
Abstract
Purpose
Soil microbial communities play critical function during nutrient cycling. However, with the increasing nutrient input into terrestrial ecosystems from human activities, the responses of soil microorganisms to the aboveground vegetation across agricultural-to-natural succession stages are still poorly understand. The aim of this study was to evaluate the changes of soil microbial communities in three typical succession stages (the cropland, the grassland, and the brushland, respectively).
Materials and methods
A field experiment was carried out in an ecological restoration region. Soil samples were collected from three succession stages (the cropland, the grassland, and the brushland) based on their well-dated successional chronosequence in July 2016. Illumina MiSeq sequencing was used to identify the bacterial community structures. The responses of soil bacterial communities and its relationships with soil physicochemical properties and enzyme activities were assessed.
Results and discussion
The results showed that soil nutrients (soil organic carbon (SOC), total N, and NH4+) and enzyme activities (β-1,4-glucosidase and phosphatase) were significantly increased across the conversion from agricultural to natural ecosystem, and the enzyme activities were significantly affected by SOC and total N. It indicated that vegetation restoration greatly improved soil quality and nutrient cycling rates mediated by microbial metabolisms. Furthermore, there were no changes in soil bacterial community structures during the three vegetation succession stages, which implied the stability and adaption of microbial communities under the vegetation succession in semiarid climate. It should be noted that Firmicutes taxa were more sensitive than other taxa during natural vegetation recovery. Structural equation model (SEM) revealed that soil nutrients (soil organic matter (SOM) and total P), element stoichiometry (SOC:total P), and extracellular enzyme activities (urease and alkaline phosphatase) were dominant factors to shape the relative abundance of Firmicutes.
Conclusions
Firmicutes can be considered as bio-indicators to monitor soil quality and nutrient turnover during natural vegetation recovery. This study presents better understanding about the connections among soil nutrient cycling, enzyme activities, and soil bacterial communities during vegetation natural restoration, especially in typical ecological critical zone.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The Loess Plateau in China is one of the most eroded regions in the world (Wang and Bennett 2008). To remediate land degradation and restore degraded soil, a policy of converting agricultural land into grass or forest land has been implemented since 2001 (Li 2002). Natural recovery without further anthropogenic disturbance is an effective means to restore degraded soil (Walker et al. 2007). This process is characterized by an initial dominant species, subsequent coexistence with other species, substitution by another dominant species, and finally the climax community adapted to local conditions, which is mainly undergone four succession stages: cropland, grassland, brushland, and forest (Turner et al. 1998). The southern Loess Plateau belongs to the semiarid climatic region. Grassland and brushland are the dominant community types during agricultural-to-natural ecosystem conversion in this region (Wang et al. 2009). The availability of nitrogen and phosphorus is one of the most common limiting factors in the establishment of vegetation communities in a degraded ecosystem (Cui et al. 2018).
Soil microorganisms transform organic substrates to mineral elements and hence strongly influence the establishment of plants during secondary succession (Miethling et al. 2000). Recent studies have showed that soil microbial diversity plays an important role in maintaining multiple ecosystem functions simultaneously in terrestrial ecosystems (Leff et al. 2015; Delgado-Baquerizo et al. 2017). Soil provides microbes with nutrients and habitats and can in turn be affected by microbes (Yuan et al. 2016). Additionally, soil microorganisms are more sensitive than aboveground vegetation, which has been widely studied to evaluate the effects of land-use change in ecosystems (Fernandez et al. 2016). Soil properties and microbial communities can be significantly affected by crop rotation, coverage patterns (Zhao et al. 2015), fertilization (Carbonetto et al. 2014; Ge et al. 2017), and vegetation types (Zhang et al. 2016). To understand the connection between vegetation recovery and soil microbial communities will provide useful information about the recovery of revegetated ecosystems, considering the role of microorganisms in soil energy transfer, nutrient cycling, and vegetation re-establishment.
Previous studies indicated that soil microbial activities and community structures can significantly change during different land-use types (Sheng et al. 2013; Xun et al. 2016). Zhang et al. (2016) found that the dominant communities of bacteria were shifted from Acidobacteria to Proteobacteria communities during 30-year succession. Bacterial diversities decreased soon (< 5 years) after abandonment compared to the farmland, but they could recover to farmland levels after 15–20 years and were much improved after continued succession. Studies also showed that the changes in bacterial diversities occurred during long-term rather than short-term land-use change (Sun et al. 2011; Ling et al. 2014). The conflict results from those studies could be caused by the difference of recovery periods or climatic region. However, the effect of natural vegetation recovery on soil microbial communities is still less known, especially in the Loess Plateau—a typical ecological critical zone. In order to understand the nutrient cycling mediated by microbes and improve the availability of soil nutrients during ecosystem succession, it is very important to explore the response of soil microbial communities to natural vegetation recovery.
The enzymes produced by microbes and plants are closely related to soil energy flows and nutrient cycling and respond rapidly to soil changes, which these enzymes have been identified as direct participants involved in C, N, and P cycling (Burke et al. 2011). For instance, β-1,4-glucosidase (BG) plays an essential role in the C cycle by hydrolysing cellulose to glucose, whereas urease and alkaline phosphatase (AP) catalyze the terminal reactions in the production of urea and organic P, respectively. The enzyme activities involved in C, N, and P cycling can be served as indicators of soil nutrient availability and microbial activities (Cui et al. 2018). Therefore, the quantification of those key enzyme activities can assist to identify the response of microbial communities to ecosystem succession.
Recently, advances in sequencing technologies have provided new tools for microbial community analysis and have significantly changed our understanding of microbial diversity in the environment (Strickland and Rousk 2010). Many studies have used these technologies to thoroughly and accurately investigate the response of soil microbial community under different cropping systems (Gomez-Montano et al. 2013; Xun et al. 2016). Therefore, we quantified soil bacterial communities via high-throughput sequencing of bacterial 16S rRNA gene obtained from three typical succession stages (the cropland, the grassland, and the brushland, respectively) in the southern region of Loess Plateau. Moreover, soil physicochemical properties and enzyme activities were also investigated. Our objectives were (1) to illuminate the response of bacterial community structure to the typical succession stages during natural vegetation recovery and (2) to evaluate the relationships between soil nutrients, enzyme activities, and soil bacterial community structures during natural vegetation recovery.
