
Abstract
This is the first in a series of two papers that presents new experimental data to extend the range of acetone photophysics to elevated pressure and temperature conditions. In this work, a flexible static and flow system is designed and characterized to study the independent as well as coupled effect of elevated pressure and temperature on acetone photophysics over pressures of 0.05‒4.0 MPa and temperatures of 295‒750 K for 282 nm excitation wavelength in nitrogen and air as bath gases. Experimental results show that at 282 nm excitation, relative fluorescence quantum yield increases with increasing pressure, decreases with increasing temperature, and that the pressure sensitivity varies weakly with elevated temperature. The previously assumed linearity of fluorescence with tracer number density is shown to only be valid over a small range. Additionally, acetone fluorescence is only moderately quenched in the presence of oxygen. The present findings yield insight into the competition between the non-radiative and collisional rates at elevated temperature and pressure, as well as provide validation datasets for an updated fluorescence model developed in the second paper.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Laser induced fluorescence (LIF) is a near-instantaneous, non-intrusive technique for illuminating a specific species in a flow in a combustion system. Planar laser induced fluorescence (PLIF) is a special form of LIF where the excitation beam has been transformed into a thin sheet of light with its thickness parallel to some flow field of interest that instantaneously illuminates a whole plane within the flow. Because properties can be easily obtained as a function of space, PLIF is inherently suited for studies of temperature and/or species mixing [1]. Also, since fluorescence lifetimes are nanoseconds or less, PLIF can “freeze out” events that occur on this timescale, with recent examples showing resolution down to the Kolmogorov length scale [2].
In units of total number of photons collected, the fluorescence equation for weak (i.e. non-saturated) excitation and single species emission is:
where an experimentally obtained fluorescence signal, S ′f , is expressed in terms of its underlying optical, thermophysical, and photophysical dependencies, and B is the inherent background noise. Here, \(\frac{\varOmega }{4\pi }\) is the collection solid angle, g is the gain of the detection system, f t and f s are the temporal and spatial filters respectively, V c is the imaging collection volume in [cm3], n is the transmission in the reference line, t l is the average laser pulse width in [s], A l is the cross-sectional area of the laser beam in [cm2], V o is the reference power level in [W], \({\raise0.7ex\hbox{${hc}$} \!\mathord{\left/ {\vphantom {{hc} \lambda }}\right.\kern-0pt} \!\lower0.7ex\hbox{$\lambda $}}\) is the energy per photon at laser wavelength λ in [J], where h is Planck’s constant and c is the speed of light, L is the absorption path length in [m], η t is the light-sensitive species number density in \(\left[ {\frac{\text{molecules}}{{{\text{cm}}^{3} }}} \right]\), σ is the tracer absorption cross section (ACS) or tracer absorption “strength” in [cm2], and ϕ is the dimensionless Stern–Volmer factor or fluorescence quantum yield (FQY). FQY ranges from 0 to 1 and is defined as the probability that a molecule will undergo de-excitation via fluorescence versus all other processes. In the case with a tracer, the number density is taken as:
where P tracer is the tracer partial pressure, k is Boltzmann’s constant, T is the thermodynamic temperature, P is the pressure, and χ tracer is the tracer mole fraction.
1.1 Tracer LIF overview
There are two primary targeted species in tracer LIF studies: naturally occurring species in flames and reaction zones, such as nitric oxide (NO) and the hydroxyl radical (OH), and seeded species studied in low to intermediate temperature systems, such as that of acetone. Usually the tracer shares similar thermophysical properties of an otherwise non-fluorescing parent component, such that the seeded tracer tracks the flow. Although most tracers are unsuitable up to combustion temperatures, the method still offers insight into overall combustion, as the mixing process of fuel and oxidizer is fundamental in controlling combustion [3]. Doped tracer PLIF diagnostics may offer more flexibility over naturally occurring species because the amount of doping and thus the measured signals of interest. An application using carbon monoxide has even widened the tracer PLIF range into the infrared regime [4].
Tracers are carefully chosen to match the properties of the parent component [5], specifically, boiling point and heat of vaporization. Although the work by [6, 7] has rejected the common assumption of co-evaporation of tracer and parent fuel on similar boiling points, and suggested that precise mixing is controlled by the relative amounts of each component in the liquid phase and only occurs near the system’s azeotrope. Nonetheless, experimentalists using tracer PLIF can extract information such as mixing, temperature, and even pressure and velocity [8, 9] through the photophysical behavior of the representative tracer. Once “calibration” data for ACS and FQY have been obtained through a full range of pressure, temperature, and excitation wavelength for a given species, the LIF signal for that species, in accordance with Eq. 1, may be used as a diagnostic tool in practical combustion environments to extract parameters of interest.
1.2 Acetone tracer LIF as a diagnostic
Acetone is one of the most popular molecules for tracer LIF diagnostics. It inherently possesses many of the required characteristics that deem it an ideal tracer [10, 11] such as low cost and high vapor pressure (30.93 kPa at 298 K) [12], low toxicity, a broad absorption spectrum (absorbs ultraviolet light 225 nm < λ < 320 nm; accessible via excimer, solid state, and dye lasers), high FQY (0.71 × 10−3 at 296 K) [13], short emission lifetimes (2 ns at room temperature and pressure) [14], and temperature and pressure signal sensitivity. Acetone is favorable over toxic tracers like NO and toluene, and 3-pentanone whose vapor pressure (13.3 kPa at 293 K) is considerably lower. On spectroscopic grounds, acetone shares a similar structure-less, broadband absorption feature and separable fluorescence spectrum as that of other ketones. Acetone FQY is moderately high compared to biacetyl [10] and acetylene, but significantly lower than toluene and 3-pentanone [13]. Only NO outperforms acetone in terms of temperature stability for a tracer; acetone pyrolyzes around 1000 K [15] while NO is chemically stable in flame temperatures.
Table 1 shows a comprehensive list of previous applications, organized chronologically, where acetone LIF has been used as a diagnostic technique. Studies are further divided into single and dual tracer LIF applications. As shown, acetone LIF has been used for imaging a wide range of flow and combustion systems.
1.3 Shift towards higher temperature and pressure systems
A cursory glance in many fields of current research in combustion demonstrates the growing interest and need for high pressure studies [57]. Tracer LIF has already proven to be a useful diagnostic technique, especially for aromatics such as naphthalene [13, 58, 59], toluene [60,61,62,63,64,65,66,67], aldehydes [68,69,70], and ketones such as acetone, 3-pentanone [5, 13, 71], and biacetyl [72,73,74,75,76]. However, the bulk of historical work has been on low-pressure or high-temperature studies, and rarely ever on coupled, elevated temperature and pressure. Based on these experiments, a basic fluorescence model was constructed and fit to available data [77]. However, the model as well as composition effects are poorly understood at elevated pressure and temperature [78]. Before acetone LIF can be extended with confidence to elevated pressure and temperature environments as a diagnostic technique, ACS and FQY calibration data must be determined a priori as a function of flow field parameters.
