
Abstract
IDH1 mutations in gliomas associate with longer survival. Prooxidant and antiproliferative effects of IDH1 mutations and its d-2-hydroxyglutarate (2-HG) product have been described in vitro, but inconsistently observed. It is also unclear whether overexpression of mutant IDH1 in wild-type cells accurately phenocopies the effects of endogenous IDH1-mutations on tumor apoptosis and autophagy. Herein we investigated the effects of 2-HG and mutant IDH1 overexpression on proliferation, apoptosis, oxidative stress, and autophagy in IDH1 wild-type glioma cells, and compared those results with patient-derived tumors. 2-HG reduced viability and proliferation of U87MG and LN18 cells, triggered apoptosis in LN18 cells, and autophagy in U87MG cells. In vitro studies and flank xenografts of U87MG cells overexpressing R132H IDH1 exhibited increased oxidative stress, including increases of both manganese superoxide dismutase (MnSOD) and p62. Patient-derived IDH1-mutant tumors showed no significant differences in apoptosis or autophagy, but showed p62 accumulation and actually trended toward reduced MnSOD expression. These data indicate that mutant IDH1 and 2-HG can induce oxidative stress, autophagy, and apoptosis, but these effects vary greatly according to cell type.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mutations in isocitrate dehydrogenase 1 and 2 (IDH1/2) are present in the majority of grades II and III astrocytomas and oligodendrogliomas, and in the 10–15 % of glioblastomas (GBMs) that develop from lower grade tumors (reviewed in [10]). The mutation is also present in a large proportion of chondroid tumors, cholangiocarcinomas, and some hematopoietic neoplasms. Gliomas with these mutations tend to be far less aggressive than their grade-matched wild-type counterparts, to the point where a grade IV GBM with an IDH1/2 mutation may actually be more favorable than a wild-type grade III astrocytoma. With rare exceptions, IDH1/2 mutations are specific to codons 132 and 172 of IDH1 and IDH2, respectively, with R132H IDH1 being the most common mutation by far. These codons normally encode arginine residues comprising the isocitrate substrate binding pocket, but when mutated, impart new enzyme kinetics. Instead of oxidizing isocitrate to alpha-ketoglutarate (α-KG), the neoenzyme reduces α-KG to d-2-hydroxyglutarate (hereafter referred to as 2-HG)—a compound whose concentration is increased 10- to 100-fold in mutant tumors [7].
The actions of mutant IDH1/2 and its 2-HG oncometabolite have therefore been the subject of intense recent investigation. Because of its structural similarity to α-KG, 2-HG can competitively inhibit any enzyme that requires α-KG as a cofactor, including Jumonji C-domain containing histone demethylases and TET DNA demethylases [10]. Consequently, IDH1/2 mutations are strongly associated with global histone and DNA hypermethylation in human tumors (though the association is less clear in transgenic mouse models). Independent of their role in tumorigenesis, however, the reasons for these mutations being relatively favorable prognostic markers are unknown.
Prior to its discovery in cancer, 2-HG was known to be elevated in d-2-hydroxyglutaric aciduria, which is a very rare germline metabolic disorder characterized by seizures, developmental delays, and dysmorphisms. Research modeling this disease showed that 2-HG induces oxidative stress (OS) in rat brain cortical slides [19]. Wild-type IDH1 and IDH2 help maintain intracellular redox status, as their oxidation of isocitrate also produces NADPH, an important reducing equivalent in the cell [1]. In contrast, mutant IDH1 consumes NADPH during catalysis [7]. Together these findings suggest that, in addition to hypermethylation, mutant IDH1 and 2-HG might promote OS and autophagy or apoptosis, which could contribute to the less aggressive behavior of this glioma subset. However, neither α-KG nor NADPH levels appear to significantly differ in mutant versus wild-type gliomas [7, 14]; to date, characterization of OS, autophagic, and apoptotic activity in IDH1-mutant gliomas have not been done. Furthermore, while recent studies have shown that overexpression of R132H IDH1 predisposes U87MG and LN229 glioma cells, which are normally wild type for IDH1, to oxidative stress [22, 27], it is unclear whether such cells might respond differently to induced overexpression of the mutation or abrupt challenge with 2-HG, as compared to when a single mutant allele arises spontaneously in a tumor progenitor cell and persists throughout that tumor’s growth. Likewise, it is not known whether responses to 2-HG and mutant IDH1 might vary between glioma cell lines.
Herein we study the effect of R132H IDH1 and its 2-HG product on oxidative stress, autophagy, and apoptosis in wild-type glioma cells versus those that evolve with a spontaneous endogenous mutation. Our data indicate that overexpression of mutant IDH1 strongly induce markers of oxidative stress and autophagy in wild-type cells, but not in patient-derived tumors. This suggests that overexpression may not model all facets of IDH1-mutant tumors accurately, and/or mutant gliomas may evolve ways of compensating for the deleterious side effects of the mutation.
Methods
Cell culture and reagents
d-2-Hydroxypentanedioic acid disodium (d-2-hydroxyglutaric acid disodium salt, d-2-HG) was acquired from PepTech Corporation (Burlington, MA) and Sigma-Aldrich (St. Louis, MO). U87MG and LN18 glioma cell lines were purchased from ATCC (Manassas, VA). All cell lines were cultured in RPMI 1640, 10 % fetal bovine serum, and 1 % penicillin streptomycin purchased from Life Technologies (Carlsbad, CA). Cells were cultured in humidified conditions at 37 °C and 5 % CO2.
To establish primary glioma cultures, resected tumor tissues from two primary GBMs (designated as 2169 and 10932) with epidermal growth factor receptor (EGFR) amplification were transported to the lab in saline and dissociated using the Papain Dissociation System Kit (Worthington Biochemical Corporation, Lakewood, NJ). Briefly, cells were cultured overnight on GelTrex LDEV-Free reduced growth factor basement membrane matrix coated plates (Life Technologies, Carlsbad, CA) in medium consisting of neurobasal A medium without phenol red (Life Technologies) containing B27 supplement without vitamin A, 20 ng/mL recombinant EGF, 20 ng/mL recombinant FGF, 1 mM sodium pyruvate, 2 mM l-glutamine, and 100 u/mL of penicillin streptomycin. After culturing overnight, red blood cells were lysed, and cells were sub-cultured on GelTrex coated plates in the full medium described above, incubated at 37 °C with 5 % CO2. To passage cells, TrypLE (Life Technologies) was used to dissociate cells from the plate.