2 Materials and methods
2.1 Study site and soil sampling
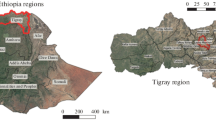
A field experiment was carried out in ecological restoration region. These study sites were located in the Weihe River valley region of the southern Loess Plateau, China (108° 04′ E, 34° 18′ N) (Fig. 1). The mean annual temperature is 12.9 °C, with a mean minimum temperature in January of − 1.2 °C and a mean maximum temperature in July of 26.1 °C. The area is characterized by a warm temperate semiarid and sub-humid climate. The mean annual precipitation is 735.1 mm, over 75% of which occurs from June to September, while the mean annual evaporation is 993.2 mm. The soil is classified as a Huang soil (a Calcaric Cambisol in the FAO classification). The sampling area was located in a farming-forestry ecotone, which has long-term cultivation history. Due to the policy of “Grain for Gree,” the grassland (abandoned arable land growing with grass) and the brushland had approximately 5- and 15-year restoring history, respectively, in this farming-forestry ecotone. According to the investigation about land-use history, it showed that the previous land-use type of the study sites was for agriculture cultivation. The sites were mainly planted with soybean, corn, and wheat under the similar fertilization management (manure and chemical fertilizer) before conversion. The substitution of space for time, a common method in ecosystem research, is an effective way to investigate the changes in soil conditions and plant communities during natural succession (Lawrencer et al. 2010; Zhang et al. 2016). We used this method to study the response of soil bacterial communities to natural recovery. Hence, three typical succession communities (the cropland, the grassland, and the brushland) were selected as the experiment sites based on their well-dated successional chronosequence.

Locations of the Loess Plateau in China (a), the study site in the southern Loess Plateau (b), and a map of the sampling sites (c). Red boxes represented the cropland, yellow boxes represented the grassland, and green boxes represented the brushland
All of the experiment sites had similar soil texture, altitude, and slope gradient and nearly the same geographical coordinates. We selected three disjunct, but closely located, maize plots as the crop land system because the amount and types of fertilizer applied are different under different crop land planting types. The cropland was fertilized with approximately 65 kg N ha−1, 8.5 kg P ha−1 synthetic fertilizers during the growth season. The properties of the sampling plots are shown in Table 1.
Three 20 × 20-m plots were established in each land type (experiment site) in July 2016. As shown in Fig. 1c. the red, yellow, and green plots represented the cropland, the grassland, and the brushland, respectively. Three 5 × 5-m quadrats were randomly selected in each plot. Ten soil cores from the topsoil (0–20 cm) were collected in each quadrat and then mixed to one composite sample. Each composite sample was passed immediately through a 2-mm sieve and then divided into three parts for future analysis. The first part was immediately stored at − 80 °C for soil genomic DNA extraction. The second part was stored at 4 °C for measurement of enzyme activities within 2 weeks. The third part was air-dried for analyzing physicochemical properties. A total of nine soil samples from each land type were separately measured to determine soil physicochemical properties and enzyme activities. Additionally, three soil samples from each land type were performed separately for DNA extraction and high-throughput sequencing.
2.2 Analysis of soil properties
Soil moisture was determined gravimetrically in fresh soils at 105 °C overnight. Soil bulk density was determined through a ring sampler weighing. Soil pH was determined with a glass electrode in a suspension with a 1:2.5 soil/water ratio (w/v). Soil organic carbon (SOC) and total N (TN) were analyzed using the standard procedure of dichromate oxidation (Kalembasa and Jenkinson 1973) and Kjeldahl digestion (Bremner 1960), respectively. Mineral N was extracted with 2 M KCl, and then NO3−-N and NH4+-N were determined using a Seal Auto Analyzer. Total P (TP) and available P (AP) were extracted with H2SO4-HClO4 (Parkinson and Allen 1975) and sodium bicarbonate (Olsen et al. 1954), respectively, and then determined by the molybdenum blue method using an ultraviolet spectrophotometer (Hitachi UV2300) at 700 nm.
2.3 Analysis of soil enzyme activities
Three soil enzyme activities (β-1,4-glucosidase, urease, and alkaline phosphatase) were assayed using a modification of method described by Tabatabai and Bremner (1969). β-1,4-glucosidase activity was measured based on the paranitrophenol concentration after the hydrolysis reaction. Briefly, 5 g of fresh soil was incubated at 37 °C for 1 h with 20 ml of pH 6.0 buffer solution and 5 ml of 25 mM p-nitrophenol glucopyranoside. At the end of incubation, 5 ml of 0.5 M CaCl2 solution and 20 ml of Tris buffer solution (pH 12.0) were added, and then soil suspension was shaken thoroughly and filtered. The concentration of paranitrophenol in filtrate was determined at 400 nm with a spectrophotometer. To measure urease activity, 5 g of fresh soil was incubated with 10 ml of urea solution (100 g L−1) and 20 ml of citrate buffer. The samples were incubated at 37.8 °C for 3 h and then diluted to 50 ml with distilled water. The suspension was filtered, and then 1 ml aliquot was treated with 4 ml of sodium phenol solution (100 ml of 6.6 M phenol solution and 100 ml of 6.8 M NaOH) and 3 ml of 0.9% sodium hypochlorite solution. The released ammonium was directly quantified using a spectrophotometer at 578 nm. Alkaline phosphatase activity was measured based on the amount of phenol released. Five grams of fresh soil was incubated with 10 ml of disodium phenyl phosphate solution (6.75 g L−1) and 10 ml of NH4Cl-NH4OH buffer solution at 37 °C for 3 h. The suspension was then filtered with 1 ml of potassium ferricyanide and 4-amino antipyrine as the color developing agent, and the concentration of phenol in filtrate was determined at 578 nm. The units of enzyme activities of β-1,4-glucosidase, urease, and alkaline phosphatase were μg paranitrophenol p-nitrophenol g−1 dry soil h−1, μg urea g−1 dry soil h−1, and μg phenol g−1 dry soil h−1, respectively.
