSP27 acts as a signaling molecule to activate NF-κB in macrophages. Cell Stress Chaperones 18(1):53–63
Kim J-Y, An Y-M, Yoo BR, Kim J-M, Han SY, Na Y et al (2018) HSP27 inhibitor attenuates radiation-induced pulmonary inflammation. Sci Rep 8(1):4189. https://doi.org/10.1038/s41598-018-22635-9
Multhoff G, Pockley AG, Schmid TE, Schilling D (2015) The role of heat shock protein 70 (Hsp70) in radiation-induced immunomodulation. Cancer Lett 368(2):179–184. https://doi.org/10.1016/j.canlet.2015.02.013
Garrido C, Brunet M, Didelot C, Zermati Y, Schmitt E, Kroemer G (2006) Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell Cycle 5(22):2592–2601. https://doi.org/10.4161/cc.5.22.3448
Wang X, Chen M, Zhou J, Zhang X (2014) HSP27, 70 and 90, anti-apoptotic proteins, in clinical cancer therapy (Review). Int J Oncol 45(1):18–30. https://doi.org/10.3892/ijo.2014.2399
Elmallah MIY, Cordonnier M, Vautrot V, Chanteloup G, Garrido C, Gobbo J (2020) Membrane-anchored heat-shock protein 70 (Hsp70) in cancer. Cancer Lett 469:134–141. https://doi.org/10.1016/j.canlet.2019.10.037
Choi S-K, Kam H, Kim K-Y, Park SI, Lee Y-S (2019) Targeting heat shock protein 27 in cancer: a druggable target for cancer treatment? Cancers 11(8):1195. https://doi.org/10.3390/cancers11081195
Sheng B, Qi C, Liu B, Lin Y, Fu T, Zeng Q (2017) Increased HSP27 correlates with malignant biological behavior of non-small cell lung cancer and predicts patient’s survival. Sci Rep 7(1):13807. https://doi.org/10.1038/s41598-017-13956-2
Jin HO, Hong SE, Kim JY, Kim MR, Chang YH, Hong YJ et al (2019) Induction of HSP27 and HSP70 by constitutive overexpression of Redd1 confers resistance of lung cancer cells to ionizing radiation. Oncol Rep 41(5):3119–3126. https://doi.org/10.3892/or.2019.7036
Tsao MN, Li YQ, Lu G, Xu Y, Wong CS (1999) Upregulation of vascular endothelial growth factor is associated with radiation-induced blood-spinal cord barrier breakdown. J Neuropathol Exp Neurol 58(10):1051–1060. https://doi.org/10.1097/00005072-199910000-00003
Langley RE, Bump EA, Quartuccio SG, Medeiros D, Braunhut SJ (1997) Radiation-induced apoptosis in microvascular endothelial cells. Br J Cancer 75(5):666–672. https://doi.org/10.1038/bjc.1997.119
Lumniczky K, Szatmári T, Sáfrány G (2017) Ionizing radiation-induced immune and inflammatory reactions in the brain. Front Immunol 8:517. https://doi.org/10.3389/fimmu.2017.00517
Kim JM, Miller JA, Kotecha R, Xiao R, Juloori A, Ward MC et al (2017) The risk of radiation necrosis following stereotactic radiosurgery with concurrent systemic therapies. J Neurooncol 133(2):357–368. https://doi.org/10.1007/s11060-017-2442-8
Klos J, van Laar PJ, Sinnige PF, Enting RH, Kramer MCA, van der Weide HL et al (2019) Quantifying effects of radiotherapy-induced microvascular injury; review of established and emerging brain MRI techniques. Radiother Oncol 140:41–53. https://doi.org/10.1016/j.radonc.2019.05.020
Vujaskovic Z, Anscher MS, Feng QF, Rabbani ZN, Amin K, Samulski TS et al (2001) Radiation-induced hypoxia may perpetuate late normal tissue injury. Int J Radiat Oncol Biol Phys 50(4):851–855. https://doi.org/10.1016/s0360-3016(01)01593-0
Li YQ, Ballinger JR, Nordal RA, Su ZF, Wong CS (2001) Hypoxia in radiation-induced blood-spinal cord barrier breakdown. Cancer Res 61(8):3348–3354
Fleckenstein K, Zgonjanin L, Chen L, Rabbani Z, Jackson IL, Thrasher B et al (2007) Temporal onset of hypoxia and oxidative stress after pulmonary irradiation. Int J Radiat Oncol Biol Phys 68(1):196–204. https://doi.org/10.1016/j.ijrobp.2006.12.056
Rabbani ZN, Mi J, Zhang Y, Delong M, Jackson IL, Fleckenstein K et al (2010) Hypoxia inducible factor 1alpha signaling in fractionated radiation-induced lung injury: role of oxidative stress and tissue hypoxia. Radiat Res 173(2):165–174. https://doi.org/10.1667/RR1816.1
Toullec A, Buard V, Rannou E, Tarlet G, Guipaud O, Robine S et al (2018) HIF-1α deletion in the endothelium, but not in the epithelium, protects from radiation-induced enteritis. Cell Mol Gastroenterol Hepatol 5(1):15–30. https://doi.org/10.1016/j.jcmgh.2017.08.001
Taniguchi CM, Wu C, Atwood T, Maxim P, Giaccia A (2012) Intestinal HIF-2 to protect against radiation-induced gastrointestinal syndrome. J Clin Oncol 30(1(15_suppl)):10629. https://doi.org/10.1200/jco.2012.30.15_suppl.10629
Graham K, Unger E (2018) Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int J Nanomed 13:6049–6058. https://doi.org/10.2147/IJN.S140462
Kaidi A, Qualtrough D, Williams AC, Paraskeva C (2006) Direct transcriptional up-regulation of cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes colorectal tumor cell survival and enhances HIF-1 transcriptional activity during hypoxia. Cancer Res 66(13):6683–6691. https://doi.org/10.1158/0008-5472.can-06-0425
Dorr W, Hendry JH (2001) Consequential late effects in normal tissues. Radiother Oncol 61(3):223–231. https://doi.org/10.1016/s0167-8140(01)00429-7