Vector, wild type, and R132H mutant IDH1 stable U87MG cell lines were generated by transfecting U87MG cells with plasmids generously gifted by Dr. Hai Yan. Colonies were selected via G418 (Sigma-Aldrich, St. Louis, MO), and single colonies were passaged and isolated from remaining colonies. Thereafter cells were cultured in regular medium (RPMI, 10 % FBS, 1 % penicillin streptomycin). U87MG cells were transiently transfected with GFP-light chain 3 (LC3), plasmid courtesy of Dr. Haining Zhu.
(3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTS) cell viability assay
Cells were seeded into a 96-well plate (12,000 cells/mL). 24 h later, cells were treated with the specified concentration of 2-HG. Six days later, the viability assay was conducted using the CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI), according to manufacturer’s instructions. Cells were incubated with the MTS solution for 1 h at 37 °C before measurements.
Flow cytometry
For cell cycle analysis, cells were seeded at 60,000 cells/mL. 24 h later, cells were treated with 2-HG at designated concentration. Six days after treatment with 2-HG, cells were incubated with 20 μM bromodeoxyuridine (BrdU) for 45 min and then trypsinized briefly to collect. Cells were fixed using 95 % ethanol. Nuclei were isolated from samples and incubated overnight at 4 °C with a BrdU-AlexaFluor 488 conjugated antibody (Invitrogen, Carlsbad, CA). Nuclei were washed and stained with propidium iodide for 30 min and then analyzed with FACS Calibur (Becton-Dickinson, San Jose, CA) and Cell Quest Software. For TUNEL analysis, cells were seeded at 60,000 cells/mL and treated with 2-HG 24 h later. Six days after treatment with 2-HG cells were collected with brief trypsinization. Then cells were cross-linked with 1 % (w/v) paraformaldehyde on ice for 15 min and then fixed with 95 % ethanol overnight. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was conducted using terminal transferase from New England Biolabs (Ipswich, MA) according to manufacturers’ instructions, and cells were incubated at 37 °C for 1 h. Cells were permeabilized for 30 s with 0.1 % TritonX-100 and 5 mg/mL BSA in PBS. Samples were incubated overnight at 4 °C with BrdU-Alexa Fluor 488 antibody then stained with propidium iodide and analyzed as above.
Caspase activity assays
For all caspase activity assays, cells were seeded at a density of 60,000 cells/mL. Cells were treated with 2-HG 24 h after cells were seeded. Six days after 2-HG treatment, Caspase 3/7, 8, and 9 activities were measured using the Caspase-Glo 3/7, Caspase 8, or Caspase 9 Assays from Promega (Madison, WI) according to the manufacturer’s instructions. Samples were incubated at room temperature in the dark for 1 h before measuring luminescence with a Molecular Devices SpectraMax2 plate reader (Sunnyvale, CA). MTS assays were conducted simultaneously with all caspase activity assays, and caspase activity data were normalized to MTS measurements to control for cell number between conditions.
Western immunoblotting
Protein expression was analyzed by seeding cells (60,000 cells/mL) and treating with 2-HG 24 h later. At the designated time, cells were harvested, centrifuged, and snap frozen. Cell pellets were lysed with tissue protein extraction reagent (T-PER, ThermoScientific, Rockford, IL) and protease inhibitor cocktail (Sigma-Aldrich). SDS-PAGE was conducted and protein was transferred to PVDF membrane. Antibodies for alpha-tubulin, beta-actin, LC3, wild-type IDH1, and BID were purchased from Cell Signaling (Danvers, MA). MnSOD antibody was purchased from Millipore (Billerica, MA). P62 antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). R132H IDH1 antibody was purchased from Dianova GmbH (Hamburg, Germany). Blots were visualized using Amersham ECL Western Blotting Detection Reagents (GE Healthcare, Pittsburgh, PA) and films were processed using a Kodak X-OMAT 2000A Processor (Rochester, NY). Patient-derived tumor samples and xenografts were weighed and homogenized in ice cold extraction buffer (100 mg/mL, T-PER). The concentration of total protein was measured using a protein assay kit (Bio-Rad Laboratories, Inc., Hercules, CA). For each sample, 5 μg of total protein was loaded per lane. Bands were quantified using Image Quant 5.2 software.
Slot blot analysis
Stably transfected cells were passaged and 72 h later were collected in PBS by scraping on ice, centrifuged gently, and snap frozen until further processing. All samples were lysed, protein concentration was determined, and slot blot analysis was conducted measuring levels of protein carbonylation, 4-hydroxynonenal, and 3-nitrotyrosine as previously reported [31, 35].
Xenografts
Six to eight-week-old female NOD.Cg-Prkdc scid Il2rg tm1Wjl/SzJ mice (Jackson Laboratory, Bar Harbor, ME) were randomized to inoculate U87MG cells expressing either GFP vector or GFP-R132H IDH1. Cells (5 × 106/100 μL) were injected subcutaneously in the right flank of individual mice. Tumors were dissected on ice and weighed when harvested 4 weeks post-injection of cells.
Patient-derived gliomas
Formalin-fixed, paraffin-embedded (FFPE) gliomas were retrieved from pathology archives. Deidentified tissue microarrays (TMAs) were constructed from the gliomas. Three 2-mm-diameter cores per tumor were obtained, with each core embedded in a separate TMA block. A total of 104 cases comprised the TMAs, including 9 nonneoplastic controls (cortical dysplasias), 9 grade II astrocytomas, 11 grade III astrocytomas, 12 anaplastic oligodendrogliomas, 16 grade II oligodendrogliomas, and 47 grade IV glioblastomas (GBMs).