2.4 DNA extraction, high-throughput sequencing, and data analysis
DNA was extracted from 0.5 g of freeze-dried soil using a FastDNA Spin Kit for Soil (MP Biomedicals, Cleveland, USA) according to the manufacturer’s instruction. The DNA extracts were assessed for quality and quantity using an automatic microplate reader (BioTek ELX 800 USA). The integrity of the DNA extracts was confirmed via 1% agarose gel electrophoresis. The primers 338F (5´-CTCCTACGGGAGGCAGCAG-3′) and 806R (5´-GGACTACHVGGGTWTCTAAT-3′) (Altschul et al. 1990) were designed to amplify the V3–V4 hypervariable regions of the bacterial 16S rRNA gene. The conditrions of PCR amplifications were 3 min at 95 °C, 35 cycles of 94 °C for 45 s, 55 °C for 45 s and 72 °C for 30 s, followed by 72 °C for 8 min. Successful PCR amplification was verified via 2% agarose gel electrophoresis. The triplicate PCR products were pooled and purified through gel extraction and then quantified using the AxyPrepDNA gel extraction kit (AXYGEN Corporation, USA) and the QuantiFluor™-ST blue fluorescence quantitative system (Promega Corporation, USA). The purified PCR products were then mixed at equimolar ratios for sequencing. Sequencing was conducted in an Illumina PE300 system (Illumina Corporation, USA) by Majorbio Bio-pharm Technology Co., Ltd. Approximately 35,000 high-quality sequences per sample with an average length of 437 to 439 bp were generated.
The primer sequences were trimmed after the raw sequences were de-noised, sorted, and distinguished using the Trimmomatic software platform (Bolger et al. 2014). The remaining sequences were filtered for redundancy, and the chimeras were removed prior to the OTU clustering. All unique sequences from each sample were then clustered into operational taxonomic units (OTUs) at a similarity of 97% using Usearch software (vsesion 7.1; Edgar 2010). In detail, OTU clustering was carried out for non-repeat sequences (excluding single sequences) according to 97% similarity, and representative sequences were obtained for OTUs. All optimization sequences were mapped to OTU representative sequences and then selecting the sequences that are more than 97% similar to OTU representative sequences to generate OTU table. The taxonomic identity of representative sequences for each OTU was determined according to the bacterial 16S rRNA Silva reference database (http://www.arb-silva.de) using the RDP-naïve Bayesian classifier (Cole et al. 2009).
For the high-throughput sequencing data, the indices of community richness (Chao1 and ACE estimators), community diversity (Shannon and Simpson indices), and an abundance-based coverage estimator were calculated. The rarefaction curves were obtained using Mothur (http://www.mothur.org/) (Schloss et al. 2009). The percentage of taxonomy was designated the relative abundance. Taxonomic alpha diversity was calculated as the estimated richness utilizing the OTU richness, and phylogenetic diversity was calculated as Faith’s phylogenetic diversity (Faith 1992).
2.5 Statistical analysis
Statistical analysis of the data was conducted using SPSS20.0 for Windows (SPSS Inc., Chicago, USA). A one-way analysis of variance (ANOVA) and a least significant difference (LSD) multiple comparisons (P < 0.05) test were used to assess the significance of differences among different sampling sites (soil properties, enzyme activities, and microbial composition and diversity). All bar graphs were drawn using OriginPro 9.0.
A Venn diagram was constructed based on 97% similarity of each sampled OTUs to visually display the overlapping sections and differences among different environmental samples. A weighted UniFrac-based hierarchical cluster dendrogram was used to visually display the evolutionary relationships among different environmental samples. Principle coordinate analysis (PCoA) was used to evaluate the overall differences in the structures of the microbial communities based on the UniFrac distance among different land types. Adonis analysis between different ecosystem types was performed to test significant differences between bacterial communities. Redundancy analysis (RDA) was used after the bacterial community data underwent Hellinger transformation and environmental factor data was standardized, in an effort to determine the effect of environmental factors on bacterial taxa using the Vegan package in R. Indicator species analysis was conducted using the multipatt function of the indicspecies package (version 1.7.4) in R. The structural equation modeling (SEM) framework was applied to investigate direct and indirect effects of environmental variables on Firmicutes. The x2 values, P values, RMSEA, and AIC were adopted to evaluate the overall goodness of structural equation model fit. Finally, we calculated the standardized total effects of SOM, total P, SOC:TP, urease, and alkaline phosphatase on Firmicutes abundance. The SEM was carried out by using the Amos 21.0 software package (SmallWaters Corporation, Chicago, IL, USA).
3 Results
3.1 Soil physicochemical properties
Soil physicochemical properties significantly changed during the three succession stages (P < 0.05) (Table 2). The cropland had the greatest values of bulk density, moisture content, and NO3−-N, TP, and AP. The lowest contents of SOC, TN, NO3−-N, and AP were observed in the grassland, whereas the soil pH in the grassland was higher than those in the other two succession stages. The brushland had the greatest contents of SOC, total N, and NH4+-N.
3.2 Soil enzymatic activities
The measured enzyme activities exhibited significant differences among the three succession stages (P < 0.05) (Fig. 2). The lowest activities of the three enzymes were observed in glassland (33.32 μg p-nitrophenol g−1 h−1, 54.56 μg urea g−1 h−1, and 24.23 μg phenol g−1 h−1) among the three succession stages. The brushland had significantly greater β-1,4-glucosidase (five to eight times) and alkaline phosphatase (two to five times) activities than the cropland and the grassland. However, the cropland had significantly greater urease activity (one to three times) than the grassland (54.6 μg urea g−1 h−1) and the brushland (109.4 μg urea g−1 h−1) (Table 3).