Bourgeois JF, Gourgou S, Kramar A, Lagarde JM, Gall Y, Guillot B (2003) Radiation-induced skin fibrosis after treatment of breast cancer: profilometric analysis. Skin Res Technol 9(1):39–42
Lombardo S, Spagnolo F, Calderazzo M, Fronda L, Gambardella P, Musolino T et al (2019) Fatal idiopathic pulmonary fibrosis exacerbation after radiotherapy. D40 non-inflammatory dplds case reports. American Thoracic Society, New York, p A6340A
Musa AE, Shabeeb D (2019) Radiation-induced heart diseases: protective effects of natural products. Medicina 55(5):126
Najafi M, Farhood B, Mortezaee K (2019) Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J Cell Biochem 120(3):2782–2790. https://doi.org/10.1002/jcb.27681
Hallett MA, Venmar KT, Fingleton B (2012) Cytokine stimulation of epithelial cancer cells: the similar and divergent functions of IL-4 and IL-13. Can Res 72(24):6338–6343
Straub JM, New J, Hamilton CD, Lominska C, Shnayder Y, Thomas SM (2015) Radiation-induced fibrosis: mechanisms and implications for therapy. J Cancer Res Clin Oncol 141(11):1985–1994
Pohlers D, Brenmoehl J, Löffler I, Müller CK, Leipner C, Schultze-Mosgau S et al (2009) TGF-β and fibrosis in different organs—molecular pathway imprints. Biochim Biophys Acta Mol Basis Dis 1792(8):746–756. https://doi.org/10.1016/j.bbadis.2009.06.004
Nevens D, Duprez F, Daisne JF, Laenen A, De Neve W, Nuyts S (2017) Radiotherapy induced dermatitis is a strong predictor for late fibrosis in head and neck cancer. The development of a predictive model for late fibrosis. Radiother Oncol 122(2):212–216
Rigo F (2017) Effects of radiotherapy on vessels and coronary arteries. Anti-cancer treatments and cardiotoxicity. Elsevier, New York, pp 87–89
Stansborough RL, Al-Dasooqi N, Bateman EH, Keefe DM, Gibson RJ (2016) Radiotherapy-induced gut toxicity: involvement of matrix metalloproteinases and the intestinal microvasculature. Int J Radiat Biol 92(5):241–248
Vallee A, Lecarpentier Y, Guillevin R, Vallee JN (2017) Interactions between TGF-beta1, canonical WNT/beta-catenin pathway and PPAR gamma in radiation-induced fibrosis. Oncotarget 8(52):90579–90604. https://doi.org/10.18632/oncotarget.21234
Martin M, Lefaix J, Delanian S (2000) TGF-beta1 and radiation fibrosis: a master switch and a specific therapeutic target? Int J Radiat Oncol Biol Phys 47(2):277–290. https://doi.org/10.1016/s0360-3016(00)00435-1
Eldabaje R, Le DL, Huang W, Yang LX (2015) Radiation-associated cardiac injury. Anticancer Res 35(5):2487–2492
Vozenin-Brotons M-C, Sivan V, Gault N, Renard C, Geffrotin C, Delanian S et al (2001) Antifibrotic action of Cu/Zn SOD is mediated by TGF-β1 repression and phenotypic reversion of myofibroblasts. Free Radical Biol Med 30(1):30–42
Kang SK, Rabbani ZN, Folz RJ, Golson ML, Huang H, Yu D et al (2003) Overexpression of extracellular superoxide dismutase protects mice from radiation-induced lung injury. Int J Radiat Oncol Biol Phys 57(4):1056–1066
Amini P, Kolivand S, Saffar H, Rezapoor S, Motevaseli E, Najafi M et al (2018) Protective effect of Selenium-l-methionine on radiation-induced acute pneumonitis and lung fibrosis in rat. Curr Clin Pharmacol. https://doi.org/10.2174/1574884714666181214101917
Aliasgharzadeh A, Farhood B, Amini P, Saffar H, Motevaseli E, Rezapoor S et al (2019) Melatonin attenuates upregulation of Duox1 and Duox2 and protects against lung injury following chest irradiation in rats. Cell J 21(3):236–242. https://doi.org/10.22074/cellj.2019.6207
Jang SS, Kim HG, Lee JS, Han JM, Park HJ, Huh GJ et al (2013) Melatonin reduces X-ray radiation-induced lung injury in mice by modulating oxidative stress and cytokine expression. Int J Radiat Biol 89(2):97–105
Lee JC, Kinniry PA, Arguiri E, Serota M, Kanterakis S, Chatterjee S et al (2010) Dietary curcumin increases antioxidant defenses in lung, ameliorates radiation-induced pulmonary fibrosis, and improves survival in mice. Radiat Res 173(5):590–601
Gauter-Fleckenstein B, Fleckenstein K, Owzar K, Jiang C, Rebouças JS, Batinic-Haberle I et al (2010) Early and late administration of MnTE-2-PyP5+ in mitigation and treatment of radiation-induced lung damage. Free Radical Biol Med 48(8):1034–1043
Calveley VL, Jelveh S, Langan A, Mahmood J, Yeung IW, Van Dyk J et al (2010) Genistein can mitigate the effect of radiation on rat lung tissue. Radiat Res 173(5):602–611
Mahmood J, Jelveh S, Zaidi A, Doctrow S, Hill R (2012) Mitigation of radiation-induced lung injury with EUK-207 and genistein: effects in adolescent rats. Radiat Res 179(2):125–134
Rabender C, Mezzaroma E, Mauro AG, Mullangi R, Abbate A, Anscher M et al (2016) IPW-5371 proves effective as a radiation countermeasure by mitigating radiation-induced late effects. Radiat Res 186(5):478–488
Ahmadi A, Najafi M, Farhood B, Mortezaee K (2019) Transforming growth factor-β signaling: tumorigenesis and targeting for cancer therapy. J Cell Physiol 234(8):12173–12187. https://doi.org/10.1002/jcp.27955