This is the first of two papers that presents new experiments to extend the range of acetone photophysics to elevated pressure and temperature conditions at the excitation wavelength near the absorption peak. In paper 2 [78], the acetone photophysical model, model predictions at elevated pressure and temperature, and then model updates based on the new data are presented. The primary objective of this paper is to present new independent as well as coupled high pressure and temperature acetone photophysical “calibration” data in terms of ACS and FQY at 282 nm excitation over a wide range of acetone number densities, pressures of 0.5‒40 atm, and temperatures of 295‒750 K in both nitrogen (N2) and air bath gases to enable acetone LIF to be applied as a diagnostic technique in high pressure and temperature systems. Through both experimental results and model improvements, this work aims to assess current and future tracer LIF applications in engine-like conditions. By providing the theoretical and experimental groundwork at elevated temperatures and pressures, the set of experiments here can easily be extended to other excitation wavelengths, bath gases, and tracers.
2 Previously reported fundamental acetone photophysical experiments
2.1 Acetone experiments
Table 2 outlines the summary of previously reported fundamental ketone photophysical experiments. Highlighted in bold are contributions from the current work. Fundamental “calibration” data for other tracers such as toluene [63, 65, 79, 80], fluoranthene [81], and fluoroketone [82] are available in the literature. Prior to the work of Thurber [77], only two absolute measurements of FQY were conducted, despite early attempts at quantification [83]. Heicklen [84] and Halpern and Ware [85] independently measured acetone FQY at 313 nm excitation (0.2 and 0.12, respectively). However, there was significant uncertainty in both measurements. The majority of the fundamental work on quantifying acetone FQY at low and room pressure, as well as high temperature and room temperature, was carried out by [77, 86, 87]. Bryant et al. [88] subsequently extended the range to include low temperature and low pressure effects at 266 nm excitation.
Trends in previously reported acetone photophysics are summarized as follows: Fluorescence signal generally increases with increased excitation energy (lower excitation wavelength), decreases with increasing temperature, increases with increasing pressure (and subsequently that signals are more sensitive to temperature than pressure). In addition, a dependence on the type of bath gas, and a quenching effect proportional to the percent oxygen in the bath gas is reported. Additional acetone gas phase studies, conducted prior to the work of Thurber [77] include [45, 70, 89, 90]. The excitation wavelength and the pressure/temperature conditions investigated in the earlier studies are listed below:
-
1.
Tait and Greenhalgh [70]; λ = 308 nm, T = 300‒700 K, P = 1 atm.
-
2.
Bazile and Stepowski [45]; λ = 284 nm, T = 300‒600 K, P = 1 atm.
-
3.
Ghandi and Felton [89]; λ = 266 nm, T = 300‒600 K, P = 1 atm.
-
4.
Yuen et al. [90]; λ = 266 nm, T = 300 K, P = 1‒8 atm.
Note that all four of the gas phase experiments are in general agreement with that of Thurber [77], but early experiments usually assumed that acetone FQY remained constant up to pyrolysis temperatures; the non-radiative intersystem crossing rate for acetone was shown to increase with temperature [91]. Studies were also conducted to obtain photophysical calibration data for acetone in the liquid phase [92,93,94].
For the coupled effect of elevated pressure and temperature on acetone photophysics, there have been four studies [14, 43, 95, 96]. There are contradictions in the trends, however. For acetone seeded in both air and oxygen at 248 nm excitation, as temperature is increased, the relative fluorescence pressure trace decreases in the data [95] (where the normalized fluorescence vs. pressure data at a specified temperature is referred to as ‘fluorescence pressure trace’). Meanwhile, after converting the data from [96] into relative fluorescence, for acetone seeded in N2 and air, as temperature is increased at increasing pressure, relative fluorescence decreases experimentally, contrary to the model which predicts an increase. The 248 nm data of Loffler et al. [43] agree qualitatively with [96] at 248 nm excitation, and the 308 nm data support the trend that increasing pressure and temperature at higher excitation wavelengths, generally increase fluorescence [77]. The abovementioned elevated pressure and temperature studies allude to competition between the non-radiative and collisional rates, and it is not clear from these reports which will dominate [78].
2.2 Current design range
In order to quantify the independent and coupled effect of elevated temperature and pressure on acetone photophysics, to clear up discrepancies in previous poorly understood compositional dependencies, and to determine the range and applicability for using acetone LIF as a combustion diagnostic, the following range of parameters was studied in this work: temperatures of 295‒750 K, pressures of 0.05‒4 MPa, tracer partial pressures of 1 Pa–23.1 kPa, and oxygen molar percentages of 0–21% in the bath gas using N2 and air. 282 nm was chosen as the excitation wavelength for this study because modeling in [78] indicated that with this excitation wavelength, elevating temperature would have minimal effect on the fluorescence signal sensitivity with pressure. It was also desired to provide the calibration data required to support the previous work of [40], where it was assumed that elevating pressure had little to no effect on the LIF temperature signals.
3 Experimental setup
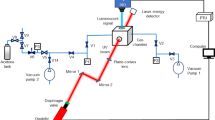
Figure 1 shows the experimental setup for studying the temperature, pressure, and composition dependences on acetone absorption and relative fluorescence. For validation purposes and increased flexibility, there are both static and flowing gas delivery systems embedded in the setup. Both systems utilize the same internal flow network with the primary difference being the method of tracer seeding. The static system was used to obtain room temperature, high pressure data, while the flow system was used to obtain all data sets.

Schematic of the current experimental setup
3.1 Static system
In the static system, the tracer is seeded in the gas phase by means of manometric filling by partial pressure. Therefore, in the static mode, both acetone number density η ace and acetone mole fraction χ ace could be fixed for a given experimental run. The back pressure control (BPC) valve was used as an isolation valve to maintain constant pressure in the test section.
3.2 Flow system
The flow system used the same high pressure components as the static configuration, with the addition of three key components: sonic nozzles, a tracer syringe pump, and a dual stage temperature controlled heating system. The mixing cell volume was used purely to enhance mixing, while the BPC valve was used as a throttling valve to control test section pressure. Bath gas flow was controlled via the upstream pressure regulator and sonic nozzle and they retained linearity up to the highest pressure and flow rates.
In most previous studies, acetone seeding configurations relied heavily on a bubbling method to seed the tracer to the desired mole fraction where the primary assumption was that the partial pressure of acetone was the vapor pressure at the bath temperature. This assumption may not hold true at high flow rates needed in this work, therefore a different seeding configuration was required. Variable acetone seeding rates were generated through the use of a positive displacement pump. Acetone was forced through a hypodermic needle orifice which was mounted parallel with seeder gas flow. Seeding was achieved through high speed flow shearing effects, similar to those used in atomizers. To ensure perfect seeding, a minimum criterion of 50:1 bath gas to tracer flow ratio by mass was established inside the atomizer. Figure 2 shows the range of flow rates utilized in the current work. As in the static cell operation, both acetone mole fraction and acetone number density could be fixed for a given experiment by varying the carrier gas flow rate.

Acetone pump flow rate regime as a function of pump setting. Lines represent linear curve fits to the calibration points. An error bar is shown but is barely distinguishable
A dual stage heating system was used to achieve elevated temperature, shown in red in Fig. 1. Stage 1 was a single 1700 W cartridge preheater that was internally mounted in series with the flow. The heaters were controlled through a feedback loop that used the central test section thermocouple as the control point. An additional second stage variable voltage external heating source was supplied to the test section in the form of heating tape to minimize temperature gradients across the absorption length and to minimize local cold zones surrounding the leg that contained the pressure gauge and thermocouple.