Deidentified fresh glioma specimens were prospectively banked via snap-freezing in liquid nitrogen at the time of surgery. Each case was annotated with the corresponding pathologic diagnosis and IDH1/2 mutation status.
Immunohistochemistry and histochemistry
Immunohistochemistry of xenografts and TMAs was performed on 4-μm-thick sections heated at 60 °C for at least 1 h. Deparaffinization, hydration and heat-induced epitope retrieval for slides stained with R132H IDH1 (1:20, Dianova, Germany), p53 (Dako, Carpinteria, CA) or Ki-67 (Dako) was performed in a Dako PT Link pre-treatment module with EnVision FLEX Target Retrieval Solution (high pH). Staining was carried out with Dako’s Envision Flex reagents on a Dako autostainer. Staining for LC3, Atg7, p62 and MnSOD was carried out manually and antigen retrieval was performed with a Biocare Medical Decloaking Chamber using high (LC3) or low pH antigen retrieval buffer from Dako. Primary antibodies were incubated for 1 h at room temperature—LC3 at 1:100 dilution (Nanotools USA, San Diego, CA); Atg7 at 1:75 dilution (Santa Cruz, Dallas, TX, sc-33211); p62 at 1:1,500 dilution (Enzo, Farmingdale, NY, BML-PW98600). MnSOD (Enzo, ADI-SOD-111) was incubated at 4 °C overnight at 1:200. Secondary antibodies used were EnVision labeled polymer-HRP (horseradish peroxidase) anti-mouse or anti-rabbit as appropriate. Staining was visualized using 3, 3′-diaminobenzidine (DAB) chromogen (Dako, CA).
For terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining, paraffin sections were deparaffinized and rehydrated, followed by TUNEL labeling according to the assay kit instructions (Millipore, Temecula, CA).
Two quantification methods were used. For LC3 and MnSOD, each TMA core was semiquantified on a relative scale from 0 to 4, with 0 = negative and 4 = strongest. (For Atg7, the scale was 0–3.) Results from all three cores were averaged together to produce a final score for a tumor. Nuclear expression of p53, ubiquitin, and Ki67/MIB-1, as well as TUNEL histochemistry, were quantified via digital imaging with an Aperio ScanScope XT whole slide scanner, followed by analysis with Aperio Spectrum Version 11.2.0.780 software (Aperio, Vista, CA). In xenografts, LC3 scoring was done manually via light microscopy, counting the number of cells per 400× field that were strongly positive for LC3. Mitoses were scored similarly, using 600× fields. 10–50 consecutive fields were scored for wild-type tumors, depending on tumor size.
Of note, 5 TMA cases were immunonegative for R132H IDH1 but were still suspected of having a less common IDH1 or IDH2 mutation based on mutation-associated variables like WHO grade and younger patient age. Of those five, a single anaplastic astrocytoma turned out to have R132S IDH1 via pyrosequencing.
Liquid chromatography–mass spectroscopy (LC–MS)
Cells were treated with exogenous 2-HG and, at various timepoints, quickly washed × 1 with PBS, scraped off, pelleted, and resuspended in 700 μL of 80 % methanol. 2-HG levels were measured as DATAN derivative using a Shimadzu UFLC coupled with an AB Sciex 4000-Qtrap hybrid linear ion trap triple quadrupole mass spectrometer in multiple reaction monitoring (MRM) mode [34]. 2-HG was analyzed using an Xterra MS C18, 3 × 100 mm, 3.5 μm column (from Waters). The mobile phase consisted of 125 mg/L ammonium formate (pH 3.48) as solvent A and acetonitrile as solvent B. d-2-HG and L-2-HG isomers were separated with a gradient elution from 2 % solvent B to 3.5 % solvent B in 3.5 min and maintaining at 3.5 min for the next 11.5 min. The column was equilibrated back to the initial conditions in 3 min. The flow rate was 0.5 mL/min with a column temperature of 30 °C. The mass spectrometer was operated in the negative electrospray ionization mode with optimal ion source settings determined by a synthetic standard of 2-HG with a declustering potential of −40 V, entrance potential of −10 V, collision energy of −16 V, collision cell exit potential of −11 V, curtain gas of 20 psi, ion spray voltage of −4,200 V, ion source gas1/gas2 of 40 psi and temperature of 550 °C. MRM transitions monitored were as follows: 362.9/146.9; 362.9/129 and 362.9/101.1.
Statistical analyses
Differences between groups were analyzed via Student’s t test or one-way ANOVA with post hoc Tukey’s test, where appropriate. Significance was reached when P < 0.05. All analyses were done via GraphPad software (La Jolla, CA).
Institutional approval
Institutional Animal Care and Use Committee (IACUC) approval was obtained prior to all animal experiments. Institutional Research Board (IRB) approval was obtained prior to collecting the archival tissues for TMA construction. For snap-frozen banked tissues, informed consent was obtained from each patient prior to surgery in a manner approved by the IRB.
Results
2-HG exposure and overexpression of mutant IDH1 decrease viability and proliferation of wild-type glioma cells
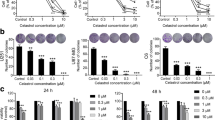
To determine whether unmodified 2-HG can enter glioma cells, both U87MG and LN18 cells were incubated with 30 mM 2-HG, a concentration within the range observed in patient-derived tumors [7]. By LC–MS, cell-associated 2-HG levels increased greatly 30 min after the pulse and, surprisingly, remained elevated through 6 days post-treatment (Fig. 1a). Viability of both U87MG and LN18 cells decreased in response to 2-HG, though such inhibition was modest (up to 10–15 % in both cell lines) and delayed, taking 6 days for an effect to manifest (Fig. 1b–d). Likewise, flank xenografts of U87MG gliomas stably expressing IDH1-R132H exhibited lower tumor mass (Fig. 1e). Regarding the cell cycle, 2-HG inhibited S-phase in both U87MG and LN18 cells (Fig. 2f, g). While U87MG cells showed no other cell cycle changes (Supplemental Figure 1a, 1b), LN18 cells showed a persistent rise in G0G1, but no lasting changes in G2M (Supplemental Figure 1c, 1d). R132H IDH1-expressing U87MG xenografts showed no differences in Mib-1 labeling (Fig. 1h) or mitoses (Supplemental Figure 1e). Similar reduction in viability by 2-HG was observed in cultured 2169 and 10932 primary IDH1 wild-type GBM cells, 6 days after 2-HG treatment (Fig. 1i, j; Supplemental Figure 1f).