Young KH, Gough MJ, Crittenden M (2015) Tumor immune remodeling by TGFβ inhibition improves the efficacy of radiation therapy. Oncoimmunology 4(3):e955696
Andarawewa KL, Paupert J, Pal A, Barcellos-Hoff MH (2007) New rationales for using TGF beta inhibitors in radiotherapy. Int J Radiat Biol 83(11–12):803–811
Groves AM, Johnston CJ, Misra RS, Williams JP, Finkelstein JN (2016) Effects of IL-4 on pulmonary fibrosis and the accumulation and phenotype of macrophage subpopulations following thoracic irradiation. Int J Radiat Biol 92(12):754–765. https://doi.org/10.1080/09553002.2016.1222094
Suzuki A, Leland P, Joshi BH, Puri RK (2015) Targeting of IL-4 and IL-13 receptors for cancer therapy. Cytokine 75(1):79–88. https://doi.org/10.1016/j.cyto.2015.05.026
Li Z, Chen L, Qin Z (2009) Paradoxical roles of IL-4 in tumor immunity. Cell Mol Immunol 6(6):415–422
Nappo G, Handle F, Santer FR, McNeill RV, Seed RI, Collins AT et al (2017) The immunosuppressive cytokine interleukin-4 increases the clonogenic potential of prostate stem-like cells by activation of STAT6 signalling. Oncogenesis 6(5):e342-e. https://doi.org/10.1038/oncsis.2017.23
Kim ES, Choi YE, Hwang SJ, Han Y-H, Park M-J, Bae IH (2016) IL-4, a direct target of miR-340/429, is involved in radiation-induced aggressive tumor behavior in human carcinoma cells. Oncotarget 7(52):86836–86856. https://doi.org/10.18632/oncotarget.13561
Chung SI, Horton JA, Ramalingam TR, White AO, Chung EJ, Hudak KE et al (2016) IL-13 is a therapeutic target in radiation lung injury. Sci Rep 6:39714. https://doi.org/10.1038/srep39714
Horton J, Hudak K, Scroggins B, Chung E, White A, Citrin D (2014) Il-13 is a critical mediator of radiation-induced pulmonary fibrosis. Int J Radiat Oncol Biol Phys 90(1):S150. https://doi.org/10.1016/j.ijrobp.2014.05.626
Nakashima H, Fujisawa T, Husain SR, Puri RK (2010) Interleukin-13 receptor α2 DNA prime boost vaccine induces tumor immunity in murine tumor models. J Transl Med 8(1):116. https://doi.org/10.1186/1479-5876-8-116
Shimato S, Natsume A, Wakabayashi T, Tsujimura K, Nakahara N, Ishii J et al (2008) Identification of a human leukocyte antigen-A24–restricted T-cell epitope derived from interleukin-13 receptor α2 chain, a glioma-associated antigen. J Neurosurg 109(1):117–122
Iwami K, Shimato S, Ohno M, Okada H, Nakahara N, Sato Y et al (2012) Peptide-pulsed dendritic cell vaccination targeting interleukin-13 receptor α2 chain in recurrent malignant glioma patients with HLA-A* 24/A* 02 allele. Cytotherapy 14(6):733–742
Terabe M, Park JM, Berzofsky JA (2004) Role of IL-13 in regulation of anti-tumor immunity and tumor growth. Cancer Immunol Immunother 53(2):79–85. https://doi.org/10.1007/s00262-003-0445-0
Kawakami K, Kawakami M, Liu Q, Puri RK (2005) Combined effects of radiation and interleukin-13 receptor-targeted cytotoxin on glioblastoma cell lines. Int J Radiat Oncol Biol Phys 63(1):230–237. https://doi.org/10.1016/j.ijrobp.2005.05.017
Vogelbaum MA, Sampson JH, Kunwar S, Chang SM, Shaffrey M, Asher AL et al (2007) Convection-enhanced delivery of cintredekin besudotox (interleukin-13-PE38QQR) followed by radiation therapy with and without temozolomide in newly diagnosed malignant gliomas: phase 1 study of final safety results. Neurosurgery 61(5):1031–1038
Robbins ME, Diz DI (2006) Pathogenic role of the renin–angiotensin system in modulating radiation-induced late effects. Int J Radiat Oncol Biol Phys 64(1):6–12. https://doi.org/10.1016/j.ijrobp.2005.08.033
Cohen EP, Fish BL, Moulder JE (1999) Angiotensin II infusion exacerbates radiation nephropathy. J Lab Clin Med 134(3):283–291. https://doi.org/10.1016/S0022-2143(99)90209-3
Moulder JE, Fish BL, Cohen EP, Bonsib SM (1996) Angiotensin II receptor antagonists in the prevention of radiation nephropathy. Radiat Res 146(1):106–110. https://doi.org/10.2307/3579403
Moulder JE, Fish BL, Regner KR, Cohen EP (2002) Angiotensin II blockade reduces radiation-induced proliferation in experimental radiation nephropathy. Radiat Res 157(4):393–401. https://doi.org/10.1667/0033-7587(2002)157[0393:AIBRRI]2.0.CO;2
Cohen EP, Fish BL, Moulder JE (2002) The renin–angiotensin system in experimental radiation nephropathy. J Lab Clin Med 139(4):251–257. https://doi.org/10.1067/mlc.2002.122279
Robbins ME, Zhao W, Garcia-Espinosa MA, Diz DI (2010) Renin–angiotensin system blockers and modulation of radiation-induced brain injury. Curr Drug Targets 11(11):1413–1422. https://doi.org/10.2174/1389450111009011413
Medhora M, Gao F, Jacobs ER, Moulder JE (2012) Radiation damage to the lung: mitigation by angiotensin-converting enzyme (ACE) inhibitors. Respirology (Carlton, Vic) 17(1):66–71. https://doi.org/10.1111/j.1440-1843.2011.02092.x
Danilczyk U, Penninger JM (2006) Angiotensin-converting enzyme II in the heart and the kidney. Circ Res 98(4):463–471
