
Abstract
Tracer based planar laser-induced fluorescence (PLIF) has emerged as a powerful in-situ measurement technique with a considerable spatial and temporal resolution for Internal combustion (IC) engines. In PLIF, the emitted fluorescence signals from a tracer molecule are processed to determine distribution of temperature, fuel, residual gases etc. However, it is imperative to have a thorough understanding of the tracer physical properties and its fluorescence intensity dependencies on excitation wavelength, pressure, temperature, and bath gas composition existing inside the combustor for accurate quantitative interpretation. This work consists of a series of two articles providing a detailed review of the existing literature of fluorescence characteristics of various molecules used as tracers in IC engine applications. Due to the overwhelming usage of organic compounds in IC engine environment, the work is restricted to them. Part A of this work is focussed on non-aromatic compounds (acetone, 3-pentanone and biacetyl) whereas part B will focus on aromatics. Due to a small energy gap between the excited singlet and triplet states of ketones, they experience rapid inter-system crossing making them far less sensitive to oxygen quenching effects than aromatic molecules. Addition of tracers to surrogate fuel can lead to difficulties related to co-evaporation, azeotrope formation and stability of tracer molecules in terms of photolysis and pyrolysis effects when subjected to intense laser irradiation and harsh engine environment. In this work, fluorescence signal variation of tracer molecules is divided into variations in absorption cross-section and fluorescence quantum yield (FQY). Absorption cross-section normally increases with temperature but is insensitive to pressure changes. FQY reduces with increase in temperature but increases with pressure for ketones for non-oxygen containing bath gases. The pressure sensitivity increases with the number of atoms in a collider molecule. FQY values decrease with decreasing laser excitation wavelength whereas the temperature and pressure sensitivity of FQY reduce with increasing wavelengths. For simultaneous high pressure and temperature conditions, the pressure sensitivity of FQY is found to reduce due to a reduction in the effective number of collisions with bath gas molecules. Among the three tracers, acetone has been widely used for marking gaseous fuels and 3-pentanone and biacetyl for liquid fuels like iso-octane. Acetone and 3-pentanone have received significant attention for fluorescence studies due to their widespread usage in IC engine applications. Biacetyl on the other hand has recently started to receive attention due to its application in high repetition rates PLIF measurements and requires more fluorescence studies to fully characterise its fluorescence behaviour and construct fluorescence models over the complete pressure and temperature range required in IC engine applications.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
1.1 Motivation
Planar laser-induced fluorescence (PLIF) is an in-situ measurement technique that uses a laser sheet to excite certain molecules in a region of interest and capture their fluorescence emission. The captured fluorescence signals are then processed to obtain several parameters of interest in complex flow situations like those found inside the cylinder of an IC engine. Being non-intrusive in nature, measurement does not interfere with the in-cylinder flow field and mixing process. Thus, very accurate measurements of the spatial distribution of fuel, exhaust residuals and temperature can be carried out. All these information can be used to understand combustion which can then be optimised to meet the increasingly stringent emission norms and fuel efficiency requirements of modern-day IC engines. The success of this method is to a large extent dependent on the behaviour of tracer molecules used. Over the years, various PLIF studies have been conducted using different tracers.
In order to be used as a tracer, a compound has to fulfil several criteria. Fluorescence signals emitted from a particular region of space is dependent on the tracer concentration, pressure, temperature and composition of the surrounding gases, most of which are unknown in a typical combustion system. It is extremely challenging to measure all of them simultaneously as there is a lack of a-priori calibration data in all these dimensions. Therefore, the key question for all tracer LIF measurements is how to reduce the complexity of the system in a manner that is suitable for obtaining the desired information.
A change in fluorescence signal intensity over a plane can be related to a corresponding change in temperature, concentration, etc. giving rise to quantification of a spatial distribution of these variables. For this, a knowledge of the physical and chemical properties of the tracers is needed and more importantly the fluorescence signal dependencies of the available compound on relevant parameters need to be properly characterised. Several studies have been conducted till date in this regard. Other than a few comparative studies of different tracer compounds [1, 2], to the best of our knowledge, the last significant review on this topic was provided by Schulz and Sick (2005) [3], but it was limited to a few molecules and to a certain regime of temperature and pressure. The literature has witnessed tremendous contributions since then. In our work, we have broadly expanded the scope by providing a detailed review of the fluorescence characteristics of several molecules and have included the combined high-pressure and high-temperature regime discussions as well (particularly relevant for IC engines and other combustor applications). Due to its extensive nature, we have divided the work into two parts: Part A discusses non-aromatic tracers (acetone, 3-pentanone and biacetyl), and Part B discusses aromatic tracers (toluene, anisole, naphthalene,1-methyl naphthalene and fluoranthene).
1.2 Background
With the advent of optical engines, optical access was now available inside the combustion chamber through transparent cylinder liner and piston window made of quartz. Pioneering works by Bowditch (1961) [4], Richman and Reynolds (1984) [5] and Bates (1988) [6] have contributed immensely to the development of modern optical engines. A thorough discussion on the development of optical engines, their advantages and shortcomings is beyond the scope of this work. Interested readers can refer to the review of Mittal and Mehta (2018) [7]. With optical access into the engine cylinder, many laser diagnostic techniques quickly became popular for in-cylinder measurements. Apart from PLIF, other measurement methodologies involving Rayleigh scattering, mie scattering, Raman scattering have also been used by several researchers. Zhao and Ladommatos (1998) [8] have provided a detailed review of such measurement methodologies. The ability of PLIF to be species specific and to provide information with a high spatial resolution at a fair signal strength, resulted in it being the most popular method for fuel distribution measurement in IC engine applications. Other than IC engines, LIF has also found applications in studies related to gas turbine combustor applications [9,10,11,12], mixing in turbulent jets [13,14,15], boundary layers [16, 17], jets [18,19,20], sprays [21,22,23,24], etc. Kohse-Hoinghaus (1994) [25] has also provided a detailed review on the application of LIF in flame studies, detection of radicals like OH, CH, NO and temperature measurements using OH radicals.
Measurements in IC engines are carried out in a linear regime where the emitted fluorescence signals are linearly proportional to the incident laser energy. Normally, pulsed lasers are preferred over continuous ones to achieve very high-powered laser beams. Many a times, depending on the tracer used, there is a reduction in fluorescence signal intensity due to collision between molecules of tracer and those of surrounding bath gas. This phenomenon is called quenching. Quenching itself has a complex temperature, pressure and gas composition dependency which are typically unknown in an engine cylinder. Therefore, it poses a significant difficulty in correcting fluorescence signal intensity. To circumvent the complexities introduced by quenching effects, other LIF methods like saturation LIF [26, 27], laser-induced predissociative fluorescence (LIPF) [28, 29] etc. have been developed over the years. However, these methods have their own limitations like low signal levels for LIPF and inability to achieve laser saturation during the total duration of laser pulse and over the entire plane of the laser in planar saturation LIF measurements. A detailed discussion about these methods is provided in Eckbreth (1996) [30] and in the review by Kohse-Hoinghaus (1994) [25]. Assuming weak excitation, fluorescence signal (in photons per unit volume) can be calculated from the following equation that was explained by Koch and Hanson (2003) [31]:
Where \(\:{\eta\:}_{\text{o}\text{p}\text{t}}\) is collection optics efficiency, \(\:\frac{hc}{\lambda\:}\) is the energy of a photon with wavelength λ (in J) that is absorbed, E is the laser fluence (J/cm2), c is the speed of light, λ is the wavelength of the laser light used, nabs is the tracer density (in molecules/cm3) which is a function of the pressure and temperature, σ is the absorption cross section (cm2/molecule) which is a function of the temperature and wavelength of light used, and ϕ (number of fluorescence photons per absorbed photon) is called the fluorescence quantum yield (FQY) which is a function of pressure, temperature, wavelength and composition of the surrounding gases. Absorption cross-section and fluorescence quantum yields are the two most important photophysical parameters concerning LIF studies. They will be discussed in the upcoming sections.
For planar measurements, the laser beam is converted into a sheet using a suitable combination of lenses. Suitable tracer compounds are added to a non-fluorescing agent (that mimics fuel) before induction into the engine cylinder so that the fluorescence emission from tracers can be used to represent the fuel concentration, equivalence ratios, temperature etc. The emitted signals are captured with the help of suitable cameras and the intensity variation over the region of interest is produced in the form of images. Such raw images are then post processed to evaluate mixture distributions, temperature variations, residual gas distribution etc. From Eq. (1), fluorescence intensity is a complex function of temperature, pressure, excitation wavelength and bath gas composition and is dependent on tracer number density. Hence, pressure, temperature and bath gas effects need to be accounted for quantitative evaluation so that a direct correlation of signal strength can be made to number densities. Different methods are used to quantitatively find out mixture distribution inside the engine cylinder - in-situ calibration using separate calibration images for each crank angle [32,33,34], fuel-air ratio laser-induced fluorescence (FARLIF) for direct determination of equivalence ratios [35,36,37,38]. A description of these methods can be found in a review by Zhao and Ladommatos (1998) [8]. Studies on mixture distribution have provided immense information on in-cylinder and cycle-to-cycle variations [33, 39,40,41], cold starting strategies [42, 43], lean burn strategies [44,45,46,47], hydrogen engines [48, 49] etc. There are studies of simultaneous liquid and vapour phase imaging using LIEF (laser induced exciplex fluorescence) in sprays [50, 51], mixture formation processes [52, 53] etc. Apart from mixture distribution, PLIF has also been used to determine exhaust gas residual distribution using NO PLIF [40], SO2 PLIF [54], negative-PLIF (N-PLIF) [55], oxygen distribution using 3-pentanone/toluene combination [56], mixture distribution using 3-pentanone/(DMA) N, N-dimethylaniline combination [44]. Equation 1 shows that temperature has a direct influence on fluorescence signals which needs to be taken into consideration when there is an appreciable temperature stratification as observed due to charge cooling in DI mode, recirculation of hot exhaust gases etc. For such reasons, in-situ temperature measurement becomes important. Methods like two tracer techniques (using single laser excitation wavelength and two tracers) with triethylamine (TEA)/3-pentanone [57] TEA/acetone [58] combinations, two line techniques (using two laser excitation wavelengths and single tracer) [59,60,61], and two colour techniques (using single tracer and excitation wavelength) [62,63,64,65] have been developed over the time. All these techniques are very important from the point of view of PLIF application and will be reviewed in part B of this work. Traditionally, due to hardware constraints, most of the studies performed using PLIF were at low speeds which is not sufficient to study a time resolved mixture formation processes. Recently, new laser systems capable of providing pulsed laser beams at higher frequencies at sufficient energy are being developed [66, 67] resulting in high speed PLIF studies [65, 68, 69].
1.3 Roadmap through the paper
It is thus very evident that PLIF is very useful particularly for engine diagnostics. In order to understand the aforementioned methodologies and develop newer ones, it is imperative to have a thorough understanding of the fluorescence characteristics of tracers used. Therefore, part A focuses on acetone, 3-pentanone and biacetyl (these three are the most widely used non-aromatic tracers in IC engines) and reviews their photophysics along with the available explanations of their fluorescence characteristics. This paper is organised as follows. After a brief summary on application of PLIF along with relevant references, the utility of PLIF and its potential in engine diagnostics has been made evident. This is followed by Sect. 2 where a detailed discussion is provided about fluorescence and other relevant photophysical processes that occur in an excited state molecule. This section will be very important for understanding the fluorescence dependencies on various photo-physical processes.
Section 3 describes tracer-based LIF study and provides a detailed discussion on aspects that needs to be considered about tracer properties when applied for PLIF studies. Various complications using tracers have also been put forward. Subsequently, the fluorescence characteristics of acetone, 3-pentanone and biacetyl as well as their usage in IC engine studies are described in separate sections. It is to be noted that, the main focus of the article is a discussion of fluorescence characteristics of organic tracer molecules. Therefore, a detailed discussion of engine-related phenomena that are studied using PLIF is not attempted here. However, for the sake of completeness, we have separately provided a brief review of various such studies at the beginning of every tracer molecule section. A detailed discussion on FQY evaluation models (like the multi-step decay model in acetone section) is not the focus of this work and therefore, it is briefly discussed. For further details about the parameters involved in the model, their calibration and other improvements, one can refer the original works referenced here. Although, some of the tracers discussed in both part A and B are used for liquid film imaging as well, the focus will be on vapour phase LIF. It is to be noted that for acetone, initially description of the fluorescence characteristics in plots is provided. The physical explanation is provided a bit later when the fluorescence multi-step decay model is introduced. However, for 3-pentanone and biacetyl, both the description of figures and their physical explanation are provided together. The article is then concluded with the discussion of several key insights obtained in this work.
2 Tracer photophysics
To acquire PLIF images in IC engine, it is preferable to replace the standard fuel with a non-fluorescing surrogate mixed with a fluorescing molecule called tracer whose fluorescence behaviour is well characterised. Over the years, organic compounds have been used almost exclusively in IC engine PLIF studies and so the current paper is restricted to fluorescence from organic compounds. Before discussing the photophysics of tracers, it is important to discuss about the absorption and emission spectra as well as the relevant photophysical processes that occur when a molecule is in its excited state once it is exposed to a suitable laser irradiation.
2.1 Energy levels of molecules
The electronic Schrödinger equation is solved using the Linear Combination of Atomic Orbitals (LCAO), resulting in Molecular Orbitals (MO). Bonding MOs, with lower energy, lead to bond formation, while anti-bonding MOs, with higher energy, inhibit it. Overlapping atomic orbitals form σ bonds (head-on overlap) and π bonds (sideways overlap). σ bonds are generally stronger than π bonds. Heteroatoms, like oxygen, may have non-bonding MOs (denoted as ‘n’), occupied by lone pairs that do not participate in bond formation. The orbital energies are illustrated in Fig. 1.

Energy level diagram of various states
Electronic energy states can be classified depending upon the degree of excitation (represented by number in subscript) and the total spin multiplicity of the state (2 S + 1) depending on the total spin ‘S’. When the electrons are paired, S = 0, the multiplicity is unity and when electrons are unpaired, S = 1 bringing the multiplicity to 3. States with unity multiplicity are called singlet states (represented by S) and with multiplicity value 3 are called triplet states (represented by T). Triplet states of a particular excitation level lie below the singlet states due to lower energy as per Hund’s rule. Upon absorption of a photon, a molecule is promoted to a higher electronic energy level. The transition normally occurs from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO). For ketones, the transition thus happens between n to π* due to the lone pair on oxygen whereas in aromatic hydrocarbons, the transition occurs between π to π*.
The Born-Oppenheimer (BO) approximation suggests that the nuclear and the electronic motions can be treated separately since the nuclei are much more massive than the electrons. Within the Born-Oppenheimer approximation, we can generate potential energy surfaces for the distinct electronic states of the molecule. Figure 2 provides a generalized illustration of the potential energy surfaces for two electronic states of a diatomic molecule. The energy level arrangement of a basic molecular system like a diatomic molecule can be elucidated using the framework of the anharmonic oscillator and non-rigid rotor model, along with corrections from the rigid-rotor approximation of the harmonic oscillator. The lowest point on each curve corresponds to the electronic energy. Figure 2 displays the quantized energies for vibration in each electronic state. For every electronic level, there exists a manifold of vibrational energy levels (v) with bounded states up to the point of the molecule’s dissociation energy. The gaps between the vibrational levels decrease with higher vibrational quantum numbers. D is the dissociation energy which is a measure of the depth of the PES from the zero vibrational quantum level and re represents the internuclear separation between two nuclei at the equilibrium position. The higher the electronic energy level, shallower is the well indicating low dissociation energy requirement of a molecule in its excited state. Till now, the discussion was for a diatomic molecule, but the results can be applied to a polyatomic molecule as well. In case of a non-linear polyatomic molecule of N atoms, the PES will be a 3 N-5-dimensional hypersurface for 3 N-6 normal vibrational modes. Each vibrational level further splits into rotational levels which are ignored in the current discussions as fine rotational lines in electronic spectrum are visible only at extremely high resolution and are not a matter of concern for the discussion on absorption or fluorescence spectra of organic molecules which will be taken up in the following sections.

(from ref [70]): Potential energy surfaces for two different excitation levels along with the vibrational energy levels at each excitation manifold as a function of internuclear separation. One of the nuclei is fixed at origin and the other is allowed to vibrate. Adapted from [70] with permission from AIP Publishing
2.2 Absorption spectrum
A molecule when excited with an incident radiation of a favourable frequency, absorbs a photon and gets promoted to a higher vibronic (combination of an electronic and vibrational) level. Any transition that occurs it is safe to assume that the molecule is in the ground vibrational level of the ground singlet state where the maximum probability of the nuclear configuration is given by the crest of the nuclear wave function as shown in the zeroth vibrational level of the ground singlet in Fig. 3. The BO approximation suggests that since the nucleus is much more massive than the electrons, the electronic transitions occur very fast so much so that the nuclear position and momenta do not change before and after the electronic transition has taken place. Thus, in a potential well diagram any transition is always represented by a straight vertical line with respect to the internuclear distance from the position of crest of the ground state nuclear wave function (Fig. 3). A sample absorption spectrum along with the electronic transition between two potential wells is shown in Fig. 3. The spectrum shows several peaks merged together. The transition is assumed to be from the ground vibrational state to various vibrational states of the excited electronic manifold. The height of the peaks varies due to the fact that some transitions are preferred more than the others due to the different extent of overlap of the orbital wave functions of the two participating vibrational levels. However, in large organic molecules like the ones discussed in this article, the high density of vibrational states nonetheless result in a broad absorption band with very weak structures.

(from ref [71]): Electronic transition with the corresponding absorption spectrum. The excited and ground potential wells are shown along with the nuclear wave functions corresponding to various vibrational levels
The probability of a transition is given by the square of the transition moment integral, Mif which is defined as [72]
Where \(\:{\varPsi\:}_{f}\) and \(\:{\varPsi\:}_{i}\) are the final and initial wave functions (the subscript ‘i’ is for initial state whereas the subscript ‘f’ is for final state), \(\:\mu\:\) is the dipole moment operator (\(\:\mu\:=e\varvec{r}\), e is the electronic charge and r is the radial vector) and \(\:d\tau\:\) is the configurational space. The wave function can be split into orbital electronic (ϕ), spin (S) and nuclear (𝛘) wave functions according to the BO approximation. \(\:d{\tau\:}_{e}\),\(\:\:{d\tau\:}_{n}\) and \(\:{d\tau\:}_{s}\)are the electronic, nuclear and spin configuration space respectively [72].
The dipole moment operator is associated only with the electronic wave functions while the nuclear and the spin wave functions remain unaffected. The magnitude of the integral determines the probability of a particular transition. If the magnitude is zero, then such a transition is called as a forbidden transition. The first integral brings about symmetry related restrictions. If the symmetry of the initial and final electronic states is not favourable, then the integral is zero and the transition is symmetry forbidden. The n to π* transition in ketones is overlap forbidden due to the poor local overlap of the non-bonding MO around oxygen atom which is located perpendicular to the antibonding π* orbital of the ketonic group. This results in a reduced magnitude of absorption cross-section for n to π* than that for the π to π* transition in aromatic hydrocarbons. However, it might so happen that the nuclear and electronic coordinates are not independent enough due to molecular vibrations resulting in a vibronic coupling. It causes a breakdown of the BO approximation leading to a departure from the perfect symmetry of the electronic orbitals. Under such circumstances, the integral is non-zero and weak vibrationally allowed transitions are observed in the absorption spectrum. The third integral is an overlap of the spin functions which brings about the spin selection rules. The two spin wave functions are orthogonal and thus overlap of non-identical waves will result in the transition integral becoming zero. Therefore, the transition is allowed only for singlet-singlet or triplet-triplet states. It is the most stringently followed selection rule. However, weak singlet to triplet transition can also be observed due to spin-orbit coupling. Due to perturbations from the magnetic field of the nucleus (heavy atom effect: heavier the nucleus, stronger is the field and thus more is the perturbation), there is a mixing of the singlet and triplet states. Thus, the overlap integral of such mixed states will result in small non-zero values.
The second integral is a mathematical statement of the Franck-Condon principle, the square of which is called the Franck-Condon factor. It determines the amplitude of the allowed transitions. The Franck-Condon factor is an overlap integral of the wave functions belonging to the initial and final vibrational energy levels. Its significance can be seen in Fig. 3 where the variation in the wave function overlaps varies the absorption spectrum peaks. The maximum peak is observed for 0–2 transition since the vibrational wave functions of both the states have a considerable magnitude along the vertical line originating from the crest of the ground vibrational energy level. It might so happen that the vibrational states are so close to each other that the features in absorption spectrum merge with each other resulting in a broadband shaped spectrum as observed for the n to π* transition in ketones. As already mentioned, the rotational structures are ignored. Absorption can be quantified by using a photo-physical parameter called absorption cross-section. This quantity was also mentioned in the fluorescence Eq. (1) in Sect. 1.2. When a sample is irradiated with light of certain frequency, a fraction of the incident radiation is absorbed. The intensity of the incident radiation (I0) and the unabsorbed fraction (I) can be measured using photodiodes. The absorption cross-section (σ) is then defined by the Beer-Lambert’s law which states
Where l is the length of the absorbing medium and n is the number density of the absorbing molecules.
2.3 Jablonski’s diagram
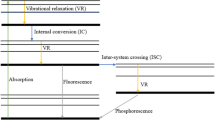
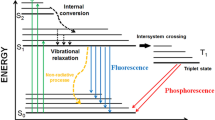
When a molecule is in its excited state, it typically returns to the ground state either through photon emission or by releasing energy through non-radiative processes. It might also undergo chemical process like photo-dissociation. In Jablonski’s diagram (Fig. 4), various processes are illustrated to dissipate the excess energy in the molecule while in the excited state: Intersystem crossing (ISC), Internal conversions (IC), vibrational relaxations (VR), fluorescence and phosphorescence. The electronic states are marked with numerical subscripts indicating the level of excitation, along with an uppercase letter ‘S’ or ‘T’ denoting singlet or triplet states. The bold horizontal line represents the ground vibrational level of an electronic set, whereas the thinner lines depict higher vibrational levels. The photophysical processes can be categorized as either intrinsic to the molecule (unimolecular) like ISC, IC or dependent on both the tracer molecule and a collider molecule (bimolecular) like VR. Furthermore, these processes can be classified as either radiative or non-radiative. Fluorescence occurs when a photon is emitted during a transition between energy states of the same multiplicity, while phosphorescence happens when the multiplicity changes. In the diagram, fluorescence (F) is depicted between S1 and S0 states, and phosphorescence occurs between T1 and S0 states. Absorption (A) is indicated by a vertical arrow between S0 and S2. Internal conversion (IC) takes place when a transition occurs between states of the same multiplicity without photon emission, and inter-system crossing (ISC) happens when multiplicities differ. In the diagram, IC is represented between S2 and S1, while ISC is shown between S1 and T1. Vibrational relaxation (VR) refers to the dissipation of energy from an excited molecule when it collides with a surrounding molecule. In the diagram, VR is illustrated by a transition to lower vibrationally excited states in the S1 state. Bimolecular quenching is not depicted here and will be discussed later.