The residence time of tracer in the heated section is:
where ρ mix is the mixture density taken from the ideal gas law, V is the total heated volume plus test section volume, and \(\dot{m}_{\text{total}}\) is the total gas flow rate. Pyrolysis modeling was used in conjunction with two flow constraints, namely 50:1 bath gas to acetone flow rate ratio and 2:1 pressure ratio across sonic nozzle, to determine required flow rates as a function of temperature and pressure to avoid the detrimental effect of tracer pyrolysis in the test section.
To run a flow cell test, a preheat flow using air was first established to slowly raise the heating length and test section to the desired temperature. The system was allowed several hours to slowly raise and stabilize to the desired temperature. Next, a matching bath gas flow rate through the acetone seeded line was instantaneously switched on. The flow rate was set in accordance to the desired test pressure as determined through flow calculations. Once gas flow re-equilibrated, the acetone injection pump line was exposed and the carrier gas was seeded to the desired partial pressure. Once the flow system was fully established at the desired temperature and pressure, the laser was fired and fluorescence and absorption were recorded.
3.3 Laser, detection, and data acquisition systems
3.3.1 Laser
The optical system was comprised of the ultraviolet (UV) laser light source, intensified charge coupled device (ICCD) camera, phototubes, and all appropriate optical elements. The incidence beam was supplied by a Q-Switched frequency doubled ND:YAG at 532 nm, 10 Hz. The output beam was used to pump a dye laser which had a maximum output power of 800 mW. To produce the required 282 nm output, the laser beam was passed through the dye cell filled with Rhodamine 590 into a frequency doubling KD*P crystal inside the UVT. This combination produced an output beam diameter of 6–7 mm at an average power of 1–100 mW (0.1–10 mJ/pulse) at 282 nm. However, the average output power used in most experiments was in the range 1–10 mW (0.1–1 mJ/pulse).
Figure 1 shows the optical configuration used to manipulate and direct the output laser beam to the appropriate detectors. The beam was directed through a 70/30 (76% transmitted, 24% reflected) polka dot beam splitter, and a split off portion was sent ~90° from incidence to the first phototube (PT). The transmitted portion of the beam passed through a 1″ spherical focusing lens, which focused the beam 500 mm downstream to the center of the test section to a calculated beam waist of 0.40–0.44 mm. The beam then passed through the exit of the test section and was terminated on a second PT. Linearity of transmission versus laser intensity were determined for each optical element.
3.3.2 Incidence and absorption measurements
To record the incident and absorption signals, two “solar blind” PTs were mounted 90° and in line with the incident laser beam, respectively. At the cost of low output signal levels, these PTs were selected due to their sub-nanosecond response time and because of the large (12 mm diameter) frontal active areas. Rejection of unwanted visible light was provided by the sharp, UV only spectral response of the PT. To produce the required 5 mV input to the data acquisition (DAQ) system, a special three-stage pre-amplifying circuit was designed to supply a transimpedance gain of 10 mV/µA to each PT signal.
For each laser pulse, the PTs allowed for both the absorption and fluorescence measurements to be normalized by a measure of the incident laser pulse. An equal number of shots were recorded by the PT, amplified, and sent to SR250 Boxcar Integrator-Averagers and the DAQ for gating and recording. A subsequent number of shots were recorded with the PT boxes blocked from the laser light to subtract for background light. Here, background light was taken as the sum total of any residual background scattering, dark current from the PT, and any internally generated noise in the circuit. Timing of the gating was controlled by the ICCD camera. Typical average gate widths were in the range 80–110 ns. Periodically, the PT was recalibrated to ensure constant operation under the saturation limits.
3.3.3 Fluorescence measurements
Fluorescence was collected perpendicular to the laser path at the center of the test section using a 1024 by 256 pixel ICCD camera to allow easy separation of the UV and visible components. A UV lens was mounted in front of the ICCD camera. A WG320 high pass filter was used to eliminate Rayleigh scattering from the incident 282 nm laser beam. No additional low pass filter was required to block visible room light since all data points were taken in darkness.
For each pulse, a fluorescence image was recorded, gated, and output to the DAQ. Here, the gate width is defined as the time during which the light was detected by the intensifier, intensified, and then applied to the CCD array. Since high pressure had a broadening effect on the fluorescence lifetime, gate widths were chosen slightly wider at ~200 ns at lower pressures also to anticipate this effect at higher pressures; expanding the gate width introduced minimal noise into the system. Immediately before or after fluorescence was recorded, an equal number of shots were taken with test section empty or flowing with just bath gas to account for background scattering light.
3.4 Test section
The entire test section was constructed out of 0.165 cm (0.065″) thick, 1.27 cm (½″) outer diameter (OD) stainless steel (SS) fittings with a total laser absorption length of 0.228 m and internal volume of 145 cm3. It was preferred to have this longer test section length to increase the sensitivity of absorption measurements by allowing for a broader absorption signal range. The OD was chosen to minimize the highest required flow rate while maximizing the total available fluorescent signal.
The central cross, as shown in Fig. 1, allowed optical access for simultaneous fluorescence, pressure, and temperature measurement where the beam was focused at the center of the test section. Two more type-K thermocouples were mounted at gas inlet and exit to assess error in the temperature measurement due to minor temperature gradients across the absorption length during high temperature experiments. The entire test section was insulated with high-temperature fiberglass tape. Test section pressure was controlled downstream by throttling the BPC valve.
Optical access was provided through three 3 mm thick, 1.27 cm (0.5″) OD optical grade sapphire laser windows. This size and type was chosen to maximize the solid angle of fluorescence collection while deterring gas leakage. Sets of 1.58 mm (1/16″) thick composite graphite/metallic gaskets were shaped into rings and mounted on either side of the glass viewing port. In this way, as the SS and glass shifted with increasing temperature, the semi-metallic gasket could absorb the thermal expansion while maintaining a seal at higher pressure. However, the semi-metallic gaskets limited the window apertures to 0.478 cm on all three sides.
4 Data reduction
4.1 Absorption cross section (ACS)
Absorption of photons in the UV populates excited electronic energy levels in the respective absorbing molecules. The Beer–Lambert law shows that for an absorbing molecule, the corresponding laser signal I(x) will exponentially decay from its initial magnitude I 0 across the distance of the absorption length x as weighted by the strength of absorption, σ, and the total number of absorbers available, also known as the tracer number density η tracer:
which can be rearranged for the unknown:
where L is the total absorption path length (length of the test section), I L is the laser light intensity at the end of the test section, n o is the dimensionless optical transmission across the test section, and χ ace is the acetone mole fraction.