Moulder JE, Fish BL, Cohen EP (2004) Impact of angiotensin II type 2 receptor blockade on experimental radiation nephropathy. Radiat Res 161(3):312–317. https://doi.org/10.1667/rr3129
Moulder JE, Fish BL, Cohen EP (1998) Radiation nephropathy is treatable with an angiotensin converting enzyme inhibitor or an angiotensin II type-1 (AT1) receptor antagonist. Radiother Oncol 46(3):307–315. https://doi.org/10.1016/S0167-8140(97)00175-8
Cohen EP, Hussain S, Moulder JE (2003) Successful treatment of radiation nephropathy with angiotensin II blockade. Int J Radiat Oncol Biol Phys 55(1):190–193. https://doi.org/10.1016/s0360-3016(02)03793-8
Moulder JE, Cohen EP, Fish BL (2014) Mitigation of experimental radiation nephropathy by renin-equivalent doses of angiotensin converting enzyme inhibitors. Int J Radiat Biol 90(9):762–768. https://doi.org/10.3109/09553002.2014.938375
Moulder JE, Cohen EP, Fish BL (2010) Captopril and losartan for mitigation of renal injury caused by single-dose total-body irradiation. Radiat Res 175(1):29–36. https://doi.org/10.1667/RR2400.1
Cohen EP, Bedi M, Irving AA, Jacobs E, Tomic R, Klein J et al (2012) Mitigation of late renal and pulmonary injury after hematopoietic stem cell transplantation. Int J Radiat Oncol Biol Phys 83(1):292–296. https://doi.org/10.1016/j.ijrobp.2011.05.081
Cohen EP, Irving AA, Drobyski WR, Klein JP, Passweg J, Talano JA et al (2008) Captopril to mitigate chronic renal failure after hematopoietic stem cell transplantation: a randomized controlled trial. Int J Radiat Oncol Biol Phys 70(5):1546–1551. https://doi.org/10.1016/j.ijrobp.2007.08.041
Datta PK, Moulder JE, Fish BL, Cohen EP, Lianos EA (2001) Induction of heme oxygenase 1 in radiation nephropathy: role of angiotensin II. Radiat Res 155(5):734–739
Ghosh SN, Zhang R, Fish BL, Semenenko VA, Li XA, Moulder JE et al (2009) Renin–angiotensin system suppression mitigates experimental radiation pneumonitis. Int J Radiat Oncol Biol Phys 75(5):1528–1536. https://doi.org/10.1016/j.ijrobp.2009.07.1743
Kma L, Gao F, Fish BL, Moulder JE, Jacobs ER, Medhora M (2012) Angiotensin converting enzyme inhibitors mitigate collagen synthesis induced by a single dose of radiation to the whole thorax. J Radiat Res 53(1):10–17
Mahmood J, Jelveh S, Zaidi A, Doctrow SR, Medhora M, Hill RP (2014) Targeting the renin–angiotensin system combined with an antioxidant is highly effective in mitigating radiation-induced lung damage. Int J Radiat Oncol Biol Phys 89(4):722–728. https://doi.org/10.1016/j.ijrobp.2014.03.048
Molthen RC, Wu Q, Fish BL, Moulder JE, Jacobs ER, Medhora MM (2012) Mitigation of radiation induced pulmonary vascular injury by delayed treatment with captopril. Respirology 17(8):1261–1268. https://doi.org/10.1111/j.1440-1843.2012.02247.x
Sun F, Sun H, Zheng X, Yang G, Gong N, Zhou H et al (2018) Angiotensin-converting enzyme inhibitors decrease the incidence of radiation-induced pneumonitis among lung cancer patients: a systematic review and meta-analysis. J Cancer 9(12):2123–2131. https://doi.org/10.7150/jca.24665
Kharofa J, Cohen EP, Tomic R, Xiang Q, Gore E (2012) Decreased risk of radiation pneumonitis with incidental concurrent use of angiotensin-converting enzyme inhibitors and thoracic radiation therapy. Int J Radiat Oncol Biol Phys 84(1):238–243. https://doi.org/10.1016/j.ijrobp.2011.11.013
Wang H, Liao Z, Zhuang Y, Xu T, Nguyen QN, Levy LB et al (2013) Do angiotensin-converting enzyme inhibitors reduce the risk of symptomatic radiation pneumonitis in patients with non-small cell lung cancer after definitive radiation therapy? Analysis of a single-institution database. Int J Radiat Oncol Biol Phys 87(5):1071–1077. https://doi.org/10.1016/j.ijrobp.2013.08.033
Bracci S, Valeriani M, Agolli L, De Sanctis V, Maurizi Enrici R, Osti MF (2016) Renin–angiotensin system inhibitors might help to reduce the development of symptomatic radiation pneumonitis after stereotactic body radiotherapy for lung cancer. Clin Lung Cancer 17(3):189–197. https://doi.org/10.1016/j.cllc.2015.08.007
Molteni A, Wolfe LF, Ward WF, Ts'ao CH, Molteni LB, Veno P et al (2007) Effect of an angiotensin II receptor blocker and two angiotensin converting enzyme inhibitors on transforming growth factor-beta (TGF-beta) and alpha-actomyosin (alpha SMA), important mediators of radiation-induced pneumopathy and lung fibrosis. Curr Pharm Des 13(13):1307–1316. https://doi.org/10.2174/138161207780618777
Moore ED, Kooshki M, Metheny-Barlow LJ, Gallagher PE, Robbins ME (2013) Angiotensin-(1–7) prevents radiation-induced inflammation in rat primary astrocytes through regulation of MAP kinase signaling. Free Radical Biol Med 65:1060–1068. https://doi.org/10.1016/j.freeradbiomed.2013.08.183
Oikawa T, Freeman M, Lo W, Vaughan DE, Fogo A (1997) Modulation of plasminogen activator inhibitor-1 in vivo: a new mechanism for the anti-fibrotic effect of renin–angiotensin inhibition. Kidney Int 51(1):164–172. https://doi.org/10.1038/ki.1997.20
