
Abstract
Mutant isocitrate dehydrogenase 1 (IDH1) is common in gliomas, and produces D-2-hydroxyglutarate (D-2-HG). The full effects of IDH1 mutations on glioma biology and tumor microenvironment are unknown. We analyzed a discovery cohort of 169 World Health Organization (WHO) grade II–IV gliomas, followed by a validation cohort of 148 cases, for IDH1 mutations, intratumoral microthrombi, and venous thromboemboli (VTE). 430 gliomas from The Cancer Genome Atlas were analyzed for mRNAs associated with coagulation, and 95 gliomas in a tissue microarray were assessed for tissue factor (TF) protein. In vitro and in vivo assays evaluated platelet aggregation and clotting time in the presence of mutant IDH1 or D-2-HG. VTE occurred in 26–30 % of patients with wild-type IDH1 gliomas, but not in patients with mutant IDH1 gliomas (0 %). IDH1 mutation status was the most powerful predictive marker for VTE, independent of variables such as GBM diagnosis and prolonged hospital stay. Microthrombi were far less common within mutant IDH1 gliomas regardless of WHO grade (85–90 % in wild-type versus 2–6 % in mutant), and were an independent predictor of IDH1 wild-type status. Among all 35 coagulation-associated genes, F3 mRNA, encoding TF, showed the strongest inverse relationship with IDH1 mutations. Mutant IDH1 gliomas had F3 gene promoter hypermethylation, with lower TF protein expression. D-2-HG rapidly inhibited platelet aggregation and blood clotting via a novel calcium-dependent, methylation-independent mechanism. Mutant IDH1 glioma engraftment in mice significantly prolonged bleeding time. Our data suggest that mutant IDH1 has potent antithrombotic activity within gliomas and throughout the peripheral circulation. These findings have implications for the pathologic evaluation of gliomas, the effect of altered isocitrate metabolism on tumor microenvironment, and risk assessment of glioma patients for VTE.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diffusely infiltrative glioma is the most common primary brain tumor in adults. For over 100 years, gliomas have been classified according to their appearance under the light microscope, and for over 60 years, necrosis has been associated with aggressive behavior [40]. The World Health Organization (WHO) grades these tumors on a progressive scale of severity from I to IV (with grade I being a distinct type of non-infiltrative tumor), and necrosis is a major diagnostic feature of grade IV malignancy, a.k.a. glioblastoma (GBM) [29]. While the advent of molecular diagnostics has extended our understanding of gliomas, there remains an important role for light microscopy, because physical differences among genetically classified gliomas can provide important clues about the effects of key mutations.
Somatic point mutations of isocitrate dehydrogenase 1 or 2 (IDH1/2) occur in a large subset of gliomas [61]. Wild-type IDH1/2 enzymes oxidize isocitrate into α-ketoglutarate (α-KG), but mutations in the isocitrate binding pocket unmask a latent oxidoreductase reaction, causing the enzyme to convert α-ketoglutarate (α-KG) into the rare metabolite D-2-hydroxyglutarate (D-2-HG) [13]. Most infiltrative gliomas with wild-type IDH1/2 are diagnosed as WHO grade IV GBMs, whereas the overwhelming majority of IDH1/2 mutant gliomas are not (Supplemental Figure 1). This suggests that IDH1/2 mutations may inhibit development of the diagnostic features of GBM. Furthermore, necrosis is far less extensive in IDH1/2-mutant GBMs compared to wild-type GBMs [27, 35]. The molecular basis for this, and for the less aggressive behavior of IDH1/2-mutant gliomas [20], is unclear. Studies on D-2-HG in gliomas and other IDH1/2 mutant cancers have mostly focused on its ability to induce histone and DNA hypermethylation via competitive inhibition of α-KG-dependent demethylases [11, 14, 30, 36, 41, 50, 60]. Yet the full extent of mutant IDH1/2 activities within the cell, and in the tumor microenvironment, are not known.
Many cancers cause aberrant blood coagulation, marked by deep venous thrombi (DVT) and pulmonary emboli (PE), together known as venous thromboemboli (VTE). Gliomas are one of the most at-risk cancers for VTE. This complication causes great suffering, can be difficult to manage, and contributes to cancer-associated mortality [37, 49, 56]. The Food and Drug Administration Oncologic Drug Advisory Committee, the American Society of Clinical Oncology, and the National Comprehensive Cancer Network have all expressed an urgent need to better identify which cancer patients are at greatest risk of VTE [15, 31, 34]. Tumor genotype has recently been shown to affect VTE risk in carcinomas of the lung and colon [1, 12], and others have studied prognostic biomarkers for VTE in brain cancer patients through blood-based testing [52, 63]. Advanced age and GBM diagnosis have been proposed as VTE risk factors [37], but to date, there is no data on whether glioma genotype alters VTE risk.
Materials and methods
Patient cohorts
The discovery cohort consisted of 169 gliomas that had been tested for IDH1/2 mutations as part of routine clinical care at the University of Kentucky (UK) from 2009 to 2014 (Table 1). This cohort included 15 grade II oligodendrogliomas, 14 grade II astrocytomas, 6 grade III oligodendrogliomas, 16 grade III astrocytomas, and 118 GBMs. (Some cases had originally been resected prior to 2009, but for clinical reasons required retrospective testing.) The validation cohort comprised 148 gliomas that had been tested for IDH1/2 mutations at New York University (NYU) (Table 1), including 5 grade II astrocytomas, 7 grade II oligodendrogliomas, 11 grade III astrocytomas, 5 grade III oligodendrogliomas, and 120 GBMs. All gliomas in both cohorts were from adult patients (18+ years). A separate prospective cohort at Northwestern Memorial Hospital (NMH) consisted of 29 patients with newly diagnosed WHO II-IV gliomas, in which preoperative arterial blood was obtained (Table 2).