Jablonski’s diagram showing various photo-physical processes
2.4 Internal conversion and inter-system crossing
The non-radiative interactions consist of transition of a molecule to lower energy levels without a photon emission. As discussed in Sect. 2.1, the energy level curve of a polyatomic molecule is a hypersurface for different vibrational modes. It is possible for such hypersurfaces to intersect or to be very close to each other. Such points are called isoenergetic points. The non-radiative transitions occur at these points. When the multiplicity of the initial and final states remains unchanged, the phenomenon is internal conversion (IC) whereas if the multiplicity changes, the phenomenon is called inter-system crossing (ISC).
From time dependent perturbation theory, the rate constant for non-radiative transition can be expressed as [73]
Where \(\:{\varPsi\:}_{f}\) and \(\:{\varPsi\:}_{i}\) are the final and initial wave functions, h is the Planck’s constant, \(\:\rho\:\)E is the density of states which is a measure of the number of vibrational energy levels in the final electronic state located around per unit energy at the electronic energy level of the initial state. \({\rm{\mathord{\buildrel{\lower3pt\hbox{$\scriptscriptstyle\frown$}}\over H} }}^*\) is a perturbation operator which become the nuclear energy operator \(({\rm{\mathord{\buildrel{\lower3pt\hbox{$\scriptscriptstyle\frown$}}\over H} }}_N^*)\) for internal conversion and under certain approximations, the expression \(\:<{\varPsi\:}_{f}\left|{\mathord{\buildrel{\lower3pt\hbox{$\scriptscriptstyle\frown$}}\over H^{*}}}\right|{\varPsi\:}_{i}>\) can be split into an electronic part (βIC) and a vibrational part \(< {\chi _f}|\,\chi _i^{} >\) which is the familiar Franck-Condon overlap integral. Thus, for IC, [73]
For inter-system crossing, the perturbation operator gets influenced by the spin-orbit coupling as a result of which, \({\rm{\mathord{\buildrel{\lower3pt\hbox{$\scriptscriptstyle\frown$}}\over H} }}^*\) can be expressed as \({\rm{\mathord{\buildrel{\lower3pt\hbox{$\scriptscriptstyle\frown$}}\over H^*_{SO}}}}\). Analogously, for ISC, [73]
The rate of non-radiative transitions depends on the density of vibrational energy levels in the final state as well as the degree of overlap of vibrational wave functions as defined by the Franck-Condon overlap integral. At the point of transition, the density of vibrational levels is greater at the lower electronic state than at the higher electronic level as expected from an anharmonic oscillator, (Fig. 5). Such close energy levels resemble a state of quasi-continuum which enhances the probability of not just non-radiative transition but also the vibrational relaxations thereafter. Therefore, the forward transition rate towards lower electronic level is much faster than the backward transition rates and is nearly irreversible (reversed ISC is observed and is discussed subsequently in Sect. 2.5). The energy difference between both the vibronic and electronic levels normally reduce at higher energy states. Thus, the hypersurfaces are closer higher up the electronic manifold. This increases the probability of finding isoenergetic points at higher electronic levels. Therefore, the probability of transitions increases above S1 excited state.

Energy level diagram of two different electronic manifolds. The upper electronic level is singlet and lower electronic level can be either singlet or triplet. The gap between the two ground levels is also indicated. The figure represents both an IC or an ISC depending on the multiplicity of the lower energy level
The greater the energy gap (ΔE) between the ground states, the higher is the value of \(\:\rho\:\)E, indicating that the rates of non-radiative transitions should increase with increasing energy gap. However, the transitions are again restricted by the vibrational wave function overlap. At the isoenergetic points, the vibronic wave functions of the molecules must overlap to a large extent for a high probability. The extent of this overlap is quantified by the Franck-Condon integral function. Generally, a high amount of wave functions overlap occurs at points normally at the extremities of a potential well because most of the energy is in the potential energy form and the wave functions have larger amplitudes. As the energy gap increases between electronic states, the overlap occurs at the centre of the potential well function [72] where most of the energy is present in the form of kinetic energy and the wave functions have low amplitudes. This reduces the value of the Franck-Condon integral with an increase in energy gap. Thus, large energy gap between S1 and S0 makes IC less probable but less energy gaps above S1 makes IC highly probable. The low energy gap between S1 and T1 states of ketonic compounds causes a high degree of ISC rates as will be seen later. In addition to the Frank-Condon overlap criteria, the symmetry and spin multiplicity rules also bring about further restrictions. Apart from the internal conversion, the molecule can also make a transition from S1 state to a triplet (Tn) state after which it can lose energy in the triplet manifold and reach T1. This type of transition is called an intersystem crossing. The triplet state acts as a metastable state and has a longer lifetime. The transition between T1 and S0 is spin forbidden, therefore the probability of such a transition is low and the lifetime in the triplet state is large. It is also observed that the presence of paramagnetic molecules like oxygen enhances the rate of ISC which is referred to as oxygen assisted inter-system crossing.
2.5 Fluorescence and phosphorescence
A molecule when excited can make a radiative transition to its ground state. This transition will emit a photon whose frequency is determined by the difference in the energy of the states between which the transition is being made. If the transition is from an excited singlet state to ground singlet state, it is called fluorescence and if it is from an excited triplet state to a ground singlet state then it is called phosphorescence.
Normally, the energy gap between the first excited (S1) and the ground (S0) singlet states is too large making the probability of internal conversion very less between them. Thus, whenever a molecule is excited from a ground singlet state, it can end up in any higher singlet state (SN) depending on the frequency of laser excitation. The molecule then undergoes several internal conversions to go into lower energy singlet states until it arrives at a high vibrational energy level of S1 singlet state, (Fig. 4). The molecule then undergoes vibration relaxations which are typically very fast as compared to fluorescence. These relaxations will bring down the molecule from an excited vibrational energy level to the ground state vibrational level of the same excited singlet state (S1). As the probability of internal conversion is very low from S1 to S0, in the absence of any intersystem crossing and bimolecular quenching effects, the only possibility of the electron is to make a radiative transition and fluorescence is obtained. Thus, the fluorescence spectrum obtained is quite independent of the excitation wavelength used for many molecules. Because of energy losses in internal conversion and vibrational relaxations, normally the fluorescence spectrum is redshifted. This is called Stokes shift. This is desirable as the frequency difference can be used to filter out the emanating fluorescence signal from the scattered incident laser radiation by using filters during fluorescence image acquisition. It is also found that, in a large number of molecules, the gap between the vibrational levels are similar for both the excited and ground electronic states, as a result of which the fluorescence spectrum of a molecule is a mirror image of its absorption spectrum with a very little overlap. The probability of transition to the lower singlet level is also dictated by Franck-Condon overlap integrals and it results in the broad structures in the fluorescence spectrum as previously discussed in the absorption spectrum.
In an excited state, the molecule can have an intersystem crossing and end up in a triplet state at a high vibrational level. It again undergoes vibrational relaxation and ends up in the lower vibrational states. It then emits a photon and ends up in the S0 state. As the probability of a transition from a triplet to a singlet state is low, the rate of transition is also low and thus the phosphorescence obtained can be observed over longer periods than fluorescence. Because of the energy losses in vibrational relaxations, phosphorescence signals are also redshifted.
The intersystem crossings can be bidirectional. It might so happen that the energy gap between the S1 state and the lowest vibration energy state of T1 (∆EST) is very small. The molecule might gain thermal energy from the surroundings and reach the isoenergetic point for the intersystem crossing. It might then cross over to S1 and then fluoresce to S0 state. This type of emission was first observed in a deoxygenated solution of eosin in glycerol and ethanol solution and is thus called E-type or eosin-type delayed fluorescence. If ∆EST is large, then there cannot be such reverse intersystem crossings. However, two triplet state molecules can interact with each other to give one excited state molecule at S1 and one non-excited state molecule at S0. The excited state molecule then fluoresces to return back to ground state. This type of fluorescence was first observed in the pyrene and phenanthrene solution and is therefore called P-Type delayed fluorescence. This is a biphotonic process because it depends on two different molecules to reach into triplet states and thus its intensity is proportional to the square of the intensity of absorbed incident radiation. Both these fluorescence processes are called delayed because they occur fairly slow and their timescales are comparable to phosphorescence. However, their spectrum is similar to normal fluorescence signals as the transition is from S1 to S0 states.
The intensity of fluorescence signals is dependent on the FQY (Fluorescence quantum yield) values as it is seen in Eq. (1). FQY is defined as the ratio of the fluorescence rate constant to the sum of the rate constants of all the processes participating in depopulating the excited singlet state. Mathematically, it is expressed as
where \(\:{k}_{f}\:\)is the fluorescence rate constant, \(\:{k}_{IC}\) is the rate constant for internal conversion, \(\:{k}_{ISC}\) is the rate constant for inter-system crossing and the final summation term represents the cumulative quenching effect from ‘i’ quenchers each of which depends on the quencher concentration \(\:{n}_{i}\:\)and the quenching rate constant \(\:{k}_{i}\).
2.6 Fluorescence quenching and vibrational relaxation
All the processes discussed till now were intrinsic to the excited molecule and required no participation from any bath gas molecule. Apart from these unimolecular processes, there exist bimolecular pathways by which an excited molecule can give up energy and avoid fluorescence. This is called fluorescence quenching. When an excited molecule and a foreign molecule approach each other, they interact in a way so that the excited molecule loses energy and ends up in the ground singlet state thus effectively reducing the fluorescence yield.
In addition, some collisions might only lead to relaxation of the excited molecules down the manifold. The vibrational energy lost due to several collisions with the surrounding bath gas molecules might manifest itself as a rise in kinetic energy of the collider species. This interaction is called vibrational relaxation and it does not eliminate fluorescence as the donor still remains in the excited singlet state unlike fluorescence quenching where the molecule ends up in the ground singlet reducing the fluorescence yield. In the later sections, it will be seen that vibrational relaxations bring about the pressure dependency of fluorescence yield and it can be positive or negative depending on the nature of the molecule and the excitation wavelength.
An important process of fluorescence quenching is the electronic energy transfer from the initially excited donor (D) molecule to the acceptor or quencher molecule (A). If the energy transfer takes place by the emission of a photon from the donor and then subsequent absorption by the quencher or the acceptor molecule then this process is called radiative transfer. The radiative process is a trivial process as it involves only emission and reabsorption. It takes place over longer distances, the intermolecular distances being typically larger than the wavelength of the interacting photon. This transfer requires an overlap of the emission spectrum of donor and the absorption spectrum of the acceptor. This might lead to radiation imprisonment resulting in a decrease of the fluorescence intensity in the overlap region and a distortion in the emission spectrum of the donor molecule. The following is the mechanism of radiative transfer where the asterisk represents an excited state molecule.
If the two molecules interact and the energy is exchanged before there is any emission then the process is called a non-radiative transfer. In such circumstances, the energy difference between the energy levels of the donor and acceptor are similar and are therefore in resonance. Therefore, it is also called resonance energy transfer. The intermolecular distances are less than the wavelength typically between 80 and 100 Å [71]. There are two mechanisms by which non-radiative transfer can be explained – a long range dipole-dipole interaction or Forster’s mechanism and a shorter-range electron exchange interaction or Dexter’s mechanism. The interaction energy can be split into a coulombic term and an exchange term. Forster’s mechanism takes place through coulombic interaction which at long ranges is dipole-dipole and at shorter ranges also includes multipole interactions. The donor molecule gets de-excited to the lower energy level whereas the energy transferred is manifested as the excitation of the acceptor molecule to a higher energy level. For, exchange interaction, electron is exchanged between the donor and acceptor during the energy transfer. This happens due to the spatial overlap of the electronic clouds making it effective in extremely short ranges. For allowed transitions of a molecule like the singlet-singlet transition, both coulombic and exchange interactions are observed but the coulombic interaction usually dominates. For forbidden transition, where a molecule shifts between triplet and singlet states, the coulombic interaction is negligible making exchange interaction the dominant player. The below equations [3] correspond to the interaction between a donor and an acceptor with ‘1’ representing singlet state and ‘3’ representing the triplet state. Asterisk represents an excited molecule. The following reactions are allowed via Forster’s mechanism-
None of the molecule undergoes a conversion from singlet to triplet or vice versa. The following reactions are allowed via Dexter’s mechanism-
Here, both singlet-singlet and singlet-triplet conversion of the quencher is seen in contrast to the Forster’s mechanism. Another frequently observed quenching is that caused by a quencher molecule whose ground state is triplet like that for oxygen molecule. In such cases, the collision between the excited donor and oxygen results in the formation of a short-lived complex such as \(\:{{\left(D,{O}_{2}\right)}^{*}}_{3}\) which then breaks apart to give the final products in which the singlet donor is transferred to a triplet state and the oxygen molecule is either transferred to an excited singlet or remains in the triplet state. The equations of this mechanism are provided here.
This forbidden transition is made possible by the spin-orbit coupling interaction and then there is exchange of charge. The exchange interaction mandates a close approach of the oxygen molecule and the excited molecule essentially making oxygen quenching diffusion controlled [72]. This process can only happen when there is a significant amount of energy gap between the singlet and triplet energy states of the donor molecule which exceeds the amount of energy needed to excite oxygen to its singlet state (0.98 eV) [3]. This is the case for aromatic molecules like toluene because of which they experience heavy oxygen quenching in an oxygenated environment. The quenching of aromatic molecules can be explained by the above mechanism. However, for ketones, the gap between excited singlet and triplet states is very small. Because of this, ketones have very fast ISC rates and experience very less quenching from oxygen molecules. However, it is observed that in the presence of oxygen, ketones show a reduced fluorescence. Nau and Scanaio (1996) [74] showed that the above mentioned mechanism for oxygen quenching has a weak influence on ketones and the resulting triplet state of a ketone due to oxygen molecule interaction is referred to as ‘oxygen assisted inter-system crossing’. The extremely small lifetimes of ketones in the excited state in comparison to the aromatics is also another factor for the much-reduced sensitivity of oxygen quenching in ketones.
In a certain bimolecular process, an excited molecule can be quenched by another molecule in a ground state of the same kind. Also, an excited molecule can be quenched by another excited molecule of the same kind. Such type of quenching is normally referred to as self-quenching. The excess energy of the excited molecules is manifested as internal energy and both the molecules remain in the ground state. The following equations represent self-quenching [72]. Self-quenching was observed for biacetyl [75], toluene [76] and naphthalene [77]. Fuhrmann et al. (2016) [76] attributed the self-quenching in toluene by collision with excited state molecules.
In Sect. 2, only a brief discussion of the underlying photophysical processes observed in an excited state molecule is provided. Most of the discussions here is referenced from [3, 71,72,73]. For more information, one can always refer these sources.
3 Tracer based LIF
Selection of a proper tracer is very crucial for the accuracy and success of LIF measurements. It is found that commercial grade gasoline does emit some fluorescence signals upon laser excitation due to the presence of several components capable of fluorescing. Hence, the fluorescence emitted is a complex mixture of signals emitted by each component. The fluorescence signals from these constituent compounds have varying dependencies on pressure, temperature, excitation wavelength and bath gas composition. The fluorescence intensity thus depends on the relative amounts of constituent compounds and the gasoline composition also differs with different batches of fuel [42]. These make it very difficult to characterize the absorption and fluorescence behaviour of commercial gasoline. Therefore, gasoline is unsuitable for quantitative PLIF measurement. However, gasoline can be used for qualitative fuel distribution visualization [78]. Gasoline was also used for semi-quantitative studies under cold engine operating conditions in the works of Weaver et al. [42] as the distillation of actual fuel becomes important in such studies. A common problem faced while using gasoline is the fouling of transparent liner which limits optical access into the engine cylinder [78, 79]. This requires frequent cleaning of the cylinder walls and piston face. To reduce fouling, Kim et al. (1997) [79] used a mixture of iso-octane as the base fuel and 10% gasoline as the fluorescing component.
For quantitative measurements, the fluorescence signal must be correctly interpreted so that it provides useful information about tracer number densities. Thus, it is attractive to use a single compound as a tracer with well-studied and documented fluorescence relationship with temperature, pressure, and excitation wavelength along with a non-emitting base like iso-octane which mimics gasoline. Ideally, a tracer should emit strong fluorescence signals that is directly proportional to the quantity of interest and independent of ambient conditions. Ideally, the tracer should also mimic completely the surrogate in which it is doped in terms of droplet formation, evaporation, convection, diffusion, reactivity, and reaction rate [3]. However, it is not possible for the tracer compounds used in PLIF studies to satisfy all the requirements. Here, we provide some information of the physical properties of iso-octane and tracers like acetone, 3-pentanone and biacetyl in Table 1 (from ref [80]).
A large Stokes shift between the absorption and fluorescence spectrum of a tracer molecule is desirable. This would allow to separate the scattered incident laser radiation from the emitted fluorescence signals using suitable filters. Also, this would prevent the reabsorption of the emitted fluorescence signals. It is good to have a tracer that has a decent value of absorption cross section at wavelengths which can be accessed easily by commercially available laser systems. If the absorption is very low, the fluorescence signal intensity will be very weak compromising the signal-to-noise ratio (SNR) of the system. If the absorption is too high, then the mixture will not be optically thin. Fujikawa et al. (1997) [32] tried to limit the laser extinction to around 3% (by limiting dopant concentration) using ketones as tracers to ensure optically thin mixture even at the compression TDC where the mixture density and hence the laser extinction will be the highest. A large amount of laser extinction is also observed in conditions where the mixture has not completely vapourised like in DI studies involving sprays in the region of interest [83], exciplex studies [84] etc. To reduce the effect of large laser extinction by liquid presence, the laser beam is split into two and the two separate beams enter the cylinder through two opposite directions [84].
Several ketonic compounds possess potential for fluorescence imaging based on their fluorescent properties. Lozano et al. (1992) [85] conducted a study comparing the fluorescence of biacetyl and acetone in a turbulent axisymmetric jet. Their findings indicated that in their experimental setup, acetone exhibited higher fluorescence than biacetyl, resulting in a superior signal-to-noise ratio. Additionally, acetone’s high volatility enables its seeding in high quantities, thereby enhancing the fluorescence signal level, making it a preferred molecule for tracing gaseous alternative fuels in IC engine application. Examples of such studies are discussed in the beginning of Sect. 4. However, in conventional internal combustion engines, surrogate fuels used are typically non-gaseous, with iso-octane commonly utilized in spark-ignited engines. Similar evaporation characteristics of tracer and iso-octane is one of the most important factors to effectively trace iso-octane. Figure 6 (from Johansson et al. [86]) compares the vaporization characteristics of acetone, biacetyl and 3-pentanone iso-octane, revealing that acetone’s volatility renders it unsuitable for tracing iso-octane. Biacetyl emerges as a better option than acetone, while 3-pentanone is deemed most suitable from this perspective. Following this, an overwhelming number of mixture distribution studies were performed using 3-pentanone/iso-octane mixtures some of which will be briefly discussed in the beginning of Sect. 5. Table 2 contains the information of photophysical properties for acetone, 3-pentanone and biacetyl.