Davis TA, Landauer MR, Mog SR, Barshishat-Kupper M, Zins SR, Amare MF et al (2010) Timing of captopril administration determines radiation protection or radiation sensitization in a murine model of total body irradiation. Exp Hematol 38(4):270–281. https://doi.org/10.1016/j.exphem.2010.01.004
McCart EA, Lee YH, Jha J, Mungunsukh O, Rittase WB, Summers TA et al (2019) Delayed captopril administration mitigates hematopoietic injury in a murine model of total body irradiation. Sci Rep 9(1):2198. https://doi.org/10.1038/s41598-019-38651-2
Rodgers KE, Espinoza T, Roda N, Meeks CJ, Hill C, Louie SG et al (2012) Accelerated hematopoietic recovery with angiotensin-(1–7) after total body radiation. Int J Radiat Biol 88(6):466–476. https://doi.org/10.3109/09553002.2012.676228
Pinter M, Jain RK (2017) Targeting the renin–angiotensin system to improve cancer treatment: implications for immunotherapy. Sci Transl Med 9(410):eaan5616. https://doi.org/10.1126/scitranslmed.aan5616
Gallagher PE, Cook K, Soto-Pantoja D, Menon J, Tallant EA (2011) Angiotensin peptides and lung cancer. Curr Cancer Drug Targets 11(4):394–404. https://doi.org/10.2174/156800911795538048
Chundury A, Rehman S, Roach M, Mullen D, DeWeese T, Bradley J et al (2015) PD-0428: radiation pneumonitis with stereotactic body radiotherapy: effects of angiotensin converting enzyme inhibitors. Radiother Oncol 115:S208–S209. https://doi.org/10.1016/S0167-8140(15)40424-4
Wei J, Wang B, Wang H, Meng L, Zhao Q, Li X et al (2019) Radiation-induced normal tissue damage: oxidative stress and epigenetic mechanisms. Oxid Med Cell Longev 2019:11. https://doi.org/10.1155/2019/3010342
Shrishrimal S, Kosmacek EA, Oberley-Deegan RE (2019) Reactive oxygen species drive epigenetic changes in radiation-induced fibrosis. Oxid Med Cell Longev 2019:27. https://doi.org/10.1155/2019/4278658
Weigel C, Schmezer P, Plass C, Popanda O (2015) Epigenetics in radiation-induced fibrosis. Oncogene 34(17):2145–2155. https://doi.org/10.1038/onc.2014.145
Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR et al (2013) Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153(2):320–334
Stock CJW, Michaeloudes C, Leoni P, Durham AL, Mumby S, Wells AU et al (2019) Bromodomain and extraterminal (BET) protein inhibition restores redox balance and inhibits myofibroblast activation. Biomed Res Int 2019:11. https://doi.org/10.1155/2019/1484736
Ding N, Hah N, Yu RT, Sherman MH, Benner C, Leblanc M et al (2015) BRD4 is a novel therapeutic target for liver fibrosis. Proc Natl Acad Sci USA 112(51):15713–15718. https://doi.org/10.1073/pnas.1522163112
Burke MA, Wakimoto H, Jiao Z, Gorham JM, DePalma SR, Conner DA et al (2018) Epigenomic control of cardiac fibrosis by bet bromodomain proteins in dilated cardiomyopathy. J Cardiac Fail 24(8):S2. https://doi.org/10.1016/j.cardfail.2018.07.011
Wang J, Zhou F, Li Z, Mei H, Wang Y, Ma H et al (2018) Pharmacological targeting of BET proteins attenuates radiation-induced lung fibrosis. Sci Rep 8(1):998. https://doi.org/10.1038/s41598-018-19343-9
Valinciute G, Weigel C, Veldwijk MR, Oakes CC, Herskind C, Wenz F et al (2017) BET-bromodomain inhibitors modulate epigenetic patterns at the diacylglycerol kinase alpha enhancer associated with radiation-induced fibrosis. Radiother Oncol 125(1):168–174. https://doi.org/10.1016/j.radonc.2017.08.028
Alqahtani A, Choucair K, Ashraf M, Hammouda DM, Alloghbi A, Khan T et al (2019) Bromodomain and extra-terminal motif inhibitors: a review of preclinical and clinical advances in cancer therapy. Future Sci OA 5(3):FSO372-FSO. https://doi.org/10.4155/fsoa-2018-0115
Mao W, Ghasemzadeh A, Freeman ZT, Obradovic A, Chaimowitz MG, Nirschl TR et al (2019) Immunogenicity of prostate cancer is augmented by BET bromodomain inhibition. J ImmunoTher Cancer 7(1):277. https://doi.org/10.1186/s40425-019-0758-y
Stathis A, Bertoni F (2018) BET Proteins as targets for anticancer treatment. Cancer Discov 8(1):24–36. https://doi.org/10.1158/2159-8290.cd-17-0605
Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ et al (2010) miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med 207(8):1589–1597. https://doi.org/10.1084/jem.20100035
Ly D, Savage JE, Shankavaram UT, Saleh AD, Mitchell JB, Soule BP et al (2009) Interactions between mir-21 and its targets in radiation-induced fibrosis. Int J Radiat Oncol Biol Phys 75(3):S537–S538. https://doi.org/10.1016/j.ijrobp.2009.07.1228
Liu Z, Liang X, Li X, Liu X, Zhu M, Gu Y et al (2019) MiRNA-21 functions in ionizing radiation-induced epithelium-to-mesenchymal transition (EMT) by downregulating PTEN. Toxicol Res (Camb) 8(3):328–340. https://doi.org/10.1039/c9tx00019d
Choi SH, Hong ZY, Nam JK, Lee HJ, Jang J, Yoo RJ et al (2015) A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin Cancer Res 21(16):3716–3726. https://doi.org/10.1158/1078-0432.ccr-14-3193