Three types of soil enzyme activities (μg g−1 h−1) (a, b, and c indicate β-1,4-glucosidase, urease, and alkaline phosphatase, respectively) in the different succession lands. Values are the means ± standard error (n = 3). Different letters indicate significant differences (P < 0.05) among the different succession lands based on one-way ANOVA followed by LSD test
3.3 Microbial communities
The ACE and Chao1 estimators indicated that the richness of bacterial community was significantly lower in the brushland than that in the cropland and the grassland. There was no significant difference in the ACE and Chao1 estimators between the cropland and the grassland (P < 0.05) (Fig. S1, Electronic Supplementary Material), and the brushland’s curve was markedly lower compared with the other two succession stages. However, the bacterial species diversity had no significant differences among the three succession stages indicated by Shannon and Simpson indices (P < 0.05), which was supported by Shannon-Wiener curve analysis (Fig. S2, Electronic Supplementary Material). All of the rarefaction curves tended to approach saturation at a similarity level of 97%, indicating that the volume of sequence data was sufficient, and the addition of a large number of reads made a small contribution to the total number of OTUs. The saturation at a similarity level of 97% indicated that the bacterial communities were adequately sampled.
A total of 325,041 high-quality sequences remained from the complete dataset after quality trimming and removal of chimeras (an average of 36,116 per sample), among which a total of 1879 OTUs were identified. The taxonomic composition of bacterial communities at the phylum level was Actinobacteria (31.5% on average), Proteobacteria (22.3%), Chloroflexi (17.1%), Acidobacteria (15.0%), Gemmatimonadetes (4.5%), Firmicutes (2.4%), Bacteroidetes (2.3%), and Cyanobacteria (1.5%) (Fig. 3).

Relative abundance of soil bacterial communities at the phylum level. The relative abundances were average from three replicate ratios between the abundance of the sequence type and the total number of sequences. Data were analyzed by one-way analysis of variance and means were compared by LSD test. Different letters indicate significant differences (P < 0.05) among the different succession lands
The relative abundance of all the phyla exhibited no significant differences among the three succession stages except for Firmicutes (Fig. 3). The relative abundance of Firmicutes in the brushland (0.64%) was significantly (P < 0.05) lower than that in the other two succession stages (3.67 and 2.90% in the cropland and the grassland, respectively). According to the further analysis of the composition of Firmicutes at the family level, the results showed the significant differences in Firmicutes microbial taxa among the different succession stages (Fig. 4). There were eight families (36 types of OTUs) found in Firmicutes, including Alicyclobacillaceae, Bacillaceae, Clostridiaceae_1, Erysipelotrichaceae, Paenibacillaceae, Peptostreptococcaceae, Peptococcaceae, and Planococcaceae. In the cropland, the Bacillaceae (0.14%), Planococcaceae (0.21%), Alicyclobacillaceae (0.22%), and Paenibacillaceae (0.13%) were the major families of Firmicutes. The major families of Firmicutes in the grassland were Bacillaceae (0.17%), Paenibacillaceae (0.16%), Peptostreptococcaceae (0.30%), Erysipelotrichaceae (0.21%), and Clostridiaceae_1 (0.26%). However, the total relative abundance of all eight families of Firmicutes at the brushland was less than 0.6%. Also, indicator species analysis showed that Actinobacteria and Firmicutes taxa (from phylum to family levels) were the main indicators of the grassland and the brushland stages, respectively (Table 4).

Relative abundance of Firmicutes at the family level. The relative abundances were averaged from three replicate ratios between the abundance of the sequence type and the total number of sequences. Data were analyzed by one-way analysis of variance and means were compared by LSD test. Different letters indicate significant differences (P < 0.05) among the different succession lands
The Venn diagram generated from OTUs showed that the three succession lands shared a majority of the OTUs (Fig. 5). Hierarchical cluster analysis and PCoA based on weighted UniFrac distances (Figs. 6 and 7) revealed that bacterial communities kept consistent composition among the three succession stages. It was also comfirmed by Adonis analysis (Table S1, Electronic Supplementary Material). However, compared with the cropland and the grassland, there were slightly more OTUs unique to the brushland, which mainly came from Firmicutes. Furthermore, SEM revealed that nutrient stoichiometry (SOC:TP) and enzyme activities (urease and alkaline phosphatase) exerted direct effect on Firmicutes (Fig. 9a). SOM play an indirect but the most important role in affecting on Firmicutes abundance (Fig. 9b).

Venn diagram of the numbers of shared and unique OTUs following the different succession lands after normalizing the number of reads. OTUs were defined at 97% sequence similarity

Hierarchical cluster dendrogram of soil bacterial communities based on the weighted UniFrac distance following the different succession lands

Principal coordinate analysis (PCoA) of soil bacterial communities following the different succession lands
4 Discussion
4.1 Effect of vegetation recovery on soil physicochemical properties and enzyme activities
Soil physicochemical properties were significantly affected by vegetation recovery. Soil nutrient contents after the succession from the cropland to the grassland decreased substantially because of the cessation of fertilizer inputs and the sustained depletion of nutrients by plants. As the development of primary producers and the accumulation of vegetation biomass, however, the level of soil nutrients gradually increased in the brushland from the decomposition of plant residues (Zhang et al. 2016). Particularly, soil TN was significantly higher in the brushland than those in the other two land types, which could be due to the nitrogen fixation of leguminous plants because the major species (Robinia pseudoacacia) in the brushland were leguminous plants (Boddey et al. 1997).