3.1 Preferential vaporization of tracers
Le Coz et al. (1994) [101] found that in a mixture of 5% (by vol.) 3-pentanone in iso-octane there is a preferential vapourization of 3-pentanone so much so that the mole fraction of 3-pentanone in vapour phase is twice than that in the liquid phase. Davy et al. (2003) [102] studied the evaporation of a 3-pentanone/iso-octane binary system and found that co-evaporation of both the components will occur if the composition resembles that of an azeotrope (32% (by vol.) 3-pentanone mixed at 293.15 K and 101 kPa). The lower the composition of 3-pentanone than the azeotropic composition, more will be the preferential evaporation of 3-pentanone. For composition greater than azeotropic there will be preferential vapourization of iso-octane. Also, with increasing temperature, the azeotropic composition shifts towards a larger volume percentage of 3-pentanone. The preferential vapourization of 3-pentanone at lower concentration is due to the repulsive interaction between major and minor components and with increasing 3-pentanone concentrations, there is more interaction among 3-pentanone molecules partly compensating the repulsive interactions [103]. Since the laser extinction will be too high, 3-pentanone is normally not used in azeotropic concentrations. Lin and Sick (2004) [103] further found that since toluene and iso-octane have similar polarity, they do not form an azeotrope and co-evaporate better in comparison to 3-pentanone. In contrast to 3-pentanone, the co-evaporation improves for toluene with iso-octane with increasing temperature. However, due to oxygen quenching in the case of toluene especially at elevated pressures in IC engines, either nitrogen has to be used instead of air or the FARLIF technique has to be used. Also, the applicability of toluene-FARLIF at simultaneous high pressure and temperature conditions in IC engines is questionable [38].
To address this issue, multi-tracer LIF methods have been proposed. In this, a system of tracers would be used to mimic the different volatile fractions. Han and Steeper (2002) [104] proposed a ternary mixture comprising of 3-pentanone, 3-hexanone and iso-octane in 1/9/90 (vol%). The idea is that since 3-hexanone evaporates slower than iso-octane, the remaining liquid mixture when devoid of 3-pentanone due to early evaporation will still show fluorescence due to 3-hexanone upon vapourization. Tong et al. (2001) [105] studied the impact of different volatility tracer/surrogate mixture on in-cylinder fuel-air mixture formation during cold start operating conditions. The injection was during intake stroke and low volatility case showed a larger spray penetration, a thicker spray core due to larger droplet sizes from poorer evaporation. This resulted in an even greater impingement on liner. The evaporation seemed to be in the batch-distillation mode [106].
Stevens et al. (2007) [108] proposed a new six component surrogate fuel for representing three different volatile groups (two fuel component each) and three tracers acetone, toluene and 1,2,4 trimethylbenzene (TMB) representing the three volatile fractions. Williams et al. (2010) [109] used the same six component fuel and three tracer combination to study the fuel-mixture formation for both early (280 CAD BTDC of compression) and late injections (140 CAD BTDC of compression). The authors found that at 60 CAD BTDC of compression stroke the mixture distribution for acetone and toluene was quite uniform as it had got a longer time to mix owing to the in-cylinder turbulence but some inhomogeneity could still be observed for TMB fluorescence image. For late injection, the fuel mixture was convected in an anti-clockwise manner due to an in-cylinder tumble motion and by 60 CAD showed significant inhomogeneity due to a lower mixing time. However, this time all the three tracer fluorescence images showed a high degree of similarity. The authors then concluded that the similarity in distribution of the three components suggest that the in-cylinder fuel vapourization was rapid ( evaporation in liquid phase diffusion limited regime [106]) and thus a single tracer could be used instead for that particular condition. Zhang and Sick (2007) [110] used acetone and p-xylene with iso-octane to represent the low and high volatile fractions and they also found similar distribution for both the tracers in compression stroke. Although, a single tracer/surrogate mixture is not a perfect representation of the multi-component fuel, nonetheless PLIF studies using them have provided significant information about mixture formation processes in IC engines. Similar multi-component tracer imaging were performed to study fuel sprays in engine relevant conditions using aromatic tracers [111], to study preferential vapourization using a pair of aromatic tracers [112], to identify optimal tracer in primary reference fuels of iso-octane and n-heptane for HCCI engine application [113] and will be discussed in part B.
3.2 Thermal stability of tracers
For the tracer to correctly represent the surrogate fuel, it is very important that the tracer molecule is thermally stable at elevated pressure and temperature conditions in IC engines compression stroke and during the flame development and propagation stages. Leininger et al. (2006) [114] reported that only 0.33% of 1-methylnaphthalene decomposed after 1 h at 10 MPa and 673 K showing its high thermal stability. Smith et al. (1944) [115] studied the thermal decomposition of acetone till 888 K in various bath gases at atmospheric pressure. Thurber et al. (1997) [59] noted pyrolysis above 1000 K at atmospheric pressure. Grossman et al. (1996) [116] observed pyrolysis of both acetone and 3-pentanone in a test cell at 573 K and concluded that after 10 min of residence time, 10% of the injected compound had dissociated. Pyrolysis usually begins with an abstraction of a hydrogen radical or a methyl radical from ketones [117] and biacetyl [118] molecules. Therefore, either acetyl radical or an acetonyl radical is formed both of which contain ketonic group that is expected to fluoresce upon laser irradiation. This becomes especially important in calibration test cells where the residence time can be much longer in seconds (1 s in [119], 0.9 s in [120] etc.) as compared to IC engines where the pressure and temperature elevation occurs typically in milliseconds. Therefore, Trost et al. (2013) [117] studied the effect of pressure and temperature in the pyrolysis of acetone, 3-pentanone and toluene and tabulated the residence times in which no pyrolysis was observed. Table 3 shows the results in seconds at various pressure and temperature regimes. <1 s in the table shows tracer pyrolysis could be avoided only for extremely short periods. From the table, it is clear that toluene is thermally more stable than the ketones. 3-pentanone shows a lesser stability than acetone.
3.3 Impact on tracer-surrogate mixture properties
Tracer addition to the surrogate fuel also changes the physical and chemical properties of the mixture. For example, Schiessl and Sommerer (2018) [121] have explained that with addition of a tracer of lower molar heat capacity, as the seeding percentage increase, the molar heat capacity of the mixture itself decreases which will result in an increase in temperature by the end of compressions troke. The increased temperature will itself cause an increased rate of chemical reactions resulting in faster combustion. Also, tracer addition might itself lead to change in chemical kinetics. Addition of tracers can lead to a change in the equivalence ratios maintained that itself may result in variation of flame speeds. Neij et al. (1994) [122] found that 2% or 5% addition of 3-pentanone to iso-octane would only change the flame speed by less than 20%. Zhang et al. (2004) [123] studied the effect of addition of toluene (3% by vol.) and 3-pentanone (5 and 15% by vol.) in iso-octane and found that for homogeneous operation in SIDI engine at least for near stoichiometric cases, there is not much impact on engine performance and flame propagation. Sick and Westbrook (2009) [118] numerically studied the mixture of 10/% biacetyl with iso-octane. They found that the spatial separation of biacetyl and iso-octane profiles is about 11 μm within a flame front at 10 bar pressure and thus for combustion with flame front propagation biacetyl is a reliable marker. However, for combustion mode that proceeds on a distributed scale like that observed in HCCI combustion early tracer consumption might introduce appreciable error in fuel distribution quantification [3].
Sick and Westbrook (2009) [118] also studied biacetyl/iso-octane mixture in HCCI combustion modes. They found that in HCCI autoignition environments, there is significant difference in the consumption rates of biacetyl and iso-octane, the trend of which might even reverse with temperature. Therefore, using biacetyl fluorescence to quantify fuel concentration might even lead to errors of about hundreds of percent without having some prior information of temperature. Fitzgerald et al. (2008) [124] studied the impact of addition of 3-pentanone and acetone in iso-octane and n-heptane from the point of view of HCCI combustion. They observed that for iso-octane, acetone addition delays the main combustion period (around 1.5 CAD) whereas 3-pentanone advances it by over 1 CAD. However, the impact of tracer addition on n-heptane was much more pronounced with a delay in combustion phasing by around 6 CAD. This was due to an early decomposition of tracer molecules which then reduce the radical populations and inhibit the low temperature heat release phase of n-heptane. Schiessl and Sommerer (2018) [121] have numerically studied the impact of toluene addition in iso-octane at low (350 K) and high (420 K) initial temperatures before compression. At the end of compression stroke, for lower temperature toluene retards the reaction due to low radical formation and its subsequent consumption of the radicals produced by iso-octane decomposition. At high temperatures, toluene itself produces radicals and thus greatly helps in accelerating the reaction. It is thus advisable to perform some baseline experiments to quantify the changes (if any) in engine performance due to tracer addition.
3.4 Photolytic dissociation of tracers
In addition to the above discussed concerns about tracer/surrogate mixtures, it is a well-documented fact that tracers like acetone, 3-pentanone and biacetyl undergo photolytic dissociation when excited by UV lasers. Ketonic photolysis was reviewed by Haas et al. (2004) [125]. Photolysis of acetone is due to the result of cleavage of α-CC bonds [126]. Biacetyl has a higher probability of photolysis at shorter wavelengths around 266 nm [3]. For longer wavelengths like that of 355 nm which is commonly used in PLIF studies in IC engines, photolysis of biacetyl is less of a concern [127]. Photolysis has normally been ignored in PLIF studies till now. However, with the advent of high speed laser (in kHz range) with high pulse energy, impact of photolysis might become more relevant [128]. From next section, fluorescence characteristics of individual tracers will be discussed in detail.
4 Acetone
Acetone as a tracer has been used by several researchers to successfully find out the distribution of fuel-air mixture, temperature and residual air [129] inside IC engines. Its high volatility makes it suitable for mixing studies in gases and SI engines using alternative fuels like hydrogen and CNG. Yip et al. (1994) [130] used combined OH and acetone PLIF to visualise combusting flows where OH-PLIF marks the zone of chemical reactions and acetone-PLIF signals mark the unburnt fuel region. Being a ketone, acetone has been extensively used in both port fuel injection (PFI) and direct injection (DI) modes of SI engines. Hiltner and Samimy (1996) [94] examined fuel-air mixture distribution in a spark-ignited natural gas PFI engine, noting significant spatial inhomogeneity during intake stroke which is found to reduce in the compression stroke due to turbulent mixing. Acetone-PLIF also sheds light on the effects of injection timings in both PFI and DI engines. In PFI engines, fuel can be injected in the intake manifold either during intake valve open (IVO) or intake valve close (IVC) conditions. Wolff et al. (1994) [131] quantitatively determined mixture distribution, revealing that IVO injection induces more inhomogeneity and higher fuel droplet content, resulting in slower flame growth. Brault et al. (1998) [132] performed acetone PLIF in a CNG version of the Honda VTEC-E lean burn engine for both IVO and IVC injections. They found that during IVC injection, since a fuel rich mixture is already present in the manifold, a rich mixture enters initially upon intake valve opening followed by a very lean mixture in the late intake stroke. During IVO injection, an initially lean mixture enters due to some fuel carry over from the previous cycle followed by a fairly rich mixture as the injection begins in IVO condition. For more detailed discussions on IVO and IVC injections in PFI engines, one can refer Nayek and Mittal (2023) [46].
Early research on injection timing in DI engines highlighted the development of homogeneous and stratified modes. Injecting fuel during the intake stroke yields a homogeneous mixture as the injected fuel has more time to evaporate and mix with the in-cylinder flow. Conversely, injecting fuel during the compression stroke allows little time for mixing, resulting in a stratified mixture. This stratification can be advantageous for low-load operation, featuring a lean mixture overall with a rich ignitable mixture near the spark plug. Itoh et al. (2006) [133] performed PLIF studies to evaluate the mixture formation in wall guided, air guided and spray guided versions of DI engine. Toyota [134] introduced an air-guided concept using helical and straight intake ports fitted with a swirl control valve (SCV) and verified fuel stratification by acetone based LIF studies [135]. Similarly, Volkswagen [136] and BMW [137] also verified their wall guided and spray guided stratified mixture formation concepts using PLIF studies. Acetone PLIF imaging has also found its utility in hydrogen DI engines. Kaiser and White (2008) [85, 93] used it to study the mixture formation process at different injection timings in a swirl plane (Fig. 7).The schematic in (Fig. 7) shows hydrogen jets, valve orientation and spark plug location. Left of the (Fig. 7) shows both instantaneous and ensemble-averaged mixture distribution images for all injection timings. Early injection displays uniform fuel distribution, while intermediate and late injections exhibit significant fuel stratification near the injector. In single shot images, intermediate injection shows some homogeneity in the fuel-rich region, while late injection results in pronounced stratification without uniform mixture distribution. Later on, Salazar and Kaiser (2009) [138] extended the study to understand the influence of various injector designs, injector locations and injection timings on fuel distribution. They found that increasing the number of injector holes dilutes fuel concentration due to enhanced mixing with ambient air, but an excessive number of holes, such as with a 13-hole injector, led to spray collapse due to jet-jet interaction In a subsequent work, Salazar and Kaiser (2010) [139] further studied the spray-flow interaction using a single hole injector.

From the discussion of the above acetone PLIF works, it is clear that acetone has been extensively used in IC engines. Next, we focus on its fluorescence characteristics.
4.1 Absorption cross-section
Acetone has a broadband absorption spectrum ranging from about wavelengths of 225 nm to 320 nm at room temperature of 25⁰ C with a peak value of 4.7 × 10− 20 cm 2 in the region between 270 nm and 280 nm in the ambient temperature [85]. Absorption occurs due to the symmetry forbidden n→𝛑* transition of an electron from a non-bonding orbital localized around oxygen to an anti-bonding molecular orbital around the ketonic group. Figure 8 shows the molecule of acetone. The ketonic group and the two methyl groups can be discerned. It also shows the absorption spectrum as a function of temperature from shock tube experiments [87]. The figure contains the comparison of results obtained from the work of Thurber [140]. The spectrum shows an apparent redshift with temperature along with an increase of the absorption cross-section values. The peak values of the spectrum is also found to occur at redshifted wavelengths.

This behaviour can be explained by the fact that with an increase in temperature, the molecule is vibrationally excited causing the electron to be in a higher vibrational energy level of the ground state electronic level. This reduces the energy required for making the transition resulting in the red shifting of spectrum. With increased vibrational energy in the ground state due to higher temperatures, the Franck-Condon overlap integrals are enhanced. This is because the molecules in the vibrationally excited ground electronic state have bending or asymmetric stretching modes which have better Franck-Condon overlap with the bent singlet and triplet excited states [141]. This increases the likelihood of transition which is reflected as the increasing cross-section values with temperature. Koch et al. (2008) [87] used a gaussian profile for the curve fitting purpose of cross-section at different temperatures between 230 nm and 330 nm. The expression developed was
where A is the amplitude, \(\:{\lambda\:}_{c}\:\text{i}\)s the centre wavelength and w is the characteristic width, all of which are a function of temperature. The previous figure describes about the overall development of the spectrum. However, for understanding fluorescence signal intensity variation, the variation of absorption cross-section at specific wavelengths with temperature needs to be studied. Thurber at al. (1998) [142] characterised the cross-section values at various wavelengths (Fig. 9). The curves are normalised with their respective values at 295 K. It is seen that the shorter wavelengths are largely temperature insensitive as compared to the longer ones which show a monotonic increase with temperature. This behaviour can also be verified from the absorption spectrum of (Fig. 8).

4.2 Fluorescence signal variation
Acetone fluorescence spectrum ranges from 350 nm to 550 nm with peaks at 445 nm [85] (Fig. 10) while an excited acetone molecule drops down from S1 singlet state to the ground S0 state. The fluorescence spectrum is spectrally red shifted from absorption spectrum making it a prospective tracer for PLIF studies. The signal throughout the spectrum domain is integrated to obtain fluorescence signal intensity. The absolute FQY of acetone is 0.00084 at 308 nm, 1.013 bar N2 and 298 K [91]. All the figures reported in this work for fluorescence signals and FQY values are obtained from studies which were performed at a constant tracer number density.

Left image (from ref [32]): Fluorescence spectrum of acetone obtained at 266 nm excitation. Adapted from [32] with permission from SAE International. Right image (from ref [142]): Normalised fluorescence signals at various wavelengths plotted as a function of temperature for which the pressure mantained was atmospheric. Adapted from [142] with permission from Optical Society of America
Fluorescence intensity is a function of temperature and pressure. For acetone, there is a very high probability of transfer to triplet state from an excited singlet state during inter-system crossing. Lifetime in the excited singlet state is therefore short which results in a low sensitivity to bath gas quenching [92]. The dependency of fluorescence signals on temperature was found to dominate over pressure dependencies in the works of Ghandhi and Felton (1996) [143]. (Figure 10) shows the variation of fluorescence signal intensity with temperature at atmospheric pressure for various wavelengths from the works of Thurber et al. (1998) [142]. The measurement was performed using a heated cell for temperatures from 295 K to 1000 K. It is found that the signal decreases with temperature for shorter wavelengths and the decrease becomes smaller as the wavelength is increased. At 320 nm, the signal is found to increase and reaches a peak value near 1000 K. The dramatic increase of signals at 320 nm and a slight increase for 300 nm can be due to the effects of increasing absorption cross-section at these wavelengths. The fluorescence signals are a product of both absorption cross-section and fluorescence quantum yield. So, a study of FQY variation with temperature becomes important. In order to remove the effects of absorption cross-section, the signals are divided with the cross-section values obtained at different temperatures. Since the cross-section values at shorter wavelengths were seen to be more or less constant, the fluorescence quantum yield values are expected to decrease following the decreasing signal trend. Variation of FQY with temperature at constant atmospheric pressure in nitrogen bath gas is shown in (Fig. 11) [142]. The fluorescence yield reduces with increasing temperature. The shorter the wavelength, the greater is the reduction. 248 nm shows a very steep reduction.

Acetone fluorescence has a pressure dependency. The dependency also varies with excitation wavelength and the bath gas. Thurber and Hanson (1999) [89] prepared a photophysical model for acetone fluorescence. This model predicted the fluorescence signals at varied pressures at ambient temperature for different excitation wavelength. The results of this model are plotted along with the experimental results obtained in a static cell experiment at room temperature (297 K) [89] (Fig. 12). Figure 12 also shows the fluorescence signal intensity variation for different wavelengths (248 nm, 266 nm and 308 nm) in a nitrogen bath gas. Plots are normalised to the data at each wavelength at atmospheric pressure. The figure shows that the fluorescence signal increases with increasing pressure. The increase is higher for shorter wavelengths and the increment reduces with increasing wavelength. 248 nm shows a significant increase whereas the 308 nm shows a slight increment in the beginning and then quickly settles down to a constant value. The solid lines show the model prediction which very closely agrees to the experimental values. For bath gas dependency, the experiment was performed with a constant wavelength of 248 nm at room temperature of 297 K for methane, helium and nitrogen bath gas. Figure 12 shows the fluorescence signals normalised to nitrogen fluorescence at 1 atm. The plots show that the increase in fluorescence signal with pressure is maximum for methane and is the least for helium.