Kwon OS, Kim KT, Lee E, Kim M, Choi SH, Li H et al (2016) Induction of MiR-21 by stereotactic body radiotherapy contributes to the pulmonary fibrotic response. PLoS ONE 11(5):e0154942. https://doi.org/10.1371/journal.pone.0154942
Bu H, Wedel S, Cavinato M, Jansen-Dürr P (2017) MicroRNA regulation of oxidative stress-induced cellular senescence. Oxid Med Cell Longev 2017:2398696. https://doi.org/10.1155/2017/2398696
He Y, Thummuri D, Zheng G, Okunieff P, Citrin DE, Vujaskovic Z et al (2019) Cellular senescence and radiation-induced pulmonary fibrosis. Transl Res 209:14–21. https://doi.org/10.1016/j.trsl.2019.03.006
Tian W, Yin X, Wang L, Wang J, Zhu W, Cao J et al (2015) The key role of miR-21-regulated SOD2 in the medium-mediated bystander responses in human fibroblasts induced by alpha-irradiated keratinocytes. Mutat Res 780:77–85. https://doi.org/10.1016/j.mrfmmm.2015.08.003
Kura B, Kalocayova B, LeBaron TW, Frimmel K, Buday J, Surovy J et al (2019) Regulation of microRNAs by molecular hydrogen contributes to the prevention of radiation-induced damage in the rat myocardium. Mol Cell Biochem 457(1–2):61–72
Anastasov N, Höfig I, Vasconcellos IG, Rappl K, Braselmann H, Ludyga N et al (2012) Radiation resistance due to high expression of miR-21 and G2/M checkpoint arrest in breast cancer cells. Radiat Oncol (London, England) 7:206. https://doi.org/10.1186/1748-717X-7-206
Liu J, Zhu H, Yang X, Ge Y, Zhang C, Qin Q et al (2014) MicroRNA-21 is a novel promising target in cancer radiation therapy. Tumour Biol 35(5):3975–3979. https://doi.org/10.1007/s13277-014-1623-8
Lin S-C, Chang I-W, Hsieh P-L, Lin C-Y, Sun D-P, Sheu M-J et al (2017) High immunoreactivity of DUOX2 is associated with poor response to preoperative chemoradiation therapy and worse prognosis in rectal cancers. J Cancer 8(14):2756
Mortezaee K, Goradel NH, Amini P, Shabeeb D, Musa AE, Najafi M et al (2019) NADPH oxidase as a target for modulation of radiation response; implications to carcinogenesis and radiotherapy. Curr Mol Pharmacol 12(1):50–60. https://doi.org/10.2174/1874467211666181010154709
Mangoni M, Sottili M, Gerini C, Desideri I, Bastida C, Pallotta S et al (2017) A PPAR-gamma agonist protects from radiation-induced intestinal toxicity. United Eur Gastroenterol J 5(2):218–226. https://doi.org/10.1177/2050640616640443
Fan P, Abderrahman B, Chai TS, Yerrum S, Jordan VC (2018) Targeting peroxisome proliferator-activated receptor γ to increase estrogen-induced apoptosis in estrogen-deprived breast cancer cells. Mol Cancer Ther 17(12):2732–2745. https://doi.org/10.1158/1535-7163.mct-18-0088
Atkinson J, Kapralov AA, Yanamala N, Tyurina YY, Amoscato AA, Pearce L et al (2011) A mitochondria-targeted inhibitor of cytochrome c peroxidase mitigates radiation-induced death. Nat Commun 2:497
Sharlow ER, Leimgruber S, Lira A, McConnell MJ, As N, Bloom GS et al (2016) A small molecule screen exposes mTOR signaling pathway involvement in radiation-induced apoptosis. ACS Chem Biol 11(5):1428–1437
Zhang S, Wang W, Gu Q, Xue J, Cao H, Tang Y et al (2014) Protein and miRNA profiling of radiation-induced skin injury in rats: the protective role of peroxiredoxin-6 against ionizing radiation. Free Radical Biol Med 69:96–107. https://doi.org/10.1016/j.freeradbiomed.2014.01.019
Bhanja P, Norris A, Gupta-Saraf P, Hoover A, Saha S (2018) BCN057 induces intestinal stem cell repair and mitigates radiation-induced intestinal injury. Stem Cell Res Ther 9(1):26
Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM (2007) WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci USA 104(2):618–623. https://doi.org/10.1073/pnas.0606599104
Romesser PB, Kim AS, Jeong J, Mayle A, Dow LE, Lowe SW (2019) Preclinical murine platform to evaluate therapeutic countermeasures against radiation-induced gastrointestinal syndrome. Proc Natl Acad Sci 116(41):20672–20678
Baranov A, Gale RP, Guskova A, Piatkin E, Selidovkin G, Muravyova L et al (1989) Bone marrow transplantation after the Chernobyl nuclear accident. N Engl J Med 321(4):205–212. https://doi.org/10.1056/nejm198907273210401
Kulkarni S, Wang TC, Guha C (2016) Stromal progenitor cells in mitigation of non-hematopoietic radiation injuries. Curr Pathobiol Rep 4(4):221–230
Kiang JG (2016) Adult mesenchymal stem cells and radiation injury. Health Phys 111(2):198–203. https://doi.org/10.1097/HP.0000000000000459
Gong W, Guo M, Han Z, Wang Y, Yang P, Xu C et al (2016) Mesenchymal stem cells stimulate intestinal stem cells to repair radiation-induced intestinal injury. Cell Death Dis 7(9):e2387. https://doi.org/10.1038/cddis.2016.276
Chang PY, Qu YQ, Wang J, Dong LH (2015) The potential of mesenchymal stem cells in the management of radiation enteropathy. Cell Death Dis 6:e1840. https://doi.org/10.1038/cddis.2015.189
Benderitter M, Caviggioli F, Chapel A, Coppes RP, Guha C, Klinger M et al (2014) Stem cell therapies for the treatment of radiation-induced normal tissue side effects. Antioxid Redox Signal 21(2):338–355. https://doi.org/10.1089/ars.2013.5652