The primary correction step for ACS was the removal of the effect of loss of the transmitted signal through the windows and focusing lens. Voltages were registered with and without absorbing tracer present such that the optical efficiency cancels:
where the subscripts L and o represent the measurements at the end of the test section and for the incidence line, respectively. By making this correction every time, the effect of a specific design on the absorption data is effectively removed, which allows measurement of σ on an absolute basis. The left hand side represents the corrected intensity ratio where the input intensities are replaced by the proportional output voltages. The final form of the equation is thus:
4.2 Fluorescence quantum yield (FQY)
The images acquired by the ICCD camera are of arbitrary units. For the present experiments, only fluorescence ratios were determined. By normalizing, assuming constant filtering and ICCD gain, assuming identical collection optics, the terms due to solid angle collection, gain characteristics, collection optics, etc., cancel, and through division of an appropriate reference value (R) and subtraction of background light (B), the measured fluorescence ratio is:
Typical fluorescence images showed exponential decay depending on the amount of absorption. The final relative FQY ratios are obtained through division of ACS, laser intensity, and tracer number density:
4.3 Uncertainty analysis
In all measurements, the laser energy fluctuated by 2%. The uncertainty in test section temperature was 1 K, but there was an additional ~2% uncertainty due to the temperature gradients across the test section at the highest temperature tested. At 750 K and 3.5 MPa, at the lowest achievable residence time (highest flow rate), the largest encountered temperature gradient from gas inlet to exit was <20 K. At 750 K and 0.1 MPa, the temperature gradient across the absorption length was as high as ~15 K. The uncertainty in test section pressure was 20 kPa. There was an additional 1–2% uncertainty in the incidence and absorption measurements due to voltage sway in the Boxcar reading. Uncertainty in static cell operation was typically 1–2%, which is less than uncertainty in flow cell operation.
The single biggest improvement for reducing uncertainty were techniques used to improve the raw signal to noise (SNR) ratio in the signals. For fluorescence images, using 400 shots, intensifying images using a smaller region of interest, and post binning images improved raw SNR by a factor of ~1000. For incidence and absorption measurements, raw SNR was improved by a factor of 3500 through use of ultra-low noise components in the amplifying circuit and by averaging 400 shots. Typical resultant root mean square errors in ACS and FQY were 4% at low pressure and temperature, and 5–6% at elevated pressure and temperature.
5 Experimental results
Eight concerns were identified during design, including leakage, material incompatibility, tracer accumulation, temperature gradients across the absorption length, verifying excitation wavelength, verifying laser operation in the optically thin regime, saturation effects, and noise. Specific details of the strategies used to mitigate these errors are provided in [101] to give tracer LIF designers insight into how to validate the experimental design and systematically remove sources of error.
5.1 Fluorescence vs. laser energy
For all subsequent figures, the reference laser energy is taken to be the laser energy incident on the test section window. Linearity of fluorescence with the number of shots integrated and recorded was verified for a wide range of laser energies and acetone number densities, results of which are shown in Fig. 3. Results show good linearity in signals. Tests were repeated for a much higher number of shots up to 1000, acetone number densities, and laser energies. 1000 shots were taken as the maximum to eliminate degradation due to photolysis.

Linearity of fluorescence with number of recorded shots. Fluorescence is in arbitrary units and taken as the total integrated signal over the number of laser shots. For all curves, P ace = 2.5 kPa, T = 297 K
From Eq. 1, the measured fluorescence signal is theoretically proportional to laser energy. Experimentally, this relation only holds true for weak excitation, since at high excitation energies, an excited acetone molecule may be pumped into a “saturated” state where the linearity may break down. At constant number density and test section pressure and temperature, the fluorescence equation should only depend on changes in the incident laser intensity: \(\frac{{S_{\text{f}} }}{{S_{\text{f,R}} }} \propto \frac{{V_{\text{o}} }}{{V_{\text{o,R}} }}\).
To further isolate the energy dependence, experiments were conducted seeding only acetone in the pure form. The test section was filled with 2.5 kPa acetone, probed with laser light at 282 nm, and 500 shots were averaged for fluorescence measurements. A maximum energy of ~0.5 mJ/pulse was chosen in accordance with detection limits on the ICCD camera and phototubes. From Fig. 4, the linearity holds true over the entire range of desired pumping energy. Results are in good agreement with [10, 77]. For all subsequent experiments, laser energies were kept in this calibrated range, and usually did not exceed 0.2 mJ/pulse.

Linearity of fluorescence with laser energy per pulse. For the static cell configuration, fluorescence is normalized to 1 with respect to the fluorescence value at the highest pumping energy of 0.5 mJ/pulse; P ace = 2.5 kPa, T = 297 K. For the flow cell configuration, fluorescence is normalized to 1 with respect to the fluorescence value at 0.68 mJ/pulse; P ace = 2.5 kPa, T = 297 K
Although not shown here, linearity was validated for P ace = 2.5 kPa diluted in 0.1 and 3 MPa N2, and as anticipated, the fluorescence signal is directly proportional to the pumping laser energy per pulse. It is worth noting that when diluted with nitrogen, more pumping power is available, in that higher excitation energies are achievable before reaching the nonlinear saturation pumping energy, relative to the pure acetone excitation energies. This is consistent with the fact that N2 is a much less efficient collider than acetone; for a given acetone number density, it is possible to pump more acetone molecules into a higher vibrational level where an acetone–acetone collision will return the molecules to lower levels much more rapidly than an acetone-N2 collision. Linearity with number of shots registered was also validated at a number of flow conditions.
5.2 Composition dependencies
In many previous studies, fluorescence was assumed linear in tracer number density. However, this relation may not hold for higher acetone pressures due to the higher collision efficiency of acetone, see [78]. Higher acetone pressure is associated with faster vibrational relaxation rate; so a small variance in P ace could alter fluorescence significantly. Upon examination of Eq. 9, one can see that the measured fluorescence signal is theoretically proportional to tracer number density. Experimentally, this relation only holds true for low acetone number density, since at high number densities, one is no longer operating in the “optically thin regime” where deviation from linearity is expected due to the exponential term in Eq. 9.
At constant incidence laser energy and test section pressure and temperature, the fluorescence equation depends on changes in tracer number density: \(\frac{{S_{\text{f}} }}{{S_{\text{f,R}} }} \propto \frac{\eta }{{\eta_{\text{R}} }}\). To isolate the composition dependence, initial experiments were conducted seeding only acetone in the undiluted form at weak excitation. Then mixtures containing N2 and O2 were addressed separately.
To examine this dependence in static cell mode, the test section was slowly filled to a desired partial pressure of acetone and 500 shots were averaged for each fluorescence measurement. Fluorescence was divided by laser energy to account for small fluctuations in laser energy per pulse over the duration of the test. Due to differing amounts of absorbing species, fluorescence intensity was corrected for absorption in the first half of the cell. Therefore, Eq. 1 becomes:
For the “uncorrected” case, Eq. 1 becomes:
In Fig. 5a, relative fluorescence is taken with respect to the “corrected” laser intensity while in Fig. 5b, it is taken with respect to the uncorrected intensity. As shown in Fig. 5a, both data and model demonstrate a nonlinearity between fluorescence and acetone partial pressure at higher tracer number densities. For the pure acetone case, one can see the linearity begins to break down in the range 3–4 kPa.

Nonlinearity of fluorescence with acetone number density for a “corrected” laser intensity and b “uncorrected” laser intensity for pure acetone vapor. For all points, laser energy is ~0.11 mJ/pulse T = 297 K. Fluorescence is normalized to 1 with respect to the fluorescence value at a partial pressure sufficiently below the saturation pressure at 297 K consistent with Ambrose et al. [12]. The dotted line is a fit to the region of maximum linearity. Model line is from [77]
Figure 5b plots the results along with a corresponding linear fit for reference. From this plot, one can clearly see that fluorescence is proportional to number density only for small values. There is a clear low-η dependence before the exponential decay term dominates. To the authors’ knowledge, only one other study of [77] examined this effect, and the results presented here are in qualitative agreement with that. Therefore, to uphold this linearity, and to remain in the optically thin regime, P ace in the mixtures were kept below 4 kPa for all subsequent static cell fluorescence measurements.