2-HG and mutant IDH1 inhibit glioma growth. Both U87MG and LN18 cells were pulsed with 30 mM exogenous unmodified 2-HG in vitro. LC–MS of the cell lysates at various timepoints after treatment showed that 2-HG rapidly accumulated in cells and persisted through 6 days. a Data represent mean ± SEM, n = 3. By phase contrast microscopy, b both U87MG and LN18 cells showed signs of toxicity in response to 2-HG. Images are 200× magnification. Both U87MG (c) and LN18 cells (d) showed similar dose-dependent sensitivity to 2-HG as measured by MTS assay. For each cell type, data are normalized to the vehicle (0 mM) condition and are expressed as mean ± SEM, n = 12. *P < 0.05; **P 0.001. e U87MG cells stably expressing R132H IDH1 were smaller than control tumors. Each dot represents a single xenograft, data are expressed as mean ± SEM, n = 11. In both U87MG (f) and LN18 cells (g), 2-HG suppressed entry into S-phase after 3 days of treatment. Data represent mean ± SEM, n = 5. *P < 0.05; **P < 0.01; ***P < 0.001. However, there was no significant difference in Mib-1 proliferation index between vector and R132H IDH1 xenografts (h), n = 11. Similar to the glioma cell lines, primary cultures of 2169 patient-derived GBM cells (IDH1 wild type) were sensitive to 6 days of 2-HG as observed by phase contrast microscopy (i ×200) and MTS assay (j)

In vitro and in vivo effects of 2-HG and mutant IDH1 on apoptosis. 2-HG suppressed caspase 3/7 activity in U87MG cells (a) while increasing activity in LN18 cells (b). Data are normalized to day 6 control and represent mean ± SEM, n = 3. *P < 0.05; **P < 0.01. While TUNEL flow cytometry revealed no change in U87MG apoptosis compared to untreated (c), LN18 cells showed a 35-fold increase in response to 2-HG after 6 days (d). Data are represented as mean fold change relative to control ± SEM, n = 5. **P < 0.01. Similar to a and b, 6 days of 2-HG inhibited caspase 9 activity in U87MG cells (e) but increased caspase 9 activity in LN18 cells (f). 2 h of 5 μM staurosporine (STS) was the positive control for both cell types. Data represent mean ± SEM, n = 6, and were normalized to control cells. **P < 0.01; ***P < 0.001. g Consistent with in vitro data, U87MG xenografts expressing R132H IDH1 showed no significant changes in apoptosis as measured by TUNEL histochemistry. Each dot represents a single xenograft, data are expressed as mean ± SEM, n = 11. h Similar to U87MG cells, primary cultures of patient-derived GBM cells (IDH1 wild type) suppressed caspase 3/7 activity in response to 6 days of 2-HG. Data represent mean ± SEM, n = 3. **P < 0.01
Apoptotic response to 2-HG is cell-type specific
Given that 2-HG reduced glioma cell viability, we sought to determine whether cells were undergoing apoptosis. Remarkably, U87MG cells showed reduced caspase 3/7 activity in response to 2-HG, with no change in apoptosis as measured by TUNEL labeling (Fig. 2a, c). In contrast, LN18 cells showed a marked increase in caspase 3/7 activity and a 35-fold increase in apoptosis (Fig. 2b, d). Similar changes in caspase activity were seen in both cell lines even when media was changed 3 days after the 2-HG pulse (Supplemental Figure 2a, 2b). Further studies showed that the caspase 9-dependent intrinsic apoptotic pathway was specifically being activated in LN18 cells (Fig. 2f). On the other hand, 2-HG suppressed caspase 9 in U87MG cells (Fig. 2e). Neither cell line showed a significant change in caspase 8-dependent extrinsic pathway activity (Supplemental Figure 2c, 2d). Similarly, TUNEL staining in flank xenografts revealed no change in apoptotic activity between control and R132H IDH1 U87MG tumors (Fig. 2g). Primary GBM cultures acted similar to U87MG cells, showing a marked decrease in caspase 3/7 activity when treated with 2-HG (Fig. 2h).
2-HG and R132H IDH1 induce oxidative stress
2-HG induced apoptosis in LN18 cells but not U87MG cells, yet viability was still reduced in U87MG cells. Because 2-HG and mutant IDH1 have been shown to promote oxidative stress and mitochondrial dysfunction in rat brain slices [18, 19], and the caspase response to 2-HG was similar in U87MG cells and primary cultured GBM cells (Fig. 2a, h), we focused on U87MG cells to study markers of oxidative stress. By 24 h of 2-HG treatment, U87MG cells showed a rapid upregulation of manganese superoxide dismutase (MnSOD), a sensitive marker of mitochondrial oxidative stress (Fig. 3a) [8]. Cells stably expressing R132H IDH1 also showed an increase in another oxidative stress marker, protein carbonylation (Fig. 3b), but no change in 3-nitrotyrosine formation compared to vector controls (Supplemental Figure 3), suggesting that the mutation produces reactive oxygen species (ROS) but not reactive nitrogen species. Similarly, no change in lipid peroxidation was seen as measured by 4-hydroxy-2-nonenal levels (Supplemental Figure 3). In vivo, U87MG cells expressing R132H IDH1 showed a marked upregulation of MnSOD expression, both by western blot (Fig. 3c) and immunohistochemistry (Fig. 3d).