Enzyme activities play an important role in soil nutrient cycling and therefore can be useful indicators to assess the level of soil fertility and microbial activity (Zhang et al. 2011). As shown in Fig. 2, the activities of β-1,4-glucosidase and alkaline phosphatase are significantly higher in the brushland than in the cropland and the grassland. It indicated that the microorganisms’ activities represented by enzyme activities were significantly affected by vegetation natural succession, which could mainly attribute to the improvement of soil quality during succession. Previous studies showed that soil enzyme activities were closely related to the decomposition of organic compounds because the transformation of important organic elements is facilitated by microorganisms (Jiang et al. 2009; Duan et al. 2018). Our results are consistent with these studies, based on the significant correlations among enzyme activities (β-1,4-glucosidase and alkaline phosphatase) and most of the nutrients (P < 0.01 or P < 0.05) (Table S2, Electronic Supplementary Material). This finding suggested that the synthesis of enzyme would be stimulated to decompose the large fraction of organic compounds due to the low nutrient level in soils. Consquently, the proliferation of soil microbial activities increases with improvements in soil physical structure and chemical properties (Bandick and Dick 1999), which in turn enchance to synthesize enzymes by microbes. However, the highest urease in the cropland was likely caused by the anthropogenic application of urea and stimulated microbes to secrete urease (Wang et al. 1991). Hence, the vegetation recovery from the cropland to natural the brushland could enhance microbial activity and nutrient cycling, thereby improve soil quality, the transition stage during this recovery such as the grassland in this study might be low nutrients and enzyme activities though.
4.2 Effect of vegetation recovery on bacterial community structures
The bacterial richness was significantly affected by natural vegetation recovery according to the results of ACE and Chao1 estimators (Table 3). Generally, soil moisture was the key driver in regulating microbial biomass and community richness (Buckley and Schmidt 2001). In this study, the soil moisture in the brushland was the lowest (Table 2), which could lead to significant change of species richness from the grassland to the brushland. Moreover, the bacterial community structures kept relatively stable during the three succession stages (Figs. 6 and 7 and Table S1, Electronic Supplementary Material). Previous study suggested that the land-use history is a stronger determinant of the composition of microbial communities than vegetation and soil properties (Jangid et al. 2011). Kamlesh et al. (2010) found that the microbial community was not fully restored over 30 years due to the strong effect of cultivation on soil microbial community structure. However, in this satudy, about 15 years of succession might be still not long to change the composition of microbial communities.
Furthermore, Allison et al. (2007) found that the soil microbial community composition in the restoration lands was mediated by the changes of environmental factors such as AP, exchangeable irons, and soil water instead of soil carbon. The SOC:TN ratios recorded in our study were relatively constant across the three land types, despite great variation in soil physicochemical properties. Previous studies demonstrated that soil characteristics more strongly influence soil bacterial communities than land-use type, especially C:N ratio and phosphate (Barber et al. 2017). Therefore, the stable C:N ratios in our study would be further explained by the similar compositions of microbial community among the three succession lands. In addition, although previous studies showed that P is more critical to soil microbes than N (Fatemi et al. 2016), the TP contents in the three land types kept relatively constant, which could weaken the effect of soil nutrients on the structures of bacterial community. Meanwhile, most of relative abundance of the bacterial taxa was not significantly correlated with soil physicochemical properties (P < 0.05) (Table S3, Electronic Supplementary Material), which indicated that the bacterial community structures were little affected by soil physicochemical properties during 15 years of natural succession.
It should be noted that the relative abundance of Firmicutes taxa was significantly lower in the brushland than that in the other two land types (P < 0.05) (Fig. 3). The correlation analysis showed that Firmicutes was significantly correlated with most soil physicochemical parameters except for NO3−-N and AP (Table S3, Electronic Supplementary Material), which indicated that bacterial taxa from Firmicutes were strongly affected by environment factors. Many Firmicutes species were endospore formers (Tamez-Hidalgo et al. 2016), which made them hardy in potentially harsh conditions (Filippidou et al. 2015). For instance, Firmicutes could be induced under anaerobic condition (Huang et al. 2016). There were higher freguency of Firmicutes under the arid, pesticides, and heavy metal pollution conditions compared with other environments (Gomez-Montano et al. 2013). But they may also be defeated by other microbial groups in good condition (Lennon et al. 2012). Quadros et al. (2016) observed the decrease of the relative abundance of Firmicutes in a restoration area of coal mine. Also, anthropogenic addition of organic matters and N fertilizers could increase the relative abundance of Firmicutes (Ramirez et al. 2012; Tamez-Hidalgo et al. 2016). Additionally, Trivedi et al. (2016) summarized from a meta-analysis that the relative abundance of Firmicutes had significant differences between agricultural and natural soils in arid and semiarid regions, which suggested that the conversion from the cropland to the grassland would greatly affect the taxa of Firmicutes in the Loess Plateau—a typical arid and semiarid region. Moreover, Firmicutes taxa (from phylum to family levels) in the bacterial communities have been identified as potential indicators that respond to the natural vegetation succession. Therefore, as one of the major bacterial phyla, Firmicutes taxa could be used as indicators to monitor the effect of vegetation natural recovery on soil quality and nutrient turnover.
4.3 The response of bacterial communities to soil properties
In the present study, the soil bacterial communities kept consistent during the three vegetation succession. However, Zhang et al. (2016) reported that the dominant bacterial communities shifted from Acidobacteria to Proteobacteria during vegetation succession in the Loess Plateau. The different results may be due to the different climatic condition and the time spans of land-use conversion. For instance, the mean annual precipitation (510 mm) was lower in the region studied by Zhang et al. (2016) than that in our study region (735 mm), which may have contributed to differences in the reported soil microbial proliferation and activities (Zeglin et al. 2013). Moreover, the time span of vegetation recovery was 30 years in Zhang et al. (2016), which was longer than that in our study (15 years). Due to the hysteresis of the soil microbial response to changes in the soil environment (Peacock et al. 2001), the 15-year recovery time could not fully show the shift of soil bacterial communities.
The correlation analysis between soil nutrient contents and soil enzyme activities (Table S2, Electronic Supplementary Material) showed that β-1,4-glucosidase, urease, and phosphatase activities were significantly positively correlated with most of the examined soil nutrients (including SOC, TN, and NH4+-N contents and SOC:TP) (P < 0.05). Soil enzymes mediate and catalyze a number of soil biochemical and nutrient cycling (Dick et al. 1996). Normally, enzyme activities increase with the increasing of soil microbial populations (Adamczyk et al. 2013). In the present study, soil enzyme activities increased greatly during vegetation natural recovery, which indicated an increase in microbial community activity during the same period. Soil microbes maintain physiologic stability and acquire required nutrients by adjusting their own metabolism and secreting extracellular enzymes (Stark et al. 2014; Cui et al. 2018). The significant correlation between enzyme activities with soil nutrients implied that the stability of soil bacterial community during vegetation restoration could greatly attribute to the adjustment of microbial metabolism. For instance, microbes may release phosphatase to hydrolyse organic phosphorus in the soils with low P availability (Waring et al. 2014). Consequently, they can maintain the stability of community structure during land-use conversion and environment change.