To further understand composition dependencies, tests were repeated for the same acetone partial pressures seeded up to a total pressure of 101 kPa in N2 and results are plotted in Fig. 6. Two trends are evident. First, as expected, in comparison to Fig. 5, both data and model predict slightly higher fluorescence yield when acetone is diluted in 101 kPa of N2. Higher overall pressures act to increase vibrational relaxation of acetone molecules to lower levels in the excited singlet where the probability of fluorescing is high. Therefore, FQY is higher. Secondly, N2 has a stabilizing effect on the linearity of fluorescence with number density, since the region of linearity is wider in Fig. 6b vs. Fig. 5b. This is also expected since the less efficient collider N2 will transfer much less energy per collision, on average.

Nonlinearity of fluorescence with acetone number density for a “corrected” laser intensity and b “uncorrected” laser intensity for acetone vapor diluted to 1 atm of N2. For all points, laser energy is ~0.1 mJ/pulse and T = 297 K. Fluorescence is normalized to 1 at the highest acetone partial pressure. The dotted line is a fit to the region of maximum linearity. Model line is from [77]
Experiments were also conducted to verify the linearity of measured fluorescence signal with acetone number density for the flow cell. Linearity was investigated for total pressures of 0.1, 1, 2, 3, and 3.5 MPa. Unlike the static cell configuration, it was not possible to seed up to the vapor pressure in the flow system configuration due to design constraints. Therefore, the minimum partial pressure was determined by camera detection limits and the maximum partial pressures were chosen in accordance with the maximum allowable seeding rates as predicted by a flow calculation. Defining the signal dynamic range as the ratio of the highest seeding tracer mole fraction to the lowest detectable mole fraction, the signal dynamic range for the flow system setup was [72]:
This corresponded to a range 1.72 kPa < P ace < 7.6 kPa. While not plotted here, data at 101 kPa do exhibit the same inherent nonlinearity as in the static case, the observed deviation from linearity at the high pressure end is somewhat suppressed. This reinforced the trend that high pressure would have a stabilizing effect on emission in flow devices where small acetone concentration gradients may be expected. As the amount of N2 is increased, nonlinearity in acetone number density diminishes due to the lower collisional energy transfer rate of N2. In the limit of very high N2 pressures, FQY is stabilized as the system approaches the complete vibrational relaxation asymptote.
5.3 Room temperature, elevated pressure experiments
Before isolating pressure dependence on FQY, it was important to verify that acetone ACS did not vary with increasing pressure. Tests were conducted with pure acetone and acetone seeded in N2 up to 40 bar. In accordance with Eq. 7, the experimentally measured room temperature ACS at 282 nm excitation and 296 K was σ = 4.71 × 10−20 cm2. Using the flow cell configuration, room temperature absorption coefficients were also measured under a wide range of pressures. No deviation from the static cell measurement was observed. Table 3 compares the room temperature ACS measured here with measurements conducted in previous work and shows that values are in very good agreement. Since measurements were conducted over a wide range of acetone number densities and total gas pressures, it was verified that acetone ACS did not change with pressure due to the closely spaced vibrational energy levels associated with ketones.
After systematic examination of each individual dependence on the fluorescence signal, isolation of the pressure dependence on FQY was made possible. Since ACS is constant for all pressures, fluorescence signal per laser energy per acetone number density will yield FQY as a function of pressure:
Experiments were conducted using the static cell to study this effect; results are plotted in Fig. 7 along with the model predictions from Thurber [77] for acetone diluted in N2.

Room temperature (T = 297 K) pressure dependence on acetone fluorescence quantum yield in N2. Each point represents an average of 500 shots. Fluorescence is normalized to 1 at 1 atm for each system. χ ace = 0.003 for all points. Solid line represents the model predictions based on Thurber [77]
Clearly, data and the model agree quite well, despite the lingering over-prediction of the pressure dependence found in all other previous work. As pressure is increased for the same temperature, acetone molecules are more rapidly returned to lower lying vibrational levels in the excited singlet where fluorescence probabilities are higher. As expected at the relatively moderate excitation wavelength of 282 nm, there is unmistakable proof of the high pressure asymptote as FQY begins to level off around 2 MPa in N2.
Tests were repeated using the flow system configuration. Acetone partial pressure was fixed for a given desired flow rate and total pressure. After N2 flow was stabilized at the highest pressure for roughly 10 min, 500 fluorescence images were recorded. Then, the carrier gas flow rate and pressure were simultaneously decreased and the measurement was repeated. Figure 7 also plots the flow cell results and compares with data from the static case. There is excellent agreement between flow and static cell data, and fluorescence behavior is clearly independent of configuration. Room temperature results here are in qualitative agreement with those of [77, 90] which showed the origin of a similar level-off in their intermediate pressure range.
By including some percentage of oxygen in the bath gas, the effect of oxygen-enhanced intersystem crossing may also be addressed experimentally. Identical experiments using the static cell and flow cell were conducted in air and results are plotted in Fig. 8. Note that the collisional constant of proportionality from [77] was taken to be the same for N2, air, and oxygen for the model prediction curve. As shown, there is discrepancy between data and model. The model predicts FQY to reach a maximum value at 6 atm and quickly decrease with increasing oxygen number density, while the data exhibit behavior similar to that of N2, where fluorescence increases with increasing pressure and then levels off at a high pressure asymptotic value of ~1.4. Identical room temperature experiments using air as a bath gas were also conducted in the flow system to examine the high pressure behavior and check for consistency in fluorescence data. Again, there is excellent agreement between the static and flow cell configurations over the pressure range 0.1‒1.5 MPa. As in the case of N2 data, each data point was independently checked several times for repeatability and consistency for both test configurations.

Room temperature (T = 296 K) pressure dependence on acetone fluorescence quantum yield in air. For all points, χ ace = 0.005. Each point represents an average of 500 shots
The data validate the fact that oxygen does have a quenching effect on both singlet state fluorescence and triplet phosphorescence that continues into the high pressure regime as evidenced by the lower high pressure asymptotic value reached in air as compared with N2 data, but the model greatly over-predicts this trend. Lower pressure results at 282 nm excitation agree well with previous studies conducted at higher excitation wavelengths where the quenching effect is more pronounced [77]. Room temperature, high pressure results are also in qualitative agreement with [14, 95] where the fluorescence curves follow similar behavior.
The trend in the data suggests a re-examination of the oxygen quenching rate in the fluorescence model, since the value was originally fit to the low pressure range 1‒8 atm [77, 90]. There, it was not clear whether or not fluorescence increased past this point. Data here indicate that the enhanced intersystem crossing rate and collisional rate for bath gases containing oxygen are somehow coupled in a way that is not currently represented in the model of Thurber [77].