(from ref [89]): Left image shows the normalised fluorescence signal intensity variation of acetone for various wavelengths with pressure in a nitrogen bath gas. Right image shows the fluorescence variation with 248 nm with pressure at different bath gas composition. Only non-quenching composition was used was comparison. Temperature was held constant at 297 K. The dotted lines are the step ladder model curves developed by Thurber and Hanson (1999) [89] which closely agree with the experimental data
Grossman et al. (1996) [116] performed experiments in a flow cell at a wavelength of 248 nm and at a temperature of 383 K. The bath gases used were nitrogen, oxygen and synthetic air. The authors found that for nitrogen, the fluorescence signal increases with pressure and levels off by 8 bar. For oxygen, after the initial increase, the fluorescence signal falls off at a rate of 17% per 10 bar. For synthetic air, the signal increases till a larger pressure value as compared to oxygen and the falls thereafter upon further pressure increase. Thus, oxygen containing bath gases lead to a fall in fluorescence signals after an early rise. The magnitude of rise and the pressure at which the subsequent decrement is found both reduce with increasing oxygen content. Thurber and Hanson (1999) [89] also studied the effects of bath gas composition which could cause fluorescence quenching. (Figure 13) shows the normalised fluorescence signal dependence with different bath gases at 248 nm and 308 nm at room temperature. The signal normalisation was done for nitrogen value at 1 atm.

(from ref [89]): Left image shows the fluorescence signal variation of acetone for 248 nm and the right image shows for 308 nm with increasing pressure and at 297 K. The bath gases selected were nitrogen, air and oxygen to compare the quenching effects with oxygen concentration. The dotted lines are the step ladder model curves developed by Thurber and Hanson (1999) [89] which closely agree with the experimental data
The result for 248 nm for oxygen and air are very similar to the works of Grossman et al. (1996) [116]. The plot for oxygen shows an early peak and decrease near about 3 atm while the air values start levelling off near 8 atm. For 308 nm, the pressure dependence reduces as can be seen in (Fig. 13). The increase in fluorescence signal for oxygen is barely visible accompanied by a very early fall in signal levels. The behaviour observed in air bath gas seems to be intermediate to the behaviour observed in oxygen and nitrogen bath gases. Similar plots were also obtained for 266 nm, but are not shown for brevity. The behaviour for 266 nm was intermediate to the plots for 248 nm and 308 nm for all the bath gases. It was discussed that the absorption cross-section remains constant with pressure and so any change in fluorescence signals with pressure will effectively be because of variation in FQY. Therefore, the pressure dependencies of fluorescence signal intensity will qualitatively mimic the FQY variation with pressure. Yuen et al. (1997) [144] used 266 nm wavelength to study the fluorescence signal dependence for different pressures at various bath gases in a flow cell experiment. The temperature was maintained at 273 K. It was found that fluorescence quantum yield value increases with pressure from 0.6 atm till 5 atm. For methane, nitrogen and helium, the fluorescence quantum yield was constant after 5 bar within experimental uncertainty. For air, the fluorescence quantum yield reduces after 5 atm of pressure. Figure 14 [89] shows the variation of FQY with partial pressures of acetone diluted with nitrogen bath gas such that the total pressure was 1 atm at different excitation wavelengths of 248 nm and 308 nm. It was found that the FQY value increases with pressures at 248 nm. For 308 nm, the increase was very slight. The dotted line compares the value obtained from the photophysical model of acetone. It is clear that the pressure dependency FQY becomes more prominent at lower wavelengths like it is in the case for temperature dependency.

(from ref [89]): Figures show the variation of FQY of acetone with pressure normalised to the value at 0.1 atm for 248 nm (top image) and 308 nm (bottom image) in a nitrogen bath gas at 297 K. FQY was found to increase with pressure like the signal intensity. The dotted lines are the step ladder model curves developed by Thurber and Hanson (1999) [89] which closely agree with the experimental data
Discussion
Fluorescence intensity variation with temperature, pressure and excitation wavelengths can be explained by considering the effects of several photo-physical processes like absorption, inter-system crossing, oxygen assisted inter-system crossing, vibrational relaxation, fluorescence etc. In Fig. 15, the excited singlet state is denoted as S1 and the ground state as S0. The thick dash lines refer to the ground state of a particular electronic energy level as well as the thermalised levels. The thin horizontal lines refer to the vibrational energy levels. The arrows show several processes like fluorescence, absorption, vibrational relaxation (VR) etc.
When acetone is excited with laser irradiation, depending upon the excitation wavelength, it gets transferred to a certain vibrational energy level of the excited S1 electronic level from the thermalised level of S0. The molecule reaches higher vibrational levels with reduced wavelengths. The vertically upward arrow in Fig. 15 shows this absorption phenomenon. Ossler and Alden (1997) [145] suggested that even intermediate size molecules like acetone and 3-pentanone behave as large molecules with many atoms where the gap between vibrational energy levels is very less as the excited molecule ends up at higher vibronic levels of the excited singlet state. Under such situations, vibrational relaxation becomes fast enough to bring down the molecule to a lower vibronic energy level of the excited singlet state. The smaller series of arrows pointing downwards in Fig. 15 show transition from the upper to lower vibronic levels of S1 and thus represent energy loss through vibrational relaxation. Hansen and Lee (1975) [92] showed that the non-radiative processes like intersystem crossing and internal conversion have very high rates. For simple ketones, these rates are about two orders of magnitude higher than rates of radiative processes. They further showed that for ketones with n→𝛑* transition, the inter-system crossing is the most dominant non-radiative process. In Fig. 15, this is shown with horizontal arrows extending from S1 manifold. Halpern and Ware (1970) [146] showed that for hexafluoroacetone, the non-radiative decay rates increase with higher vibronic levels. The fluorescence rates remain constant with increasing vibronic levels in S1 singlet state while the non-radiative rates increase with the vibronic levels for a number of ketonic compounds as shown by Breuer and Lee (1971) [147]. Therefore, at higher vibronic levels in the excited state, the probability of a molecule to fluoresce is less yielding low fluorescence quantum yield values. With decreasing inter-system crossing rates at lower vibronic levels, the fluorescence yields increase.
The aforementioned works significantly improved the understanding of ketone photophysics leading to the development of several models that have incorporated the various physical insights provided by the works discussed in the previous paragraph. Yuen et al. (1997) [144] proposed a 4-level model to explain the pressure dependency of acetone they found in their work. They further concluded that at atmospheric pressures, the vibrational collisions are not fast enough to completely thermalize the vibrational populations.

Thurber et al. (1998) [142] proposed a multi-step decay model to explain both the temperature and pressure dependencies of acetone fluorescence. The model has since then become the most widely used to study acetone photophysics and will be the one that will be discussed in this section. Various other researchers have brought in modifications to optimise several parameters in it to extend the applicability to extended regimes of combined high pressures and temperatures. This type of decay model has also been proposed for 3-pentanone fluorescence [148]. The relevant parameters had to be reoptimized separately for 3-pentanone by performing relevant calibration experiments. However, it has to be noted that such a multi-step decay model is yet to be seen for biacetyl. The model is schematically represented in Fig. 15. There are a few assumptions used in this model. Only three photophysical processes are considered here and their rates are denoted by: fluorescence (\(\:{k}_{f}\)), vibrational relaxation (\(\:{k}_{\text{coll}}\)) and non-radiative processes (\(\:{k}_{\text{N}\text{R}}\)). Since for ketones the ISC rates is significantly much faster than the IC rates due to collisional quenching, the non-radiative rates is typically assumed to be the ISC rate although at higher laser excitation energy, photodissociation and internal conversions might become appreciable. Also, the ISC is considered to be irreversible. Due to the broad distribution of the fluorescence spectrum and the dense spacings of the vibrational energy modes in the temperature ranges considered, the distribution of excited state levels is considered to be continuous. Also, the model assumes only energy loss through collisions which is approximated by a downward cascade to the vibrational level where the molecule sits in thermal equilibrium (ΔEthermal). However, it is to be noted that only the broad physical description of the model will be provided here. For a detailed discussion of the parameters used in this model, the empirical relationships of the rate constants with the vibrational energy levels, one can refer to the original works [89, 142].
In order to evaluate FQY (\(\:\varphi\:\)), the model uses Eq. 20 to calculate the sum over all N vibrational levels in the excited singlet state occupied by an average molecule as it decays. The decay of the molecules from higher vibrational levels is considered to be a multi-step process as shown in (Fig. 15). Level 1 represents the initially excited vibrational level in the excited state manifold and level N is a vibrational level that is very close to the thermalised level (ΔEthermal) at which the summation is stopped. For a certain vibrational level E, contribution to the total FQY is calculated by the effective FQY from that level, which shows the probability of fluorescence in contrast to other decay pathways, multiplied by the likelihood of the molecule decaying to level E before fluorescing or undergoing intersystem crossing. In Eq. 20, the first term represents contribution from the vibrational level N, the last term accounts for the contribution from the thermalized level, and the summation term consists of contribution from the various intermediate levels.
At higher temperatures, the acetone molecule in S1 singlet state is located at higher vibrational energy levels. Thus, higher the temperature, higher is the probability of inter system crossing which consequently lowers the FQY value. This trend can be observed in (Fig. 11) where the FQY monotonically decreases with temperature. With lower excitation wavelengths, the molecule will be excited to higher vibrational energy levels owing to higher energy content per photon. This trend can also be seen in (Fig. 11) where 248 nm excitation shows the largest drop in FQY and 320 nm shows the least drop. To understand the behaviour of fluorescence signal, it is important to keep in mind that it is a product of both FQY and absorption cross-section. In (Fig. 10), there is a drop in signal levels with temperature for shorter wavelengths and increase in signals for longer wavelengths. It can be seen from (Fig. 9) that the absorption cross-section for shorter wavelengths are constant with increasing temperature. So, for shorter wavelengths, the drop in signal value is due to the drop in FQY with increasing temperature. But for longer wavelengths, absorption cross-section increase with temperature and the drop in FQY is less. Thus, the increase in absorption cross-section more than compensates for the drop in FQY values giving an increase in signal strengths.
To understand pressure dependencies, we need to keep in mind that the absorption cross-section values are pressure independent. Any dependency of fluorescence signal with pressure is solely from the result of pressure dependency of FQY. With higher pressures, the intermolecular vibrational relaxation rates increase due to increased collisions. Upon collision, the molecule in its excited state loses energy and reaches the lower vibronic levels of S1 state. At lower vibronic levels, the inter-system crossing rates are lower increasing the FQY value. Since larger pressures result in increased collisions and increased vibrational relaxation, this causes the increase of FQY values with increasing pressures. This trend can be seen in (Fig. 12) for non-oxygen containing bath gases. Upon using different excitation wavelengths, the lower wavelengths excite the molecule to a higher vibronic level of S1 state. This makes the system more sensitive to vibrational relaxations. Therefore, the fluorescence signal values show a greater increase with pressure upon excitation with shorter wavelengths. Longer wavelengths like 308 nm, excite the molecule to vibronic levels very close to the thermalized level of S1 state. Because of this a very small increase is seen in signal levels for 308 nm. This trend can be seen in (Fig. 12). At very high pressures, the collisions are large enough to completely bring the excited acetone molecule to the thermalised level of the excited electronic state. After this, any further increase in pressure is not going to increase the fluorescence signal. So, for bath gases not containing oxygen, the fluorescence signals will keep on increasing until it saturates at a high-pressure limit value. Thurber and Hanson (1999) [89] plotted the model predicted absolute values of FQY with pressure variation in a nitrogen bath gas as shown in (Fig. 16).

Figure 16 shows that the absolute FQY values keep on increasing from 248 nm to 308 nm as already explained. All the curves monotonically increase with pressure and merge with each other at a high-pressure limit. This justifies the fact that the plots for 248 nm show a maximum increase and for 308 nm show a minimum increase with pressure in (Fig. 12) and (Fig. 14). The plot for 266 nm is intermediate to 248 and 308 nm.
Just like the high-pressure limit of FQY values, the model also predicts an existence of a low-pressure limit. At very low pressures, the vibrational relaxation will be negligible and the fluorescence and inter-system crossing will be from a particular vibronic level of the first excited state which the electron occupies when excited by a suitable laser frequency. This vibronic energy level is determined by the laser wavelength and temperature. Hence, the low-pressure limit will be dependent on excitation wavelength and temperature. The high-pressure limit is however dependent only on temperature. The low-pressure limit for FQY is smaller for smaller wavelengths.
The collisions with bath gas molecules resulting in the vibrational relaxation also depend on the nature of the collider molecule. For excited polyatomic molecules, the effectiveness of collisions increase with the number of atoms in the collider molecule [89]. So, in (Fig. 12), it is seen that the fluorescence signal increases with pressure at the highest rate for methane which is polyatomic followed by nitrogen (diatomic) and then helium (monoatomic). For excitation at the same wavelength, a collider with higher collisional effectiveness will make the fluorescence signal approach its high-pressure limit sooner i.e. at lower pressures. For bath gases containing oxygen, apart from vibrational relaxation with increasing pressures, at high enough pressures, oxygen interacts with tracer molecules to form a complex. This complex formation enhances inter-system crossing [74]. Thus, oxygen presence brings an additional deactivation channel which is popularly known as oxygen quenching in the literature.
In oxygen containing bath gases, upon increasing pressure, there is a competition between oxygen quenching and vibrational relaxation. At 308 nm excitation, the molecule is almost vibrationally relaxed. Thus, vibrational relaxation has a very limited ability to increase FQY whereas the added pressure increases oxygen quenching. As a result, in (Fig. 13), a slight increase in signal followed by a rapid decrease in fluorescence signal with pressure is seen. At 248 nm, since the molecule is excited to a higher vibronic level, there is a significant amount of vibrational relaxation. Thus, upon increasing pressure, a comparatively larger amount of signal increase is obtained. This behaviour is observed in (Fig. 13). The effect of quenching will be more evident for longer wavelengths excitation. Quenching is dependent on collision with oxygen molecules. Thus, higher concentration increases the probability of collision. The difference in pure oxygen and air bath gas is the amount of quenching. Since the concentration of oxygen is lesser in air as compared to pure oxygen bath gas, it shows a lower amount of quenching. This explains the lower peak signal value and the appearance of this peak at a lower pressure in oxygen bath gas (Fig. 13).
Till now, all these discussions were focussed on either room temperature high pressure or ambient pressure high temperature behaviour. Inside IC engines, the compression stroke produces very high pressures and temperatures. Extrapolation of cell data to combined high pressure and high temperature conditions by simple multiplication of the temperature and pressure effects is erroneous. This is because the temperature and the pressure effects are not multiplicative and cannot be separated from each other [149]. Upon increasing the temperature or decreasing excitation wavelength, the effect of pressure on fluorescence behaviour will be more pronounced as now the molecule which is located at higher vibronic levels in the excited state, will have more probability for vibrational relaxations due to collisions, the magnitude of which depends on the surrounding pressure. Now, at longer excitation wavelengths and lower temperature, the molecule will be located in lower vibronic levels of the excited state. Here, there will be less room for collisional relaxation showing less pressure dependence. Therefore, high temperature dependency is accompanied by high pressure dependency and vice-versa. Because of this, it becomes important to use fluorescence models to predict high temperature high pressure fluorescence signal variations. However, it was observed that models optimised from the cell data with high pressure low temperature and low-pressure high temperature data like the step decay models of [142] for acetone and [148] for 3-pentanone are unable to capture the fluorescence characteristics at simultaneous high-pressure high temperature data [120, 149, 150]. Hence, model re-optimisation with high pressure and high temperature data is required. In the subsequent section, the literature available for fluorescence characteristics at simultaneous high temperature high pressure conditions is reviewed.
4.3 Combined high-pressure and high-temperature regime
Ossler and Alden (1997) [145] studied the effective lifetime of first excited electronic state of acetone at various pressures and temperatures using a picosecond laser of 266 nm wavelength. They measured the effective lifetimes and presented their results in the form of isotherms with increasing pressures for nitrogen and air as bath gases. Since effective lifetimes correlate with the FQY values, the plots also represented the nature of absolute FQY variations. They found that isotherms for nitrogen show an increase in effective lifetime values with increasing pressures and the rate of increase slowed down at higher pressures. Thus, nitrogen has a stabilising effect on fluorescence. Isotherms for air also show an increasing trend with pressure. No reduction in lifetime values were observed in this work. Loffler et al. (2010) [119] studied the behaviour of acetone fluorescence in elevated temperature and pressure. They plotted the intensity of fluorescence signals at wavelengths of 248 nm and 308 nm (Fig. 17) in a nitrogen bath gas. The plots are normalised at 1 bar and 295 K. The range covers a pressure from 0.2 bar to 20 bar and a temperature range from 295 K to 728 K.

As the absorption cross-section is independent of pressure, the shape of every isotherm is strictly determined by the FQY pressure dependence. So, for both 248 nm and 308 nm, the shape of a particular fluorescence signal isotherm is very close to the shape of a FQY isotherm at that temperature and pressure ranges. The temperature effects of absorption cross-section do influence the curves. At 248 nm, we have seen that the absorption cross-section remains constant with temperature but increases with temperature at 308 nm. So, for 248 nm, the plots are dictated solely by the FQY behaviour. Hence, for 248 nm, fluorescence signal isotherms should look very similar to FQY isotherms when plotted on an absolute scale. From (Fig. 17), it is observed that for 248 nm, fluorescence signals increase along with pressure and the amount of increase decreases at higher pressures. Fluorescence signals reduce with increasing temperature as high temperature isotherms are found to be in progressively low signal regions of the plot. At lower temperature like 295 K, an approach to a high-pressure saturation limit is visible. At higher temperatures like 728 K, the approach to saturation is yet to be seen. This behaviour was also observed by Braeur et al. (2006) [120] where carbon dioxide was used as a bath gas.
It is witnessed that for 308 nm in (Fig. 17), fluorescence signals increase both with increasing temperature and pressures. High temperature isotherms are located in progressively higher signal regions of the plot. The increase of signal for 308 nm is found to be very less with pressure in comparison to that for 248 nm. This suggests that 248 nm excitation makes the system more pressure sensitive than 308 nm excitation. This difference in sensitivity at varied wavelengths was also seen in the works of Thurber and Hanson (1999) [89] in (Fig. 12). Since the behaviour of FQY isotherms can be directly observed from 248 nm excitation, the nature of the FQY isotherms become very similar to that found in the earlier work by [145].
Hartwig et al. (2017-a) [150] studied the FQY characteristics of acetone in a test cell for conditions of combined high pressure and high temperature. They studied the pressure effects on FQY in a nitrogen bath gas at high temperatures and produced plots for 400, 500, 600 and 700 K. In (Fig. 18), only 400 K and 700 K data are shown for comparison. Each isotherm is normalised to its respective FQY values at 0.101 MPa. Therefore, the curves cannot be visually compared on an absolute basis rather the plots compare the relative shapes. The sharpness by which the curves rise show the pressure sensitivity at that temperature. The black dotted lines refer to the curve fitted data at 295 K whereas the bold coloured curves are predicted by model proposed by Thurber and Hanson (1999) [89]. It is observed that Thurber’s model shows significant deviation from the experimental data especially at high temperature and high-pressure regimes. A similar over prediction of pressure effects by this model was also observed by Braeuer et al. (2006) [120] when they studied acetone fluorescence in a carbon-dioxide bath gas in a 248 nm laser excitation.

Hartwig et al. found that the FQY value increases with pressure. This was expected due to the increasing vibrational relaxation at increased pressures. Since the work was extended to even higher pressures of about 35 bar, the high-pressure saturation limits in nitrogen bath gas was observed even at high temperatures of 500 K (not shown here). This type of saturation was not observed in the previous figure (Fig. 17) as the curves were plotted till 20 bar against 35 bar of current study. Hence, it is expected that the saturation is yet to reach in Fig. 17. The curve for 400 K overshoots the curve at 295 K suggesting an increased pressure sensitivity. The curve for 500 K was found to be slightly above 295 K but within the experimental uncertainty. The curve for 700 K was found to lie below 295 K. Therefore, in moving from 295 K to higher temperatures, the pressure sensitivity begins to increase, reaches a maximum value near about 400 K and then begins to reduce.
Till now, non-quenching bath gas was discussed. For bath gas containing oxygen, an additional quenching effect is expected. Loffler et al. (2009) [119] obtained fluorescence signal plots for air at 248 nm and 308 nm (Fig. 19). For 248 nm excitation, it was found that at low temperature, with increase in pressure, there appears to be an increase in fluorescence signal until a certain pressure after which there is a fall with increasing pressure. This behaviour was seen in the previous works of Grossman et al. (1996) [116]. This decrease with higher pressures was found to vanish at high temperatures where the signal is found to monotonically increase with increasing pressures. This behaviour was also observed from the works of Ossler and Alden (1997) [145]. Just like nitrogen bath gas, fluorescence signal reduces with increasing temperature. The isotherms at higher temperatures give progressively low fluorescence signal values. For 308 nm, the fluorescence signal increases with temperature, a trend that is opposite to 248 nm and also seen in the corresponding nitrogen figure (Fig. 17).