2-HG and R132H IDH1 induce oxidative stress. a U87MG cells upregulate MnSOD expression in response to 24 h of 2-HG treatment. b Slot blot analysis showed an elevation of protein carbonylation in U87MG cells stably expressing R132H IDH1 compared to cells overexpressing vector or wild-type IDH1, 72 h after passaging. Data represent mean ± SEM, n = 3. *P < 0.05. U87MG xenografts expressing R132H IDH1 also showed increased MnSOD, both by western blot (c) and MnSOD immunohistochemistry (d). Those same xenografts consistently showed markedly reduced vacuolation compared to control tumors (e upper panels) and depletion of lipid stores as indicated by Oil Red O staining (e lower panels). d and e are ×400. Each column in c represents a separate xenograft tumor, n = 3 per group
An incidental and unexpected finding was that, while control U87MG xenografts showed abundant large vacuolar structures and a rounder, more epithelioid morphology, tumors expressing R132H IDH1 showed a near complete loss of vacuoles with a more spindly morphology (Fig. 3e, upper panels). Lipid stores were markedly lower in mutant tumors, as measured by Oil Red O staining (Fig. 3e, lower panels).
Autophagic response to 2-HG and mutant IDH1
U87MG cells showed signs of reduced viability and elevated oxidative stress in response to 2-HG or R132H IDH1 (Figs. 1, 3), yet did not undergo apoptosis (Fig. 2). Because prior work has shown that oxidative stress can induce autophagy, and that this may lead to autophagic cell death in other diseases and contexts [4, 16], we explored the effects of 2-HG and R132H IDH1 on autophagy. 2-HG increased autophagosome formation in U87MG cells expressing GFP-LC3 (Fig. 4a, b), which is a dynamic marker of autophagosome formation [16]. Likewise, a 2.3-fold increase in conversion of LC3-I to LC3-II was observed 24 h after treatment of nontransfected U87MG cells with 2-HG (Fig. 4c) as well as in R132H IDH1-expressing U87MG xenografts (Fig. 4d), but not in LN18 cells (Supplemental Figure 4a). Immunohistochemical analysis of the xenografts showed increased LC3 staining in mutant tumors (Fig. 4e, f). However, mutant xenografts also showed marked p62 accumulation (Fig. 4d).

2-HG and R132H IDH1 induce autophagy. a U87MG cells transiently transfected with GFP-LC3 showed increased autophagosome formation after 24 h of 2-HG treatment, quantified in b. Data represent mean ± SEM, n = 60 cells/group, ×600 magnification. Western blot analysis (c) showed increased conversion of LC3-I to LC3-II in U87MG cells treated with 2-HG or rapamycin (positive control) for 24 h. d U87MG xenografts expressing R132H IDH1 also showed increased LC3-I to LC3-II conversion, as well as p62 accumulation. Each column in c represents a separate xenograft tumor, n = 3 per group. e Immunohistochemical staining of LC3 showed increased numbers of R132H IDH1 xenograft cells with LC3 aggregation (×400), quantified in f. Each dot represents a single xenograft, data are expressed as mean ± SEM, n = 11
Analysis of patient-derived gliomas
To investigate whether these observed effects of 2-HG and mutant IDH1 are also present in actual IDH1-mutant tumors, patient-derived snap-frozen gliomas and FFPE TMAs were studied. Western blots from snap-frozen grades III and IV astrocytomas revealed no significant differences in MnSOD or LC3I-II levels between wild type and IDH1 mutant tumors (Fig. 5a), although there was a surprising trend toward reduced MnSOD in IDH1-mutant tumors revealed by densitometry analysis (P = 0.07) that was confirmed in the Cancer Genome Atlas dataset (Supplemental Figure 4b). We observed p62 accumulation in mutant IDH1 tumors (Fig. 5a, b), consistent with our observations in mutant U87MG xenografts (Fig. 4d). Immunohistochemical analysis of glioma TMAs revealed an increase in apoptosis, MnSOD, and LC3 expression with increasing WHO grade (Fig. 5c, e, g), but no significant differences when sorting grades III and IV gliomas according to IDH1 status (Fig. 5d, f, h). Similarly, Mib-1 proliferation index increased according to glioma grade, but not by IDH1 status (Supplemental Figure 4b, 4c). No significant differences existed between grade-matched astrocytic and oligodendroglial tumors (not shown).

Quantification of autophagy, apoptosis, MnSOD, and LC3 in patient-derived IDH1-mutant gliomas. In snap-frozen patient-derived grades III and IV gliomas (a), there was no significant change in LC3-I and LC3-II according to IDH1 mutation status, although p62 was higher in the mutant tumors. Lanes 1 and 2 represent R132S IDH1 mutant tumors, lanes 3–6 are R132H IDH1. Densitometric analysis of a showed p62 accumulation to be significantly increased in the mutant tumors b. Data represent mean ± SEM. TMA analysis of grades II–IV gliomas showed increasing apoptosis (c), MnSOD (e) and LC3 aggregation (g) with increasing WHO grade. However, there were no significant differences among pooled grades III and IV tumors according to IDH1 status in any of those parameters (d, f, h). In all scatterplots, each data point represents the average score from 3 separate TMA cores in a given tumor; data are expressed as mean ± SEM. Control brain tissues are surgical resections of nonneoplastic epileptic foci
In characterizing autophagy in FFPE tissues, it has been recommended to also evaluate Atg7 and ubiquitin expression in addition to LC3 [17]. As seen with LC3, Atg7 increased with increasing WHO grade, but showed no association with IDH1 mutations among high-grade gliomas (Supplemental Figure 4d, 4e). Conversely, nuclear localization of ubiquitin decreased with increasing glioma grade, but was also not associated with IDH1 mutations (Supplemental Figure 4f, 4g).
Discussion
Although it is now clear that the 2-HG product of mutant IDH1 strongly influences epigenetics, specifically by promoting hypermethylation of histones and DNA [10], these mutations likely have a broader range of effects. Prior work indicated a prooxidant activity of 2-HG and mutant IDH1 in both nonneoplastic tissues and glioma cell lines in vitro [18, 19, 22, 27], but to date such changes have not been compared to in vivo tumors or evaluated in patient biopsy materials. We found that wild-type glioma cells challenged with either 2-HG or R132H IDH1 overexpression do exhibit signs of oxidative stress and undergo either apoptosis or autophagy, but patient-derived mutant gliomas generally showed no differences in those processes. This suggests that responses to 2-HG and mutant IDH1 depend greatly on other accompanying genetic alterations, as well as whether the cells are abruptly forced to overexpress the mutation or have evolved along with a spontaneous endogenous mutation.