Rasmusson I (2006) Immune modulation by mesenchymal stem cells. Exp Cell Res 312(12):2169–2179. https://doi.org/10.1016/j.yexcr.2006.03.019
Zachman DK, Leon RP, Das P, Goldman DC, Hamlin KL, Guha C et al (2013) Endothelial cells mitigate DNA damage and promote the regeneration of hematopoietic stem cells after radiation injury. Stem Cell Res 11(3):1013–1021. https://doi.org/10.1016/j.scr.2013.07.001
Piryani SO, Jiao Y, Kam AYF, Liu Y, Vo-Dinh T, Chen BJ et al (2019) Endothelial cell-derived extracellular vesicles mitigate radiation-induced hematopoietic injury. Int J Radiat Oncol Biol Phys 104(2):291–301. https://doi.org/10.1016/j.ijrobp.2019.02.008
Kim A, Shim S, Kim M-J, Myung JK, Park S (2018) Mesenchymal stem cell-mediated Notch2 activation overcomes radiation-induced injury of the hematopoietic system. Sci Rep 8(1):9277. https://doi.org/10.1038/s41598-018-27666-w
Tao X, Sun M, Chen M, Ying R, Su W, Zhang J et al (2019) HMGB1-modified mesenchymal stem cells attenuate radiation-induced vascular injury possibly via their high motility and facilitation of endothelial differentiation. Stem Cell Res Ther 10(1):92. https://doi.org/10.1186/s13287-019-1197-x
Weyemi U, Redon CE, Aziz T, Choudhuri R, Maeda D, Parekh PR et al (2015) Inactivation of NADPH oxidases NOX4 and NOX5 protects human primary fibroblasts from ionizing radiation-induced DNA damage. Radiat Res 183(3):262–270. https://doi.org/10.1667/rr13799.1
Deng W, Abdel-Mageed A, Connors R, Pietryga D, Senagore A (2015) Successful mitigation of radiation injuries in mice using mesenchymal stem cells genetically modified to secrete extracellular superoxide dismutase. J Stem Cell Res Ther 5(288):2
Gan J, Meng F, Zhou X, Li C, He Y, Zeng X et al (2015) Hematopoietic recovery of acute radiation syndrome by human superoxide dismutase-expressing umbilical cord mesenchymal stromal cells. Cytotherapy 17(4):403–417. https://doi.org/10.1016/j.jcyt.2014.11.011
Patwardhan RS, Sharma D, Checker R, Sandur SK (2014) Mitigation of radiation-induced hematopoietic injury via regulation of cellular MAPK/phosphatase levels and increasing hematopoietic stem cells. Free Radical Biol Med 68:52–64. https://doi.org/10.1016/j.freeradbiomed.2013.11.004
Cline JM, Dugan G, Bourland JD, Perry DL, Stitzel JD, Weaver AA et al (2018) Post-irradiation treatment with a superoxide dismutase mimic, MnTnHex-2-PyP(5+), mitigates radiation injury in the lungs of non-human primates after whole-thorax exposure to ionizing radiation. Antioxidants (Basel, Switzerland) 7(3):40. https://doi.org/10.3390/antiox7030040
Long W, Zhang G, Dong Y, Li D (2018) Dark tea extract mitigates hematopoietic radiation injury with antioxidative activity. J Radiat Res 59(4):387–394. https://doi.org/10.1093/jrr/rrx072
Zanoni M, Cortesi M, Zamagni A, Tesei A (2019) The role of mesenchymal stem cells in radiation-induced lung fibrosis. Int J Mol Sci 20(16):3876. https://doi.org/10.3390/ijms20163876
Ortiz LA, Gambelli F, McBride C, Gaupp D, Baddoo M, Kaminski N et al (2003) Mesenchymal stem cell engraftment in lung is enhanced in response to bleomycin exposure and ameliorates its fibrotic effects. Proc Natl Acad Sci USA 100(14):8407–8411. https://doi.org/10.1073/pnas.1432929100
Klein D, Steens J, Wiesemann A, Schulz F, Kaschani F, Rock K et al (2017) Mesenchymal stem cell therapy protects lungs from radiation-induced endothelial cell loss by restoring superoxide dismutase 1 expression. Antioxid Redox Signal 26(11):563–582. https://doi.org/10.1089/ars.2016.6748
Wei L, Zhang J, Yang ZL, You H (2017) Extracellular superoxide dismutase increased the therapeutic potential of human mesenchymal stromal cells in radiation pulmonary fibrosis. Cytotherapy 19(5):586–602. https://doi.org/10.1016/j.jcyt.2017.02.359
Li B, Li C, Zhu M, Zhang Y, Du J, Xu Y et al (2017) Hypoxia-induced mesenchymal stromal cells exhibit an enhanced therapeutic effect on radiation-induced lung injury in mice due to an increased proliferation potential and enhanced antioxidant ability. Cell Physiol Biochem 44(4):1295–1310. https://doi.org/10.1159/000485490
Yao Y, Zheng Z, Song Q (2018) Mesenchymal stem cells: a double-edged sword in radiation-induced lung injury. Thorac Cancer 9(2):208–217. https://doi.org/10.1111/1759-7714.12573
Cohen EP, Pais P, Moulder JE (2010) Chronic kidney disease after hematopoietic stem cell transplantation. Semin Nephrol 30(6):627–634. https://doi.org/10.1016/j.semnephrol.2010.09.010
Medhora M, Gao F, Wu Q, Molthen RC, Jacobs ER, Moulder JE et al (2014) Model development and use of ACE inhibitors for preclinical mitigation of radiation-induced injury to multiple organs. Radiat Res 182(5):545–555. https://doi.org/10.1667/RR13425.1
Heylmann D, Rödel F, Kindler T, Kaina B (2014) Radiation sensitivity of human and murine peripheral blood lymphocytes, stem and progenitor cells. Biochim Biophys Acta Rev Cancer 1846(1):121–129