Unlike 248 nm, here a steady decline in signal levels can be observed after an initial rise. The initial rise period and the corresponding relative peak signal value attained reduces with increasing temperature and the pressures at which this peak is obtained also diminishes with increasing temperature so much so that at 698 K, there is hardly any rise in signal levels with a steady decline starting approximately from atmospheric pressures. At high pressure regions, the isotherms appear to diverge. The authors also measured the fluorescence signal variation for high temperature and high pressure at 248 nm and 308 nm excitation for a 1:1 mixture of air and burned iso-octane (plots not shown for brevity). Burnt iso-octane was used to simulate EGR mixture. The results could be used for calibration while measuring temperature and intake air fraction distribution inside the combustion chamber of an IC engine. The mixture consisted of 74.2% nitrogen, 11.4% oxygen, 7.9% carbon-dioxide, and 6.5% water vapor. It was found that at 308 nm, the behaviour of fluorescence signal intensity was very close to the behaviour in air as a bath gas. This was because of the presence of oxygen in lower concentration in the mixture like air. At 248 nm, it was found that the signal was stronger than nitrogen and air bath gases at lower pressures due to stronger vibrational relaxation from carbon dioxide owing to its polyatomic nature. At higher pressures, the influence of oxygen was identified as the signal was weaker than nitrogen but stronger than air as a bath gas.
Braeuer et al. (2006) [120] carried out an experiment to study fluorescence signal behaviour of acetone in air bath gas for 248 nm excitation. The pressure was varied till 20 bars and the temperature till 693 K. The results are normalised and shown in (Fig. 20). It is found that the fluorescence curve for 293 K rises with increasing pressure, reaches a maximum at 12.5 bars and then starts to decrease. This decrease was due to the increased quenching from oxygen due to higher collisions at higher pressures. However, this behaviour was not observed for isotherms at higher temperatures. This was also observed in the works of Loffler et al. (2009) [119] and Ossler and Alden (1997) [145]. Each isotherm is normalised to the fluorescence signal at atmospheric pressure at the temperature of the isotherm. Since the reference point is different for the isotherms, such plots compare the relative shape of each curve. At low temperature, there is drop in signal after reaching a peak value. At higher temperature, no drop in signal was observed. Curve for 474 K overshoots the curve at 293 K and the curve at 690 K remains under the 474 K curve. This trend shows that the isotherms in air bath gas become sharper with increasing temperature from 293 K, reach a peak value at 400 K and then keep on getting flatter on further increase of temperature. This same trend is observed in (Fig. 19) where a relatively flat curve can be seen for 748 K as compared to 295 K.

(from ref [120]): Normalised fluorescence signal curves at 248 nm in an air bath gas at various pressures and temperatures. Each curve is an isotherm. Data from Thurber and Hanson (1999) [89] have also been plotted for comparison. Reprinted from [120] with permission from the Optical Society of America
Discussion
In the studies so far, for simultaneous pressure and temperature variations, two types of plots were presented for fluorescence signal intensities and FQY variations- one, where the isotherms are normalised with respect to a fixed reference point (at a fixed pressure and fixed temperature), second, where each isotherm is normalised with respect to a changing reference point (at a fixed pressure and at the temperature of that particular isotherm). In the first case, the normalised isotherms closely resemble isotherms when plotted on an absolute scale. In the second case, the normalised isotherms can only be used to compare the shapes (relative steepness or flatness) of isotherms when plotted on an absolute scale. Thus, plots in the second case will demonstrate the pressure sensitivity of the plotted isotherms.
Since it was found that the fluorescence model developed by Thurber deviated strongly from the experimental data at high-temperature and pressure regime, Hartwig et al. (2017-b) [151] tried to modify the model by re-optimization of various parameters with high pressure, high temperature fluorescence data. They hypothesized that at increased temperatures, the collisions might become less effective resulting in a lower pressure sensitivity. A similar reasoning was also given by [120] who suggested that at high temperatures, the density dependent probability of collisions decreases due to a decreasing number density of the bath gas at high temperatures. Both [120] and [151] modified the oxygen quenching rate by expressing it as a product of probability of quenching and the collision frequency. The probability of quenching was given an exponential dependence on vibrational energy levels suggesting that with increased temperature or shorter wavelength where a molecule is pumped to higher vibrational levels of the excited singlet states, the efficiency of quenching after collision with oxygen will increase but the probability of collision with oxygen itself will decrease due to reduced number densities with temperature. Based on these improvements, Hartwig et al. (2017-b) [151] computed a plot for absolute values of FQY as a function of pressure, temperature and excitation wavelengths (Fig. 21) in a nitrogen bath gas. Isotherms at three different temperatures (295 K, 600 K and 900 K), two excitation wavelengths (320 nm and 225 nm) were plotted along a very large pressure range.
Like (Fig. 16), two different pressure limits- high pressure and low pressure can be identified for FQY. The low-pressure limit is wavelength dependent whereas the high-pressure limit is wavelength independent. The low-pressure limit is determined by the vibronic level in which the electron is excited from the ground state. The excited state vibronic level is determined by temperature and energy content of the photon. Therefore, low pressure limit changes for different combinations of temperature and excitation wavelength. But high-pressure limit is found to vary only with temperature. This limit for FQY is achieved when fluorescence arises from the vibrationally relaxed level of excited singlet state. The vibrationally relaxed level in turn is determined by the surrounding temperature. Higher the temperature, higher is the energy of the vibrationally relaxed level because of the modified Boltzmann distribution in the excited electronic state. Therefore, high pressure limit changes with temperature. Since non-radiative decay rates are faster from the vibronic levels which are higher in energy; with increasing temperature, the high-pressure limit decreases as can be seen from (Fig. 21). Hartwig et al. suggested that even at high pressures where the collisional relaxation process dominates over non-radiative decay rates, it can never completely eliminate the effect of non-radiative decay processes.
In (Fig. 17), the fluorescence signal plots for 248 nm excitation will be independent of absorption cross-section and will solely be determined by the FQY behaviour. At high temperatures, the FQY high pressure limits are lower and at a particular pressure, the FQY value reduces monotonically with increasing temperature. Hence, the isotherms for fluorescence signals are shifted progressively downward with temperature. In (Fig. 21), high temperature isotherms show asymptotic behaviour at very high pressures. Thus, in (Fig. 17), the low temperature isotherms show an asymptotic behaviour within 20 bars but there is an interesting possibility that the high temperature isotherms which do not show saturation within 20 bars might need still higher pressures to display asymptotic behaviour.

(from ref [151]): Model predicted FQY values for acetone at different wavelengths for varying pressure and temperature. The different high pressure and low-pressure limits can be clearly discerned
In (Fig. 17), the isotherms at 308 nm will have an effect of increasing absorption cross-section which shows an increase with temperature. Hence, in spite of the reducing FQY values, the increasing cross-section more than compensates the reduction and as a result the fluorescence signals increases with temperature at a constant pressure. So, the fluorescence signal isotherms are positioned progressively higher for higher temperatures. In (Fig. 21), the isotherms for longer wavelengths are quite flat unlike for 225 nm. This flatness shows a reduced sensitivity to pressure increase. Since at longer wavelengths, the excited state vibronic level occupied lies very close to vibrationally relaxed level, there is a very little room for collisional relaxation. This causes poor sensitivity to pressure increase. Hence in (Fig. 17), the isotherms for 308 nm are flatter than those for 248 nm. The probability of collision reduces with increasing temperature [119, 120]. This reduces the pressure dependencies at higher temperatures. As a result, isotherms normally remain flat with respect to pressure changes at higher temperatures and remain steep at low temperatures. This can be very well discerned by comparing the steepness between isotherms at 295 K and at 798 K where the isotherm for 798 K shows little rise with pressure in contrast to that of 295 K. Although, the curves are steep at low temperatures, strictly speaking, this steepness of curves doesn’t reduce monotonically with increasing temperatures as already seen for the nitrogen bath gas at 248 nm in the studies of Hartwig et al.(2017-a) [150]. The authors observed a peak in pressure dependency at a temperature of 400 K for nitrogen bath gas. With increasing temperatures, the molecule is in a higher energy vibronic level which increase the pressure sensitivity due to increased probability of collisional relaxation. This increasing sensitivity competes with the decreasing pressure sensitivity due to poor collisional probability at higher temperatures and as a result an increase in collisional relaxation is observed with temperature increase from room temperature followed by a peak in collisional processes and finally a decline with further temperature increase.
(Figure 19) shows a trend reversal in fluorescence intensity isotherms for 248 and 308 nm with temperature due to absorption cross-section effects as already discussed in the nitrogen case. In air bath gas, there is an additional quenching effect by oxygen which gives a very different behaviour than oxygen devoid bath gases. It was proposed by Braeuer et al. (2006) [120] that apart from the collisional dependence, oxygen quenching rates also depend on the vibronic energy level with a higher quenching probability at a higher vibrational energy level. Quenching due to collisions is only affected by pressure whereas the temperature contribution for quenching will remain same for a particular isotherm. So, any visible quenching at higher pressures is due to increased collisions. In (Figs. 18) and 19, at 248 nm there is a significant room for collisional relaxations. Vibrational relaxation and oxygen quenching compete with each other making quenching effects less visible. At lower temperatures, a rise in signal followed by a peak and then constant drop is observed for air bath gas. The reduction in signal at higher pressures is due to increased quenching rates. At higher temperature, this drop in signal vanishes with the isotherm showing a monotonic increase in fluorescence signals. This is due to the fact that collisions become lower at higher temperatures reducing the quenching rates. In order to see quenching effects, a still higher pressure should be needed where the decreased quenching due to lesser collisions at higher temperature can be compensated by more collisions at higher pressures.
The maximum pressure sensitivity at 400 K and then subsequent fall at higher temperatures in air bath gas in (Fig. 20) can be explained as the initial rise was due to the competition between higher vibrational relaxation and the later fall was due to efficient oxygen quenching at higher temperatures [120]. In (Fig. 19), for 308 nm, the quenching effects are clearer due to negligible vibrational relaxations. Hence, a certain rise in signals followed by a peak and then monotonic reduction is seen in all the isotherms. With increasing temperatures, owing to higher quenching efficiency and less efficient vibrational relaxations, the rise becomes progressively smaller and the quenching process becomes dominant than the collisional relaxations at lower pressures. This shows a shift of the peaks to the lower pressures at higher temperatures so much so that at 748 K, there is effectively no rise in signal level owing to very less collisions at high temperatures with a significant quenching due to the vibronic energy dependence.
5 3-Pentanone
In order to perform PLIF studies using iso-octane, a tracer with similar vapourization characteristics is required. Acetone has a lower boiling point and is thus suitable to mark gaseous fuels. Hence, 3-pentanone has grown very popular for PLIF in SI engines using gasoline owing to its similar boiling characteristics with iso-octane. Johansson et al. (1995) [86] were one of the first to use 3-pentanone fluorescence for imaging fuel distribution in a PFI engine. They could relate the cycle-to-cycle variation in combustion to a variation in mixture distribution at the spark plug. Quantitative studies were also performed by Berckmuller et al. (1997) [40] in a lean burn PFI engine and found that cycles with richer mixtures at the spark plug produced higher combustion pressures. To further study mixture formation and its CCV, Hokimoto et al. (2017) [33] used simultaneous PLIF and TR-PIV (time resolved-particle image velocimetry) and determined the instantaneous fuel mixture distribution alongwith the velocity fields obtained from PIV, (Fig. 22) (adapted from ref [33]). The area near the spark plug, where the flame is believed to develop is presented by a dotted circle in the − 7 CAD ATDC images. It was observed that the equivalence ratio at low loads cycles is quite lean inside the dotted circles which led to slower flame growth. Another interesting phenomenon observed for lowest load cycles is a vortex structure opposing the main tumble flow. This opposing flow is represented with a red vector. The effect of this reverse flow is to restrict the fuel to the right side of the spark plug causing poor mixing. Thus, the dotted area becomes increasingly devoid of fuel mixtures and such negative flows are detrimental to mixing process and increase the cycle-to-cycle variation in combustion. Haramiishi et al. (2020) [96] also performed simultaneous PLIF and TR-PIV and found that CCV in combustion is due to CCV in mixture distribution and turbulence kinetic energy (TKE) distribution (affected by flow field). In their study, the variation in TKE was caused due to variation in tumble vortex centre and was more important than variation in fuel concentration from the point of view of combustion CCV as in PFI engines, the fuel gets more time to mix with air leading to lower CCV in fuel distribution. However, CCV in mixture distribution are expected to be more important for DI engines due to lesser mixing times and a greater dependence on spray morphology. Recently, small-bore PFI engines have garnered attention due to their prevalence in two and three-wheeler segments in the developing world. Pradheep et al. (2016) [34] and Garg et al. (2020) [152] have conducted PLIF studies in such engines.

Kampanis et al. (2006) [97] investigated fuel distribution in a small-bore DI engine with a 5-valve arrangement, varied intake injection timing, and swirl flows. They observed significant piston surface impingement leading to rich regions nearby. At 1500 rpm, a weak tumble directed the rich mixture cloud upwards, while at 3000 rpm, increased spray dispersion slightly reduced inhomogeneity. High swirl conditions significantly reduced fuel film and enhanced vaporization, reducing inhomogeneity. The impact of in-cylinder flow fields on mixture distribution was further studied by Kim et al. (2013) [83]. They found that for early injection, the mixture distribution developed differently but by the late compression stroke was quite similar for both tumble and swirl flows in engine heated up conditions. For late injection, the tumble flow was found to be slightly more effective than swirl for stratified charge formation. Also, the swirl flow had a more pronounced impact on fuel sprays than the tumble case. A notable distinction of DI engines from PFI ones operating with liquid fuels is the significant charge cooling effect. Liquid droplets vaporize, absorb heat from the in-cylinder charge and cool it. Attar et al. (2014) [153] conducted PLIF imaging for fuel distribution and two-line PLIF thermometry to measure in-cylinder charge temperature, estimating a 34 K temperature drop for a stratified fuel-air mixture at 250 CAD ATDC intake. Charge cooling increased with higher fuel injection quantities. Temperature measurement is crucial for HCCI engines since the temperature stratification has implication on controlled autoignition. Rothamer et al. (2008) [55] used two-line technique and developed a negative PLIF or N-PLIF method to quantify residual hot exhaust gas distribution in HCCI engines. They observed high EGR presence near exhaust valves during early intake stroke and noted a correlation between high-temperature regions and EGR presence, with EGR temperatures reaching up to 550 K near the exhaust valve. These studies highlight the widespread use of 3-pentanone for PLIF studies with liquid fuel, leading to the discussion of its fluorescence characteristics.
5.1 Absorption cross-section
Figure 23 shows the schematic of 3-pentanone molecule. 3-pentanone belongs to the same ketonic family as acetone. It contains two ethyl groups on both sides of the ketonic functional group. Therefore, most of the photo physics that has already been discussed for acetone is also applicable for 3-pentanone.

Schematic diagram of a 3-pentanone molecule
Figure 24 [87] shows the absorption spectrum of 3-pentanone as a function of temperature. A broadband absorption spectrum is obtained like that for the acetone molecule due to densely spaced vibronic levels in excited electronic level. It varies from about 230 nm to 320 nm with a maximum value of 5𝚡10⁻²⁰ cm² at 275 nm under ambient conditions [87]. The spectrum is found to get redshifted with increasing temperatures. The cross-section values also increase due to better vibronic coupling. Figure 24 plots the cross-section values at 4 different wavelengths. Just like acetone, at shorter wavelengths like 248 and 266 nm absorption cross-section values show little variation with temperature whereas at longer wavelength like 308 nm and 284 nm, cross-section values increase with temperature. A similar curve fit was developed for 3-pentanone like the one for acetone. A similar reasoning as that for acetone can be given for explaining 3-pentanone absorption spectrum behaviour.

(from ref [87]): Left: Shows the change in the absorption spectrum of 3-pentanone with temperature. Comparison with the work of Koch and Hanson (2003) [31] is also provided. Right: Absorption cross-section of 3-pentanone values are plotted with temperature for a number of wavelengths. Adapted from [87] with permission from Elsevier
5.2 Fluorescence signal variation
Fluorescence signal emitted from excited 3-pentanone has a broadband shape ranging from 300 to 550 nm [32] with a peak at 420 nm [90]. Koch (2005) [154] in his thesis reported the change in 3-pentanone fluorescence spectrum with pressure and wavelength. With increasing pressure and wavelength, the spectrum became narrower. The pressure effect can also be seen in (Fig. 25) where a reduction in FWHM (Full width at half maximum) was found for spectrums obtained at different pressures and at a constant temperature of 333 K with an excitation wavelength of 266 nm. The slight narrowing of spectrum can be explained due to the narrow energy distribution of emitting molecules and greater vibrational relaxations at higher pressures [145]. However, the excitation wavelengths have no effect on the peak of the fluorescence spectrum [31]. The absolute FQY value of 3-pentanone is 0.0011 at 308 nm, 1.013 bar N2 and 298 K [91].

(from ref [145]): Evolution of fluorescence spectra of 3-pentanone at 1) constant temperature and varying pressures (left image) and at varying temperature (right image) for 266 nm laser excitation
A variation of the fluorescence spectrum with temperature (Fig. 25) were also found in the works of Ossler and Alden (1997) [145]. A frequency quadrupled Nd-YAG laser was used. Peaks were found in the emitted signals due to the scattering of 266 nm and the second harmonic 532 nm. With increasing temperature, the spectrum gets shifted to the shorter wavelengths. The authors found the biggest change in spectrum at high temperature and high-pressure regimes. Different bath gases like air and nitrogen produced similar changes. With change in pressure, the fluorescence spectra grow in terms of signal strength [90] and with increasing temperature, the spectra diminish [145]. These variations are not seen in (Fig. 25) as the spectra are normalised to their respective maximum value thus only showing their contraction and shifting. Modica et al. (2007) [90] showed the variation in fluorescence intensity with pressure with the help of fluorescence spectrums in absolute scales. They obtained spectrums in a nitrogen bath gas at a fixed temperature of 473 K for three different pressures (0.1, 1 and 3 MPa). With increasing pressures, the peak value of spectrum increased indicating stronger fluorescence emissions. The peak of the spectrum varied between 415 and 430 nm depending on the ambient pressures. The authors [90] also found that the spectrum changes with excitation wavelengths. For 248 nm, the spectrum is between 315 and 575 nm; for 266 nm, between 350 and 550 nm and for 308 nm, between 350 and 580 nm.
Relative fluorescence signals were measured for 3-pentanone with three excitation wavelengths (248, 277 and 312 nm) in synthetic air in a temperature range of 383–573 K at a constant pressure of 1 bar by Grossman et al. (1996) [116]. They found that for 248 nm excitation, the fluorescence signals reduced by 35% per 100 K. For 277 nm, the signals reduced by 15% per 100 K and for 312 nm, the signals increased by 80%. The authors confirmed that the decrease in fluorescence signals at shorter wavelengths was not due to decomposition of 3-pentanone by gas chromatographic measurements. They attributed the variation in fluorescence signal with temperature to absorption cross-section variations with temperature. Fluorescence signal plots on a per molecule basis are shown for three different wavelengths in (Fig. 26) from the work of Koch and Hanson (2003) [31]. The plot shows a variation of fluorescence signal intensity with temperature till 900 K and the intensity plotted is normalised to the respective intensity at 297 K. All the curves thus originate from unity at 297 K. Results from previous works of Fujikawa et al. (1997) [32], Tait and Greenhalgh (1993) [155], Ossler and Alden (1997) [145], Wolff et al. (1995) [156] and Grossman et al. (1996) [116] were compared. The signal behaviour is found to be very similar to acetone. For shorter wavelengths like 248 and 266 nm, there is a steady decline in fluorescence signals. Whereas, for longer wavelengths like 308 nm, the signal first increases, reaches a peak value and then decreases with further increase in temperature.