Because 2-HG is small and relatively polar, some prior studies have used esterified 2-HG to improve in vitro cell permeability [6, 24, 38]. However, the current data showed that unmodified exogenous 2-HG can enter glioma cells (Fig. 1a). This is consistent with a recent study showing that unmodified 2-HG can cross the plasma membrane via a sodium-dependent di- or tricarboxylate transporter, NaCT [3]. In the current study, exogenous 2-HG elicited the same effect on MnSOD as did endogenous expression of R132H IDH1 (Fig. 3a, c). Likewise, nonesterified 2-HG has been shown to produce metabolic changes comparable to those achieved by overexpression of R132H IDH1 in vitro [32]. Since 2-HG is readily released from mutant tumor cells, this has implications regarding possible effects of 2-HG on admixed nonneoplastic cells like tumor-associated macrophages and endothelial cells [10].
The divergent apoptotic responses between U87MG and LN18 cells to exogenous 2-HG were striking, with suppression of caspase activity in U87MG cells but stimulation in LN18 cells (Fig. 2). This underscores an important aspect of mutant IDH1—its effects in a particular tumor will likely depend on other mutations that are present in that tumor. Considering that IDH1 is the earliest known mutation in the secondary pathway of gliomagenesis, it is possible that, in addition to epigenetic modifications, mutant IDH1 may promote gliomagenesis simply by acting as a selective pressure in favor of specific additional modifications, such as p53 mutations in astrocytomas or 1p/19q codeletion and CIC mutations in oligodendrogliomas [10]. In LN18 cells, 2-HG greatly induces caspase activity and apoptosis; however, in U87MG cells it inhibits caspase activity and does not promote apoptosis. These cell-type-specific effects of 2-HG may be the product of the differential genetic backgrounds of the cell types. For example, LN18 cells have mutant p53 and normal PTEN, whereas U87MG cells have normal p53, lack PTEN, and express higher levels of antiapoptotic Bcl-2 and XIAP than LN18 cells [11, 12]. Furthermore, while 2-HG and R132H IDH1 clearly inhibited glioma in vitro and in vivo growth (Fig. 1), the same mutation actually promoted growth in a BRAF-mutant melanoma cell line [33]. Our data therefore highlight the importance of considering the genetic background when modeling IDH1 mutations.
MnSOD is located in the mitochondria and is upregulated in response to oxidative stress [8]. This data showed a direct correlation between MnSOD and WHO grade (Fig. 5e), consistent with previous reports in gliomas [9] and consistent with the general pattern seen in other cancers, wherein more aggressive and metastatic tumors tend to have elevated ROS and antioxidant enzyme production [8]. Therefore, while the trend toward lower MnSOD in IDH-mutant high-grade gliomas (Fig. 5a, f; Supplemental Figure 4b) differs from the known prooxidative properties of 2-HG, it matches the tendency for such mutant tumors to be less aggressive than their wild-type counterparts. Exactly what mechanism(s) might contribute to a suppression of oxidative stress in IDH1 mutant gliomas is/are unclear as of yet, but the fact that culturing IDH1-mutant cells is quite difficult, especially in standard ambient oxygen levels [30], suggests that such compensation can easily be overwhelmed. Furthermore, since mitochondria are normally a key source of ROS [23], and 2-HG can impair mitochondrial metabolism [18], decreased MnSOD production could actually be a sign that mitochondria are too damaged to be a significant source of ROS in IDH1-mutant tumors. Another possibility is that the lower MnSOD levels represent a confounding variable of necrosis, since IDH1-wild type glioblastomas are far more likely to contain significant necrosis than their mutant counterparts [10], and necrosis can trigger oxidative stress. While care was taken to avoid sampling necrotic areas for analysis, some confounding effect may have been present, particularly in the snap-frozen tissues. However, separate analyses comparing only grades II and III tumors, in which necrosis is by definition absent, still showed no differences in MnSOD or LC3 by IDH1 status (not shown).
The morphologic differences elicited by mutant IDH1 in U87MG xenografts were particularly striking, to the point where the absence of large vacuoles was just as reliable a discriminator between control and mutant tumors as R132H IDH1 immunostaining (Fig. 3e, upper panels). The fact that overexpression of the mutant correlated with a precipitous drop in lipid stores (Fig. 3e, lower panels) suggests the mutation exerts a profound impact on tumor metabolism, specifically in lipids. Indeed, recent work has shown that, during hypoxia or mitochondrial dysfunction, wild-type IDH1 and IDH2 synthesize citrate via reductive carboxylation, which helps export mitochondrial acetyl CoA into the cytosol where it is used to build fatty acids [26, 28, 37]. In contrast, cell-free assays showed that mutant IDH1 is incapable of performing reductive carboxylation [21]. Since IDH1 mutations are nearly always heterozygous [10], and the mutant heterodimerizes to wild-type IDH1 [14], it is possible that mutant IDH1 impairs the ability of wild-type IDH1 to synthesize citrate, as has been suggested previously [32]. Experiments addressing these issues are ongoing.
Autophagy is a lysosomal pathway for recycling damaged macromolecules and organelles, including mitochondria, and is a typical response to OS [4]. Although believed to act as a tumor suppressor in nonneoplastic cells, once oncogenesis has taken place, autophagy helps cancer cells withstand the stress caused by rapid proliferation and uneven blood supply [5]. In our study, U87MG cells exposed to 2-HG or mutant IDH1 showed increased autophagosome formation (Fig. 4), concordant with elevated markers of OS seen in the same cells (Fig. 3). In patient-derived gliomas, however, while LC3 and Atg7 aggregates increased with glioma grade as expected, they showed no difference according to IDH1 status (Fig. 5g, h; Supplemental Figures 4d, 3e). The reasons for this are not clear, but the one finding that was consistent between IDH1-mutant U87MG cells and patient-derived mutant tumors was upregulation of p62 (Figs. 4d, 5a). P62 targets ubiquitinated proteins for autophagic degradation, and its accumulation is frequently a sign of dysfunctional mitochondria and autophagy that is not going to completion [16, 29]. Newer data also indicate a role for p62 in combating OS via upregulation of antioxidant enzymes, including MnSOD [29]. [Although speculative, it is interesting to note that IDH1-mutant tumors with the highest levels of p62 on western blot also had the highest levels of MnSOD (Fig. 5a).] But the effects of p62 are clearly complex, as other work has also suggested roles in activating mitosis and prosurvival pathways as well as facilitating apoptosis [29].