Mutation screening was done via R132H IDH1 immunohistochemistry (Dianova, Hamburg). Immunonegative tumors were tested for less common IDH1/2 mutations via pyrosequencing (UK) or by 450 K methylation array profiling (NYU) as described previously [50]. Survival data was obtained for 166 cases in the UK cohort via the Kentucky Cancer Registry, but was not available for NYU cases. In the discovery cohort, the median follow-up time for IDH1/2 wild-type gliomas was 7.6 months (with 74 % having died of disease at the time of study), whereas IDH1/2 mutant gliomas had a median follow-up time of 31 months (12 % died of disease at the time of study). Data on coagulation history were available by chart review for 162 and 136 patients at UK and NYU, respectively. At least 6 months of follow-up time was obtained on each case (in the validation cohort, the median amount of tracking time in the patient charts was 9 months for IDH1/2 wild-type gliomas and 14 months for mutant gliomas). For the prospective NMH cohort, coagulation history was also determined by chart review, with an average length of follow-up of 3.5 months (median 3.9 months, range 0.3–6.7 months), and an average time from surgery to first VTE of 0.71 months (median 0.45 months). The glioma tissue microarray (TMA) cohort containing 95 grade II–IV gliomas has been previously described [28]. Institutional Research Board approval was obtained at UK, NYU, and NMH prior to study initiation.
Platelet free plasma (PFP) collection and preparation
Blood was collected from an arterial line immediately prior to surgery and placed in a vacutainer (Becton–Dickinson, Franklin Lakes, New Jersey) containing 3.2 % sodium citrate (Fisher Scientific, Waltham, MA, USA). Whole blood was centrifuged at 2500×g for 15 min at room temperature. The supernatant was transferred to a new 2 mL tube (Eppendorf, Hauppauge, NY) and spun under the same previous conditions. Supernatant (PFP) was transferred into cryovials and stored at −80 °C until use.
Tissue factor microparticle (TF-MP) procoagulant activity assay
Circulating TF-MP procoagulant activity was measured using the chromogenic reporter substrate (Pefachrome FXa 8595, Enzyme Research Laboratories, South Bend, IN). Briefly, 250 µL of human platelet free plasma was diluted with 1 mL of HBSA (137 mM NaCl, 5.4 mM KCl, 5.6 mM Glucose, 10 mM HEPES, 0.1 % bovine serum albumin, pH 7.4) and centrifuged at 21,000×g for 15 min at 4 °C to pellet MPs. Pelleted MPs were washed once with HBSA and resuspended in 160 µL HBSA. 50 µL of MP suspensions were then incubated in 50µL of HBSA containing 73.2 nM FX (Enzyme Research Laboratories), 2.4 nM FVIIa (Enzyme Research Laboratories), and 10 mM CaCl2 for 2 h at 37 °C in a 96-well plate. The reaction was stopped with 25 µL of HBSA containing 25 mM EDTA. Then, 25 µL of FXa substrate (Pefachrome FXa, 4.0 mM) was added to the reaction and incubated for 15 min at 37 °C, and OD 405 nm recorded using Synergy 2 Multi-Mode Microplate Reader (BioTek). To calculate TF-MP procoagulant activity, a standard curve was generated using recombinant relipidated TF (0–30 pg/mL, Haematologic Technologies, Essex Junction, VT).
Pathologic evaluation
All pathologic slides on each case in the discovery (UK) and validation (NYU) cohorts were collected and analyzed for this study. Cases were scored by light microscopic examination while blinded to IDH1/2 status (CH at UK, MS and CT at NYU) for key histologic features, including necrosis, microvascular proliferation (MVP), and microthrombi. Necrosis and MVP were scored as present or absent, in accordance with standard WHO grading. Microthrombi were scored according to the average number of microthrombi per tissue block. Cutoff Finder was used for finding the optimal cutoff value to discriminate between mutant IDH1/2 and wild-type IDH1/2 gliomas in the discovery cohort [5].
Tissue factor (TF) immunohistochemistry (IHC) was performed on 4 µm-thick sections cut from a separate cohort of formalin fixed paraffin embedded TMA slides according to routine histology protocols [28]. Anti-human TF antibody (catalog #4509, Sekisui, Stamford, CT) was diluted 1:500 in TBS-Tween and applied to slides at room temperature for 1 h. TMA cores were evaluated while blinded to IDH1/2 status according to previously published methods [28]. Briefly, each core was scored on a semiquantitative scale as follows: 0 = negative, 1 = weak, 2 = moderate, 3 = strong. Three cores per tumor were randomly scattered on the TMA blocks and were averaged to produce a final score for that tumor.
Statistical evaluation
Statistical significance of differences between groups was determined via two-sample t test, one-way ANOVA with post hoc Tukey’s test, Fisher’s exact test, or log-rank test as appropriate using GraphPad software (La Jolla, CA). The Bonferroni method was used to adjust for multiple comparisons in the analysis of the association between F3 promoter methylation and IDH1/2 status. Odds ratios were calculated based on exact logistic regression. Hazard ratios were calculated based on Cox proportional hazards models or competing risk regression. Multivariable analyses were performed using competing risk regression based on Fine and Gray’s proportional subhazards model with Firth’s correction [24], logistic regression models with Firth’s correction or exact logistic regression, as appropriate, when there were rare or no events in one group, using SAS 9.4 software. Model fit parameter C index for Cox proportional hazards models and competing risk regression, or C statistics for logistic regression, were provided where applicable. Optimal cutoffs for TF IHC and F3 mRNA from TMA and TCGA datasets were objectively determined using recursive partitioning analysis and Cutoff Finder [5], respectively. In all analyses, the significance target was P ≤ 0.05.
See the Electronic Supplementary Data for additional methods.