(from ref [31]): Relative fluorescence signal intensity of 3-pentanone plotted for varying temperature, normalised to signal levels at 297 K,1 atm pressure in nitrogen. Three different excitation wavelengths are used- 248 nm, 266 nm and 308 nm
Figure 27 [31] shows the variation of fluorescence quantum yield (FQY) with temperature. The plots are normalised with the respective FQY values at 297 K. Hence, all the curves originate from unity at 297 K. There is a steady decline in FQY values with temperature. The lower the wavelength, higher is the temperature sensitivity.

(from ref [31]): Relative FQY of 3-pentanone plotted for varying temperature, normalised at 297 K, 1 atm pressure in nitrogen. Three different excitation wavelengths are used- 248 nm, 266 nm and 308 nm
Figure 28 shows the dependence of fluorescence signal intensity with pressure for air and oxygen bath gas from the work of Grossman et al. (1996) [116]. The normalised plots are taken at 248 nm laser excitation with squared curve representing air and circled plot representing oxygen bath gas. For nitrogen bath gas (not shown in the figure), the signal increases with pressure and levels off at 5 bar. For air bath gas, the signal increases with pressure, reaches a peak at 25 bar after which the signal intensity reduced by 10% per 10 bar. For oxygen bath gas, a slight increase in signal with a further decrease of 17% per 10 bar was observed.

(from ref [116]): Fluorescence signal intensity of 3-pentanone dependence with pressure is plotted. Left image shows the effect of different bath gases at 248 nm excitation at 383 K- square curve for air and circle curve for pure oxygen. Right image shows the effect of wavelength at 473 K- square curve is for 312 nm and the circle curve is for 248 nm
Figure 28 also shows the normalised fluorescence signals plotted for increasing pressures at a constant temperature of 473 K. Two different excitation wavelengths were used- 248 nm(circle) and 312 nm(square) in synthetic air bath gas. At 248 nm, increase in signal is found with increasing pressure whereas for 312 nm, there is a decrease in signal right from the beginning with no apparent signal rise. Similar studies were performed for fluorescence studies at 277 nm and 312 nm. At 383 K, for 277 nm, there is a slight increase in signals with pressures up to 2 bar and then there is a decrease of about 40% at 50 bar. For 312 nm, no initial increase in fluorescence signal levels were found. Similar results were obtained for 473 and 573 K. Furthermore, fluorescence signals were also measured for a mixture of 80%:20% of iso-octane and 3-pentanone from a point of view toward IC engine application. It was found that the signals increase with pressure till 5 bar and then remains constant till 25 bar. Figure 29 (Koch PhD thesis (2005) [154]) shows the variation of relative FQY with pressures at a constant temperature of 296 K at three different wavelengths of 248, 266 and 308 nm. High pressure limit was taken as the reference value. As the reference data was same for all the plots, these curves resemble plots for absolute FQY. The plot also includes experimental data from the work of Koch et al. (2004) [91]. The bath gas used is nitrogen. Plots show the FQY values increase with pressure. Increase of FQY is found to be maximum for 248 nm and least for 308 nm. Shorter wavelength excitation produces lower FQY as also seen in the works of acetone and previous plots found for temperature dependency of 3-pentanone. This signifies that pressure dependency increases with shorter wavelengths. Furthermore, all the plots seem to converge at higher pressures. This shows the existence of a high-pressure limit which is wavelength independent just like acetone.

Figure 30 shows the variation of FQY with pressure in 3 different bath gases (air, nitrogen and pure oxygen) at a fixed temperature of 296 K. Plots were normalised taking the high-pressure limit value for nitrogen as reference. Data is shown for 248 nm and 308 nm. The behaviour in nitrogen is similar to Fig. 29 and has already been discussed. However, for oxygen containing bath gases like air, the behaviour is markedly different than nitrogen. Oxygen is found to have dual effect- it contributes to vibrational relaxations as well as quenching for shorter wavelengths and only quenching in longer wavelength. At 6 bars, FQY is dependent on the amount of oxygen in bath gas but it shows wavelength independence as FQY in pure oxygen bath gas is 65% of high-pressure limit for both the wavelengths. For air as a bath gas, FQY values are almost same as nitrogen bath gas till 1 bar. But at 6 bar, the signal in air decreased about 10%. At 308 nm, only quenching effect of oxygen is evident. Behaviour at 266 nm excitation is intermediate to both 248 and 308 nm and hence, the curves are not shown here for brevity.

(from ref [154]): FQY values of 3-pentanone normalised to the high-pressure limit is plotted for varying pressures at 296 K with the effect of different bath gases. Left image is for 248 nm excitation and right is for 308 nm excitation. Adapted with permission
Fluorescence signals are a product of absorption cross-section and FQY. Therefore, in Fig. 26, the decrease in intensity at shorter wavelengths is mainly due to the decrease of FQY with temperature since the absorption cross-section at shorter wavelengths were found to be more or less constant at 248 nm and slightly increasing at 266 nm with temperature in Fig. 24. For 308 nm, the increase in fluorescence intensity at lower temperatures is due to the increase in absorption cross-section that more than compensates the effect of decreasing FQY. At still higher temperatures, the drop in FQY value is large enough to cause a net decreasing effect in the fluorescence signal. Owing to the similarities between acetone and 3-pentanone fluorescence behaviour, Koch and Hanson (2003) [148] proposed a photophysical model for 3-pentanone in line with the acetone photophysical model proposed by Thurber et al. (1998) [142]. With low excitation wavelength or increasing temperature, a 3-pentanone molecule will be excited to a higher vibronic level of the excited singlet state. At higher vibronic levels, the non-radiative decay mode becomes faster and as a result the FQY reduces. So, low excitation wavelengths and high temperatures reduce FQY as can be seen in Fig. 27. The wavelength dependence can be seen in the Figs. 29 and 30. On comparison, it is clearly seen that shorter wavelengths give lower FQY in all the bath gases.
The pressure dependency on fluorescence intensity can be explained on a reasoning similar to that for acetone. With increasing pressure, collisions become more probable resulting in faster vibrational relaxation. This causes the molecule to decay into lower vibronic levels of the excited singlet state. Since the non-radiative decay rates are slower for lower levels, FQY increases with pressure. This can be seen in all the previous figures where FQY increases with pressure for various bath gases. Just like acetone, there is also a wavelength independent high-pressure limit as seen in Fig. 29. Oxygen containing bath gases introduce quenching effect at higher pressures. These effects are more pronounced for lower wavelengths as was the case for acetone. A similar explanation can be given for 3-pentanone also. Since 3-pentanone fluorescence was studied particularly from an engine point of view, behaviour at simultaneous high pressures and high temperatures are desired for different bath gases. The model proposed by Koch and Hanson (2003) [148] was used to predict fluorescence signals in an IC engine. Koban et al. (2005) [149] compared the fluorescence signal as calculated from the predicted FQY value by the model at 248 nm along with the fluorescence signal experimentally obtained from the engine. It was found that the model greatly overestimates the fluorescence signal. Thus, further insight into high pressure high temperature fluorescence characteristics of 3-pentanone was needed. For this, several experiments were performed to characterize the fluorescence behaviour in temperature and pressures relevant to the compression stroke of IC engines. These studies will be the subject matter of review in the following sub-section.
5.3 Combined high-pressure and high-temperature regime
Ossler and Alden (1997) [145] measured the effective fluorescence lifetimes of 3-pentanone excitation using a 266 nm picosecond laser at various temperature and pressures in nitrogen and air bath gas. They found that for nitrogen bath gas, the lifetimes increase with increasing pressure. Largest variations were found in low temperature low pressure regions. This increasing trend was shifted to higher pressures at higher temperatures. At lower temperatures, a high-pressure asymptotic value was found whereas for higher temperatures, the signal was still found to increase in the studied pressure ranges. At 323 K, the lifetime reached an asymptotic value at 0.2 MPa. Lifetime values were found to decrease with increasing temperature. For air bath gas, the behaviour was very similar to nitrogen at low pressures below 0.1 MPa. However, at lower temperatures, a drop in lifetime was observed at higher pressures. Figure 31 shows the variation of normalised fluorescence signals reported in the work of Modica et al. (2007) [90] for a temperature range of 373 to 573 K and for a pressure range of 0.1 to 4 MPa on a per molecule basis in a nitrogen bath gas. Since each isotherm is normalised to its fluorescence signal at 0.1 MPa, the reference value is variable and thus, the curves can only represent relative pressure sensitivities.

(from ref [90]): Normalised fluorescence signal intensity of 3-pentanone value isotherms are plotted for varying pressures and temperatures in nitrogen bath gas at 266 nm excitation
The authors reported a steady increase in fluorescence signals with pressure between 0.1 and 1 MPa which then reaches a saturation at 4 MPa for different temperatures. At high temperatures, the pressure effect on signal is stronger with an increment of 41% at 523 K and 66% at 573 K. The plots emphasize that the pressure sensitivity increases with temperature. The authors suggested that this increased sensitivity at higher temperatures is due to molecules occupying higher vibronic levels which will have a greater tendency to drop down to vibrationally relaxed levels at higher pressures. Apparently, the effect of reduced collisions at increased temperature is not yet visible unlike acetone where a peak in pressure sensitivity was observed. The reduction in pressure sensitivity might occur at temperatures larger than that used in the study. The high-pressure limit in nitrogen is also evident from the isotherms. Similar behaviour is seen in the works of Grossman et al. (1996) [116] where in 248 nm excitation in nitrogen at 383 K, the signals increase in between 0.1 and 0.5 MPa and saturate at 2.5 MPa. Further, Modica et al. found that fluorescence signals increase slightly with temperature, reach a peak and then start decreasing on further increase in temperature at pressures beyond 1 MPa. The increase was found in the temperature range of 373 to 473 K. The authors have suggested a reverse inter-system crossing from triplet to singlet state which occurs at low temperatures at low energy gaps between singlet and triplet states, reducing the net non-radiative rate. The phenomenon of delayed fluorescence thus increases the signal values. However, with increase in temperatures, signals are expected to monotonically decrease irrespective of the pressure at short wavelengths, a behaviour found in acetone and also found by [145] and [119]. Loffler et al. (2009) [119] didn’t find such increase of signals at lower temperatures and thus suggested that this increase might be due to a slight increase in absorption cross-section at 266 nm with temperature. They also suggested that since the camera integration time for signals was much longer in the works of Modica et al. (2007), it might be possible that the increase in signals was due to capture of phosphorescence in low oxygen environments [157]. This explanation seems plausible, as the delayed fluorescence proposed by Modica et al. has a time scale comparable to phosphorescence.

(from ref [90]): Normalised fluorescence signal intensity of 3-pentanone value isotherms are plotted for varying pressures and temperatures in carbon-dioxide, air and pure oxygen as bath gas at 266 nm excitation
Modica et al. (2007) [90] also carried out similar experiments in carbon-dioxide bath gas. They found that like nitrogen bath gas, high pressure limits can be observed for carbon-dioxide and the signal intensity is also found to increase with pressure. It is observed that the fluorescence saturation is obtained at lower pressures in carbon-dioxide than nitrogen bath gas. This suggests a more efficient vibrational relaxation in carbon-dioxide. Similar results were also seen in acetone. Thus, polyatomic bath gases are more efficient in vibrational relaxation than molecules containing lesser number of atoms. The authors compiled the results for different bath gases and plotted them after normalisation of the curves with their respective values for 0.1 MPa in Fig. 32. Figure 32 compares the variation of normalised fluorescence signal in carbon dioxide, air and oxygen with increasing pressures at 3 different temperatures on a per molecule basis. These 3 bath gases were selected to understand the effect of exhaust gas residuals on fluorescence signals in IC engines. Since each isotherm is normalised with respect to its value at 0.1 MPa, only the relative shapes of isotherms can be compared. Excitation wavelength used was 266 nm. It was found that carbon dioxide behaves like nitrogen and the curves get saturated at higher pressures. In air bath gas, fluorescence signals increase between 0.1 and 1 MPa. After this, the signals were found to either decrease slightly or remined constant. The most drastic effect of quenching is observed in pure oxygen bath gas where almost no rise in signals is evident.

(from ref [158]): Absolute FQY value isotherms for 3-pentanone are plotted for varying pressures and temperatures in nitrogen bath gas at 248 nm excitation (left image) and 308 nm excitation (right image). Solid lines represent values predicted by Cheung’s model and dotted lines represent values predicted by Koch’s model [154]
As it was observed that Koch’s model for FQY prediction failed to provide accurate results at high-pressure and high-temperature regimes, Cheung et al. (2012-b) [159] tried to optimise the model by using data at such regimes. Figure 33 shows the behaviour of FQY in an absolute scale at various temperatures and low pressures at 248 and 308 nm excitation obtained in the work of Cheung and Hanson (2012-a) [158]. At very low pressures, vibrational relaxation is very limited due to fewer collisions at low pressures. FQY is then solely determined by inter-system crossings. Such low-pressure data are important for determination of inter-system decay rates as a function of vibronic energy levels governed by excitation wavelengths and temperatures for preparing photophysical model. Error bars in Fig. 33 represent 6% uncertainty. It is found that FQY values become lower with increasing temperature and with decreasing excitation wavelengths. Data for 266 and 277 nm excitation show similar behaviour intermediate to 248 and 308 nm and are not shown here. As expected, due to fewer collisions at such low-pressure regimes, the pressure dependency is significantly reduced giving rise to straight line behaviour parallel to pressure axis. With increasing pressure, only a slight increase was found for 248 and 266 nm at 295 K. This pressure independence suggests the existence of a low-pressure limit similar to like that for acetone. As expected, the low-pressure limit is dependent on vibronic energy level and thus is dependent on both temperature and excitation wavelengths unlike the high-pressure limit which is wavelength independent as seen previously in Fig. 29. With increasing temperature and decreasing wavelength, the low-pressure limit value decreases.
In a subsequent work, Cheung and Hanson (2012-b) [159], experimentally determined the FQY values for combined high-pressure and temperature regimes. Figure 34 shows the data for nitrogen bath gas, Fig. 35 shows the data for air bath gas. Figure 34 [159] shows the behaviour of FQY at simultaneous high pressures and temperatures in nitrogen bath gas in an absolute scale at 248 and 308 nm excitation. It is observed that at 248 nm with increasing pressure, there is a monotonic increase in FQY as observed for acetone. This increase with pressure is due to the vibrational relaxation from high to lower vibronic levels where the FQY values are higher owing to slower inter-system crossing rates. With increasing temperature, FQY values reduce due to faster inter-system crossing at higher vibronic levels. At 248 nm, saturation vanishes at higher temperature also reported in the works of Ossler and Alden (1997) [145]. At higher temperature, the high-pressure saturation effect is expected to show at higher pressures because with increasing temperatures, the collision frequency reduces. Therefore, very high pressures are needed so that the increase in collisions due to pressure can outweigh the reduction in collisions at higher temperatures.

(from ref [159]): Absolute FQY value isotherms for 3-pentanone are plotted for varying pressures and temperatures in nitrogen bath gas at 248 nm excitation (left image) and 308 nm excitation (right image). Solid lines represent values predicted by Cheung’s model and dotted lines represent values predicted by Koch’s model [148]
At 308 nm, just like acetone, the FQY plots are found to be very insensitive to rising pressure because at longer wavelengths, molecules occupy vibronic levels very close to vibrationally relaxed levels. Since FQY at 308 nm do not increase much with pressure, data at 308 nm should be very close to the high-pressure limit. Thus, 308 nm data at various temperatures can be used to compare the variation in high pressure limits with temperature. From Fig. 34, it is clear that with increasing temperature, the high-pressure limit should fall as was also seen in Fig. 21 for acetone. Therefore, even at very high pressures, increased vibrational relaxations cannot completely eliminate the effect of increased inter-system crossings at higher temperature. For other bath gases like carbon-dioxide, helium, methane etc.; behaviour is expected to be more or less same with nitrogen since the low- and high-pressure limits are bath gas independent. The only difference will be observed in the sharpness of curves and the pressures at which saturation is reached. Bath gases with molecules containing higher number of atoms are expected to show a sharper rise in FQY showing an earlier departure from low pressure limit. High pressure limits will also be reached at lower pressures for such bath gases. Similar studies were conducted for air bath gas (Fig. 36). Figure 36 [159] shows the variation of FQY in absolute scales at 248 and 308 nm excitation. Plots for 277 and 266 nm show an intermediate behaviour and are not presented for brevity. The bath gas used is air. Pressure and temperature dependencies on FQY can be clearly understood from these plots. Error bars indicate 7% uncertainty. Comparison with other works are also provided for 248 nm.

(from ref [159]): Absolute FQY value isotherms for 3-pentanone are plotted for varying pressures and temperatures in air bath gas at 248 nm excitation (left image) and 308 nm excitation (right image). Solid lines represent values predicted by Cheung’s model and dotted lines represent values predicted by Koch’s model [154]. Data from the work of Grossman [116], Koch [154], Braeur [120] are also plotted for comparison at 248 nm excitation
It is observed that for 248 nm, there is effect of both vibrational relaxations and oxygen quenching. At lower temperatures, a rise in signal followed by a peak and monotonic decrease thereafter with increasing pressures is observed like in the previous works. At higher temperatures, the fall in signal with pressure for air bath gas vanishes for 248 nm excitation. This trend was also observed in previous works. Since with increasing temperature, the probability of collisions become lower as already discussed for acetone, we believe that the fall in signals should be observed at still higher pressures exceeding those covered in the experiment. For a particular temperature and pressure, FQY reduces with reducing wavelength. At 308 nm excitation, the molecule occupies a vibronic energy level very close to relaxed levels. Thus, only quenching effect is visible due to no apparent signal increase with pressures. At longer wavelengths, since there is little scope for vibrational relaxations, FQY values at such wavelengths are very close to the high-pressure asymptotic values. Thus, isotherms at low wavelengths should merge with isotherms at longer wavelengths at high pressures. Therefore, unlike nitrogen, there should not be a high-pressure limit in air. We believe that from Fig. 35, 248 nm isotherms should merge with 308 nm curves at sufficiently high pressures.
6 Biacetyl
Another tracer molecule that has found application in PLIF studies in IC engines is biacetyl. However, in comparison to acetone and 3-pentanone its use has been limited. Baritaud and Heinz (1992) [160] used biacetyl to perform PLIF and determined fuel distribution in a PFI engine. The authors could evaluate the dependence of in-cylinder spatial inhomogeneities and CCV of mixture distribution on engine speed and injection timing. Deschamps et al. (1994) [45] carried out laser doppler velocimetry (LDV) and PLIF in a swirl plane close to the spark plug to characterise the flow field and the mixture distribution. From the results they could successfully augment combustion by optimising spark plug location and using charge stratification such that combustion lean limit was extended. Lately, the development in DI engines revolve around spray guided mode where one of the spray plumes of a multi-hole injector is targeted towards the spark plug. This mode has the potential of application over wider range of loads and speeds. Unlike PFI engines, in stratified DI mode, high speed (kHz) Nd: YAG lasers are needed to study the spray behaviour and subsequent mixture formation. As the laser intensity per shot reduces with higher frequency and harmonics, a third order harmonic (355 nm) is found to possess pulse energy close to millijoules unlike fourth harmonics (266 nm) that possess in order of microjoules level [161]. Thus, biacetyl is very helpful for high speed PLIF studies at 355 nm. Smith and Sick (2005) [161] studied the fuel distribution in a spark ignited direct ignition engine with images being taken at an interval of 1 CAD. With such a high temporal resolution, the authors could track the motion of fuel plumes inside the cylinder in a particular engine cycle. Smith and Sick (2007) [162] measured the fuel mixture distribution in late injection for stratified mode and found significant mixture inhomogeneity around the spark plug.
Recently, biacetyl has begun to receive attention as high-speed PLIF studies have been used to investigate the rare misfires and partial burning events found in the stratified mode. Smith and Sick (2006) [99] studied the impact of spark plug orientation (spray plume facing the ground electrode (case C), ground electrode away from the spray plume (case A) and spark plug perpendicular to the plume (case B)) on combustion using mixture distribution images shown in Fig. 36. They observed biacetyl fluorescence in the two-spray plume region, showing different behaviours when interacting with the spark plug. For case A, the plume disperses around the ground electrode, creating a low-velocity fuel-rich region that minimizes spark stretching. In case B, most of the spray passes through the spark plug unhindered, stretching the spark in the flow direction and minimizing heat loss. Case C restricts the spray plume, forming a low-velocity region and causing highly stratified fuel mixture accumulation, resulting in the shortest burn duration but the highest misfire rate. When the spray plumes straddle the spark plug instead of directly hitting it, the misfires were found to greatly increase. Furthermore, Peterson et al. (2014) [100] studied misfire, well-burnt, and partial-burnt cycles in stratified mode combustion using simultaneous PLIF and PIV. They found that spark energy was sufficient for flame kernel formation in all cases. Well-burnt cycles showed rapid flame propagation, while partial-burnt cycles exhibited delayed interaction with the flammable mixture, leading to incomplete fuel consumption. Misfire cycles showed hindered flame propagation due to surrounding leaner mixture and unfavourable flow fields near the spark plug, resulting in flame extinguishment.