5.4 Room pressure, elevated temperature experiments
For all subsequent figures, data are collected solely through the flow system configuration. With prior knowledge of the total fluorescence signal per molecule, it was possible to separately examine the temperature dependence of the two individual components. First, experiments were conducted to study ACS. To maximize signal sensitivity across the test section absorbing length, acetone was introduced at the highest allowable seeding rates into the carrier gas flow. For simplicity, tests were conducted in total pressure of 101 kPa, constant number density, and the highest allowable gas residence times. Therefore, to validate the assumption that ACS was not a function of seeding rate or experimental apparatus, multiple tests were conducted using different acetone number densities. In order to accurately deduce FQY, ACS was measured at the same temperatures as the fluorescence per molecule data.
As shown in Fig. 9, ACS increases linearly with temperature at 282 nm excitation, reaching a value of 1.5 times the room temperature value at 750 K. Data agree well with [77]. Data from the current work also support the previously reported trend that absorption strength increases with temperature for all excitation wavelengths as the absorption spectrum is slowly shifted towards the red [11, 95].

Temperature dependence of the absorption cross section at 282 nm excitation. Data of [77] are also reported for comparison. The line represents a best fit to the data of the current work
Figure 10 plots resultant FQY as a function of temperature, along with the model predictions from [77]. It is shown that FQY decreases linearly with temperature at 282 nm excitation and implies that the non-radiative rate is indeed a function of increasing vibrational energy in the excited singlet. The results shown in Fig. 10 are consistent with the reported trends in the literature [77], wherein it was shown that the fluorescence increases with decreasing temperature at longer excitation wavelengths, begins to level off at intermediate excitation wavelengths, and then decreases with increasing temperature at shorter excitation wavelength.

Temperature dependence of fluorescence quantum yield at 282 nm excitation. Relative fluorescence is taken to be 1 at 0.125 MPa and 295 K. Data from [77] are also plotted for comparison. The line represents model predictions
5.5 Coupled elevated pressure and temperature experiments
High pressure experiments were conducted over a range of temperatures at 100 K intervals to carefully examine the competition between the collisional and non-radiative rates, identify the existence of an isotherm where fluorescence was a maximum, and hopefully address discrepancies in previously reported results. Experiments here were conducted in the same manner as in Sect. 5.3 where temperature was fixed for a given measurement and the full range of pressure was cycled through at constant acetone number density. As temperature was increased for a given experiment, appropriate residence times were selected to ensure the elimination of thermal destruction of the tracer. As before, linearity with laser energy and acetone number density were checked at various combined elevated pressures and temperatures. When operating within the calibrated room temperature values of number density and pumping energy, no deviation was observed.
Results are plotted in Fig. 11a–d for N2 as a bath gas and in Fig. 12a, b for air as a bath gas. In each graph, a curve fit to the original room temperature data is included for purposes of comparing with the evolving temperature data. For both carrier gases, the fluorescence pressure trace at 400 K lies above the 295 K trace, especially at higher pressures, indicating an increase in relative fluorescence at 400 K in comparison to 295 K. The increase in magnitude is outside experimental uncertainty of the 295 K data at this excitation wavelength, so one can confidently deduce that an increase in fluorescence here is a result of an overall higher vibrational de-excitation path with increased pressure.

Temperature dependence of fluorescence quantum yield at 282 nm excitation, a 400 K, b 500 K, c 600 K, and d 700 K in an N2 bath gas. For 400 and 500 K, P ace = 4.46 kPa; for 600 K and 700 K, P ace = 5.67 kPa. Fluorescence is normalized to 1 for each respective isotherm

Temperature dependence of fluorescence quantum yield at 282 nm excitation, a 400 K and b 500 K in an air bath gas. For all data points, P ace = 0.044 atm. Fluorescence is normalized to 1 for each respective isotherm
For N2 bath gas at 500 K, the experimental fluorescence pressure trace is slightly higher than the 295 K trace, but within the experimental uncertainty of 295 K. The fluorescence pressure trace at 700 K falls below the 295 K trace for all pressures. In going from 400 to 700 K, it is clear that the experimental fluorescence pressure trace shows a decreasing trend. Overall, the experimental fluorescence pressure trace first rises from 295 to 400 K and, thereafter, decreases. Experimental trends here thus suggest a saturation limit on the effect of the collisional vibrational de-excitation path that is dependent on the initial energy level of an excited molecule in the first excited singlet state.
Contrary to the experimental data, the fluorescence model of Thurber [77] predicts higher fluorescence pressure trace even at 700 K, and a discrepancy between the experimental data and model is most noticeable at this temperature. This discrepancy suggests that the model of Thurber [77] fails to capture the effect of coupled temperature and pressure variation with N2 as the bath gas.
With air as the bath gas, the experimental fluorescence pressure trace rises with increase in temperature. The model also shows an increasing trend with temperature. However, as mentioned previously, the model has a fundamental flaw of predicting very high oxygen quenching effect at high pressure which is contrary to the experimental data.
Overall, the model of [77] is in disagreement with fluorescence predictions for both carrier gases. For N2, the model predicts a weak, but steady increase in the fluorescence pressure trace with increasing temperature, while data show strong evidence for the existence of an isotherm at which the fluorescence pressure trace is maximum at 282 nm excitation for acetone. For air, the model predicts an initial quenching behavior where FQY briefly peaks in the intermediate pressure range, followed by a steady decrease in fluorescence with increasing pressure, whereas data indicate that fluorescence gradually increases with pressure and temperature. These observations necessitate a refinement in the fluorescence model, which will be presented in a sequel to this experimental paper.
5.6 Conclusions
In this work, high temperature and pressure static and flow systems were designed, rigorously validated, and tested to investigate acetone LIF in the range 295–750 K, 0.05–4 MPa with 282 nm excitation in both N2 and air over a wide range of tracer number densities and laser energies. This paper extends the range and applicability of acetone for use as a diagnostic in tracer LIF applications. Calibration data in terms of ACS and FQY have been provided to the end user wishing to use acetone LIF in elevated pressure and temperature studies.
Experimental results for both the static and flow cell systems are conclusive. At sufficiently high enough pressures, for all temperatures, acetone fluorescence reaches the fully vibrationally relaxed asymptote in the intermediate pressure range for N2. In the presence of oxygen, fluorescence is shown to also asymptotically relax, reaching a moderately lower relative FQY. At the intermediate excitation energy of 282 nm, acetone relative fluorescence pressure trace rises with increasing temperature up to a maximum isotherm of ~425 K; for all subsequently higher temperatures, acetone fluorescence pressure traces decrease. At elevated temperatures, acetone fluorescence is weakly quenched in the presence of oxygen. For adequately high oxygen collision rates which occur at high enough initial energies in the excited singlet level, at high enough temperatures, fluorescence levels off and decreases only minimally in an air bath gas. At this time it is unclear as to whether or not there exists a similar maximum fluorescence isotherm in the presence of oxygen.
Elevated temperature and pressure experimental data in this work and recent studies contradict the existing model assumption that fluorescence continually increases at higher pressures. Instead, there is evidence that collisional effects are in fact saturated at high enough temperatures such that the non-radiative rate causes sufficient crossover to the triplet, even at pressures in excess of 35 atm for acetone fluorescence.