Results
We observed systemic coagulation (including DVT and PE) in 30/117 (26 %) IDH1/2 wild-type glioma patients from the discovery cohort (Table 1), consistent with results reported by others [37]. In striking contrast, none of the 45 (0 %) patients with mutant IDH1/2 glioma presented with or developed any VTE (P < 0.001). The validation cohort produced similar findings, wherein 31/105 (30 %) IDH1/2 wild-type glioma patients developed VTE but none of the 31 (0 %) IDH1/2 mutant glioma patients did (P < 0.001). Of the patients who had VTE, the median time from surgery to first VTE episode was 1.0 months in the discovery cohort (Q1 = 0.1 months, Q3 = 3.0 months) and 3.5 months in the validation cohort (Q1 = 0.3 months, Q3 = 8.0 months). Univariable and multivariable analysis of clinical variables often associated with VTE risk, including GBM diagnosis, KPS score, and length of hospital stay, showed a potential link between IDH1/2 status and VTE in the discovery and validation cohorts (OR = 0.043, 95 % CI 0.002–0.78, P = 0.033 in the discovery cohort; OR = 0.052, 95 % CI 0.003–0.91, P = 0.043 in the validation cohort) (Supplemental Table 1). A competing risk analysis of VTE with death in the discovery cohort showed a trend toward mutant IDH1/2 as an independent prognosticator for VTE (HR 0.059, 95 % CI 0.003–1.1, P = 0.059) (Supplemental Table 2). Preoperative prothrombin and partial thromboplastin times showed no difference in mutant versus wild-type IDH1/2 patients (Supplemental Figure 2), suggesting no functional differences in circulating clotting factors I, II, V, VII, VIII, IX, X, XI, & XII.
Microscopic analysis of tissue sections in the discovery cohort showed that a cutoff of 0.15 microthrombi/block optimally discriminated between mutant IDH1/2 and wild-type IDH1/2 gliomas (AUC 0.92, P < 0.001) This was slightly adjusted to a working cutoff of 0.25 microthrombi per block, so as to improve its universal practicality for routine neuropathological examination. The adjusted cutoff only altered assignment of one case, yielding similar overall results (AUC 0.92, P < 0.001). Gliomas with microthrombi were nearly always IDH1/2 wild-type (100 of 117 = 85.5 %, see Table 1; Fig. 1, and Supplemental Figure 3). In contrast, only a single IDH1/2-mutant glioma displayed microthrombi (1 of 52 = 1.9 %) (P < 0.001, Fisher’s exact test). This inverse relationship between IDH1/2 mutations and microthrombi held across all glioma grades in discovery and validation cohorts (Table 1; Fig. 1a–f). Others have suggested that necrosis in a glioma may be caused by microthrombi [42]. Yet in our discovery cohort, whereas 96 % (94 of 98) of necrotic IDH1/2 wild-type tumors also contained microthrombi, only 10 % (1 of 10) of necrotic IDH1/2-mutant gliomas did. The necrotic gliomas in the validation cohort showed similar results, with microthrombi in 92 % (97 of 106) and 8 % (1 of 13) of wild-type and mutant tumors, respectively (P < 0.001 for each cohort, Fisher’s exact test, Fig. 1g–h).

Microthrombi and IDH1/2 mutations in gliomas. Frequency of intratumoral microthrombi according to IDH1/2 mutation status in discovery (left) and validation (right) cohorts, in all WHO grade II–IV gliomas (a, b), grade II–III (c, d), grade IV GBM (e, f), and necrotic gliomas (g, h). P < 0.001 in a, b and e–h; P = 0.02 in c and 0.0034 in d. All P values were calculated by Fisher’s exact test
Receiver operating characteristic (ROC) analysis showed that, although all variables produced statistically significant areas under the curve (AUCs), the presence of microthrombi (AUC 0.92, 95 % CI 0.88–0.95, P < 0.001) was superior to advanced age (AUC 0.83, 95 % CI 0.77–0.89, P < 0.001), necrosis (AUC 0.82, 95 % CI 0.76–0.89, P < 0.001), and MVP (AUC 0.82, 95 % CI 0.88-0.95, P < 0.001) as a predictor of IDH1/2 mutation status in the discovery cohort (microthrombi versus age: χ 2 = 5.02, P = 0.025; versus necrosis: χ 2 = 10.21, P = 0.001; versus MVP: χ 2 = 8.06, P = 0.0045.) (Supplemental Figure 4). The validation cohort confirmed this, as all variables had significant AUCs (microthrombi AUC 0.92, 95 % CI 0.87–0.97, P < 0.001; patient age AUC 0.71, 95 % CI 0.62–0.80, P < 0.001; necrosis AUC 0.77, 95 % CI 0.69–0.86, P < 0.001; MVP AUC 0.72, 95 % CI 0.63–0.81, P < 0.001), but the microthrombi AUC was stronger than the others (microthrombi versus age: χ 2 = 20.01, P < 0.001; versus necrosis: χ 2 = 9.23, P = 0.024; versus MVP: χ 2 = 20.20, P < 0.001). On a multivariable analysis of key variables correlating with IDH1/2 status, microthrombi remained a significant independent predictor in both cohorts (Supplemental Table 3).
In the past, microthrombi had been proposed as another diagnostic criterion for GBM, in addition to necrosis and MVP [38, 51]. For the tumor series examined here, univariate analysis revealed that the presence of microthrombi (log-rank P < 0.001; HR 5.1, 95 % CI 2.9–9) was comparable to necrosis (log-rank P < 0.001; HR 4.0, 95 % CI 2.3–6.9) and MVP (log-rank P < 0.001; HR 6.4, 95 % CI 3.3–12) in prognostic stratification, and predicted a much worse prognosis for grade III gliomas (log-rank P = 0.008; HR 9.0, 95 % CI 1.2–65) (Supplemental Figure 5). On multivariable analysis of these histologic variables, only microthrombi (HR 2.7, 95 % CI 1.2–5.8, P = 0.01) and MVP (HR 4.2, 95 % CI 1.6–10.7, P = 0.003) proved to be significant, not necrosis (Supplemental Table 4).
We next sought to identify possible mechanisms for the inverse relationship between mutant IDH1/2 and thrombosis in gliomas. In The Cancer Genome Atlas (TCGA), mRNA levels of 35 coagulation-associated genes were examined with respect to glioma grade and IDH1/2 status. Among procoagulant genes, F3 showed the strongest inverse association with tumor IDH1/2 mutation status. The mean transcript level for F3, encoding Factor III [also known as Tissue Factor (TF)], was 75 % lower in mutant gliomas across all tumor grades (95 % CI 67–84, P = 1.1 × 10−52) (Fig. 2 and Supplemental Figure 6). TF is a transmembrane glycoprotein that is expressed in many tumors [2, 16]. In its membrane-bound form, TF initiates coagulation. Mutant IDH1/2 suppresses expression of many genes by inducing hypermethylation [11, 14, 30, 36, 41, 50, 58, 60], so we investigated F3 promoter methylation among TCGA gliomas. Of 17 CpG sites within the F3 promoter, 8 had significantly higher levels of methylation in IDH1/2 mutant tumors, compared to wild-type tumors, across all glioma grades (Fig. 3a, Supplemental Table 6). TF immunoreactivity was lower in IDH1/2 mutant gliomas compared to wild-type gliomas (Fig. 3b, c), consistent with the TCGA F3 mRNA data. In wild-type gliomas there was a greater range of TF expression, and in those tumors, low TF expression correlated with longer survival (log-rank P = 0.049; HR 2.0, 95 % CI 0.99–4.0) (Supplemental Figure 7), a finding independently confirmed by others [17].