Biacetyl, thus has become an important fluorescent tracer lately especially for the high speed PLIF applications. Therefore, we now discuss its fluorescence characteristics in the upcoming section.
6.1 Absorption cross-section
Biacetyl is a diketone and its chemical structure can be seen from Fig. 37. The absorption spectrum can be divided into two regions as obtained from the works of Horowitz et al. (2001) [88]: a broadband spectrum in the range of 210 and 325 nm and a second region between 350 and 480 nm. The second region shows some number of features as compared to the first region. The largest cross-section is found to occur at 417 nm with a value of about 7.62×\(\:{10}^{-20}\) cm2 at 298 K. Plots in Fig. 37 are also provided for the works of Calvert and Pitts (1966) [163] and Plum et al. (1983) [164] for comparison. The data were found to be in good agreement.

With increasing temperature, the absorption cross-section is presented for two different excitation wavelengths at 266 nm and 355 nm (Fig. 38) [165]. For 266 nm, the absorption cross-section increases with temperature whereas for 355 nm, the cross-section value remains constant within the experimental scatter.

Guibert et al. (2006) [75] studied the changes in absorption cross-section of the longer wavelength band of Biacetyl. The absorption spectrum was plotted with temperature (Fig. 39). It was found that the spectrum grows slightly with temperature and has a slight broadening as compared to the ketonic counterparts. The largest cross-section of 3-pentanone increases by 25% in the temperature range of 300 and 540 K [154] whereas that of biacetyl increases by about 7% in the range of 373 to 573 K. The slight increase in cross-section of the shorter wavelength band was also observed in the previous work of Wermuth and Sick (2005) [163, 165] where cross-section changed slightly at 355 nm wavelength. In contrast to acetone and 3-pentanone, the absorption cross-section values for shorter wavelengths are found to increase with temperature whereas for longer wavelengths, the absorption cross-section values are constant. The reason for this behaviour is not clear.

(from ref [75]): Variation of absorption spectrum of biacetyl with temperature
6.2 Fluorescence signal variation
Guibert et al. (2005) [75] studied the fluorescence characteristics of biacetyl when excited by an argon ion laser excitation. They observed that the resulting fluorescence spectrum spanned from 440 to 510 nm with a peak at 485 nm and the phosphorescence spectrum spanned from 510 to 600 nm with a peak at 517 nm. The fluorescence spectrum they obtained at 523 K and 2 MPa is shown in Fig. 40. The authors found that the fluorescence signal from biacetyl increases with increasing mole fraction, reaches a peak and then starts to decrease after a further increase in mole fraction. This behaviour was explained due to self-quenching as well as fluorescence radiation reabsorption by the ground state biacetyl molecule. Since the measurements were done in the laser regimes near 355 nm (352.9 nm to 365.2 nm using Ar++ laser), the change in fluorescence signal is solely due to the change in FQY as the absorption cross-section is pressure independent and the temperature dependence is meagre around the wavelength range used. They studied the fluorescence signal dependencies of biacetyl with pressure and temperature in a nitrogen bath gas. The obtained results is shown in Fig. 41. Each curve in the figure is an isobar with the values ranging from 0.1 to 4 MPa. Therefore, the figure is essentially providing information at simultaneous high temperature and high-pressure regimes. The plots depict fluorescence signal in an absolute scale.

(from ref [75]): Fluorescence spectrum of biacetyl at 523 K and 2 MPa using laser wavelength around 352.9 to 365.2 nm

(from ref [75]): Fluorescence signal variation of biacetyl with temperature and pressure in nitrogen bath gas
It is observed that with increasing temperature, the signal values first increase, reach a peak and then starts decreasing. The temperature range considered is between 373 and 573 K. With increasing pressure, the signal is found to increase. At very high pressures above 2 MPa, it is seen that the isobars overlap indicating the advent of the high-pressure limit region as observed in the ketonic compounds. Similar plots were developed by the authors for fluorescence signals as a function of pressure and temperature where absolute fluorescence signals were plotted as isotherms in a nitrogen bath gas at a constant tracer concentration. The pressure range covered is between 0.1 and 4 MPa. It was observed that the signals increase with pressure and reach a stagnation beyond 2.5 MPa. Furthermore, studies related to fluorescence signal dependencies on bath gas was carried out. Figure 42 [75] shows the bath gas dependence of fluorescence signals at a constant tracer concentration. The figure contains the plots of isotherms at three different temperatures with two bath gases (nitrogen and air) along with pressure in the X-axis. Since the fluorescence signal is normalised, only the relative shapes of the curves can be compared. It is observed that the nitrogen bath gas upon pressure increment gives rise to an increasing fluorescing signal which later becomes constant after a certain pressure. The air bath gas shows an increase in signal with increasing pressure till 1 MPa, after which a reduction in fluorescence signals is observed with further pressure rise. Thus, decrease is attributed to oxygen quenching which was also observed in the previous ketonic compounds.