These data highlight the difficulty in modeling IDH1-mutant tumors in vitro and in vivo. For example, one limitation to this study is the absence of cultures from gliomas containing spontaneous endogenous IDH1 mutations. Our attempts at culturing IDH1-mutant gliomas have been unsuccessful thus far, which has been a problem in the field [15]. One study described the successful establishment of an IDH1-mutant oligodendroglioma cell line [25], but even then, the tumor cells eventually deleted the wild-type IDH1 gene during in vitro passaging (http://www.atcc.org/products/all/ACS-1018.aspx), which greatly reduces 2-HG production [13]. As an alternative, we and many others induced overexpression of mutant IDH1 in normally wild-type cells using powerful cytomegalovirus promoters [2, 14, 20, 22, 32, 36, 39]. But since heterozygosity is the typical IDH1 mutation pattern, only one copy of the mutant gene is present in a spontaneously occurring tumor, raising the question of whether multiple copies of mutant may have differing effects compared to a single copy. In the current data, while mutant IDH1 expression did appear to be stronger versus wild-type IDH1, the overall levels were broadly comparable (Fig. 3c, which were done at identical exposures on the same membranes). And as mentioned above, mutant IDH1 requires wild-type IDH1 for maximal efficacy [13]; so from a functional perspective, wild-type IDH1 levels could act to dampen the effects of mutant IDH1 overexpression, at least as far as 2-HG production is concerned. Furthermore, the effects on MnSOD and autophagy in the xenografts matched what was observed via exogenous treatment with a physiologic concentration of 2-HG (Figs. 3, 4). Nevertheless, this is a nontrivial consideration, especially if mutant IDH1 is found to have other effects on cell biology besides 2-HG production. Also important to note is that these in vivo xenografts were in the flank, which generated adequate tissue to thoroughly assess intrinsic tumor activities like oxidative stress and autophagy via histologic and western blot analyses. But for studying other facets of glioma behavior, such as invasion or potential therapies that would need to cross the blood–brain barrier, orthotopic xenografts are of course preferred.
In summary, this study is the first to examine the effects of mutant IDH1 on oxidative stress, apoptosis, and autophagy by comparing results from artificial in vitro and in vivo models to actual patient-derived tissues via rigorous tissue-based analyses. Our data suggest that mutant IDH1 can have profound effects on oxidative stress, autophagy, and metabolism, but their nature and extent depend greatly on other coexisting genetic alterations and whether the IDH1 mutation develops spontaneously or is overexpressed in wild-type cells. This does not obviate the use of induced models, but rather indicates the need to compare their data with actual patient tumors as much as possible. Further studies exploring the effects of 2-HG action will provide great insight into how to maximize and tailor adjuvant therapies.
References
Bleeker FE, Atai NA, Lamba S et al (2010) The prognostic IDH1(R132) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol 119(4):487–494
Bralten LB, Kloosterhof NK, Balvers R et al (2011) IDH1 R132H decreases proliferation of glioma cell lines in vitro and in vivo. Ann Neurol 69(3):455–463
Brauburger K, Burckhardt G, Burckhardt BC (2011) The sodium-dependent di- and tricarboxylate transporter, NaCT, is not responsible for the uptake of d-, L-2-hydroxyglutarate and 3-hydroxyglutarate into neurons. J Inherit Metab Dis 34(2):477–482
Cherra SJ 3rd, Dagda RK, Chu CT (2010) Review: autophagy and neurodegeneration: survival at a cost? Neuropathol Appl Neurobiol 36(2):125–132
Choi AM, Ryter SW, Levine B (2013) Autophagy in human health and disease. N Engl J Med 368(7):651–662
Chowdhury R, Yeoh KK, Tian YM et al (2011) The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 12(5):463–469
Dang L, White DW, Gross S et al (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462(7274):739–744
Dhar SK, St Clair DK (2012) Manganese superoxide dismutase regulation and cancer. Free Radic Biol Med. 52(11–12):2209–2222
Haapasalo H, Kylaniemi M, Paunul N, Kinnula VL, Soini Y (2003) Expression of antioxidant enzymes in astrocytic brain tumors. Brain Pathol 13(2):155–164
Horbinski C (2013) What do we know about IDH1/2 mutations so far, and how do we use it? Acta Neuropathol 125(5):621–636
Ishii N, Maier D, Merlo A et al (1999) Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol 9(3):469–479
Jane EP, Premkumar DR, Pollack IF (2011) Bortezomib sensitizes malignant human glioma cells to TRAIL, mediated by inhibition of the NF-κB signaling pathway. Mol Cancer Therap 10(1):198–208
Jin G, Reitman ZJ, Duncan CG et al (2013) Disruption of wild-type IDH1 suppresses d-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res 73(2):496–501
Jin G, Reitman ZJ, Spasojevic I et al (2011) 2-Hydroxyglutarate production, but not dominant negative function, is conferred by glioma-derived NADP-dependent isocitrate dehydrogenase mutations. PloS One 6(2):e16812
Klink B, Miletic H, Stieber D et al (2013) A novel, diffusely infiltrative xenograft model of human anaplastic oligodendroglioma with mutations in FUBP1, CIC, and IDH1. PLoS ONE 8(3):e59773
Klionsky DJ, Abeliovich H, Agostinis P et al (2008) Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 4(2):151–175
Ladoire S, Chaba K, Martins I et al (2012) Immunohistochemical detection of cytoplasmic LC3 puncta in human cancer specimens. Autophagy. 8(8):1175–1184