Coagulation-associated genes according to IDH1/2 status in gliomas. Expression of key genes known to be involved in clotting and glioma-associated thrombosis [21, 22, 33, 45, 47, 62] (Supplemental Table 5) was assessed via Illumina HiSeq 2000 RNA Sequencing platform in grade II-IV TCGA gliomas. Each row represents a single tumor, columns indicate either IDH1/2 status (aqua IDH2 mutant, red IDH1 mutant, black IDH1/2 wild-type); WHO grade (peach II, green III, purple IV); or gene expression as a ratio of the individual tumor mRNA relative to the mean of all IDH1/2 wild-type tumors for that gene (blue downregulated, yellow upregulated, white no change). Genes in the pink shaded area are procoagulant; genes in the light blue shaded area are anti-coagulant. N = 196 IDH1/2 wild-type and 234 mutant IDH1/2 gliomas

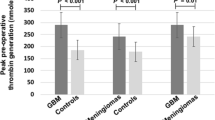
TF by IDH1/2 status in gliomas. a Mean increase in methylation beta value at each CpG site in the F3 promoter of mutant IDH1/2 gliomas, relative to wild-type gliomas. CpG sites are grouped according to their location within and around the CpG island of the F3 promoter. *P < 0.01 in all 3 grades of glioma. See Supplemental Table 6 for additional details. b Representative H&E photomicrographs of IDH1/2 wild-type and mutant GBMs from the TMA cohort, and their corresponding tissue factor IHC. Scale bar in lower right panel = 50 µm. c TF IHC scores according to IDH1/2 mutation status in high-grade (III-IV) gliomas. d Preoperative TF-MP activity, measured in pg Factor Xa generation per ml in platelet-free plasma, obtained from the arterial blood of patients with IDH1/2 wild-type and mutant gliomas (see Table 2). Dotted line represents the cutoff for “strong TF activity” in cancer patients, as published previously [19]. In c and d, each point represents a single tumor, and red triangles in d represent patients who developed VTE. Bars mean ± SD
Many cancers release TF-containing microparticles (TP-MPs) into the circulation, an event which has been repeatedly linked to cancer-induced VTE [3, 7, 10, 32, 43–45, 55]. To further establish a link between IDH1/2 mutations, TF, and VTE in gliomas, we analyzed circulating TF-MP activity in platelet-free plasma, isolated from preoperative arterial blood in a prospective cohort of 29 newly-diagnosed glioma patients (Table 2). Average circulating TF-MP activity, quantified as picograms of Factor Xa generated per ml plasma, was elevated in patients whose gliomas were IDH1/2 wild-type, compared to patients with IDH1/2 mutant gliomas (1.2 ± 0.21 versus 0.49 ± 0.19 pg/ml, P = 0.036, Fig. 3d). Furthermore, there was a positive correlation between the degree of preoperative circulating TF-MP activity and development of VTE (Spearman coefficient ρ(27) = 0.45, 95 % CI 0.089–0.71, P = 0.014) (Table 2).
The major metabolic consequence of IDH1/2 mutation is elevated production of D-2-HG [13]. The effect of this metabolite on platelet aggregation, a key event in coagulation, has not yet been investigated. By adding D-2-HG to human platelets isolated from whole blood, we observed a rapid, potent, and concentration-dependent suppression of platelet aggregation (Fig. 4a, b, Supplemental Figures 8a and b). D-2-HG inhibited iCa++ accumulation in platelets by 72 % (Fig. 4c, Supplemental Figure 8c), and this inhibitory effect was prevented by adding the calcium ionophore A23187 to platelet-D-2-HG mixtures (Fig. 4d, Supplemental Figure 8d). D-2-HG directly bound iCa++ (K d = 48.9 ± 3.5 mM, R 2 = 0.99913) (Supplemental Figure 9), reduced the levels of free Ca++ in human serum (Fig. 4e), and delayed clotting of human blood (Fig. 4f). Finally, glioma xenografts expressing R132H IDH1 lowered free serum Ca++ in mice by 28 % (Fig. 4g) and markedly delayed clotting after tail snip (Fig. 4h, Supplemental Figure 10).

Platelet activity, calcium, and clotting time in the presence of mutant IDH1 and D-2-HG. Washed human platelets (2 × 108/mL) from 3 distinct donors were stimulated by a 1 μg/mL collagen or b 0.1 U/mL thrombin in the presence or absence of D-2-HG. Platelet aggregation was measured by turbidity (Chronolog) with stirring (1000 rpm). c Platelets were labeled with Fura2-AM and iCa++ levels were determined via 340 nm/380 nm excitation ratio under the specified conditions. d Washed human platelets were stimulated by thrombin or the calcium ionophore A23187 in the presence or absence of D-2-HG. e Human serum levels of free Ca++ in the presence of D-2-HG (note, SD bars small and not visible). f Clotting time of human blood in the presence of D-2-HG. Data are expressed as clotting time beyond matched untreated controls (mean baseline = 190 s) ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. g Free Ca++ levels in serum from mice bearing flank xenografts of human gliomas with and without R132H IDH1. h Tail vein bleeding times for mice bearing flank xenografts of human gliomas with and without R132H IDH1
Discussion
To date, studies of mutant IDH1/2 have been tumor cell-centric: i.e., they have focused on what the mutation does inside the tumor cell. Our data suggest this mutation also exerts profound effects outside the tumor cell, and not only within the immediate tumor microenvironment, but systemically. This greatly advances our understanding of the significance of this mutation in gliomas, and accounts for some puzzling features of mutant IDH1/2. For example, most mutant IDH1/2 gliomas do not present with necrosis, and even when they do, it is much less extensive than in wild-type tumors [27, 35]. For the first time, our demonstration of a relationship between mutant IDH1/2, reduced TF expression, impaired platelet activity, and the absence of microthrombi within mutant tumors provides a plausible explanation for the striking paucity of necrosis in this subset of gliomas.