The redundancy analysis and Pearson’s correlation analysis showed that the bacterial community was not affected by change of soil physicochemical properties during this 15-year period of vegetation restoration (Fig. 8 and Table S3, Electronic Supplementary Material). However, the phylum of Firmicutes was significantly correlated with most of the measured soil properties (P < 0.05) (Tables S3 and S4, Electronic Supplementary Material). The significant positive correlations were observed between Firmicutes with SOC and SOC:TP. Furthermore, SEM revealed that SOM as the most important factor indirectly shaped the bacterial groups of Firmicutes (Fig. 9). Previous studies showed that the Firmicutes played an important role in the decomposition of litter (Lee et al. 2011; Nuccio et al. 2013). Due to the dramatic decrease of Firmicutes from the cropland to the brushland, the residues or litters from the brushland may reduce the processing of decomposition and thus increase the accumulation of SOC and TN in the brushland. Firmicutes also had significant positive correlation with TP in our study, which may due to Firmicutes that belong to a part of phosphate-solubilizing bacteria (Kumar and Rai 2017). Moreover, SOC:TP, urease, and AP collectively exerted direct effect on Firmicutes (Fig. 9). Previous studies indicated that nutrient ratios are more important than single nutrient characteristics in regulating microbial metabolism and decomposition of organic matter due to relative constant ratios of microbial biomass C, N, and P on a global scale (Sinsabaugh et al. 2009; Kirkby et al. 2014). Inorganic P also played more important role than N in regulating soil enzyme activities and microbial nutrient acquisition (Fatemi et al. 2016). Wei et al. (2017) reported that phosphorus content can affect the abundance of nitrogen-cycle genes and thus could affect urease activity and nitrogen acquisition of Firmicutes taxa. In this study, our results suggested that the fate of Firmicutes phyla was affected by soil nutrients (TP and SOM), nutrient stoichiometry (C:P ratio), and extracellular enzyme activities (AP and urease).

Redundancy analysis (RDA) showing the relationship between soil bacterial communities and (at phylum level) and soil physiochemical parameters. SOC, soil organic carbon; TN, total nitrogen; MN, NO3−-N+NH4+-N; TP, total phosphorus; AP, available phosphorus; BD, bulk density; Moisture, moisture content

The structural equation model (SEM) examining the multivariate effects on Firmicutes abundance. RMSEA = 0.07, AIC = 53.1, the width of the arrows indicates the strength of the standardized path coefficient. The solid lines indicate positive path coefficients and dashed lines indicate negative path coefficients, R2 values represent the proportion of the variance explained for each endogenous variable. SOM, soil organic matter; Total P, total phosphorus; AP, alkaline phosphatase
5 Conclusions
This study demonstrated that the vegetation succession from agricultural to natural ecosystems could greatly improve soil microbial activities (enzyme activities related to C and P cycling). However, compared with the changes of cover vegetation and soil properties, soil bacterial community structures displayed high stability during the natural vegetation recovery under 15 years. Moreover, Firmicutes taxa were more sensitive to soil condition changes than other taxa during the natural vegetation recovery. Thus, they can be considered as bio-indicators to monitor the effects of vegetation natural recovery on soil quality and nutrient turnover. Furthermore, SEM revealed that the relative abundance of Firmicutes was collectively determined by SOC, TP, C:P ratio, AP, and urease activities. Those findings would help to understand the mechanisms of nutrient cycling with microbial activities and bacterial community structures during agricultural-to-natural ecosystem conversion.
References
Adamczyk B, Kilpeläinen P, Kitunen V, Smolander A (2013) Potential activities of enzymes involved in N, C, P and S cycling in boreal forest soil under different tree species. Pedobiologia 57(2):97–102
Allison VJ, Yermakov Z, Miller RM, Jastrow JD, Matamala R (2007) Using landscape and depth gradients to decouple the impact of correlated environmental variables on soil microbial community composition. Soil Biol Biochem 39(2):505–516
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410
Bandick AK, Dick RP (1999) Field management effects on soil enzyme activities. Soil Biol Biochem 31(11):1471–1479
Barber NA, Chantos-Davidson KM, Peralta RA, Sherwood JP, Swingley WD (2017) Soil microbial community composition in tallgrass prairie restorations coverge with remnants across a 27-year chronosequence. Environ Microbiol 19:3118–3131
Boddey RM, Jcdemoraes S, Bjr A, Urquiaga S (1997) The contribution of biological nitrogen fixation for sustainable agricultural systems in the tropics. Soil Biol Biochem 29(5–6):787–799
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30(15):2114–2120
Bremner JM (1960) Determination of nitrogen in soil by the Kjeldahl method. J Agric Sci 55(1):11–33
Buckley DH, Schmidt TM (2001) The structure of microbial communities in soil and the lasting impact of cultivation. Microb Ecol 42(1):11–12
Burke DJ, Weintraub MN, Hewins CR, Kalisz S (2011) Relationship between soil enzyme activities, nutrient cycling and soil fungal communities in a northern hardwood forest. Soil Biol Biochem 43(4):795–803
Carbonetto B, Rascovan N, Alvarez R, Mentaberry A, Vazquez MP (2014) Structure, composition and metagenomic profile of soil microbiomes associated to agricultural land use and tillage systems in Argentine Pampas. PLoS One 9:e99949