Latini A, da Silva CG, Ferreira GC et al (2005) Mitochondrial energy metabolism is markedly impaired by d-2-hydroxyglutaric acid in rat tissues. Mol Genet Metab 86(1–2):188–199
Latini A, Scussiato K, Rosa RB et al (2003) d-2-hydroxyglutaric acid induces oxidative stress in cerebral cortex of young rats. Eur J Neurosci 17(10):2017–2022
Lazovic J, Soto H, Piccioni D et al (2012) Detection of 2-hydroxyglutaric acid in vivo by proton magnetic resonance spectroscopy in U87 glioma cells overexpressing isocitrate dehydrogenase-1 mutation. Neuro Oncol. 14(12):1465–1472
Leonardi R, Subramanian C, Jackowski S, Rock CO (2012) Cancer-associated isocitrate dehydrogenase mutations inactivate NADPH-dependent reductive carboxylation. The J Biol Chem 287(18):14615–14620
Li S, Chou AP, Chen W et al (2013) Overexpression of isocitrate dehydrogenase mutant proteins renders glioma cells more sensitive to radiation. Neuro Oncol. 15(1):57–68
Li X, Fang P, Mai J, Choi ET, Wang H, Yang XF (2013) Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol. 6:19
Losman JA, Looper R, Koivunen P et al (2013) (R)-2-Hydroxyglutarate Is sufficient to promote leukemogenesis and its effects are reversible. Science 339(6127):1621–1625
Luchman HA, Stechishin OD, Dang NH et al (2012) An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro-oncology 14(2):184–191
Metallo CM, Gameiro PA, Bell EL et al (2011) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481(7381):380–384
Mohrenz IV, Antonietti P, Pusch S et al (2013) Isocitrate dehydrogenase 1 mutant R132H sensitizes glioma cells to BCNU-induced oxidative stress and cell death. Apoptosis 18(11):1416–1425
Mullen AR, Wheaton WW, Jin ES et al (2011) Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481(7381):385–388
Nezis IP, Stenmark H (2012) p62 at the interface of autophagy, oxidative stress signaling, and cancer. Antioxid Redox Signal 17(5):786–793
Piaskowski S, Bienkowski M, Stoczynska-Fidelus E et al (2011) Glioma cells showing IDH1 mutation cannot be propagated in standard cell culture conditions. Br J Cancer 104(6):968–970
Poon HF, Castegna A, Farr SA et al (2004) Quantitative proteomics analysis of specific protein expression and oxidative modification in aged senescence-accelerated-prone 8 mice brain. Neuroscience 126(4):915–926
Reitman ZJ, Jin G, Karoly ED et al (2011) Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci USA 108(8):3270–3275
Shibata T, Kokubu A, Miyamoto M, Sasajima Y, Yamazaki N (2011) Mutant IDH1 confers an in vivo growth in a melanoma cell line with BRAF mutation. Am J Pathol 178(3):1395–1402
Struys EA, Jansen EE, Verhoeven NM, Jakobs C (2004) Measurement of urinary d- and L-2-hydroxyglutarate enantiomers by stable-isotope-dilution liquid chromatography-tandem mass spectrometry after derivatization with diacetyl-L-tartaric anhydride. Clinical Chem 50(8):1391–1395
Sultana R, Butterfield DA (2008) Slot-blot analysis of 3-nitrotyrosine-modified brain proteins. Methods Enzymol 440:309–316
Valadez JG, Grover VK, Carter MD et al. (2012) Identification of Hedgehog pathway responsive glioblastomas by isocitrate dehydrogenase mutation. Cancer Lett 328(2):297–306
Wise DR, Ward PS, Shay JE et al (2011) Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci USA 108(49):19611–19616
Xu W, Yang H, Liu Y et al (2010) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. cancer cell 19(1):17–30
Zhu J, Cui G, Chen M et al (2012) Expression of R132H mutational IDH1 in human U87 glioblastoma cells affects the SREBP1a pathway and induces cellular proliferation. J Mol Neurosci 50(1):165–171
Acknowledgments
C.H. was supported by K08 CA155764 (National Cancer Institute), 2P20 RR020171 COBRE pilot grant (National Institute of General Medical Sciences), The Peter and Carmen Lucia Buck Training Program in Translational Clinical Oncology, and the University of Kentucky College of Medicine Physician Scientist Program. The Markey Biospecimen and Tissue Procurement (BSTP) Shared Resource Facility facilitated the construction of tissue microarrays and immunohistochemical studies. Special thanks to Dana Napier for her excellent histologic expertise. Study data were collected and managed using REDCap electronic data capture tools hosted at the University of Kentucky. This research was also supported by the Markey Cancer Center Free Radical Biology in Cancer (FRBC) Shared Resource Facility. Flow cytometry and cell sorting was carried out at the University of Kentucky Flow Cytometry and Cell Sorting (FCCS) Core Facility, which is supported in part by the Office of the Vice President for Research, the Markey Cancer Center and a grant from the NIH Shared Instrument Program (S10 RR026827-01A1). The BSTP, FRBC, and FCCS Shared Resource Facilities are all supported by the University of Kentucky Markey Cancer Center (P30CA177558). We thank Dr. Hai Yan of Duke University Medical Center for supplying us with pEGFP-N1-IDH1 and pEGFP-N1-IDH1R132H plasmids. We thank Dr. Haining Zhu and Dr. Jozsef Gal, Department of Molecular and Cellular Biochemistry of the University of Kentucky, for providing the GFP-LC3 plasmid. We thank Dr. Daret St. Clair, Department of Toxicology, for her excellent suggestions involving MnSOD experiments. We also thank Drs. Jeremy Rich and Monica Venere from the Cleveland Clinic Foundation for their generous help and training with the patient-derived glioma cultures.
Conflict of interest
None of the authors have any conflicts of interest pertaining to the data in this study.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Gilbert, M.R., Liu, Y., Neltner, J. et al. Autophagy and oxidative stress in gliomas with IDH1 mutations. Acta Neuropathol 127, 221–233 (2014). https://doi.org/10.1007/s00401-013-1194-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-013-1194-6