Although mutant IDH1/2 clearly promotes gliomagenesis, it may also limit the development of the histologic characteristics of GBM. While histologic features are obviously the result of many diverse mechanisms, blocking the development of intratumoral thrombi, and its resultant necrosis, potentially could help explain why most mutant IDH1/2 gliomas are not grade IV, whereas most wild-type gliomas are (Supplemental Figure 1). We also show that microthrombi may be comparable to MVP and necrosis in prognostic stratification, though additional prospective data are needed to verify this. Indeed, the entire WHO grading system can be regarded as a set of histologic features that act as surrogate markers for specific underlying gene alterations. It is little surprise, then, that molecular data usually provide more accurate diagnostic and prognostic information for gliomas than traditional histotyping [39]. Diffusely infiltrative gliomas that lack mutant IDH1/2 have the same aggressive behavior as GBM, even when histologic criteria for a GBM diagnosis are not met [4, 39]. Because the presence of microthrombi is the strongest histologic predictor that an infiltrative glioma lacks mutant IDH1/2 (Supplemental Figure 4, Supplemental Table 3), and because it is an even stronger correlate of shorter survival than necrosis (Supplemental Figure 5, Supplemental Table 4), microthrombi may prove to be a useful histologic criterion for GBM. Although upfront screening for mutant IDH1/2 is routine in more developed parts of the world, access to advanced molecular diagnostics is challenging in many other areas. Furthermore, even in more developed parts of the world, cost containment is a growing concern in medicine. When a glioma is immunonegative for the most common IDH1/2 mutation, R132H IDH1, screening for microthrombi could help decide when it is worthwhile to employ additional tests to detect less frequent IDH1/2 mutations.
VTE risk is higher in patients diagnosed with a GBM or those of advanced age, compared to patients with grade II–III gliomas [9, 48]. Our data suggest that this may be due to the fact that IDH1/2 mutations are less common in older patients and in GBMs. A recent paper showed that KRAS mutations increase TF expression and VTE risk in colorectal carcinomas [1], but the current study is the first example, in any cancer, of a specific mutation conferring reduced VTE risk. This could therefore help stratify glioma patients into low- and high-risk groups for prospective trials, potentially avoiding unnecessary thromboprophylaxis in patients at very low risk of VTE.
Thus far, our data suggest two complementary mechanisms whereby mutant IDH1/2 suppresses thrombosis (Fig. 5). The first is methylation-based, wherein mutant IDH1/2 causes F3 promoter hypermethylation [57], leading to reduced F3 mRNA transcription and TF protein expression. Indeed, the original study that showed global CpG hypermethylation in mutant IDH1/2 GBMs included F3 among the top 50 hypermethylated and downregulated genes in that subset of GBMs [36]. Many cancers release significant amounts of biologically active TF into the systemic circulation in the form of membrane-bound vesicles, called TF-containing microparticles (TF-MPs), which have been repeatedly linked to cancer-induced VTE [3, 7, 10, 32, 43–45, 55]. Such a connection has been equivocal in gliomas, as some studies have shown elevated circulating TF-MPs in glioma patients with VTE [44, 45], but not others [52–54]. Our data supports a role for TF-MPs in glioma-induced VTE (Fig. 3d; Table 2). Differences in methods and sampling may account for these conflicting reports and, as has been previously observed, makes comparisons between studies difficult [19]. In contrast to prior work, we analyzed TF-MP activity in peripheral arterial plasma rather than venous plasma. Other groups often included patients with recurrent and treated tumors, whereas we only studied preoperative blood obtained from new, untreated glioma patients. Our data therefore suggest that a relative lack of TF-MP production and activity could contribute not only to the absence of microthrombi within a mutant IDH1/2 tumor (Figs. 1, 2, 3), but also to the absence of distal VTE (Fig. 3d; Tables 1, 2).

Postulated mechanism of mutant IDH1/2 suppression of local and systemic thrombosis. a Wild-type IDH1/2 glioma cells produce tissue factor-containing microparticles (TF-MPs, blue dots), which promote microthrombus formation within the tumor (yellow and brown objects = platelets). TF-MPs from the glioma also circulate throughout the body, increasing the risk of DVT and PE. b In contrast, mutant IDH1/2 gliomas make very little TF, and consequently release very little TF-MPs, but do produce D-2-HG (pink triangles). In the local tumor vascular bed, where permeability increases in higher-grade tumors (arrows), the reduced numbers of TF-MPs, plus the antiplatelet activity of D-2-HG, combine to prevent intratumoral thrombosis. In the systemic circulation, D-2-HG is not present at sufficient concentrations to inhibit peripheral platelet aggregation, but the lack of glioma-derived TF-MPs still results in low risk for VTE
The second potential mechanism is a novel, rapid, calcium-dependent inhibition of human platelet aggregation and clotting by D-2-HG (Fig. 4). Homogenates of mutant IDH1/2 gliomas contain up to 30 mM D-2-HG [13], and cells expressing mutant IDH1 release D-2-HG into culture medium [13, 18]. A precise estimate of the concentration of D-2-HG in the immediate tumor microenvironment is difficult to calculate, but concentrations exceeding 100 µM D-2-HG have recently been reported in the circulating cerebrospinal fluid of mutant IDH1/2 glioma patients [23], meaning that the levels of D-2-HG within the immediate tumor microenvironment may frequently be in the millimolar range. Glioma xenografts expressing R132H IDH1 showed severely reduced coagulation following tail snip of host mice, thereby suggesting that an intact tumor can release sufficient D-2-HG to affect clotting. Of note, in the model shown in Fig. 4, R132H IDH1 did not alter TF expression in U87MG cells (not shown) or significantly alter circulating TF-MP activity in the periphery (Supplemental Figure 11). R132H IDH1 typically requires sustained expression (>30 cell passages) to induce DNA hypermethylation [57]. This is much longer than what was done for the current model (<10 passages), which was specifically designed to study the effect of R132H IDH1 on in vivo platelet activity, not TF expression and thrombosis. In higher-grade gliomas, where the blood–brain barrier breaks down and is most conducive to thrombosis, D-2-HG could enter the blood and exert a local suppressive effect on platelet aggregation within the tumor vascular bed (Fig. 5). However, mutant IDH1/2 gliomas only achieve low micromolar concentrations of D-2-HG in the serum of patients [8], so platelet activity and coagulation are unlikely to be significantly affected in the periphery. To our knowledge, no data exists on the level of D-2-HG that is reached in the interstitial and intravascular compartments within a mutant IDH1/2 glioma, though it is likely to be much higher than that observed in the peripheral circulation. D-2-HG could also exert paracrine effects on other nonneoplastic cells in and around the tumor, including another hallmark of GBM, endothelial cell proliferation (Supplemental Figure 12).