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM (2009) The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37(Database issue):D141–D145
Cui YX, Fang LC, Guo XB, Wang X, Zhang YJ, Li PF, Zhang XC (2018) Ecoenzymatic stoichiometry and microbial nutrient limitation in rhizosphere soil in the arid area of the northern Loess Plateau, China. Soil Biol Biochem 116(2018):11–21
Delgado-Baquerizo M, Eldridge DJ, Ochoa V, Gozalo B, Singh BK, Maestre FT (2017) Soil microbial communities drive the resistances of ecosystem multifunctionality to global change in drylands across the globe. Ecol Lett 20(10):1295–1305
Dick RP, Breakwell DP, Turco RF (1996) Soil enzyme activities and biodiversity measurements as integrative microbiological indicators. In: Doran JW, Jones AJ (eds) Methods for assessing soil quality. SSSA Spec Publ 49. SSSA, Madison, pp 247–271
Duan CJ, Fang LC, Yang CL, Chen WB, Cui YX, Li SQ (2018) Reveal the response of enzyme activities to heavy metals through in situ zymography. Ecotoxicol Environ Saf 156:106–115
Edgar RC (2010) Search and clustering orders of magnitude faster than blast. Bioinformatics 26(19):2460–2461
Faith DP (1992) Conservation evaluation and phylogenetic diversity. Biol Conserv 61(1):1–10
Fatemi FR, Fernandez IJ, Simon KS, Dail DB (2016) Nitrogen and phosphorus regulation of soil enzyme activities in acid forest soils. Soil Biol Biochem 98:171–179
Fernandez AL, Sheaffer CC, Wyse DL, Staley C, Gould TJ, Sadowsky MJ (2016) Associations between soil bacterial community structure and nutrient cycling functions in long-term organic farm soils following cover crop and organic fertilizer amendment. Sci Total Environ 566-567:949–959
Filippidou S, Thomas J, Tina W, Chien-Chi L, Po-E L, Patrick CS, Pilar J (2015) Under-detection of endospore-forming Firmicutes in metagenomic data. Comput Struct Biotechnol J 13:299–306
Ge TD, Li B, Zhu Z, Hu Y, Yuan H, Dorodnikov M, Jones DL, Wu J, Kuzyakov Y (2017) Rice rhizodeposition and its utilization by microbial groups depends on N fertilization. Biol Fertil Soils 53(1):37–48
Gomez-Montano L, Jumpponen A, Gonzales MA, Cusicanquid J, Valdiviae C, Motavallif PP, Hermanb M, Garrett KA (2013) Do bacterial and fungal communities in soils of the Bolivian Altiplano change under shorter fallow periods? Soil Biol Biochem 65(5):50–59
Huang X, Liu L, Wen T, Zhang J, Wang F, Cai Z (2016) Changes in the soil microbial community after reductive soil disinfestation and cucumber seedling cultivation. Appl Microbiol Biotechnol 100(12):1–13
Jangid K, Williams MA, Franzluebbers AJ, Schmidt TM, Coleman DC, Whitman WB (2011) Land-use history has a stronger impact on soil microbial community composition than aboveground vegetation and soil properties. Soil Biol Biochem 43(10):2184–2193
Jiang JP, Xiong YC, Jiang HM, Ye DY, Song YJ, Li FM (2009) Soil microbial activity during secondary vegetation succession in semiarid abandoned lands of Loess Plateau. Pedosphere 19(6):735–747
Kalembasa SJ, Jenkinson DS (1973) A comparative study of titrimetric and gravimetric methods for the determination of organic carbon in soil. J Sci Food Agric 24(9):1085–1090
Kamlesh J, Marka W, Alanj F, Blair JM, Coleman DC, Whitman WB (2010) Development of soil microbial communities during tallgrass prairie restoration. Soil Biol Biochem 42(2):302
Kirkby CA, Richardson AE, Wade LJ, Passioura JB, Batten GD, Blanchard C, Kirkegaard JA (2014) Nutrient availability limits carbon sequestration in arable soils. Soil Biol Biochem 68(1):402–409
Kumar A, Rai LC (2017) Soil organic carbon and availability of soil phosphorus regulate abundance of culturable phosphate solubilizing bacteria in paddy fields of the Indo-Gangetic plain. Pedoshpere 10:1002–1016
Lawrencer W, Davida W, Richardd B, Bruced C (2010) The use of chronosequences in studies of ecological succession and soil development. J Ecol 98(4):725–736
Lee CG, Watanabe T, Sato Y, Murase J, Asakawa S, Kimura M (2011) Bacterial populations assimilating carbon from 13C-labeled plant residue in soil: analysis by a DNA-SIP approach. Soil Biol Biochem 43(4):814–822
Leff JW, Jones SE, Prober SM, Barberán AB, Borer ET, Firn LF et al (2015) Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe. PNAS 112(35):10967–10972
Lennon JT, Aanderud ZT, Lehmkuhl BK, Jr SD (2012) Mapping the niche space of soil microorganisms using taxonomy and traits. Ecology 93(8):1867–1879
Li S (2002) The regulation of conversion of cultivated land back into forestry and structure of eco-environment. Forest Economy (China) 12:25–35
Ling N, Sun Y, Ma J, Guo J, Zhu P, Peng C (2014) Response of the bacterial diversity and soil enzyme activity in particle-size fractions of mollisol after different fertilization in a long-term experiment. Biol Fertil Soils 50(6):901–911
Miethling R, Wieland G, Backhaus H, Tebbe CC (2000) Variation of microbial rhizosphere communities in response to crop species, soil origin, and inoculation with Sinorhizobium meliloti L33. Microb Ecol 40(1):43–56
Nuccio EE, Hodge A, Pett-Ridge J, Herman DJ, Weber P, Firestone MK (2013) An arbuscular mycorrhizal fungus modifies the soil microbial community and nitrogen cycling during litter decomposition. Environ Microbiol 15(6):1870–1881
Olsen SR, Cole CV, Watanabe FS, Dean LA (1954) Estimation of available phosphorus in soils by extraction with sodium biocarbonate. US Dept. Agric. Circ. 939, Washington, DC
Parkinson JA, Allen SE (1975) A wet oxidation procedure suitable for the determination of nitrogen and mineral nutrients in biological material. Commun Soil Sci Plan 52(1):730–733