Examination of Table 2 shows that future work should include elevated temperature and pressure tests with pure oxygen, since it is unclear if fluorescence decreases with increasing oxygen number density as originally predicted by the model for both 248 and 282 nm excitation. Therefore, it would be highly desirable to obtain fluorescence data using CO2, O2, or H2O as bath gases for these as well as other excitation wavelengths in high pressure conditions and temperatures up to ~500 K. Only then may the quenching behavior at elevated pressure be adequately assessed. Furthermore, tests could be conducted using other bath gases such as methane, helium, etc. to assess the temperature dependence of the collisional constants in the fluorescence model from [104,105,106] to determine if collisional efficiency is indeed a function of increasing molecular complexity throughout a broader range of temperatures. Lastly, using a similar experimental design, experiments could easily be done for other tracers, such as 3-pentanone, biacetyl, toluene, acetaldehyde, etc. Elevated temperature and pressure studies using toluene or benzene would be especially interesting due to the existence of these substances in commercially available transportation-relevant fuels and to study the role of oxygen quenching efficiency for extracting fuel/air ratio from practical systems. By providing the theoretical and experimental groundwork at elevated temperatures and pressures, this work may be easily extended to other excitation wavelengths, bath gases, and tracers.
References
R.K. Hanson, J.M. Seitzman, P.H. Paul, Appl. Phys. B 50, 441–454 (1990)
C.N. Markides, E. Mastorakos, Chem. Eng. Sci. 61, 2835–2842 (2006)
C. Schulz, J. Phys. Conf. Ser. 45, 27 (2006)
B.J. Kirby, R.K. Hanson, Appl. Phys. B 69, 505–507 (1999)
H. Neij, B. Johansson, M. Alden, Combust. Flame 99, 449–457 (1994)
M. Davy, P. Williams, D. Han, R. Steeper, Exp. Fluids 35, 92–99 (2003)
R. Zhang, V. Stanislav, V. Bohac, V. Sick, Exp. Fluids 40, 161–163 (2006)
B. Hiller, R.K. Hanson, Appl. Opt. 27, 33–48 (1998)
J. Sakakibara, K. Hishida, M. Maeda, International Journal of Heat Mass Transfer 40, 3163–3176 (1997)
A. Lozano, PhD dissertation, Stanford University, 1992
C. Schulz, V. Sick, Prog. Energy Combust. Sci. 31, 75–121 (2005)
D. Ambrose, C.H.S. Sprake, R. Townsend, J. Chem. Thermodyn. 6, 693–700 (1974)
J.D. Koch, R.K. Hanson, W. Koban, C. Schulz, Appl. Opt. 43, 5901–5910 (2004)
F. Ossler, M. Alden, Appl. Phys. B 64, 493–502 (1997)
J. Ernst, K. Spindler, H.G. Wagner, Berichte der Bunsen-Gesellschaft für Physikalische Chemie 80, 645–650 (1976)
W. Lawrenz, J. Kohler, F. Meier, W. Stolz, R. Wirth, W.H. Bloss, Technical Paper Series SAE-922320 (1992)
V.G. McDonell, G.S. Samuelsen, Meas. Sci. Technol. 11, 870–886 (2000)
F. Samouda, J.J. Brandner, C. Barrot, S. Colin, 1st European Conference on Gas Micro Flows. in Journal of Physics: Conference Series, vol. 362, p. 012026 (2000)
D. Wolff, H. Schluter, V. Beushausen, P. Andresen, Berichte der Bunsen-Gesellschaft für Physikalische Chemie 97, 1738–1741 (1993)
S.H. Smith, M.G. Mungal, J. Fluid Mech. 357, 83–122 (1998)
C.J. Bourdon, J.C. Dutton, Phys. Fluids 15, 499–510 (2003)
B.H. Failor, S. Chantrenne, P.L. Coleman, J.S. Levine, Y. Song, H.M. Sze, Rev. Sci. Instrum. 74, 1070–1076 (2002)
S.A. Filatyev, M.P. Thariyan, R.P. Lucht, J.P. Gore, Combust. Flame 150, 201–209 (2007)
J.Z. Reid, D. Wall, K. Lynch, B.S. Thurow, in AIAA-2009-4295, 39th AIAA Fluid Dynamics Conference, San Antonio, June 22–25 2009
K. Hatanaka, M. Hirota, T. Saito, Y. Nakamura, Y. Suzuki, T. Koyaguchi, in 28th International Symposium on Shock Waves, pp. 179–184 (2012)
A. Lakshmanarao, M.W. Renfro, G.B. King, N.M. Laurendeau, Exp. Fluids 30, 595–596 (2001)
H. Hu, M.M. Koochesfahani, Exp. Fluids 33, 202–209 (2002)
T. Rossman, M.G. Mungal, R.K. Hanson, Acetone PLIF and schlieren imaging of high compressibility mixing layers. in AIAA-2001-0290, 39th AIAA Aerospace Sciences Meeting, Reno, January 8–11 2001
L.M. Pickett, J.B. Ghandhi, Phys. Fluids 14, 985–998 (2002)
B.D. Collins, J.W. Jacobs, J. Fluid Mech. 464, 113–136 (2002)
N. Qi, B.H. Failor, J. Banister, J.S. Levine, H.M. Sze, D. Lojewski, IEEE Trans. Plasma Sci. 33, 752–762 (2005)
B.D. Ritchie, PhD thesis, Georgia Tech University, 2006
L. Ben, G. Charnay, R. Bazile, B. Ferret, Exp. Fluids 43, 77–88 (2007)
R.L. Gordon, C. Heeger, A. Dreizler, Appl. Phys. B 96, 745–748 (2009)
J.K. Yoon, K.J. Myong, J. Senda, H. Fujimoto, J. Mech. Sci. Technol. 23, 2565–2573 (2009)
B. Williams, P. Ewart, X. Wang, R. Stone, H. Ma, H. Walmsley, R. Cracknell, R. Stevens, D. Richardson, H. Fu, S. Wallace, Combust. Flame 157, 1866–1878 (2010)
T. Handa, M. Masuda, M. Kashitani, Y. Yamaguchi, Exp. Fluids 50, 1685–1694 (2011)
M.C. Thurber, F. Grisch, R.K. Hanson, Opt. Lett. 22, 251–253 (1997)
J. Clarkson, J.F. Griffiths, J.P. Macnamara, B.J. Whitaker, Combust. Flame 125, 1162–1175 (2001)
G. Mittal, C.-J. Sung, Combust. Flame 145, 160–180 (2006)
O. Degardin, B. Renou, A.M. Boukhalfa, Exp. Fluids 40, 452–463 (2006)
C. Pfeifer, S. Kress, D. Kuhn, Heat Mass Transf 46, 747–755 (2010)
M. Loffler, F. Beyrau, A. Leipertz, Appl. Opt. 49, 37–49 (2010)
B. Bork, T. Konig, F. Weckenmann, G. Lamanna, A. Dreizler, in 10th International Conference on Laser-Light and Interactions with Particles, Marseille, August 25–29 2014