The antiplatelet activity of D-2-HG cannot be due to methylation, because the effect is too rapid, and because platelets lack DNA. The most well-studied mechanism of D-2-HG activity is competitive inhibition of demethylase dioxygenases that require alpha-ketoglutarate as a cofactor [20]. But in platelets the only alpha-ketoglutarate -dependent dioxygenases are procollagen-lysine, 2-oxoglutarate 5-dioxygenases 1 and 3 [6]. These enzymes crosslink collagen chains to each other and are involved in neither aggregation nor methylation. 12/15 lipoxygenase is involved in platelet aggregation, but does not depend on alpha-ketoglutarate. Our data therefore suggest an entirely new mechanism of action by D-2-HG, and its perturbation of calcium signaling could affect numerous pathways and activities in and around mutant IDH1/2 glioma cells.
Our discovery that mutant IDH1/2 and D-2-HG have anticoagulant properties is consistent with prior studies. First, well before the discovery of IDH1/2 mutations, Kondziolka et al. reported that intratumoral hemorrhage was significantly more frequent in grade II-III gliomas (18/135 cases, 13.3 %) than GBMs (17/264, 6.4 %) (P = 0.025 via Fisher’s exact test) [25]. Furthermore, they observed the highest levels of intratumoral hemorrhages in oligodendrogliomas, which are now known to be almost invariably driven by IDH1/2 mutations. Second, brain bleeding can occur in patients with the rare metabolic disorder D-2-HG aciduria [26, 59]. Third, mice constitutively expressing mutant IDH1 in the brain die of perinatal intracerebral hemorrhage [46].
In summary, our study has new implications for several aspects of glioma research and clinical practice, including understanding the histologic and biologic differences between mutant IDH1/2 and wild-type IDH1/2 gliomas, demonstrating a novel methylation-independent mechanism of action by mutant IDH1/2, and more effectively identifying glioma patients at low risk of VTE. This creates many new opportunities to advance our understanding of the relationships between glioma mutations and tumor biology, and to ultimately improve personalized medicine in glioma patients.
References
Ades S, Kumar S, Alam M, Goodwin A, Weckstein D, Dugan M, Ashikaga T, Evans M, Verschraegen C, Holmes CE (2015) Tumor oncogene (KRAS) status and risk of venous thrombosis in patients with metastatic colorectal cancer. J Thromb Haemost 13:998–1003. doi:10.1111/jth.12910
Anand M, Brat DJ (2012) Oncogenic regulation of tissue factor and thrombosis in cancer. Thromb Res 129(Suppl 1):S46–S49. doi:10.1016/s0049-3848(12)70015-4
Bastida E, Ordinas A, Escolar G, Jamieson GA (1984) Tissue factor in microvesicles shed from U87MG human glioblastoma cells induces coagulation, platelet aggregation, and thrombogenesis. Blood 64:177–184
Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, Morozova O et al (2015) Comprehensive, Integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 372:2481–2498. doi:10.1056/NEJMoa1402121
Budczies J, Klauschen F, Sinn BV, Gyorffy B, Schmitt WD, Darb-Esfahani S, Denkert C (2012) Cutoff finder: a comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS One 7:e51862. doi:10.1371/journal.pone.0051862
Burkhart JM, Vaudel M, Gambaryan S, Radau S, Walter U, Martens L, Geiger J, Sickmann A, Zahedi RP (2012) The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 120:e73–e82. doi:10.1182/blood-2012-04-416594
Butenas S, Bouchard BA, Brummel-Ziedins KE, Parhami-Seren B, Mann KG (2005) Tissue factor activity in whole blood. Blood 105:2764–2770. doi:10.1182/blood-2004-09-3567
Capper D, Simon M, Langhans CD, Okun JG, Tonn JC, Weller M, von Deimling A, Hartmann C, German Glioma N (2011) 2-Hydroxyglutarate concentration in serum from patients with gliomas does not correlate with IDH1/2 mutation status or tumor size. Int J Cancer 131:766–768. doi:10.1002/ijc.26425
Chaichana KL, Pendleton C, Jackson C, Martinez-Gutierrez JC, Diaz-Stransky A, Aguayo J, Olivi A, Weingart J, Gallia G, Lim M et al (2013) Deep venous thrombosis and pulmonary embolisms in adult patients undergoing craniotomy for brain tumors. Neurol Res 35:206–211. doi:10.1179/1743132812y.0000000126
Chou J, Mackman N, Merrill-Skoloff G, Pedersen B, Furie BC, Furie B (2004) Hematopoietic cell-derived microparticle tissue factor contributes to fibrin formation during thrombus propagation. Blood 104:3190–3197. doi:10.1182/blood-2004-03-0935
Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M et al (2011) The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 12:463–469. doi:10.1038/embor.2011.43
Corrales-Rodriguez L, Soulieres D, Weng X, Tehfe M, Florescu M, Blais N (2014) Mutations in NSCLC and their link with lung cancer-associated thrombosis: a case-control study. Thromb Res 133:48–51. doi:10.1016/j.thromres.2013.10.042
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC et al (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462:739–744
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF et al (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18:553–567. doi:10.1016/j.ccr.2010.11.015
Food and Drug Administration Oncologic Drug Advisory Committee Wednesday, June 20, 2012 morning session http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM314708.pdf. Accessed July 10, 2015
Garnier D, Milsom C, Magnus N, Meehan B, Weitz J, Yu J, Rak J (2010) Role of the tissue factor pathway in the biology of tumor initiating cells. Thromb Res 125(Suppl 2):S44–S50. doi:10.1016/s0049-3848(10)70012-8