Chen JJ, Gao Y, Tian Q, Liang YM, Yang L (2014) Platelet factor 4 protects bone marrow mesenchymal stem cells from acute radiation injury. Br J Radiol 87(1040):20140184. https://doi.org/10.1259/bjr.20140184
Lemon JA, Taylor K, Verdecchia K, Phan N, Boreham DR (2014) The influence of Trp53 in the dose response of radiation-induced apoptosis, DNA repair and genomic stability in murine haematopoietic cells. Dose Response Publ Int Hormesis Soc 12(3):365–385. https://doi.org/10.2203/dose-response.14-008.Lemon
Farhood B, Goradel NH, Mortezaee K, Khanlarkhani N, Salehi E, Nashtaei MS et al (2019) Intercellular communications-redox interactions in radiation toxicity; potential targets for radiation mitigation. J Cell Commun Signal 13(1):3–16. https://doi.org/10.1007/s12079-018-0473-3
Azzam EI, Jay-Gerin J-P, Pain D (2012) Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett 327(1–2):48–60. https://doi.org/10.1016/j.canlet.2011.12.012
Brown SL, Kolozsvary A, Liu J, Jenrow KA, Ryu S, Kim JH (2010) Antioxidant diet supplementation starting 24 hours after exposure reduces radiation lethality. Radiat Res 173(4):462–468. https://doi.org/10.1667/RR1716.1
Cohen EP, Fish BL, Moulder JE (2015) Late-onset effects of radiation and chronic kidney disease. Lancet 386(10005):1737–1738
Tsoutsou PG, Koukourakis MI (2006) Radiation pneumonitis and fibrosis: mechanisms underlying its pathogenesis and implications for future research. Int J Radiat Oncol Biol Phys 66(5):1281–1293
Chen Y, Chou C, Shun C, Wei M, Kuo S (2016) The expression of CXCL16 during lung irradiation may lead to radiation pneumonitis and fibrosis through inducing neutrophil and macrophage infiltration in lung tissue. Int J Radiat Oncol Biol Phys 96(2):S65–S66
Sieber F, Muir SA, Cohen EP, North PE, Fish BL, Irving AA et al (2009) High-dose selenium for the mitigation of radiation injury: a pilot study in a rat model. Radiat Res 171(3):368–373. https://doi.org/10.1667/0033-7587-171.3.368
Christofidou-Solomidou M, Tyagi S, Tan KS, Hagan S, Pietrofesa R, Dukes F et al (2011) Dietary flaxseed administered post thoracic radiation treatment improves survival and mitigates radiation-induced pneumonopathy in mice. BMC Cancer 11:269. https://doi.org/10.1186/1471-2407-11-269
Pietrofesa R, Turowski J, Tyagi S, Dukes F, Arguiri E, Busch TM (2013) Radiation mitigating properties of the lignan component in flaxseed. BMC Cancer. https://doi.org/10.1186/1471-2407-13-179
Lee JC, Krochak R, Blouin A, Kanterakis S, Chatterjee S, Arguiri E et al (2009) Dietary flaxseed prevents radiation-induced oxidative lung damage, inflammation and fibrosis in a mouse model of thoracic radiation injury. Cancer Biol Ther 8(1):47–53
Williams JP, Brown SL, Georges GE, Hauer-Jensen M, Hill RP, Huser AK et al (2010) Animal models for medical countermeasures to radiation exposure. Radiat Res 173(4):557–578
Naeeji A, Mozdarani H, Monfared AS, Faeghi F, Ahmadi A, Gholami M et al (2017) Oral administration of vitamin C, cimetidine and famotidine on micronuclei induced by low dose radiation in mouse bone marrow cells. J Biomed Phys Eng 7(2):117
Haydont V, Gilliot O, Rivera S, Bourgier C, François A, Aigueperse J et al (2007) Successful mitigation of delayed intestinal radiation injury using pravastatin is not associated with acute injury improvement or tumor protection. Int J Radiat Oncol Biol Phys 68(5):1471–1482
Gedon NKY, Mueller RS (2018) Atopic dermatitis in cats and dogs: a difficult disease for animals and owners. Clin Transl Allergy 8(1):41
Moulder JE, Fish BL (1989) Late toxicity of total body irradiation with bone marrow transplantation in a rat model. Int J Radiat Oncol Biol Phys 16(6):1501–1509. https://doi.org/10.1016/0360-3016(89)90955-3
Stewart FA, Luts A, Lebesque JV (1989) The lack of long-term recovery and reirradiation tolerance in the mouse kidney. Int J Radiat Biol 56(4):449–462. https://doi.org/10.1080/09553008914551601
van Rongen E, Kuijpers WC, Madhuizen HT, van der Kogel AJ (1988) Effects of multifraction irradiation on the rat kidney. Int J Radiat Oncol Biol Phys 15(5):1161–1170. https://doi.org/10.1016/0360-3016(88)90199-x
Molteni JM, Cohen EF, Ward WF, Fish BL, Taylor JM, Wolfe LF, Brizio-Molteni L, Veno PA (2000) Control of radiation-induced pneumopathy and lung fibrosis by angiotensin-converting enzyme inhibitors and an angiotensin II type 1 receptor blocker. Int J Radiat Biol 76(4):523–532
van der Veen SJ, Ghobadi G, de Boer RA, Faber H, Cannon MV, Nagle PW et al (2015) ACE inhibition attenuates radiation-induced cardiopulmonary damage. Radiother Oncol 114(1):96–103
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest, financial or otherwise.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
khodamoradi, E., Hoseini-Ghahfarokhi, M., Amini, P. et al. Targets for protection and mitigation of radiation injury. Cell. Mol. Life Sci. 77, 3129–3159 (2020). https://doi.org/10.1007/s00018-020-03479-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-020-03479-x