R. Bazile, D. Stepowski, Exp. Fluids 20, 1–9 (1995)
C. Maqua, V. Depredurand, G. Castanet, M. Wolff, F. Lemoine, Exp. Fluids 43, 979–992 (2007)
K. Ammigan, H.L. Clack, 2007 Fall Meeting of the Western States Section of the Combustion Institute, Sandia National Laboratories, Livermore, Paper #07F-47 (2007)
B. Yip, M.F. Miller, A. Lozano, R.K. Hanson, Exp. Fluids 17, 330–336 (1994)
M.F. Miller, C.T. Bowman, M.G. Mungal, J. Fluid Mech. 356, 25–64 (1998)
S.H. Starner, J. Gounder, A.R. Masri, Combust. Flame 143, 420–432 (2005)
X. Mercier, M. Orain, F. Grisch, Appl. Phys. B 88, 151–160 (2007)
D. Galley, S. Ducruix, F. Lacas, D. Veynante, Combust. Flame 158, 155–171 (2011)
G.F. King, R.P. Lucht, J.C. Dutton, Opt. Lett. 22, 633–635 (1997)
T.R. Meyer, R.C. Dutton, R.P. Lucht, Appl. Phys. B 11, 3401 (1999)
T.R. Meyer, R.C. Dutton, R.P. Lucht, Phys. Fluids 11, 3401–3415 (1999)
T.R. Meyer, G.F. King, G.C. Martin, R.P. Lucht, F.R. Schauer, J.C. Dutton, Exp. Fluids 32, 603–611 (2002)
D.K. Manley, A. McIlroy, C.A. Taatjes, Phys. Today 61, 47–52 (2008)
F. Ossler, T. Metz, M. Alden, Appl. Phys. B 72, 479–489 (2001)
T. Kim, M.S. Beckman, P.V. Farrell, J.B. Ghandhi, SAE Technical Paper Series 2002-01-0219 (2002)
D. Frieden, V. Sick, J. Gronki, C. Schulz, Appl. Phys. B 75, 137–141 (2002)
W. Koban, J.D. Koch, R.K. Hanson, C. Schulz, Appl. Phys. B 80, 777–784 (2005)
A. Hoffman, F. Zimmermann, H. Scharr, S. Kromker, C. Schulz, Appl. Phys. B 80, 125–131 (2005)
W. Koban, J.D. Koch, R.K. Hanson, C. Schulz, Appl. Phys. B 80, 147–150 (2005)
M. Luong, R. Zhang, C. Schulz, V. Sick, Appl. Phys. B 91, 669–675 (2008)
B.H. Cheung, R.K. Hanson, Appl. Phys. B 98, 581–591 (2010)
M. Cundy, P. Trunk, A. Dreizler, V. Sick, Exp. Fluids 51, 1169–1176 (2011)
K. Mohri, M. Luong, G. Vanhove, T. Dreler, C. Schulz, Appl. Phys. B 103, 707–715 (2011)
A. Arnold, H. Becker, R. Suntz, P. Monkhouse, J. Wolfrum, R. Maly, W. Pfister, Opt. Lett. 15, 831–833 (1992)
B.A. Williams, J.W. Fleming, Appl. Phys. B 75, 883–890 (2002)
N.P. Tait, D.A. Greenhalgh, in Twenty-Fourth Symposium (International) on Combustion, Sydney (The Combustion Institute, 1992), pp. 1621–1628
S. Einecke, C. Schulz, V. Sick, Appl. Phys. B 71, 717–723 (2000)
I. van Cruyningen, A. Lozano, R.K. Hanson, Exp. Fluids 10, 41–49 (1990)
T.A. Baritaud, T.A. Heinze, Technical Paper Series, SAE-922355 (1992)
P. Guibert, W. Perrard, C. Morin, J. Fluids Eng. 124, 512–522 (2002)
J.D. Smith, V. Sick, Proc. Combust. Inst. 31, 747–755 (2007)
M. Cundy, T. Schuchtm, O. Thiele, V. Sick, Appl. Opt. 48, B94–B104 (2009)
M.C. Thurber, PhD thesis, Stanford University, 1999
J.W. Hartwig, M. Raju, C.J. Sung, Appl. Phys. B (2017, under review)
W. Koban, J.D. Koch, V. Sick, N. Wermuth, R.K. Hanson, C. Schulz, Proc. Combust. Inst. 30, 1545–1553 (2005)
R. Devillers, G. Bruneaux, C. Schulz, Appl. Phys. B 96, 735–739 (2009)
M. Kuhnl, C. Morin, P. Guibert, Appl. Phys. B 102, 659–671 (2011)
A. Roy, J.P.R. Gustavsson, C. Segal, Exp. Fluids 51, 1455–1463 (2011)
G.W. Luckey, W.A. Noyes Jr., J. Chem. Phys. 19, 227–231 (1951)
J. Heicklen, J. Am. Chem. Soc. 81, 3863–3866 (1958)
A.M. Halpern, W.R. Ware, J. Chem. Phys. 34, 1271–1276 (1971)
M.C. Thurber, F. Grisch, B.J. Kirby, M. Votsmeier, R.K. Hanson, Appl. Opt. 37, 4963–4978 (1998)
M.C. Thurber, R.K. Hanson, Appl. Phys. B 69, 229–240 (1999)
R.A. Bryant, J.M. Donbar, J.F. Driscoll, Exp. Fluids 28, 471–476 (2000)
J.B. Ghandhi, P.G. Felton, Exp. Fluids 21, 143–144 (1996)
L.S. Yuen, J.E. Peters, R.P. Lucht, Appl. Opt. 36, 3271–3277 (1997)
D.A. Hansen, E.K.C. Lee, Journal of Chemical Physics 62, 183–189 (1975)
T. Tran, Y. Kochar, J. Seitzman, in AIAA Paper 2005-0827 43rd Aerospace Sciences Meeting and Exhibit, Reno, January 10–13 2005
T. Tran, Y. Kochar, J. Seitzman, in AIAA-2006-831, 44th AIAA Aerospace Sciences Meeting, Reno, January 9–12 2006
T. Tran, PhD thesis, Georgia Institute of Technology, 2008
F. Grossman, P.B. Monkhouse, M. Ridder, V. Sick, J. Wolfrum, Appl. Phys. B 62, 249–253 (1996)
A. Braeuer, F. Beyrau, A. Leipertz, Appl. Opt. 45, 4982–4989 (2006)
J.D. Koch, R.K. Hanson, Appl. Phys. B 76, 319–324 (2003)
J.D. Koch, PhD thesis, Stanford University, 2005
V. Modica, C. Morin, P. Guibert, Appl. Phys. B 87, 193–204 (2007)
P. Guibert, V. Modica, C. Morin, Exp. Fluids 40, 245–256 (2006)
J.W. Hartwig, M.S. Thesis, Case Western Reserve University, 2009
A.J. Hynes, E.A. Kenyon, A.J. Pounds, P.H. Wine, Spectrochim. Acta A 48, 1235–1242 (1992)
T. Gierczak, J.B. Burkholder, S. Bauerle, A.R. Ravishankara, Chem. Phys. 231, 229–244 (1998)
J. Troe, J. Chem. Phys. 77, 3485–3492 (1982)
J. Troe, J. Phys. Chem. 87, 1800–1804 (1983)
T.C. Brown, J.A. Taylor, K.D. King, R.G. Gilbert, J. Phys. Chem. 87, 5214–5219 (1983)
Acknowledgements
The authors wish to acknowledge Ed Burwell and Dave Conger for their assistance in designing and calibrating the photodiodes and heating system, respectively.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hartwig, J., Mittal, G., Kumar, K. et al. Acetone photophysics at 282 nm excitation at elevated pressure and temperature. I: absorption and fluorescence experiments. Appl. Phys. B 123, 191 (2017). https://doi.org/10.1007/s00340-017-6774-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00340-017-6774-z