Gerber NK, Goenka A, Turcan S, Reyngold M, Makarov V, Kannan K, Beal K, Omuro A, Yamada Y, Gutin P et al (2014) Transcriptional diversity of long-term glioblastoma survivors. Neuro Oncol 16:1186–1195. doi:10.1093/neuonc/nou043
Gilbert MR, Liu Y, Neltner J, Pu H, Morris A, Sunkara M, Pittman T, Kyprianou N, Horbinski C (2014) Autophagy and oxidative stress in gliomas with IDH1 mutations. Acta Neuropathol 127:221–233. doi:10.1007/s00401-013-1194-6
Hisada Y, Alexander W, Kasthuri R, Voorhees P, Mobarrez F, Taylor A, McNamara C, Wallen H, Witkowski M, Key NS et al (2016) Measurement of microparticle tissue factor activity in clinical samples: a summary of two tissue factor-dependent FXa generation assays. Thromb Res 139:90–97. doi:10.1016/j.thromres.2016.01.011
Horbinski C (2013) What do we know about IDH1/2 mutations so far, and how do we use it? Acta Neuropathol 125:621–636. doi:10.1007/s00401-013-1106-9
Hu J, Yan J, Rao G, Latha K, Overwijk WW, Heimberger AB, Li S (2014) The duality of fgl2—secreted immune checkpoint regulator versus membrane-associated procoagulant: therapeutic potential and implications. Int Rev Immunol 2014:26
Isaka T, Yoshimine T, Maruno M, Kuroda R, Ishii H, Hayakawa T (1994) Altered expression of antithrombotic molecules in human glioma vessels. Acta Neuropathol 87:81–85
Kalinina J, Ahn J, Devi NS, Wang L, Li W, Olson JJ, Glantz M, Smith T, Kim EL, Giese A et al. (2016) Selective detection of the D-enantiomer of 2-Hydroxyglutarate in the CSF of glioma patients with mutated isocitrate dehydrogenase. Clin Cancer Res (In Press)
Kohl M, Plischke M, Leffondre K, Heinze G (2015) PSHREG: a SAS macro for proportional and nonproportional subdistribution hazards regression. Comput Methods Programs Biomed 118:218–233. doi:10.1016/j.cmpb.2014.11.009
Kondziolka D, Bernstein M, Resch L, Tator CH, Fleming JF, Vanderlinden RG, Schutz H (1987) Significance of hemorrhage into brain tumors: clinicopathological study. J Neurosurg 67:852–857. doi:10.3171/jns.1987.67.6.0852
Kwong KL, Mak T, Fong CM, Poon KH, Wong SN, So KT (2002) D-2-hydroxyglutaric aciduria and subdural haemorrhage. Acta Paediatr 91:716–718
Lai A, Kharbanda S, Pope WB, Tran A, Solis OE, Peale F, Forrest WF, Pujara K, Carrillo JA, Pandita A et al (2011) Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol 29:4482–4490
Liu Y, Gilbert MR, Kyprianou N, Rangnekar VM, Horbinski C (2014) The tumor suppressor prostate apoptosis response-4 (Par-4) is regulated by mutant IDH1 and kills glioma stem cells. Acta Neuropathol 128:723–732. doi:10.1007/s00401-014-1334-7
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (2007) WHO Classification of Tumors of the Central Nervous System. In: Ohgaki H (ed) World Health Organization Classification of Tumors 4th edn. IARC, City
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A et al (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483:474–478. doi:10.1038/nature10860
Lyman GH, Bohlke K, Khorana AA, Kuderer NM, Lee AY, Arcelus JI, Balaban EP, Clarke JM, Flowers CR, Francis CW et al (2015) Venous thromboembolism prophylaxis and treatment in patients with cancer: american society of clinical oncology clinical practice guideline update 2014. J Clin Oncol 33:654–656. doi:10.1200/jco.2014.59.7351
Milsom C, Yu J, May L, Meehan B, Magnus N, Al-Nedawi K, Luyendyk J, Weitz J, Klement P, Broze G et al (2007) The role of tumor-and host-related tissue factor pools in oncogene-driven tumor progression. Thromb Res 120(Suppl 2):S82–S91. doi:10.1016/s0049-3848(07)70135-4
Nadir Y, Vlodavsky I, Brenner B (2008) Heparanase, tissue factor, and cancer. Semin Thromb Hemost 34:187–194
National Comprehensive Cancer Network Cancer-Associated Venous Thromboembolic Disease (version 2.2014) http://www.nccn.org/professionals/physician_gls/f_guidelines.asp. Accessed Aug 2 2015
Nobusawa S, Watanabe T, Kleihues P, Ohgaki H (2009) IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res 15:6002–6007. doi:10.1158/1078-0432.CCR-09-0715
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP et al (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522
Perry JR (2012) Thromboembolic disease in patients with high-grade glioma. Neuro Oncol 14(Suppl 4): iv73-80.doi:10.1093/neuonc/nos197
Prayson NF, Koch P, Angelov L, Prayson RA (2011) Microscopic thrombi in anaplastic astrocytoma predict worse survival? Ann Diagn Pathol 15:389–393. doi:10.1016/j.anndiagpath.2011.05.002
Reuss DE, Sahm F, Schrimpf D, Wiestler B, Capper D, Koelsche C, Schweizer L, Korshunov A, Jones DT, Hovestadt V et al (2015) ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an “integrated” diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol 129:133–146. doi:10.1007/s00401-014-1370-3
Ringertz N (1950) Grading of gliomas. Acta Pathol Microbiol Scand 27:51–64
Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E et al (2013) An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340:626–630
Rong Y, Durden DL, Van Meir EG, Brat DJ (2006) ‘Pseudopalisading’ necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol 65:529–539
Rong Y, Post DE, Pieper RO, Durden DL, Van Meir EG, Brat DJ (2005) PTEN and hypoxia regulate tissue factor expression and plasma coagulation by glioblastoma. Cancer Res 65:1406–1413. doi:10.1158/0008-5472.can-04-3376