Peacock AD, Macnaughton SJ, Cantu JM, Dale VH, White DC (2001) Soil microbial biomass and community composition along an anthropogenic disturbance gradient within a long-leaf pine habitat. Ecol Indic 1(2):113–121
Quadros PDD, Zhalnina K, Davis-Richardson AG, Jennifer CD, Fátima BM, Flávio A, De OC, Eric WT (2016) Coal mining practices reduce the microbial biomass, richness and diversity of soil. Appl Soil Ecol 98:195–203
Ramirez KS, Craine JM, Fierer N (2012) Consistent effects of nitrogen amendments on soil microbial communities and processes across biomes. Glob Chang Biol 18(6):1918–1927
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75(23):7537–7541
Sheng R, Delong M, Minna W, Hongjie D, Hongling Q, Wenxue W (2013) Effect of agricultural land use change on community composition of bacteria and ammonia oxidizers. J Soils Sediments 13(7):1246–1256
Sinsabaugh RL, Hill BH, Follstad Shah JJ (2009) Ecoenzymatic stoichiometry of microbial organic nutrient acquisition in soil and sediment. Nature 462(7274):795–799
Stark S, Männistö MK, Eskelinen A (2014) Nutrient availability and pH jointly constrain microbial extracellular enzyme activities in nutrient-poor tundra soils. Plant Soil 383:373–385
Strickland MS, Rousk J (2010) Considering fungal:bacterial dominance in soils—methods, controls, and ecosystem implications. Soil Biol Biochem 42(9):1385–1395
Sun B, Dong ZX, Zhang XX, Li Y, Cao H, Cui ZL (2011) Rice to vegetables: short- versus long-term impact of land-use change on the indigenous soil microbial community. Microb Ecol 62(2):474–485
Tabatabai MA, Bremner JM (1969) Use of p-nitrophenyl phosphate for assay of soil phosphatase activity. Soil Biol Biochem 1(4):301–307
Tamez-Hidalgo P, Christensen BT, Lever MA, Elsgaard L, Lomstein BA (2016) Endospores, prokaryotes, and microbial indicators in arable soils from three long-term experiments. Biol Fertil Soils 52(1):101–112
Trivedi P, Delgado-Baquerizo M, Anderson IC, Singh BK (2016) Response of soil properties and microbial communities to agriculture: implications for primary productity and soil health indicators. Front Plant Sci 2016(e53649):7
Turner MG, Baker WL, Peterson CJ, Peet RK (1998) Factors influencing succession: lessons from large, infrequent natural disturbances. Ecosystems 1(6):511–523
Walker L, Walker J, Hobbs RJ (2007) Linking restoration and ecological succession. Springer, New York
Wang X, Bennett J (2008) Policy analysis of the conversion of crop land to forest and grassland program in China. Environ Econ Policy Stud 9(2):119–143
Wang Z, Cleemput OV, Demeyer P, Baert L (1991) Effect of urease inhibitors on urea hydrolysis and ammonia volatilization. Biol Fertil Soils 11(1):43–47
Wang GL, Liu GB, Xu MX (2009) Above- and belowground dynamics of plant community succession following abandonment of farmland on the Loess Plateau, China. Plant Soil 316(1–2):227–239
Waring BG, Weintraub SR, Sinsabaugh RL (2014) Ecoenzymatic stoichiometry of microbial nutrient acquisition in tropical soils. Biogeochemistry 117:101–113
Wei XM, Hu YJ, Peng PQ, Zhu ZK, Atere CT, O’Donnell AG, Wu JS, Ge TD (2017) Effect of P stoichiometry on the abundance of nitrogen-cycle genes in phosphorus-limited paddy soil. Biol Fertil Soils 53(7):767–776
Xun W, Xu ZH, Li W, Ren Y, Huang T, Ran W, Wang BR, Shen QR, Zhang RF (2016) Long-term organic-inorganic fertilization ensures great soil productivity and bacterial diversity after natural-to-agricultural ecosystem conversion. J Microbiol 54(9):611–617
Yuan H, Zhu Z, Liu S, Ge TD, Jing H, Li B, Liu Q, Lynn TM, Wu J, Kuzyakov Y (2016) Microbial utilization of rice root exudates: 13C labeling and plfa composition. Biol Fertil Soils 52(5):615–627
Zeglin LH, Bottomley PJ, Jumpponen A, Rice CW, Arango M, Lindsley A, McGowan A, Mfombep P, Myrold DD (2013) Altered precipitation regime affects the function and composition of soil microbial communities on multiple time scales. Ecology 94(10):2334–2345
Zhang C, Liu GB, Xue S, Wang GL (2016) Soil bacterial community dynamics reflect changes in plant community and soil properties during the secondary succession of abandoned farmland in the Loess Plateau. Soil Biol Biochem 97:40–49
Zhang C, Xue S, Liu GB, Song ZL (2011) A comparison of soil qualities of different revegetation types in the Loess Plateau, China. Plant Soil 347(1–2):163–178
Zhao D, Luo J, Wang J, Huang R, Guo K, Li Y, Wu QL (2015) The influence of land use on the abundance and diversity of ammonia oxidizers. Curr Microbiol 70(2):282–289
Funding
This work was financially supported by the National Natural Sciences Foundation of China (41571314 and 41201226), CAS “Light of West China” Program (XAB2016A03), and State Key Research & Development Plan Project (2017YFC0504504).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Yuan Ge
Electronic Supplementary Material
The supplementary information includes additional tables and figures showing the results of Adonis analysis, the correlations among soil physicochemical properties, soil enzyme activities, and bacterial communities and the similarity of the bacterial community from the different sampling areas.
ESM 1
(DOCX 225 kb)
Rights and permissions
About this article

Cite this article
Cui, Y., Fang, L., Guo, X. et al. Responses of soil bacterial communities, enzyme activities, and nutrients to agricultural-to-natural ecosystem conversion in the Loess Plateau, China. J Soils Sediments 19, 1427–1440 (2019). https://doi.org/10.1007/s11368-018-2110-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11368-018-2110-4