(from ref [75]): Normalised fluorescence signal isotherm variation of biacetyl with pressure in nitrogen and air bath gas. Each isotherm is normalised to the corresponding value at atmospheric pressure
Similar studies were conducted by Smith and Sick (2007) [162] to study dependence of biacetyl fluorescence on temperature and pressure in air. The authors used an optical engine setup and a wavelength of 355 nm for excitation. The temperature range studied was from 450 to 750 K whereas the pressure ranged from 4 bar to 16 bar. The authors found that fluorescence signal decreases with increasing temperature. At lower temperatures, the signal is more or less temperature independent until a certain threshold temperature beyond which a steady decline is observed is signals with temperature. For lower pressures, a significant drop in signals is observed beyond 550 K whereas the value is 600 K at higher pressures. Furthermore, over the investigated temperature range, the drop in signals is lower at lower pressures (20%) and larger at higher pressures (35%). The decreasing trend in signals in air is consistent with the previous work of Guibert et al. (2005) [75]. Error bars indicated about 10% uncertainty. The decreasing signal strength at increasing pressures is due to oxygen quenching. Since the behaviour of biacetyl fluorescence is found to be very similar to the ketonic counterparts, it is plausible that the previously provided explanations for acetone and 3-pentanone fluorescence characteristics can be extended to biacetyl as well. Thus, the increasing signal with pressure in non-oxygen containing bath gases is due to the increased vibrational relaxations to the lower vibronic levels which have lower non-radiative decay rates. Similarly, the decrease of signal with temperature is due to the higher non-radiative rates at higher vibronic levels in the excited singlet state.
Guibert et al. (2005) [75] found a slight increase in signal followed by a decrease with increasing temperature in nitrogen bath gas. This is something that is not observed for ketones where a monotonic decrease is found. The reason for this discrepancy can be due to extremely long integration times of 2500 ms as opposed to Smith and Sick (2005) [161] where they have used an exposure time of 200 ns. There is a possibility of phosphorescence being captured in the oxygen deficient (nitrogen) environment leading to a non-monotonic temperature dependence. Guibert et al. suggested that at lower temperature, there is a reverse inter-system crossing between the T1 triplet and the S1 singlet state due to low energy gap between the two. This effectively reduces the inter-system crossing rate and thus increases the FQY values. At higher temperatures, the inter-system crossing is from S1 to T2 with no reverse inter-system crossing. This leads to a lower fluorescence rate resulting in decreasing signals at very high temperatures. The fluorescence resulting from reverse inter-system crossing (delayed fluorescence) has a very large time scale and may not be captured by the short exposure times in a typical LIF experiment. Thus, it is quite possible that the slight signal increase at lower temperature may not be seen in a LIF experiment. Smith and Sick (2007) [162] found a temperature independence at lower temperature followed by a decrease at higher temperature. The reason for temperature independence at lower temperature in their work is apparently not clear. Data related to signal dependence on different excitation wavelengths is sparsely present and thus is not covered in the current article. To the best of our knowledge, no fluorescence model for biacetyl has been developed till date. For this, more experimental data are needed.
7 Conclusion
This paper presents an overview of the ketonic fluorescent tracer compounds that have found wide applications pertaining to the non-intrusive diagnostics of IC engines using PLIF. A lot of studies have been carried out by various researchers which have greatly augmented the understanding about fluorescence characteristics of these compounds especially in the conditions of extreme temperature and pressure normally encountered in the compression stroke of an IC engine. Today, a multitude of organic compounds exist as a possible choice for tracer with a vast literature base on their fluorescence characteristics. Therefore, it becomes imperative to provide a thorough review about the fluorescence characteristics of tracer molecules. Having a good understanding of the fluorescence characteristics will also aid in developing new diagnostic techniques. The work is divided into two parts. Part A discusses ketonic tracers whereas part B of this work discusses aromatic tracers.
In order to understand the fluorescence signal intensity, the basic strategy is to decompose it into absorption cross-section and FQY and individually study their variations. Absorption cross-section is dependent on excitation wavelength and temperature and is relatively simpler to quantify its variation. However, FQY depends on excitation wavelength, temperature, pressure and the mixture composition of the bath gas and thus, is complicated to quantify the variation and model it. The following insights were obtained about the behaviour of the discussed molecules-.
-
Among the carbonyl compounds, acetone is generally preferred to mark gaseous fuels like hydrogen, CNG owing to its lower boiling point (56.1 ⁰C). On the contrary, in order to mark liquid fuels like iso-octane with boiling point (99.2 ⁰C), 3-pentanone (boiling point- 102 ⁰C) and biacetyl (boiling point- 88 ⁰C) are used. 3-pentanone is the most preferred due to its very similar boiling point with iso-octane and a higher fluorescence yield than acetone. Biacetyl has also come to use because it can be excited with 355 nm laser wavelength that is available in commercial pulsed kHz laser with millijoules order energy per pulse.
-
However, there are issues of preferential vapourization of 3-pentanone in a mixture with iso-octane due to its formation of an azeotrope. Aromatic tracers like toluene do not form azeotropes with iso-octane due to the non-polar nature of both the molecules. To achieve perfect co-evaporation of iso-octane and 3-pentanone, the blending of 3-pentanone should be an azeotrope for all the temperatures something that is not possible as azeotrope composition itself varies with temperature. Also, azeotrope composition of 3-pentanone and iso-octane (32% by vol of 3-pentanone at ambient conditions) lead to excessive laser attenuation. Therefore, studies have used in the range of 15–25 vol % 3-pentanone to reduce laser sheet extinction and minimize preferential evaporation of 3-pentanone. Remaining typically used vol % for acetone and biacetyl are mentioned in Table 2. To capture the behaviour of multi-component fuel like gasoline, a mixture of surrogate fuels at the different vapourization regimes and specific tracer to mark the different surrogates have been used for studying mixture formation using PLIF. Tracer combinations like acetone/p-xylene and acetone/toluene/TMB are discussed. Some variations in spatial distribution of different boiling fractions were found during the intake stroke whereas the differences were negligible during the compression stroke due to high in-cylinder temperature.
-
Pyrolysis of tracer molecules are not much of a concern in IC engines due to their lower residence times (in milliseconds). But in calibration test cells, the residence times are much longer (in seconds). Table 3 is provided to compare the typical duration of tracers before pyrolysis occurs for acetone, toluene and 3-pentanone under high temperature and pressure conditions. Toluene was found to be the most stable and 3-pentanone the least stable out of the three. Addition of tracers may change the effective equivalence ratios, physical and chemical properties of the mixture. It was found that addition of 5 and 15% 3-pentanone, 10% biacetyl and 3% toluene by vol. into iso-octane doesn’t much alter the combustion process in SI engines. However, for HCCI engines, tracers like toluene, acetone and 3-pentanone do have some measurable effect on combustion phasing due to their direct alteration of chemical mechanism for autoignition. It is thus advisable to perform some baseline experiments with the required vol % of tracers to study any possible variation in engine performance especially for HCCI engines before performing PLIF studies. Other than this, biacetyl is found to show appreciable photolysis at shorter wavelengths and thus is mostly used with longer 355 nm excitation. The photolysis effect also becomes more important especially for high speed PLIF studies using biacetyl.
-
A major difference between the aromatic and ketonic compounds is the very less oxygen quenching in ketonic compounds. Ketones have a smaller energy gap between the first excited (S1) and the ground (S0) singlet states as compared to the energy gap between excited singlet and ground triplet state for oxygen. Also, the lifetime of the excited states is very less in S1 state due to very fast ISC rates. Together, these effects make ketones very insensitive to oxygen quenching. However, a minute reduction in fluorescence signals in oxygen containing bath gases at elevated pressures are referred to as oxygen assisted ISC. As temperature increases, absorption spectrum of the tracer molecules broadens towards longer wavelengths, and the absorption cross-section values increase. Values of absorption cross-section and spectrum range is provided in Table 2 for all the three tracers discussed. To calculate FQY, a step ladder model was first introduced for the case of acetone and then later on extended to 3-pentanone as well. The fluorescence rates are assumed to be constant in the model with respect to different vibrational levels in the excited state manifold. Non-radiative rates increase with the vibrational energy levels. Literature values of absolute FQY is provided for acetone and 3-pentanone whereas the lifetime values are provided for all the three tracers.
-
Changes in excitation wavelength or temperature affect the molecule’s excitation to higher vibrational levels of the excited singlet state. Since fluorescence rates have a weak dependency on vibrational energy levels, variations in non-radiative decay rates predominantly influence FQY. Decreased excitation wavelength and increased temperature lead to a decreasing trend in FQY. Excited molecule undergoes several collisions with the bath gas molecules. In the step ladder model, this is assumed to be a multi-step decay process. Vibrational relaxations increase fluorescence intensity as emissions occur from lower vibrational levels with high FQY values (decreased rates of non-radiative processes like ISC and IC). FQY increases with pressure in the presence of non-quenching bath gases. More atoms in the collider result in a faster relaxation and a faster rise in FQY value.
-
At very low pressures (around 10 mbar), the collisions become rare and the fluorescence emanates from a vibrational level which is determined by the combination of temperature and excitation wavelength. This low value of FQY is the low-pressure limit. On increase of pressure, the collisions become significant and the FQY value deviates from the low-pressure limit. It then rises with pressure and at very high-pressure levels (typically around 20 bars), reaches a high- pressure limit. This is due to the molecule being relaxed to the thermalized level of the excited electronic state. The high-pressure limit is only dependent on temperature and the low-pressure limit is dependent on both temperature and excitation wavelength. However, both these limits are independent of the bath gas composition. If the bath gas contains molecules which show a quenching behaviour like oxygen, then with increase in pressure, the FQY values reduce due to greater collision and greater deactivation of the excited molecule. This type of behaviour was observed for all the three tracers discussed in this article.
-
Another important domain of simultaneous high-pressure and high-temperature was discussed in this review article. Earlier, the models for acetone and 3-pentanone were prepared from the works of ambient temperature and high pressure or high temperature and ambient pressure. Therefore, the models proved to be ineffective to predict the FQY values at simultaneous high pressure and high temperature like in an IC engine environment. It is normally expected that at high temperature, since the molecule is located at very high vibronic levels in comparison to the thermalised levels, it has got a greater probability for vibrational relaxation and thus should exhibit an enhanced pressure sensitivity. This led to the models overestimating the pressure effects at very high temperatures. However, it was later observed from the cell experiments that at very high temperatures, the collision frequency suffers due to the decreased number density of the bath gas molecules. Therefore, the pressure sensitivity decreases.
Although, there has been a significant progress in the understanding of fluorescence characteristics of acetone and 3-pentanone, there are a lot of gaps in understanding of biacetyl fluorescence characteristics. The absorption cross-section of shorter wavelength (266) in biacetyl shows increment with temperature rather than the longer wavelength of 355 nm in a direct contrast to acetone and 3-pentanone. The reason for this behaviour is not yet clear. Also, there is not enough parametric studies related to biacetyl fluorescence dependency on variations in pressure, temperature and bath gas composition. Because of this, at present there is a dearth of the ketonic type phenomenological models for biacetyl. The understanding of biacetyl fluorescence is important since it can be excited at 355 nm. Also, biacetyl as a tracer started gaining importance due to the emergence of diode-pumped solid state (DPSS) lasers which had the capacity of laser repetition rates of around 4.8 to 12 kHz with a pulse energy of around 0.5 mJ. Fajardo et al. (2006) [166] and Peterson and Sick (2009) [68] have used such DPSS Nd-YAG laser at 355 nm for PLIF studies using biacetyl. Due to the low pulse energy, it necessitated the usage of high-speed intensified CMOS cameras.
However, the use of high-speed image intensifiers might result in blurring of measured scalar gradients, degradation of spatial resolution and might also introduce some additional noise [15, 165]. Gordon et al. (2009) [15] also recommended to avoid the use of high-speed intensifiers wherever possible. For ketonic tracers showing fluorescence in the visible wavelength spectrum, with the increase in laser fluence, the need for an image-intensifier can be circumvented as the PLIF signal strength is proportional to the laser pulse energy. This is made possible by the emergence of pulse burst laser systems that can have high pulse energy at kHz rates (15 mJ/pulse at 266 nm at 10 kHz for acetone PLIF by Miller et al. (2013) [67], 145 mJ/pulse at 266 nm at 10 kHz by Papageorge and Sutton (2017) [128]). For low repetition lasers, the volumetric element of tracer fluid has a single interaction with laser probe but that is not the case for high repetition laser systems which at such high energies might have unwanted photolytic or saturation effects on the tracer element. Brushnan et al. (2013) [168] found an appreciable surface heating due to the usage of high speed DPSS Nd: YLF laser at 2.4 kHz which raises the possibility of higher surface heating by pulse burst laser systems thus altering the flow properties in boundary layer. Michael et al. (2015) [169] found that it was necessary to use lower laser fluence while performing laser-induced incandescence (LII) measurements as LII signals were sensitive to soot characteristics which might change at high laser fluences. Papageorge and Sutton (2017) [128] also observed changes in fluorescence signals from acetone, 3-pentanone and biacetyl as a function of laser pulse numbers. Therefore, the ‘non-intrusive’ assumption may not hold for high energy high repetition rates laser measurement systems and further studies are required in this regard.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
J. Trost, L. Zigan, A. Leipertz, D. Sahoo, P.C. Miles, Appl. Opt. 52, 8001 (2013)
S. Faust, M. Goschütz, S.A. Kaiser, T. Dreier, C. Schulz, Appl. Phys. B 117, 183 (2014)
C. Schulz, V. Sick, Prog Energy Combust. Sci. 31, 75 (2005)
F.W. Bowditch, SAE Tech. Paper 610002 (1961)
R.M. Richman, W.C. Reynolds, SAE Tech. Paper 840379 (1984)
S.C. Bates, SAE Tech. Paper 880520 (1988)
M. Mittal, P. Mehta, SAE Technical Paper 2018-01-1775 (2018)
H. Zhao, N. Ladommatos, Prog Energy Combust. Sci. 24, 297 (1998)
V.G. McDonell, G.S. Samuelsen, Meas. Sci. Technol. 11, 870 (2000)
D.K. Sharaborin, A.G. Savitskii, G.Y. Bakharev, A.S. Lobasov, L.M. Chikishev, V.M. Dulin, Exp. Fluids 62, (2021)
L. Voigt, J. Heinze, T. Aumeier, T. Behrendt, F. di Mare, Ceramics; Controls, Diagnostics and Instrumentation; Education; Manufacturing Materials and Metallurgy, vol. 6 (American Society of Mechanical Engineers, 2017)
R. Sadanandan, M. Stöhr, W. Meier, Appl. Phys. B 90, 609 (2008)
A. Lakshmanarao, M.W. Renfro, G.B. King, N.M. Laurendeau, Exp. Fluids. 30, 595 (2001)
H. Hu, M. Koochesfahani, Exp. Fluids. 33, 202 (2002)
R.L. Gordon, C. Heeger, A. Dreizler, Appl. Phys. B 96, 745 (2009)
S.T. Seitz, Thermal Boundary Layer Measurements Using Planar Laser Induced Fluorescence, 2018
T. Lacassagne, M.E.L. Hajem, J.-Y. Champagne, S. Simoëns, Exp. Fluids 64, (2023)
T. Sakurai, T. Handa, S. Koike, K. Mii, A. Nakano, J. Vis. 18, 511 (2015)
T.R. Meyer, J.C. Dutton, R.P. Lucht, Phys. Fluids. 11, 3401 (1999)
T.R. Meyer, G.F. King, G.C. Martin, R.P. Lucht, F.R. Schauer, J.C. Dutton, Exp. Fluids. 32, 603 (2002)
J. Trost, L. Zigan, A. Leipertz, Proc. Combust. Inst. 34, 3645 (2013)
L. Zigan, J. Trost, A. Leipertz, Appl. Opt. 55, 1453 (2016)
G. Tea, G. Bruneaux, J.T. Kashdan, C. Schulz, Proc. Combust. Inst. 33, 783 (2011)
B. Peterson, E. Baum, B. Böhm, V. Sick, A. Dreizler, Proc. Combust. Inst. 35, 2923 (2015)
K. Kohse-Höinghaus, Prog Energy Combust. Sci. 20, 203 (1994)
R.P. Lucht, D.W. Sweeney, N.M. Laurendeau, Combust. Flame. 50, 189 (1983)
J.T. Salmon, R.P. Lucht, D.W. Sweeney, N.M. Laurendeau, Symp. Combust. 20, 1187 (1985)
P. Andresen, A. Bath, W. Gröger, H.W. Lülf, G. Meijer, J.J. Ter Meulen, Appl. Opt. 27, 365 (1988)
M.P. Lee, P.H. Paul, R.K. Hanson, Opt. Lett. 12, 75 (1987)
A.C. Eckbreth, Laser Diagnostics for Combustion Temperature and Species (Taylor & Francis, London, England, 1996)
J.D. Koch, R.K. Hanson, Appl. Phys. B 76, 319 (2003)
T. Fujikawa, Y. Hattori, K. Akihama, SAE Tech. Paper Ser. 972944 (1997)
S. Hokimoto, T. Kuboyama, Y. Moriyoshi, M. Iida, T. Watanabe, SAE Technical Paper 2017-01-2213 (2017)
R. Pradheep, M. Mittal, P.S. Mehta, V. Balaji, Others, in 18th Annual Conference on Liquid Atomization and Spray Systems (ILASS-Asia 2016) (2016)
J. Reboux, D. Puechberty, F. Dionnet, SAE Tech. Paper 941988 (1994)
J. Scholz, T. Wiersbinski, V. Beushausen, SAE Technical Paper 2007-01-0645 (2007)
J.C. Sacadura, L. Robin, F. Dionnet, D. Gervais, P. Gastaldi, A. Ahmed, SAE Technical Paper 2000-01-1794 (2000)
W. Koban, J.D. Koch, R.K. Hanson, C. Schulz, Appl. Phys. B 80, 147 (2005)
M. Ritter, L.-M. Malbec, O. Laget, SAE Int. J. Adv. Curr. Prac Mobil. 3, 95 (2020)
M. Berckmüller, N.P. Tait, D.A. Greenhalgh, SAE Tech. Paper 970826 (1997)
J. Yang, M. Xu, D.L.S. Hung, Q. Wu, X. Dong, Energy Convers. Manag. 138, 565 (2017)
C. Weaver, S. Wooldridge, S. Johnson, V. Sick, G. Lavoie, SAE Technical Paper 2003-01-3236 (2003)
P.O. Witze, R.M. Green, SAE Tech. Paper 970866 (1997)
Y. Li, H. Zhao, B. Leach, T. Ma, N. Ladommatos, SAE Technical Paper 2004-01-1354 (2004)
B. Deschamps, R. Snyder, T. Baritaud, SAE Tech. Paper 941993 (1994)
S. Nayek, M. Mittal, J. Eng. Gas Turbine Power 1 (2023)
B. Peterson, D.L. Reuss, V. Sick, Proc. Combust. Inst. 33, 3089 (2011)
S.R. Engel, P. Koch, A. Braeuer, A. Leipertz, Appl. Opt. 48, 6643 (2009)
T. Blotevogel, M. Hartmann, H. Rottengruber, A. Leipertz, Appl. Opt. 47, 6488 (2008)
I. Düwel, W. Koban, F.P. Zimmermann, T. Dreier, C. Schulz, Appl. Phys. B 97, 909 (2009)
G. Zhang, M. Xu, Y. Zhang, W. Zeng, SAE Technical Paper 2011-01-1879 (2011)
J.B. Ghandhi, P.G. Felton, B.F. Gajdeczko, F.V. Bracco, SAE Tech. Paper 940394 (1994)
P. Wieske, S. Wissel, G. Grunefeld, M. Graf, S. Pischinger, SAE Technical Paper 2006-01-3378 (2006)
K. Fukui, T. Fujikawa, M. Tohyama, Y. Hattori, K. Akihama, SAE Int. J. Engines. 6, 289 (2013)
D.A. Rothamer, J.A. Snyder, R.K. Hanson, R.R. Steeper, SAE Int. J. Fuels Lubr. 1, 520 (2008)
W. Koban, J. Schorr, C. Schulz, Appl. Phys. B 74, 111 (2002)
A. Kakuho, M. Nagamine, Y. Amenomori, T. Urushihara, T. Itoh, SAE Technical Paper 2006-01-1202 (2006)
S. Lind, J. Trost, L. Zigan, A. Leipertz, S. Will, Proc. Combust. Inst. 35, 3783 (2015)
M.C. Thurber, F. Grisch, R.K. Hanson, Opt. Lett. 22, 251 (1997)
M.C. Thurber, R.K. Hanson, Exp. Fluids. 30, 93 (2001)
S. Einecke, C. Schulz, V. Sick, A. Dreizler, R. Schießl, U. Maas, SAE Tech. Paper 982468 (1998)
R.P.C. Zegers, M. Yu, C. Bekdemir, N.J. Dam, C.C.M. Luijten, L.P.H. de Goey, Appl. Phys. B 112, 7 (2013)
M. Luong, R. Zhang, C. Schulz, V. Sick, Appl. Phys. B 91, 669 (2008)
U. Retzer, W. Fink, T. Will, S. Will, L. Zigan, Appl. Phys. B 125, (2019)
B. Peterson, E. Baum, B. Böhm, V. Sick, A. Dreizler, Proc. Combust. Inst. 34, 3653 (2013)
J. Hult, M. Richter, J. Nygren, M. Aldén, A. Hultqvist, M. Christensen, B. Johansson, Appl. Opt. 41, 5002 (2002)
J.D. Miller, J.B. Michael, M.N. Slipchenko, S. Roy, T.R. Meyer, J.R. Gord, Appl. Phys. B 113, 93 (2013)
B. Peterson, V. Sick, Appl. Phys. B 97, 887 (2009)
U. Retzer, H. Ulrich, S. Will, L. Zigan, Appl. Phys. B 128, (2022)
S.S. Harilal, B.E. Brumfield, N.L. LaHaye, K.C. Hartig, M.C. Phillips, Appl. Phys. Rev. 5, 021301 (2018)
B. Valeur, M.N. Berberan-Santos, Molecular Fluorescence, 2nd edn. (Wiley-VCH, Weinheim, Germany, 2013)
K. Rohatgi, Fundamentals of Photochemistry (New Age International, New Delhi, India, 2014)
M. Klessinger, J. Michl, Excited States and Photochemistry of Organic Molecules (Wiley-VCH, Weinheim, Germany, 1995)
W.M. Nau, J.C. Scaiano, J. Phys. Chem. 100, 11360 (1996)
P. Guibert, V. Modica, C. Morin, Exp. Fluids. 40, 245 (2006)
D. Fuhrmann, T. Benzler, S. Fernando, T. Endres, T. Dreier, S.A. Kaiser, C. Schulz, Proc. Combust. Inst. 36, 4505 (2017)
B. Stevens, P.J. McCartin, Mol. Phys. 8, 597 (1964)
T.D. Fansler, D.T. French, M.C. Drake, SAE Tech. Paper 950110 (1995)
K.S. Kim, M.S. Choi, C.H. Lee, W.T. Kim, SAE Tech. Paper 970509 (1997)
H. Neij, Development of Laser-Induced Fluorescence for Precombustion Diagnostics in Spark-Ignition Engines (Lund University, 1998)
D.R. Lide, (2004).S. Kim, Y. Yan, J. M. Nouri, and C. Arcoumanis, Fuel (Lond.) 106, 737 (2013)
C. Yaws, Handbook of transport property data: Viscosity, thermal conductivity, and diffusion coefficients of liquids and gases, (1995)
S. Kim, Y. Yan, J.M. Nouri, C. Arcoumanis, Fuel (Lond). 106, 737 (2013)
R. Shimizu, S. Matumoto, S. Furuno, M. Murayama, S. Kojima, SAE Tech. Paper 922356 (1992)
A. Lozano, B. Yip, R.K. Hanson, Exp. Fluids. 13, 369 (1992)
B. Johansson, H. Neij, M. Alden, G. Juhlin, SAE Tech. Paper 950108 (1995)
J.D. Koch, J. Gronki, R.K. Hanson, J. Quant. Spectrosc. Radiat. Transf. 109, 2037 (2008)
A. Horowitz, R. Meller, G.K. Moortgat, J. Photochem. Photobiol Chem. 146, 19 (2001)
M.C. Thurber, R.K. Hanson, Appl. Phys. B 69, 229 (1999)
V. Modica, C. Morin, P. Guibert, Appl. Phys. B 87, 193 (2007)
J.D. Koch, R.K. Hanson, W. Koban, C. Schulz, Appl. Opt. 43, 5901 (2004)
D.A. Hansen, E.K.C. Lee, J. Chem. Phys. 63, 3272 (1975)
S. Kaiser, C.M. White, SAE Int. J. Engines. 1, 657 (2008)
J. Hiltner, M. Samimy, SAE Tech. Paper 961102 (1996)
B. Williams, P. Ewart, R. Stone, H. Ma et al., SAE Technical Paper 2008-01-1073 (2008)
S. Haramiishi, T. Watanabe, M. Iida, M and, S. Hokimoto, SAE Technical Paper 2019-32-0552 (2019)
N. Kampanis, C. Arcoumanis, S. Kometani, R. Kato, H. Kinoshita, Int. J. Engine Res. 7, 143 (2006)
M. Berckmüller, N.P. Tait, D.A. Greenhalgh, SAE Tech. Paper 961929 (1996)
J.D. Smith, V. Sick, SAE Technical Paper 2006-01-1264 (2006)
B. Peterson, D.L. Reuss, V. Sick, Combust. Flame. 161, 240 (2014)
J.-F. Le Coz, T. Baritaud, Developments in Laser Techniques and Applications to Fluid Mechanics (Springer, Berlin Heidelberg, Berlin, Heidelberg, 1996), pp. 115–131
M. Davy, P. Williams, D. Han, R. Steeper, Exp. Fluids. 35, 92 (2003)
M.-T. Lin, V. Sick, SAE Technical Paper Series 2004-01-1355 (2004)
D. Han, R.R. Steeper, SAE Technical Paper 2002-01-0837 (2002)
K. Tong, B.D. Quay, J.V. Zello, D.A. Santavicca, in SAE Technical Paper 2001-01-3650 (2001)
A. Makino, C.K. Law, Combust. Flame. 73, 331 (1988)
B.D. Smith, R. Srivastava, Thermodynamic Data for Pure Compounds. Part A, Hydrocarbons and Ketones
R.E. Stevens, H. Ma, C.R. Stone, H.L. Walmsley, R. Cracknell, Proc. Inst. Mech. Eng. Pt. D: J. Automobile Eng. 221, 713 (2007)
B. Williams, P. Ewart, X. Wang, R. Stone, H. Ma, H. Walmsley, R. Cracknell, R. Stevens, D. Richardson, H. Fu, S. Wallace, Combust. Flame. 157, 1866 (2010)
R. Zhang, V. Sick, SAE Technical Paper 2007-01-1826 (2007)
L.M. Itani, G. Bruneaux, A. Di Lella, C. Schulz, Proc. Combust. Inst. 35, 2915 (2015)
P. Kranz, S.A. Kaiser, Proc. Combust. Inst. 37, 1365 (2019)
R. Steeper, S. De Zilwa, A. Fayoux, SAE Technical Paper 2005-01-0111 (2005)
J.-P. Leininger, F. Lorant, C. Minot, F. Behar, Energy Fuels. 20, 2518 (2006)
J.R.E. Smith, D. Phil., C.N. Hinshelwood, Proc. R. Soc. Lond. 183, 33 (1944)
F. Grossmann, P.B. Monkhouse, M. Ridder, V. Sick, J. Wolfrum, Appl. Phys. B 62, 249 (1996)
J. Trost, L. Zigan, S.C. Eichmann, T. Seeger, A. Leipertz, Appl. Opt. 52, 6300 (2013)
V. Sick, C.K. Westbrook, Proc. Combust. Inst. 32, 913 (2009)
M. Löffler, F. Beyrau, A. Leipertz, Appl. Opt. 49, 37 (2010)
F. Braeuer, Beyrau, A. Leipertz, Appl. Opt. 45, 4982 (2006)
R.A. Schiessl, J. Sommerer, SAE Technical Paper 2018-01-1784 (2018)
H. Neij, B. Johansson, M. Aldén, Combust. Flame. 99, 449 (1994)
R. Zhang, N. Wermuth, V. Sick, SAE Technical Paper 2004-01-2975 (2004)
R.P. Fitzgerald, R.R. Steeper, J.A. Snyder, SAE Int. J. Fuels Lubr. 1, 1491 (2008)
Y. Haas, Photochem. Photobiol Sci. 3, 6 (2004)
M.A. Blitz, D.E. Heard, M.J. Pilling, J. Phys. Chem. A 110, 6742 (2006)
W.E. Bell, F.E. Blacet, J. Am. Chem. Soc. 76, 5332 (1954)
M. Papageorge, J.A. Sutton, Exp. Fluids 58, (2017)
P. Koch, M.G. Löffler, M. Wensing, A. Leipertz, Int. J. Engine Res. 11, 455 (2010)
B. Yip, M.F. Miller, A. Lozano, R.K. Hanson, Exp. Fluids. 17, 330 (1994)
D. Wolff, V. Beushausen, H. Schlüter, P. Andresen, W. Hentschel, P. Manz, S. Arndt, Stroke. 86, 4 (1994)
J.M. Brault, D.S. Maymir, M. Samimy, M. Matsuki, SAE Tech. Paper 981387 (1998)
T. Itoh, A. Kakuho, K. Hiraya, E. Takahashi, T. Urushihara, Int. J. Engine Res. 7, 423 (2006)
J. Harada, T. Tomita, H. Mizuno, Z. Mashiki, Y. Ito, SAE Tech. Paper 970540 (1997)
T. Fujikawa, Y. Hattori, M. Koike, K. Akihima, T. Kobayashi, S. Matsushita, JSME Int. J. Ser. B 42, 760 (1999)
W. Hentschel, B. Block, T. Hovestadt, H. Meyer, G. Ohmstede, V. Richter, B. Stiebels, A. Winkler, SAE Technical Paper 2001-01-3648 (2001)
C. Schwarz, E. Schünemann, B. Durst, J. Fischer, A. Witt, SAE Technical Paper 2006-01-1265 (2006)
V.M. Salazar, S.A. Kaiser, SAE Int. J. Engines. 2, 119 (2009)
V.M. Salazar, S.A. Kaiser, SAE Int. J. Engines. 3, 309 (2010)
M.C. Thurber, Acetone Laser-Induced Fluorescence for Temperature and Multiparameter Imaging in Gaseous Flows, ui.adsabs.harvard.edu, 1999
A. Spielfiedel, N. Feautrier, C. Cossart-Magos, G. Chambaud, P. Rosmus, H.-J. Werner, P. Botschwina, J. Chem. Phys. 97, 8382 (1992)
M.C. Thurber, F. Grisch, B.J. Kirby, M. Votsmeier, R.K. Hanson, Appl. Opt. 37, 4963 (1998)
J.B. Ghandhi, P.G. Felton, Exp. Fluids. 21, 143 (1996)
L.S. Yuen, J.E. Peters, R.P. Lucht, Appl. Opt. 36, 3271 (1997)
F. Ossler, M. Aldén, Appl. Phys. B 64, 493 (1997)
A.M. Halpern, W.R. Ware, J. Chem. Phys. 54, 1271 (1971)
G.M. Breuer, E.K.C. Lee, J. Phys. Chem. 75, 989 (1971)
J. Koch, R. Hanson, 41st Aerospace Sciences Meeting and Exhibit (American Institute of Aeronautics and Astronautics, Reston, Virigina, 2003)
W. Koban, J.D. Koch, V. Sick, N. Wermuth, R.K. Hanson, C. Schulz, Proc. Combust. Inst. 30, 1545 (2005)
J. Hartwig, G. Mittal, K. Kumar, C.-J. Sung, Appl. Phys. B 123, 191 (2017)
J. Hartwig, M. Raju, C.-J. Sung, Appl. Phys. B 123, 193 (2017)
S. Garg, M. Mittal, S. Sahu, V. Lakshminarasimhan, SAE Technical Paper 2020-01-0786 (2020)
M. Anbari Attar, M.R. Herfatmanesh, H. Zhao, A. Cairns, Exp. Therm. Fluid Sci. 59, 96 (2014)
J. Koch, Fuel Tracer Photophysics for Quantitative Planar Laser -Induced Fluorescence (Stanford University, 2005)
N.P. Tait, D.A. Greenhalgh, Ber Bunsenges Phys. Chem. 97, 1619 (1993)
D. Wolff, V. Beushausen, P. Andresen, in, Thirtythird Japanese Symposium on Combustion, M. Löffler, K. Kröckel, P. Koch, F. Beyrau, A. Leipertz, S. Grasreiner, A. Heinisch, SAE Technical Paper 2009-01-0656 (2009)
M. Löffler, K. Kröckel, P. Koch, F. Beyrau, A. Leipertz, S. Grasreiner, and A. Heinisch. 01–0656 (2009)
B.H. Cheung, R.K. Hanson, Appl. Phys. B 106, 741 (2012)
B.H. Cheung, R.K. Hanson, Appl. Phys. B 106, 755 (2012)
T.A. Baritaud, T.A. Heinze, SAE Tech. Paper 922355 (1992)
J.D. Smith, V. Sick, Appl. Phys. B 81, 579 (2005)
J.D. Smith, V. Sick, Proc. Combust. Inst. 31, 747 (2007)
J.G. Calvert, and J. N. Pitts, Photochemistry (Wiley, New York, 1967 1966)
C.N. Plum, E. Sanhueza, R. Atkinson, W.P. Carter, J.N. Pitts, Environ. Sci. Technol. 17, 479 (1983)
N. Wermuth, V. Sick, SAE Technical Paper 2005-01-2090 (2005)
C.M. Fajardo, J.D. Smith, V. Sick, Appl. Phys. B 85, 25 (2006)
M.J. Papageorge, T.A. McManus, F. Fuest, J.A. Sutton, Appl. Phys. B 115, 197 (2014)
M. Brusnahan, L. Lu, V. Sick, Appl. Phys. B 111, 651 (2013)
J.B. Michael, P. Venkateswaran, C.R. Shaddix, T.R. Meyer, Appl. Opt. 54, 3331 (2015)
Acknowledgements
The authors would like to acknowledge several researchers working in the field of tracer based LIF and their fluorescence characterisation. Their relentless efforts over the years have led to a significant improvement in the understanding of photophysics behind fluorescent tracer molecules and their phenomenological modelling.
Author information
Authors and Affiliations
Contributions
S.N wrote the main manuscript and M.M reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nayek, S., Mittal, M. Fluorescence characteristics of organic tracer molecules for planar laser-induced fluorescence studies in internal combustion engines, part A: non-aromatics. Appl. Phys. B 130, 164 (2024). https://doi.org/10.1007/s00340-024-08272-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00340-024-08272-3