Sartori MT, Della Puppa A, Ballin A, Campello E, Radu CM, Saggiorato G, d’Avella D, Scienza R, Cella G, Simioni P (2013) Circulating microparticles of glial origin and tissue factor bearing in high-grade glioma: a potential prothrombotic role. Thromb Haemost 110:378–385. doi:10.1160/th12-12-0957
Sartori MT, Della Puppa A, Ballin A, Saggiorato G, Bernardi D, Padoan A, Scienza R, d’Avella D, Cella G (2011) Prothrombotic state in glioblastoma multiforme: an evaluation of the procoagulant activity of circulating microparticles. J Neurooncol 104:225–231
Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio II, Cairns RA, McCracken S, Wakeham A, Haight J et al (2012) D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev 26:2038–2049. doi:10.1101/gad.198200.112
Sciacca FL, Ciusani E, Silvani A, Corsini E, Frigerio S, Pogliani S, Parati E, Croci D, Boiardi A, Salmaggi A (2004) Genetic and plasma markers of venous thromboembolism in patients with high grade glioma. Clin Cancer Res 10:1312–1317
Smith TR, Lall RR, Graham RB, McClendon J Jr, Lall RR, Nanney AD, Adel JG, Zakarija A, Chandler JP (2014) Venous thromboembolism in high grade glioma among surgical patients: results from a single center over a 10 year period. J Neurooncol 120:347–352. doi:10.1007/s11060-014-1557-4
Sorensen HT, Mellemkjaer L, Olsen JH, Baron JA (2000) Prognosis of cancers associated with venous thromboembolism. N Engl J Med 343:1846–1850. doi:10.1056/nejm200012213432504
Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437. doi:10.1016/j.ccr.2012.08.024
Tehrani M, Friedman TM, Olson JJ, Brat DJ (2008) Intravascular thrombosis in central nervous system malignancies: a potential role in astrocytoma progression to glioblastoma. Brain Pathol 18:164–171. doi:10.1111/j.1750-3639.2007.00108.x
Thaler J, Ay C, Kaider A, Reitter EM, Haselbock J, Mannhalter C, Zielinski C, Marosi C, Pabinger I (2014) Biomarkers predictive of venous thromboembolism in patients with newly diagnosed high-grade gliomas. Neuro Oncol 16:1645–1651. doi:10.1093/neuonc/nou106
Thaler J, Ay C, Mackman N, Bertina RM, Kaider A, Marosi C, Key NS, Barcel DA, Scheithauer W, Kornek G et al (2012) Microparticle-associated tissue factor activity, venous thromboembolism and mortality in pancreatic, gastric, colorectal and brain cancer patients. J Thromb Haemost 10:1363–1370. doi:10.1111/j.1538-7836.2012.04754.x
Thaler J, Preusser M, Ay C, Kaider A, Marosi C, Zielinski C, Pabinger I, Hainfellner JA (2013) Intratumoral tissue factor expression and risk of venous thromboembolism in brain tumor patients. Thromb Res 131:162–165. doi:10.1016/j.thromres.2012.09.020
Thomas GM, Panicot-Dubois L, Lacroix R, Dignat-George F, Lombardo D, Dubois C (2009) Cancer cell-derived microparticles bearing P-selectin glycoprotein ligand 1 accelerate thrombus formation in vivo. J Exp Med 206:1913–1927. doi:10.1084/jem.20082297
Timp JF, Braekkan SK, Versteeg HH, Cannegieter SC (2013) Epidemiology of cancer-associated venous thrombosis. Blood 122:1712–1723. doi:10.1182/blood-2013-04-460121
Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS et al. (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483(7390):479–483
Wang P, Dong Q, Zhang C, Kuan PF, Liu Y, Jeck WR, Andersen JB, Jiang W, Savich GL, Tan TX et al. (2013) Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 32(25):3091–3100
Wang X, Jakobs C, Bawle EV (2003) D-2-Hydroxyglutaric aciduria with absence of corpus callosum and neonatal intracranial haemorrhage. J Inherit Metab Dis 26:92–94
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT, Liu LX et al (2010) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19:17–30
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773
Zerrouqi A, Pyrzynska B, Brat DJ, Van Meir EG (2014) P14ARF suppresses tumor-induced thrombosis by regulating the tissue factor pathway. Cancer Res 74:1371–1378
Zwicker JI, Liebman HA, Bauer KA, Caughey T, Campigotto F, Rosovsky R, Mantha S, Kessler CM, Eneman J, Raghavan V et al (2013) Prediction and prevention of thromboembolic events with enoxaparin in cancer patients with elevated tissue factor-bearing microparticles: a randomized-controlled phase II trial (the Microtec study). Br J Haematol 160:530–537. doi:10.1111/bjh.12163
Acknowledgments
CH was supported by the National Cancer Institute (K08CA155764). The authors thank Michael Gallagher for Fig. 5, Bernice Slone from the Kentucky Cancer Registry for outcome data, Dana Napier for histotechnical support, Sarah Langford for assistance with ionized calcium measurements, Dr. Sidney Whiteheart for advice on tail vein bleeding assays, Dr. Andrew Lane for the NMR experiment and manuscript comments, Kathleen McCortney for collecting the blood samples used in Fig. 3d and Table 2, and Dr. C. David James for manuscript comments. The University of Kentucky Biospecimen and Tissue Procurement, Biostatistics, and Redox Metabolism Shared Resource Facilities are supported by the Markey Cancer Center (P30CA177558). The Northwestern Nervous System Tumor Bank is supported by the Department of Neurological Surgery. Methylation profiling of brain tumors at NYU is supported by The Friedberg Charitable Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
D. Unruh and S. R. Schwarze contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Unruh, D., Schwarze, S.R., Khoury, L. et al. Mutant IDH1 and thrombosis in gliomas. Acta Neuropathol 132, 917–930 (2016). https://doi.org/10.1007/s00401-016-1620-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-016-1620-7